 Nelmari Ruiz-Otero1*
Nelmari Ruiz-Otero1* Rejji Kuruvilla2
Rejji Kuruvilla2- 1Division of Endocrinology, Diabetes & Metabolism, Department of Medicine, Johns Hopkins University School of Medicine, Baltimore, MD, United States
- 2Department of Biology, Johns Hopkins University, Baltimore, MD, United States
Cell-cell interactions are necessary for optimal endocrine functions in the pancreas. β-cells, characterized by the expression and secretion of the hormone insulin, are a major constituent of functional micro-organs in the pancreas known as islets of Langerhans. Cell-cell contacts between β-cells are required to regulate insulin production and glucose-stimulated insulin secretion, which are key determinants of blood glucose homeostasis. Contact-dependent interactions between β-cells are mediated by gap junctions and cell adhesion molecules such as E-cadherin and N-CAM. Recent genome-wide studies have implicated Delta/Notch-like EGF-related receptor (Dner) as a potential susceptibility locus for Type 2 Diabetes in humans. DNER is a transmembrane protein and a proposed Notch ligand. DNER has been implicated in neuron-glia development and cell-cell interactions. Studies herein demonstrate that DNER is expressed in β-cells with an onset during early postnatal life and sustained throughout adulthood in mice. DNER loss in adult β-cells in mice (β-Dner cKO mice) disrupted islet architecture and decreased the expression of N-CAM and E-cadherin. β-Dner cKO mice also exhibited impaired glucose tolerance, defects in glucose- and KCl-induced insulin secretion, and decreased insulin sensitivity. Together, these studies suggest that DNER plays a crucial role in mediating islet cell-cell interactions and glucose homeostasis.
Introduction
Novel genomic and proteomic analyses have allowed researchers to identify hundreds of candidates of genes involved in glucose homeostasis (1–4). However, understanding the molecular role and cellular expression, regulation, and overall function of identified genes is crucial to properly diagnose and treat diseases leading to glucose homeostasis defects, such as diabetes. Genome-wide studies in two independent populations have implicated Delta/Notch-like EGF-related receptor (DNER) as a potential susceptibility loci for Type 2 diabetes in humans (5, 6). DNER is a single-pass transmembrane protein with ten EGF-like repeats and a short cytoplasmic tail (7). Interestingly, the sequence and protein structure of DNER shows similarities with conserved Notch ligands, suggesting a role for DNER in Notch signaling (8, 9). DNER contains ten EGF-like domains, which are often implicated in protein-protein interactions, often at the plasma membrane where DNER is localized (7, 9–12). Moreover, previous studies have characterized a crucial role for DNER in mediating neuron-glia interactions (8, 13). DNER was first identified in the olfactory bulb, hippocampus and Purkinje cells in the cerebellum (7, 9). Studies in mice suggest that DNER induces the morphological differentiation of Bergmann glia, which are polarized astrocytes located in the cerebellar cortex (8). Mice with global deletion of DNER showed delayed maturation of the cerebellum, motor coordination deficits, and impaired glutamate removal from synapses (14). Importantly, DNER-Notch signaling is critical in the development and maturation of the central nervous system (8, 11, 13–15).
While DNER expression was initially believed to be restricted to the nervous system, studies have found that DNER is expressed in pancreatic tissues (16). Specifically, DNER expression was observed in mature β-cells using RNA-sequencing analysis (17). The onset of DNER expression in β-cells coincides with the period of β-cell maturation, when β-cells begin to coordinate their insulin release in response to glucose (17). Notably, β-cell maturation is characterized by increases in junctional protein content, where adhesive proteins and secretory machinery begin to assemble at the cell periphery for proper β-cell function (18–20). Furthermore, DNER was identified to be up-regulated upon glucose and cAMP stimulation in an insulin secreting cell line (3). Together, these findings suggest a potential role for DNER in blood glucose homeostasis. Here, we describe studies examining DNER function in the pancreatic β-cells and its role in blood glucose homeostasis in mice.
Materials and methods
Animals
All procedures relating to animal care and treatment conformed to Johns Hopkins University Animal Care and Use Committee (ACUC) and NIH guidelines. Animals were under direct supervision of the staff veterinarian and were monitored daily by Animal Care Staff. In addition to the sterile environment and weekly cage changes, mice were kept in a temperature-controlled room at 22°C and in a 12-hour light/12-hour dark cycle. Mice were maintained on a C57BL/6 background, or mixed C57BL/6J and 129P, or C57BL/6J and FVB backgrounds. Both sexes were used for analyses.
Dnertm3a KO first allele (C57BL/6NTac-Dnertm3a(EUCOMM)Hmgu/Ieg) mice were purchased from EMMA Repository (strain ID: EM:08389). Dnertm3a mice were crossed to a ROSA26-FLPe mouse (Jackson strain number: 003946) to knock out the LacZ and neo cassette to generate the conditional Dnertm3c allele (Dner-floxed) mice, which were then mated to Pdx1-CreER to generate Pdx1-CreER; Dnerf/f (β-Dner cKO) mice.
Three-week old mice were weaned and introduced to control (TD.07570; Envigo) or tamoxifen diet (TD.130856; Envigo with 250 mg/kg diet) for 10 days with a 2-day break on standard chow after the fifth day. At eight weeks of age, control- and tamoxifen-fed animals were subjected to metabolic assays and pancreata were dissected for further morphological examination.
Metabolic assays
Glucose tolerance test
Mice were housed individually and fasted overnight (16 hours) before glucose administration. Mice were administered 2g/kg of glucose by gavage feeding needle. Blood glucose levels were measured with a OneTouch Ultra 2 glucometer (Lifescan) before glucose administration and over a two-hour period. The area under the curve was determined for glucose tolerance for each animal using Graphpad Prism, version 9.
In vivo insulin secretion assay
Mice were housed individually and fasted overnight before glucose administration. Mice were given 3g/kg of glucose by gavage feeding needle. During a one-hour period, blood was collected in EDTA coated tubes and centrifuged. Insulin was measured from the plasma fraction collected using the Ultrasensitive Insulin ELISA (Crystal Chem, 90080).
Insulin sensitivity assay
Mice were individually housed with food overnight before being given an intraperitoneal (IP) insulin injection containing 0.75U/kg (Novolin-R, Novo Nordisk diluted in sodium chloride; Sigma-Aldrich). Blood glucose levels were assessed using a OneTouch Ultra 2 glucometer.
Islet isolation
Islets from adult animals were isolated as previously described (21). Pancreata were distended by injecting Collagenase P, from Clostridium histolyticum (Sigma-Aldrich 1124900200: 0.28mg/mL) diluted in Hanks’ Balanced Salt Solution (HBSS) (Gibco, 14175103) through the bile duct. Tissues were then incubated at 37°C, washed in 0.1% BSA in HBSS and subjected to discontinuous gradient using histopaque (6:5 Histopaque 1119: Histopaque 1077; Sigma-Aldrich 11191 and 10771) and HBSS. Islet layers were handpicked for further assays.
Ex vivo insulin secretion assay
Isolated islets were cultured in RPMI-1640 5%FBS 5U/L penicillin-streptomycin, (Gibco, 11875093) overnight to allow recovery. Islets were washed three times and were left pre-incubating for one hour in low glucose (2.8mM) Krebs Ringer Buffer (KRB). Groups of 5-10 islets were incubated in 500μL of either in low or high (16.7mM) glucose KRB for 30 minutes. After stimulation, the supernatant was collected and islets were lysed in acid ethanol (0.4N HCl, 75% EtOH) overnight. Islets were neutralized with Tris base (0.885M) and insulin was measured from the cell and supernatant fractions using the Ultrasensitive Insulin ELISA (Crystal Chem, 90080).
Total insulin content was measured by ELISA, normalized by the total amount of islets per sample, and averaged from at least two replicates per animal.
Immunohistochemistry
Cryo-preserved tissues
Pancreata were dissected and fixed in 4% paraformaldehyde in PBS (PFA, Sigma) at 4°C overnight (16 hours). Tissues were cryo-protected in 30% sucrose in PBS for 24 hours and then equilibrated in 1:1 30%sucrose: OCT (Sakura Finetek) before embedding in OCT. Tissue was collected in 50μm thick sections.
Slides were washed in PBS, permeabilized in 1% Triton X-100 in PBS, and blocked for one hour at room temperature using 5% donkey serum 2%BSA in 0.5% Triton X-100 in PBS. Sections were then incubated for 24 hours at 4°C with primary antibodies (see Table 1). Following PBS washes, sections were incubated with corresponding secondary antibodies and DAPI for one hour at room temperature, washed, and mounted using Fluoromount Aqueous Mounting Medium.
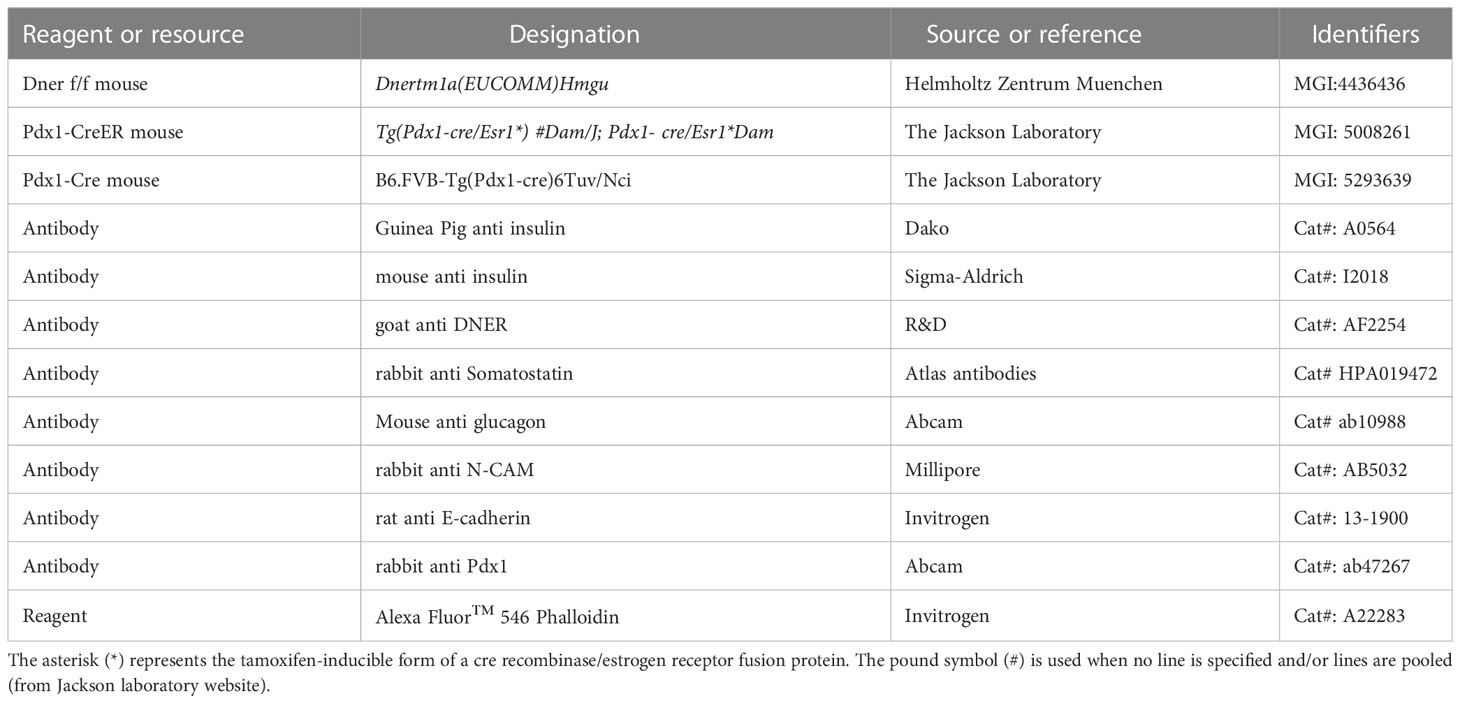
Table 1 Reagents and resources.
Paraffin embedded tissues
Pancreata were fixed in Bouin’s solution (Sigma-Aldrich, HT10132) at room temperature for four hours and left in 70% ethanol overnight. The pancreata were later dehydrated using increasing concentrations of ethanol, then incubated in a series of xylene and warm paraffin washes before being embedded in paraffin. Tissues were collected in 6μm thick sections.
Slides were washed in xylene and decreasing concentrations of ethanol to rehydrate. Then slides were washed with water and incubated in hot sodium citrate solution for antigen retrieval for 10 minutes. After cooling, slides were washed in PBS, incubated with glycine (0.1M), washed, and blocked for one hour on 5% goat serum 2%BSA in 0.5% Triton X-100 in PBS. Sections were then incubated overnight at 4°C with primary antibodies. Following PBS washes, sections were incubated with secondary antibodies and DAPI for one hour at room temperature, washed and mounted using Fluoromount Aqueous Mounting Medium.
Imaging and image processing
Z-stacks were obtained using Zeiss LSM 700 microscopes equipped with 405, 488, 555, and 633 lasers. Images were processed using FIJI software. To measure the fluorescence intensity, an outline of the islet was made using the Fiji software to create the ROI. The area and integrated density of the desired channel was measured in each ROI. A second area was selected to serve as background and the mean grey value was measured. The corrected fluorescence intensity was calculated as: integrated density – (Islet area* mean grey fluorescence background readings).
qRT-PCR
RNA was isolated from freshly isolated islets using Trizol Reagent (Invitrogen, 15596018) phenol-chloroform extraction and ethanol precipitation. RNA was then reverse transcribed using SuperScript IV First-Strand Synthesis System (Invitrogen, 18091050). Quantitative RT PCR was performed using Power SYBR Green PCR Master Mix (Applied Biosystems, 4367659) and the primer sets listed on Table 2 on StepOnePlus Real-Time PCR System (ThermoFisher). Using 18S as endogenous control for normalization, the fold change was calculated using the 2(-ΔΔCt) method.
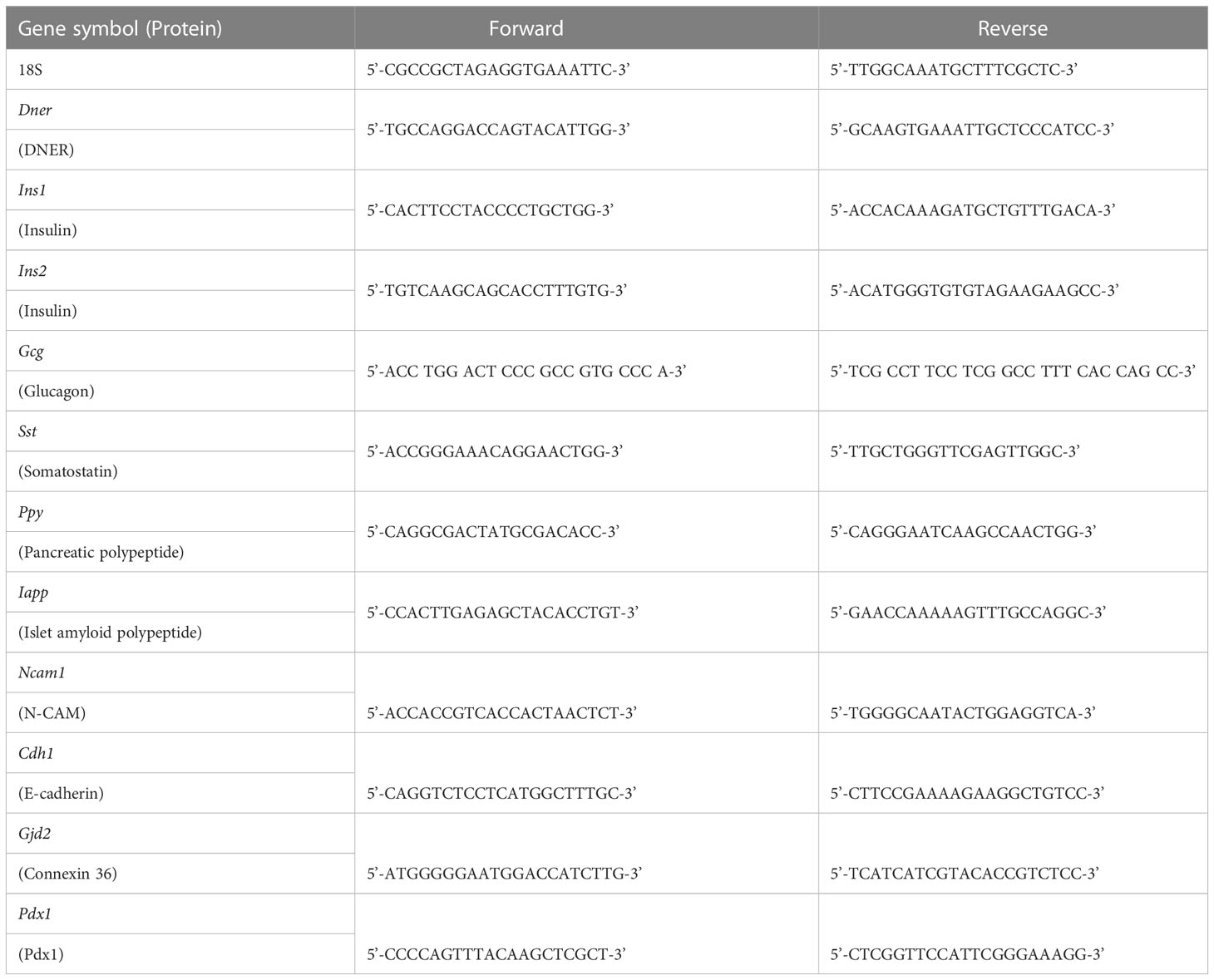
Table 2 Primers used for qRT-PCR.
Quantifications and statistical analysis
Sample sizes were calculated based on power analysis and similar to those reported in previous publications (22–24). All analyses were performed in a semi-blinded manner, such that the investigator was not aware of the genotypes of each specific sample. All Student’s t-tests were performed assuming Gaussian distribution, two- tailed, unpaired, and with a confidence interval of 95%, except for normalized data (qRT-PCR and insulin sensitivity), which were done using a One sample t-test. Statistical analyses were based on at least 3 independent experiments and described in the figure legends.
Results
DNER expression in the pancreas
To define when and where DNER was expressed in the pancreas, we performed immunohistochemistry using an anti-DNER antibody in mice. DNER protein expression was mostly absent from the islets at perinatal periods, but after postnatal day seven (P7) and throughout adulthood, it was found mainly in insulin-producing β-cells with some limited expression in non-insulin expressing islet cells (Figure 1). Importantly, adult islets also show co-expression of DNER with glucagon (Supplemental Figure 1) These findings, together with recent evidence of transcriptional expression of DNER in mature β-cells (17) and its regulation by glucose in an insulin-producing cell line (3) suggested that DNER may play a functional role in mature β-cells.
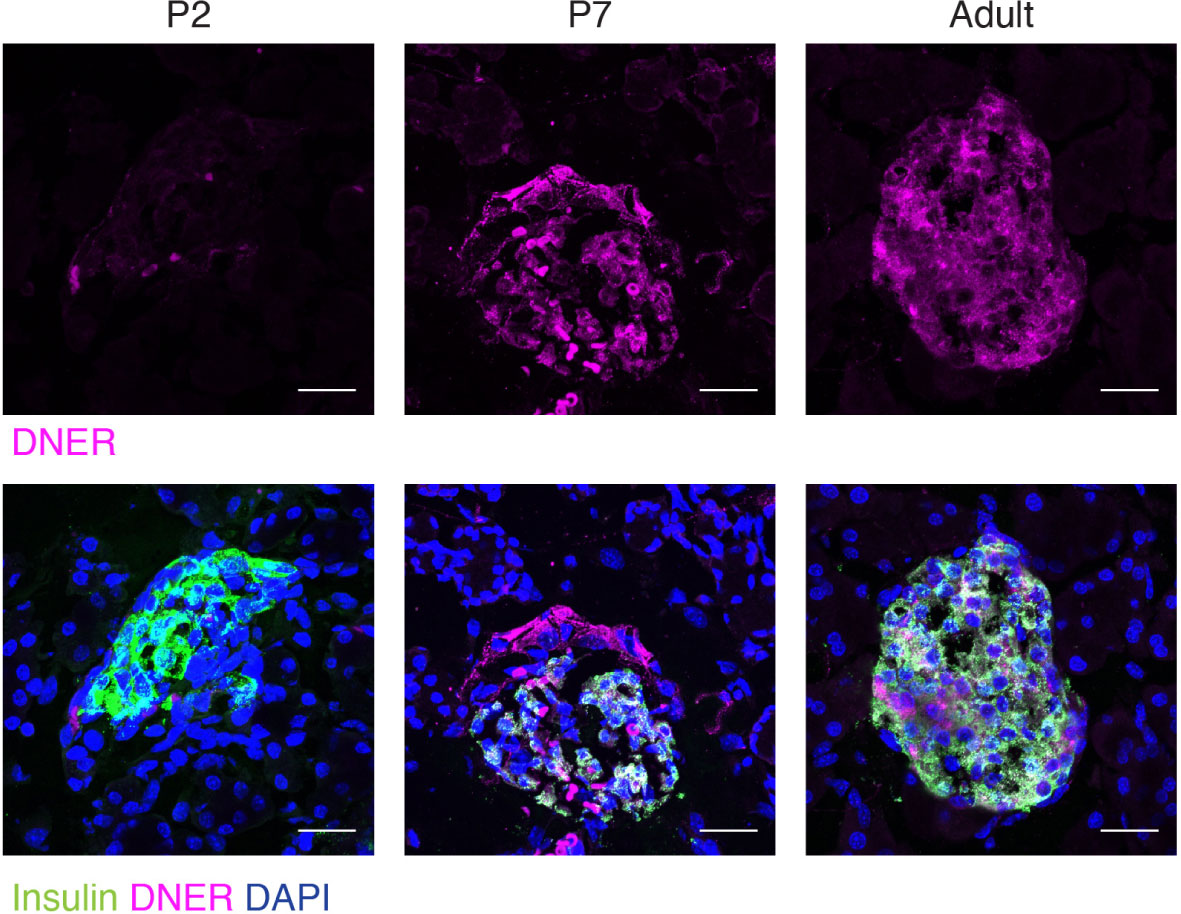
Figure 1 DNER expression in β-cells corresponds to the onset of β-cell maturation. Immunohistochemistry in wild-type tissues shows DNER (magenta) expression co-localizing with insulin (green) after P7. Scale bars: 25μm. Representative images from 3 independent experiments.
Functional characterization of mice lacking DNER in β-cells
To understand the role of DNER in mature β-cells, Pdx1-CreER (25) expressing mice were crossed with DNER floxed allele-containing mice (Dnerf/f) (EUCOMM) (Supplemental Figure 2A). In the mature pancreas, Pdx1 is primarily expressed by the β-cells, although a small proportion of α-, PP- and δ-cells express Pdx1 (26, 27). Juvenile Pdx1-CreER; Dnerf/f mice (P21) were introduced to either a control (TD.07570; Envigo) or tamoxifen diet (TD.130856; Envigo; 250 mg of tamoxifen/kg diet) for ten days to generate control or β-Dner cKO mice respectively. Pdx1-CreER; Dnerf/f animals were born at expected Mendelian frequencies (from a Pdx1-CreER; Dnerf/f to Dner f/f cross 52.38% of the progeny was Dnerf/f and 47.62% were Pdx1-CreER; Dnerf/f mice; N= 6 litters and 42 animals), they did not display gross anatomical or morphological abnormalities and survived to adulthood. DNER loss in islets was confirmed using immunohistochemistry (Supplemental Figure 2B). Quantitative Real Time Polymerase Chain Reaction (qRT-PCR) analysis from isolated islets also showed a decrease in DNER transcripts (73% decrease) in β-Dner cKO islets relative to control islets (Supplemental Figure 2C), indicating significant deletion of DNER following induction with tamoxifen.
To assess whether DNER plays a role in blood glucose homeostasis, glucose tolerance tests were performed in β-Dner cKO mice and litter-mate controls. β-Dner cKO animals exhibited impaired glucose tolerance at eight-weeks of age (Figures 2A, B). Defects in glucose tolerance typically arise due to impaired insulin secretion, lack of insulin sensitivity or a combination of both, although insulin-independent mechanisms may also influence glucose physiology (28, 29). To first determine whether insulin secretion was affected in mice lacking DNER in β-cells, a glucose-stimulated insulin secretion (GSIS) assay was performed in vivo. Adult β-Dner cKO mice had lower concentrations of circulating insulin after glucose stimulus (Figure 2C). However, levels of blood glucose and circulating insulin when fed ad libitum where similar in β-Dner cKO and littermate controls (Figures 2D, E). In addition, β-Dner cKO animals responded poorly to exogenously administered insulin, showing no significant decreases in glucose levels (Figure 2F). The insulin sensitivity defects were surprising given that DNER depletion in Pdx1-expressing cells is not expected to directly affect insulin-responsive tissues such as the liver or adipose tissue. Insulin resistance often arises from obesity, but female β-Dner cKO mice had similar body weight to controls, and male β-Dner cKO mice had decreased body weight than controls (Supplemental Figure 3A). However, males had a similar trend to glucose intolerance as females, although not statistically significant (Supplemental Figures 3B, C). Markedly, in vivo insulin secretion is significantly dampened in females but not in males, although a trend to a modest decreased insulin secretion is also present (Supplemental Figures 3D, E). Together, these results suggest a novel role for DNER in regulating blood glucose homeostasis.
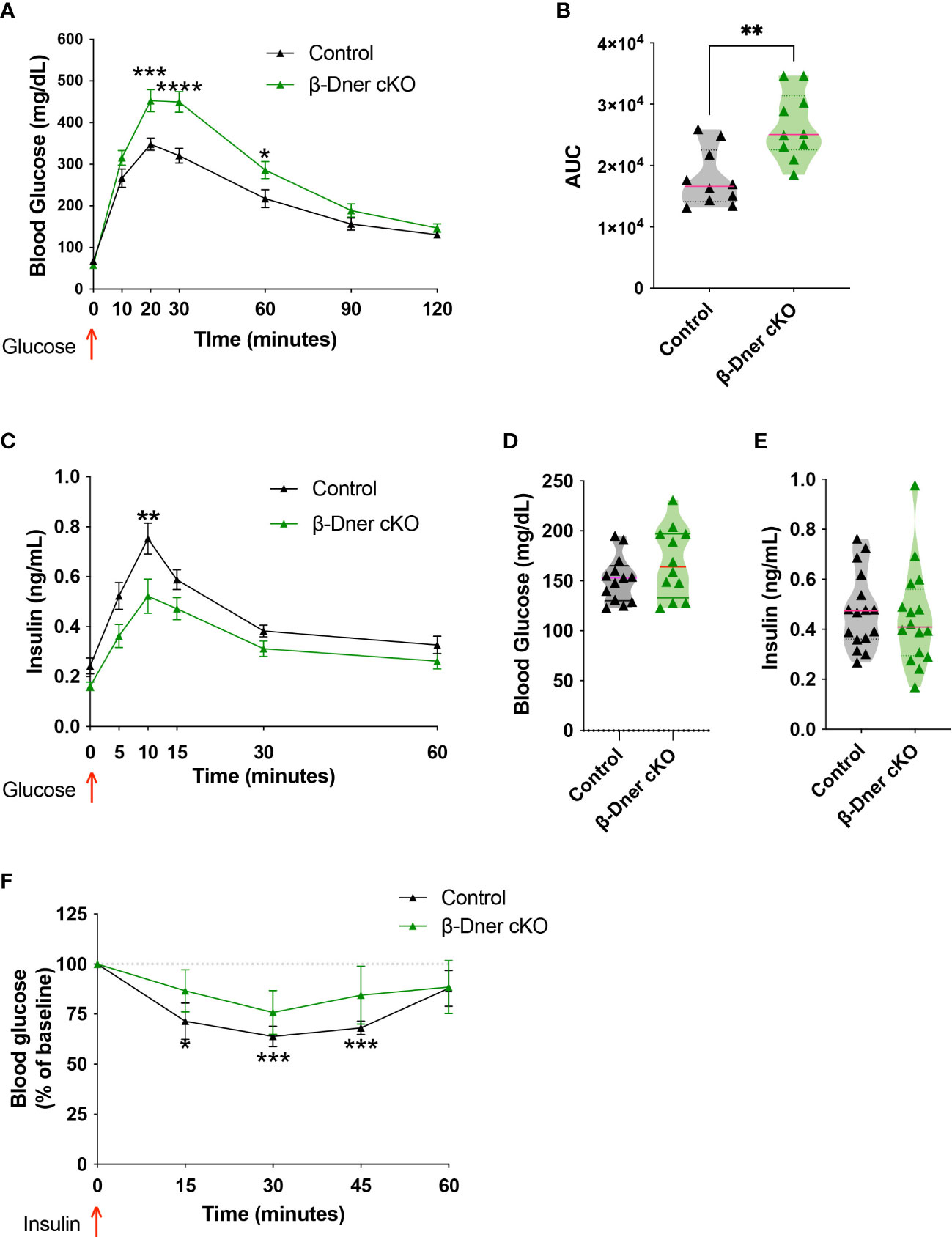
Figure 2 Loss of DNER from β-cells results in impaired glucose tolerance, decreased insulin secretion and loss of insulin sensitivity in vivo. (A) Glucose tolerance test (Means ± SEM for N = 10 animals per group: 2-way ANOVA *p < 0.05, ***p < 0.001, ****p < 0.0001). (B) Area under the curve (AUC) for glucose tolerance (t-test, **p<0.01). (C) Glucose-stimulated insulin secretion in vivo (Means ± SEM for N= 17 animals per group: 2-way ANOVA **p < 0.01). (D, E) Fed ad libidum levels of glucose and circulating insulin (F) Insulin sensitivity test (Means ± SEM for N= 6 controls and 7 β-Dner cKO animals: One sample t-test relative to baseline: *p < 0.05, ***p< 0.001).
To determine if the insulin secretion defects observed in β-Dner cKO mice were intrinsic to the islets, we performed glucose-stimulated insulin secretion assays using isolated islets from β-Dner cKO or control mice. As expected, a high concentration of glucose (16.7mM) elicited enhanced insulin secretion respective to basal glucose levels (2.8mM) in control islets. However, islets isolated from β-Dner cKO mice showed no difference in insulin secretion between low and high glucose concentrations (Figure 3A). Regulation of glucose-stimulated insulin release can occur at multiple levels including glucose sensing, metabolism, β-cell depolarization and insulin granule mobilization and exocytosis (30–33). To assess which steps in this pathway contributed to the diminished insulin secretion in β-Dner cKO islets, we treated isolated islets with a known insulin secretagogue, potassium chloride (KCl). High extracellular K+ depolarizes the β-cell plasma membrane, which by-passes the need for glucose metabolism to promote insulin secretion. Specifically, elevated potassium chloride (KCl, 30 mM) treatment results in the opening of voltage-dependent Ca2+ channels to promote exocytosis of secretion-ready insulin granules (34). We found that β-Dner cKO islets did not secrete insulin in response to high KCl in contrast to controls (Figure 3B), suggesting secretory defects independent of glucose entry and metabolism.
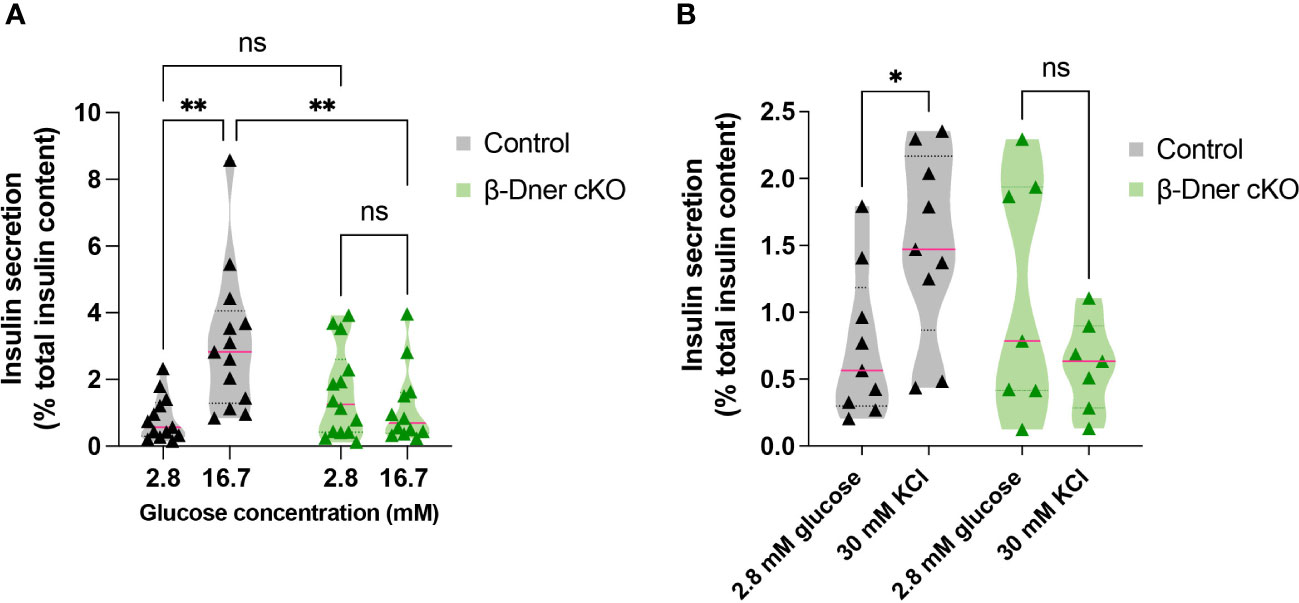
Figure 3 β-Dner cKO islets have intrinsic insulin secretion deficits. (A) Glucose-stimulated insulin secretion in isolated islets (Means ± SEM for islets from N= 10-12 animals per group: 2-way ANOVA **p < 0.01, ns p>0.05). (B) Depolarization-induced insulin secretion using KCl in isolated islets (Means ± SEM for islets from N= 7-9 animals per group: Two-way ANOVA *p < 0.05). ns or not significant: p>0.05.
Lack of DNER leads to low insulin levels
Given the crucial role of islet morphology to its function, we decided to visualize control and DNER-depleted tissues using immunohistochemistry against islet hormones. We observed decreased insulin immunoreactivity (Figure 4A). Consistent with the decreased immunostaining, insulin protein content was reduced in β-Dner cKO islets (Figure 4B). Next, to determine whether insulin protein levels were decreased because of reduced RNA expression, we performed qRT-PCR on cDNA from isolated islets. Unlike humans, rodents contain two insulin coding genes: a functional transposon-mediated duplication Ins1 and the ancestral gene Ins2. The two transcripts are localized in different chromosomes and differ in some regulatory elements, including the exclusion of an intron in Ins1, and the regulation through FoxO1 of Ins2 but not Ins1 leading to differential modulation of their levels in certain conditions (35–37). Only Ins1, but not Ins2 nor any other endocrine transcript tested were downregulated in DNER depleted islets (Figure 4C). The decrease observed in insulin was not due to loss of β-cells, as the number of β- cells per islet was unchanged versus control (Figure 4D). We also assessed α-cell number using glucagon immunostaining (Supplemental Figures 4A, B) and observed a decrease in glucagon+ α-cells. However, islet numbers were similar among control and β-Dner cKO tissues (Supplemental Figure 4C). Therefore, it is possible that DNER also acts regulating other islet endocrine cells. Taken together, these data indicate DNER in regulates insulin content in islets from adult mice.
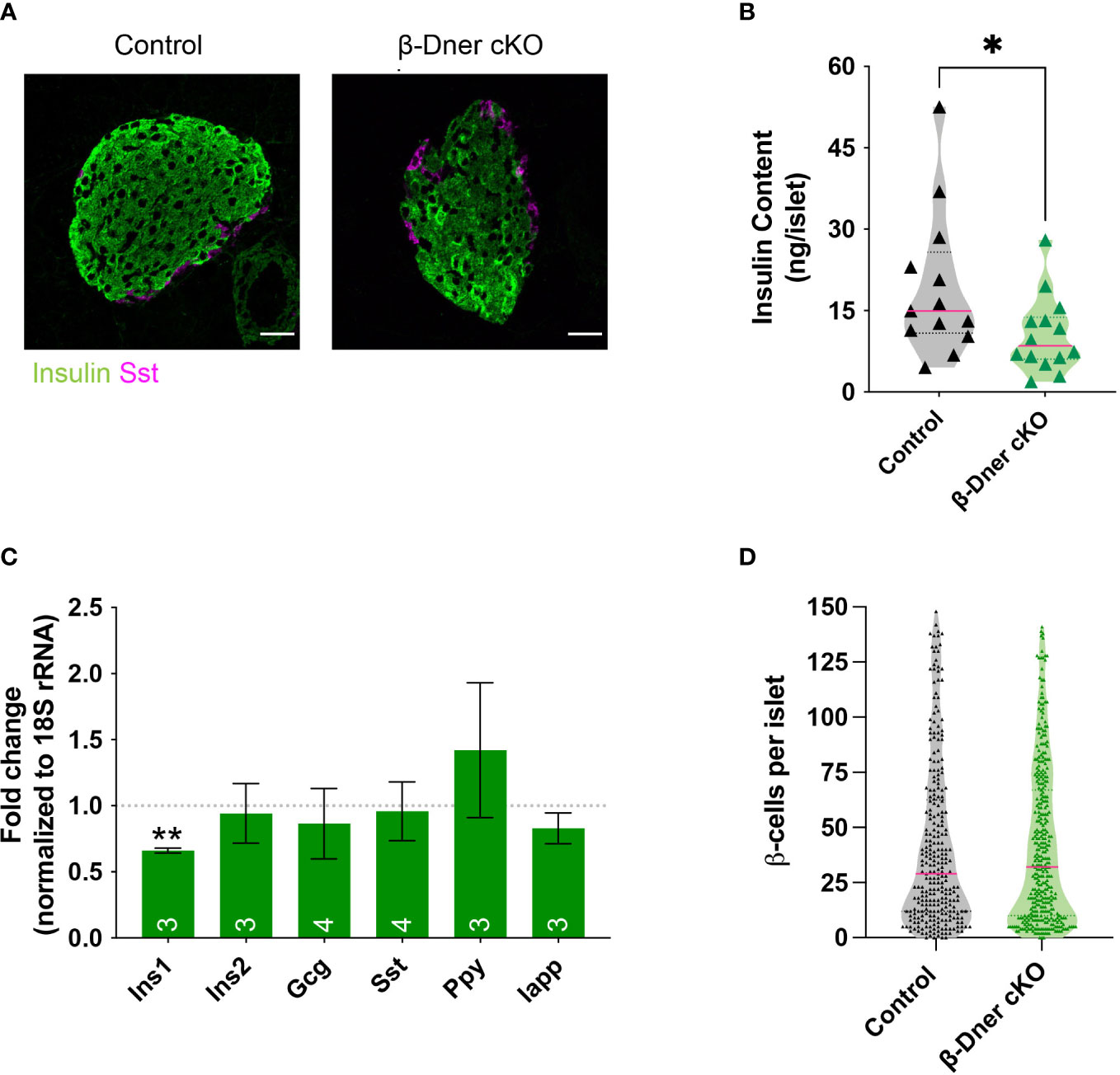
Figure 4 β-Dner cKO animals have lower insulin levels than littermate controls. (A) Immunohistochemistry of pancreatic islets stained for insulin (green) and somatostatin (magenta). Scale bars: 25μm. Representative images from 4 independent experiments. (B) Insulin protein content measured by ELISA (N=9-10 animals per group: t-test *p<0.05). (C) qRT-PCR analyses for endocrine markers, normalized by 18S and shown as relative to controls (Means ± SEM for islets from N= 3-4 animals per group: One sample t-test **p< 0.01). (D) β-cell number quantifications (N=4-5 animals per group; data shown as cells per islet).
DNER mediates proteins involved in β-cell-cell interactions
Since DNER regulates neuron-glia interactions in the CNS, we hypothesized that DNER might similarly regulate cell-cell interactions between islet cell types, either among β-cells and/or neighboring cells. To examine this possibility immunohistochemistry was used to visualize E-cadherin, a key mediator of β-cell-cell contacts. E-cadherin is expressed in multiple cell types in the pancreas, but plays an important role in promoting Ca2+-dependent intra-islet homophilic adhesions between β-cells (38–40). Interestingly, DNER deletion in β-Dner cKO mice resulted in decreased immunoreactivity of E-cadherin in islet cells (Figures 5A, B). This result was specific to islet cells, as exocrine pancreatic tissue near the islets did not show changes in E-cadherin levels (Figures 5A, B). These results provide evidence supporting of a role for DNER in regulating E-cadherin in pancreatic islets.
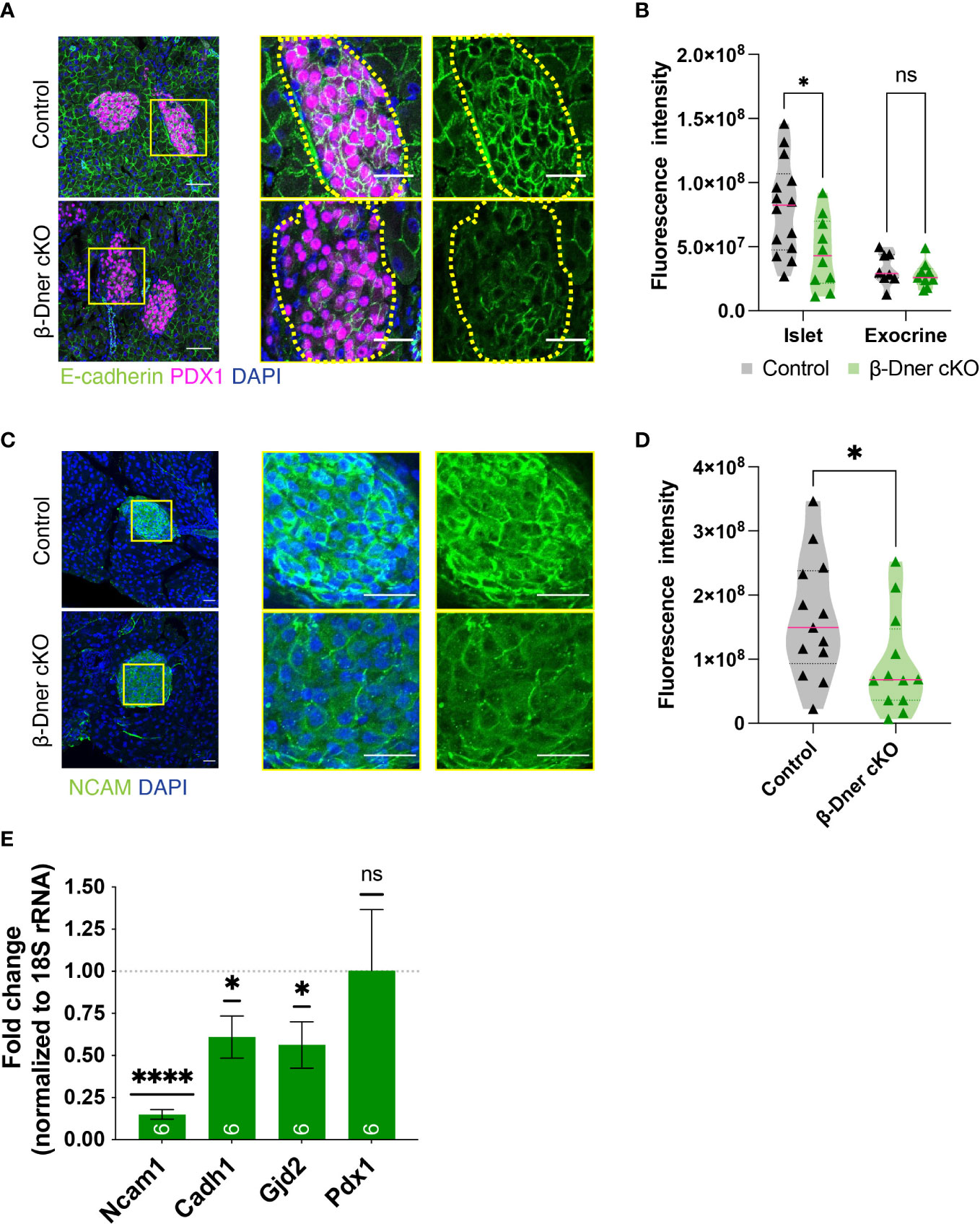
Figure 5 β-cell-cell contacts are disturbed in β-Dner cKO islets due to decreased expression of cell adhesion markers. (A) Immunohistochemistry for E-cadherin (green) and Pdx1 (magenta). Scale bars: 25μm. Representative images from 3 independent experiments. (B) Fluorescence intensity quantifications (arbitrary units) of E-cadherin immunohistochemistry in islets identified by Pdx1-positive immunohistochemistry and exocrine cells identified by DAPI-based nuclear morphology. (N= 3-4 animals per group, 2≥ islets per animal: t-test *p<0.05, ns p>0.05). (C) Immunohistochemistry for NCAM (green) shown in islets and innervating axons on the pancreas. Scale bars: 25μm. Representative images from 3 independent experiments. (D) Fluorescence intensity quantifications (arbitrary units) of NCAM immunohistochemistry. (N=3 animals per group, 3≥ islets per animal: t-test *p<0.05). (E) Levels of transcripts for NCAM (Ncam1), E-cadherin (Cdh1), Connexin 36 (Gjd2), and Pdx1 (Pdx1) measured by qRT-PCR, normalized by 18S and shown relative to controls (Means ± SEM for islets from N= 5-6 animals per group: One sample t-test ****p< 0.0001, * p<0.05). ns or not significant: p>0.05.
To further investigate the role of DNER in regulating β-cell-cell association, we assessed the levels of Neural Cell Adhesion Molecule (N-CAM), a transmembrane protein involved in Ca2+-independent cell-cell interactions and expressed in adult endocrine islets (41). N-CAM is necessary for segregation of cells within the mouse islet, specifically, the localization of α-cells to the islet periphery and the β-cells to the interior of the islets (41). Importantly, N-CAM also modulates exocytosis in both α- and β-cells (42). N-CAM immunoreactivity was largely reduced within β-Dner cKO islets (Figures 5C, D). We next performed qRT-PCR analyses to assess the levels of Ncam1, Cadh1 and Gjd2, which encode for N-CAM, E-cadherin and Connexin 36, respectively (Figure 5E). Connexin 36 is a gap junction protein expressed by β-cells and allows for synchronous Ca2+ waves to coordinate insulin secretion within islets (43). qRT-PCR analyses showed an 85% reduction of Ncam1, and approximately 40% reduction each for Cadh1 and Gjd2. This was not a result of global suppression of transcription as Pdx1, also expressed by β-cells, showed no decrease in expression in β-Dner cKO islets. These results suggest that DNER loss results in the downregulation of genes involved in β-cell-cell interactions and adhesion.
DNER regulates F-actin in β-cells
Upon DNER loss, islets showed decreased N-CAM and E-cadherin expression. N-CAM contains an intracellular domain, which is thought to bind the actin cytoskeleton and influence its remodeling (41, 42). Moreover, similar to our observation in β-Dner cKO mice, NCAM-/- mice showed glucose intolerance and defective insulin secretion, which were attributed, at least in part, to impaired actin remodeling (42). Remodeling of cortical actin in response to a glucose stimulus allows docked insulin granules to access the plasma membrane and fuse and provides actin tracks for myosin-mediated transport of insulin granules that are stored deeper in cytoplasm (42, 44, 45). Additionally, re-arrangement of F-actin upon glucose exposure results in an increase in focal adhesions between β-cells, which are essential for optimal insulin secretion (46–48). Thus, we examined whether the glucose homeostasis defects in β-Dner cKO mice might be related to impaired actin remodeling, similar to that in NCAM-/- mice. To determine if F-actin levels were altered in response to a glucose stimulus in β-Dner cKO β-cells, we stimulated islets with low (2.8mM) or high (16.7mM) glucose, and stained the islets with the F-actin binding dye, Phalloidin conjugated to 546-Alexa Fluor. As expected, in control islets, F-actin levels significantly decreased (an average of 45% reduction) after high glucose stimulation. In contrast, DNER depleted β-cells had significantly reduced F-actin levels at low glucose, which were significantly altered after the high glucose stimulation (Figures 6A, B). Together, these results suggest that DNER is required to maintain F-actin levels in β-cells, which is crucial for the regulated secretion of insulin granules in response to glucose.
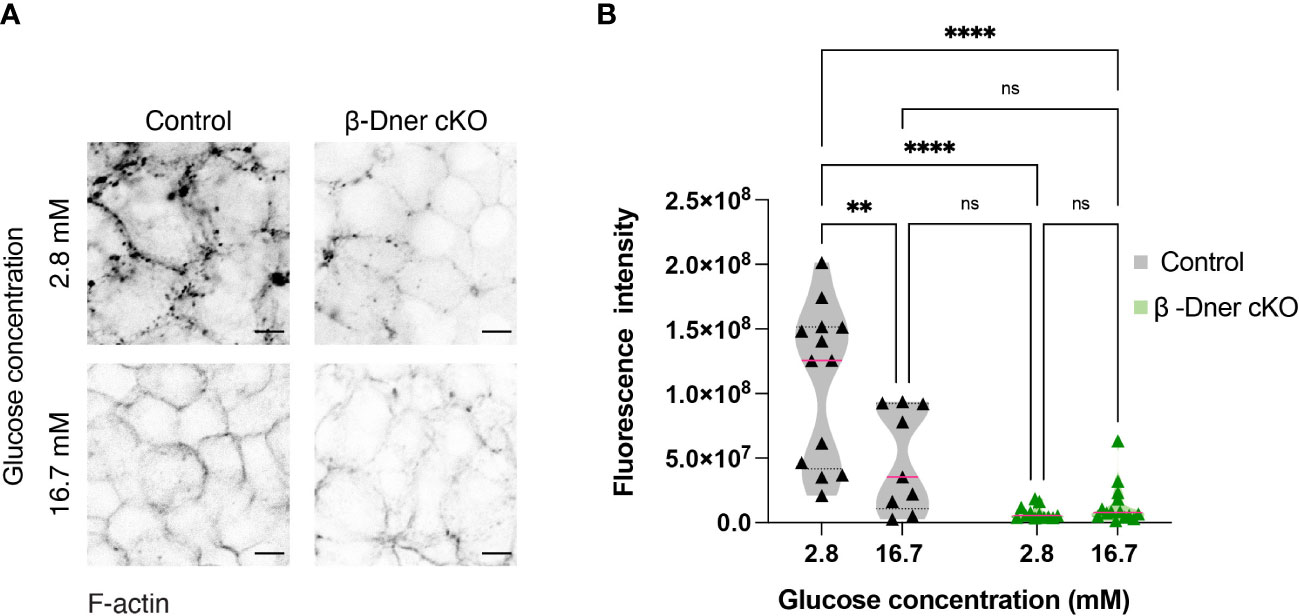
Figure 6 β-cells of β-Dner cKO have lower overall F-actin levels and impaired actin remodeling. (A) Immunohistochemistry of isolated islets incubated in 2.8mM or 16.7mM. Representative images from 3 independent experiments. (B) Fluorescence quantifications of isolated islets at different glucose concentrations (N=3 animals per group: 2-Way ANOVA **p<0.01, ****<0.0001). ns or not significant: p>0.05.
Discussion
Here, we have established that DNER is expressed within pancreatic β-cells, and that loss of DNER leads to glucose intolerance, decreased insulin secretion and insulin resistance in adult mice. Furthermore, loss of DNER resulted in decreased islet insulin content and diminished expression of genes critical for cell-cell interactions (Ncam1, Cadh1 and Gjd2). Together, these findings support a physiological role for DNER in regulating blood glucose homeostasis, likely via regulating β-cell-cell association.
Glucose intolerance arises from defects in glucose-stimulated insulin secretion from the pancreatic β-cells, dampened responses to insulin in insulin-sensitive tissues such as the liver, adipose tissues or skeletal muscles, or a combination of defects in both insulin secretion and sensitivity. Moreover, blood glucose uptake can be regulated by insulin-independent mechanisms, including a combination of autonomic and neuroendocrine factors, as well as a well-described involvement of the CNS in postprandial glucose regulation (49, 50). β-Dner cKO mice had impaired glucose-stimulated insulin secretion, which at least in part, may result from defective β-cell-cell communication and actin dynamics. Decreased insulin secretion in β-Dner cKO mice can be explained by dampened levels of islet insulin, and also by defects in the secretory capacity of β-cells. Our experiments in isolated islets showed intrinsic defects in glucose stimulated insulin release, and defects downstream of β-cell depolarization, as seen by the lack of response to KCl in mutant islets. Importantly, diminished insulin secretion in response to KCl indicates secretory defects independent of glucose entry and metabolism. Future studies will be required to determine whether DNER’s activity in the β-cells directly modulates insulin secretion, or whether the defects observed are an indirect consequence of DNER loss.
Depletion of DNER in β-cells resulted in insulin resistance. Insulin resistance often arises from hyperinsulinemia or obesity (4, 51, 52). Neither hyperinsulinemia (Figures 2C, E) nor increased body weight was observed in β-Dner cKO animals (Supplemental Figures 2A, B). It is possible that defects in insulin sensitivity could arise due to DNER ablation in some Pdx1-positive cells outside of the pancreas (53–55). Some studies using Pdx1-Cre or Pdx1-CreER animals crossed to genetic reporter lines indicated recombination in cells present in the medial preoptic area, raphe nucleus, inferior olivary nucleus, lateral hypothalamus, arcuate nucleus, and dorsomedial hypothalamus (53–55). The hypothalamus is a non-classical insulin-responsive brain region in which astrocytes promote glucose availability in the CNS and systemic glucose metabolism (56–59). Moreover, DNER loss in the CNS has previously been linked to defects in neuron- glia interactions, crucial for their development and function (8, 13). Therefore, loss of DNER expression in Pdx1-positive hypothalamic neurons could potentially contribute to defects in insulin sensitivity and glucose homeostasis in β-Dner cKO mice. Future studies using other pancreas-specific or hypothalamic-specific Cre lines are necessary to examine DNER regulation of insulin-sensitive tissues. Nevertheless, experiments performed in isolated islets lacking DNER in vitro showed insulin release defects demonstrating that the insulin secretory defect is at least in part intrinsic to the islet. Thus, these results demonstrate that DNER function is critical to the normal physiological function of pancreatic β-cells in regulating insulin secretion and blood glucose.
Surprisingly, β-cells from β-Dner cKO animals had reduced expression of N-CAM and E-cadherin, both at the protein and the transcript level. Furthermore, the transcript for Gjd2, which encodes for connexin 36 was also downregulated. Connexin 36 is the major gap junction component expressed in β-cells, and it’s loss leads to defects in calcium signaling and coordination of insulin release within islets (60–62). Therefore, we speculate that reduction of connexin 36 and consequently a dysregulation of calcium dynamics within the islets might be, at least in part, responsible for the insulin secretory defects observed in β-Dner cKO mice.
Our results showing DNER expression in postnatal β-cells as well as previous reports of its enrichment in mature β-cells in sequencing studies (17), indicate a role for DNER in the functionally mature β-cells. Importantly, maturation of β-cells correlates with increased expression and functional localization of connexins, secretory machinery, ion channels and other proteins localized in inter-cellular spaces, including N-CAM and E-cadherin (17–20, 63). Our results suggest that DNER is necessary for the expression, and perhaps, localization of N-CAM and E-cadherin in the plasma membrane in mature β-cells.
Prior studies demonstrated N-CAM was required for F-actin remodeling in islets upon glucose stimulation (42). Like β-Dner cKO animals, NCAM-/- mice displayed defects in insulin secretion. Given the decreased N-CAM expression in DNER depleted β-cells, we investigated whether F-actin levels were affected in β-Dner cKO islets. In contrast to NCAM-/- islets, where a denser submembrane actin network was observed even in high glucose concentrations (15mM or 30mM) (42), we observed decreased levels of F-actin in β-Dner cKO isolated islets, irrespective of glucose concentrations. Whether the reduced F-actin is due to defective actin assembly, decreased actin protein levels, or a combination of these factors requires further investigation. Upon glucose stimulation, cortical F-actin is remodeled to allow insulin granule access to the plasma membrane, provide myosin-mediated transport of insulin granules from the cytoplasm to the plasma membrane, and to form focal adhesions which provide a structural and molecular link between the extracellular matrix (ECM), the cytoskeleton and signaling molecules (44–48). Glucose stimulation promotes the formation of these contact sites downstream of Ca2+ entry. Furthermore, p-FAK and paxillin regulate the localization of SNAP-25 and syntaxin-1, which are crucial for insulin granule exocytosis (47, 48). If β-Dner cKO islets are unable to form focal adhesions and localize exocytic machinery, that could be a potential mechanism to link the loss of F-actin with the dampened insulin secretion. How DNER modulates the expression of adhesive molecules and regulates the actin cytoskeleton remains to be answered. Whether DNER physically interacts with adhesive proteins or if it directly modulates F-actin levels are open questions for future studies. Nonetheless, this study shows that DNER plays a novel role in maintaining β-cell-cell contacts and maintaining islet morphology. Specifically, our findings suggest that defects in insulin secretion arise, in part, from dysregulation of the actin cytoskeleton and decreased expression of adhesive markers in β-Dner cKO islets.
DNER alleles in humans have been correlated with insulin resistance phenotypes in Type 2 Diabetes patients (5, 6). However, the specific cell types expressing DNER and the molecular pathways influenced by DNER to elicit disease pathology were not evaluated. We anticipate that the mechanisms we have defined in this study will provide a foundation for future studies to fully elucidate the role of DNER in the healthy pancreas, and to assess whether DNER mutations or polymorphisms contribute to diabetes risk. Targeting of DNER or other cell-cell signaling pathways may represent a novel therapeutic strategy for diabetes patients.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics statement
The animal study was reviewed and approved by Johns Hopkins University Animal Care and Use Committee (ACUC).
Author contributions
NR-O contributed to study design, investigations, data analyses, and writing and editing the manuscript. RK contributed to writing/editing the manuscript and funding acquisition. All authors contributed to the article and approved the submitted version.
Funding
NIH R01 awards, NS073751 and NS107342, to RK.
Acknowledgments
We would like to thank the JHU Mouse Tri-Lab for their helpful comments on this project. Specifically, Dr. Aurelia Mapps for her continued support in the preparation of this manuscript. We would also like to thank Dr. Ronadip Banerjee for his support in the process of this submission.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2023.1161085/full#supplementary-material
Supplementary Figure 1 | DNER is expressed in endocrine α-cells. (A) Immunohistochemistry of adult wild-type pancreatic tissues shows DNER (red) and glucagon (green). Scale bars: 25μm. Representative images from 3 independent experiments.
Supplementary Figure 2 | DNER deletion from adult β-cells in β-Dner cKO animals. (A) Schematic of Dnerf/f (Dnertm3c) mouse generation and β-Dner cKO from Dnertm3a (EUCOMM) [adapted from Groza et al. (64)]. (B) Immunohistochemistry of β-Dner cKO islets (outlined) shows reduced DNER (magenta) expression. Scale bars: 25μm. Representative images from 3 independent experiments. (C) qRT-PCR for DNER transcripts, normalized by 18S and shown as relative to controls (N=7 animals per group: One sample t-test ****p < 0.0001).
Supplementary Figure 3 | β-Dner cKO animals are not obese. β-Dner cKO males and females have similar changes in glucose tolerance and insulin secretion. (A) Weight of mice at 8-weeks of age (N= 10-12 animals per group: t-test comparison per sex: *p<0.05). (B) and (C) Glucose tolerance test of males and females respectively (Means ± SEM for N = 5 animals per group: 2-way ANOVA *p < 0.05, *** p < 0.001, ****p < 0.0001). (D) and (E) Glucose-stimulated insulin secretion in vivo of males and females respectively (Means ± SEM for N= 7-10 animals per group: 2-way ANOVA **p < 0.01).
Supplementary Figure 4 | β-Dner cKO have normal islet endocrine organization and decreased α-cell number (A) Immunohistochemistry of pancreatic islets stained for glucagon (green) and Pdx1 (magenta). Scale bars: 50μm. Representative images from 3 independent experiments. (B) α-cell number quantifications (N=4-5 animals per group; data shown as cells per islet, t-test **p<0.01). (C) Islet number per pancreatic section (N=4-5 animals per group). Pancreatic sections were quantified every 100μm. (D) Levels of transcripts measured by qRT-PCR, normalized by 18S and shown relative to controls (Means ± SEM for islets from N ≥ 2 animals per group).
References
1. Bao W, Hu FB, Rong S, Rong Y, Bowers K, Schisterman EF, et al. Predicting risk of type 2 diabetes mellitus with genetic risk models on the basis of established genome-wide association markers: a systematic review. Am J Epidemiol (2013) 178:1197–207. doi: 10.1093/aje/kwt123
2. Fuchsberger C, Flannick J, Teslovich TM, Mahajan A, Agarwala V, Gaulton KJ, et al. The genetic architecture of type 2 diabetes. Nature (2016) 536:41–7. doi: 10.1038/nature18642
3. Glauser DA, Brun T, Gauthier BR, Schlegel W. Transcriptional response of pancreatic beta cells to metabolic stimulation: large scale identification of immediate-early and secondary response genes. BMC Mol Biol (2007) 8:54. doi: 10.1186/1471-2199-8-54
4. Parks BW, Sallam T, Mehrabian M, Psychogios N, Hui ST, Norheim F, et al. Genetic architecture of insulin resistance in the mouse. Cell Metab (2015) 21:334–46. doi: 10.1016/j.cmet.2015.01.002
5. Deng Z, Shen J, Ye J, Shu Q, Zhao J, Fang M, et al. Association between single nucleotide polymorphisms of Delta/Notch-like epidermal growth factor (EGF)-related receptor (DNER) and delta-like 1 ligand (DLL 1) with the risk of type 2 diabetes mellitus in a Chinese han population. Cell Biochem Biophys (2015) 71:331–5. doi: 10.1007/s12013-014-0202-3
6. Hanson RL, Muller YL, Kobes S, Guo T, Bian L, Ossowski V, et al. A genome-wide association study in American indians implicates DNER as a susceptibility locus for type 2 diabetes. Diabetes (2014) 63:369–76. doi: 10.2337/db13-0416
7. Eiraku M, Hirata Y, Takeshima H, Hirano T, Kengaku M. Delta/Notch-like epidermal growth factor (EGF)-related receptor, a novel EGF-like repeat-containing protein targeted to dendrites of developing and adult central nervous system neurons. J Biol Chem (2002) 277:25400–7. doi: 10.1074/jbc.M110793200
8. Eiraku M, Tohgo A, Ono K, Kaneko M, Fujishima K, Hirano T, et al. DNER acts as a neuron-specific notch ligand during bergmann glial development. Nat Neurosci (2005) 8:873–80. doi: 10.1038/nn1492
9. Nishizumi H, Komiyama T, Miyabayashi T, Sakano S, Sakano H. BET, a novel neuronal transmembrane protein with multiple EGF-like motifs. Neuroreport (2002) 13:909–15. doi: 10.1097/00001756-200205070-00035
10. Fehon RG, Kooh PJ, Rebay I, Regan CL, Xu T, Muskavitch MA, et al. Molecular interactions between the protein products of the neurogenic loci notch and delta, two EGF-hoologouse genes in drosophila. Cell (1990) 61:523–34. doi: 10.1016/0092-8674(90)90534-L
11. Fukazawa N, Yokoyama S, Eiraku M, Kengaku M, Maeda N. Receptor type protein tyrosine phosphatase -pleiotrophin signaling controls endocytic trafficking of DNER that regulates neuritogenesis. Mol Cell Biol (2008) 28:4494–506. doi: 10.1128/MCB.00074-08
12. Rebay I, Fleming RJ, Fehon RG, Cherbas L, Cherbas P, Artavanis-Tsakonas S. Specific EGF repeats of notch mediate interactions with delta and serrate: implications for notch as a multifunctional receptor. Cell (1991) 67:687–99. doi: 10.1016/0092-8674(91)90064-6
13. Hsieh F-Y, Ma T-L, Shih H-Y, Lin S-J, Huang C-W, Wang H-Y, et al. Dner inhibits neural progenitor proliferation and induces neuronal and glial differentiation in zebrafish. Dev Biol (2013) 375:1–12. doi: 10.1016/j.ydbio.2013.01.007
14. Tohgo A, Eiraku M, Miyazaki T, Miura E, Kawaguchi S, Nishi M, et al. Impaired cerebellar functions in mutant mice lacking DNER. Mol Cell Neurosci (2006) 31:326–33. doi: 10.1016/j.mcn.2005.10.003
15. Kurisu J, Fukuda T, Yokoyama S, Hirano T, Kengaku M. Polarized targeting of DNER into dendritic plasma membrane in hippocampal neurons depends on endocytosis: endocytosis mediates dendritic sorting of DNER. J Neurochem (2010) 113:1598–610. doi: 10.1111/j.1471-4159.2010.06714.x
16. Hald J, Galbo T, Rescan C, Radzikowski L, Sprinkel AE, Heimberg H, et al. Pancreatic islet and progenitor cell surface markers with cell sorting potential. Diabetologia (2012) 55:154–65. doi: 10.1007/s00125-011-2295-1
17. Qiu W-L, Zhang Y-W, Feng Y, Li L-C, Yang L, Xu C-R. Deciphering pancreatic islet β cell and α cell maturation pathways and characteristic features at the single-cell level. Cell Metab (2017) 25:1194–1205.e4. doi: 10.1016/j.cmet.2017.04.003
18. Santos-Silva JC, Carvalho CP de F, de Oliveira RB, Boschero AC, Collares-Buzato CB. Cell-to-cell contact dependence and junctional protein content are correlated with in vivo maturation of pancreatic beta cells. Can J Physiol Pharmacol (2012) 90:837–50. doi: 10.1139/y2012-064
19. Carvalho CPF, Barbosa HCL, Britan A, Santos-Silva JCR, Boschero AC, Meda P, et al. Beta cell coupling and connexin expression change during the functional maturation of rat pancreatic islets. Diabetologia (2010) 53:1428–37. doi: 10.1007/s00125-010-1726-8
20. Bader E, Migliorini A, Gegg M, Moruzzi N, Gerdes J, Roscioni SS, et al. Identification of proliferative and mature β-cells in the islets of langerhans. Nature (2016) 535:430–4. doi: 10.1038/nature18624
21. Wollheim CB, Meda P, Halban PA. “[13] isolation of pancreatic islets and primary culture of the intact microorgans or of dispersed islet cells. In: Methods in enzymology. USA and UK: Elsevier (1990). p. 188–223. doi: 10.1016/0076-6879(90)92071-K
22. Borden P, Houtz J, Leach SD, Kuruvilla R. Sympathetic innervation during development is necessary for pancreatic islet architecture and functional maturation. Cell Rep (2013) 4:287–301. doi: 10.1016/j.celrep.2013.06.019
23. Ceasrine AM, Lin EE, Lumelsky DN, Iyer R, Kuruvilla R. Adrb2 controls glucose homeostasis by developmental regulation of pancreatic islet vasculature. eLife (2018) 7:e39689. doi: 10.7554/eLife.39689
24. Houtz J, Borden P, Ceasrine A, Minichiello L, Kuruvilla R. Neurotrophin signaling is required for glucose-induced insulin secretion. Dev Cell (2016) 39:329–45. doi: 10.1016/j.devcel.2016.10.003
25. Gu G, Dubauskaite J, Melton DA. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development (2002) 129:2447–57. doi: 10.1242/dev.129.10.2447
26. Gannon M, Gamer LW, Wright CVE. Regulatory regions driving developmental and tissue-specific expression of the essential pancreatic gene pdx1. Dev Biol (2001) 238:185–201. doi: 10.1006/dbio.2001.0359
27. Guz Y, Montminy MR, Stein R, Leonard J, Gamer LW, Wright CVE, et al. Expression of murine STF-1, a putative insulin gene transcription factor, in β cells of pancreas, duodenal epithelium and pancreatic exocrine and endocrine progenitors during ontogeny. Development (1995) 121:11–8. doi: 10.1242/dev.121.1.11
28. Andrikopoulos S, Blair AR, Deluca N, Fam BC, Proietto J. Evaluating the glucose tolerance test in mice. Am J Physiology-Endocrinol Metab (2008) 295:E1323–32. doi: 10.1152/ajpendo.90617.2008
29. Pacini G, Mari A. Methods for clinical assessment of insulin sensitivity and β-cell function. Best Pract Res Clin Endocrinol Metab (2003) 17:305–22. doi: 10.1016/S1521-690X(03)00042-3
30. Hou JC, Min L, Pessin JE. Chapter 16 insulin granule biogenesis, trafficking and exocytosis. In: Vitamins & hormones. Elsevier (2009). p. 473–506. doi: 10.1016/S0083-6729(08)00616-X
31. Komatsu M, Takei M, Ishii H, Sato Y. Glucose-stimulated insulin secretion: a newer perspective. J Diabetes Invest (2013) 4:511–6. doi: 10.1111/jdi.12094
32. Rorsman P, Braun M. Regulation of insulin secretion in human pancreatic islets. Annu Rev Physiol (2013) 75:155–79. doi: 10.1146/annurev-physiol-030212-183754
33. Rorsman P, Renström E. Insulin granule dynamics in pancreatic beta cells. Diabetologia (2003) 46:1029–45. doi: 10.1007/s00125-003-1153-1
34. Hatlapatka K, Willenborg M, Rustenbeck I. Plasma membrane depolarization as a determinant of the first phase of insulin secretion. Am J Physiology-Endocrinol Metab (2009) 297:E315–22. doi: 10.1152/ajpendo.90981.2008
35. Deltour L, Leduque P, Blume N, Madsen O, Dubois P, Jami J, et al. Differential expression of the two nonallelic proinsulin genes in the developing mouse embryo. Proc Natl Acad Sci USA (1993) 90:527–31. doi: 10.1073/pnas.90.2.527
36. Wentworth BM, Schaefer IM, Villa-Komaroff L, Chirgwin JM. Characterization of the two nonallelic genes encoding mouse preproinsulin. J Mol Evol (1986) 23:305–12. doi: 10.1007/BF02100639
37. Meur G, Qian Q, da Silva Xavier G, Pullen TJ, Tsuboi T, McKinnon C, et al. Nucleo-cytosolic shuttling of FoxO1 directly regulates mouse Ins2 but not Ins1 gene expression in pancreatic beta cells (MIN6). J Biol Chem (2011) 286:13647–56. doi: 10.1074/jbc.M110.204248
38. Bosco D, Rouiller DG, Halban PA. Differential expression of e-cadherin at the surface of rat -cells as a marker of functional heterogeneity. J Endocrinol (2007) 194:21–9. doi: 10.1677/JOE-06-0169
39. Carvell M, Marsh P, Persaud S, Jones P. E-cadherin interactions regulate β-cell proliferation in islet-like structures. Cell Physiol Biochem (2007) 20:617–26. doi: 10.1159/000107545
40. Dahl U, Sjödin A, Semb H. Cadherins regulate aggregation of pancreatic β-cells. Vivo Dev (1996) 122:2895–902. doi: 10.1242/dev.122.9.2895
41. Esni F, Täljedal I-B, Perl A-K, Cremer H, Christofori G, Semb H. Neural cell adhesion molecule (N-CAM) is required for cell type segregation and normal ultrastructure in pancreatic islets. J Cell Biol (1999) 144:325–37. doi: 10.1083/jcb.144.2.325
42. Olofsson CS, Håkansson J, Salehi A, Bengtsson M, Galvanovskis J, Partridge C, et al. Impaired insulin exocytosis in neural cell adhesion molecule –/– mice due to defective reorganization of the submembrane f-actin network. Endocrinology (2009) 150:3067–75. doi: 10.1210/en.2008-0475
43. Ravier MA, Guldenagel M, Charollais A, Gjinovci A, Caille D, Sohl G, et al. Loss of Connexin36 channels alters -cell coupling, islet synchronization of glucose-induced Ca2+ and insulin oscillations, and basal insulin release. Diabetes (2005) 54:1798–807. doi: 10.2337/diabetes.54.6.1798
44. Arous C, Halban PA. The skeleton in the closet: actin cytoskeletal remodeling in β-cell function. Am J Physiology-Endocrinol Metab (2015) 309:E611–20. doi: 10.1152/ajpendo.00268.2015
45. Kalwat MA, Thurmond DC. Signaling mechanisms of glucose-induced f-actin remodeling in pancreatic islet β cells. Exp Mol Med (2013) 45:e37–7. doi: 10.1038/emm.2013.73
46. Rondas D, Tomas A, Soto-Ribeiro M, Wehrle-Haller B, Halban PA. Novel mechanistic link between focal adhesion remodeling and glucose-stimulated insulin secretion. J Biol Chem (2012) 287:2423–36. doi: 10.1074/jbc.M111.279885
47. Rondas D, Tomas A, Halban PA. Focal adhesion remodeling is crucial for glucose-stimulated insulin secretion and involves activation of focal adhesion kinase and paxillin. Diabetes (2011) 60:1146–57. doi: 10.2337/db10-0946
48. Yang S-Y, Lee J-J, Lee J-H, Lee K, Oh SH, Lim Y-M, et al. Secretagogin affects insulin secretion in pancreatic β-cells by regulating actin dynamics and focal adhesion. Biochem J (2016) 473:1791–803. doi: 10.1042/BCJ20160137
49. DiCostanzo CA, Dardevet DP, Neal DW, Lautz M, Allen E, Snead W, et al. Role of the hepatic sympathetic nerves in the regulation of net hepatic glucose uptake and the mediation of the portal glucose signal. Am J Physiology-Endocrinol Metab (2006) 290:E9–E16. doi: 10.1152/ajpendo.00184.2005
50. Mirzadeh Z, Faber CL, Schwartz MW. Central nervous system control of glucose homeostasis: a therapeutic target for type 2 diabetes? Annu Rev Pharmacol Toxicol (2022) 62:55–84. doi: 10.1146/annurev-pharmtox-052220-010446
51. Czech MP. Insulin action and resistance in obesity and type 2 diabetes. Nat Med (2017) 23:804–14. doi: 10.1038/nm.4350
52. Skovsø S, Panzhinskiy E, Kolic J, Cen HH, Dionne DA, Dai X-Q, et al. Beta-cell specific insr deletion promotes insulin hypersecretion and improves glucose tolerance prior to global insulin resistance. Nat Commun (2022) 13:735. doi: 10.1038/s41467-022-28039-8
53. Honig G, Liou A, Berger M, German MS, Tecott LH. Precise pattern of recombination in serotonergic and hypothalamic neurons in a Pdx1-cre transgenic mouse line. J Biomed Sci (2010) 17:82. doi: 10.1186/1423-0127-17-82
54. Song J, Xu Y, Hu X, Choi B, Tong Q. Brain expression of cre recombinase driven by pancreas-specific promoters. genesis (2010) 48:628–34. doi: 10.1002/dvg.20672
55. Wicksteed B, Brissova M, Yan W, Opland DM, Plank JL, Reinert RB, et al, et al. Conditional gene targeting in mouse pancreatic ß-Cells: analysis of ectopic Cre transgene expression in the brain. Diabetes (2010) 59(12):3090–8. doi: 10.2337/db10-0624
56. Chan O, Sherwin RS. Hypothalamic regulation of glucose-stimulated insulin secretion. Diabetes (2012) 61:564–5. doi: 10.2337/db11-1846
57. Chen W, Balland E, Cowley MA. Hypothalamic insulin resistance in obesity: effects on glucose homeostasis. Neuroendocrinology (2017) 104:364–81. doi: 10.1159/000455865
58. García-Cáceres C, Quarta C, Varela L, Gao Y, Gruber T, Legutko B, et al. Astrocytic insulin signaling couples brain glucose uptake with nutrient availability. Cell (2016) 166:867–80. doi: 10.1016/j.cell.2016.07.028
59. Ono H. Molecular mechanisms of hypothalamic insulin resistance. IJMS (2019) 20:1317. doi: 10.3390/ijms20061317
60. Benninger RKP, Head WS, Zhang M, Satin LS, Piston DW. Gap junctions and other mechanisms of cell-cell communication regulate basal insulin secretion in the pancreatic islet: cell-cell communication in basal insulin secretion. J Physiol (2011) 589:5453–66. doi: 10.1113/jphysiol.2011.218909
61. Benninger RKP, Hutchens T, Head WS, McCaughey MJ, Zhang M, Le Marchand SJ, et al. Intrinsic islet heterogeneity and gap junction coupling determine spatiotemporal Ca2+ wave dynamics. Biophys J (2014) 107:2723–33. doi: 10.1016/j.bpj.2014.10.048
62. Carvalho CPF, Oliveira RB, Britan A, Santos-Silva JC, Boschero AC, Meda P, et al. Impaired β-cell-β-cell coupling mediated by Cx36 gap junctions in prediabetic mice. Am J Physiology-Endocrinol Metab (2012) 303:E144–51. doi: 10.1152/ajpendo.00489.2011
63. Stolovich-Rain M, Enk J, Vikesa J, Nielsen FC, Saada A, Glaser B, et al. Weaning triggers a maturation step of pancreatic β cells. Dev Cell (2015) 32:535–45. doi: 10.1016/j.devcel.2015.01.002
Keywords: DNER, β-cell, pancreatic islets, cell-adhesion, N-CAM
Citation: Ruiz-Otero N and Kuruvilla R (2023) Role of Delta/Notch-like EGF-related receptor in blood glucose homeostasis. Front. Endocrinol. 14:1161085. doi: 10.3389/fendo.2023.1161085
Received: 07 February 2023; Accepted: 18 April 2023;
Published: 08 May 2023.
Edited by:
Jean Buteau, University of Alberta, CanadaReviewed by:
Wataru Nishimura, International University of Health and Welfare School of Medicine, JapanMostafa Bakhti, Helmholtz Center München, Helmholtz Association of German Research Centres (HZ), Germany
Copyright © 2023 Ruiz-Otero and Kuruvilla. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nelmari Ruiz-Otero, bnJ1aXpvdDFAamh1LmVkdQ==