 Venkatram Yellapragada1,2
Venkatram Yellapragada1,2 Xiaonan Liu3,4
Xiaonan Liu3,4 Carina Lund1,2
Carina Lund1,2 Johanna Känsäkoski2
Johanna Känsäkoski2 Kristiina Pulli1,2Sanna Vuoristo5Karolina Lundin5Timo Tuuri5Markku Varjosalo3,4†
Kristiina Pulli1,2Sanna Vuoristo5Karolina Lundin5Timo Tuuri5Markku Varjosalo3,4† Taneli Raivio1,2,6*†
Taneli Raivio1,2,6*†- 1Stem Cells and Metabolism Research Program, Faculty of Medicine, University of Helsinki, Helsinki, Finland
- 2Department of Physiology, Faculty of Medicine, University of Helsinki, Helsinki, Finland
- 3Molecular Systems Biology Research Group, Institute of Biotechnology & HiLIFE, University of Helsinki, Helsinki, Finland
- 4Proteomics Unit, Institute of Biotechnology, University of Helsinki, Helsinki, Finland
- 5Department of Obstetrics and Gynecology, Helsinki University Hospital, HUH, Helsinki, Finland
- 6New Children's Hospital, Pediatric Research Center, Helsinki University Hospital, HUH, Helsinki, Finland
Paternally-inherited loss-of-function mutations in makorin ring finger protein 3 gene (MKRN3) underlie central precocious puberty. To investigate the puberty-related mechanism(s) of MKRN3 in humans, we generated two distinct bi-allelic MKRN3 knock-out human pluripotent stem cell lines, Del 1 and Del 2, and differentiated them into GNRH1-expressing neurons. Both Del 1 and Del 2 clones could be differentiated into neuronal progenitors and GNRH1-expressing neurons, however, the relative expression of GNRH1 did not differ from wild type cells (P = NS). Subsequently, we investigated stable and dynamic protein-protein interaction (PPI) partners of MKRN3 by stably expressing it in HEK cells followed by mass spectrometry analyses. We found 81 high-confidence novel protein interaction partners, which are implicated in cellular processes such as insulin signaling, RNA metabolism and cell-cell adhesion. Of the identified interactors, 20 have been previously implicated in puberty timing. In conclusion, our stem cell model for generation of GNRH1-expressing neurons did not offer mechanistic insight for the role of MKRN3 in puberty initiation. The PPI data, however, indicate that MKRN3 may regulate puberty by interacting with other puberty-related proteins. Further studies are required to elucidate the possible mechanisms and outcomes of these interactions.
Introduction
The onset of puberty is dependent on the reactivation of hypothalamic GnRH secretion, which leads to increased gonadotropin and sex steroid secretion, secondary sex characteristics and the attainment of adult height and fertility (1, 2). In patients with central precocious puberty (CPP), puberty starts before the age of 8 years in girls (Tanner breast stage 2) and 9 years in boys (Tanner genital stage G2) (1, 3, 4). Premature central activation of GnRH secretion may be pathological due to intracranial lesions, or idiopathic when no underlying cause can be identified (4). In girls, the annual incidence of CPP varied from 15 to 29 per 1,00,000 (5), and the overall risk and incidence is higher in girls than in boys (2, 3, 6).
The genetic etiology of idiopathic CPP is not completely understood, but the clinically most relevant gene underlying CPP is makorin ring finger protein 3 gene (MKRN3) (7–11). MKRN3 is a single-exon, maternally imprinted gene, which is expressed only from the paternal allele (12). Consequently, only paternally-inherited loss-of-function mutations in MKRN3 cause CPP (7, 11). MKRN3 belongs to the makorin family of ubiquitin ligases together with MKRN1 and MKRN2. MKRN3 is a close relative of MKRN1, being an intronless retrocopy of the latter (12, 13). Similar to other makorin family members, MKRN3 too consists of three zinc finger domains (C3H), one zinc RING finger domain (C3HC4) and one MKRN3-specific Cys-His domain (CH) and is expressed ubiquitously in human fetal and adult tissues (13, 14). Based on its structure, MKRN3 is predicted to function as a putative E3-ubiquitin ligase and it potentially affects gene expression, targeted protein degradation and protein function modulation via its E3 ligase activity (12–15). Interestingly, MKRN3 is expressed in the mouse and human hypothalamus (7, 8), but the mechanistic processes by which a paternally inherited loss-of-function MKRN3 mutation causes CPP are currently unclear.
Human pluripotent stem cells (hPSCs) have the indefinite capability of self-renewal and they can be differentiated into specialized cell types (16). To this end, we have recently described a protocol for the differentiation of GNRH1-expressing neurons from hPSCs (17), which offers a possibility to investigate the role of genetic factors in the regulation of GNRH1 in humans. Clustered Regularly Inter Spaced Palindromic Repeats (CRISPR) and CRISPR-associated protein 9 (CRISPR/Cas9) has become a prevailing technology in the area of gene editing (18). In brief, the CRISPR/Cas9 approach comprises of a short guide RNA (crRNA) fused to bacterial-specific trans-activating crRNA called tracrRNA which processes the crRNA, the fusion of crRNA and tracrRNA forms tracr:cr RNA complex which directs the Cas9 enzyme to a specific locus on DNA, generating double strand breaks (18). These double strand breaks are naturally repaired by an error-prone non-homologous end joining in the absence of a donor template (19). We utilized this technique to generate bi-allelic MKRN3 deletions in hPSCs and differentiated the knock-out (KO) cell lines into GNRH1-expressing neurons by employing our recently described protocol comprising dual SMAD inhibition, FGF8 treatment and Notch inhibition (17).
Most cellular proteins do not function in isolation, but perform their biological functions by interacting with each other (20). Protein-protein interactions (PPIs) form a network that operates in a coordinative manner to bring up physiological activities within a living cell (21). Mass spectrometry (MS) has evolved into an efficient technique in analyzing and characterizing PPIs (20, 22). To further investigate the putative mechanisms by which MKRN3 may affect the timing of puberty, we investigated MKRN3 PPI partners in HEK cells by employing our recently described MS-based approach, which allows the detection of stable and transient interaction partners for the protein of interest (20). This cell line was selected as a model, since HEK cells share certain characteristics of neurons (23), are widely available to researchers, and are frequently employed in biomedical research.
Materials and Methods
Human Pluripotent Stem Cells
Human pluripotent stem cell line H9 [46, XX, (Wicell)] was used in this study (24). hPSCs were maintained on Matrigel® (BD Biosciences) coated dishes with StemPro (Thermo Fisher Scientific) or Essential 8 medium (Thermo Fisher Scientific). During maintenance, the cells were split with 0.5 mM EDTA (Thermo Fisher Scientific, MA) and plated in 1:3–1:8 ratios. Medium was changed daily and before the differentiation, cells were plated on Geltrex-coated dishes (Thermo Fisher Scientific) and grown until >90% confluency.
Guide RNA Design and Production
CRISPR guides were designed using https://benchling.com/ to target the only exon of MKRN3. Based on their off-target and specificity scores, two pairs of the best possible guides were selected. Guide RNA DNA templates (gRNAs) were prepared by PCR (gRNA-PCR) amplification based on a method previously published (25). In brief, gRNA templates having 19 bp overhangs on 5′ and 3′ ends were fused to U6 promoter and terminator sequences (tracr) using PCR mentioned above with Phusion polymerase (Thermo Fisher Scientific). Further information on the gRNA-PCR is given in the Supplementary Material. All the CRISPR related oligos and guide sequences have been listed in Supplementary Table 1.
Generating CRISPR/Cas9 Based MKRN3 KO Cell Lines
For generating MKRN3 KO in hPSCs, two gRNAs targeting different locations of MKRN3 and a plasmid encoding wild type (WT) Streptococcus pyogenes Cas9 (SpCas9, referred as Cas9 hereafter), Green fluorescent protein (GFP) and puromycin resistance gene were electroporated to two million H9 cells with the Neon transfection system according to the manufacturer's instructions (Thermo Fisher Scientific). A total of 4 μg plasmid DNA and 250 ng of each gRNA was used per electroporation, and the electroporated cells were plated on Matrigel®-coated dishes and supplemented with 10 μM ROCK inhibitor (Y-27632 2HCl, Selleckchem) to enhance survival of hPSCs by inhibiting dissociation-induced apoptosis (26). Culture medium was changed every 24 h, with transient selection of surviving clones using 0.12 μg/ml puromycin (Sigma-Aldrich) starting after 48 or 72 h.
Colony-Picking or Fluorescence Activated Cell Sorting (FACS)
After 48 h of puromycin selection, emerging colonies were either manually picked or single cell sorted using a flow sorter (Sony Biotechnology Inc.). Manual picking of the colonies was carried out using a 10 μl pipette and Stereozoom®S4E light microscope (Leica microsystems). The individual colonies were identified under microscope and manually detached. Colonies were plated in a single well of a Matrigel® coated 96-well tissue culture plate (Sarstedt) containing E8 cell culture medium, supplemented with 10 μM ROCK inhibitor.
For cell sorting, Sony SH800 flow sorter was used to sort GFP positive (indicating successful entry of Cas9 plasmid) single cells, which were then plated on each well of the 96 well-plates containing E8 cell culture medium supplemented with 10 μM ROCK inhibitor. During both manual picking and cell sorting, medium was refreshed every 48 h (without ROCK inhibitor and puromycin) and the colonies were grown until they reached 70–80% confluency.
PCR-Based Screening of All the Surviving Clones
Genomic DNA (gDNA) from all the surviving colonies originating from single colonies or cells, were isolated using Direct cell PCR lysis buffer (Viagen Biotech) supplemented with 20 μg/ml of Proteinase K (Thermo Fisher Technologies). The gDNA served as a template to identify cell lines with bi-allelic or mono-allelic deletion using a specific primer pair-based PCR screening with AmpliTaq gold DNA polymerase (Thermo Fisher Scientific). Conditions for screening PCR's are provided in Supplementary Material, primers used are listed in Supplementary Table 2.
Targeted Sequencing of MKRN3 KO Cell Lines
gDNA from WT and the MKRN3 KO cell lines were PCR amplified with primers 200 bps upstream and downstream of MKRN3 and the product was purified using A'SAP PCR clean up kit (Arcticzymes) according to the manufacturer's instructions. The purified PCR products were subjected to Next generation sequencing using Nextera DNA library preparation kit (Illumina Technologies) performed at the Institute for Molecular Medicine Finland (FIMM, Helsinki). The targeted sequencing data was analyzed with the Integrative Genomics Viewer (IGV) tool from Broad institute (27). The primers used for targeted sequencing of MKRN3 are listed in Supplementary Table 2.
Differentiation of hPSCs to GNRH1-Expressing Neurons
At >90% confluency, both WT and MKRN3 KO hPSCs were differentiated into GNRH1-expressing neurons as described in Lund et al. (17). Briefly, the cells were treated with 2 μM Dorsomorphin (Selleckchem) and 10 μM SB431542 (Sigma-Aldrich) for 10 days to produce neuro-ectoderm, a further 10 days with 100 ng/ml FGF8 (Peprotech), and differentiation into post-mitotic neurons was stimulated by Notch inhibition using 20 μM DAPT (Sigma-Aldrich). The successful differentiation into GNRH1-expressing neurons was confirmed by morphological analysis, antibody staining to identify GnRH expressing cells and qPCR.
RNA Isolation and Reverse Transcription
RNA extraction (Nucleospin RNA, Machery-Nagel) and cDNA synthesis by reverse transcription (iScript™ cDNA kit, BIO-RAD) were performed according to the manufacturer's instructions except that DNase treatment during RNA extraction was performed separately with RQ1 DNase (Promega), 1U/μg RNA, in presence of RNase inhibitor (Promega). The reverse transcription was performed in a regular thermal cycler with 1 μg of total RNA using iScript™ mix containing a blend of oligo (dT) and random hexamer primers. Conditions for RT PCR are provided in the Supplementary Material.
Analysis of GNRH1 Expression
The expression levels of GNRH1 gene were measured by qPCR, normalized to Cyclophilin G (PPIG) and compared to expression in undifferentiated PSCs, as previously described (17). The normal range for GNRH1 mRNA expression in WT cells was determined based on nine differentiation experiments (n = 9). Day 25, was used to characterize GNRH1 expression in our protocol (17), and the same day was selected here for comparison of WT (n = 9) and MKRN3 KO cell lines (n = 6) in all the experiments. Conditions for qPCR are provided in the Supplementary Material, the primers used in qPCR are listed in Supplementary Table 2.
Immunocytochemistry
The cells were fixed using 4% paraformaldehyde for 15 min at room temperature (RT) and permeabilized for 7 min in 0.5% Triton X-100 (Sigma-Aldrich) diluted in PBS. Unspecific binding was blocked with UltraVision Protein Block (Thermo Fisher Scientific) for 10 min. Primary antibody incubation was performed overnight at 4°C and secondary antibody for 1 h at RT. Antibodies were diluted in PBS containing 0.1% Tween 20 (Sigma-Aldrich). Slides were mounted using VECTASHIELD® Mounting Medium with DAPI (Vector laboratories) for counterstaining the nuclei and microscopic images were obtained using ZEISS Axio Imager.Z2 upright epifluorescent microscope (Zeiss) using 10x/NA 0.3 and 20x/NA 0.8 EC PL APO CS2 objectives (Biomedicum Imaging Unit) and analyzed with ImageJ (NIH). All the antibodies and dilutions are listed in Supplementary Table 3.
Generation of Stable Expression Cell Line and Cell Culture for Protein-Protein Interaction Studies in HEKs
For the generation of stable and inducible cell lines, gateway cloning (28) was employed to create MKRN3 expression vector. MKRN3 PCR-product with flanking site specific attachment sites (attB) and donor vector with site specific attachment sites (attP) were recombined by BP reaction to get the gateway compatible entry clone. Recombination between attB and attP sites results in attL and attR sites which are recombined by LR reaction between the entry clones and the MAC-C destination vector (20) to generate the MAC-tagged MKRN3 expression vector. Flp-In™ 293 T-REx cell lines (Invitrogen, Life Technologies, R78007) were co-transfected with the expression vector and the pOG44 vector (Invitrogen) using the DreamFect™ reagent (Oz Biosciences). Two days after transfection, culture medium was changed to DMEM/F12 (Thermo Fisher Scientific) supplemented with 10% FBS, 50 mg/mL penicillin, 50 mg/mL streptomycin (Sigma-Aldrich) and hygromycin B (Thermo Fisher Scientific) (100 μg/mL). The hygromycin B selection for isogenic clones was performed for 2 weeks.
The stable cell line was expanded to 30 × 150 mm plates. When cells reached 80% confluency, 1 μg/ml tetracycline (Sigma-Aldrich) was added for 24 h to express MAC-tagged MKRN3. For biotinylation labeling (BioID), 50 μM additional biotin (Thermo Fisher Scientific) was added at the same time with tetracycline. Cells deriving from 5 × 150 mm fully confluent dishes (approximately 5 × 107 cells) were pelleted as one biological replicate. Three biological replicates for each condition and in total six biological replicates for two different conditions (with and without biotin) were collected. Samples were snap frozen at −80°C until further use.
Affinity Purification of Protein Complexes and Mass Spectrometry (MS)
The cells with only tetracycline induction were used for affinity purification (29). The cells treated with tetracycline and biotin were applied for BioID purification as previously described (20). Purified protein complexes (AP-MS)/proteins (BioID) were processed and digested to peptides for MS analysis. The analysis was performed on Qrbitrap Elite hybrid mass spectrometer using Xcalibur version 2.0.7 SP1 (Thermo Scientific) coupled with an EASY nLC 1000-reverse phase HPLC system via an electrospray ionization sprayer (Thermo Fisher Scientific). MS was performed in data-dependent acquisition mode using Fourier transform mass spectrometry full scan (300–1,700 m/z) resolution of 60,000 and collision-induced dissociation scan of top 20 most abundant ions.
Data Processing
For protein identification, Thermo.RAW files were searched against reviewed selected human UniProtKB/SwissProt database (http://www.uniprot.org/, version 2017-02) with SEQUEST search engine. The decoy database was the reverse of the target database. All data were reported based on 95% confidence for protein identification, as determined by the false discovery rate (FDR) ≤5%. Contaminant Repository for Affinity Purification (CRAPome, http://www.crapome.org/) (30) database and in-house GFP samples database were used as controls with a cut-off frequency of 20% (411 runs from CRAPome database and 100 runs of in house GFP control samples) for identification of high-confidence interactions (HCIs). HCIs data were imported into Cytoscape 3.4.0 to construct protein interaction network. The known prey-prey interaction data were obtained from IMEx database (http://www.imexconsortium.org/) (31). Gene ontology classification analysis was based on DAVID bioinformatics resource (https://david.ncifcrf.gov/) (32). We also compared the HCI partners of MKRN3 with proteins encoded by 371 genes, that are associated with puberty timing according to large population based studies (33, 34). In addition, we investigated the HCIs to proteins encoded by 37 genes implicated in congenital hypogonadotropic hypogonadism (CHH) (35–40).
Statistics
The relative GNRH1 expression in WT H9 and MKRN3 KO cell lines Del 1 and Del 2 was tested with unpaired t-test after logarithmic transformation to normalize the distributions. P-value < 0.05 was accepted to indicate statistical significance.
Results
CRISPR/Cas9 Based Generation of MKRN3 KO Cell Lines in hPSCs
To investigate the impact of MKRN3 deficiency in the differentiation of GNRH1-expressing neurons, we generated two hPSC lines with bi-allelic deletions of MKRN3 using CRISPR/Cas9. Bi-allelic KO cell lines were generated, as heterozygous MKRN3 mutations cause CPP only when inherited from the father, and we could not selectively target the desired deletion to the paternal allele. The schematic indicates the guide targeting areas and deletions in these cell lines (Figure 1A). In brief, two pairs of RNA guides spanning different lengths on the only exon of MKRN3 together with Cas9 resulted in deletions of either −174 or −901 bps. For further studies, we chose two apparently homozygous KO lines Del 1 (small deletion, −174 bps), and Del 2 (large deletion, −901 bp), which were validated to carry desired deletions by PCR (Figure 1B) and targeted sequencing Supplementary Figures 1, 2). The absence of functional MKRN3 expression was further validated by performing PCR with cDNAs from the WT and MKRN3 KO cell lines as template and with primers flanking the deletion regions (Figure 2A). The length of the transcripts produced from the KO clones corresponded to the size of deletions seen at DNA level (Figure 2B), consistent with the bi-allelic deletion in the coding region of MKRN3.
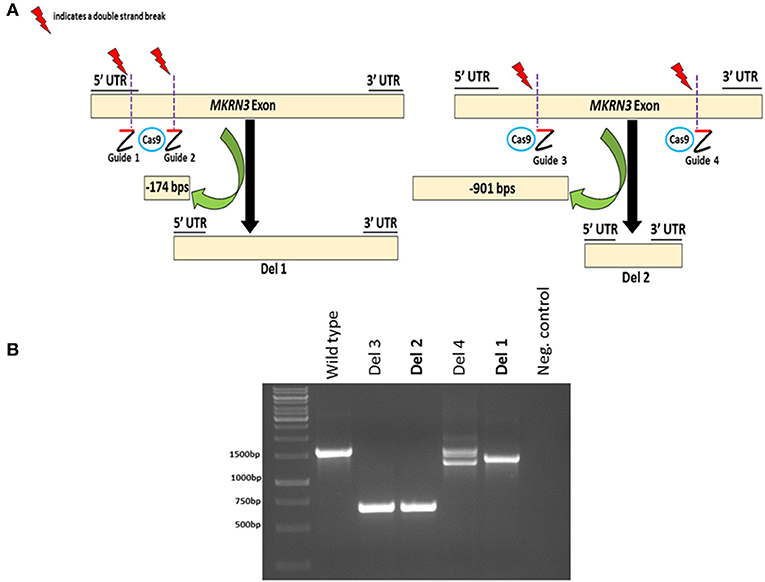
Figure 1. CRISPR/Cas9-based generation of two bi-allelic MKRN3 KO cell lines (Del 1 and Del 2) in human pluripotent stem cell line H9 and verification of CRISPR/Cas9 induced deletions at genomic DNA level. (A) Left panel, Del 1 has a deletion of 174 bps, translating to a deletion of 51 amino acids (11% of protein) including the translation start site. Right panel, Del 2 has a deletion of 901 bps, translating to 300 amino acid deletion (60% of protein). (B) The presence or absence of CRISPR/Cas9-induced deletions in MKRN3 was verified by PCR amplification of the region of interest from genomic DNA of WT human pluripotent H9 cells, and in CRISPR/Cas9 edited cells. Four examples of edited cell lines are shown (Del 1–Del 4). The same pair of guide RNAs, designed to delete ~900 bps, was used for editing Del 2 and Del 3 cell lines, whereas two different gRNA pairs were used to edit Del 1 and Del 4 cell lines. The PCR products were visualized on a 1.5% agarose gel.
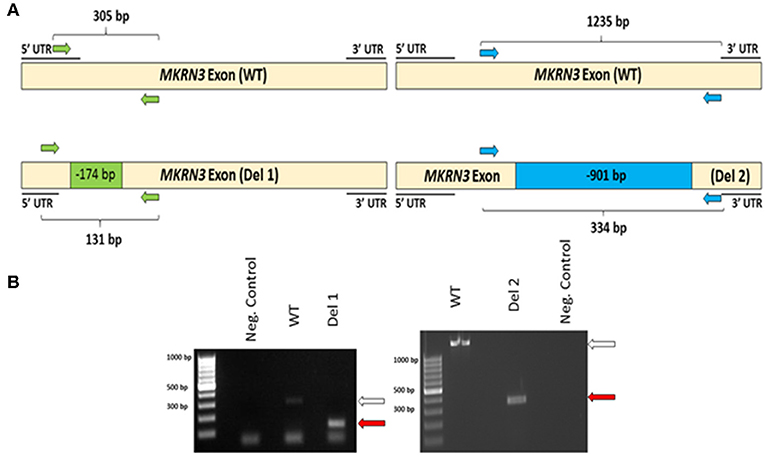
Figure 2. (A) Schematic of MKRN3 cDNA and the two pairs of primers (green and blue arrows) employed in detection of WT, Del 1, (left panel) and Del 2, (right panel). cell lines. In WT cells, the expected product sizes are 305 and 1,235 bp, in Del 1 cells 131 bp and in Del 2 cells 334 bp. (B) MKRN3 amplification from the cDNA of the two bi-allelic MKRN3 deletion cell lines Del 1, (left panel) and Del 2, (right panel). White arrow indicates the expected wild type (WT) product sizes (305 and 1,235 bp, respectively) and the red arrows indicate the expected sizes (131 and 334 bp, respectively) for the PCR products from Del 1 and Del 2 cell lines. The cDNA used is from day 25 of the protocol generating GNRH1-expressing neurons. The PCR products were visualized on a 1.5% agarose gel.
Differentiation of MKRN3 KO and WT hPSCs to GNRH1- Expressing Neurons
We next differentiated MKRN3 KO cell lines Del 1 and Del 2 into GNRH1-expressing neurons, according to our recently described protocol (Figure 3A) (17). Both cell lines could be differentiated into neuronal progenitors and further into GNRH1-expressing neurons (Figure 3B). At day 25 of differentiation the relative GNRH1 mRNA expression between the MKRN3 KO cell lines and WT H9 cells (Figure 3C), did not differ (P = NS). The mRNA expression levels of OTX2, a transcriptional regulator of GNRH1 (41), together with OTX1, both of which are enriched in GnRH-neurons of mice (42) were not significantly different between WT and Del 2 (n = 3) cells at day 25 of the differentiation protocol (Supplementary Figure 3).
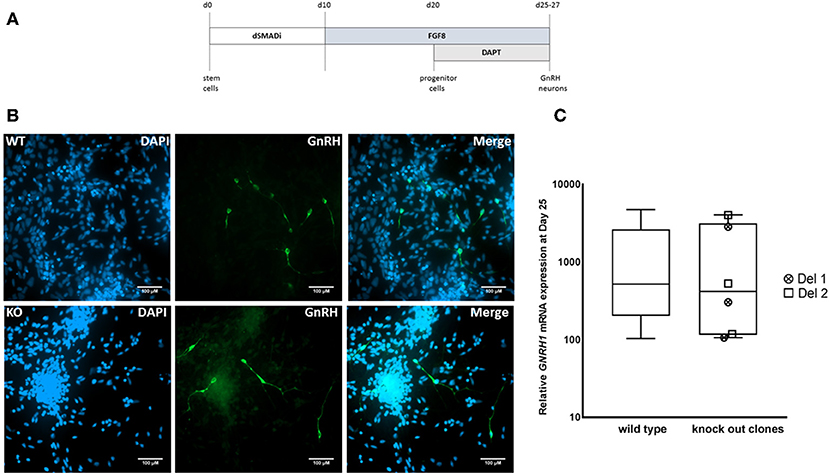
Figure 3. (A) Schematic for the generation of GNRH1-expressing neurons from hPSCs (17). In brief, dual SMAD inhibition by Dorsomorphin and SB431542 was applied for the first 10 days, followed by 10 days of FGF8 treatment and final stage of neuronal maturation for 5–7 days by combining FGF8 with Notch inhibition by DAPT. This differentiation methodology has been adapted from a previously published protocol and the figure has been modified from the original publication by Lund et al. (17). (B) Immunocytochemistry in WT and Del 2 derived day 25 GnRH-positive neurons (green) nuclear stained with DAPI (blue), scale bars represent 100 μM. (C) The relative expression of GNRH1 in WT H9 cells (n = 9) and in two bi-allelic MKRN3 KO cell lines (Del 1 and Del 2) following three independent repeats (n = 6) of differentiation into GNRH1-expressing neurons (17). Expression levels are relative to day 0 hPSCs.
MKRN3 and Its Protein Interaction Partners
Using MS analysis, we identified 104 HCIs including both transient and stable interactions for MKRN3 (Supplementary Table 4). These 104 HCIs total for 81 unique HCIs (Figure 4), were classified into functional groups according to their annotation categories involved in various pathways regulating cellular processes, mainly the regulation of insulin secretion, cell-cell adhesion, TP53-regulated cell metabolism, transcriptional repression and RNA activity related interactors (32). Importantly, our MS data revealed that MKRN3 physically interacts with as many as 19 proteins (including LIN28B) encoded by genes, which have been previously associated with the age at menarche (33, 34). In addition, MS data identified OTUD4, a protein implicated in CHH (43), as HCI partner for MKRN3.
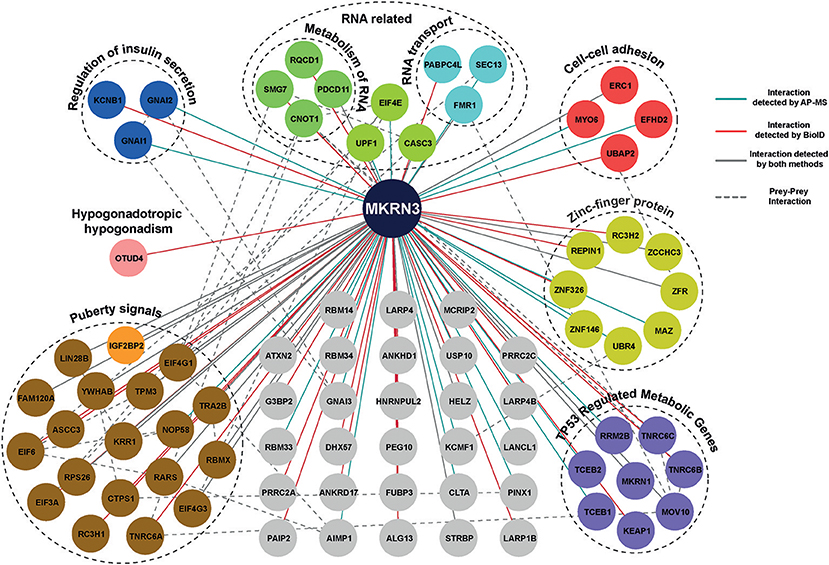
Figure 4. Protein-protein interaction map of MKRN3. The network represents PPIs of MKRN3 detected by different purification methods. Blue edges represent experimentally validated interaction by AP-MS; Red lines represent interactions detected by BioID purification; Overlap of two purification methods is shown with gray color, while a dashed line describes prey-prey interactions. The colored nodes represent the different clusters of prey proteins.
Discussion
In mice, expression of Mkrn3 decreases during postnatal development, and in humans, circulating MKRN3 levels decrease toward the onset of puberty in both sexes (44, 45). However, investigation of peripheral MKRN3 levels does not allow for mechanistic explanation of the role of MKRN3 in regulation of puberty in humans. Therefore, we employed our recently described differentiation protocol generating GNRH1-expressing neurons (17) and genetically engineered hPSCs to investigate if MKRN3 is required for GnRH neuron differentiation in this model. In addition, we investigated PPI partners of MKRN3 in HEK cells and paid special attention to possible interactors encoded by puberty-related genes.
Although several studies have reported that paternally inherited mutations in MKRN3 cause CPP (46), the underlying mechanisms still remain to be explained (1, 2). In our study, we first examined the role of MKRN3 on GNRH1 expression by using two bi-allelic MKRN3 KO clonal cell lines (Del 1 and Del 2), generated with CRISPR/Cas9 technology. These cell lines were subsequently differentiated into GNRH1-expressing neurons through our recently described differentiation protocol, which is based on dual SMAD inhibition, FGF8 treatment and Notch inhibition (17). Our results show that both the Del 1 and Del 2 cell lines could be successfully differentiated into GNRH1-expressing neurons, suggesting that MKRN3 is dispensable for the differentiation. Intriguingly, GNRH1 expression of Del 1 and Del 2 cell lines did not differ from the controls, which suggests that MKRN3 does not directly alter GNRH1 expression. However, there are many environmental and metabolic signals postnatally, that may regulate further maturation and function of GnRH neurons (47, 48). While MKRN3 does not affect the GnRH neuron differentiation or their ability to express GNRH1 in our model, there are other potential mechanisms by which MKRN3 may regulate GnRH neuron function. For example, major regulation of GnRH neuron activity is carried out by Kisspeptin and KNDy neurons in the hypothalamus, and putative role of MKRN3 on these neurons is not known. The possibility of other MKRNs compensating for the loss of MKRN3 in Del 1 and Del 2 cell lines cannot be ruled out. On the other hand, hPSC-based disease modeling is the closest available approach to model human neuronal disorders (49), and with regard to MKRN3, murine models have not yet been reported. What is known so far, however, is that MKRN3 is not significantly enriched or depleted in mouse GnRH neurons (42).
Given that we could not find a mechanistic cue for the role of MKRN3 in GNRH1 expression in our stem cell model, we considered alternative approaches to gain insight on how this putative E3-ligase might affect puberty timing. In brief the method investigating PPIs employs a MAC-tag system that contains affinity tags for affinity purification-mass spectrometry (AP-MS) as well as the biotin ligase BirA* for BioID. Therefore, a single construct is sufficient to identify both stable and transient interactions (20). We did not carry out PPI studies in our stem cell model, since the efficient cloning of all different tagged MKRN3 constructs to hPSCs was beyond the scope of this work. While this is a potential caveat, it is important to note that HEK cells, one of the most widely employed human cell lines in biomedical research (50), are not unsuitable for our purpose since they exhibit some neuronal properties (23). In addition, our laboratory is extremely experienced in using HEK cell line-based platform, which allows rapid and efficient generation of expression constructs and subsequent data analysis. Based on the list of HCI proteins for MKRN3, we found that MKRN3 might be involved in cell-cell adhesion, RNA metabolism and insulin secretion. Insulin has been suggested to modify GnRH neuron function in humans, and insulin sensitivity is thought to be an important factor in the initiation of puberty (1, 51, 52). MKRN3 interacts with a group of proteins encoded by metabolic genes, which in turn, are regulated by TP53, a tumor suppressor gene involved in several cancers and implicated in puberty (53). Other PPI partners of MKRN3 are involved in RNA transport and metabolism, which remains less explored areas in terms of puberty.
In addition, we found that MKRN3 interacts with 20 proteins encoded by puberty timing-associated genes of which LIN28B is one of the most established puberty-related genes in genetic studies (54, 55) and OTUD4 represents the only puberty-related disease gene (43). Although LIN28B has been associated with the age at menarche, adult height and childhood growth (56–60), and is known to be a negative regulator of let-7 class of microRNAs (61), the mechanism by which it regulates puberty timing is unknown (1, 62, 63). In rats and non-human primates Lin28b expression declines in the hypothalamus at puberty (64), and in mice, expression of both Lin28b and Mkrn3 is reduced prior to puberty (7, 65). Our findings provide evidence of physical interaction between human LIN28B and MKRN3 and it is tempting to speculate that they may act in concert to regulate the timing of puberty. We also examined MKRN3 interactions among 37 genes implicated in CHH (35–40), and found that MKRN3 interacts with OTUD4. OTUD4 is a deubiquitinase coding gene (43), and it is intriguing to hypothesize that it may regulate the timing of puberty by counteracting the effects of MKRN3. Further studies are required to confirm this hypothesis. Finally, MKRN3 interacts with other zinc finger genes (ZNFs) which are transcriptional repressors and have an important role during puberty (66), however, we could not observe MKRN3's interaction with ZNFs critical for puberty onset, such as ZNF573 and GATAD1 (66). Even so, the interactions observed invite the possibility of MKRN3 being an epigenetic transcriptional repressor involved in puberty timing.
In conclusion, our results suggest that MKRN3 is dispensable for the GnRH neuron differentiation process and GNRH1 expression during differentiation from human pluripotent stem cells. Although the mechanism by which MKRN3 regulates humans sexual maturation remains unclear, our results suggest that MKRN3 may act in concert with previously identified puberty-related proteins, and it is enticing to hypothesize that this interaction is translated to functional cooperation in terms of puberty timing. Further studies are required before we understand how and why the HPG axis activates prematurely in children with a paternally inherited loss-of-function mutations in MKRN3.
Author Contributions
VY, SV, TT, and TR planned the project. TT, MV, and TR supervised the project. VY, CL, KP, and KL carried out the stem cell work. XL and MV designed the protein interaction study. XL and JK carried out the protein interaction work. VY wrote the manuscript and TR edited the manuscript.
Funding
This work was supported by the Academy of Finland, Foundation for Pediatric Research, Sigrid-Juselius Foundation, Novo Nordisk Foundation, Jalmari Ja Rauha Ahokkaan Säätiö, University of Helsinki, and Helsinki University Central Hospital.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Jere Weltner, Diego Balboa, and Biomedicum Stem Cell Center for their support. We thank Biomedicum Imaging Unit for providing the microscopy facility. We thank Annika Tarkkanen for proofreading the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2019.00048/full#supplementary-material
Abbreviations
CPP, central precocious puberty; MKRN3, makorin ring finger protein 3; hPSCs, human pluripotent stem cells; WT, wild type; KO, knock-out; CRISPR, clustered regularly interspaced palindromic repeats; Cas9, CRISPR associated protein 9; bps, base pairs; gRNA, guide RNA DNA template; GFP, green fluorescent protein; PPI, protein-protein interaction; HCI, high-confident interactions; CHH, congenital hypogonadotropic hypogonadism.
References
1. Abreu AP, Kaiser UB. Pubertal development and regulation. Lancet Diabetes Endocrinol. (2016) 4:254–64. doi: 10.1016/S2213-8587(15)00418-0
2. Latronico AC, Brito VN, Carel JC. Causes, diagnosis, and treatment of central precocious puberty. Lancet Diabetes Endocrinol. (2016) 4:265–74. doi: 10.1016/S2213-8587(15)00380-0
3. Neely EK, Crossen SS. Precocious puberty. Curr Opin Obstetr Gynecol. (2014) 26:332–8. doi: 10.1097/GCO.0000000000000099
4. Carel J-C, Léger J. Precocious puberty. N Engl J Med. (2008) 358:2366–77. doi: 10.1056/NEJMcp0800459
5. Fuqua JS. Treatment and outcomes of precocious puberty: an update. J Clin Endocrinol Metabol. (2013) 98:2198–207. doi: 10.1210/jc.2013-1024
6. Soriano-Guillén L, Corripio R, Labarta JI, Cañete R, Castro-Feijóo L, Espino R, et al. Central precocious puberty in children living in spain: incidence, prevalence, and influence of adoption and immigration. J Clin Endocrinol Metabol. (2010) 95:4305–13. doi: 10.1210/jc.2010-1025
7. Abreu AP, Dauber A, Macedo DB, Noel SD, Brito VN, Gill JC, et al. Central precocious puberty caused by mutations in the imprinted gene MKRN3. N Engl J Med. (2013) 368:2467–75. doi: 10.1056/NEJMoa1302160
8. Känsäkoski J, Raivio T, Juul A, Tommiska J. A missense mutation in MKRN3 in a danish girl with central precocious puberty and her brother with early puberty. Pediatric Res. (2015) 78:709–11. doi: 10.1038/pr.2015.159
9. Simon D, Ba I, Mekhail N, Ecosse E, Paulsen A, Zenaty D, et al. Mutations in the maternally imprinted gene MKRN3 are common in familial central precocious puberty. Eur J Endocrinol. (2016) 174:1–8. doi: 10.1530/EJE-15-0488
10. Bessa DS, Macedo DB, Brito VN, França MM, Montenegro LR, Cunha-Silva M, et al. High frequency of MKRN3 mutations in male central precocious puberty previously classified as idiopathic. Neuroendocrinology (2017) 105:17–25. doi: 10.1159/000446963
11. Macedo DB, Abreu AP, Reis AC, Montenegro LR, Dauber A, Beneduzzi D, et al. Central precocious puberty that appears to be sporadic caused by paternally inherited mutations in the imprinted gene makorin ring finger 3. J Clin Endocrinol Metabol. (2014) 99: E1097–103. doi: 10.1210/jc.2013-3126
12. Abreu AP, Macedo DB, Brito VN, Kaiser UB, Latronico AC. A new pathway in the control of the initiation of puberty: the MKRN3 Gene. J Mol Endocrinol. (2015) 54:R131–9. doi: 10.1530/JME-14-0315
13. Böhne A, Darras A, D'Cotta H, Baroiller JF, Galiana-Arnoux D, Volff JN. The vertebrate makorin ubiquitin ligase gene family has been shaped by large-scale duplication and retroposition from an Ancestral Gonad-Specific, Maternal-Effect Gene. BMC Genomics (2010) 11:721. doi: 10.1186/1471-2164-11-721
14. Jong MT, Gray TA, Ji Y, Glenn CC, Saitoh S, Driscoll DJ, et al. A novel imprinted gene, encoding a RING zinc-finger protein, and overlapping antisense transcript in the prader-Willi syndrome critical region. Hum Mol Genet. (1999) 8:783–93. doi: 10.1093/hmg/8.5.783
15. Liu H, Kong X, Chen F. Mkrn3 functions as a novel ubiquitin E3 ligase to inhibit Nptx1 during puberty initiation. Oncotarget (2017) 8:85102–9. doi: 10.18632/oncotarget.19347
16. Tabar V, Studer, L. Pluripotent stem cells in regenerative medicine: challenges and recent progress. Nat Rev Genetics (2014) 15:82–92. doi: 10.1038/nrg3563
17. Lund C, Pulli K, Yellapragada V, Giacobini P, Lundin K, Vuoristo S, et al. Development of gonadotropin-releasing hormone-secreting neurons from human pluripotent stem cells. Stem Cell Rep. (2016) 7:149–57. doi: 10.1016/j.stemcr.2016.06.007
18. Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 System. Nat Protoc. (2013) 8:2281–308. doi: 10.1038/nprot.2013.143
19. Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Ann Rev Biochem. (2010) 79:181–211. doi: 10.1146/annurev.biochem.052308.093131
20. Liu X, Salokas K, Tamene F, Jiu Y, Weldatsadik RG, Öhman T, et al. An AP-MS- and BioID-compatible MAC-Tag enables comprehensive mapping of protein interactions and subcellular localizations. Nat Commun. (2018) 9:1188. doi: 10.1038/s41467-018-03523-2
21. Herce HD, Deng W, Helma J, Leonhardt H, Cardoso MC. Visualization and targeted disruption of protein interactions in living cells. Nat Commun. (2013) 4:2660. doi: 10.1038/ncomms3660
22. Dunham WH, Mullin M, Gingras AC. Affinity-purification coupled to mass spectrometry: basic principles and strategies. Proteomics (2012) 12:1576–90. doi: 10.1002/pmic.201100523
23. Shaw G, Morse S, Ararat M, Graham FL. Preferential transformation of human neuronal cells by human adenoviruses and the origin of HEK 293 cells. FASEB J. (2002) 16:869–71. doi: 10.1096/fj.01-0995fje
24. Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, et al. Embryonic stem cell lines derived from human blastocysts. Science (1998) 282:1145 LP−7.
25. Balboa D, Weltner J, Eurola S, Trokovic R, Wartiovaara K, Otonkoski T. Conditionally stabilized DCas9 activator for controlling gene expression in human cell reprogramming and differentiation. Stem Cell Rep. (2015) 5:448–59. doi: 10.1016/j.stemcr.2015.08.001
26. Watanabe K, Ueno M, Kamiya D, Nishiyama A, Matsumura M, Wataya T, et al. A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nat Biotechnol. (2007) 25:681. doi: 10.1038/nbt1310
27. Thorvaldsdóttir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Briefings Bioinform. (2013) 14:178–92. doi: 10.1093/bib/bbs017
28. Hartley JL, Temple GF, Brasch MA. DNA cloning using in vitro site-specific recombination. Genome Res. (2000) 10: 1788–95. doi: 10.1101/gr.143000.that
29. Varjosalo M, Sacco R, Stukalov A, van Drogen A, Planyavsky M, Hauri S, et al. Interlaboratory reproducibility of large-scale human protein-complex analysis by standardized AP-MS. Nat Methods (2013) 10:307–14. doi: 10.1038/nmeth.2400
30. Mellacheruvu D, Wright Z, Couzens AL, Lambert JP, St-Denis NA, Li T, et al. The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nat Methods (2013) 10:730–6. doi: 10.1038/nmeth.2557
31. Orchard S, Kerrien S, Abbani S, Aranda B, Bhate J, Bidwell S, et al. Protein interaction data curation: the international molecular exchange (IMEx) consortium. Nat Methods (2012) 9:345–50. doi: 10.1038/nmeth.1931
32. Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. (2009) 4:44–57. doi: 10.1038/nprot.2008.211
33. Perry JR, Day F, Elks CE, Sulem P, Thompson DJ, Ferreira T, et al. Parent-of-origin-specific allelic associations among 106 genomic loci for age at menarche. Nature (2014) 514:92–7. doi: 10.1038/nature13545
34. Day FR, Thompson DJ, Helgason H, Chasman DI, Finucane H, Sulem P, et al. Genomic analyses identify hundreds of variants associated with age at menarche and support a role for puberty timing in cancer risk. Nat Genet. (2017) 49:834–41. doi: 10.1038/ng.3841
35. Boehm U, Bouloux PM, Dattani MT, de Roux N, Dodé C, Dunkel L, et al. European consensus statement on congenital hypogonadotropic hypogonadism—pathogenesis, diagnosis and treatment. Nat Rev Endocrinol. (2015) 11:547–64. doi: 10.1038/nrendo.2015.112
36. Bouilly J, Messina A, Papadakis G, Cassatella D, Xu C, Acierno JS, et al. DCC/NTN1 complex mutations in patients with congenital hypogonadotropic hypogonadism impair GnRH neuron development. Hum Mol Genet. (2018) 27:359–72. doi: 10.1093/hmg/ddx408
37. Howard SR, Guasti L, Ruiz-Babot G, Mancini A, David A, Storr HL, et al. IGSF10 mutations dysregulate gonadotropin-releasing hormone neuronal migration resulting in delayed puberty. EMBO Mol Med. (2016) 8:626–42. doi: 10.15252/emmm.201606250
38. Xu C, Messina A, Somm E, Miraoui H, Kinnunen T, Acierno J, et al. KLB Encoding ?-Klotho, is mutated in patients with congenital hypogonadotropic hypogonadism. EMBO Mol Med (2017) 9:1397–79. doi: 10.15252/emmm.201607376
39. Richards MR, Plummer L, Chan, Y.-M, Lippincott MF, Quinton R, Kumanov P, et al. Phenotypic spectrum of POLR3B mutations: isolated hypogonadotropic hypogonadism without neurological or dental anomalies. J Med Genetics (2017) 54: 25–19. doi: 10.1136/jmedgenet-2016-104064
40. Marcos S, Monnier C, Rovira X, Fouveaut C, Pitteloud N, Ango F, et al. Defective signaling through plexin-A1 compromises the development of the peripheral olfactory system and neuroendocrine reproductive axis in mice. Hum Mol Genet. (2017) 26:2006–17. doi: 10.1093/hmg/ddx080
41. Kelley CG, Lavorgna G, Clark ME, Boncinelli E, Mellon PL. The Otx2 Homeoprotein regulates expression from the gonadotropin-releasing hormone proximal promoter. Mol Endocrinol. (2000) 14:1246–56. doi: 10.1210/mend.14.8.0509
42. Burger LL, Vanacker C, Phumsatitpong C, Wagenmaker ER, Wang L, Olson DP, et al. Identification of genes enriched in GnRH neurons by translating ribosome affinity purification and RNAseq in mice. Endocrinology (2018) 159:1922–40. doi: 10.1210/en.2018-00001
43. Margolin DH, Kousi M, Chan YM, Lim ET, Schmahmann JD, Hadjivassiliou M, et al. Ataxia, dementia, and hypogonadotropism caused by disordered ubiquitination. N Engl J Med. (2013) 368:1992–2003. doi: 10.1056/NEJMoa1215993
44. Hagen CP, Sørensen K, Mieritz MG, Johannsen TH, Almstrup K, Juul A. Circulating MKRN3 Levels decline prior to pubertal onset and through puberty: a longitudinal study of healthy girls. J Clin Endocrinol Metabol. (2015) 100:1920–6. doi: 10.1210/jc.2014-4462
45. Varimo T, Dunkel L, Vaaralahti K, Miettinen PJ, Hero M, Raivio T. Circulating makorin ring-finger protein-3 (MKRN3) levels decline in boys before the clinical onset of puberty. Eur J Endocrinol. (2016) 174, 785–90. doi: 10.1530/EJE-15-1193
46. Shin YL. An update on the genetic causes of central precocious puberty. Ann Pediatr Endocrinol Metab. (2016) 21:66–9. doi: 10.6065/apem.2016.21.2.66
47. Millar RP. New insights into GnRH neuron development, programming and regulation in health and disease. Neuroendocrinology (2015) 102:181–83. doi: 10.1159/000441115
48. Skorupskaite K, George JT, Anderson RA. The kisspeptin-GnRH pathway in human reproductive health and disease. Hum Reprod Update (2014) 20:485–500. doi: 10.1093/humupd/dmu009
49. Telias M, Ben-Yosef D. Modeling neurodevelopmental disorders using human pluripotent stem cells. Stem Cell Rev Rep. (2014) 10:494–511. doi: 10.1007/s12015-014-9507-2
50. Lin YC, Boone M, Meuris L, Lemmens I, Van Roy N, Soete A, et al. Genome dynamics of the human embryonic kidney 293 lineage in response to cell biology manipulations. Nat Commun. (2014) 5:4767. doi: 10.1038/ncomms5767
51. Sørensen K, Mouritsen A, Mogensen SS, Aksglaede L, Juul A. Insulin sensitivity and lipid profiles in girls with central precocious puberty before and during gonadal suppression. J Clin Endocrinol Metabol. (2010) 95:3736–44. doi: 10.1210/jc.2010-0731
52. Moret M, Stettler R, Rodieux F, Gaillard RC, Waeber G, Wirthner D, et al. Insulin modulation of luteinizing hormone secretion in normal female volunteers and lean polycystic ovary syndrome patients. Neuroendocrinology (2009) 89:131–39. doi: 10.1159/000160911
53. Roth CL, Mastronardi C, Lomniczi A, Wright H, Cabrera R, Mungenast AE, et al. Expression of a tumor-related gene network increases in the mammalian hypothalamus at the time of female puberty. Endocrinology (2007) 148:5147–61. doi: 10.1210/en.2007-0634
54. Elks CE, Perry JR, Sulem P, Chasman DI, Franceschini N, He C, et al. Thirty new loci for age at menarche identified by a meta-analysis of genome-wide association studies. Nat Genetics (2010) 42:1077–85. doi: 10.1038/ng.714
55. Avendaño MS, Vazquez MJ, Tena-Sempere M. Disentangling Puberty: novel neuroendocrine pathways and mechanisms for the control of mammalian puberty. Hum Reproduc Update (2017) 23:737–63. doi: 10.1093/humupd/dmx025
56. Perry JR, Stolk L, Franceschini N, Lunetta KL, Zhai G, McArdle PF, et al. Meta-analysis of genome-wide association data identifies two loci influencing age at menarche. Nat Genetics (2009) 41:648–50. doi: 10.1038/ng.386
57. Sulem P, Gudbjartsson DF, Rafnar T, Holm H, Olafsdottir EJ, Olafsdottir GH, et al. Genome-wide association study identifies sequence variants on 6q21 associated with age at menarche. Nat Genet. (2009) 41:734–8. doi: 10.1038/ng.383
58. He C, Kraft P, Chen C, Buring JE, Paré G, Hankinson SE, et al. Genome-wide association studies identify loci associated with age at menarche and age at natural menopause. Nat Genet. 41:724–8. doi: 10.1038/ng.385
59. Ong KK, Elks CE, Li S, Zhao JH, Luan J, Andersen LB, et al. Genetic variation in LIN28B is associated with the timing of puberty. Nat Genetics (2009) 41:729–33. doi: 10.1038/ng.382
60. Cousminer DL, Berry DJ, Timpson NJ, Ang W, Thiering E, Byrne EM, et al. Genome-wide association and longitudinal analyses reveal genetic loci linking pubertal height growth, pubertal timing and childhood adiposity. Hum Mol Genetics (2013) 22:2735–47. doi: 10.1093/hmg/ddt104
61. Heo I, Joo C, Cho J, Ha M, Han J, Kim VN. Lin28 Mediates the terminal uridylation of Let-7 precursor microRNA. Mol Cell (2008) 32:276–84. doi: 10.1016/j.molcel.2008.09.014
62. Gajdos ZK, Henderson KD, Hirschhorn JN, Palmert MR. Genetic determinants of pubertal timing in the general population. Mol Cell Endocrinol. (2010) 324:21–9. doi: 10.1016/j.mce.2010.01.038
63. Corre C, Shinoda G, Zhu H, Cousminer DL, Crossman C, Bellissimo C, et al. Sex-specific regulation of weight and puberty by the Lin28/Let-7 axis. J Endocrinol. (2016) 228:179–91. doi: 10.1530/JOE-15-0360
64. Sangiao-Alvarellos S, Manfredi-Lozano M, Ruiz-Pino F, Navarro VM, Sánchez-Garrido MA, Leon S, et al. Changes in hypothalamic expression of the Lin28/Let-7 system and related microRNAs during postnatal maturation and after experimental manipulations of puberty. Endocrinology (2013) 154:942–55. doi: 10.1210/en.2012-2006
65. Grieco A, Rzeczkowska P, Alm C, Palmert MR. Investigation of peripubertal expression of Lin28a and Lin28b in C57BL/6 Female Mice. Mol Cell Endocrinol. (2013) 365:241–8. doi: 10.1016/j.mce.2012.10.025
Keywords: MKRN3, puberty timing, protein interaction, GNRH1, CRISPR/Cas9
Citation: Yellapragada V, Liu X, Lund C, Känsäkoski J, Pulli K, Vuoristo S, Lundin K, Tuuri T, Varjosalo M and Raivio T (2019) MKRN3 Interacts With Several Proteins Implicated in Puberty Timing but Does Not Influence GNRH1 Expression. Front. Endocrinol. 10:48. doi: 10.3389/fendo.2019.00048
Received: 12 October 2018; Accepted: 21 January 2019;
Published: 08 February 2019.
Edited by:
Gustavo M. Somoza, Institute of Biotechnological Research (IIB-INTECH), ArgentinaReviewed by:
Stephanie Constantin, National Institutes of Health (NIH), United StatesMarina Olga Fernandez, Instituto de Biología y Medicina Experimental (IBYME), Argentina
Copyright © 2019 Yellapragada, Liu, Lund, Känsäkoski, Pulli, Vuoristo, Lundin, Tuuri, Varjosalo and Raivio. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Taneli Raivio, dGFuZWxpLnJhaXZpb0BoZWxzaW5raS5maQ==
†These authors have contributed equally to this work