 David C. D. Hope
David C. D. Hope Matthew L. Vincent
Matthew L. Vincent Tricia M. M. Tan
Tricia M. M. Tan- Division of Diabetes, Endocrinology and Metabolism, Department of Metabolism, Digestion and Reproduction, Imperial College London, London, United Kingdom
Obesity and Type 2 diabetes represent global health challenges, and there is an unmet need for long-lasting and effective pharmacotherapies. Although long-acting glucagon-like peptide-1 (GLP-1) analogues are now in routine use for diabetes and are now being utilised for obesity per se, the need for ever better treatments has driven the development of co-agonists, with the theoretical advantages of improved efficacy by targeting multiple pathways and reduced adverse effects. In this review, we highlight the past and present progress in our understanding and development of treatments based on GLP-1/glucagon co-agonism. We also reflect on the divergent effects of varying the GLP-1:glucagon activity and ratio in the context of pre-clinical and human clinical trial findings. In particular, the multiple metabolic actions of glucagon highlight the importance of understanding the contributions of individual hormone action to inform the safe, effective and tailored use of GLP-1/glucagon co-agonists to target weight loss and metabolic disease in the future.
Introduction – Glucagon as an Anti-Obesity Agent
Obesity is a leading cause of global morbidity and death. It is a driver of multiple co-morbidities such as type 2 diabetes (T2D), non-alcoholic fatty liver disease (NAFLD), hypertension, hypercholesterolaemia, cardiovascular disease and cancer (1). In 2016, 1.9 billion adults were classified as overweight and 650 million as obese according to the World Health Organisation in 2016. The current COVID-19 pandemic has also highlighted the strong link between obesity and poorer outcomes (2, 3). There is a growing health and socioeconomic burden of obesity, with an increasing demand for effective anti-obesity drugs ideally comparable to bariatric surgery, the current gold-standard for obesity treatment which offers highly effective, long-lasting and life-extending results (4). The gut hormone glucagon-like peptide-1 (GLP-1) and its analogues, which have been in clinical use for diabetes for over a decade, have useful appetite-suppressive effects and are now licensed for obesity. Despite the undeniable success of the GLP-1 analogues, there remains a ‘gap’ between the efficacy of GLP-1 analogues and that of bariatric surgery. To plug this gap, researchers have pursued the ‘co-agonist’ strategy by combining GLP-1 with related hormones from the proglucagon family and related peptides, including GIP and glucagon itself (5). By combining hormones in this way, the dose of individual hormones can be reduced, widening the therapeutic window and avoiding toxicity. In this mini-review, we highlight past and present progress in the translational research of GLP-1 and glucagon co-agonism. We also highlight the importance of striking a balance between GLP-1 and glucagon agonism, to allow for maximal drug efficacy whilst minimising potential risks.
Oxyntomodulin, a Natural GLP-1 and Glucagon Co-Agonist
The journey to discovery of the glucagon family of peptides and an endogenous GLP-1/glucagon co-agonist, oxyntomodulin (OXM) is an example of a concerted effort from many dedicated research groups. A pivotal point early on in this research was the use of the known peptide sequence of glucagon to facilitate the discovery of other ‘glucagon-like peptides’ in the gastrointestinal tract with the help of the radioimmunoassay method (6, 7). The search for ‘glucagon-like reactivity (GLI)’ in the gut revealed a partial peptide sequence for a peptide named ‘Glicentin’, later to be fully characterised as a 69-amino acid peptide containing a 30-amino acid ‘Glicentin-related pancreatic polypeptide’ (GRPP), the full sequence of glucagon, and an 8-amino acid c-terminal extension (8–10). The 8-amino acid extended glucagon fragment, ‘Glucagon-37’ was isolated from porcine jejuno-ileum, characterised and shown to be the bioactive ‘enteroglucagon’ due to its ability to bind to and stimulate glucagon receptors in liver membrane extracts (11–13). Due to the potent effect on oxyntic cell signalling, bioactive enteroglucagon/Glucagon-37 was named oxyntomodulin (14). As the primary structure of glicentin was discovered, evidence also emerged for post-translational processing of proglucagon to form glucagon and glicentin related pancreatic peptide, secreted ‘synchronously’ from the pancreatic alpha cell (10). Beyond the protein-based methodologies used to characterise glicentin, GRPP and oxyntomodulin, the increased capability to sequence genes at the time led to the first sequence of mammalian preproglucagon (15). This revealed two further glucagon-like polypeptides, now known as GLP-1 and GLP-2, and confirmed earlier reports suggesting that the MW of proglucagon is much larger than that of glicentin alone (16). The post-translational processing of pro-glucagon is now known to be differentially regulated in pancreas and gut (17, 18). Alternative processing of proglucagon leads to the formation of glucagon, GRPP and major pro-glucagon fragment (MPGF) in the pancreas, whereas in the gut and brain glicentin, GRPP, oxyntomodulin, GLP-1 and GLP-2 are formed (Figure 1). The endogenous gut hormone oxyntomodulin is therefore formed through specific splicing of the proglucagon gene and includes the full sequence of glucagon along with the 8-amino acid sequence named IP-1. This process occurs in the intestinal L cells of the gastrointestinal tract leading to the co-secretion of GLP-1 and oxyntomodulin in response to nutrient sensing (19, 20).
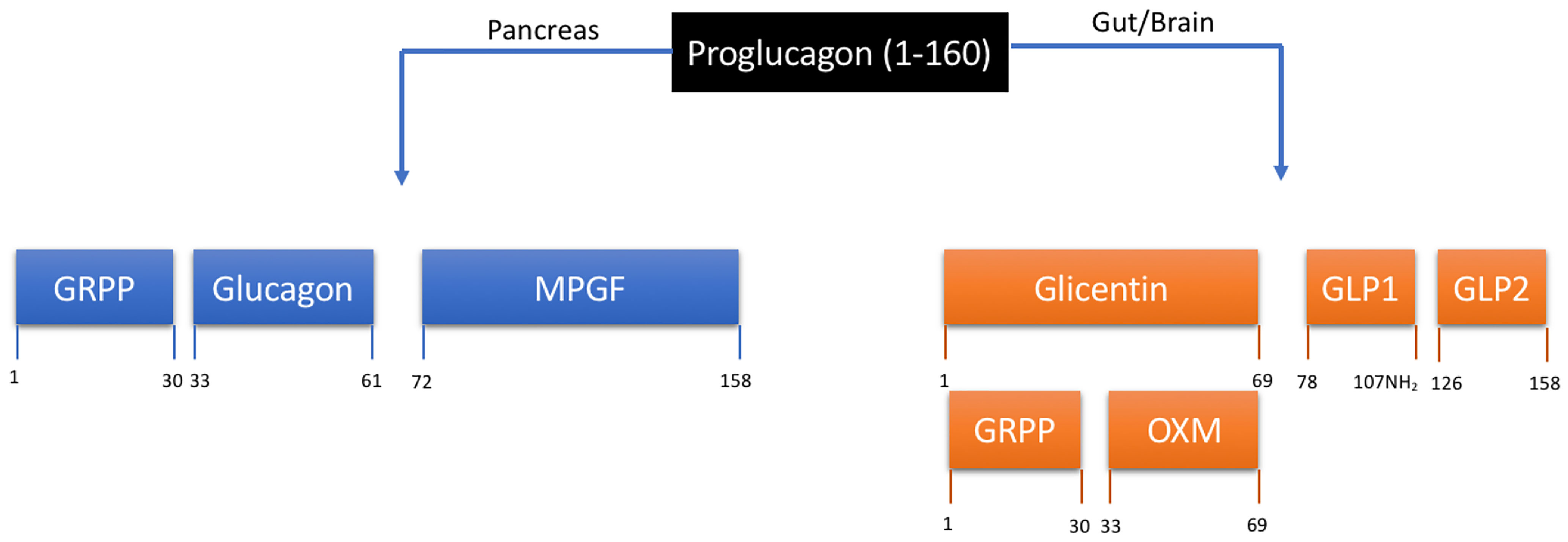
Figure 1 Tissue-specific processing of proglucagon. Proteolytic cleavage is tissue specific and regulated by prohormone convertases (PC) 1 and 2. In the pancreas, PC2 results in the formation of glucagon, glicentin-related pancreatic peptide (GRPP) and the inactive fusion protein major proglucagon fragment (MPGF). In the gut and brain, PC1/3 results in the formation of glicentin, GLP-1 and GLP-2. Glicentin is further processed to form GRPP and oxyntomodulin (OXM). Numerical annotations represent amino acid positions within the 160 amino acid proglucagon peptide.
Whilst much attention was focused on GLP-1 in the 1980s and 90s, following the discovery of its incretin effect in humans (21), little was known about the physiological role of oxyntomodulin in humans at the time (21, 22). Infusion studies in humans demonstrated a potent effect of high levels of OXM infusion on gastric emptying (23). Furthermore, changes in OXM were observed following intestinal bypass surgery suggesting anatomical changes influenced intestinal secretion of the peptide (24, 25). In the early 2000s, its potential role in weight loss was investigated following research showing that GLP-1 and glucagon inhibit food intake when administered intracerebroventricularly (ICV) in rodents (26, 27). ICV injection of OXM led to a significant food intake reduction up to 4 hours after injection, comparable to GLP-1 infusion (28). This effect was inhibited by the GLP-1 antagonist exendin (9–39), suggesting that OXM may act through the GLP-1 receptor (GLP-1R) to regulate food intake. Daily ICV injections of OXM over 7 days led to food intake reduction and increased weight loss compared with saline treated controls (29). Importantly, OXM-treated rats had increased weight loss compared to pair-fed controls and this was associated with a 0.5°C increase in core body temperature during the seven-day treatment period, suggesting for the first time an energy expenditure effect of OXM that is independent of its anorectic effects. Peripherally administered OXM led to a dose-dependent food intake reduction and increased weight loss in rats, and these animals lost significantly more body weight and white adipose tissue than pair-fed controls. ICV injection of exendin-9-39 attenuated the anorectic effect of peripheral OXM, further suggesting that OXM-mediated anorexia is produced through a central GLP-1R-dependent mechanism (30).
Concurrent to the pre-clinical studies, the first human study of oxyntomodulin was carried out in a small double-blind placebo-controlled crossover study (31). Thirteen healthy participants were given intravenous infusions of OXM at a dose of 3 pmol/kg/min for 90 minutes, with matched controls being given saline. During this infusion period, the ad-libitum food intake was significantly reduced in participants receiving OXM compared to saline control and a cumulative caloric intake reduction was observed up to 12 hours later. Later, a 28-day randomized controlled trial examined the effect of prolonged OXM injections on body weight loss and energy intake. Healthy overweight volunteers were randomised to receive pre-prandial subcutaneous injections with either saline or 400 nmol OXM three times daily, coincident with meals, for 4 weeks. OXM-treated subjects were found to have decreased energy intake on Days 2 (660 vs 508 kJ) and 29 (711 vs 428 kJ). At the end of the 28-day period, study subjects experienced significant weight loss (2.3 kg vs 0.5 kg) compared to saline treated controls in addition to concurrent changes in leptin and adiponectin suggesting reduction in adiposity (32).
Oxyntomodulin Activates Both GLP-1 and Glucagon Receptors Controlling Energy Balance and Glycaemia
Given OXM contains the entire glucagon sequence, it is no surprise that this peptide activates the glucagon receptor (13, 33). However, the potent effect on reducing weight gain in pre-clinical studies led to research efforts to determine the CNS binding site of OXM. OXM was shown to increase cAMP production in Baby Hamster Kidney (BHK) cells transfected with rodent GLP-1R and GCGR, suggesting meaningful signalling through both receptors but was less potent than Exendin-4 at the GLP-1R (34). The anorectic effects of centrally administered OXM were also abolished in Glp-1r-/- knock-out mice and not in Gcgr-/- mice, suggesting that the anorectic effects of OXM are mediated through GLP-1R. Consistent with these findings, previous in vitro studies showed that OXM is a full agonist at the GCGR and GLP-1R but is 3-fold less potent at the GCGR than native glucagon and 100-fold less potent at the GLP-1R than native GLP-1, in terms of activating cAMP accumulation (13).
Despite convincing data suggesting the central role of GLP-1R signalling in the food intake reduction effects of OXM, there were still some unexplained effects of the gut hormone including increased energy expenditure, suggesting a GLP-1R independent mechanism of action (35). The stimulatory effect of glucagon on thermogenesis in brown adipose tissue in vitro in addition to enhanced metabolic rate in humans had been previously shown (36, 37). Kosinski and colleagues constructed a variant of OXM, OXMQ3E, that was unable to activate the GCGR by changing the third amino acid residue from neutral glutamine to acidic glutamate (38). OXMQ3E produced less weight loss compared with OXM in diet-induced obese (DIO) mice despite similar food intake reduction. GCGR antagonism also reduced the weight loss effect of OXM, suggesting a role for GCGR mediated energy expenditure (38). Moreover, the OXMQ3E peptide was unable to produce any weight loss in Glp1r-/- mice, suggesting that both GLP-1R and GCGR are required for weight loss to occur. Importantly, OXM treatment led to several beneficial metabolic effects not seen in OXMQ3E treated mice including a decrease in plasma triglycerides and plasma cholesterol in addition to an increase in adiponectin. More recently, metabolic cage studies have confirmed that the increased oxygen consumption and energy expenditure effect of OXM is mediated through glucagon receptor signalling (29, 39, 40).
To characterise the effects of OXM on glucose metabolism, Du and colleagues performed hyperglycaemic clamp studies in mice treated with OXM and OXMQ3E (41). Both OXM and OXMQ3E improved glucose tolerance when given to DIO mice. The authors found the glucose infusion rate (GIR) during the clamp study decreased with OXM compared with OXMQ3E in wild-type mice, likely due to increased hepatic glucose output from GCGR agonism with OXM. Therefore, concurrent agonism at the GLP-1R and GCGR is important for the glucose-lowering effect of OXM: GLP-1R activation offsets the hyperglycaemia associated with GCGR activation. As we will discuss later, this further justified the development of synthetic GLP-1/glucagon co-agonists as promising weight loss therapeutics, particularly in the context of avoiding unwanted hyperglycaemia in obesity-associated type 2 diabetes.
Rationale for GLP-1/Glucagon Combination Therapy for Weight Loss
Although significant progress was made in understanding the potential use of the endogenous GLP-1/glucagon co-agonist OXM as a weight loss therapeutic in the early 2000s, evidence from various pre-clinical and human studies prior to this demonstrated the distinct mechanisms of weight loss afforded by these hormones. GLP-1, secreted from the intestinal L cells postprandially, acts both centrally and peripherally to exert its anorectic and metabolic effects. ICV GLP-1 reduces food intake in rodents, and this effect is blocked with Exendin (9–39). It is now known that GLP-1 receptors are widely expressed in the hypothalamus, hindbrain and amygdala with a neuronal link between the periphery and CNS to regulate GLP-1 action (42). In addition to the effect on food intake reduction and glucose-stimulated insulin secretion, GLP-1 also acts to delay gastric emptying therefore aiding satiety and glycaemic control (42). Given at higher doses than in diabetes treatment, GLP-1 analogues are now routinely licensed for the treatment of obesity, for example high-dose Liraglutide 3 mg daily (43) and Semaglutide 2.4 mg weekly (44). However, despite the progress in the development of GLP-1 analogues, the overall efficacy of GLP-1 analogues for weight loss is still limited by gastrointestinal side effects, in particular nausea at higher doses (45).
Glucagon is typically stimulated by the fall in insulin during fasting in concert with low glucose levels, and responds to hypoglycaemia by mobilising hepatic glucose through the stimulation of glycogenolysis and gluconeogenesis (46). In T2D, loss of the suppressive effects of glucagon and insulin is associated with hyperglucagonaemia and this is thought to contribute to hyperglycaemia (47–50). Based on evidence for the contribution of hyperglucagonemia to hyperglycaemia in T2D, research demonstrated the use of glucagon antagonism to improve glycaemic control. In their seminal study, Peterson and Sullivan utilised a novel non-peptide glucagon receptor antagonist (Bay 27-9955) in healthy, lean males to investigate effects on glycaemia (51). Following oral administration of Bay 27-9955 at two doses, a significant blunting of acute hyperglycaemia was observed following a glucagon infusion, in the absence of clinical side effects. Following the promising results of Bay 27-9955, several glucagon receptor antagonists have been developed however when administered over a longer period, have been met with several adverse effects including increased plasma alanine aminotransferase (ALT), LDL-cholesterol in addition to increased blood pressure and body weight (52). Furthermore, treatment with the glucagon antagonist, LY2409021 over 6 months led to a significant increase in both ALT and hepatic fat fraction (HFF) compared to sitagliptin and placebo groups (53). The development of hepatic steatosis is likely due to blockade of glucagon’s lipolytic properties in the liver, therefore making GCGR antagonism an untenable strategy. Despite its glucose mobilising effect, the broad catabolic and thermogenic nature of glucagon receptor agonism adds to its attractive portfolio as a weight loss therapeutic. The enhanced metabolic rate observed with glucagon administration was shown early on in rodents and humans (36, 40). Glucagon-induced brown adipose tissue (BAT) thermogenesis has been demonstrated in rodents (37, 54). However this seems to be species specific: BAT activation is not observed in humans with a glucagon infusion (55). Futile cycling of glucose has also been suggested to confer glucagon’s energy expenditure effects, whereby glucagon stimulates opposing pathways of hepatic glucose production and consumption (56). Circulating FGF-21 has also been implicated in energy expenditure effects with chronic glucagon agonism, as Fgf21-/- knock-out mice are protected from these effects (57). Despite several postulated theories of glucagon induced energy expenditure, the mechanism is likely multi-faceted and the precise contribution to these facets remains uncertain (58). Glucagon is also known to act peripherally to enhance lipolysis in white adipose tissue and improve whole body lipid metabolism (59, 60). In particular, glucagon has firmly been demonstrated to enhance hepatic lipid metabolism, Hepatic GCGR agonism leads to upregulation of lipid catabolism pathways in the hepatocyte where a number of key regulatory transporters and enzymes facilitate beta oxidation of fatty acids (61). Furthermore, exogenous administration of glucagon was shown early on to inhibit food intake in humans and rodents (62, 63). Interestingly, this effect seems to be mediated through hepatic glucagon signalling, as infusion into the portal vein induced a satiating response whereas infusion into the inferior vena cava did not (64). The combination of GLP-1 and glucagon administration in rodents has been shown to increase c-Fos expression in appetite regulating centres and polypharmacy with these hormones leads to a synergistic effect on food intake reduction over single hormone administration (65).
Given the pre-existing data on GLP-1 and glucagon, we designed a double-blinded randomised cross-over study, in which we gave volunteers who were overweight short-term intravenous infusions of glucagon (50 ng/kg/min or 14 pmol/kg/min), GLP-1 (0.8 pmol/kg/min), combination of glucagon + GLP-1 at the same doses, or placebo over 45 minutes, and demonstrated that there was a significant increase in resting energy expenditure with glucagon alone which was preserved with the combination of glucagon + GLP-1, whereas GLP-1 did not affect energy expenditure (66). As expected, glucagon infusion caused an increase in glucose which was largely neutralised by co-infusion with GLP-1. In a follow up study with lower, sub-anorectic doses of GLP-1 (0.4 pmol/kg/min), glucagon (2.8 pmol/kg/min), GLP-1 + glucagon combination at the same doses, or placebo for 120 minutes we showed that there was a 13% decrease in food intake after the combination, with the individual infusions having no significant effect (67), further supporting the concept of co-agonism with these hormones. Another study did not show any differences with the hormone combination on glucose and food intake reduction or enhanced energy expenditure but used far lower doses of GLP-1 (1 pmol/kg/min) and glucagon (0.86 pmol/kg/min) (68). Overall, these physiological studies support the notion that GLP-1 and glucagon possess dose-dependent synergism, leading to enhanced suppression of food intake, plus increased resting energy expenditure.
Pre-Clinical Studies of Synthetic GLP-1/Glucagon Co-Agonists
Concurrent to research investigating combination treatments with individual GLP-1 and glucagon infusions, efforts were also focused on optimising the peptide chemistry of native oxyntomodulin, glucagon or GLP-1 in view of designing receptor potent and long-acting synthetic GLP-1/GCG co-agonists. While the results from studies using native OXM in rodents and humans were promising, the short half-life in vivo and the large amount of peptide required to produce an effect made it a poor choice as a treatment in humans. As such, synthetic GLP-1/GCG co-agonists resistant to dipeptidyl peptidase-4 (DPP-4) proteolysis became an attractive target for anti-obesity treatments. Due to the uncertainty regarding the optimum balance of GLP-1R and GCGR agonism in a unimolecular co-agonist, development of novel peptides varied from sequence modification and enhancing stability of native OXM, sequence modification of the glucagon peptide to confer increased GLP-1R potency or modification of GLP-1 peptide to confer increased GCGR potency; the natural advantage of this concept was due to the sequence similarity across the glucagon family of peptides.
By modifying the OXM peptide sequence with an amino acid-peptide substitution at position 2 to prevent DPP-4 action, in addition to a cholesterol-peptide conjugate, Pocai and colleagues were the first group to show that synthetic dual-agonists were an effective treatment strategy in an animal model. They showed that administration of their modified long-acting OXM peptide led to a decrease in food intake and increased weight loss in addition to improved metabolic profile, superior to GLP-1 alone over 14 days in diet-induced obese (DIO) mice (69). Further studies by a variety of groups demonstrated structural modifications of OXM to enhance the longevity of the native peptide through modification with polyethylene glycol (PEGylation), fatty acid conjugation and amino acid substitutions (70–73). By modifying the primary sequence of OXM and linking to the constant region of human IgG4, Jung and colleagues demonstrated their peptide HM12525A had a potent and balanced activity at both GLP-1 and glucagon receptors in vitro and led to body weight loss of 30% in DIO mice over 14 days compared to liraglutide treated mice (74). Enhanced energy expenditure was also observed with HM12525A in addition to reduced adiposity and improved liver function in a NASH model in db/db mice. HM12525A has been variously re-designated as JNJ-64565111 and now efinopegdutide, and this drug has been taken forward into Phase 2 trials (see below).
Day and colleagues were the first group to use a series of modifications to the C-terminal portion of the glucagon sequence to increase GLP-1R potency, combined with PEGylation to generate two PEGylated peptides with adequate co-agonist properties, one unbalanced towards GLP-1R, and the other a ‘near-balanced’ agonist at both GLP-1R and GCGR. The near-balanced peptide was shown to be ~2-fold less potent at GLP-1R and ~10 fold less potent at GCGR than native ligands in cAMP synthesis (75). When given to DIO mice, both peptides led to significant weight reduction, improved glucose tolerance, increased energy expenditure, and reductions in plasma cholesterol and liver steatosis. These effects were more dramatic with the balanced agonist and interestingly the beneficial metabolic effects occurred with no change in oral nutrient intake in peptide vs control groups. This study demonstrated the importance of combined GLP-1R and GCGR signalling, as administration of the balanced co-agonist to Glp1r-/- mice led to hyperglycaemia, further demonstrating the necessity of GLP-1R and GCGR co-agonism to minimise this predicted side-effect. The same group further assessed various ratios of GCGR to GLP-1R potency of their peptides, on the extent of weight loss while minimising hyperglycaemia (76). Importantly, the authors demonstrated that the peptides which were most able to produce weight loss without hyperglycaemia demonstrated balanced potency at the GLP-1 and glucagon receptors.
Other groups have also modified the primary sequence of glucagon to confer increased GLP-1R potency. MEDI0832 (now known as cotadutide) is a balanced co-agonist based on the peptide sequence of glucagon, modified at specific amino acid positions in addition to a palmitic fatty acid side chain to prolong activity. The novel peptide is biased towards GLP-1R agonism versus GCGR with a 3-4 fold reduced potency at the GLP-1R compared to native GLP-1 and around an 8-fold reduced potency at the GCGR compared to native glucagon. Chronic daily administration of cotadutide over 27 days in obese mice was associated with food intake reduction, decreased adiposity, improved fasting glucose, and increased energy expenditure. Consistent with previous studies, cotadutide outperformed GLP-1 alone (40 nmol/kg/day liraglutide) in achieving maximal weight loss in obese mice, and this was attributed to the increased energy expenditure (77). Cotadutide has been taken forward in clinical trial development (see below).
An alternative approach in the design of effective synthetic co-agonists has been to modify GLP-1 analogues to confer increased GCG activity. By engineering the C-terminal portion of Exendin-4 to include amino acid sequences from glucagon in addition to a fatty acid side chain, Evers and colleagues developed a potent balanced co-agonist ‘peptide 14’. This was shown to be around 10 times less potent at the human GLP-1R and approximately equivalent potency at GCGR in comparison to the native peptides at eliciting cAMP response in vitro. Daily administration of peptide 14 in DIO mice over 32 days led to a 30% body weight loss above that seen for liraglutide alone at 15% body weight loss. In db/db mice, peptide 14 prevented a 1.5% increase in HbA1C over 32 days seen in the vehicle control treated group (78). Based on these pre-clinical findings, a lead candidate, SAR425899 with similar receptor potencies was taken forward in Phase 1 and Phase 2 trials (see below).
Novel strategies have also been employed to enhance the pharmacokinetic profile of GLP-1/glucagon analogues in view of prolongation of drug effect. Recently, a surfactant conjugated co-agonist peptide ‘17’ was shown to have a half-life of 52 hours in vivo and at a higher dose led to 40% body weight loss in obese rats over 27 days (79). This has subsequently been taken forward into Phase 1 clinical trials as ALT-801 (Table 1).
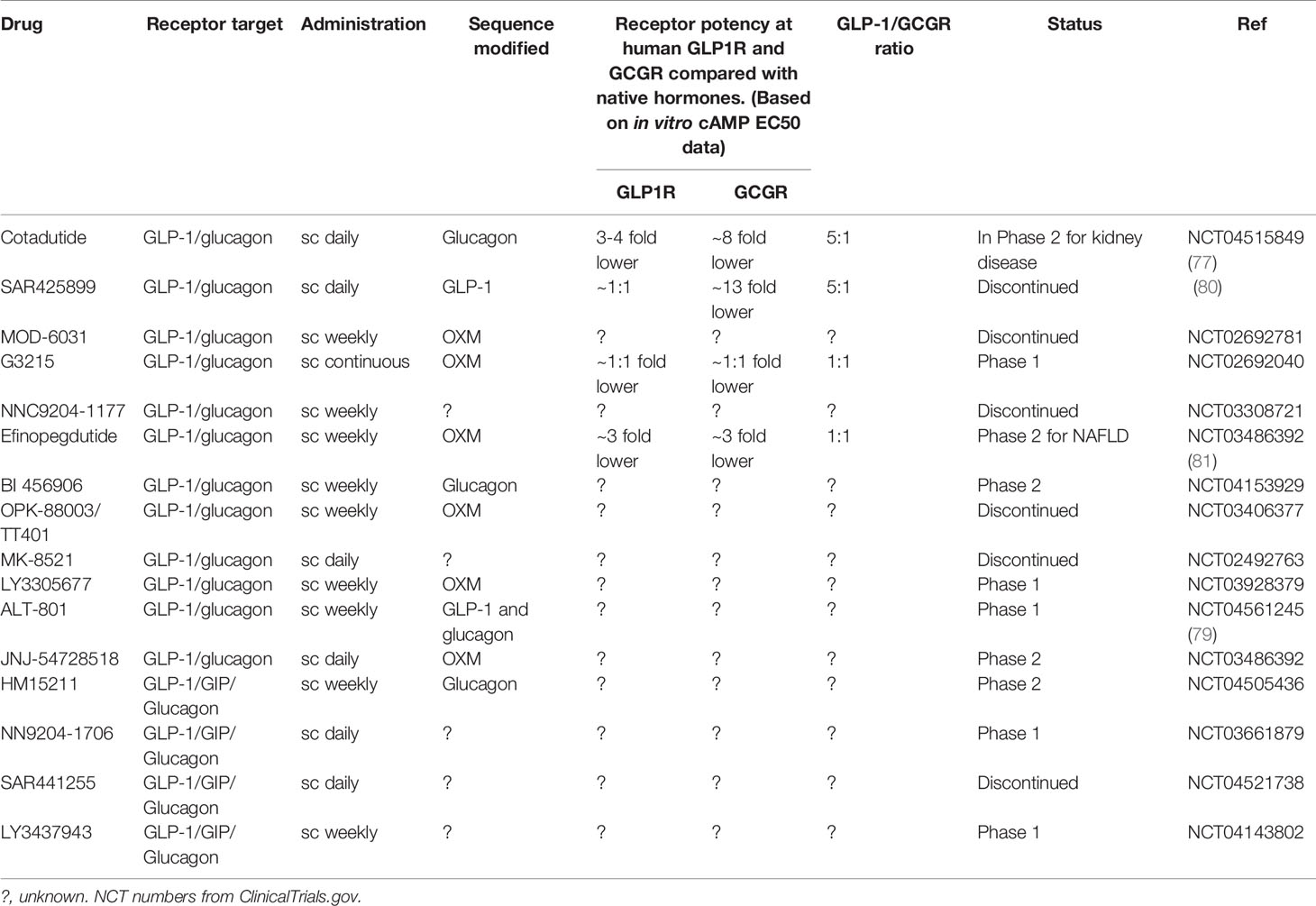
Table 1 Current glucagon containing anti-obesity drugs in development.
Lessons From GLP-1/Glucagon Co-Agonists in Clinical Development
The first GLP-1/glucagon co-agonist to advance to human clinical trials was cotadutide (MEDI0832). In a randomised, placebo-controlled double blinded phase 1 study of ascending single doses in healthy overweight humans, cotadutide was shown to be safe and, in common with the GLP-1 analogues, to be associated with dose-dependent gastrointestinal adverse events especially nausea and vomiting. As an exploratory outcome, single doses of cotadutide led to dose-dependent improvement in glucose excursions post meals within 24 hours and food intake reduction from a single dose of 100 μg. Doses as low as 10 μg had a beneficial effect on post meal glucose excursions and there was no evidence of glucagon-induced hyperglycaemia (82). In a combined multiple ascending dose (MAD) and Phase 2a study in people with type 2 diabetes over 41 days, the safety of cotadutide was confirmed (83). Over 41 days, daily doses of cotadutide of up to 200 μg led to improved glucose AUC0-4h after a mixed meal in comparison to placebo as well as fasting and post prandial glucose levels. Cotadutide also led to 2.1 kg body weight loss relative to placebo. In a separate follow up Phase 2a study over 49 days, the mechanism of improved glycaemia was shown to be a combination of enhanced insulin secretion and delayed gastric emptying (84).
An important exploratory outcome measured in the initial 41-day Phase 2a study was the effect of cotadutide treatment on liver fat reduction as measured by MRI, with a 39.12% reduction was observed in the treatment group compared to 19.51% in placebo group (83). This significant reduction in liver fat content with cotadutide is likely to be due to hepatic glucagon signalling and subsequent upregulation of fatty acid oxidation. In a Phase 2b study in people with type 2 diabetes and obesity over 54 weeks, treatment with cotadutide at doses of 100-300 μg daily improved some non-invasive markers of NAFLD such as transaminase levels, the FIB-4 index, fatty liver disease fibrosis score (NFS) and fatty liver index (FLI) but fatty liver disease was not assessed directly with liver biopsy. This study also included an open-label comparator arm where participants took liraglutide 1.8 mg. Improvements in HbA1c were shown to be similar with reductions of 1.03-1.19% with cotadutide vs 1.17% with liraglutide. With respect to body weight, cotadutide at 200 μg led to weight loss of 3.22 kg on average versus 3.33 kg with liraglutide, but the 300 μg dose led to weight loss of 5.02 kg albeit with far higher gastrointestinal adverse event rates than liraglutide (85).
Although cotadutide has progressed well through clinical trials, other GLP-1/glucagon co-agonists have shown mixed results. A recent Phase 2 randomised placebo-controlled trial tested efinopegdutide in people with obesity over a 26-week period, where participants were randomly assigned weekly treatment with either placebo, 5 mg, 7.4 mg, 10 mg efinopegdutide, or 3 mg liraglutide daily. Participants given efinopegdutide showed a dose-dependent increase in body weight loss of 6.7 to 10.0% (placebo subtracted). Participants taking liraglutide achieved a placebo subtracted weight loss of 5.8% in line with clinical experience. Although there was no significant improvement in glycaemia with efinopegdutide, this was explicable given that the participants had normal glycaemia at baseline. However, up to 89% of participants taking efinopegdutide experienced gastrointestinal adverse events (mostly nausea, vomiting and diarrhoea) relative to 28% taking placebo and 60% taking liraglutide (86). This may be because the efinopegdutide arms did not have a dose titration phase unlike the liraglutide arm (86). The original development partner, Janssen Pharmaceuticals, has handed back the license for efinopegdutide to the original developer, Hanmi Pharmaceutical, but the license has been taken up by Merck to be developed as a once weekly treatment for NAFLD.
SAR425899 (Sanofi) is another example of a GLP-1/glucagon co-agonist which has been tested in Phase 1 trials. When tested as a once-daily injection in single and multiple doses varying up to 0.18 mg in healthy normal to overweight volunteers, gastrointestinal adverse events (nausea, diarrhoea, constipation, vomiting) were encountered, but the drug was described as well tolerated. The multiple-dose regimen, given for up to 4 weeks, led to a dose-dependent weight loss between 2.87 and 5.46 kg, compared to 2.37 kg for placebo. In a small group with T2D, SAR425899 improved fasting glucose and glucose tolerance after a mixed meal (80). Results from Phase 2 trials were subsequently halted due to excessive rates of gastrointestinal adverse events leading to participant withdrawals. It is unlikely therefore this drug will proceed further in development.
Several other multi-agonists capable of co-agonism of the GLP-1 and glucagon receptors are currently in development including BI 456906 (Boehringer Ingelheim), LY3305677 (Lilly) LY4347943 (Lilly), JNJ-54729518 (J&J), HM15211 (Hanmi), NNC9204-1706 (Novo), Alt-801 (Altimmune) and G3215 (Imperial College/Zihipp Ltd.) – see Table 1. Led by the results of cotadutide, GLP-1/glucagon co-agonists have been shown to have promising weight loss and glycaemic effects in these early phase clinical results however data from later phase clinical trials are expected later this year/early next year. Data from longer-term treatment will therefore be eagerly awaited to determine the extent of body weight loss and metabolic outcomes and whether this is comparable to the latest GLP-1 mono-agonist therapies.
Striking the Balance of GLP-1 and Glucagon Agonism to Minimise Potential Risks
Based on outcomes from safety and pharmacokinetic studies with GLP-1 analogues, the most common side effect observed with GLP-1 agonism is dose-dependent nausea and vomiting (45). A theoretical advantage of a co-agonist approach is the ability to reduce the dose of GLP-1 whilst also enhancing glucagon’s weight loss effects. As the current development leader, cotadutide has still not been able to escape the spectre of dose-dependent gastrointestinal adverse events (82). Fortunately, a tolerated treatment dose window of 150 μg daily or less of cotadutide was associated with fewer adverse effects and this facilitated its progress to Phase 2 where its marked beneficial metabolic effects were observed.
Even with careful engineering of receptor balance, potency and pharmacokinetics, problems may still crop up, as illustrated by SAR425899’s unexpected development failure due to excessive gastrointestinal adverse events during its Phase 2 trials. A follow up study using radio-ligand PET technology to measure receptor occupancy demonstrated a high degree of GLP-1R occupancy but no detectable GCGR occupancy suggesting that in vivo SAR425899 may be acting in effect as a GLP-1 analogue and not a co-agonist (87). The higher frequency of gastrointestinal side effects also observed with efinopegutide in Phase 2 trials may also be explained by a relatively high receptor potency of at GCGR and GLP-1R, within 3-fold of the native ligands (81). Further data from other drug candidates (Table 1) in early phase trials is awaited to determine any divergent effects on gastrointestinal side effects.
A key initial concern of using glucagon within a co-agonist was unwanted hyperglycaemia. So far, the co-agonist drugs which have reached phase 2 trials have demonstrated an improved glycaemic profile with chronic administration. There is recent evidence that hepatic glucagon receptor stimulation may improve insulin stimulated glucose disposal. In a series of acute studies using euglycemic clamps, Kim and colleagues demonstrated that a glucagon agonist IUB288 leads to improved glucose tolerance by augmenting insulin action with evidence of increased hepatic AKT phosphorylation (88). Low dose glucagon agonism may therefore enhance insulin sensitivity which is in keeping with pre-prandial physiology in the fasted setting where the body is prepared to metabolise essential nutrients.
With chronic administration of glucagon-containing co-agonists, the catabolism of lean mass (i.e. protein and amino acids) becomes an important consideration. Direct evidence for the effect of glucagon on lean mass is seen from clinical situations of glucagon excess, in the glucagonoma syndrome (89). Despite the demonstrable importance of surveillance of lean mass during testing of obesity drug candidates, it is common for only fat mass loss or total body weight loss to be measured or presented in pre-clinical and early clinical trials. Furthermore, plasma amino acid levels are not routinely measured or reported. Exceptionally, in a Phase 2a study of cotadutide, individual plasma amino acid profiles were reported; after 49 days of treatment a significant reduction in plasma alanine was observed (84). However, with stronger glucagon receptor stimulation, it would be important for further pre-clinical studies to characterise the effects of lead drug candidates, on plasma amino acids and lean mass. Furthermore, with the increasing prominence of sarcopenic obesity, preserving lean mass in any weight loss strategy is important and this will be an area of increasing clinical and research interest.
Future Perspectives/Conclusions
In the search for an anti-obesity pharmacotherapeutic which can rival the weight loss effects of bariatric surgery, research and development of gut hormone co-agonists is gaining momentum. GLP-1/glucagon co-agonists such as cotadutide and efinopegdutide offer the promise of increased efficacy whilst minimising side effects. Furthermore, with enhanced glucagon action the GLP-1/glucagon co-agonist has the advantage of being tailored to treat NAFLD directly.
However, the field is not staying still, and recently published data from the high-dose semaglutide STEP Phase 3 trials (44) and the SURPASS Phase 3 trials of the GLP-1/glucose-dependent insulinotropic peptide (GIP) co-agonist tirzepatide (90) have set the efficacy bar high in terms of weight loss and glycaemic improvement. We therefore conclude that before the place of GLP-1/glucagon co-agonists within the therapeutic armamentarium can be defined, future research efforts need to address the following outstanding questions:
1. What is the optimal balance and receptor potency of GLP-1 and glucagon in a long-acting co-agonist, to minimise adverse effects and to optimise efficacy (Figure 2)?
2. Does GLP-1/glucagon co-agonism offer long-lasting and enhanced efficacy for lowering blood glucose over that of GLP-1 analogues alone?
3. What are the optimal drug characteristics of a GLP-1/glucagon co-agonist for the treatment of NAFLD via hepatic GCGR agonism?
4. Are there long-term effects on lean mass with the co-agonists, and if so, can this be prevented?
5. Will the co-agonists inherit the favourable effects of GLP-1 analogues on prevention of cardiovascular events and progression of kidney disease?
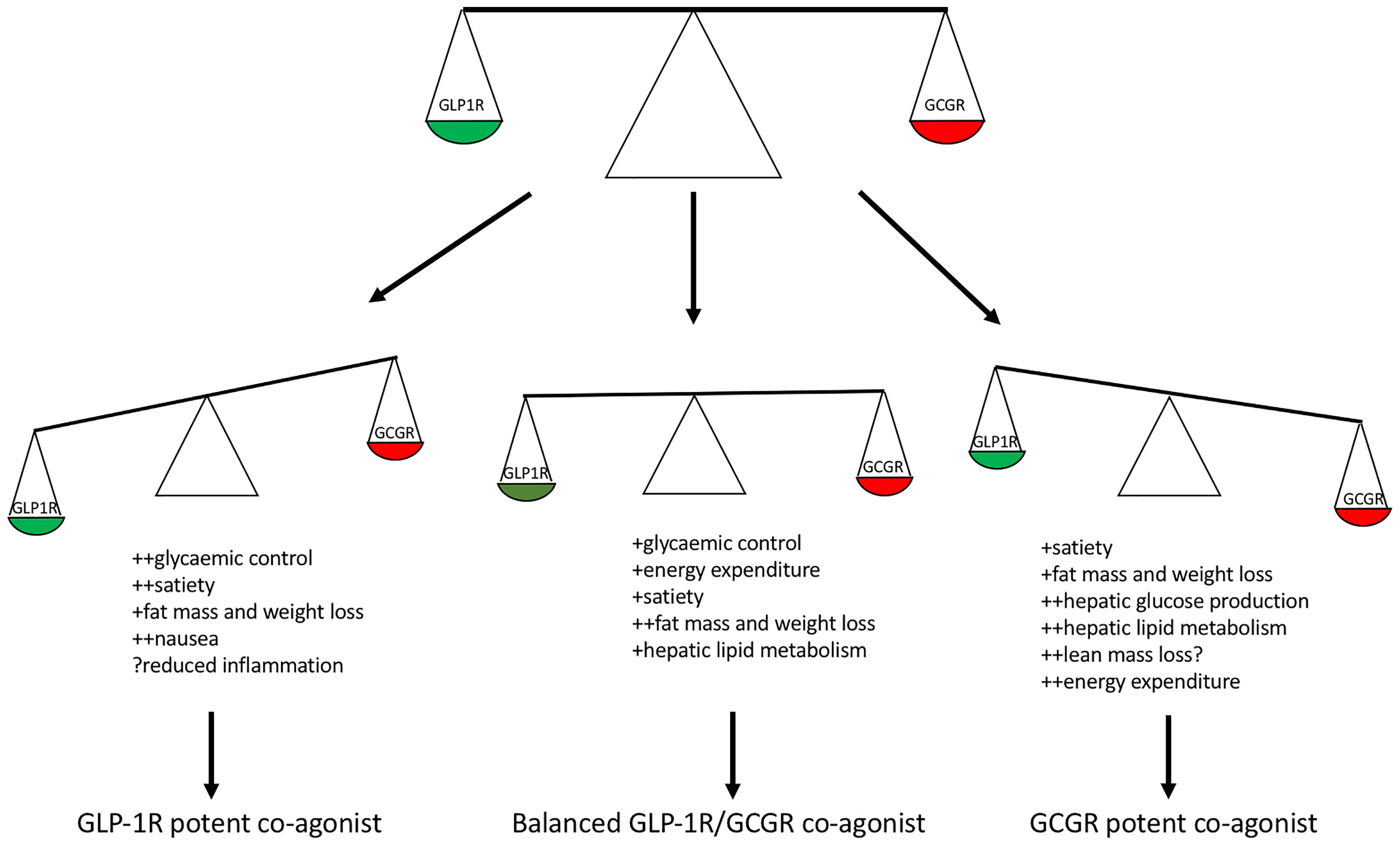
Figure 2 Balance of GLP-1R and GCGR potency within a co-agonist and predicted clinical effects. Glucagon receptor potent co-agonists predicted to result in enhanced weight loss due to increased energy expenditure however possible loss of lean mass and increased hepatic glucose production. GLP-1R potent co-agonists predicted to confer enhanced weight loss and glycaemic control with risk of gastrointestinal side effects. A balanced GLP-1R/GCGR co-agonist with respect to in vitro cAMP stimulation predicted to enhance glycaemic control and healthy fat mass loss.
Author Contributions
All authors contributed to the article and approved the submitted version.
Funding
DCDH is funded by a Medical Research Council Clinical Research Training Fellowship (MR/S02171X/1). MLV is supported by the NIHR Biomedical Research Centre.
Conflict of Interest
TMMT is a shareholder and consultant for Zihipp Ltd., which is developing gut hormone analogues for treatment of metabolic disease.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Bluher M. Obesity: Global Epidemiology and Pathogenesis. Nat Rev Endocrinol (2019) 15(5):288–98. doi: 10.1038/s41574-019-0176-8
2. Liu D, Zhang T, Wang Y, Xia L. The Centrality of Obesity in the Course of Severe COVID-19. Front Endocrinol (Lausanne) (2021) 12:620566. doi: 10.3389/fendo.2021.620566
3. Popkin BM, Du S, Green WD, Beck MA, Algaith T, Herbst CH, et al. Individuals With Obesity and COVID-19: A Global Perspective on the Epidemiology and Biological Relationships. Obes Rev (2020) 21(11):e13128. doi: 10.1111/obr.13128
4. Maciejewski ML, Arterburn DE, Van Scoyoc L, Smith VA, Yancy WS Jr., Weidenbacher HJ, et al. Bariatric Surgery and Long-Term Durability of Weight Loss. JAMA Surg (2016) 151(11):1046–55. doi: 10.1001/jamasurg.2016.2317
5. Sanchez-Garrido MA, Brandt SJ, Clemmensen C, Muller TD, DiMarchi RD, Tschop MH. GLP-1/Glucagon Receptor Co-Agonism for Treatment of Obesity. Diabetologia (2017) 60(10):1851–61. doi: 10.1007/s00125-017-4354-8
6. Unger RH, Ohneda A, Valverde I, Eisentraut AM, Exton J. Characterization of the Responses of Circulating Glucagon-Like Immunoreactivity to Intraduodenal and Intravenous Administration of Glucose. J Clin Invest (1968) 47(1):48–65. doi: 10.1172/JCI105714
7. Unger RH, Eisentraut AM, Mc CM, Madison LL. Glucagon Antibodies and an Immunoassay for Glucagon. J Clin Invest (1961) 40:1280–9. doi: 10.1172/JCI104357
8. Holst JJ. Evidence That Glicentin Contains the Entire Sequence of Glucagon. Biochem J (1980) 187(2):337–43. doi: 10.1042/bj1870337
9. Sundby F, Jacobsen H, Moody AJ. Purification and Characterization of a Protein From Porcine Gut With Glucagon-Like Immunoreactivity. Horm Metab Res (1976) 8(5):366–71. doi: 10.1055/s-0028-1093615
10. Moody AJ, Holst JJ, Thim L, Jensen SL. Relationship of Glicentin to Proglucagon and Glucagon in the Porcine Pancreas. Nature (1981) 289(5797):514–6. doi: 10.1038/289514a0
11. Bataille D, Jarrousse C, Kervran A, Depigny C, Dubrasquet M. The Biological Significance of “Enteroglucagon”. Present Status. Peptides (1986) 7:37–42. doi: 10.1016/0196-9781(86)90161-0
12. Bataille D, Tatemoto K, Coudray AM, Rosselin G, Mutt V. Bioactive “Enteroglucagon” (Oxyntomodulin): Evidence for a C-Terminal Extension of the Glucagon Molecule. C R Seances Acad Sci III (1981) 293(6):323–8.
13. Bataille D, Coudray AM, Carlqvist M, Rosselin G, Mutt V. Isolation of Glucagon-37 (Bioactive Enteroglucagon/Oxyntomodulin) From Porcine Jejuno-Ileum. Isolation of the Peptide. FEBS Lett (1982) 146(1):73–8. doi: 10.1016/0014-5793(82)80708-4
14. Bataille D, Gespach C, Coudray AM, Rosselin G. “Enterolglucagon’: A Specific Effect on Gastric Glands Isolated From the Rat Fundus. Evidence for an “Oxyntomodulin” Action. Biosci Rep (1981) 1(2):151–5. doi: 10.1007/BF01117012
15. Bell GI, Santerre RF, Mullenbach GT. Hamster Preproglucagon Contains the Sequence of Glucagon and Two Related Peptides. Nature (1983) 302(5910):716–8. doi: 10.1038/302716a0
16. Patzelt C, Tager HS, Carroll RJ, Steiner DF. Identification and Processing of Proglucagon in Pancreatic Islets. Nature (1979) 282(5736):260–6. doi: 10.1038/282260a0
17. Mojsov S, Heinrich G, Wilson IB, Ravazzola M, Orci L, Habener JF. Preproglucagon Gene Expression in Pancreas and Intestine Diversifies at the Level of Post-Translational Processing. J Biol Chem (1986) 261(25):11880–9. doi: 10.1016/S0021-9258(18)67324-7
18. Orskov C, Holst JJ, Knuhtsen S, Baldissera FG, Poulsen SS, Nielsen OV. Glucagon-Like Peptides GLP-1 and GLP-2, Predicted Products of the Glucagon Gene, Are Secreted Separately From Pig Small Intestine But Not Pancreas. Endocrinology (1986) 119(4):1467–75. doi: 10.1210/endo-119-4-1467
19. Wewer Albrechtsen NJ, Hornburg D, Albrechtsen R, Svendsen B, Torang S, Jepsen SL, et al. Oxyntomodulin Identified as a Marker of Type 2 Diabetes and Gastric Bypass Surgery by Mass-Spectrometry Based Profiling of Human Plasma. EBioMedicine (2016) 7:112–20. doi: 10.1016/j.ebiom.2016.03.034
20. Dhanvantari S, Seidah NG, Brubaker PL. Role of Prohormone Convertases in the Tissue-Specific Processing of Proglucagon. Mol Endocrinol (1996) 10(4):342–55. doi: 10.1210/mend.10.4.8721980
21. Kreymann B, Williams G, Ghatei MA, Bloom SR. Glucagon-Like Peptide-1 7-36: A Physiological Incretin in Man. Lancet (1987) 2(8571):1300–4. doi: 10.1016/S0140-6736(87)91194-9
22. Holst JJ. Enteroglucagon. Annu Rev Physiol (1997) 59:257–71. doi: 10.1146/annurev.physiol.59.1.257
23. Schjoldager B, Mortensen PE, Myhre J, Christiansen J, Holst JJ. Oxyntomodulin From Distal Gut. Role in Regulation of Gastric and Pancreatic Functions. Dig Dis Sci (1989) 34(9):1411–9. doi: 10.1007/BF01538078
24. Holst JJ, Sorensen TI, Andersen AN, Stadil F, Andersen B, Lauritsen KB, et al. Plasma Enteroglucagon After Jejunoileal Bypass With 3:1 or 1:3 Jejunoileal Ratio. Scand J Gastroenterol (1979) 14(2):205–7. doi: 10.3109/00365527909179871
25. Sarson DL, Scopinaro N, Bloom SR. Gut Hormone Changes After Jejunoileal (JIB) or Biliopancreatic (BPB) Bypass Surgery for Morbid Obesity. Int J Obes (1981) 5(5):471–80.
26. Turton MD, O’Shea D, Gunn I, Beak SA, Edwards CM, Meeran K, et al. A Role for Glucagon-Like Peptide-1 in the Central Regulation of Feeding. Nature (1996) 379(6560):69–72. doi: 10.1038/379069a0
27. Tang-Christensen M, Larsen PJ, Goke R, Fink-Jensen A, Jessop DS, Moller M, et al. Central Administration of GLP-1-(7-36) Amide Inhibits Food and Water Intake in Rats. Am J Physiol (1996) 271(4 Pt 2):R848–56. doi: 10.1152/ajpregu.1996.271.4.R848
28. Dakin CL, Gunn I, Small CJ, Edwards CM, Hay DL, Smith DM, et al. Oxyntomodulin Inhibits Food Intake in the Rat. Endocrinology (2001) 142(10):4244–50. doi: 10.1210/endo.142.10.8430
29. Dakin CL, Small CJ, Park AJ, Seth A, Ghatei MA, Bloom SR. Repeated ICV Administration of Oxyntomodulin Causes a Greater Reduction in Body Weight Gain Than in Pair-Fed Rats. Am J Physiol Endocrinol Metab (2002) 283(6):E1173–7. doi: 10.1152/ajpendo.00233.2002
30. Dakin CL, Small CJ, Batterham RL, Neary NM, Cohen MA, Patterson M, et al. Peripheral Oxyntomodulin Reduces Food Intake and Body Weight Gain in Rats. Endocrinology (2004) 145(6):2687–95. doi: 10.1210/en.2003-1338
31. Cohen MA, Ellis SM, Le Roux CW, Batterham RL, Park A, Patterson M, et al. Oxyntomodulin Suppresses Appetite and Reduces Food Intake in Humans. J Clin Endocrinol Metab (2003) 88(10):4696–701. doi: 10.1210/jc.2003-030421
32. Wynne K, Park AJ, Small CJ, Patterson M, Ellis SM, Murphy KG, et al. Subcutaneous Oxyntomodulin Reduces Body Weight in Overweight and Obese Subjects: A Double-Blind, Randomized, Controlled Trial. Diabetes (2005) 54(8):2390–5. doi: 10.2337/diabetes.54.8.2390
33. Baldissera FG, Holst JJ, Knuhtsen S, Hilsted L, Nielsen OV. Oxyntomodulin (Glicentin-(33-69)): Pharmacokinetics, Binding to Liver Cell Membranes, Effects on Isolated Perfused Pig Pancreas, and Secretion From Isolated Perfused Lower Small Intestine of Pigs. Regul Pept (1988) 21(1-2):151–66. doi: 10.1016/0167-0115(88)90099-7
34. Baggio LL, Huang Q, Brown TJ, Drucker DJ. Oxyntomodulin and Glucagon-Like Peptide-1 Differentially Regulate Murine Food Intake and Energy Expenditure. Gastroenterology (2004) 127(2):546–58. doi: 10.1053/j.gastro.2004.04.063
35. Pocai A. Action and Therapeutic Potential of Oxyntomodulin. Mol Metab (2014) 3(3):241–51. doi: 10.1016/j.molmet.2013.12.001
36. Nair KS. Hyperglucagonemia Increases Resting Metabolic Rate in Man During Insulin Deficiency. J Clin Endocrinol Metab (1987) 64(5):896–901. doi: 10.1210/jcem-64-5-896
37. Billington CJ, Bartness TJ, Briggs J, Levine AS, Morley JE. Glucagon Stimulation of Brown Adipose Tissue Growth and Thermogenesis. Am J Physiol (1987) 252(1 Pt 2):R160–5. doi: 10.1152/ajpregu.1987.252.1.R160
38. Kosinski JR, Hubert J, Carrington PE, Chicchi GG, Mu J, Miller C, et al. The Glucagon Receptor Is Involved in Mediating the Body Weight-Lowering Effects of Oxyntomodulin. Obes (Silver Spring) (2012) 20(8):1566–71. doi: 10.1038/oby.2012.67
39. Scott R, Minnion J, Tan T, Bloom SR. Oxyntomodulin Analogue Increases Energy Expenditure via the Glucagon Receptor. Peptides (2018) 104:70–7. doi: 10.1016/j.peptides.2018.04.008
40. Davidson IW, Salter JM, Best CH. Calorigenic Action of Glucagon. Nature (1957) 180(4595):1124. doi: 10.1038/1801124a0
41. Du X, Kosinski JR, Lao J, Shen X, Petrov A, Chicchi GG, et al. Differential Effects of Oxyntomodulin and GLP-1 on Glucose Metabolism. Am J Physiol Endocrinol Metab (2012) 303(2):E265–71. doi: 10.1152/ajpendo.00142.2012
42. Sandoval DA, D’Alessio DA. Physiology of Proglucagon Peptides: Role of Glucagon and GLP-1 in Health and Disease. Physiol Rev (2015) 95(2):513–48. doi: 10.1152/physrev.00013.2014
43. Pi-Sunyer X, Astrup A, Fujioka K, Greenway F, Halpern A, Krempf M, et al. A Randomized, Controlled Trial of 3.0 Mg of Liraglutide in Weight Management. N Engl J Med (2015) 373(1):11–22. doi: 10.1056/NEJMoa1411892
44. Wilding JPH, Batterham RL, Calanna S, Davies M, Van Gaal LF, Lingvay I, et al. Once-Weekly Semaglutide in Adults With Overweight or Obesity. N Engl J Med (2021) 384(11):989. doi: 10.1056/NEJMoa2032183
45. Filippatos TD, Panagiotopoulou TV, Elisaf MS. Adverse Effects of GLP-1 Receptor Agonists. Rev Diabetes Stud (2014) 11(3-4):202–30. doi: 10.1900/RDS.2014.11.202
46. Habegger KM, Heppner KM, Geary N, Bartness TJ, DiMarchi R, Tschop MH. The Metabolic Actions of Glucagon Revisited. Nat Rev Endocrinol (2010) 6(12):689–97. doi: 10.1038/nrendo.2010.187
47. Unger RH, Orci L. The Essential Role of Glucagon in the Pathogenesis of Diabetes Mellitus. Lancet (1975) 1(7897):14–6. doi: 10.1016/S0140-6736(75)92375-2
48. Cryer PE. Minireview: Glucagon in the Pathogenesis of Hypoglycemia and Hyperglycemia in Diabetes. Endocrinology (2012) 153(3):1039–48. doi: 10.1210/en.2011-1499
49. Haedersdal S, Lund A, Knop FK, Vilsboll T. The Role of Glucagon in the Pathophysiology and Treatment of Type 2 Diabetes. Mayo Clin Proc (2018) 93(2):217–39. doi: 10.1016/j.mayocp.2017.12.003
50. Magnusson I, Rothman DL, Katz LD, Shulman RG, Shulman GI. Increased Rate of Gluconeogenesis in Type II Diabetes Mellitus. A 13C Nuclear Magnetic Resonance Study. J Clin Invest (1992) 90(4):1323–7. doi: 10.1172/JCI115997
51. Petersen KF, Sullivan JT. Effects of a Novel Glucagon Receptor Antagonist (Bay 27-9955) on Glucagon-Stimulated Glucose Production in Humans. Diabetologia (2001) 44(11):2018–24. doi: 10.1007/s001250100006
52. Cheng C, Jabri S, Taoka BM, Sinz CJ. Small Molecule Glucagon Receptor Antagonists: An Updated Patent Review (2015-2019). Expert Opin Ther Pat (2020) 30(7):509–26. doi: 10.1080/13543776.2020.1769600
53. Guzman CB, Zhang XM, Liu R, Regev A, Shankar S, Garhyan P, et al. Treatment With LY2409021, a Glucagon Receptor Antagonist, Increases Liver Fat in Patients With Type 2 Diabetes. Diabetes Obes Metab (2017) 19(11):1521–8. doi: 10.1111/dom.12958
54. Joel CD. Stimulation of Metabolism of Rat Brown Adipose Tissue by Addition of Lipolytic Hormones In Vitro. J Biol Chem (1966) 241(4):814–21. doi: 10.1016/S0021-9258(18)96838-9
55. Salem V, Izzi-Engbeaya C, Coello C, Thomas DB, Chambers ES, Comninos AN, et al. Glucagon Increases Energy Expenditure Independently of Brown Adipose Tissue Activation in Humans. Diabetes Obes Metab (2016) 18(1):72–81. doi: 10.1111/dom.12585
56. Miyoshi H, Shulman GI, Peters EJ, Wolfe MH, Elahi D, Wolfe RR. Hormonal Control of Substrate Cycling in Humans. J Clin Invest (1988) 81(5):1545–55. doi: 10.1172/JCI113487
57. Habegger KM, Stemmer K, Cheng C, Muller TD, Heppner KM, Ottaway N, et al. Fibroblast Growth Factor 21 Mediates Specific Glucagon Actions. Diabetes (2013) 62(5):1453–63. doi: 10.2337/db12-1116
58. Kleinert M, Sachs S, Habegger KM, Hofmann SM, Muller TD. Glucagon Regulation of Energy Expenditure. Int J Mol Sci (2019) 20(21):5407. doi: 10.3390/ijms20215407
59. Perea A, Clemente F, Martinell J, Villanueva-Penacarrillo ML, Valverde I. Physiological Effect of Glucagon in Human Isolated Adipocytes. Horm Metab Res (1995) 27(8):372–5. doi: 10.1055/s-2007-979981
60. Slavin BG, Ong JM, Kern PA. Hormonal Regulation of Hormone-Sensitive Lipase Activity and mRNA Levels in Isolated Rat Adipocytes. J Lipid Res (1994) 35(9):1535–41. doi: 10.1016/S0022-2275(20)41151-4
61. Galsgaard KD, Pedersen J, Knop FK, Holst JJ, Wewer Albrechtsen NJ. Glucagon Receptor Signaling and Lipid Metabolism. Front Physiol (2019) 10:413. doi: 10.3389/fphys.2019.00413
62. Schulman JL, Carleton JL, Whitney G, Whitehorn JC. Effect of Glucagon on Food Intake and Body Weight in Man. J Appl Physiol (1957) 11(3):419–21. doi: 10.1152/jappl.1957.11.3.419
63. Geary N, Smith GP. Pancreatic Glucagon and Postprandial Satiety in the Rat. Physiol Behav (1982) 28(2):313–22. doi: 10.1016/0031-9384(82)90081-6
64. Geary N, Le Sauter J, Noh U. Glucagon Acts in the Liver to Control Spontaneous Meal Size in Rats. Am J Physiol (1993) 264(1 Pt 2):R116–22. doi: 10.1152/ajpregu.1993.264.1.R116
65. Parker JA, McCullough KA, Field BC, Minnion JS, Martin NM, Ghatei MA, et al. Glucagon and GLP-1 Inhibit Food Intake and Increase C-Fos Expression in Similar Appetite Regulating Centres in the Brainstem and Amygdala. Int J Obes (Lond) (2013) 37(10):1391–8. doi: 10.1038/ijo.2012.227
66. Tan TM, Field BC, McCullough KA, Troke RC, Chambers ES, Salem V, et al. Coadministration of Glucagon-Like Peptide-1 During Glucagon Infusion in Humans Results in Increased Energy Expenditure and Amelioration of Hyperglycemia. Diabetes (2013) 62(4):1131–8. doi: 10.2337/db12-0797
67. Cegla J, Troke RC, Jones B, Tharakan G, Kenkre J, McCullough KA, et al. Coinfusion of Low-Dose GLP-1 and Glucagon in Man Results in a Reduction in Food Intake. Diabetes (2014) 63(11):3711–20. doi: 10.2337/db14-0242
68. Bagger JI, Holst JJ, Hartmann B, Andersen B, Knop FK, Vilsboll T. Effect of Oxyntomodulin, Glucagon, GLP-1, and Combined Glucagon +GLP-1 Infusion on Food Intake, Appetite, and Resting Energy Expenditure. J Clin Endocrinol Metab (2015) 100(12):4541–52. doi: 10.1210/jc.2015-2335
69. Pocai A, Carrington PE, Adams JR, Wright M, Eiermann G, Zhu L, et al. Glucagon-Like Peptide 1/Glucagon Receptor Dual Agonism Reverses Obesity in Mice. Diabetes (2009) 58(10):2258–66. doi: 10.2337/db09-0278
70. Santoprete A, Capito E, Carrington PE, Pocai A, Finotto M, Langella A, et al. DPP-IV-Resistant, Long-Acting Oxyntomodulin Derivatives. J Pept Sci (2011) 17(4):270–80. doi: 10.1002/psc.1328
71. Bianchi E, Carrington PE, Ingallinella P, Finotto M, Santoprete A, Petrov A, et al. A PEGylated Analog of the Gut Hormone Oxyntomodulin With Long-Lasting Antihyperglycemic, Insulinotropic and Anorexigenic Activity. Bioorg Med Chem (2013) 21(22):7064–73. doi: 10.1016/j.bmc.2013.09.016
72. Druce MR, Minnion JS, Field BC, Patel SR, Shillito JC, Tilby M, et al. Investigation of Structure-Activity Relationships of Oxyntomodulin (Oxm) Using Oxm Analogs. Endocrinology (2009) 150(4):1712–22. doi: 10.1210/en.2008-0828
73. Kerr BD, Flatt PR, Gault VA. (D-Ser2)Oxm[mPEG-PAL]: A Novel Chemically Modified Analogue of Oxyntomodulin With Antihyperglycaemic, Insulinotropic and Anorexigenic Actions. Biochem Pharmacol (2010) 80(11):1727–35. doi: 10.1016/j.bcp.2010.08.010
74. Jung SY, Park YH, Kang JH, Trautmann M, Hompesch M, Kwon SC. Lipolytic and Insulinotropic Effects of HM12525A, A Novel Long-Acting GLP-1/Glucagon Dual Agonist. (2014). Presented at EASD Meeting, 2014. Available at: https://www.easd.org/virtualmeeting/home.html#!resources/lipolytic-and-insulinotropic-effects-of-hm12525a-a-novel-long-acting-glp-1-glucagon-dual-agonist (Accessed 30 Aug 2021).
75. Day JW, Ottaway N, Patterson JT, Gelfanov V, Smiley D, Gidda J, et al. A New Glucagon and GLP-1 Co-Agonist Eliminates Obesity in Rodents. Nat Chem Biol (2009) 5(10):749–57. doi: 10.1038/nchembio.209
76. Day JW, Gelfanov V, Smiley D, Carrington PE, Eiermann G, Chicchi G, et al. Optimization of Co-Agonism at GLP-1 and Glucagon Receptors to Safely Maximize Weight Reduction in DIO-Rodents. Biopolymers (2012) 98(5):443–50. doi: 10.1002/bip.22072
77. Henderson SJ, Konkar A, Hornigold DC, Trevaskis JL, Jackson R, Fritsch Fredin M, et al. Robust Anti-Obesity and Metabolic Effects of a Dual GLP-1/Glucagon Receptor Peptide Agonist in Rodents and non-Human Primates. Diabetes Obes Metab (2016) 18(12):1176–90. doi: 10.1111/dom.12735
78. Evers A, Haack T, Lorenz M, Bossart M, Elvert R, Henkel B, et al. Design of Novel Exendin-Based Dual Glucagon-Like Peptide 1 (GLP-1)/Glucagon Receptor Agonists. J Med Chem (2017) 60(10):4293–303. doi: 10.1021/acs.jmedchem.7b00174
79. Nestor JJ, Zhang X, Jaw-Tsai S, Parkes DG, Becker CK. Design and Characterization of a Surfactant-Conjugated, Long-Acting, Balanced GLP-1/Glucagon Receptor Dual Agonist. Pept Sci (2021) n/a(n/a):e24221. doi: 10.1002/pep2.24221
80. Tillner J, Posch MG, Wagner F, Teichert L, Hijazi Y, Einig C, et al. A Novel Dual Glucagon-Like Peptide and Glucagon Receptor Agonist SAR425899: Results of Randomized, Placebo-Controlled First-in-Human and First-in-Patient Trials. Diabetes Obes Metab (2019) 21(1):120–8. doi: 10.1111/dom.13494
81. Jung SY, Kim JS, Choi IY, Lee GH, Kim YH, Kang JH, et al. The Novel Long Acting GLP-1/Glucagon Dual Agonist HM12525A Reduces Body Weight and Improves Glycermic Control in Rodent Models. (2013). Presented at Scientific Sessions of the ADA Meeting (2013). Available at: http://www.hanmi.co.kr/ehanmi/rnd/HM12525A/1.2013%20ADA_Poster_HM12525A.pdf (Accessed 30 Aug 2021).
82. Ambery PD, Klammt S, Posch MG, Petrone M, Pu W, Rondinone C, et al. MEDI0382, A GLP-1/Glucagon Receptor Dual Agonist, Meets Safety and Tolerability Endpoints in a Single-Dose, Healthy-Subject, Randomized, Phase 1 Study. Br J Clin Pharmacol (2018) 84(10):2325–35. doi: 10.1111/bcp.13688
83. Ambery P, Parker VE, Stumvoll M, Posch MG, Heise T, Plum-Moerschel L, et al. MEDI0382, a GLP-1 and Glucagon Receptor Dual Agonist, in Obese or Overweight Patients With Type 2 Diabetes: A Randomised, Controlled, Double-Blind, Ascending Dose and Phase 2a Study. Lancet (2018) 391(10140):2607–18. doi: 10.1016/S0140-6736(18)30726-8
84. Parker VER, Robertson D, Wang T, Hornigold DC, Petrone M, Cooper AT, et al. Efficacy, Safety, and Mechanistic Insights of Cotadutide, a Dual Receptor Glucagon-Like Peptide-1 and Glucagon Agonist. J Clin Endocrinol Metab (2020) 105(3):803–20. doi: 10.1210/clinem/dgz047
85. Nahra R, Wang T, Gadde KM, Oscarsson J, Stumvoll M, Jermutus L, et al. Effects of Cotadutide on Metabolic and Hepatic Parameters in Adults With Overweight or Obesity and Type 2 Diabetes: A 54-Week Randomized Phase 2b Study. Diabetes Care (2021) 44(6):1433–42. doi: 10.2337/figshare.14272955.v1
86. Alba M, Yee J, Frustaci ME, Samtani MN, Fleck P. Efficacy and Safety of Glucagon-Like Peptide-1/Glucagon Receptor Co-Agonist JNJ-64565111 in Individuals With Obesity Without Type 2 Diabetes Mellitus: A Randomized Dose-Ranging Study. Clin Obes (2021) 11(2):e12432. doi: 10.1111/cob.12432
87. Eriksson O, Haack T, Hijazi Y, Teichert L, Tavernier V, Laitinen I, et al. Receptor Occupancy of Dual Glucagon-Like Peptide 1/Glucagon Receptor Agonist SAR425899 in Individuals With Type 2 Diabetes. Sci Rep (2020) 10(1):16758. doi: 10.1038/s41598-020-73815-5
88. Kim T, Holleman CL, Nason S, Arble DM, Ottaway N, Chabenne J, et al. Hepatic Glucagon Receptor Signaling Enhances Insulin-Stimulated Glucose Disposal in Rodents. Diabetes (2018) 67(11):2157–66. doi: 10.2337/db18-0068
89. Mallinson CN, Bloom SR, Warin AP, Salmon PR, Cox B. A Glucagonoma Syndrome. Lancet (1974) 2(7871):1–5. doi: 10.1016/S0140-6736(74)91343-9
90. Rosenstock J, Wysham C, Frias JP, Kaneko S, Lee CJ, Fernandez Lando L, et al. Efficacy and Safety of a Novel Dual GIP and GLP-1 Receptor Agonist Tirzepatide in Patients With Type 2 Diabetes (SURPASS-1): A Double-Blind, Randomised, Phase 3 Trial. Lancet (2021) 398(10295):143–55. doi: 10.1016/S0140-6736(21)01324-6
Keywords: glucagon, GLP-1, obesity, co-agonist, weight loss
Citation: Hope DCD, Vincent ML and Tan TMM (2021) Striking the Balance: GLP-1/Glucagon Co-Agonism as a Treatment Strategy for Obesity. Front. Endocrinol. 12:735019. doi: 10.3389/fendo.2021.735019
Received: 01 July 2021; Accepted: 18 August 2021;
Published: 08 September 2021.
Edited by:
Peter Flatt, Ulster University, United KingdomReviewed by:
Victor Alan Gault, Ulster University, United KingdomSteven Patterson, Glasgow Caledonian University, United Kingdom
Copyright © 2021 Hope, Vincent and Tan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tricia M. M. Tan, dC50YW5AaW1wZXJpYWwuYWMudWs=