 He Cai
He Cai Yunxia Liu
Yunxia Liu Hongbo Men
Hongbo Men- The Cardiovascular Center, First Hospital of Jilin University, Changchun, China
Physiological reactive oxygen species (ROS) are important regulators of intercellular signal transduction. Oxidative and antioxidation systems maintain a dynamic balance under physiological conditions. Increases in ROS levels destroy the dynamic balance, leading to oxidative stress damage. Oxidative stress is involved in the pathogenesis of aging-related cardiovascular diseases (ACVD), such as atherosclerosis, myocardial infarction, and heart failure, by contributing to apoptosis, hypertrophy, and fibrosis. Oxidative phosphorylation in mitochondria is the main source of ROS. Increasing evidence demonstrates the relationship between ACVD and humanin (HN), an endogenous peptide encoded by mitochondrial DNA. HN protects cardiomyocytes, endothelial cells, and fibroblasts from oxidative stress, highlighting its protective role in atherosclerosis, ischemia–reperfusion injury, and heart failure. Herein, we reviewed the signaling pathways associated with the HN effects on redox signals, including Kelch-like ECH-associated protein 1 (Keap1)/nuclear factor erythroid 2-related factor 2 (Nrf2), chaperone-mediated autophagy (CMA), c-jun NH2 terminal kinase (JNK)/p38 mitogen-activated protein kinase (p38 MAPK), adenosine monophosphate-activated protein kinase (AMPK), and phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt)-Janus kinase 2 (JAK2)/signal transducer and activator of transcription 3 (STAT3). Furthermore, we discussed the relationship among HN, redox signaling pathways, and ACVD. Finally, we propose that HN may be a candidate drug for ACVD.
Introduction
With the increase of an aging population, aging-related cardiovascular diseases (ACVDs) confer a heavy economic burden on society (1, 2). Oxidative stress is involved in the pathogenesis of ACVD (3–5), such as atherosclerosis (6), myocardial infarction (7), and heart failure (8), by contributing to apoptosis, hypertrophy, and fibrosis. Oxidative phosphorylation in mitochondria is the main source of reactive oxygen species (ROS). Increased ROS levels destroy the dynamic balance between oxidative and antioxidation systems, leading to oxidative stress damage. Thus, suppressing ROS generation is a potential strategy for the treatment of ACVD.
Humanin (HN), an endogenous active peptide encoded by mitochondrial DNA, has been shown to be related to ACVD (9) (1): the serum HN level negatively correlates with age (10, 11); (2) HN reduced H2O2-induced oxidative stress damage in myocardial cells and isolated myocardial mitochondria by promoting the expression of antioxidant defense system proteins (12) and inhibiting the activity of complexes I and III of the electron transport chain (13); (3) HN reduced ROS production, protecting endothelial cells from oxidative stress damage induced by abnormal glycolipid metabolism (14–16); (4) HN restored chaperone-mediated autophagy (CMA) by regulating heat shock protein 90 (Hsp90) and decreasing ROS production, thereby protecting cardiomyocytes and fibroblasts from oxidative stress damage (17); (5) HN upregulates the expression of antioxidant enzymes, preserving the heart function after myocardial infarction in an ischemia–reperfusion injury model by reducing myocardial cell death and the area of myocardial infarction (18–20). Of note, there are only a few studies that investigated the redox signaling pathways associated with HN. Moreover, there is no detailed review about the redox signaling pathways involving HN in ACVD. Herein, we reviewed the regulation of HN expression and the main downstream signaling pathways involved in oxidative stress and discussed the relationship among HN, the redox signaling pathways, and ACVD. Finally, we propose that HN may be a candidate drug for ACVD.
The Origin and Functions of HN
Mitochondrial DNA encodes mitochondria-derived peptides (MDPs). MDPs include HN, mitochondrial ORF of the twelve S c (MOTS-c), and small humanin-like peptides (SHLPs) 1–6. HN was first discovered in patients with Alzheimer’s disease (AD). HN suppresses neuronal cell death, suggesting it may be a candidate drug for AD (21, 22). HN is transcribed from a 75-bp open reading frame sequence of the large 16S mitochondrial ribosomal RNA (rRNA) in the cytoplasm, generating a 24-amino-acid peptide with the sequence, Met-Ala-Pro-Arg-Gly-Phe-Ser-Cys-Leu-Leu-Leu-Leu-Thr-Ser-Glu-Ile-Asp-Leu-Pro-Val-Lys-Arg-Arg-Ala (22–24). However, HN mRNA is translated into a 21-amino-acid peptide in mitochondria, without the last three amino acid residues found in the HN translated in the cytoplasm. Notably, both variants contain basic amino acids in the N-terminal and C-terminal with similar functions (21, 25, 26). HN mediates a variety of intracellular and extracellular signaling pathways and plays multiple protection functions. It inhibits the translocation of proapoptotic proteins, such as Bax, Bid, and tBid, into mitochondria by binding to them. Furthermore, HN suppresses cytochrome C release and the formation of apoptotic bodies, thereby inhibiting mitochondria-dependent apoptosis (27, 28). The Golgi apparatus and endoplasmic reticulum are required for HN release (22), and released HN binds to two kinds of receptors on the cell membrane, the cell membrane trimer comprising CNTFR, WSX-1, and the 130-kDa gp130, and formyl peptide receptor-like 1 (FPRL1) (29, 30). After binding to the trimer receptor: (1) HN activates AMP-activated protein kinase (AMPK), further suppressing the mammalian target of rapamycin (mTOR) and nuclear factor kappa B (NF-κB) signaling pathways; (2) HN activates the phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT)-Janus kinase 2 (JAK2)/signal transducer and activator of transcription 3 (STAT3) signaling pathway; and (3) HN inhibits the c-jun NH2 terminal kinase (JNK)/p38 mitogen-activated protein kinase (MAPK) signaling pathways, protecting cellular and mitochondrial functions (31–37). Moreover, HN activates FPRL1 receptor and extracellular signal-regulated kinases (ERK1/2) (30). HN has many protective functions, such as anti-aging, inhibition of myocardial fibrosis, regulation of mitochondrial homeostasis, anti-inflammation, improving metabolism, regulation of the redox system, and autophagy promotion. It protects the retinal segment epithelium from oxidative stress-induced aging (38) and inhibits myocardial fibrosis in aged mice (39). HN promotes mitochondrial biogenesis and regulates mitochondrial homeostasis (40) and it decreases the expression of tumor necrosis factor alpha (TNF-α), interleukin (IL)-1β, and IL-6, to inhibit inflammation (41). Additionally, HN has potential in the treatment of diabetes by improving β-cell survival (32), promoting insulin secretion (42), and improving insulin resistance (10). HN also promotes chaperone-mediated autophagy (17), decreases ROS production, promotes the expression of antioxidant proteins and maintains the redox system balance (12).
HN and Oxidative Stress
Regulation of HN Expression Under Oxidative Stress
Oxidative stress contributes to ischemia–reperfusion injury (43). The expression of HN is increased after ischemia–reperfusion injury in mice, indicating the association of HN expression with oxidative stress (44). HN levels in the skeletal muscle and in the plasma of humans and mice negatively correlate with the increase in age (10, 45). HN levels in the peripheral blood are regulated by insulin-like growth factor (IGF) and IGF-binding protein (IGFBP). IGFBP-3 is the main component of IGFBP in the peripheral blood, with high affinity to HN (46, 47). The decrease in HN in the peripheral blood of AD patients suggests that HN in the peripheral blood may have protective effects on the nervous system (48). IGFBP-3 may transport HN through the blood–brain barrier, thereby reducing ROS production and further protecting nerve cells (49, 50). Growth hormone down-regulates HN levels in the peripheral blood through high expression of IGF-1 (51). Mitochondrial stressors, such as serum deprivation and chemotherapeutic drugs, can increase HN expression (52–54). In contrast, antiapoptotic factors decrease HN expression. Tripartite motif protein 11 (TRIM11) degrades intracellular HN through the proteasome pathway (49, 55). Therefore, HN levels under oxidative stress may be regulated by increasing IGFBP-3 levels or inhibiting IGF-1 levels (Figure 1).
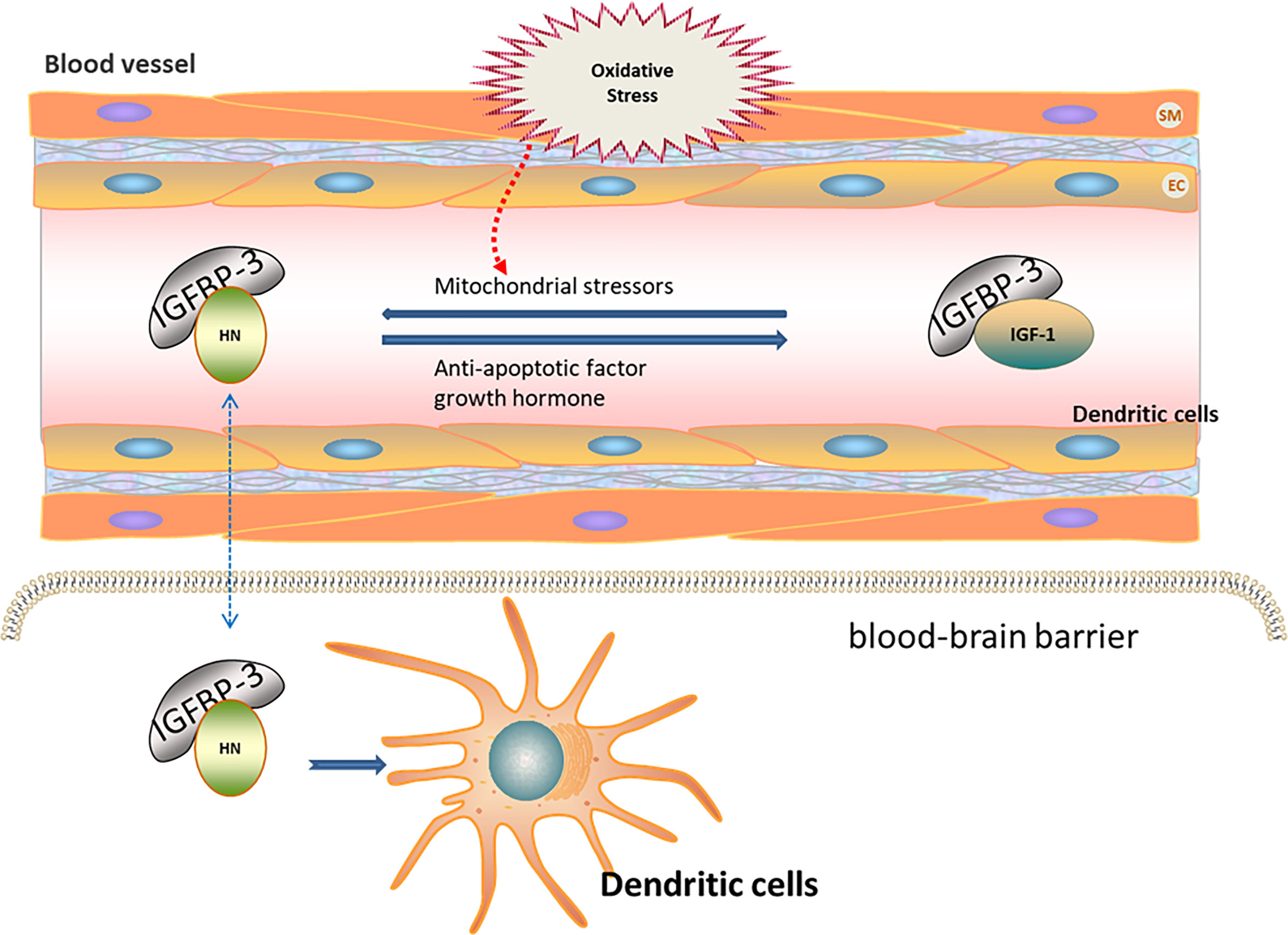
Figure 1 Regulation of HN expression under oxidative stress. HN levels in peripheral blood are regulated by IGF and IGFBP. IGFBP-3 is the main component of IGFBP in peripheral blood, with high affinity to HN. IGFBP-3 may transport HN through the blood–brain barrier (Blue dotted arrow), thereby reducing ROS and further protecting nerve cells. Growth hormone down-regulates HN levels in peripheral blood through the high expression of IGF-1. Mitochondrial stressors, such as serum deprivation and chemotherapeutic drugs, increase HN expressions. Anti-apoptotic factors decrease HN expressions. HN levels under oxidative stress may be regulated by increasing IGFBP-3 levels or inhibiting IGF-1 levels.
HN Regulates Redox Signaling Pathways
HN promotes the expression of antioxidant enzymes that inhibit ROS production through intracellular and/or extracellular pathways (56, 57). Intracellularly, (1) HN protects mitochondrial function by inhibiting electron transport chain complexes I and III and decreasing ROS formation; (2) HN activates the Kelch-like ECH-associated protein 1 (Keap1)/nuclear factor erythroid 2-related factor 2 (Nrf2) signaling pathway and the expression of antioxidant stress elements of nuclear genes through the reverse signal transduction between mitochondria and nuclei; (3) HN activates CMA through HSP90, promoting the absorption of oxidation products and further reducing ROS production. Extracellularly, HN binds to receptors on the cell membrane, triggering downstream signaling pathways, including the JNK/p38 MAPK, AMPK, and PI3K/AKT-JAK2/STAT3 signaling pathways, thereby promoting autophagy, reducing ROS production, and protecting the function of cells and mitochondria (Table 1). Understanding these signaling pathways is essential for understanding the antioxidant effect of HN in ACVD
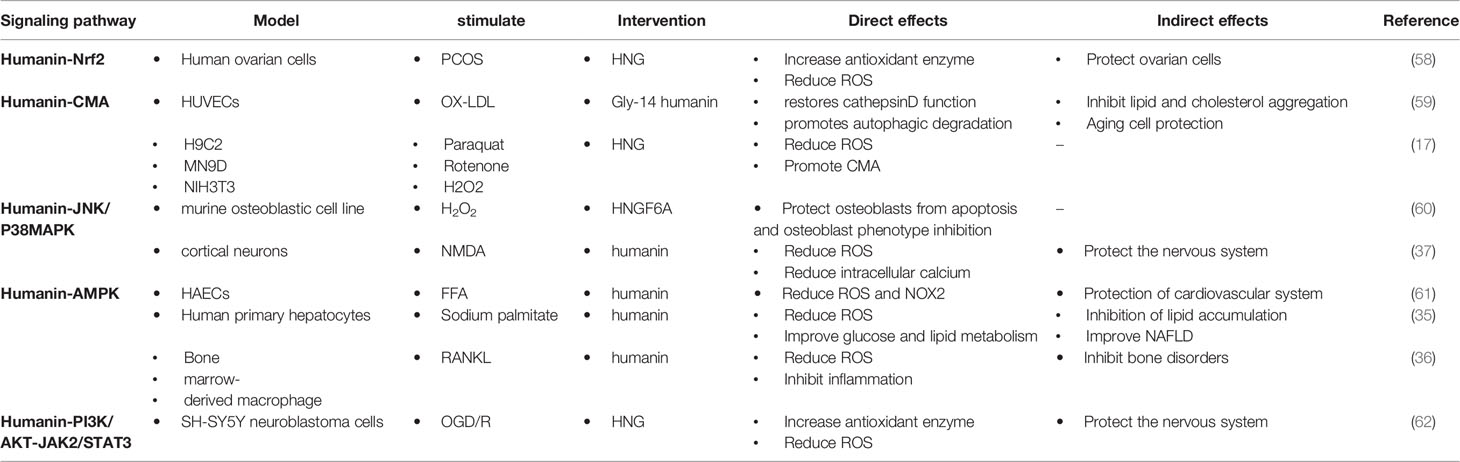
Table 1 Regulation and biological effect of HN signaling pathways on oxidative stress.
HN Regulates the Expression of the Keap1/Nrf2 Signaling Pathway Through Mitochondria-Nuclear Retrograde Signal Transduction
Nrf2 is a redox-sensitive transcription regulator found in various cells. Under physiological conditions, Keap1 promotes the ubiquitination and proteasome degradation of Nrf2. Under oxidative stress, the conformation of Keap1 is changed by cysteine sulfhydryl modification. Moreover, autophagic degradation of Keap1 is promoted by autophagy-related proteins, thereby increasing free Nrf2 levels in the cytoplasm. After being transferred to the nucleus, Nrf2 binds to antioxidant response elements (AREs), enhancing the expression of antioxidant genes (63, 64). Of note, increased age is associated with decreased Nrf2 stability and reduced antioxidant capacity under oxidative stress (65, 66). Additionally, mitochondria can regulate the expression of nuclear genes through the mitochondria-nuclear retrograde signal transduction pathway. In response to oxidative stress, mitochondria adapt by changing protein expression, resulting in the mitochondrial unfolded protein response (UPRmt) (67, 68). UPRmt regulates the expression of nuclear antioxidant genes through the mitochondria-nuclear retrograde signal transduction pathway. UPRmt can be triggered by excessive protein misfolding (67), inhibition of mitochondrial transcription and translation (69), impaired electron transport chain activity, and increased ROS levels (70). Under physiological conditions, the N-terminal of activating transcription factor associated with stress 1 (ATFS-1)/activating transcription factor 5 (ATF5), which is a mitochondria-targeting signal, efficiently mediates the import of ATF5 into the mitochondria. In contrast, the ATF5 import efficiency of mitochondria decreases when they are under stress. The C-terminal of ATFS-1/ATF5 acts as nuclear localization signal, enhancing the ATFS-1/ATF5 import into the nucleus, which promotes the expression of antioxidant enzyme genes in the nucleus, the synthesis of mitochondrial polypeptides, and the recovery of mitochondrial function (71, 72). MOTS-c was the first mitochondrial polypeptide found to regulate nuclear gene expression through a retrograde signal transduction pathway (73). A recent study revealed that HN activates the expression of nuclear antioxidant genes through the Keap1/Nrf2 signaling pathway. In this study, patients with polycystic ovary syndrome were investigated, and a rat model of polycystic ovary syndrome was established. Compared with the control group, the expression of superoxide dismutase (SOD), catalase (CAT), heme oxygenase 1 (HO-1), NADPH quinine oxidoreductase 1 (NQO1), and Nrf2 in the serum, ovarian tissue, and a human ovarian cell line increased; the level of ROS and Keap1 decreased in the group that was treated with the HN analog, HNG (58). Moreover, in a mouse AD model, HNG reduced p62 expression, upregulated autophagy-activating kinase 1, restored the function of cathepsin D, and promoted autophagy in mouse hippocampus tissues. Because Keap1 is ubiquitinated in a p62-dependent manner, it is speculated that HN does not degrade Keap1 by promoting autophagy. Whether HN activates Nrf2 by affecting the conformation of Keap1 needs further study (74, 75) (Figure 2).
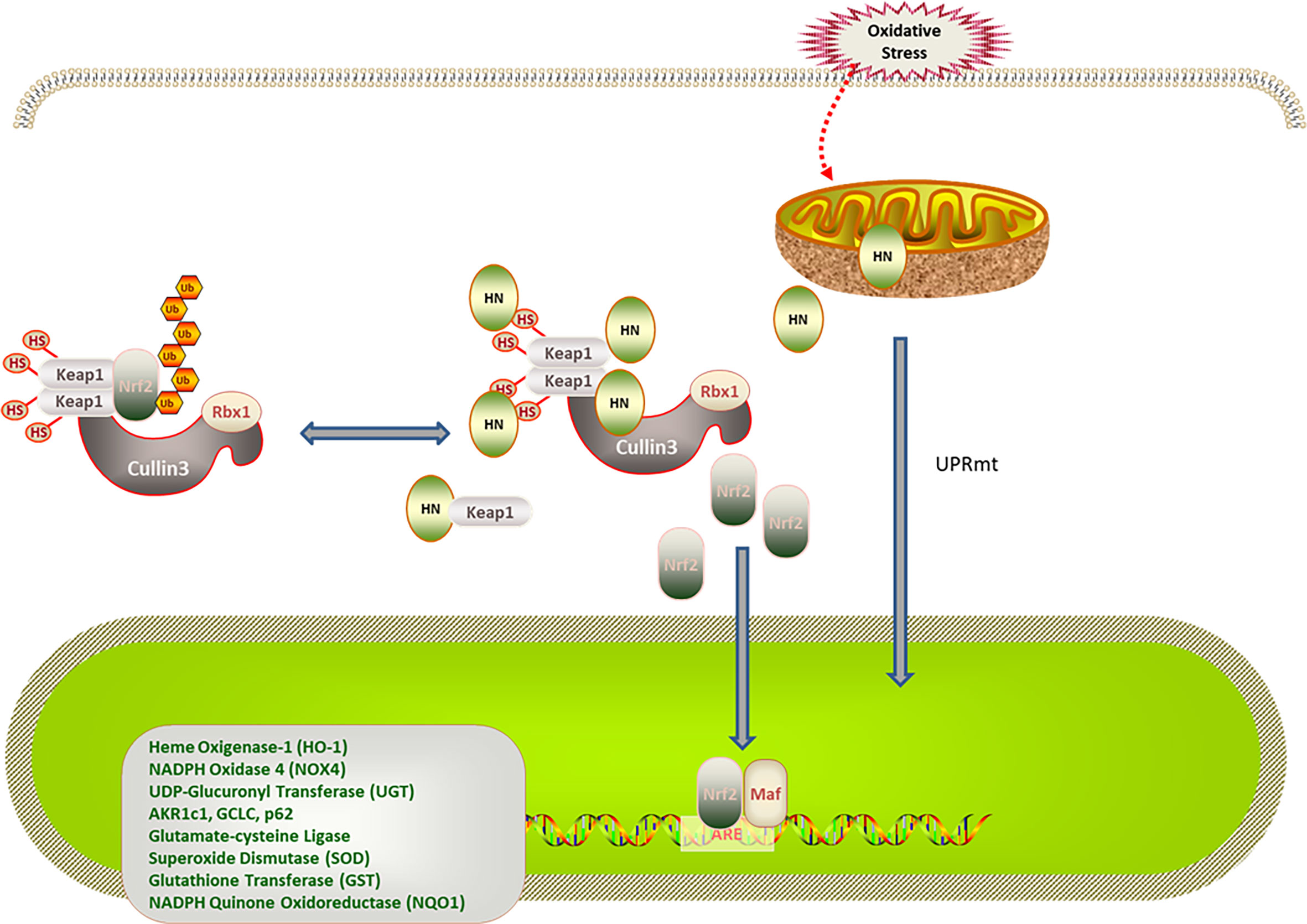
Figure 2 Regulation of HN on Keap1/Nrf2 signaling pathway under oxidative stress. HN promotes Keap1 degradation and release of Nrf2. Under stress conditions, HN inhibits the expression of Keap1, promotes the dissociation of Nrf2 and Keap1, activates the expression of antioxidant genes (SOD, CAD, HO-1, NQO1), and protects mitochondria from oxidative stress damage. Stress also triggers UPRmt, promotes the expression of antioxidant genes and the recovery of mitochondrial function, further promoting the formation of HN.
HN and Autophagy Signaling Pathways
Autophagy depends on lysosomal catabolism, which is one of the degradation processes for products of oxidative stress. Autophagy is classified into macroautophagy, microautophagy, and CMA (76, 77). In CMA, the cytosolic heat shock cognate chaperone of 70 kD (HSC70) is involved in recognizing substrate proteins containing a pentapeptide motif, forming a substrate–chaperone complex. The complex is then recognized by lysosome-associated membrane protein type 2A (LAMP-2A), contributing to the transformation of single-span LAMP-2A into a multimeric translocation complex (78, 79). HSP90 at the cytosolic side of the lysosomal membrane facilitates the substrate binding, enhancing the stability of LAMP-2A in the transformation from the monomeric to the multimeric form (78, 79). The luminal chaperone, Lys-HSC70, assists the delivery of the substrates into the lysosome after formation of the translocation complex. Interestingly, oxidative stress and hypoxia are the classical stimulators of oxidation-induced CMA activation that removes oxidized proteins to restore cell homeostasis (80). Impaired CMA leads to the accumulation of oxidative products, increasing oxidative stress damage (81, 82). However, CMA function decreases with age, suggesting a negative association between aging and the antioxidative stress ability (83). Thus, improving the CMA function may be a strategy for treating oxidative stress-related diseases in older adults.
Intriguingly, HN is an endogenous activator of CMA in a dose-dependent manner. One study found that HN protected cells from oxidative stress. This study used NIH3T3 mouse fibroblasts, H9C2 cardiomyoblasts, and MN9D dopaminergic neuronal cells. When oxidative stress was induced, Hsc70 recognized the oxidized protein (substrate), transported it to the lysosomal membrane, and bound to LAMP-2A receptor on the lysosomal membrane. Furthermore, endogenous HN, which is located on the cytoplasmic side of the lysosomal membrane, stabilized the binding of the substrate and the lysosome through Hsp90. With the help of lysosomal cavity chaperone (Lys-Hsc70), the substrate was transported to the lysosomal body and the oxidized protein was removed to maintain cell stability. Thereby, the cell damage caused by oxidative stress was reduced. Moreover, exogenous HNG enhanced the Hsp90-mediated binding of the substrate to the lysosome, upregulated CMA, and reduced oxidative stress damage (17).
Cathepsin D is also implicated in the HN involvement in autophagic degradation. Cathepsin D, as an intracellular lysosomal restriction inhibitor, is an aspartic protease and cysteine cathepsin. Gly-14 HN restores the activity of cathepsin D through FPRL1, promotes the autophagic degradation of oxidized low-density lipoprotein (ox-LDL) in endothelial cells, reduces the ox-LDL-induced oxidative stress injury of endothelial cells, and decreases the lipid and cholesterol accumulation in endothelial cells (59) (Figure 3).
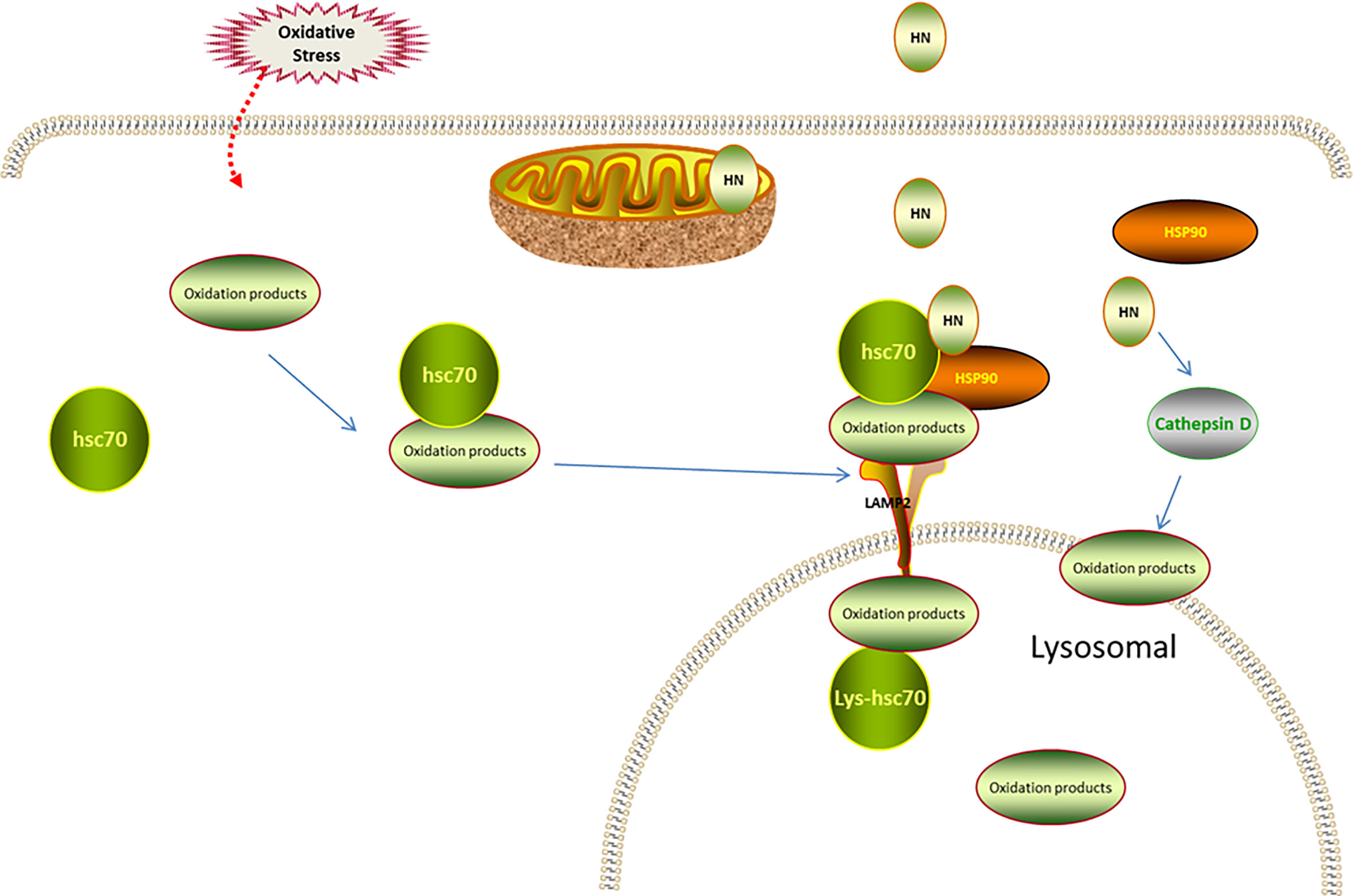
Figure 3 Regulation of HN on CMA under oxidative stress. Under oxidative stress, Hsc70 recognizes the oxidized protein (substrate), transports it to lysosomal membrane, and binds to LAMP-2 receptor on lysosomal membrane. The endogenous HN located in lysosomal membrane cytoplasmic assay stabilizes the binding of substrate and lysosome through Hsp90, and transports the substrate to lysosomal body with the assistance of Lys-Hsc70 to remove oxidized protein. Exogenous supplement of HNG enhances the binding of substrate and lysosome mediated by Hsp90, up-regulates CMA and further reduces oxidative stress damage. HN restores the activity of cathepsin D through FPRL1, promotes autophagy and reduces the production of ROS.
HN and the JNK/p38 MAPK Signaling Pathways
Mitogen-activated protein kinases (MAPKs), including ERK1/2, JNK, and p38 MAPK, are evolutionarily conserved enzymes that connect cell surface receptors and intracellular regulatory targets (84, 85). MAPK inhibition protects cells from oxidative stress (86). Previous studies have found that HN protected neurons by inhibiting JNK and p38 MAPK (87–89). Furthermore, H2O2 was used to establish an oxidative stress damage model in the murine osteoblast cell line, MC3T3-E1; in this model the HN analog, HNGF6A, decreased ROS production and cell damage caused by oxidative stress by inhibiting JNK and p38 MAPK phosphorylation (60). In another study using the N-methyl-D-aspartate (NMDA)-mediated excitotoxicity model of cortical neurons in vitro, HN reduced the release of lactate dehydrogenase, reduced the level of intracellular calcium, inhibited the activation of JNK and p38 MAPK, reduced ROS production by 45.7%, and reduced the oxidative stress injury. The antioxidant mechanism of HN depended on the level of intracellular calcium. Calcium overload leads to increased ROS production, which aggravates the calcium overload. HN weakens the intracellular Ca2+ influx, inhibiting the calcium overload and promoting cell survival, indicating that reducing intracellular calcium levels is also required for the HN regulation of JNK and p38 MAPK (37) (Figure 4).
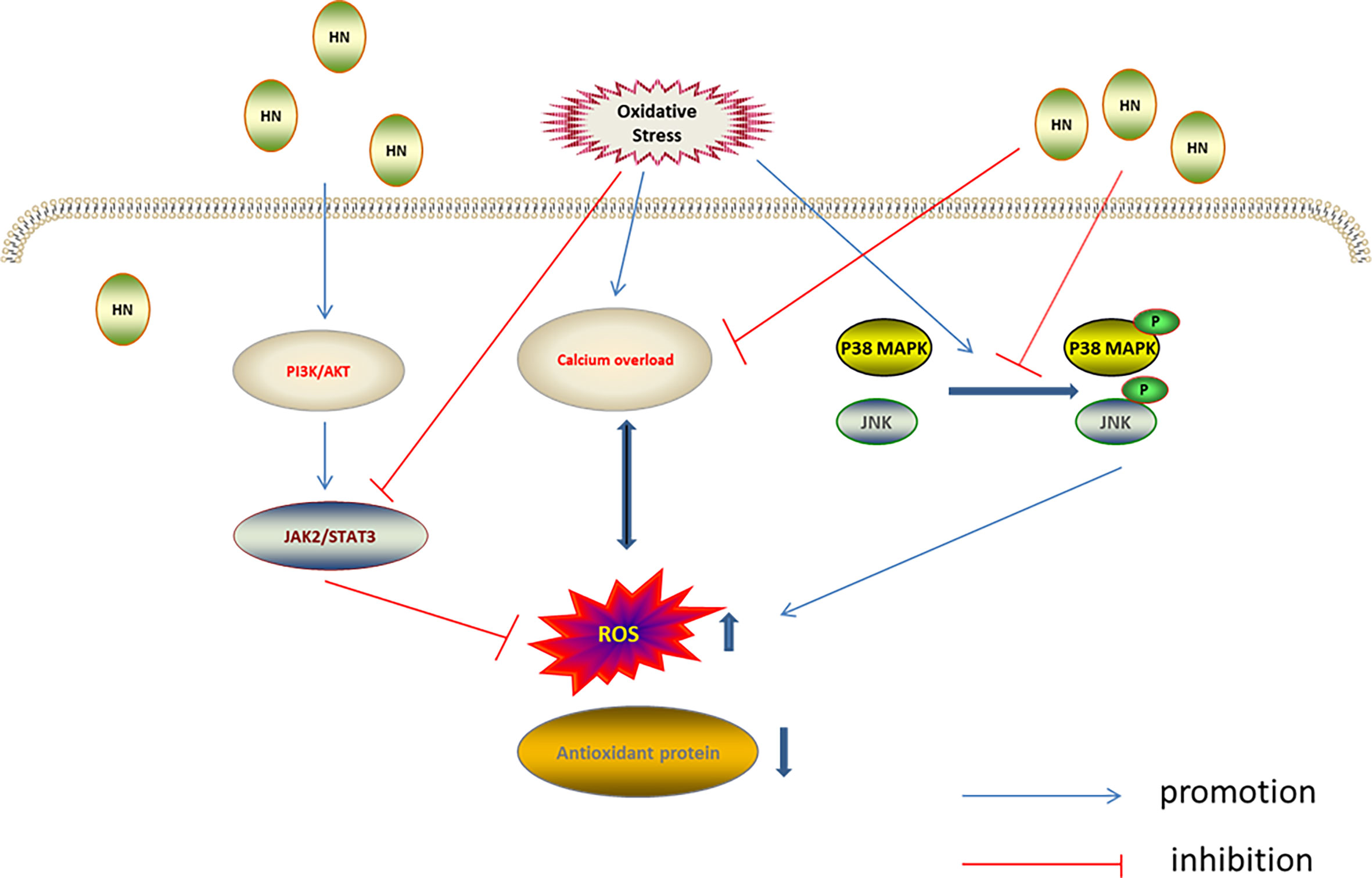
Figure 4 Regulation of HN on MAPKs, PI3K/AKT, and JAK2/STAT3 under oxidative stress. HN inhibits intracellular Ca2+ influx, inhibits ROS formation, activates MAPKs inhibitors, inhibits the activation of JNK and p38 MAPK, and reduces oxidative stress damage; HN activates the PI3K/Akt signaling pathway, which further activates the downstream JAK2/STAT3 signaling pathway, promotes the expression of antioxidant protein, reduces the level of ROS, and reduces the oxidative stress damage.
HN and the AMPK Signaling Pathway
AMPK is involved in the antioxidation effect of HN. AMPK is one of the cell energy sensors in eukaryotic organisms (90); decreased ATP levels activate AMPK, upregulating energy metabolism (90, 91). AMPK keeps cellular metabolic homeostasis by regulating mitochondrial ROS production (92). Interestingly, mitochondria-derived ROS activate AMPK indirectly (93). Compared with wild type mice, increased superoxide and mitochondrial superoxide levels were observed in the aorta of AMPK-knockout mice (94). Receptor activator of NF-κB ligand (RANKL) induced differentiation of mouse bone marrow cells, leading to decreased AMPK phosphorylation and increased ROS levels. It has been shown that HN increased AMPK phosphorylation and inhibited the NF-κB pathway, decreasing ROS production and enhancing cell activity (36). HN also inhibited oxidative stress damage in aortic endothelial cells induced by high free fatty acids by activating AMPK. Furthermore, HN reduced the expression of NADPH oxidase 2 (NOX2) and ROS production, inhibited the activation of the inflammatory body, Nod‐like receptor family protein 3 (NLRP3), and thereby protected endothelial cells from oxidative stress damage. NOX2 is the enzyme dedicated to ROS production. It has been demonstrated that HN inhibited ROS production by inhibiting NOX2 (61). AMPK and mTOR play a key role in cell energy metabolism and cell survival. mTOR is a highly conserved serine/threonine kinase, which is involved in regulating cell survival and cell metabolism. mTOR has been reported to play a pathogenic role in insulin resistance and adipogenesis in several cell types. By increasing AMPK phosphorylation, HN inhibited mTOR and regulatory element binding protein 1 (SREBP1) to improve insulin resistance and reduce ROS production (35) (Figure 5).
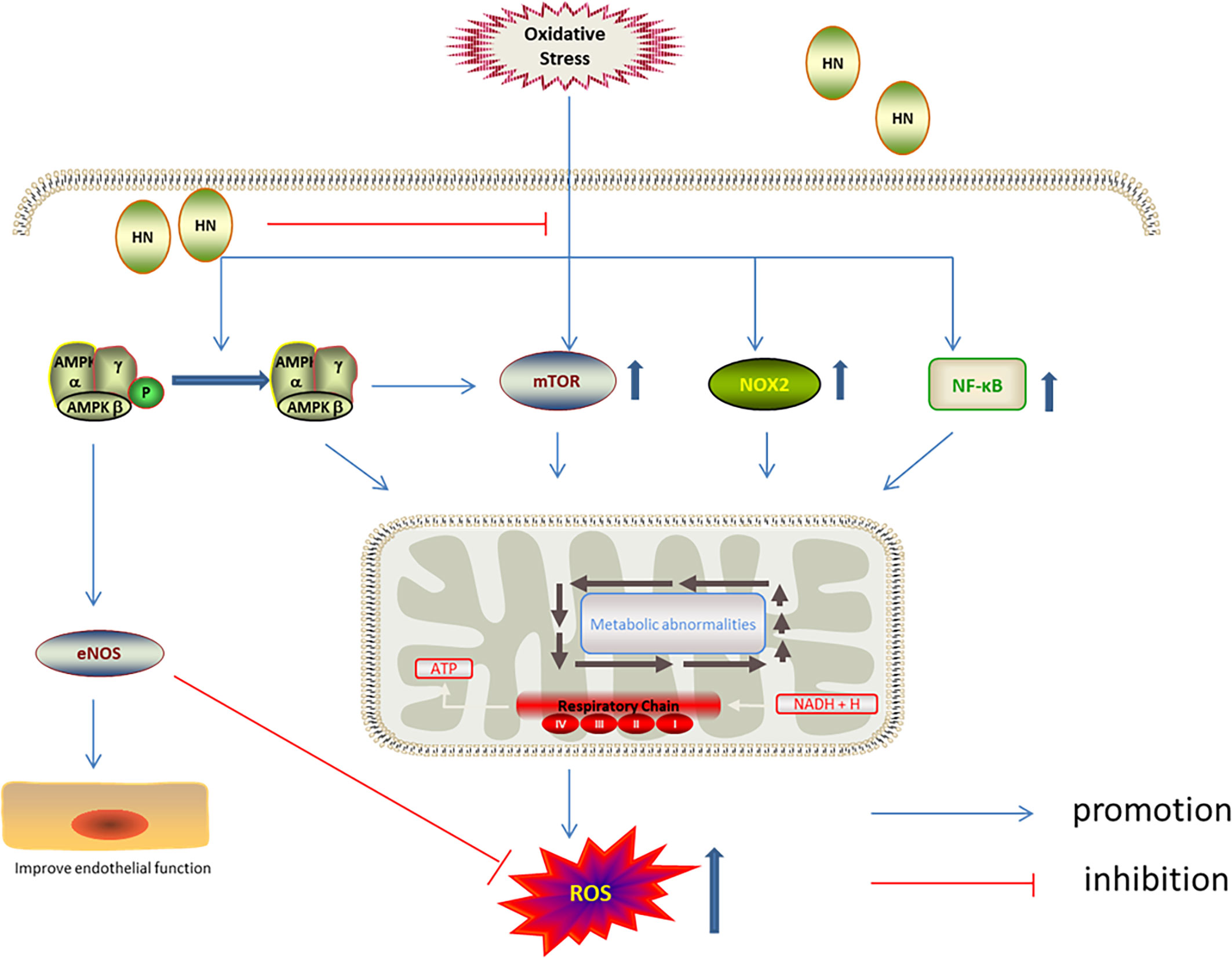
Figure 5 Regulation of HN on AMPK/mTOR under oxidative stress. HN up-regulates the phosphorylation of AMPK, activates eNOS, inhibits mTOR, NOX2 and NF-κB, to reduce the production of ROS, improve endothelial function.
HN and the PI3K/AKT, JAK2/STAT3 Signaling Pathways
The JAK2/STAT3 cascade, which is regulated by PI3K/AKT, plays a key role in cell proliferation, anti-apoptosis, anti-aging, cancer development and migration (33, 95, 96). Under oxidative stress, JAK2/STAT3 activity decreases and ROS production increases, leading to increased mitochondrial membrane permeability and cell apoptosis (97). Previous studies have found that HN reactivates the JAK2/STAT3 signaling pathway through the PI3K/Akt pathway, which plays a neuroprotective role (38, 98). Recently, HN has also been shown to reactivate JAK2/STAT3 through the PI3K/Akt signaling pathway to reduce oxidative stress. Furthermore, the neuroblastoma cell line, SH-SY5Y, was used to establish an oxygen/glucose deprivation/reoxygenation (OGD/R) model in vitro. In this model, the JAK2/STAT3 signaling pathway was inhibited, the intracellular malondialdehyde (MDA) level increased, and the SOD level decreased. After HNG intervention, the JAK2/STAT3 signaling pathway was activated, the SOD level increased, and the MDA level decreased. Application of HNG + PI3K/Akt inhibitor decreased the levels of JAK2 and STAT3, indicating that the PI3K/Akt inhibitor completely counteracted the activation of the JAK2/STAT3 signaling pathway by HN and suppressed the protective effect of HN (62) (Figure 4).
HN Regulation of the Redox System in ACVD
HN has protective effects against a variety of cardiovascular diseases, including atherosclerosis (99, 100), acute myocardial infarction, myocardial ischemia–reperfusion injury (45, 99, 101), and myocardium aging (10, 39, 98). The mechanisms of these protective effects all involve oxidative stress (Figure 6).
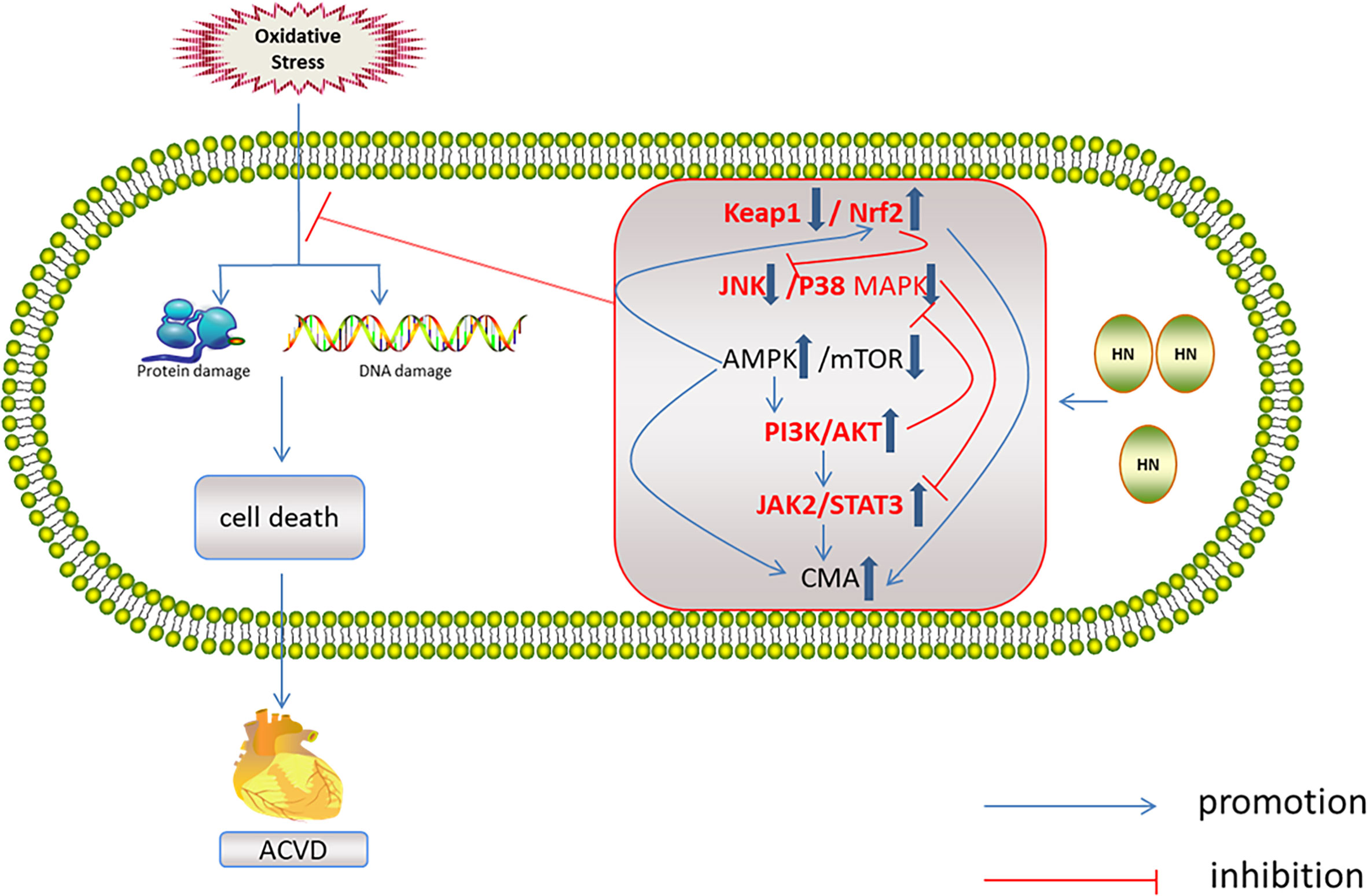
Figure 6 Regulation of HN on redox system in ACVD. HN activates AMPK and PI3K/AKT, JAK2/STAT3 signaling pathways, induces CMA, promotes Nrf2 release, and inhibits JNK/p38MAPK pathway, inhibits oxidative stress damage and ACVD (inside the box: the red font represents the future research direction, the blue thick arrow represents the influence of HN on the five signal pathways, and other signs form a signal network).
Arteriosclerosis
Atherosclerosis is an age-related disease. HN and its potent analogs have beneficial effects against age-related diseases (23). Endothelial dysfunction contributes to atherosclerosis (102). Importantly, HN improves endothelial dysfunction through antioxidation, because (1) HN inhibits NOX2, thereby decreasing mitochondrial ROS production; (2) NLRP3 inflammasome activated by mitochondrial ROS leads to endothelial injury; however, HN inhibits the activation of NLRP3 inflammasome by activating AMPK (61).
Hypercholesterolemia is involved in atherosclerosis because ox-LDL infiltrates the subendothelium to form atherosclerotic plaques after endothelial cell injury (103). However, HN prevents the progression of atherosclerotic plaques in hypercholesterolemic mice with apolipoprotein E (APOE) deficiency by reducing the level of nitrotyrosine (NT) and increasing the expression of endothelial nitric oxide synthase (eNOS), which are involved in oxidative stress (14). Ox-LDL is formed by ROS-related oxidation of LDL, ultimately promoting the formation and progression of atherosclerotic plaques by increasing lipid and cholesterol accumulation. Ox-LDL increases the expression of p62 and LC3-II and inhibits the function of cathepsin D activity. HN inhibits the ox-LDL-induced lipid and cholesterol accumulation by decreasing the LC3-II and p62 levels and restoring the ox-LDL-induced cathepsin D functional impairment, thereby reducing the formation of atherosclerotic plaques (45, 104). Lectin-like oxidized low-density lipoprotein-1 (LOX-1) is the main receptor involved in absorption of ox-LDL by endothelial cells. LOX-1 mediates the binding, internalization, and proteolytic degradation of ox-LDL by endothelial cells (105, 106). The HNG-induced decrease in LOX-1 protein expression also contributes to the inhibition of the formation and progression of atherosclerotic plaques (15).
High glucose levels are also implicated in atherosclerosis, leading to endothelial dysfunction. High glucose increases ROS production and the expression of pro-inflammatory factors (tumor necrosis factor-α and IL-1β), further promoting endothelial cells to produce vascular cell adhesion molecule-1 (VCAM-1) and E-selectin. VCAM-1 and E-selectin mediate the adhesion of circulating leukocytes to the endothelium, leading to atherosclerosis development. Kruppel-like factor 2 (KLF2) is involved in endothelial dysfunction induced by high glucose. HN upregulates the KLF2 gene expression, inhibiting monocyte adhesion to endothelial cells (107).
Coronary Heart Disease and Heart Failure
Oxidative stress is also involved in the pathogenesis of acute myocardial infarction and ischemia–reperfusion injury. HN protects cardiomyocytes from apoptosis through the antioxidation pathway, reducing the myocardial infarction size and improving cardiac function (108). HN also reduces the necrosis area of myocardial infarction and improves the cardiac function after myocardial infarction by reducing ROS production, which protects the function of myocardial mitochondria (18, 19). HN has been shown to protect isolated myocardial mitochondria from H2O2-induced oxidative stress. HN increased the levels of GSH, GPX, and SOD, reversing myocardial ischemia–reperfusion injury (12, 20). HNG upregulated the Akt/glycogen synthase kinase-3β pathway and inhibited myocardial fibrosis in aged mice (39). It has been found that Nrf2 and Keap1 are necessary for increasing the expression of SOD, CAT, GPX, and GSH. HN may promote the activation of Nrf2 by inhibiting the expression of Keap1 during myocardial infarction (58). In addition, the population study found that compared with normal people, the level of humanin in patients with coronary heart disease decreased and the level of lactic acid increased, suggesting that the protective effect of humanin on cardiovascular system is through antioxidant effect (100). Humanin is positively correlated with coronary artery endothelial function, which may be a target for the treatment of coronary heart disease in the future (99).
Heart failure is the most common complication of myocardial infarction. HN has been shown to decrease the incidence rate of heart failure by inhibiting myocardial hypertrophy (108). Endonuclease G deficiency induces cardiomyocyte hypertrophy by increasing ROS production. Intriguingly, HN has been demonstrated to inhibit cardiomyocyte hypertrophy induced by endonuclease G deficiency (109).
Relationship Between Redox Signaling Pathways and ACVD
HN reduces oxidative stress through the five above mentioned signaling pathways. These signaling pathways interact with each other and form a network, which is related to ACVD (Figure 6). One study has found that Nrf2 deficiency led to aging of human aortic endothelium and mouse aortic endothelium. The aging process was related to autophagy damage. Upregulation of Nrf2 by inhibiting Keap1 activated autophagy and inhibited aging (110). Furthermore, Keap1 upregulation and of Nrf2 inhibition led to oxidative stress damage and aging of vascular smooth muscle cells (111). In a D-galactose-induced mouse aging model, Keap1 expression increased, Nrf2 expression decreased, and ROS production increased (112). Nrf2 inhibits JNK phosphorylation, stabilizes mitochondrial function integrity, and reduces oxidative stress damage (113). In the aging heart, the activation of AMPK and autophagy is impaired. Activation of AMPK induces autophagy, inhibits cardiomyocyte aging, and protects aging myocardium from oxidative stress (114, 115). AMPK activates autophagy by inhibiting mTOR or phosphorylated UKL1 (116), and it activates Nrf2 and protects the myocardium from oxidative stress induced by high glucose (117). AMPK also reduces oxidative stress injury by activating AKT2/Nrf2 (118). Activation of PI3K/AKT by AMPK protects the myocardium from ischemia–reperfusion (I–R) injury (119). Therefore, AMPK is another target for inhibiting myocardial aging (114). Activation of the JAK2/STAT3 signaling pathway inhibits ventricular remodeling after myocardial infarction (120, 121), and it protects the aging heart from I–R injury (122). The JAK2/STAT3 signaling pathway also inhibits cardiomyocyte apoptosis by activating autophagy (123). JNK is the upstream regulator of JAK2/STAT3. Inhibition of JNK activates JAK2/STAT3 and protects the myocardium from oxidative stress induced by high free fatty acids (124). Upregulation of p38 MAPK/JNK phosphorylation promoted NF-κB translocation to the nucleus, induced aging, and aggravated myocardial injury (125). Inhibition of p38 MAPK and JNK phosphorylation protected the heart from oxidative stress injury in aged rats (126). PI3K/Akt inhibits MAPK and NF-κB activation, thereby protecting cardiomyocytes from injury (127, 128). PI3K/Akt inhibits mTOR and protects the myocardium from oxidative stress (129). However, there are few studies on the mechanism of HN and cardiac aging. It is necessary to explore whether HN exerts its anti-aging effects on the cardiovascular system through the above signaling pathways’ network.
Concluding Remarks and Prospect
Herein, we reviewed the signaling pathways associated with the HN effects against oxidative stress, including the Keap1/Nrf2, the autophagy, the JNK/p38 MAPK, the AMPK, the PI3K/Akt, and the JAK2/STAT3 signaling pathways. We then summarized the relationship among HN, the redox signaling pathways, and ACVD, and pointed out the future research direction for HN and ACVD. Finally, HN may be the target for ACVD treatment by reducing oxidative stress.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Michal Bell, PhD, from Liwen Bianji, Edanz Editing China (www.liwenbianji.cn/ac), for editing a draft of this manuscript.
References
1. Roth GA, Abate D, Abate KH, Abay SM, Abbafati C, Abbasi N, et al. Global, Regional, and National Age-Sex-Specific Mortality for 282 Causes of Death in 195 Countries and Territories, 1980-2017: A Systematic Analysis for the Global Burden of Disease Study 2017. Lancet (2018) 392:1736–88. doi: 10.1016/S0140-6736(18)32203-7
2. Yao SM, Zheng PP, Wan YH, Dong W, Miao GB, Wang H, et al. Adding High-Sensitivity C-reactive Protein to Frailty Assessment to Predict Mortality and Cardiovascular Events in Elderly Inpatients With Cardiovascular Disease. Exp Gerontol (2021) 146:111235. doi: 10.1016/j.exger.2021.111235
3. Lakatta EG. Arterial and Cardiac Aging: Major Shareholders in Cardiovascular Disease Enterprises: Part III: Cellular and Molecular Clues to Heart and Arterial Aging. Circulation (2003) 107:490–7. doi: 10.1161/01.CIR.0000048894.99865.02
4. Kwak HB. Aging, Exercise, and Extracellular Matrix in the Heart. J Exerc Rehabil (2013) 9:338–47. doi: 10.12965/jer.130049
5. Hipkiss AR. Mitochondrial Dysfunction, Proteotoxicity, and Aging: Causes or Effects, and the Possible Impact of NAD+-controlled Protein Glycation. Adv Clin Chem (2010) 50:123–50. doi: 10.1016/S0065-2423(10)50007-6
6. Marchio P, Guerra-Ojeda S, Vila JM, Aldasoro M, Victor VM, Mauricio MD. Targeting Early Atherosclerosis: A Focus on Oxidative Stress and Inflammation. Oxid Med Cell Longev (2019) 2019:8563845. doi: 10.1155/2019/8563845
7. Liu Y, Wang M, Liang Y, Wang C, Naruse K, Takahashi K. Treatment of Oxidative Stress With Exosomes in Myocardial Ischemia. Int J Mol Sci (2021) 22:1729. doi: 10.3390/ijms22041729
8. Wang W, Kang PM. Oxidative Stress and Antioxidant Treatments in Cardiovascular Diseases. Antioxid (Basel) (2020) 9:1292. doi: 10.3390/antiox9121292
9. Yen K, Lee C, Mehta H, Cohen P. The Emerging Role of the Mitochondrial-Derived Peptide Humanin in Stress Resistance. J Mol Endocrinol (2013) 50:R11–9. doi: 10.1530/JME-12-0203
10. Muzumdar RH, Huffman DM, Atzmon G, Buettner C, Cobb LJ, Fishman S, et al. Humanin: A Novel Central Regulator of Peripheral Insulin Action. PloS One (2009) 4:e6334. doi: 10.1371/journal.pone.0006334
11. Yen K, Mehta HH, Kim SJ, Lue Y, Hoang J, Guerrero N, et al. The Mitochondrial Derived Peptide Humanin is a Regulator of Lifespan and Healthspan. Aging (Albany NY) (2020) 12:11185–99. doi: 10.18632/aging.103534
12. Klein LE, Cui L, Gong Z, Su K, Muzumdar R. A Humanin Analog Decreases Oxidative Stress and Preserves Mitochondrial Integrity in Cardiac Myoblasts. Biochem Biophys Res Commun (2013) 440:197–203. doi: 10.1016/j.bbrc.2013.08.055
13. Thummasorn S, Shinlapawittayatorn K, Khamseekaew J, Jaiwongkam T, Chattipakorn SC, Chattipakorn N. Humanin Directly Protects Cardiac Mitochondria Against Dysfunction Initiated by Oxidative Stress by Decreasing Complex I Activity. Mitochondrion (2018) 38:31–40. doi: 10.1016/j.mito.2017.08.001
14. Oh YK, Bachar AR, Zacharias DG, Kim SG, Wan J, Cobb LJ, et al. Humanin Preserves Endothelial Function and Prevents Atherosclerotic Plaque Progression in Hypercholesterolemic ApoE Deficient Mice. Atherosclerosis (2011) 219:65–73. doi: 10.1016/j.atherosclerosis.2011.06.038
15. Ding Y, Feng Y, Zhu W, Zou Y, Xie Y, Wang F, et al. [Gly14]-Humanin Prevents Lipid Deposition and Endothelial Cell Apoptosis in a Lectin-Like Oxidized Low-density Lipoprotein Receptor-1-Dependent Manner. Lipids (2019) 54:697–705. doi: 10.1002/lipd.12195
16. Xie Y, Liu ZH, Li XY, Zhou YD, Xu X, Hu LF, et al. Protection Effect of [Gly14]-Humanin From Apoptosis Induced by High Glucose in Human Umbilical Vein Endothelial Cells. Diabetes Res Clin Pract (2014) 106:560–6. doi: 10.1016/j.diabres.2014.09.020
17. Gong Z, Tasset I, Diaz A, Anguiano J, Tas E, Cui L, et al. Humanin is an Endogenous Activator of Chaperone-Mediated Autophagy. J Cell Biol (2018) 217:635–47. doi: 10.1083/jcb.201606095
18. Thummasorn S, Shinlapawittayatorn K, Chattipakorn SC, Chattipakorn N. High-Dose Humanin Analogue Applied During Ischemia Exerts Cardioprotection Against Ischemia/Reperfusion Injury by Reducing Mitochondrial Dysfunction. Cardiovasc Ther (2017) 35:12289. doi: 10.1111/1755-5922.12289
19. Kumfu S, Charununtakorn ST, Jaiwongkam T, Chattipakorn N, Chattipakorn SC. Humanin Exerts Neuroprotection During Cardiac Ischemia-Reperfusion Injury. J Alzheimers Dis (2018) 61:1343–53. doi: 10.3233/JAD-170708
20. Thummasorn S, Apaijai N, Kerdphoo S, Shinlapawittayatorn K, Chattipakorn SC, Chattipakorn N. Humanin Exerts Cardioprotection Against Cardiac Ischemia/Reperfusion Injury Through Attenuation of Mitochondrial Dysfunction. Cardiovasc Ther (2016) 34:404–14. doi: 10.1111/1755-5922.12210
21. Hashimoto Y, Niikura T, Ito Y, Sudo H, Hata M, Arakawa E, et al. Detailed Characterization of Neuroprotection by a Rescue Factor Humanin Against Various Alzheimer's Disease-Relevant Insults. J Neurosci (2001) 21:9235–45. doi: 10.1523/JNEUROSCI.21-23-09235.2001
22. Hashimoto Y, Niikura T, Tajima H, Yasukawa T, Sudo H, Ito Y, et al. A Rescue Factor Abolishing Neuronal Cell Death by a Wide Spectrum of Familial Alzheimer's Disease Genes and Abeta. Proc Natl Acad Sci USA (2001) 98:6336–41. doi: 10.1073/pnas.101133498
23. Gong Z, Tas E, Muzumdar R. Humanin and Age-Related Diseases: A New Link. Front Endocrinol (Lausanne) (2014) 5:210. doi: 10.3389/fendo.2014.00210
24. Zapała B, Kaczyński Ł, Kieć-Wilk B, Staszel T, Knapp A, Thoresen GH, et al. Humanins, the Neuroprotective and Cytoprotective Peptides With Antiapoptotic and Anti-Inflammatory Properties. Pharmacol Rep (2010) 62:767–77. doi: 10.1016/S1734-1140(10)70337-6
25. Lee C, Yen K, Cohen P. Humanin: A Harbinger of Mitochondrial-Derived Peptides. Trends Endocrinol Metab (2013) 24:222–8. doi: 10.1016/j.tem.2013.01.005
26. Yamagishi Y, Hashimoto Y, Niikura T, Nishimoto I. Identification of Essential Amino Acids in Humanin, a Neuroprotective Factor Against Alzheimer's Disease-Relevant Insults. Peptides (2003) 24:585–95. doi: 10.1016/S0196-9781(03)00106-2
27. Zhai D, Luciano F, Zhu X, Guo B, Satterthwait AC, Reed JC. Humanin Binds and Nullifies Bid Activity by Blocking its Activation of Bax and Bak. J Biol Chem (2005) 280:15815–24. doi: 10.1074/jbc.M411902200
28. Ma ZW, Liu DX. Humanin Decreases Mitochondrial Membrane Permeability by Inhibiting the Membrane Association and Oligomerization of Bax and Bid Proteins. Acta Pharmacol Sin (2018) 39:1012–21. doi: 10.1038/aps.2017.169
29. Hashimoto Y, Kurita M, Aiso S, Nishimoto I, Matsuoka M. Humanin Inhibits Neuronal Cell Death by Interacting With a Cytokine Receptor Complex or Complexes Involving CNTF Receptor Alpha/WSX-1/Gp130. Mol Biol Cell (2009) 20:2864–73. doi: 10.1091/mbc.e09-02-0168
30. Ying G, Iribarren P, Zhou Y, Gong W, Zhang N, Yu ZX, et al. Humanin, a Newly Identified Neuroprotective Factor, Uses the G Protein-Coupled Formylpeptide Receptor-Like-1 as a Functional Receptor. J Immunol (2004) 172:7078–85. doi: 10.4049/jimmunol.172.11.7078
31. Matsuoka M, Hashimoto Y. Humanin and the Receptors for Humanin. Mol Neurobiol (2010) 41:22–8. doi: 10.1007/s12035-009-8090-z
32. Hoang PT, Park P, Cobb LJ, Paharkova-Vatchkova V, Hakimi M, Cohen P, et al. The Neurosurvival Factor Humanin Inhibits Beta-Cell Apoptosis Via Signal Transducer and Activator of Transcription 3 Activation and Delays and Ameliorates Diabetes in Nonobese Diabetic Mice. Metabolism (2010) 59:343–9. doi: 10.1016/j.metabol.2009.08.001
33. Hashimoto Y, Suzuki H, Aiso S, Niikura T, Nishimoto I, Matsuoka M. Involvement of Tyrosine Kinases and STAT3 in Humanin-mediated Neuroprotection. Life Sci (2005) 77:3092–104. doi: 10.1016/j.lfs.2005.03.031
34. Kim SJ, Guerrero N, Wassef G, Xiao J, Mehta HH, Cohen P, et al. The Mitochondrial-Derived Peptide Humanin Activates the ERK1/2, AKT, and STAT3 Signaling Pathways and has Age-Dependent Signaling Differences in the Hippocampus. Oncotarget (2016) 7:46899–912. doi: 10.18632/oncotarget.10380
35. Kwon C, Sun JL, Jeong JH, Jung TW. Humanin Attenuates Palmitate-Induced Hepatic Lipid Accumulation and Insulin Resistance Via AMPK-mediated Suppression of the mTOR Pathway. Biochem Biophys Res Commun (2020) 526:539–45. doi: 10.1016/j.bbrc.2020.03.128
36. Kang N, Kim KW, Shin DM. Humanin Suppresses Receptor Activator of Nuclear Factor-κb Ligand-Induced Osteoclast Differentiation Via AMP-activated Protein Kinase Activation. Korean J Physiol Pharmacol (2019) 23:411–7. doi: 10.4196/kjpp.2019.23.5.411
37. Yang X, Zhang H, Wu J, Yin L, Yan LJ, Zhang C. Humanin Attenuates Nmda-Induced Excitotoxicity by Inhibiting ROS-Dependent JNK/p38 Mapk Pathway. Int J Mol Sci (2018) 19:2982. doi: 10.3390/ijms19102982
38. Sreekumar PG, Ishikawa K, Spee C, Mehta HH, Wan J, Yen K, et al. The Mitochondrial-Derived Peptide Humanin Protects RPE Cells From Oxidative Stress, Senescence, and Mitochondrial Dysfunction. Invest Ophthalmol Vis Sci (2016) 57:1238–53. doi: 10.1167/iovs.15-17053
39. Qin Q, Mehta H, Yen K, Navarrete G, Brandhorst S, Wan J, et al. Chronic Treatment With the Mitochondrial Peptide Humanin Prevents Age-Related Myocardial Fibrosis in Mice. Am J Physiol Heart Circ Physiol (2018) 315:H1127–1127H1136. doi: 10.1152/ajpheart.00685.2017
40. Qin Q, Jin J, He F, Zheng Y, Li T, Zhang Y, et al. Humanin Promotes Mitochondrial Biogenesis in Pancreatic MIN6 β-Cells. Biochem Biophys Res Commun (2018) 497:292–7. doi: 10.1016/j.bbrc.2018.02.071
41. Zhao ST, Zhao L, Li JH. Neuroprotective Peptide Humanin Inhibits Inflammatory Response in Astrocytes Induced by Lipopolysaccharide. Neurochem Res (2013) 38:581–8. doi: 10.1007/s11064-012-0951-6
42. Kuliawat R, Klein L, Gong Z, Nicoletta-Gentile M, Nemkal A, Cui L, et al. Potent Humanin Analog Increases Glucose-Stimulated Insulin Secretion Through Enhanced Metabolism in the β Cell. FASEB J (2013) 27:4890–8. doi: 10.1096/fj.13-231092
43. Misra MK, Sarwat M, Bhakuni P, Tuteja R, Tuteja N. Oxidative Stress and Ischemic Myocardial Syndromes. Med Sci Monit (2009) 15:RA209–19.
44. Muzumdar RH, Huffman DM, Calvert JW, Jha S, Weinberg Y, Cui L, et al. Acute Humanin Therapy Attenuates Myocardial Ischemia and Reperfusion Injury in Mice. Arterioscler Thromb Vasc Biol (2010) 30:1940–8. doi: 10.1161/ATVBAHA.110.205997
45. Bachar AR, Scheffer L, Schroeder AS, Nakamura HK, Cobb LJ, Oh YK, et al. Humanin is Expressed in Human Vascular Walls and has a Cytoprotective Effect Against Oxidized LDL-Induced Oxidative Stress. Cardiovasc Res (2010) 88:360–6. doi: 10.1093/cvr/cvq191
46. Santer FR, Bacher N, Moser B, Morandell D, Ressler S, Firth SM, et al. Nuclear Insulin-Like Growth Factor Binding Protein-3 Induces Apoptosis and is Targeted to Ubiquitin/Proteasome-Dependent Proteolysis. Cancer Res (2006) 66:3024–33. doi: 10.1158/0008-5472.CAN-05-2013
47. Ikonen M, Liu B, Hashimoto Y, Ma L, Lee KW, Niikura T, et al. Interaction Between the Alzheimer's Survival Peptide Humanin and Insulin-Like Growth Factor-Binding Protein 3 Regulates Cell Survival and Apoptosis. Proc Natl Acad Sci U.S.A. (2003) 100:13042–7. doi: 10.1073/pnas.2135111100
48. Conte M, Sabbatinelli J, Chiariello A, Martucci M, Santoro A, Monti D, et al. Disease-Specific Plasma Levels of Mitokines FGF21, GDF15, and Humanin in Type II Diabetes and Alzheimer’s Disease in Comparison With Healthy Aging. Geroscience (2020) 43:985–1001. doi: 10.1007/s11357-020-00287-w
49. Niikura T, Hashimoto Y, Tajima H, Ishizaka M, Yamagishi Y, Kawasumi M, et al. A Tripartite Motif Protein TRIM11 Binds and Destabilizes Humanin, a Neuroprotective Peptide Against Alzheimer's Disease-Relevant Insults. Eur J Neurosci (2003) 17:1150–8. doi: 10.1046/j.1460-9568.2003.02553.x
50. Xiao J, Kim SJ, Cohen P, Yen K. Humanin: Functional Interfaces With IGF-I. Growth Horm IGF Res (2016) 29:21–7. doi: 10.1016/j.ghir.2016.03.005
51. Lee C, Wan J, Miyazaki B, Fang Y, Guevara-Aguirre J, Yen K, et al. IGF-I Regulates the Age-Dependent Signaling Peptide Humanin. Aging Cell (2014) 13:958–61. doi: 10.1111/acel.12243
52. Bodzioch M, Lapicka-Bodzioch K, Zapala B, Kamysz W, Kiec-Wilk B, Dembinska-Kiec A. Evidence for Potential Functionality of Nuclearly-Encoded Humanin Isoforms. Genomics (2009) 94:247–56. doi: 10.1016/j.ygeno.2009.05.006
53. Guo B, Zhai D, Cabezas E, Welsh K, Nouraini S, Satterthwait AC, et al. Humanin Peptide Suppresses Apoptosis by Interfering With Bax Activation. Nature (2003) 423:456–61. doi: 10.1038/nature01627
54. Jia Y, Lue YH, Swerdloff R, Lee KW, Cobb LJ, Cohen P, et al. The Cytoprotective Peptide Humanin is Induced and Neutralizes Bax After Pro-Apoptotic Stress in the Rat Testis. Andrology (2013) 1:651–9. doi: 10.1111/j.2047-2927.2013.00091.x
55. Ishikawa H, Tachikawa H, Miura Y, Takahashi N. TRIM11 Binds to and Destabilizes a Key Component of the Activator-Mediated Cofactor Complex (ARC105) Through the Ubiquitin-Proteasome System. FEBS Lett (2006) 580:4784–92. doi: 10.1016/j.febslet.2006.07.066
56. Zhao J, Zeng Y, Wang Y, Shi J, Zhao W, Wu B, et al. Humanin Protects Cortical Neurons From Calyculin A-induced Neurotoxicities by Increasing PP2A Activity and SOD. Int J Neurosci (2021), 131:527–35. doi: 10.1080/00207454.2020.1769617
57. Zhao ST, Huang XT, Zhang C, Ke Y. Humanin Protects Cortical Neurons From Ischemia and Reperfusion Injury by the Increased Activity of Superoxide Dismutase. Neurochem Res (2012) 37:153–60. doi: 10.1007/s11064-011-0593-0
58. Wang Y, Li N, Zeng Z, Tang L, Zhao S, Zhou F, et al. Humanin Regulates Oxidative Stress in the Ovaries of Polycystic Ovary Syndrome Patients Via the Keap1/Nrf2 Pathway. Mol Hum Reprod (2020) 27:gaaa081. doi: 10.1093/molehr/gaaa081
59. Ding Y, Feng Y, Zou Y, Wang F, Liu H, Liu C, et al. [Gly14]-humanin Restores Cathepsin D Function Via FPRL1 and Promotes Autophagic Degradation of Ox-LDL in Huvecs. Nutr Metab Cardiovasc Dis (2020) 30:2406–16. doi: 10.1016/j.numecd.2020.07.022
60. Zhu X, Zhao Z, Zeng C, Chen B, Huang H, Chen Y, et al. Hngf6a Inhibits Oxidative Stress-Induced Mc3t3-E1 Cell Apoptosis and Osteoblast Phenotype Inhibition by Targeting Circ_0001843/miR-214 Pathway. Calcif Tissue Int (2020) 106:518–32. doi: 10.1007/s00223-020-00660-z
61. Li W, Zhang D, Yuan W, Wang C, Huang Q, Luo J. Humanin Ameliorates Free Fatty Acid-Induced Endothelial Inflammation by Suppressing the NLRP3 Inflammasome. ACS Omega (2020) 5:22039–45. doi: 10.1021/acsomega.0c01778
62. Gao GS, Li Y, Zhai H, Bi JW, Zhang FS, Zhang XY, et al. Humanin Analogue, S14G-humanin, has Neuroprotective Effects Against Oxygen Glucose Deprivation/Reoxygenation by Reactivating Jak2/Stat3 Signaling Through the PI3K/AKT Pathway. Exp Ther Med (2017) 14:3926–34. doi: 10.3892/etm.2017.4934
63. Suzuki T, Motohashi H, Yamamoto M. Toward Clinical Application of the Keap1-Nrf2 Pathway. Trends Pharmacol Sci (2013) 34:340–6. doi: 10.1016/j.tips.2013.04.005
64. Warabi E, Takabe W, Minami T, Inoue K, Itoh K, Yamamoto M, et al. Shear Stress Stabilizes NF-E2-related Factor 2 and Induces Antioxidant Genes in Endothelial Cells: Role of Reactive Oxygen/Nitrogen Species. Free Radic Biol Med (2007) 42:260–9. doi: 10.1016/j.freeradbiomed.2006.10.043
65. Oishi T, Matsumaru D, Ota N, Kitamura H, Zhang T, Honkura Y, et al. Activation of the NRF2 Pathway in Keap1-Knockdown Mice Attenuates Progression of Age-Related Hearing Loss. NPJ Aging Mech Dis (2020) 6:14. doi: 10.1038/s41514-020-00053-4
66. Silva-Palacios A, Königsberg M, Zazueta C. Nrf2 Signaling and Redox Homeostasis in the Aging Heart: A Potential Target to Prevent Cardiovascular Diseases. Ageing Res Rev (2016) 26:81–95. doi: 10.1016/j.arr.2015.12.005
67. Zhao Q, Wang J, Levichkin IV, Stasinopoulos S, Ryan MT, Hoogenraad NJ. A Mitochondrial Specific Stress Response in Mammalian Cells. EMBO J (2002) 21:4411–9. doi: 10.1093/emboj/cdf445
68. Fiorese CJ, Haynes CM. Integrating the UPR(mt) Into the Mitochondrial Maintenance Network. Crit Rev Biochem Mol Biol (2017) 52:304–13. doi: 10.1080/10409238.2017.1291577
69. Houtkooper RH, Mouchiroud L, Ryu D, Moullan N, Katsyuba E, Knott G, et al. Mitonuclear Protein Imbalance as a Conserved Longevity Mechanism. Nature (2013) 497:451–7. doi: 10.1038/nature12188
70. Durieux J, Wolff S, Dillin A. The Cell-non-Autonomous Nature of Electron Transport Chain-Mediated Longevity. Cell (2011) 144:79–91. doi: 10.1016/j.cell.2010.12.016
71. Fiorese CJ, Schulz AM, Lin YF, Rosin N, Pellegrino MW, Haynes CM. The Transcription Factor ATF5 Mediates a Mammalian Mitochondrial Upr. Curr Biol (2016) 26:2037–43. doi: 10.1016/j.cub.2016.06.002
72. Quirós PM, Prado MA, Zamboni N, D’Amico D, Williams RW, Finley D, et al. Multi-Omics Analysis Identifies ATF4 as a Key Regulator of the Mitochondrial Stress Response in Mammals. J Cell Biol (2017) 216:2027–45. doi: 10.1083/jcb.201702058
73. Yong C, Tang BL. A Mitochondrial Encoded Messenger at the Nucleus. Cells (2018) 7:105. doi: 10.3390/cells7080105
74. Han K, Jia N, Zhong Y, Shang X. S14G-Humanin Alleviates Insulin Resistance and Increases Autophagy in Neurons of APP/PS1 Transgenic Mouse. J Cell Biochem (2018) 119:3111–7. doi: 10.1002/jcb.26452
75. Kageyama S, Gudmundsson SR, Sou YS, Ichimura Y, Tamura N, Kazuno S, et al. p62/SQSTM1-droplet Serves as a Platform for Autophagosome Formation and Anti-Oxidative Stress Response. Nat Commun (2021) 12:16. doi: 10.1038/s41467-020-20185-1
76. Agarraberes FA, Dice JF. A Molecular Chaperone Complex at the Lysosomal Membrane is Required for Protein Translocation. J Cell Sci (2001) 114:2491–9. doi: 10.1242/jcs.114.13.2491
77. Kaushik S, Bandyopadhyay U, Sridhar S, Kiffin R, Martinez-Vicente M, Kon M, et al. Chaperone-Mediated Autophagy at a Glance. J Cell Sci (2011) 124:495–9. doi: 10.1242/jcs.073874
78. Bandyopadhyay U, Kaushik S, Varticovski L, Cuervo AM. The Chaperone-Mediated Autophagy Receptor Organizes in Dynamic Protein Complexes at the Lysosomal Membrane. Mol Cell Biol (2008) 28:5747–63. doi: 10.1128/MCB.02070-07
79. Bandyopadhyay U, Sridhar S, Kaushik S, Kiffin R, Cuervo AM. Identification of Regulators of Chaperone-Mediated Autophagy. Mol Cell (2010) 39:535–47. doi: 10.1016/j.molcel.2010.08.004
80. Kiffin R, Christian C, Knecht E, Cuervo AM. Activation of Chaperone-Mediated Autophagy During Oxidative Stress. Mol Biol Cell (2004) 15:4829–40. doi: 10.1091/mbc.e04-06-0477
81. Zhang C, Cuervo AM. Restoration of Chaperone-Mediated Autophagy in Aging Liver Improves Cellular Maintenance and Hepatic Function. Nat Med (2008) 14:959–65. doi: 10.1038/nm.1851
82. Schneider JL, Villarroya J, Diaz-Carretero A, Patel B, Urbanska AM, Thi MM, et al. Loss of Hepatic Chaperone-Mediated Autophagy Accelerates Proteostasis Failure in Aging. Aging Cell (2015) 14:249–64. doi: 10.1111/acel.12310
83. Cuervo AM, Dice JF. Age-Related Decline in Chaperone-Mediated Autophagy. J Biol Chem (2000) 275:31505–13. doi: 10.1074/jbc.M002102200
84. Chander Y, Kumar R, Khandelwal N, Singh N, Shringi BN, Barua S, et al. Role of p38 Mitogen-Activated Protein Kinase Signalling in Virus Replication and Potential for Developing Broad Spectrum Antiviral Drugs. Rev Med Virol (2021) 15:2217. doi: 10.1002/rmv.2217
85. Choo AM, Geddes-Klein DM, Hockenberry A, Scarsella D, Mesfin MN, Singh P, et al. NR2A and NR2B Subunits Differentially Mediate MAP Kinase Signaling and Mitochondrial Morphology Following Excitotoxic Insult. Neurochem Int (2012) 60:506–16. doi: 10.1016/j.neuint.2012.02.007
86. Kong L, Zhu L, Yi X, Huang Y, Zhao H, Chen Y, et al. Betulinic Acid Alleviates Spleen Oxidative Damage Induced by Acute Intraperitoneal Exposure to T-2 Toxin by Activating Nrf2 and Inhibiting Mapk Signaling Pathways. Antioxid (Basel) (2021) 10:258. doi: 10.3390/antiox10020158
87. Takeshita Y, Hashimoto Y, Nawa M, Uchino H, Matsuoka M. SH3-Binding Protein 5 Mediates the Neuroprotective Effect of the Secreted Bioactive Peptide Humanin by Inhibiting C-Jun NH2-Terminal Kinase. J Biol Chem (2013) 288:24691–704. doi: 10.1074/jbc.M113.469692
88. Wang D, Li H, Yuan H, Zheng M, Bai C, Chen L, et al. Humanin Delays Apoptosis in K562 Cells by Downregulation of P38 MAP Kinase. Apoptosis (2005) 10:963–71. doi: 10.1007/s10495-005-1191-x
89. Hashimoto Y, Tsuji O, Niikura T, Yamagishi Y, Ishizaka M, Kawasumi M, et al. Involvement of C-Jun N-Terminal Kinase in Amyloid Precursor Protein-Mediated Neuronal Cell Death. J Neurochem (2003) 84:864–77. doi: 10.1046/j.1471-4159.2003.01585.x
90. Mihaylova MM, Shaw RJ. The AMPK Signalling Pathway Coordinates Cell Growth, Autophagy and Metabolism. Nat Cell Biol (2011) 13:1016–23. doi: 10.1038/ncb2329
91. Ming W, Lu G, Xin S, Huanyu L, Yinghao J, Xiaoying L, et al. Mitochondria Related Peptide MOTS-c Suppresses Ovariectomy-Induced Bone Loss Via AMPK Activation. Biochem Biophys Res Commun (2016) 476:412–9. doi: 10.1016/j.bbrc.2016.05.135
92. Rabinovitch RC, Samborska B, Faubert B, Ma EH, Gravel SP, Andrzejewski S, et al. Ampk Maintains Cellular Metabolic Homeostasis Through Regulation of Mitochondrial Reactive Oxygen Species. Cell Rep (2017) 21:1–9. doi: 10.1016/j.celrep.2017.09.026
93. Hinchy EC, Gruszczyk AV, Willows R, Navaratnam N, Hall AR, Bates G, et al. Mitochondria-Derived ROS Activate AMP-activated Protein Kinase (AMPK) Indirectly. J Biol Chem (2018) 293:17208–17. doi: 10.1074/jbc.RA118.002579
94. Wang Q, Zou MH. Measurement of Reactive Oxygen Species (ROS) and Mitochondrial ROS in AMPK Knockout Mice Blood Vessels. Methods Mol Biol (2018) 1732:507–17. doi: 10.1007/978-1-4939-7598-3_32
95. Lu Y, Zhou J, Xu C, Lin H, Xiao J, Wang Z, et al. JAK/STAT and PI3K/AKT Pathways Form a Mutual Transactivation Loop and Afford Resistance to Oxidative Stress-Induced Apoptosis in Cardiomyocytes. Cell Physiol Biochem (2008) 21:305–14. doi: 10.1159/000129389
96. Wu S, Xue J, Yang Y, Zhu H, Chen F, Wang J, et al. Isoliquiritigenin Inhibits Interferon-γ-Inducible Genes Expression in Hepatocytes Through Down-Regulating Activation of JAK1/STAT1, IRF3/Myd88, ERK/MAPK, JNK/MAPK and PI3K/Akt Signaling Pathways. Cell Physiol Biochem (2015) 37:501–14. doi: 10.1159/000430372
97. Ten VS, Starkov A. Hypoxic-Ischemic Injury in the Developing Brain: The Role of Reactive Oxygen Species Originating in Mitochondria. Neurol Res Int (2012) 2012:542976. doi: 10.1155/2012/542976
98. Cobb LJ, Lee C, Xiao J, Yen K, Wong RG, Nakamura HK, et al. Naturally Occurring Mitochondrial-Derived Peptides are Age-Dependent Regulators of Apoptosis, Insulin Sensitivity, and Inflammatory Markers. Aging (Albany NY) (2016) 8:796–809. doi: 10.18632/aging.100943
99. Widmer RJ, Flammer AJ, Herrmann J, Rodriguez-Porcel M, Wan J, Cohen P, et al. Circulating Humanin Levels are Associated With Preserved Coronary Endothelial Function. Am J Physiol Heart Circ Physiol (2013) 304:H393–7. doi: 10.1152/ajpheart.00765.2012
100. Zhloba AA, Subbotina TF, Molchan NS, Polushin YS. The Level of Circulating Humanin in Patients With Ischemic Heart Disease. Klin Lab Diagn (2018) 63:466–70. doi: 10.18821/0869-2084-2018-63-8-466-470
101. Zacharias DG, Kim SG, Massat AE, Bachar AR, Oh YK, Herrmann J, et al. Humanin, a Cytoprotective Peptide, is Expressed in Carotid Atherosclerotic [Corrected] Plaques in Humans. PloS One (2012) 7:e31065. doi: 10.1371/journal.pone.0031065
102. Zhao TC, Wang Z, Zhao TY. The Important Role of Histone Deacetylases in Modulating Vascular Physiology and Arteriosclerosis. Atherosclerosis (2020) 303:36–42. doi: 10.1016/j.atherosclerosis.2020.04.020
103. Robichaux WG 3rd, Mei FC, Yang W, Wang H, Sun H, Zhou Z, et al. Epac1 (Exchange Protein Directly Activated by cAMP 1) Upregulates Lox-1 (Oxidized Low-Density Lipoprotein Receptor 1) to Promote Foam Cell Formation and Atherosclerosis Development. Arterioscler Thromb Vasc Biol (2020) 40:e322–322e335. doi: 10.1161/ATVBAHA.119.314238
104. Zhu WW, Wang SR, Liu ZH, Cao YJ, Wang F, Wang J, et al. Gly[14]-Humanin Inhibits ox-LDL Uptake and Stimulates Cholesterol Efflux in Macrophage-Derived Foam Cells. Biochem Biophys Res Commun (2017) 482:93–9. doi: 10.1016/j.bbrc.2016.10.138
105. Kume N, Murase T, Moriwaki H, Aoyama T, Sawamura T, Masaki T, et al. Inducible Expression of Lectin-Like Oxidized LDL Receptor-1 in Vascular Endothelial Cells. Circ Res (1998) 83:322–7. doi: 10.1161/01.RES.83.3.322
106. Lu J, Yang JH, Burns AR, Chen HH, Tang D, Walterscheid JP, et al. Mediation of Electronegative Low-Density Lipoprotein Signaling by LOX-1: A Possible Mechanism of Endothelial Apoptosis. Circ Res (2009) 104:619–27. doi: 10.1161/CIRCRESAHA.108.190116
107. Wang X, Wu Z, He Y, Zhang H, Tian L, Zheng C, et al. Humanin Prevents High Glucose-Induced Monocyte Adhesion to Endothelial Cells by Targeting KLF2. Mol Immunol (2018) 101:245–50. doi: 10.1016/j.molimm.2018.07.008
108. Sharp TE 3rd, Gong Z, Scarborough A, Goetzman ES, Ali MJ, Spaletra P, et al. Efficacy of a Novel Mitochondrial-Derived Peptide in a Porcine Model of Myocardial Ischemia/Reperfusion Injury. JACC Basic Transl Sci (2020) 5:699–714. doi: 10.1016/j.jacbts.2020.04.015
109. Blasco N, Cámara Y, Núñez E, Beà A, Barés G, Forné C, et al. Cardiomyocyte Hypertrophy Induced by Endonuclease G Deficiency Requires Reactive Oxygen Radicals Accumulation and is Inhibitable by the Micropeptide Humanin. Redox Biol (2018) 16:146–56. doi: 10.1016/j.redox.2018.02.021
110. Kopacz A, Kloska D, Targosz-Korecka M, Zapotoczny B, Cysewski D, Personnic N, et al. Keap1 Governs Ageing-Induced Protein Aggregation in Endothelial Cells. Redox Biol (2020) 34:101572. doi: 10.1016/j.redox.2020.101572
111. Wei R, Enaka M, Muragaki Y. Activation of KEAP1/NRF2/P62 Signaling Alleviates High Phosphate-Induced Calcification of Vascular Smooth Muscle Cells by Suppressing Reactive Oxygen Species Production. Sci Rep (2019) 9:10366. doi: 10.1038/s41598-019-46824-2
112. Feng Q, Li Y, Lu X, Yu Y, Yuan G, Sun J, et al. Agaricus Blazei Polypeptide Exerts a Protective Effect on D-galactose-Induced Aging Mice Via the Keap1/Nrf2/ARE and P53/Trim32 Signaling Pathways. J Food Biochem (2021) 45:e13555. doi: 10.1111/jfbc.13555
113. Zhang L, Wang Y, Li C, Shao C, Zhou H, Yang J, et al. Dan Hong Injection Protects Against Cardiomyocytes Apoptosis by Maintaining Mitochondrial Integrity Through Keap1/Nuclear Factor Erythroid 2-Related Factor 2/Jnk Pathway. Front Pharmacol (2020) 11:591197. doi: 10.3389/fphar.2020.591197
114. Li C, Yu L, Xue H, Yang Z, Yin Y, Zhang B, et al. Nuclear AMPK Regulated CARM1 Stabilization Impacts Autophagy in Aged Heart. Biochem Biophys Res Commun (2017) 486:398–405. doi: 10.1016/j.bbrc.2017.03.053
115. Abdellatif M, Sedej S, Carmona-Gutierrez D, Madeo F, Kroemer G. Autophagy in Cardiovascular Aging. Circ Res (2018) 123:803–24. doi: 10.1161/CIRCRESAHA.118.312208
116. Alers S, Löffler AS, Wesselborg S, Stork B. Role of AMPK-mTOR-Ulk1/2 in the Regulation of Autophagy: Cross Talk, Shortcuts, and Feedbacks. Mol Cell Biol (2012) 32:2–11. doi: 10.1128/MCB.06159-11
117. Li X, Wu D, Tian Y. Fibroblast Growth Factor 19 Protects the Heart From Oxidative Stress-Induced Diabetic Cardiomyopathy Via Activation of AMPK/Nrf2/HO-1 Pathway. Biochem Biophys Res Commun (2018) 502:62–8. doi: 10.1016/j.bbrc.2018.05.121
118. Yang H, Feng A, Lin S, Yu L, Lin X, Yan X, et al. Fibroblast Growth factor-21 Prevents Diabetic Cardiomyopathy Via AMPK-Mediated Antioxidation and Lipid-Lowering Effects in the Heart. Cell Death Dis (2018) 9:227. doi: 10.1038/s41419-018-0307-5
119. Sun Y, Jiang C, Jiang J, Qiu L. Dexmedetomidine Protects Mice Against Myocardium Ischaemic/Reperfusion Injury by Activating an AMPK/PI3K/Akt/eNOS Pathway. Clin Exp Pharmacol Physiol (2017) 44:946–53. doi: 10.1111/1440-1681.12791
120. Wang W, Ye S, Zhang L, Jiang Q, Chen J, Chen X, et al. Granulocyte Colony-Stimulating Factor Attenuates Myocardial Remodeling and Ventricular Arrhythmia Susceptibility Via the JAK2-STAT3 Pathway in a Rabbit Model of Coronary Microembolization. BMC Cardiovasc Disord (2020) 20:85. doi: 10.1186/s12872-020-01385-5
121. Geng Z, Fan WY, Zhou B, Ye C, Tong Y, Zhou YB, et al. FNDC5 Attenuates Obesity-Induced Cardiac Hypertrophy by Inactivating JAK2/STAT3-associated Inflammation and Oxidative Stress. J Transl Med (2019) 17:107. doi: 10.1186/s12967-019-1857-8
122. Li L, Li M, Li Y, Sun W, Wang Y, Bai S, et al. Exogenous H2S Contributes to Recovery of Ischemic Post-Conditioning-Induced Cardioprotection by Decrease of ROS Level Via Down-Regulation of NF-κb and JAK2-STAT3 Pathways in the Aging Cardiomyocytes. Cell Biosci (2016) 6:26. doi: 10.1186/s13578-016-0090-x
123. Zhang J, Sun Z, Lin N, Lu W, Huang X, Weng J, et al. Fucoidan From Fucus Vesiculosus Attenuates Doxorubicin-Induced Acute Cardiotoxicity by Regulating JAK2/STAT3-Mediated Apoptosis and Autophagy. BioMed Pharmacother (2020) 130:110534. doi: 10.1016/j.biopha.2020.110534
124. Liu J, Fu H, Chang F, Wang J, Zhang S, Caudle Y, et al. Sodium Orthovanadate Suppresses Palmitate-Induced Cardiomyocyte Apoptosis by Regulation of the JAK2/STAT3 Signaling Pathway. Apoptosis (2016) 21:546–57. doi: 10.1007/s10495-016-1231-8
125. Feng W, Xu X, Zhao G, Zhao J, Dong R, Ma B, et al. Increased Age-Related Cardiac Dysfunction in Bradykinin B2 Receptor-Deficient Mice. J Gerontol A Biol Sci Med Sci (2016) 71:178–87. doi: 10.1093/gerona/glu210
126. Wei C, Zhao Y, Wang L, Peng X, Li H, Zhao Y, et al. H2 S Restores the Cardioprotection From Ischemic Post-Conditioning in Isolated Aged Rat Hearts. Cell Biol Int (2015) 39:1173–6. doi: 10.1002/cbin.10507
127. Li M, Ye J, Zhao G, Hong G, Hu X, Cao K, et al. Gas6 Attenuates Lipopolysaccharide−Induced TNF−α Expression and Apoptosis in H9C2 Cells Through NF−κb And MAPK Inhibition Via the Axl/PI3K/Akt Pathway. Int J Mol Med (2019) 44:982–94. doi: 10.3892/ijmm.2019.4275
128. Li L, Hao J, Jiang X, Li P, Sen H. Cardioprotective Effects of Ulinastatin Against Isoproterenol-Induced Chronic Heart Failure Through the PI3K−Akt, P38 MAPK and NF-κb Pathways. Mol Med Rep (2018) 17:1354–60. doi: 10.3892/mmr.2017.7934
Keywords: humanin, oxidative stress, aging-related cardiovascular diseases, redox signaling pathways, metabolic abnormalities
Citation: Cai H, Liu Y, Men H and Zheng Y (2021) Protective Mechanism of Humanin Against Oxidative Stress in Aging-Related Cardiovascular Diseases. Front. Endocrinol. 12:683151. doi: 10.3389/fendo.2021.683151
Received: 20 March 2021; Accepted: 21 May 2021;
Published: 10 June 2021.
Edited by:
Isaias Glezer, Federal University of São Paulo, BrazilReviewed by:
Jamal Bouitbir, Universität Basel, SwitzerlandKelvin Yen, University of Southern California, United States
Copyright © 2021 Cai, Liu, Men and Zheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yang Zheng, emhlbmd5YW5nQGpsdS5lZHUuY24=