 Matthew R. Brown
Matthew R. Brown Aleksey V. Matveyenko
Aleksey V. Matveyenko- 1Department of Physiology and Biomedical Engineering, Mayo Clinic College of Medicine and Science, Rochester, MN, United States
- 2Division of Endocrinology, Metabolism, Diabetes, and Nutrition, Department of Medicine, Mayo Clinic College of Medicine and Science, Rochester, MN, United States
Our ever-changing modern environment is a significant contributor to the increased prevalence of many chronic diseases, and particularly, type 2 diabetes mellitus (T2DM). Although the modern era has ushered in numerous changes to our daily living conditions, changes in “what” and “when” we eat appear to disproportionately fuel the rise of T2DM. The pancreatic islet is a key biological controller of an organism’s glucose homeostasis and thus plays an outsized role to coordinate the response to environmental factors to preserve euglycemia through a delicate balance of endocrine outputs. Both successful and failed adaptation to dynamic environmental stimuli has been postulated to occur due to changes in the transcriptional and epigenetic regulation of pathways associated with islet secretory function and survival. Therefore, in this review we examined and evaluated the current evidence elucidating the key epigenetic mechanisms and transcriptional programs underlying the islet’s coordinated response to the interaction between the timing and the composition of dietary nutrients common to modern lifestyles. With the explosion of next generation sequencing, along with the development of novel informatic and –omic approaches, future work will continue to unravel the environmental-epigenetic relationship in islet biology with the goal of identifying transcriptional and epigenetic targets associated with islet perturbations in T2DM.
Introduction
Type 2 diabetes mellitus (T2DM) is one of the major health challenges facing today’s society and projected to afflict nearly 1 in 3 people by year 2050 (1). T2DM is associated with a drastic increase in population morbidity/mortality, and more recently, has been shown to exacerbate adverse outcomes associated with COVID-19 (2). For these reasons, it is imperative to improve our understanding of the molecular mechanisms driving the increasing prevalence of T2DM which can lead to the development of novel therapeutic and preventative approaches. The pathophysiology of T2DM is mediated by complex interactions among diverse environmental and genetic susceptibilities (3), which ultimately culminate in the development of pancreatic islet failure characterized mainly by compromised β-cell insulin secretory function and loss of β-cell numbers. Although underlying genetics contribute to the pathogenesis of T2DM, environmental and epigenetic factors appear to be the primary drivers of this disease (4). Specifically, recent evidence suggests that modern changes in diet macronutrient composition (i.e., increased intake of saturated fats and refined sugars) along with disrupted circadian timing of food intake due to misalignment of circadian light/dark and fasting/feeding cycles has contributed to the induction of pancreatic islet failure and an overall increase in the predisposition to T2DM.
Indeed, pancreatic islets are constantly challenged by alterations in the timing and macronutrient composition of our diets to maintain adequate hormonal outputs for proper regulation of systemic glucose homeostasis. Specifically, the islet is required to integrate signals related to nutrient and energy status, circadian timing, and incoming neurohormones – all which can directly tune epigenetic, transcriptional, and physiological outputs. Glucose, for instance, directly suppresses (via increased ATP : AMP ratio) the AMP-activated protein kinase (AMPK) nutrient sensing pathway which controls transcriptional effectors critical to islet function such as the hepatocyte nuclear factor 4 alpha (HNF4α) (5, 6). During fasting and glucose restriction, AMPK-mediated phosphorylation/suppression of HNF4α prevents ectopic insulin secretion, in part, by thwarting transcriptional activation of the ATP-sensitive potassium channel, Kir6.2, required for depolarization-induced insulin secretion (7). Glucose-mediated activation of the mammalian target of rapamycin (mTOR) signaling pathway, meanwhile, initiates epigenetic/transcriptional programs that accelerate insulin processing and β-cell proliferation to meet the increased insulin demand (8). Hyperactivation of both mTOR and AMPK signaling can lead to β-cell failure highlighting the need for rhythmic nutrient cycles in order to maintain β-cell function (9, 10). Several metabolic intermediates such as acetyl-CoA and NAD+ are also co-factors required for histone acetylation and deacetylation, respectively, which in turn controls transcription factor recruitment and gene transcription (11). NAD+ mediated activation of the histone deacetylase sirtuin 1 (SIRT1), for example, is required in islets to transcriptionally inhibit uncoupling protein 2 (UCP2) via inactivation of the UCP2 promoter (12). UCP2 suppression is required to maintain GSIS given its role in uncoupling oxidative glucose metabolism (12), highlighting the tight link between metabolism, epigenetics, and islet function.
The circadian clock mediates the interaction between diet and the epigenome both at the organismal level through neurohormonal control over feeding behavior (13) and at the cellular level through direct transcriptional/epigenetic modulation (14). Molecularly, the circadian clock is encoded by a transcriptional-translation feedback loop (TTFL) which is driven by heterodimeric binding of the clock’s core transcription factors brain and muscle ARNT-like 1 (BMAL1) and circadian locomotor output cycles kaput (CLOCK) to palindromic E-box DNA binding motifs (14). Transcriptionally, BMAL1:CLOCK directly controls rhythmic patterns in gene expression of negative clock elements (Per) and cryptochrome (Cry), secondary transcription factors (i.e., D site of albumin promoter), and tissue specific functional effectors (15). Nutritional status and feeding time can shift transcriptional activation of this process via nutrient-stimulated signaling (16). For example, activation of insulin signaling in the liver during feeding has been shown to reset the circadian clock via mTOR-dependant Period activation, highlighting how the circadian clock is required to interpret and respond to incoming nutritional signals (17). Given this, genetic and environmental (i.e., shift work, inflammation) disruption of the β-cell circadian clock results in impaired GSIS and β-cell dysfunction due, in part, to impairments in rhythmic transcription of insulin secretion/exocytotic machinery (18–22). Outside of its direct transcriptional role, BMAL1 and CLOCK can also modify the epigenome both by acting as a pioneer transcription factor to recruit histone acetyltransferases (i.e., p300, CREB-binding protein), histone methyltransferases (i.e., mixed lineage leukemia 1), histone demethylase lysine-specific demethylase 1A (LSD1), and other transcription factors which cooperatively work together to promote gene activation (23) and by acting as histone acetyltransferase itself (CLOCK) (24). Taken together, the circadian clock acts a biological controller that integrates and responds to nutritional and behavioral input by precisely timing transcriptional/epigenetic programs to meet dynamic cellular demands.
Failed adaptation to this dynamic environment results in islet dysfunction due to impairments in these fundamental cellular processes (i.e., circadian rhythms, nutrient metabolism, mitochondrial function) which control the epigenome, undoubtably contributing to the development T2DM (25–28). Elucidating the physiological and molecular events that underly the islet’s response to various macronutrients and the overall timing of food intake has guided the chase for targeted T2DM therapies and novel preventative strategies; however, the progress has been limited because of the complex gene regulatory networks underlying these processes (29). The rapid development and adoption of genomic and epigenomic techniques are beginning to alter this narrative and are providing novel mechanistic insights into the regulatory mechanisms underlying the islet’s adaptation to various nutritional stimuli (30). As such, this review will examine the transcriptional and epigenetic mechanisms underlying the β-cell and islet response to the timing and composition of dietary macronutrients specifically overviewing current literature investigating islet adaptation to high-fat (HF) diets, high-sugar (HS) diets, ketogenic diets, low-protein diets, and time-restricted feeding/fasting-type diets (Table 1).
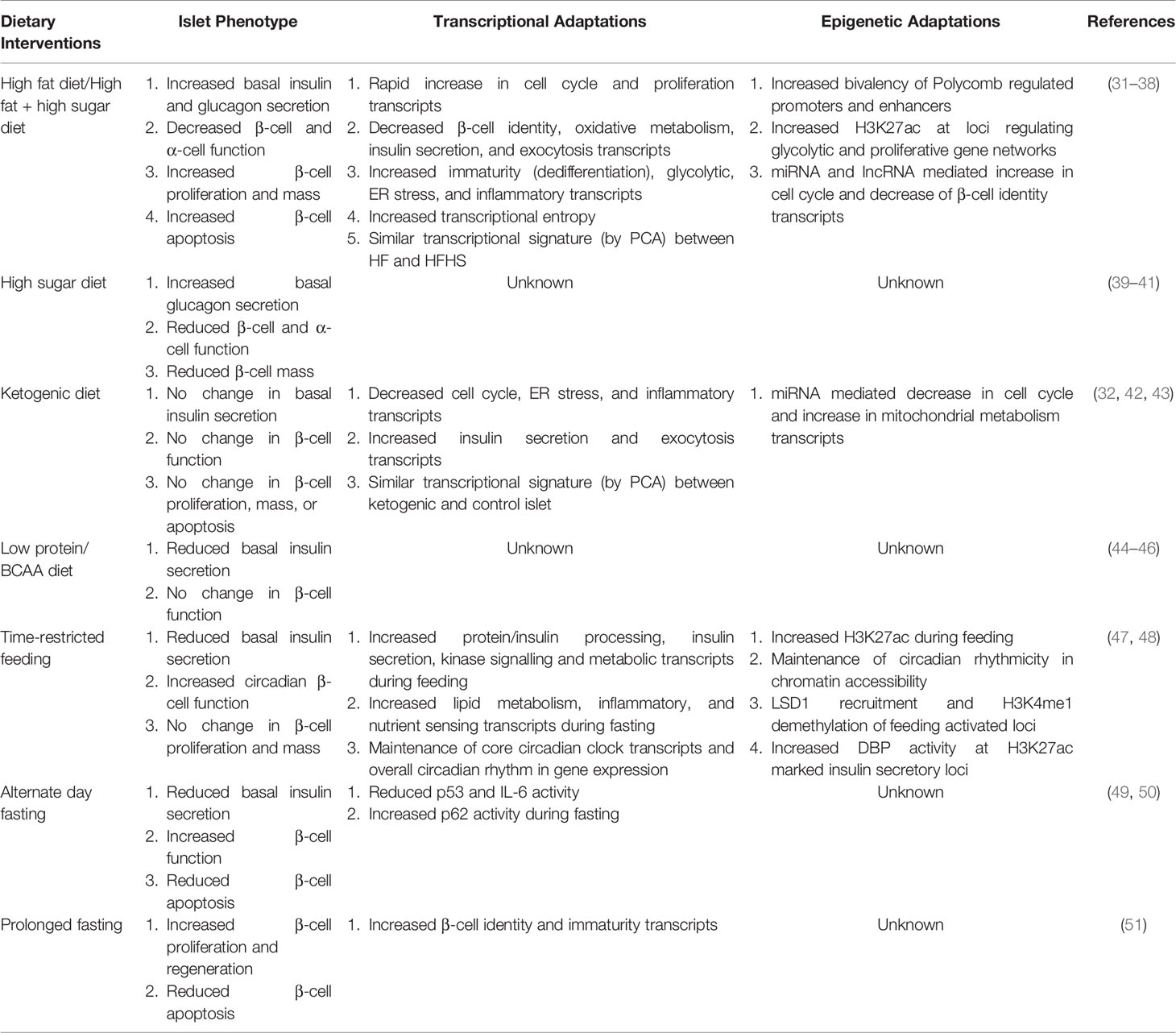
Table 1 Overview and comparison of the islet’s physiological, transcriptional, and epigenetic response to dietary interventions.
Islet Functional and Epigenetic Response to High-Fat, High-Sugar Diets
The world’s current nutritional environment is dominated by inexpensive and easily accessible high saturated fat and high refined sugar/carbohydrate, energy dense, foods (52, 53). Both excess refined sugars (i.e., sucrose and fructose) and saturated fats have been implicated as causal factors in the development of obesity and T2DM; however, their relative contribution to pancreatic islet failure in T2DM is still under investigation (54–56). Despite differing perspectives on the dietary etiology of T2DM, long-term ingestion of a diet rich in saturated fatty acids and/or refined sugars has been clearly demonstrated to lead to increased visceral adiposity (57, 58), reduced energy expenditure (59, 60), peripheral insulin resistance (61, 62), and pancreatic islet dysfunction (39, 63–66). Enumerating the physiological, molecular, and genetic consequences of these dietary stressors on peripheral metabolic tissues may allow us to develop targeted therapies to reverse the effects of diet-induced metabolic dysfunction. While we will specifically focus on the islet’s response to HF and HS diets, please refer to Imamura et al. for an overview of the systemic effects of these diets (67).
The physiological mechanisms underlying islet adaptation to HF and HS diets have been thoroughly investigated over the last 40 years. It is important to note that the vast majority of rodent studies on this topic utilized only male mice partly due to the attenuation of deleterious metabolic effects of HF diets in female rodents (68). From rodents to canines and humans, prolonged HF feeding (typically containing 40-60% fat and ~20% carbohydrates) leads to a significant increase in basal insulin secretion (basal hyperinsulinemia) which demonstrates a positive correlation with saturated fatty acid intake and the extent of insulin resistance (69–72). Along with basal hyperinsulinemia, chronic HF diet has been also shown to reduce glucose-stimulated insulin secretion (GSIS) despite an absolute increase in insulin release and islet insulin content (31, 63, 64). The functional effects on GSIS with HF diet occur in parallel to adaptive increases in β-cell mass and β-cell proliferation potentially accounting for the noted increases in absolute insulin secretion and content in rodents (73, 74). Importantly, the observed decline in β-cell function and increase in β-cell mass with HF diet in rodents is not accelerated or significantly altered upon the replacement of carbohydrates with 20% sucrose in a typical HF, high sugar (HFHS) diet (32). HS diets (10-60% sucrose or fructose, 3-10% fat) have also been demonstrated to induce β-cell dysfunction due to impairments in first-phase GSIS (39–41) and increased β-cell apoptosis (39, 75). Other endocrine cell types of the islet, particularly the pancreatic α-cell, are also affected by long-term HF and HS diets as highlighted by the development of basal hyperglucagonemia and impaired glucose-induced suppression of glucagon secretion with both dietary interventions (31, 76, 77). As highlighted below, our understanding of the physiological, transcriptional, and epigenetic changes of the non β islet cells are currently lacking and additional studies will be needed to understand their contribution to the homeostatic response to dietary interventions associated with T2DM.
The effects of HF and HFHS diets on islet function and mass are associated with distinct changes in the islet’s transcriptional programs. At the initial onset of HF diet, the islet transcriptionally activates compensatory proliferative and protein biosynthesis pathways to increase overall insulin production and secretion (31). Indeed, within one-week of HF diet transcripts associated with cell proliferation are upregulated (e.g. cyclin dependent kinase 1 [Cdk1], cell division cycle associated 2 [Cdca2], cyclin B1 [Ccnb1]) to increase β-cell mass (33). This early transcriptional response requires both demethylation of E-box binding motifs and recruitment of MYC proto-oncogene (Myc), and likely other factors, to promoter/enhancer regions of genes regulating cell replication. Indeed, the circadian transcription factor BMAL1 which canonically binds to E-box motifs has also been demonstrated to be required for compensatory β-cell expansion in response to HF diet (19). Although there is an overall increase in β-cell number and insulin transcriptional levels with HF diet, the islets are characterized by an immature transcriptional signature highlighted by declines in identity factors such as neuronal differentiation 1 (Neurod1) and forkhead box A2 (Foxa2) along with impairments in glucose oxidation (i.e., glucokinase) and mitochondrial function (31). These are features also observed in failing, dedifferentiating β-cells in T2DM which results in decoupling of glucose intake from insulin secretion (25, 78). In fact, Dhawan et al. observed hypermethylation of the promoters of glucokinase (Gck) and urocortin 3 (Ucn3) with concurrent hypomethylation of glycolytic enzymes/dedifferentiation markers hexokinase 1 and 2 (Hk1/2) and lactate dehydrogenase (Ldha) in immature β-cells mirroring the early transcriptional changes with HF diet (79).
Continued long-term (> 10 weeks) HF-diet feeding eventually leads to islet transcriptional decompensation and eventual failure with enrichment for pathways regulating inflammatory and adaptive immune response, such as CXC chemokine receptor (CXCR) signaling, which has previously reported as a key feature of long-term vs short-term HF diet administration in other metabolic tissues due, in part, to increased immune cell infiltration and activation (31, 80). Importantly, activation of CXCR signaling has recently been demonstrated to facilitate β-cell functional failure and may contribute to β-cell demise with long-term HF diet ingestion (81). As previously noted, it is well documented that female rodents are protected from the long-term effects of HF feeding (82, 83). Consistent with this notion, transcriptional analysis of purified control and HF β-cells revealed that more than 50% of differential transcripts were sex dependant (82). HF-fed male β-cells were characterized by suppression of genes critical for oxidative phosphorylation and enrichment for islet amyloid polypeptide (Iapp), both features of failing β-cells in humans with T2DM (25, 65, 84). In contrast, female β-cells exhibited strong sex-specific differential expression of insulin synthesis and ER-stress related transcripts including activation of the endoplasmic reticulum oxidoreductase 1 beta (Ero1b) which is required for proinsulin processing and is directly correlated with the islet’s insulin secretory capacity (85). Lastly, the addition of dietary sucrose to a HF diet was found to not significantly alter the islet’s gene signature (by principal component analysis) and pathway activation/suppression relative to a control, low fat low sugar diet, likely underlying the similarities observed in islet function and mass between HF and HFHS diets (68). Although there are also likely to be similarities between the islet’s transcriptome in response to a HS diet versus HF diet, we currently lack any insight into this area and is an important avenue for future investigation.
Bulk islet analysis prevents identification of cell specific changes, including from β-cells, in response to different islet stressors such as diet. The recent advent of single cell technologies (86), endocrine sorting protocols (87), and fluorescent reporter models (88) have begun to address this challenge, providing a window into cell-specific transcriptome within the islet. Single-cell RNA-sequencing of short-term (1-week) HF fed islets identified 11 subpopulations of β-cells, along with populations of other islet cell types including α-cells (89). Amongst the captured β-cells, more than two-thirds (67%) of β-cells exhibited minimal differences in gene expression compared to their control counterparts, highlighting that the majority β-cells are initially ‘agnostic’ to HF diet – a feature lost with bulk analysis. This relative proportion of this agnostic β-cell group may contribute to the heterogeneous islet response to HF diet observed in rodents (90). The remaining β-cell populations exhibited downregulation of genes important for ER stress response with activation of inflammatory pathways, while minor populations exhibited activation of β-cell proliferative pathways and suppression of immune activation which likely drives the differential analyses observed in bulk findings (90). The α-cell transcriptional signature, previously unstudied in the context of HF feeding, was similarly resistant to transcriptional changes by HF diet with nearly half of captured α-cells not responding. Amongst the remaining ‘responders’, a major subpopulation of HF diet exposed α-cells had an immature islet transcriptional signature with increased Neurogenin 3 (Ngn3) and reduced Ero1b levels (91, 92). Although the functional significance and origin of this subpopulation remains unclear, it may precipitate the α-cell dysfunction and hyperglucagonemia typically observed with HF diet interventions (31, 76). As such, additional studies are urgently needed to reveal and target subpopulations of endocrine cell types driving metabolic dysfunction in response to HF-feeding.
Underlying the transcriptional dynamics associated with HF and HFHS feeding are highly active cis-regulatory elements (i.e., promoters, enhancers, repressors), a dynamic chromatin landscape, and single nucleotide polymorphisms (SNPs) which synergize to promote activation and repression of genetic networks (93). By integrating bulk and single-cell transcriptomics of β-cells from mice fed a HF diet with genome-wide islet chromatin states, investigators are beginning to elucidate how reprogramming of the epigenome contributes to HF diet-induced β-cell dysfunction (34). Using this approach, a subpopulation of dedifferentiated HF diet fed β-cells were determined to contain significantly more bivalent genetic loci compared to control β-cells specifically in Polycomb regulated domains. Bivalent regions are characterized by open regions of chromatin with both activator (histone H3 lysine 4 trimethylation; H3K4me3) and repressor (histone H3 lysine 27 trimethylation; H3K27me3) marks with low transcriptional activity indicated by the absence of RNA polymerase 2 signal in part due to Polycomb mediated silencing. This suggests that a population of HF diet-exposed β-cells have high transcriptional entropy which signals that a cell-type has lost its functional identity and may underly β-cell dedifferentiation process commonly associated with β-cell failure in T2DM (94, 95). Ectopic hyperacetylation of histone H3 lysine 27 (H3K27ac), a histone modification associated with active enhancer and promoter regions, within bivalent, Polycomb repressed loci has been proposed as a causal factor driving β-cell dedifferentiation (94). Supporting this notion, it was recently identified that bivalent promoter and enhancer regions of the β-cell dedifferentiation marker aldehyde dehydrogenase 1 member a3 (Aldh1a3) was found to be significantly hyperacetylated with HF feeding and directly correlated to its transcriptional ‘de-repression’ (35). Globally, HF diet fed islets exhibit several-fold more hyperacetylated loci compared to hypoacetylated loci. This may be rooted in impairments to the histone deacetylase SIRT1 which is attenuated with inflammation and lipotoxicity common to HF feeding (20, 96). Importantly, a majority of the hyperacetylated regions from the HF islets were in proximal active enhancer regions (<5kb from transcriptional start site [TSS]), particularly in promoter regions of glycolytic and proliferative genes permitting recruitment and activation of transcription factors and other transcriptional regulators required for β-cell expansion with HF diet (19, 33, 35, 36, 97, 98). The current evidence has begun to enumerate how HF feeding directly rewires the islet transcriptional and epigenetic environment toward an immature, dedifferentiated phenotype; however, future work is required to establish a direct link between epigenetic modifications and islet dedifferentiation/failure in T2DM.
A majority of single nucleotide polymorphisms (SNPs) lie in cis-regulatory regions and can significantly impact chromatin accessibility and architecture (99). In a series of studies, the Attie group aimed to enumerate expression quantitative trait loci (eQTL) which drive transcriptional, translational, and phenotypic response to a HFHS diet (45% fat, 34% sucrose) in an outbred mouse colony modeling human genetic diversity (100–102). By simultaneously capturing in vivo and ex vivo insulin secretory phenotypes, the islet’s transcriptome/proteome, and the 150,000 SNPs across ~500 outbred mice, the studies revealed several potential genetic drivers of HFHS induced β-cell dysfunction. They determined that several insulin secretory traits (basal insulin secretion, GSIS, insulin content) were highly driven by genetics involving several SNPs/loci which correlate with genome wide association study (GWAS) hits associated with T2DM in humans. For example, Keller et al. found that there was an eQTL hotspot at ~30 Mb on chromosome 1 associated with basal insulin secretion, among other secretory traits (100). This was mapped to protein tyrosine phosphatase non-receptor type 18 (Ptpn18) which exhibited a positive correlation between its expression and total insulin secretion. Genetic inactivation of Ptpn18 prevented HFHS induced basal hyperinsulinemia leading to improved glucose tolerance and insulin sensitivity. These findings illustrate how the underlying genetic landscape directly controls both the transcriptional and phenotypic response of pancreatic islets due to diet-induced stress.
There is accumulating evidence that non-coding RNAs (ncRNA), including micro RNAs (miRNA) and long non-coding RNAs (lncRNA), play a critical role in the maintenance of islet function and identity (as reviewed by Eliasson and Esguerra (103)) through regulation of the islet’s transcriptional and epigenetic landscape. miRNAs are 20-24 nucleotide, single stranded RNAs that can modify gene expression through direct silencing and interference with post-transcriptional RNA molecules (104). Indeed, differential activation of the miRNA-ome has been demonstrated to be a contributing factor in the islet’s response to HF diet (37, 105). miR-802, the top-most upregulated miRNAs with HF diet, for instance was found to repress the β-cell identity transcription factor neuronal differentiation 1 (Neurod1) and was postulated to play a key role mediating HF-induced β-cell dedifferentiation (105). Several miRNAs have also been implicated as causal factors driving β-cell expansion with HF diet (37, 106–108). For example, Jacovetti et al. found that suppression of miR-338-3p is required for activation of survivin (Birc5) which is necessary for β-cell expansion due to its multifaceted role as an inhibitor of cell death and positive regulator of cell division (109). Similar to miRNAs, lncRNAs (>200 nucleotides) also have been found to modulate β-cell function and survival through control of epigenetic, transcription and post-transcriptional landscape (110). Mirroring the role of miRNAs in β-cell dedifferentiation due to HF diet, PDX1 Associated lncRNA (PLUTO), which is required for Pdx1 activation (111) and 1810019D21Rik (ROIT) which is required for NK6 Homeobox 1 (Nkx6.1) transcriptional activation were identified as two of the top-most downregulated lncRNAs with HF feeding (38). LncRNAs such as XLOC_010971 (β long intergenic noncoding RNA 2, βlinc2) and XLOC_013310 (β long intergenic noncoding RNA 3, βlinc3), have also been identified as potential mediators of β-cell decompensation with HF feeding. Respective gain- and loss-of-function studies of βlinc2 and βlinc3 highlighted previously unrecognized role of these lncRNAs in the negative regulation of β-cell survival via activation of nuclear factor-κB’s (NF-κB) nuclear translocation, a key step in β-cell apoptosis due to diabetogenic stress (112). This suggests that HF-induced changes to lncRNAs expression may contribute to β-cell demise and decompensation with long-term HF feeding. In summary, the islet’s lncRNA landscape is significantly perturbed with diet-induced metabolic dysfunction which can potentially modulate adaptation in both pancreatic β-cell function and mass.
Islet Response to the Low-Carbohydrate, High-Fat Ketogenic Diet
The ketogenic diet, consisting of ~70-80% fat, 1-10% carbohydrates and 10-20% protein, has recently re-emerged as a promising dietary therapy for T2DM nearly 100 years since it was first coined by Mayo Clinic physician Dr. Russel Wilder in 1921 as a therapy for epilepsy and later diabetes (113, 114). Modern clinical studies for the most part have reproduced the metabolic benefits of inducing a nutritional state of ketosis by demonstrating that a ketogenic diet effectively reduces body weight, lowers glycemia, and attenuates hyperinsulinemia in obese, T2DM subjects (115–117). For instance, Michalczyk et al. found that hyperinsulinemic, obese women who followed a ketogenic diet for 12-weeks exhibited a 54% reduction in fasting insulin with a 20% lowering of fasting glucose relative to baseline (117). Spurred by this clinical phenotype, several groups have investigated the islet’s physiological and genetic adaptation to a ketogenic diet in both basal and diabetic states (32, 118–121). Compared to a control low-fat, high-carbohydrate diet, the ketogenic diet induced glucose intolerance due to prevailing hypoinsulinemia and significantly reduced β-cell (and α-cell) mass (118, 119, 122). When compared to a traditional HF or HFHS diet; however, Her et al. demonstrated that 12-weeks of ketogenic diet attenuated hyperinsulinemia, enhanced β-cell secretory function, and in turn restored euglycemia (32). Similar effects have been observed for other models of diabetes including the leptin deficient ob/ob model (120), the Akita model of impaired insulin folding (121), and even the type 1 diabetes, streptozotocin (STZ) model (123).
Based on the noted effects of a ketogenic diet on islet function and mass, Her et al. also compared the islet’s transcriptional signature in response to a ketogenic diet relative to HF and HFHS diets (32). Using PCA analysis, the transcriptional signature of ketogenic islets was found to be more closely related to control islets compared to HF or HFHS islets with only 15% of genes were commonly regulated between ketogenic and HF/HFHS diets. Moreover, gene-set enrichment analysis revealed that ketogenic feeding significantly attenuated transcripts associated with cell division and activated genes regulating insulin secretion, relative to HF and HFHS diet islets mirroring the phenotypic features of the ketogenic islet. Ketogenic islets were also characterized by significant reduction in the expression of genes critical for immune activation and ER stress relative to control, low-fat diet highlighting its potential role in protecting the islet from chronic inflammation and the unfolded protein response common to obesity and T2DM (26, 124). In line with these findings, Tattikota et al. found that 4-5 weeks of ketogenic diet attenuated β-cell proliferation in ob/ob islets, in part, due to restoration (20-fold) of miR-184 expression, which is a potent inhibitor of cellular proliferation by disrupting Myc signaling (42, 125). miRNAs have also been suggested to play a key role in enhanced mitochondrial metabolism with ketogenic diet, a key step in GSIS which is dysfunctional with HF diet (43, 126). Specifically, miR-125b was shown to be significantly downregulated with ketogenic diet and was demonstrated to directly target mitochondrial fission process 1 (Mtfp1), among other mitochondrial and lysosomal transcripts. Inhibition of Mtfp1 has been shown to result in mitochondrial fragmentation, a phenotypic trait of the T2DM islet (127), and therefore suppression of miR-125b may contribute to the maintenance of glucose oxidation and insulin secretion observed with ketogenic diets (128). Despite limited insights into the epigenetic effects of ketogenic diets on the islet, these initial studies have begun to provide potential mechanistic insights underlying the therapeutic benefits of ketogenic diet in T2DM.
Islet Response to Protein- and Branched-Chain Amino Acid (BCAA) Restricted Diets
Amino acids play a multifactorial role in maintenance of islet survival and function by acting as building blocks for islet machinery, serving as signaling molecules for nutrient sensing pathways, and by directly inducing nutrient-stimulated insulin and glucagon secretion (129). As such, dietary protein and amino acid intake can tremendously impact both the function and development of the endocrine pancreas, depending on the cellular need for amino acids. Specifically, protein restriction during gestation and maturation can have long-term detrimental effects on the islet (130–132). In contrast with the need for dietary protein in normal β-cell development and maturation, an over-supply of protein and amino acids, particularly branched-chain amino acids (BCAA) leucine, isoleucine, and valine, is associated with the development of T2DM (133). In turn, diets with reduced BCAA content have been demonstrated to restore euglycemia mainly through improvements in insulin sensitivity in both obese/diabetic rodents and humans (44, 45). This is accompanied by a reduction in β-cell insulin hypersecretion with Karusheva et al. reporting a 30% decrease in post-prandial insulin secretion in T2DM subjects following 4-weeks of a diet lacking BCAA (134). Consistently, it was found that rodents fed a BCAA and amino acid (AA) low diet for 3-weeks had reduced overall insulin secretion due to a decrease in β-cell metabolic flux and corresponding increased glucose-stimulated Ca2+ oscillatory efficiency (44). More recently, Cummings et al. extended this work and observed a ~2-fold decrease in plasma insulin 5-weeks following a switch from a HFHS diet to either a BCAA or AA deficient diet which was associated with reduced β-cell mitochondrial membrane potential (a marker of basal hyperinsulinemia) (45, 46). Although the islet’s genetic and epigenetic adaptations to a BCAA/AA low diet remain largely unexplored, these phenotypic studies suggest the need to understand the molecular mechanisms underlying improvements in islet function associated with BCAA/AA-restricted diets.
Islet Response to Time-Dependent Regulation of Food Intake
Periodic fasting and, in turn, fasting mimicking diets have gained rapid popularity as potential therapeutic strategies for a wide array of metabolic and degenerative diseases. In contrast with absolute caloric restriction (CR) which limits daily nutritional energy by as much as 50% without altering meal timing or frequency, periodic fasting-type diets consists of extended periods ranging from 12 h to weeks without any caloric intake with subsequent ‘caloric-blind’ feeding periods lasting as little as a few hours (135). Although caloric restriction has been demonstrated as a therapeutically beneficial approach to restore islet function in both diabetic mice and humans (136–138), adherence to prolonged periods of caloric restriction has been challenging in humans (139). In support of fasting-type diets, Gil and Panda demonstrated that erratic and extended feeding patterns in humans significantly contribute to obesity which when normalized with prolonged fasting resulted in reduced body weight and improved health (140). Fasting can be divided into two distinct subtypes: intermittent fasting (IF) and periodic fasting (PF). IF is defined as 12 h to 2 days of fasting followed by ad libitum feeding periods that last between a few hours and 5 days. As such, IF is comprised of several fasting regimens such as alternative day fasting (ADF) (24 h fasting: 24 h feeding), 5:2 diet (2 days fasting: 5 days feeding), and time-restricted feeding (tRF) (12 to 18 h fasting: 6 to 12 h feeding). Meanwhile, PF is defined by 2 or more days of fasting, lasting up to several weeks, with long ad libitum refeeding intervals spanning at least 1-week. PF also encompasses fasting mimicking diets (FMD) which are made up of ~10-50% typical calories and consist of high fat (>50%) with low protein (<10%) and carbohydrate (<40%) content. In line with isocaloric PF, FMD last 2 or more days and are interspersed with at least 7 days of ad libitum feeding. The therapeutic interest in IF and PF is not an entirely new concept with Otto Folin and W. Denis demonstrating in 1915 that prolonged fasting ameliorated obesity and metabolic syndrome (141). Although recent studies have re-emphasized the therapeutic benefits of fasting, the molecular mechanisms underlying beneficial effects of these diets on the islet are only starting to be elucidated more than 100 years later.
While IF and PF have been clearly shown to ameliorate metabolic dysfunction across several tissues (i.e., liver, adipose, brain), we will focus specifically on the pancreatic islet’s physiological and, in turn, epigenetic response to fasting. For a comprehensive overview of the effects of IF and PF on whole organism physiology, please refer to Longo et al. (142). Prolonged fasting-type diets have been demonstrated to enhance β-cell health, primarily by limiting chronic insulin demand during metabolic stress. The benefits of acutely reducing insulin secretion on β-cell health and function for T2DM gained prominence in the 1970s when Greenwood, Mahler, and Hales suggested that β-cell overstimulation leads to impaired GSIS when they found that diazoxide treatment, a potent inhibitor of insulin secretion, restored β-cell function in T2DM subjects (143). This was followed by a series of studies demonstrating that “β-cell rest” or acute inhibition of insulin secretion, could restore β-cell function (first phase insulin secretion), insulin/Ca2+ pulsatility, and insulin content in obesity and T2DM (144–148). Chronic β-cell stimulation common to high fat feeding/obesity (149) and circadian disruption (21, 22) result in both β-cell dysfunction and apoptosis which have been shown to be reversed by ADF (49, 150), tRF (47, 151, 152), and a FMD (153). Specifically, a 12-week ADF regimen protected mice from HF diet induced β-cell dysfunction and failure (49), which was associated with enhanced GSIS, restored islet insulin content, and reduced apoptosis contingent on ADF activation of the autophagy-lysosome pathway. Although fewer studies have examined the physiological effects of PF on islet function, Kolakowski et al. observed in 1970 that 7-day PF followed by 3-days of tRF resulted in a 2.5-times increase in insulin secretory capacity (154). PF via FMD has also been demonstrated to modulate functional β-cell mass in insulin-deficient and HF diet models of diabetes (51, 153); however, the ability to translate these findings are confounded by the limited replicative capacity of human β-cells (155). Taken together, the physiological characterization of fasting-type diets has demonstrated their ability to promote β-cell rest, restore islet function, and potentially regenerate β-cell mass, highlighting the importance of cyclic fasting-feeding to maintain β-cell health.
Underlying the noted physiological effects of IF and PF on pancreatic islet function and survival in T2DM are dramatic changes to the islet’s transcriptional and epigenetic identity. Under control conditions, islets isolated from C57B/6J mice following 6-days of tRF exhibit clear transcriptional and epigenetic differences during periods of feeding versus fasting (48). Specifically, fasted islets exhibit enrichment for genes associated with lipid/fatty acid/carbohydrate metabolism and nutrient sensing (Forkhead box O [FoxO] and mTOR signaling). Indeed, FoxO signaling is activated in the β-cell upon glucose and nutrient restriction and is required for maintaining diurnal metabolic flexibility between carbohydrates and lipids (156, 157). In contrast, fed islets are significantly enriched for genes associated with protein processing/export, kinase signaling, and amino acid metabolism, all which are required for GSIS. This is consistent with circadian analysis of the mouse islet’s transcriptome which similarly found significant enrichment for protein processing/export, insulin secretion and amino acid metabolism during the animals active, feeding period and corresponding activation of nutrient sensing (mTOR), inflammatory signaling (tumor necrosis factor, cytokine receptor, nuclear factor kappa B [NFκB]) and fatty acid metabolism during inactive, fasting periods suggesting that rhythmic feeding/fasting cycles drive the islet’s transcriptional identity (158). In line with the noted changes in gene expression, Wortham et al. (48) found that nearly half of all active enhancers and promoters (~19,000 by H3K27ac) were differentially acetylated (active) during fasting vs. feeding, with 80% hyperacetylated with feeding. Although not directly compared, the hyperacetylation exhibited in the HF diet islet may be partially driven by disruption in the circadian feed-fast cycle which occur with HF feeding (159). The leptin receptor deficiency model of T2DM (Leprdb/db), which also exhibits impaired circadian food intake rhythms (160), similarly exhibits increased H3K27ac and histone H3 lysine 4 monomethylation (H3K4me1; activating chromatin mark which co-occurs with H3K27ac) signal in regions associated with feeding but not in regions associated with fasting. Importantly, regions hyperacetylated are associated with LSD1 binding where it acts as a brake on transcription by demethylating H3K4, likely contributing to β-cell rest (161). Consistently, β-cell specific LSD1 deletion physiologically and epigenetically phenocopies the HF diet state: basal hyperinsulinemia, impaired GSIS and activation of a ‘stressed/immature’ gene signature (i.e., hexokinase1 [Hk1], 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 [Pfkfb3], jun proto-oncogene [Jun], fos proto-oncogene [Fos], nuclear factor, interleukin 3 regulated [Nfil3]). Future studies will likely be needed to unravel whether mistimed feeding, nutrient overabundance, or both are factors driving hyperacetylation and β-cell dedifferentiation with HF diet and T2DM.
Circadian disruption and shift work are environmental stressors characteristic of impaired feeding/fasting rhythms and have been directly linked to the development of islet failure in T2DM (22, 162). Pancreatic islets isolated from circadian disrupted (via constant light) mice exhibit complete ablation of circadian rhythms in both gene expression and chromatin accessibility (47). Providing further evidence that rhythmic feeding/fasting cycles drive islet transcription, tRF was shown to rescue transcriptional and epigenetic (chromatin accessibility) rhythms, despite global circadian disruption, of genes/loci annotated to pathways regulating circadian rhythms along with β-cell function (i.e., insulin secretion and exocytosis) and rest pathways (i.e., FoxO signaling, fatty acid metabolism) active during feeding and fasting, respectively. The interaction of nutrient sensing pathways with the circadian clock has been shown to be a key regulator underlying feeding/fasting driven rhythms in transcription and chromatin accessibility. Under circadian disruption conditions, where feeding cycles are disrupted, the circadian transcription factor D site of albumin promoter (Dbp) is highly suppressed (~20-fold at peak). During extended fasting periods with tRF, Dbp is activated by FoxO1 (163), and likely other factors (164), where it begins to bind active promoter and enhancer regions (~75% overlap with H3K27ac) of genes associated with insulin processing and secretion to prepare the β-cell (i.e., β-cell rest) for subsequent feeding periods. Histone modifying factors such as LSD1 likely also play a role in this process and have been shown to be required for circadian transcription factor activation in the liver due to diurnal differences in nutrient availability and could play a similar role in the islet; however, future studies will be required to establish this direct link (165).
Fasting type diets can also directly modulate transcriptional mechanisms underlying β-cell proliferation and survival. Indeed, ADF impeded HF diet induced β-cell apoptosis due in part to suppression of interleukin-6 and tumor protein p53, two key regulators of DNA damage response, and activation of autophagic/lysosomal flux (49, 50). Given that PF has been found to be preserve β-cell mass in several models of β-cell loss, it seems likely that extended fasting protects the β-cell from p53-mediated DNA damage associated with diabetogenic stress (51, 166). Subsequent refeeding following a prolonged fasting period results in a mixed islet transcriptional phenotype: upregulation of β-cell identity genes such as MAF bZIP transcription factor A (Mafa), pancreatic and duodenal homeobox 1 (Pdx1), insulin 2 (Ins2), and glucose transporter 2 (Glut2) with a parallel induction of pluripotency/immaturity markers Nanog homeobox (Nanog), DNA methyltransferase 3 beta (Dnmt3b), SRY-Box transcription factor 17 (Sox17), Neurogenin 3 (Ngn3) and GATA-binding factor 6 (Gata6) (51). Chronic activation of these pluripotency and secretory factors due to constant feeding, common to both obesity and circadian disruption (22, 140), may play a role in precipitating β-cell dedifferentiation and dysfunction in T2DM. Together, this provides further evidence supporting the importance of extended fasting periods to maintain β-cell health; however, future transcriptional and epigenetic studies are still needed to fully characterize islet cell specific changes in response to modified feeding/fasting cycles which may underly β-cell failure in T2DM.
Conclusion
The interaction between the pancreatic islet and dietary inputs has long been appreciated as a key piece in the etiology of islet failure in T2DM. More recent studies suggest that the temporal regulation of food intake is another critical regulator of islet function, and when perturbed, contributes to the development of T2DM. Indeed, lifestyle-driven changes in diet composition and daily patterns of food intake appear to be primary environmental factors contributing to ever-increasing rise in the incidence of the metabolic diseases worldwide. This review outlined the current literature highlighting the complexity in gene regulatory networks dictating the islet’s response to changes in the composition of dietary macronutrients, specifically proteins, carbohydrates, and fatty acids, and the timing of their consumption (i.e., intermittent fasting, time-restricted feeding) throughout life. The current evidence underlying successful versus failed islet adaptation to dietary interventions points to the importance of the minimalist motto: “less is more”. Less basal insulin secretion, less proliferation, less transcriptional entropy, less bivalent promoters and enhancers, less acetylation of H3K27, and less feeding time are all common threads associated with maintaining robust meal-stimulated insulin secretion and mature beta cell phenotype and genotype. A diet and lifestyle that allows the β-cell and likely other islet cell types time to “rest and repair” mediated by proper alignment of fasting/feeding cycles with internal circadian clocks appears to be a critical factor in preventing β-cell hyperactivation and eventual demise. With the ongoing development of additional low-input and single-cell epigenetic techniques (i.e., CUT&RUN, SHARE-seq) (167, 168) and further adoption of current technologies (i.e., RNA-seq, ATAC-seq, ChIP-seq), future studies will likely provide additional insights into causal transcriptional and epigenetic mechanisms underlying how each islet cell type responds to the timing and composition of their diet, their contribution to T2DM development, and their potential as a therapy to ameliorate islet dysfunction in T2DM.
Author Contributions
MB performed the systematic literature search and preparation of the manuscript. AM oversaw the literature review and wrote the manuscript with MB. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We acknowledge funding support from the National Institutes of Health (R01DK098468 and R01DK128844 to AM, F99DK123834 to MB), the Center for Regenerative Medicine (Mayo Clinic, Rochester, MN), and the Mayo Clinic Graduate School of Biomedical Sciences (Biomedical Engineering and Physiology Graduate Program).
References
1. Boyle JP, Thompson TJ, Gregg EW, Barker LE, Williamson DF. Projection of the Year 2050 Burden of Diabetes in the US Adult Population: Dynamic Modeling of Incidence, Mortality, and Prediabetes Prevalence. Popul Health Metr (2010) 8:29. doi: 10.1186/1478-7954-8-29
2. Feldman EL, Savelieff MG, Hayek SS, Pennathur S, Kretzler M, Pop-Busui R. COVID-19 and Diabetes: A Collision and Collusion of Two Diseases. Diabetes (2020) 69:2549–65. doi: 10.2337/dbi20-0032
3. Smushkin G, Vella A. Genetics of Type 2 Diabetes. Curr Opin Clin Nutr Metab Care (2010) 13:471–7. doi: 10.1097/MCO.0b013e32833a558d
4. Franks PW, McCarthy MI. Exposing the Exposures Responsible for Type 2 Diabetes and Obesity. Science (2016) 354:69–73. doi: 10.1126/science.aaf5094
5. Leclerc I, Lenzner C, Gourdon L, Vaulont S, Kahn A, Viollet B. Hepatocyte Nuclear Factor-4α Involved in Type 1 Maturity-Onset Diabetes of the Young is a Novel Target of AMP-Activated Protein Kinase. Diabetes (2001) 50:1515–21. doi: 10.2337/diabetes.50.7.1515
6. Rourke JL, Hu Q, Screaton RA. AMPK and Friends: Central Regulators of β Cell Biology. Trends Endocrinol Metab (2018) 29:111–22. doi: 10.1016/j.tem.2017.11.007
7. Gupta RK, Vatamaniuk MZ, Lee CS, Flaschen RC, Fulmer JT, Matschinsky FM, et al. The MODY1 Gene HNF-4α Regulates Selected Genes Involved in Insulin Secretion. J Clin Invest (2005) 115:1006–15. doi: 10.1172/JCI200522365
8. Blandino-Rosano M, Barbaresso R, Jimenez-Palomares M, Bozadjieva N, Werneck-de-Castro JP, Hatanaka M, et al. Loss of Mtorc1 Signalling Impairs β-Cell Homeostasis and Insulin Processing. Nat Commun (2017) 8:1–15. doi: 10.1038/ncomms16014
9. Kim W-H, Lee JW, Suh YH, Lee HJ, Lee SH, Oh YK, et al. AICAR Potentiates ROS Production Induced by Chronic High Glucose: Roles of AMPK in Pancreatic β-Cell Apoptosis. Cell Signalling (2007) 19:791–805. doi: 10.1016/j.cellsig.2006.10.004
10. Yuan T, Rafizadeh S, Gorrepati KDD, Lupse B, Oberholzer J, Maedler K, et al. Reciprocal Regulation of mTOR Complexes in Pancreatic Islets From Humans With Type 2 Diabetes. Diabetologia (2017) 60:668–78. doi: 10.1007/s00125-016-4188-9
11. Katada S, Imhof A, Sassone-Corsi P. Connecting Threads: Epigenetics and Metabolism. Cell (2012) 148:24–8. doi: 10.1016/j.cell.2012.01.001
12. Bordone L, Motta MC, Picard F, Robinson A, Jhala US, Apfeld J, et al. Sirt1 Regulates Insulin Secretion by Repressing UCP2 in Pancreatic β Cells. PloS Biol (2006) 4:e31. doi: 10.1371/journal.pbio.0040031
13. Bechtold DA, Loudon AS. Hypothalamic Clocks and Rhythms in Feeding Behaviour. Trends Neurosci (2013) 36:74–82. doi: 10.1016/j.tins.2012.12.007
14. Takahashi JS. Transcriptional Architecture of the Mammalian Circadian Clock. Nat Rev Genet (2017) 18:164–79. doi: 10.1038/nrg.2016.150
15. Beytebiere JR, Trott AJ, Greenwell BJ, Osborne CA, Vitet H, Spence J, et al. Tissue-Specific BMAL1 Cistromes Reveal That Rhythmic Transcription is Associated With Rhythmic Enhancer–Enhancer Interactions. Genes Dev (2019) 33:294–309. doi: 10.1101/gad.322198.118
16. Stokkan KA, Yamazaki S, Tei H, Sakaki Y, Menaker M. Entrainment of the Circadian Clock in the Liver by Feeding. Science (2001) 291:490–3. doi: 10.1126/science.291.5503.490
17. Crosby P, Hamnett R, Putker M, Hoyle NP, Reed M, Karam CJ, et al. Insulin/IGF-1 Drives PERIOD Synthesis to Entrain Circadian Rhythms With Feeding Time. Cell (2019) 177:896–909. e20. doi: 10.1016/j.cell.2019.02.017
18. Perelis M, Marcheva B, Ramsey KM, Schipma MJ, Hutchison AL, Taguchi A, et al. Pancreatic β Cell Enhancers Regulate Rhythmic Transcription of Genes Controlling Insulin Secretion. Science (2015) 350:aac4250. doi: 10.1126/science.aac4250
19. Rakshit K, Hsu TW, Matveyenko AV. Bmal1 is Required for Beta Cell Compensatory Expansion, Survival and Metabolic Adaptation to Diet-Induced Obesity in Mice. Diabetologia (2016) 59:734–43. doi: 10.1007/s00125-015-3859-2
20. Javeed N, Brown MR, Rakshit K, Her T, Sen SK, Matveyenko AV. Pro-Inflammatory Cytokine Interleukin 1β Disrupts β Cell Circadian Clock Function and Regulation of Insulin Secretion. Endocrinology (2020) 162(1):bqaa084. doi: 10.1210/endocr/bqaa084
21. Qian J, Yeh B, Rakshit K, Colwell CS, Matveyenko AV. Circadian Disruption and Diet-Induced Obesity Synergize to Promote Development of β-Cell Failure and Diabetes in Male Rats. Endocrinology (2015) 156:4426–36. doi: 10.1210/en.2015-1516
22. Qian J, Block GD, Colwell CS, Matveyenko AV. Consequences of Exposure to Light at Night on the Pancreatic Islet Circadian Clock and Function in Rats. Diabetes (2013) 62:3469–78. doi: 10.2337/db12-1543
23. Kim YH, Lazar MA. Transcriptional Control of Circadian Rhythms and Metabolism: A Matter of Time and Space. Endocrine Rev (2020) 41:707–32. doi: 10.1210/endrev/bnaa014
24. Doi M, Hirayama J, Sassone-Corsi P. Circadian Regulator CLOCK is a Histone Acetyltransferase. Cell (2006) 125:497–508. doi: 10.1016/j.cell.2006.03.033
25. Haythorne E, Rohm M, van de Bunt M, Brereton MF, Tarasov AI, Blacker TS, et al. Diabetes Causes Marked Inhibition of Mitochondrial Metabolism in Pancreatic β-Cells. Nat Commun (2019) 10:1–17. doi: 10.1038/s41467-019-10189-x
26. Huang C-J, Lin C-Y, Haataja L, Gurlo T, Butler AE, Rizza RA, et al. High Expression Rates of Human Islet Amyloid Polypeptide Induce Endoplasmic Reticulum Stress–Mediated β-Cell Apoptosis, a Characteristic of Humans With Type 2 But Not Type 1 Diabetes. Diabetes (2007) 56:2016–27. doi: 10.2337/db07-0197
27. Camunas-Soler J, Dai X-Q, Hang Y, Bautista A, Lyon J, Suzuki K, et al. Patch-Seq Links Single-Cell Transcriptomes to Human Islet Dysfunction in Diabetes. Cell Metab (2020) 31:1017–31. doi: 10.1016/j.cmet.2020.04.005
28. Rakshit K, Qian J, Colwell CS, Matveyenko AV. The Islet Circadian Clock: Entrainment Mechanisms, Function and Role in Glucose Homeostasis. Diabetes Obes Metab (2015) 17:115–22. doi: 10.1111/dom.12523
29. Khursheed R, Singh SK, Wadhwa S, Kapoor B, Gulati M, Kumar R, et al. Treatment Strategies Against Diabetes: Success So Far and Challenges Ahead. Eur J Pharmacol (2019) 862:172625. doi: 10.1016/j.ejphar.2019.172625
30. Kim H, Kulkarni RN. Epigenetics in β-Cell Adaptation and Type 2 Diabetes. Curr Opin Pharmacol (2020) 55:125–31. doi: 10.1016/j.coph.2020.10.008
31. Gao R, Fu Q, Jiang H-M, Shen M, Zhao R-L, Qian Y, et al. Temporal Metabolic and Transcriptomic Characteristics Crossing Islets and Liver Reveal Dynamic Pathophysiology in Diet-Induced Diabetes. iScience (2021) 24:102265. doi: 10.1016/j.isci.2021.102265
32. Her TK, Lagakos WS, Brown MR, LeBrasseur NK, Rakshit K, Matveyenko AV. Dietary Carbohydrates Modulate Metabolic and β-Cell Adaptation to High-Fat Diet-Induced Obesity. Am J Physiol-Endocrinol Metab (2020) 318:E856–65. doi: 10.1152/ajpendo.00539.2019
33. Rosselot C, Kumar A, Lakshmipathi J, Zhang P, Lu G, Katz LS, et al. Myc Is Required for Adaptive β-Cell Replication in Young Mice But Is Not Sufficient in One-Year-Old Mice Fed With a High-Fat Diet. Diabetes (2019) 68:1934–49. doi: 10.2337/db18-1368
34. Lu TT-H, Heyne S, Dror E, Casas E, Leonhardt L, Boenke T, et al. Pospisilik, The Polycomb-Dependent Epigenome Controls β Cell Dysfunction, Dedifferentiation, and Diabetes. Cell Metab (2018) 27:1294–1308.e7. doi: 10.1016/j.cmet.2018.04.013
35. López-Pérez A, Norlin S, Steneberg P, Remeseiro S, Edlund H, Hörnblad A. Pan-AMPK Activator O304 Prevents Gene Expression Changes and Remobilisation of Histone Marks in Islets of Diet-Induced Obese Mice. Sci Rep (2021) 11:1–13. doi: 10.1038/s41598-021-03567-3
36. Nammo T, Udagawa H, Funahashi N, Kawaguchi M, Uebanso T, Hiramoto M, et al. Genome-Wide Profiling of Histone H3K27 Acetylation Featured Fatty Acid Signalling in Pancreatic Beta Cells in Diet-Induced Obesity in Mice. Diabetologia (2018) 61:2608–20. doi: 10.1007/s00125-018-4735-7
37. Nesca V, Guay C, Jacovetti C, Menoud V, Peyot M-L, Laybutt DR, et al. Identification of Particular Groups of microRNAs That Positively or Negatively Impact on Beta Cell Function in Obese Models of Type 2 Diabetes. Diabetologia (2013) 56:2203–12. doi: 10.1007/s00125-013-2993-y
38. Zhang FF, Liu YH, Wang DW, Liu TS, Yang Y, Guo JM, et al. Obesity-Induced Reduced Expression of the lncRNA ROIT Impairs Insulin Transcription by Downregulation of Nkx6. 1 methylation Diabetologia (2020) 63:811–24. doi: 10.1007/s00125-020-05090-y
39. Maiztegui B, Borelli MI, Raschia MA, Del Zotto HH, Gagliardino JJ. Islet Adaptive Changes to Fructose-Induced Insulin Resistance: β-Cell Mass, Glucokinase, Glucose Metabolism, and Insulin Secretion. J Endocrinol 200 (2009) 200(2):139–49. doi: 10.1677/JOE-08-0386
40. Chicco A, D'Alessandro M, Karabatas L, Pastorale C, Basabe JC, et al. Muscle Lipid Metabolism and Insulin Secretion Are Altered in Insulin-Resistant Rats Fed a High Sucrose Diet. J Nutr (2003) 133:127–33. doi: 10.1093/jn/133.1.127
41. Ferreira M, Lombardo YB, Chicco A. β– Cell Adaptation/Dysfunction in an Animal Model of Dyslipidemia and Insulin Resistance Induced by the Chronic Administration of a Sucrose-Rich Diet. Islets (2010) 2:367–73. doi: 10.4161/isl.2.6.13869
42. Tattikota SG, Rathjen T, McAnulty SJ, Wessels H-H, Akerman I, Van De Bunt M, et al. Argonaute2 Mediates Compensatory Expansion of the Pancreatic β Cell. Cell Metab (2014) 19:122–34. doi: 10.1016/j.cmet.2013.11.015
43. Cheung R, Pizza G, Chabosseau P, Rolando D, Tomas A, Burgoyne T, et al. Glucose-Dependent miR-125b is a Negative Regulator of β-Cell Function. bioRxiv (2021) 444559. doi: 10.1101/2021.05.17.444559
44. Fontana L, Cummings NE, Apelo SIA, Neuman JC, Kasza I, Schmidt BA, et al. Decreased Consumption of Branched-Chain Amino Acids Improves Metabolic Health. Cell Rep (2016) 16:520–30. doi: 10.1016/j.celrep.2016.05.092
45. Cummings NE, Williams EM, Kasza I, Konon EN, Schaid MD, Schmidt BA, et al. Restoration of Metabolic Health by Decreased Consumption of Branched-Chain Amino Acids. J Physiol (2018) 596:623–45. doi: 10.1113/JP275075
46. Corkey BE, Deeney JT, Merrins MJ. What Regulates Basal Insulin Secretion and Causes Hyperinsulinemia? Diabetes (2021) 70:2174–82. doi: 10.2337/dbi21-0009
47. Brown MR, Sen S, Mazzone A, Her TK, Xiong Y, Lee J-H, et al. Time-Restricted Feeding Prevents Deleterious Metabolic Effects of Circadian Disruption Through Epigenetic Control of Beta Cell Function. Sci Adv (2021) 7(51):eabg6856. doi: 10.1126/sciadv.abg6856
48. Wortham M, Liu F, Fleischman JY, Wallace M, Mulas F, Vinckier NK, et al. Nutrient Regulation of the Islet Epigenome Controls Adaptive Insulin Secretion. BioRxiv (2019), 742403. doi: 10.1101/742403
49. Liu H, Javaheri A, Godar RJ, Murphy J, Ma X, Rohatgi N, et al. Intermittent Fasting Preserves Beta-Cell Mass in Obesity-Induced Diabetes via the Autophagy-Lysosome Pathway. Autophagy (2017) 13:1952–68. doi: 10.1080/15548627.2017.1368596
50. de Souza Marinho T, Borges CC, Aguila MB, Mandarim-de-Lacerda CA. Intermittent Fasting Benefits on Alpha-and Beta-Cell Arrangement in Diet-Induced Obese Mice Pancreatic Islet. J Diabetes its Complications (2020) 34:107497. doi: 10.1016/j.jdiacomp.2019.107497
51. Cheng C-W, Villani V, Buono R, Wei M, Kumar S, Yilmaz OH, et al. Fasting-Mimicking Diet Promotes Ngn3-Driven β-Cell Regeneration to Reverse Diabetes. Cell (2017) 168:775–88. doi: 10.1016/j.cell.2017.01.040
52. Collaborators GO. Health Effects of Overweight and Obesity in 195 Countries Over 25 Years. New Engl J Med (2017) 377:13–27. doi: 10.1056/NEJMoa1614362
53. Zimmet P, Alberti K, Shaw J. Global and Societal Implications of the Diabetes Epidemic. Nature (2001) 414:782–7. doi: 10.1038/414782a
54. Mozaffarian D, Hao T, Rimm EB, Willett WC, Hu FB. Changes in Diet and Lifestyle and Long-Term Weight Gain in Women and Men. New Engl J Med (2011) 364:2392–404. doi: 10.1056/NEJMoa1014296
55. Prinz P. The Role of Dietary Sugars in Health: Molecular Composition or Just Calories? Eur J Clin Nutr (2019) 73:1216–23. doi: 10.1038/s41430-019-0407-z
56. Gross LS, Li L, Ford ES, Liu S. Increased Consumption of Refined Carbohydrates and the Epidemic of Type 2 Diabetes in the United States: An Ecologic Assessment. Am J Clin Nutr (2004) 79:774–9. doi: 10.1093/ajcn/79.5.774
57. Doucet E, Almeras N, White M, Despres J, Bouchard C, Tremblay A. Dietary Fat Composition and Human Adiposity. Eur J Clin Nutr (1998) 52:2–6. doi: 10.1038/sj.ejcn.1600500
58. Jürgens H, Haass W, Castaneda TR, Schürmann A, Koebnick C, Dombrowski F, et al. Consuming Fructose-Sweetened Beverages Increases Body Adiposity in Mice. Obes Res (2005) 13:1146–56. doi: 10.1038/oby.2005.136
59. Cox CL, Stanhope KL, Schwarz JM, Graham JL, Hatcher B, Griffen SC, et al. Consumption of Fructose-Sweetened Beverages for 10 Weeks Reduces Net Fat Oxidation and Energy Expenditure in Overweight/Obese Men and Women. Eur J Clin Nutr (2012) 66:201–8. doi: 10.1038/ejcn.2011.159
60. Storlien L, James D, Burleigh K, Chisholm D, Kraegen E. Fat Feeding Causes Widespread In Vivo Insulin Resistance, Decreased Energy Expenditure, and Obesity in Rats. Am J Physiol-Endocrinol Metab (1986) 251:E576–83. doi: 10.1152/ajpendo.1986.251.5.E576
61. Stanhope KL, Schwarz JM, Keim NL, Griffen SC, Bremer AA, Graham JL, et al. Consuming Fructose-Sweetened, Not Glucose-Sweetened, Beverages Increases Visceral Adiposity and Lipids and Decreases Insulin Sensitivity in Overweight/Obese Humans. J Clin Invest (2009) 119:1322–34. doi: 10.1172/JCI37385
62. Boden G. Role of Fatty Acids in the Pathogenesis of Insulin Resistance and NIDDM. Diabetes (1997) 46:3–10. doi: 10.2337/diab.46.1.3
63. Gargani S, Thévenet J, Yuan J, Lefebvre B, Delalleau N, Gmyr V, et al. Adaptive Changes of Human Islets to an Obesogenic Environment in the Mouse. Diabetologia (2013) 56:350–8. doi: 10.1007/s00125-012-2775-y
64. Dobbins RL, Szczepaniak LS, Myhill J, Tamura Y, Uchino H, Giacca A, et al. The Composition of Dietary Fat Directly Influences Glucose-Stimulated Insulin Secretion in Rats. Diabetes (2002) 51:1825–33. doi: 10.2337/diabetes.51.6.1825
65. Blencowe M, Furterer A, Wang Q, Gao F, Rosenberger M, Pei L, et al. IAPP-Induced Beta Cell Stress Recapitulates the Islet Transcriptome in Type 2 Diabetes. Diabetologia (2021) 65(1):1–15. doi: 10.1007/s00125-021-05569-2
66. Del Zotto H, Massa L, Gómez Dumm CL, Gagliardino JJ. Changes Induced by Sucrose Administration Upon the Morphology and Function of Pancreatic Islets in the Normal Hamster. Diabet/Metabolism Res Rev (1999) 15:106–12. doi: 10.1002/(SICI)1520-7560(199903/04)15:2<106::AID-DMRR18>3.0.CO;2-2
67. Imamura F, Micha R, Wu JH, de Oliveira Otto MC, Otite FO, Abioye AI, et al. Effects of Saturated Fat, Polyunsaturated Fat, Monounsaturated Fat, and Carbohydrate on Glucose-Insulin Homeostasis: A Systematic Review and Meta-Analysis of Randomised Controlled Feeding Trials. PloS Med (2016) 13:e1002087. doi: 10.1371/journal.pmed.1002087
68. Mauvais-Jarvis F, Clegg DJ, Hevener AL. The Role of Estrogens in Control of Energy Balance and Glucose Homeostasis. Endocrine Rev (2013) 34:309–38. doi: 10.1210/er.2012-1055
69. Surwit RS, Feinglos MN, Rodin J, Sutherland A, Petro AE, Opara EC, et al. Differential Effects of Fat and Sucrose on the Development of Obesity and Diabetes in C57BL/6J and AJ Mice. Metabolism (1995) 44:645–51. doi: 10.1016/0026-0495(95)90123-X
70. Feskens EJ, Loeber JG, Kromhout D. Diet and Physical Activity as Determinants of Hyperinsulinemia: The Zutphen Elderly Study. Am J Epidemiol (1994) 140:350–60. doi: 10.1093/oxfordjournals.aje.a117257
71. Rocchini AP, Marker P, Cervenka T. Time Course of Insulin Resistance Associated With Feeding Dogs a High-Fat Diet. Am J Physiol-Endocrinol And Metab (1997) 272:E147–54. doi: 10.1152/ajpendo.1997.272.1.E147
72. Bergman RN, Phillips LS, Cobelli C. Physiologic Evaluation of Factors Controlling Glucose Tolerance in Man: Measurement of Insulin Sensitivity and Beta-Cell Glucose Sensitivity From the Response to Intravenous Glucose. J Clin Invest (1981) 68:1456–67. doi: 10.1172/JCI110398
73. Stamateris RE, Sharma RB, Hollern DA, Alonso LC. Adaptive β-Cell Proliferation Increases Early in High-Fat Feeding in Mice, Concurrent With Metabolic Changes, With Induction of Islet Cyclin D2 Expression. Am J Physiol-Endocrinol Metab (2013) 305:E149–59. doi: 10.1152/ajpendo.00040.2013
74. Mosser RE, Maulis MF, Moullé VS, Dunn JC, Carboneau BA, Arasi K, et al. High-Fat Diet-Induced β-Cell Proliferation Occurs Prior to Insulin Resistance in C57Bl/6J Male Mice. Am J Physiol-Endocrinol Metab (2015) 308:E573–82. doi: 10.1152/ajpendo.00460.2014
75. Román CL, Flores LE, Maiztegui B, Raschia MA, Del Zotto H, Gagliardino JJ. Islet NADPH Oxidase Activity Modulates β-Cell Mass and Endocrine Function in Rats With Fructose-Induced Oxidative Stress. Biochim Biophys Acta (BBA)-General Subj (2014) 1840:3475–82. doi: 10.1016/j.bbagen.2014.09.011
76. Kellard JA, Rorsman NJG, Hill TG, Armour SL, van de Bunt M, Rorsman P, et al. Reduced Somatostatin Signalling Leads to Hypersecretion of Glucagon in Mice Fed a High-Fat Diet. Mol Metab (2020) 40:101021. doi: 10.1016/j.molmet.2020.101021
77. Asghar ZA, Cusumano A, Yan Z, Remedi MS, Moley KH. Reduced Islet Function Contributes to Impaired Glucose Homeostasis in Fructose-Fed Mice. Am J Physiol-Endocrinol Metab (2017) 312:E109–16. doi: 10.1152/ajpendo.00279.2016
78. Talchai C, Xuan S, Lin HV, Sussel L, Accili D. Pancreatic β Cell Dedifferentiation as a Mechanism of Diabetic β Cell Failure. Cell (2012) 150:1223–34. doi: 10.1016/j.cell.2012.07.029
79. Dhawan S, Tschen S-I, Zeng C, Guo T, Hebrok M, Matveyenko A, et al. DNA Methylation Directs Functional Maturation of Pancreatic β Cells. J Clin Invest (2015) 125:2851–60. doi: 10.1172/JCI79956
80. Lee YS, Li P, Huh JY, Hwang IJ, Lu M, Kim JI, et al. Inflammation is Necessary for Long-Term But Not Short-Term High-Fat Diet–Induced Insulin Resistance. Diabetes (2011) 60:2474–83. doi: 10.2337/db11-0194
81. Javeed N, Her TK, Brown MR, Vanderboom P, Rakshit K, Egan AM, et al. Pro-Inflammatory β Cell Small Extracellular Vesicles Induce β Cell Failure Through Activation of the CXCL10/CXCR3 Axis in Diabetes. Cell Rep (2021) 36:109613. doi: 10.1016/j.celrep.2021.109613
82. Liu G, Li Y, Zhang T, Li M, Li S, He Q, et al. Mouse Single Islet β Cell Transcriptomic Reveals Sexually Dimorphic Transcriptome and Type 2 Diabetes Genes. Genomics Proteomics Bioinf (2021) 19(3):408–22. doi: 10.1101/2020.09.22.307421
83. Le May C, Chu K, Hu M, Ortega CS, Simpson ER, Korach KS, et al. Estrogens Protect Pancreatic β-Cells From Apoptosis and Prevent Insulin-Deficient Diabetes Mellitus in Mice. Proc Natl Acad Sci (2006) 103:9232–7. doi: 10.1073/pnas.0602956103
84. Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. β-Cell Deficit and Increased β-Cell Apoptosis in Humans With Type 2 Diabetes. Diabetes (2003) 52:102–10. doi: 10.2337/diabetes.52.1.102
85. Ottosson-Laakso E, Krus U, Storm P, Prasad RB, Oskolkov N, Ahlqvist E, et al. Glucose-Induced Changes in Gene Expression in Human Pancreatic Islets: Causes or Consequences of Chronic Hyperglycemia. Diabetes (2017) 66:3013–28. doi: 10.2337/db17-0311
86. Wang YJ, Kaestner KH. Single-Cell RNA-Seq of the Pancreatic Islets—-a Promise Not Yet Fulfilled? Cell Metab (2019) 29:539–44. doi: 10.1016/j.cmet.2018.11.016
87. Hrvatin S, Deng F, O'Donnell CW, Gifford DK, Melton DA. MARIS: Method for Analyzing RNA Following Intracellular Sorting. PloS One (2014) 9:e89459. doi: 10.1371/journal.pone.0089459
88. DiGruccio MR, Mawla AM, Donaldson CJ, Noguchi GM, Vaughan J, Cowing-Zitron C, et al. Comprehensive Alpha, Beta and Delta Cell Transcriptomes Reveal That Ghrelin Selectively Activates Delta Cells and Promotes Somatostatin Release From Pancreatic Islets. Mol Metab (2016) 5:449–58. doi: 10.1016/j.molmet.2016.04.007
89. Piñeros AR, Gao H, Wu W, Liu Y, Tersey SA, Mirmira RG. Single-Cell Transcriptional Profiling of Mouse Islets Following Short-Term Obesogenic Dietary Intervention. Metabolites (2020) 10:513. doi: 10.3390/metabo10120513
90. Sims EK, Hatanaka M, Morris DL, Tersey SA, Kono T, Chaudry ZZ, et al. Divergent Compensatory Responses to High-Fat Diet Between C57BL6/J and C57BLKS/J Inbred Mouse Strains. Am J Physiol-Endocrinol Metab (2013) 305:E1495–511. doi: 10.1152/ajpendo.00366.2013
91. Gu G, Dubauskaite J, Melton DA. Direct Evidence for the Pancreatic Lineage: NGN3+ Cells are Islet Progenitors and are Distinct From Duct Progenitors. Development (2002) 129(10):2447–57. doi: 10.1242/dev.129.10.2447
92. Awazawa M, Futami T, Sakada M, Kaneko K, Ohsugi M, Nakaya K, et al. Deregulation of Pancreas-Specific Oxidoreductin ERO1β in the Pathogenesis of Diabetes Mellitus. Mol Cell Biol (2014) 34:1290–9. doi: 10.1128/MCB.01647-13
93. Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, et al. Mapping and Analysis of Chromatin State Dynamics in Nine Human Cell Types. Nature (2011) 473:43–9. doi: 10.1038/nature09906
94. Grün D, Muraro MJ, Boisset J-C, Wiebrands K, Lyubimova A, Dharmadhikari G, et al. De Novo Prediction of Stem Cell Identity Using Single-Cell Transcriptome Data. Cell Stem Cell (2016) 19:266–77. doi: 10.1016/j.stem.2016.05.010
95. Cinti F, Bouchi R, Kim-Muller JY, Ohmura Y, Sandoval PR, Masini M, et al. Evidence of β-Cell Dedifferentiation in Human Type 2 Diabetes. J Clin Endocrinol Metab (2016) 101:1044–54. doi: 10.1210/jc.2015-2860
96. Wu L, Zhou L, Lu Y, Zhang J, Jian F, Liu Y, et al. Activation of SIRT1 Protects Pancreatic β-Cells Against Palmitate-Induced Dysfunction. Biochim Biophys Acta (BBA)-Molecular Basis Dis (2012) 1822:1815–25. doi: 10.1016/j.bbadis.2012.08.009
97. Xu EE, Sasaki S, Speckmann T, Nian C, Lynn FC. SOX4 Allows Facultative β-Cell Proliferation Through Repression of Cdkn1a. Diabetes (2017) 66:2213–9. doi: 10.2337/db16-1074
98. Lee G, Jang H, Kim YY, Choe SS, Kong J, Hwang I, et al. SREBP1c-PAX4 Axis Mediates Pancreatic β-Cell Compensatory Responses Upon Metabolic Stress. Diabetes (2019) 68:81–94. doi: 10.2337/db18-0556
99. Lee D, Gorkin DU, Baker M, Strober BJ, Asoni AL, McCallion AS, et al. A Method to Predict the Impact of Regulatory Variants From DNA Sequence. Nat Genet (2015) 47:955–61. doi: 10.1038/ng.3331
100. Keller MP, Rabaglia ME, Schueler KL, Stapleton DS, Gatti DM, Vincent M, et al. Gene Loci Associated With Insulin Secretion in Islets From Nondiabetic Mice. J Clin Invest (2019) 129:4419–32. doi: 10.1172/JCI129143
101. Keller MP, Gatti DM, Schueler KL, Rabaglia ME, Stapleton DS, Simecek P, et al. Genetic Drivers of Pancreatic Islet Function. Genetics (2018) 209:335–56. doi: 10.1534/genetics.118.300864
102. Mitok KA, Freiberger EC, Schueler KL, Rabaglia ME, Stapleton DS, Kwiecien NW, et al. Islet Proteomics Reveals Genetic Variation in Dopamine Production Resulting in Altered Insulin Secretion. J Biol Chem (2018) 293:5860–77. doi: 10.1074/jbc.RA117.001102
103. Eliasson L, Esguerra JLS. Role of Non-Coding RNAs in Pancreatic Beta-Cell Development and Physiology. Acta Physiol (2014) 211:273–84. doi: 10.1111/apha.12285
104. Calderari S, Diawara MR, Garaud A, Gauguier D. Biological Roles of microRNAs in the Control of Insulin Secretion and Action. Physiol Genomics (2017) 49:1–10. doi: 10.1152/physiolgenomics.00079.2016
105. Zhang F, Ma D, Zhao W, Wang D, Liu T, Liu Y, et al. Obesity-Induced Overexpression of miR-802 Impairs Insulin Transcription and Secretion. Nat Commun (2020) 11:1822. doi: 10.1038/s41467-020-15529-w
106. Jacovetti C, Abderrahmani A, Parnaud G, Jonas J-C, Peyot M-L, Cornu M, et al. MicroRNAs Contribute to Compensatory β Cell Expansion During Pregnancy and Obesity. J Clin Invest (2012) 122:3541–51. doi: 10.1172/JCI64151
107. Shen Z, Jiang H, Hsu H-T, Qian L, Fu Q, Shen M, et al. MicroRNA-127 Inhibits Cell Proliferation via Targeting Kif3b in Pancreatic β Cells. Aging (Albany NY) (2019) 11:1342. doi: 10.18632/aging.101835
108. Li Y, Deng S, Peng J, Wang X, Essandoh K, Mu X, et al. MicroRNA-223 is Essential for Maintaining Functional β-Cell Mass During Diabetes Through Inhibiting Both FOXO1 and SOX6 Pathways. J Biol Chem (2019) 294:10438–48. doi: 10.1074/jbc.RA119.007755
109. Jiang Y, Nishimura W, Devor-Henneman D, Kusewitt D, Wang H, Holloway MP, et al. Postnatal Expansion of the Pancreatic β-Cell Mass is Dependent on Survivin. Diabetes (2008) 57:2718–27. doi: 10.2337/db08-0170
110. Singer RA, Sussel L. Islet Long Noncoding RNAs: A Playbook for Discovery and Characterization. Diabetes (2018) 67:1461–70. doi: 10.2337/dbi18-0001
111. Akerman I, Tu Z, Beucher A, Rolando DMY, Sauty-Colace C, Benazra M, et al. Human Pancreatic β Cell lncRNAs Control Cell-Specific Regulatory Networks. Cell Metab (2017) 25:400–11. doi: 10.1016/j.cmet.2016.11.016
112. Sarkar S, Kutlu B, Velmurugan K, Kizaka-Kondoh S, Lee C, Wong R, et al. Cytokine-Mediated Induction of Anti-Apoptotic Genes That are Linked to Nuclear Factor Kappa-B (NF-κb) Signalling in Human Islets and in a Mouse Beta Cell Line. Diabetologia (2009) 52:1092–101. doi: 10.1007/s00125-009-1331-x
114. Wilder RM. Optimal Diets for Diabetic Patients. J Am Med Assoc (1924) 83:733–7. doi: 10.1001/jama.1924.02660100007003
115. Hussain TA, Mathew TC, Dashti AA, Asfar S, Al-Zaid N, Dashti HM. Effect of Low-Calorie Versus Low-Carbohydrate Ketogenic Diet in Type 2 Diabetes. Nutrition (2012) 28:1016–21. doi: 10.1016/j.nut.2012.01.016
116. Dashti HM, Mathew TC, Khadada M, Al-Mousawi M, Talib H, Asfar SK, et al. Beneficial Effects of Ketogenic Diet in Obese Diabetic Subjects. Mol Cell Biochem (2007) 302:249–56. doi: 10.1007/s11010-007-9448-z
117. Michalczyk MM, Klonek G, Maszczyk A, Zajac A. The Effects of a Low Calorie Ketogenic Diet on Glycaemic Control Variables in Hyperinsulinemic Overweight/Obese Females. Nutrients (2020) 12:1854. doi: 10.3390/nu12061854
118. Ellenbroek JH, Dijck LV, Töns HA, Rabelink TJ, Carlotti F, Ballieux BEPB, et al. Long-Term Ketogenic Diet Causes Glucose Intolerance and Reduced β- and α-Cell Mass But No Weight Loss in Mice. Am J Physiol-Endocrinol Metab (2014) 306:E552–8. doi: 10.1152/ajpendo.00453.2013
119. Bielohuby M, Sisley S, Sandoval D, Herbach N, Zengin A, Fischereder M, et al. Impaired Glucose Tolerance in Rats Fed Low-Carbohydrate, High-Fat Diets. Am J Physiol-Endocrinol Metab (2013) 305:E1059–70. doi: 10.1152/ajpendo.00208.2013
120. Badman MK, Kennedy AR, Adams AC, Pissios P, Maratos-Flier E. A Very Low Carbohydrate Ketogenic Diet Improves Glucose Tolerance in Ob/Ob Mice Independently of Weight Loss. Am J Physiol-Endocrinol Metab (2009) 297:E1197–204. doi: 10.1152/ajpendo.00357.2009
121. Fujita Y, Atageldiyeva KK, Takeda Y, Yanagimachi T, Makino Y, Haneda M. A Low-Carbohydrate Diet Improves Glucose Metabolism in Lean Insulinopenic Akita Mice Along With Sodium-Glucose Cotransporter 2 Inhibitor. Front Endocrinol (2020) 11:929. doi: 10.3389/fendo.2020.601594
122. Wang Y, Miura Y, Kaneko T, Li J, Qin L-Q, Wang P-Y, et al. Glucose Intolerance Induced by a High-Fat/Low-Carbohydrate Diet in Rats. Endocrine (2002) 17:185–91. doi: 10.1385/ENDO:17:3:185
123. Al-Khalifa A, Mathew TC, Al-Zaid NS, Mathew E, Dashti H. Low Carbohydrate Ketogenic Diet Prevents the Induction of Diabetes Using Streptozotocin in Rats. Exp Toxicologic Pathol (2011) 63:663–9. doi: 10.1016/j.etp.2010.05.008
124. Eguchi K, Nagai R. Islet Inflammation in Type 2 Diabetes and Physiology. J Clin Invest (2017) 127:14–23. doi: 10.1172/JCI88877
125. Zhen Y, Liu Z, Yang H, Yu X, Wu Q, Hua S, et al. Tumor Suppressor PDCD4 Modulates miR-184-Mediated Direct Suppression of C-MYC and BCL2 Blocking Cell Growth and Survival in Nasopharyngeal Carcinoma. Cell Death Dis (2013) 4:e872–2. doi: 10.1038/cddis.2013.376
126. Brown MR, Holmes H, Rakshit K, Javeed N, Her TK, Stiller AA, et al. Electrogenic Sodium Bicarbonate Cotransporter NBCe1 Regulates Pancreatic β Cell Function in Type 2 Diabetes. J Clin Invest 131 (2021) 131(17):e142365. doi: 10.1172/JCI142365
127. Montemurro C, Nomoto H, Pei L, Parekh VS, Vongbunyong KE, Vadrevu S, et al. IAPP Toxicity Activates HIF1α/PFKFB3 Signaling Delaying β-Cell Loss at the Expense of β-Cell Function. Nat Commun (2019) 10:1–17. doi: 10.1038/s41467-019-10444-1
128. Morita M, Prudent J, Basu K, Goyon V, Katsumura S, Hulea L, et al. mTOR Controls Mitochondrial Dynamics and Cell Survival via MTFP1. Mol Cell (2017) 67:922–35. doi: 10.1016/j.molcel.2017.08.013
129. Newsholme P, Brennan L, Bender K. Amino Acid Metabolism, β-Cell Function, and Diabetes. Diabetes (2006) 55:S39–47. doi: 10.2337/db06-S006
130. Alejandro EU, Gregg B, Wallen T, Kumusoglu D, Meister D, Chen A, et al. Maternal Diet–Induced microRNAs and mTOR Underlie β Cell Dysfunction in Offspring. J Clin Invest (2014) 124:4395–410. doi: 10.1172/JCI74237
131. Alejandro EU, Jo S, Akhaphong B, Llacer PR, Gianchandani M, Gregg B, et al. Maternal Low-Protein Diet on the Last Week of Pregnancy Contributes to Insulin Resistance and β-Cell Dysfunction in the Mouse Offspring. Am J Physiology-Regulatory Integr Comp Physiol (2020) 319:R485–96. doi: 10.1152/ajpregu.00284.2019
132. Petrik J, Reusens B, Arany E, Remacle C, Coelho C, Hoet JJ, et al. A Low Protein Diet Alters the Balance of Islet Cell Replication and Apoptosis in the Fetal and Neonatal Rat and Is Associated With a Reduced Pancreatic Expression of Insulin-Like Growth Factor-Ii1. Endocrinology (1999) 140:4861–73. doi: 10.1210/endo.140.10.7042
133. Lynch CJ, Adams SH. Branched-Chain Amino Acids in Metabolic Signalling and Insulin Resistance. Nat Rev Endocrinol (2014) 10:723–36. doi: 10.1038/nrendo.2014.171
134. Karusheva Y, Koessler T, Strassburger K, Markgraf D, Mastrototaro L, Jelenik T, et al. Short-Term Dietary Reduction of Branched-Chain Amino Acids Reduces Meal-Induced Insulin Secretion and Modifies Microbiome Composition in Type 2 Diabetes: A Randomized Controlled Crossover Trial. Am J Clin Nutr (2019) 110:1098–107. doi: 10.1093/ajcn/nqz191
135. Longo VD, Mattson MP. Fasting: Molecular Mechanisms and Clinical Applications. Cell Metab (2014) 19:181–92. doi: 10.1016/j.cmet.2013.12.008
136. Corezola do Amaral ME, Kravets V, Dwulet JM, Farnsworth NL, Piscopio R, Schleicher WE, et al. Caloric Restriction Recovers Impaired β-Cell-β-Cell Gap Junction Coupling, Calcium Oscillation Coordination, and Insulin Secretion in Prediabetic Mice. Am J Physiol-Endocrinol Metab (2020) 319:E709–20. doi: 10.1152/ajpendo.00132.2020
137. Lim EL, Hollingsworth K, Aribisala BS, Chen M, Mathers J, Taylor R. Reversal of Type 2 Diabetes: Normalisation of Beta Cell Function in Association With Decreased Pancreas and Liver Triacylglycerol. Diabetologia (2011) 54:2506–14. doi: 10.1007/s00125-011-2204-7
138. Kanda Y, Hashiramoto M, Shimoda M, Hamamoto S, Tawaramoto K, Kimura T, et al. Dietary Restriction Preserves the Mass and Function of Pancreatic β Cells via Cell Kinetic Regulation and Suppression of Oxidative/ER Stress in Diabetic Mice. J Nutr Biochem (2015) 26:219–26. doi: 10.1016/j.jnutbio.2014.10.007
139. Heymsfield SB, Harp JB, Reitman ML, Beetsch JW, Schoeller DA, Erondu N, et al. Why do Obese Patients Not Lose More Weight When Treated With Low-Calorie Diets? A Mechanistic Perspective. Am J Clin Nutr (2007) 85:346–54. doi: 10.1093/ajcn/85.2.346
140. Gill S, Panda S. A Smartphone App Reveals Erratic Diurnal Eating Patterns in Humans That can be Modulated for Health Benefits. Cell Metab (2015) 22:789–98. doi: 10.1016/j.cmet.2015.09.005
141. Folin O, Denis W. On Starvation and Obesity, With Special Reference to Acidosis. J Biol Chem (1915) 21:183–92. doi: 10.1016/S0021-9258(18)88204-7
142. Longo VD, Di Tano M, Mattson MP, Guidi N. Intermittent and Periodic Fasting, Longevity and Disease. Nat Aging (2021) 1:47–59. doi: 10.1038/s43587-020-00013-3
143. Greenwood R, Mahler R, Hales C. Improvement in Insulin Secretion in Diabetes After Diazoxide. Lancet (1976) 307:444–7. doi: 10.1016/S0140-6736(76)91473-2
144. Ritzel RA, Hansen JB, Veldhuis JD, Butler PC. Induction of β-Cell Rest by a Kir6. 2/SUR1-Selective KATP-Channel Opener Preserves β-Cell Insulin Stores and Insulin Secretion in Human Islets Cultured at High (11 Mm) Glucose. J Clin Endocrinol Metab (2004) 89:795–805. doi: 10.1210/jc.2003-031120
145. Laedtke T, Kjems L, Pørksen N, Schmitz O, Veldhuis J, Kao PC, et al. Overnight Inhibition of Insulin Secretion Restores Pulsatility and Proinsulin/Insulin Ratio in Type 2 Diabetes. Am J Physiol-Endocrinol And Metab (2000) 279:E520–8. doi: 10.1152/ajpendo.2000.279.3.E520
146. Jansson L, Kullin M, Karlsson FA, Bodin B, Hansen JB, Sandler S. KATP Channels and Pancreatic Islet Blood Flow in Anesthetized Rats: Increased Blood Flow Induced by Potassium Channel Openers. Diabetes (2003) 52:2043–8. doi: 10.2337/diabetes.52.8.2043
147. Rizzo MA, Magnuson MA, Drain PF, Piston DW. A Functional Link Between Glucokinase Binding to Insulin Granules and Conformational Alterations in Response to Glucose and Insulin. J Biol Chem (2002) 277:34168–75. doi: 10.1074/jbc.M112478200
148. Grill V, Björklund A. Overstimulation and Beta-Cell Function. Diabetes (2001) 50:S122. doi: 10.2337/diabetes.50.2007.S122
149. Kohsaka A, Laposky AD, Ramsey KM, Estrada C, Joshu C, Kobayashi Y, et al. High-Fat Diet Disrupts Behavioral and Molecular Circadian Rhythms in Mice. Cell Metab (2007) 6:414–21. doi: 10.1016/j.cmet.2007.09.006
150. Kim YH, Lee JH, Yeung JL-H, Das E, Kim RY, Jiang Y, et al. Thermogenesis-Independent Metabolic Benefits Conferred by Isocaloric Intermittent Fasting in Ob/Ob Mice. Sci Rep (2019) 9:1–10. doi: 10.1038/s41598-019-39380-2
151. Chaix A, Zarrinpar A, Miu P, Panda S. Time-Restricted Feeding is a Preventative and Therapeutic Intervention Against Diverse Nutritional Challenges. Cell Metab (2014) 20:991–1005. doi: 10.1016/j.cmet.2014.11.001
152. Chellappa SL, Qian J, Vujovic N, Morris CJ, Nedeltcheva A, Nguyen H, et al. Daytime Eating Prevents Internal Circadian Misalignment and Glucose Intolerance in Night Work. Sci Adv (2021) 7:eabg9910. doi: 10.1126/sciadv.abg9910
153. Wei S, Han R, Zhao J, Wang S, Huang M, Wang Y, et al. Intermittent Administration of a Fasting-Mimicking Diet Intervenes in Diabetes Progression, Restores β Cells and Reconstructs Gut Microbiota in Mice. Nutr Metab (2018) 15:1–12. doi: 10.1186/s12986-018-0318-3
154. Kolakowski J, Pizarro M, Gasparo M.d., Desmecht P, Harvengt C, Crabbé J. Influence of Fasting on Adrenocortical and Pancreatic Islet Response to Glucose Loads in the Obese. Eur J Clin Invest (1970) 1:25–31. doi: 10.1111/j.1365-2362.1970.tb00593.x
155. Butler PC, Meier JJ, Butler AE, Bhushan A. The Replication of β Cells in Normal Physiology, in Disease and for Therapy. Nat Clin Pract Endocrinol Metab (2007) 3:758–68. doi: 10.1038/ncpendmet0647
156. Martinez SC, Cras-Méneur C, Bernal-Mizrachi E, Permutt MA. Glucose Regulates Foxo1 Through Insulin Receptor Signaling in the Pancreatic Islet β-Cell. Diabetes (2006) 55:1581–91. doi: 10.2337/db05-0678
157. Kim-Muller JY, Zhao S, Srivastava S, Mugabo Y, Noh H-L, Kim YR, et al. Metabolic Inflexibility Impairs Insulin Secretion and Results in MODY-Like Diabetes in Triple FoxO-Deficient Mice. Cell Metab (2014) 20:593–602. doi: 10.1016/j.cmet.2014.08.012
158. Rakshit K, Qian J, Ernst J, Matveyenko AV. Circadian Variation of the Pancreatic Islet Transcriptome. Physiol Genomics (2016) 48:677–87. doi: 10.1152/physiolgenomics.00019.2016
159. Turek FW, Joshu C, Kohsaka A, Lin E, Ivanova G, McDearmon E, et al. Obesity and Metabolic Syndrome in Circadian Clock Mutant Mice. Science (2005) 308:1043–5. doi: 10.1126/science.1108750
160. Hou T, Su W, Duncan MJ, Olga VA, Guo Z, Gong MC. Time-Restricted Feeding Protects the Blood Pressure Circadian Rhythm in Diabetic Mice. Proc Natl Acad Sci (2021) 118. doi: 10.1073/pnas.2015873118
161. Forneris F, Binda C, Battaglioli E, Mattevi A. LSD1: Oxidative Chemistry for Multifaceted Functions in Chromatin Regulation. Trends Biochem Sci (2008) 33:181–9. doi: 10.1016/j.tibs.2008.01.003
162. Mason IC, Qian J, Adler GK, Scheer FA. Impact of Circadian Disruption on Glucose Metabolism: Implications for Type 2 Diabetes. Diabetologia (2020) 63:462–72. doi: 10.1007/s00125-019-05059-6
163. Kitamura T, Nakae J, Kitamura Y, Kido Y, Biggs WH, Wright CV, et al. The Forkhead Transcription Factor Foxo1 Links Insulin Signaling to Pdx1 Regulation of Pancreatic β Cell Growth. J Clin Invest (2002) 110:1839–47. doi: 10.1172/JCI200216857
164. Stratmann M, Suter DM, Molina N, Naef F, Schibler U. Circadian Dbp Transcription Relies on Highly Dynamic BMAL1-CLOCK Interaction With E Boxes and Requires the Proteasome. Mol Cell (2012) 48:277–87. doi: 10.1016/j.molcel.2012.08.012
165. Nam HJ, Boo K, Kim D, Han D-H, Choe HK, Kim CR, et al. Phosphorylation of LSD1 by Pkcα is Crucial for Circadian Rhythmicity and Phase Resetting. Mol Cell (2014) 53:791–805. doi: 10.1016/j.molcel.2014.01.028
166. Tornovsky-Babeay S, Dadon D, Ziv O, Tzipilevich E, Kadosh T, Haroush RS-B, et al. Type 2 Diabetes and Congenital Hyperinsulinism Cause DNA Double-Strand Breaks and P53 Activity in β Cells. Cell Metab (2014) 19:109–21. doi: 10.1016/j.cmet.2013.11.007
167. Skene PJ, Henikoff S. An Efficient Targeted Nuclease Strategy for High-Resolution Mapping of DNA Binding Sites. Elife (2017) 6:e21856. doi: 10.7554/eLife.21856
Keywords: epigenetics, pancreatic islet, type 2 diabetes mellitus, high-fat diet, ketogenic diet, low-protein diet, intermittent fasting, time-restricted feeding
Citation: Brown MR and Matveyenko AV (2022) It’s What and When You Eat: An Overview of Transcriptional and Epigenetic Responses to Dietary Perturbations in Pancreatic Islets. Front. Endocrinol. 13:842603. doi: 10.3389/fendo.2022.842603
Received: 23 December 2021; Accepted: 07 February 2022;
Published: 10 March 2022.
Edited by:
Senta Georgia, Children’s Hospital of Los Angeles, United StatesReviewed by:
Jeffery Sivert Tessem, Brigham Young University, United StatesSangeeta Dhawan, City of Hope National Medical Center, United States
Copyright © 2022 Brown and Matveyenko. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aleksey V. Matveyenko, bWF0dmV5ZW5rby5hbGVrc2V5QG1heW8uZWR1; Matthew R. Brown, YnJvd24ubWF0dGhld0BtYXlvLmVkdQ==