 Anastasiya V. Snezhkina1*
Anastasiya V. Snezhkina1* Vladislav S. Pavlov1
Vladislav S. Pavlov1 Dmitry V. Kalinin2
Dmitry V. Kalinin2 Elena A. Pudova1
Elena A. Pudova1 George S. Krasnov1
George S. Krasnov1 Asiya F. Ayupova1
Asiya F. Ayupova1 Anastasiya A. Kobelyatskaya1
Anastasiya A. Kobelyatskaya1 Alexey A. Dmitriev1
Alexey A. Dmitriev1 Dmitrii A. Atiakshin3
Dmitrii A. Atiakshin3 Maria S. Fedorova1
Maria S. Fedorova1 Anna V. Kudryavtseva1
Anna V. Kudryavtseva1- 1Engelhardt Institute of Molecular Biology, Russian Academy of Sciences, Moscow, Russia
- 2Vishnevsky Institute of Surgery, Ministry of Health of the Russian Federation, Moscow, Russia
- 3Scientific and Educational Resource Center “Innovative Technologies of Immunophenotyping, Digital Spatial Profiling and Ultrastructural Analysis”, RUDN University, Moscow, Russia
Vagal paraganglioma (VPGL) is a rare neuroendocrine tumor that originates from the paraganglion associated with the vagus nerve. VPGLs present challenges in terms of diagnostics and treatment. VPGL can occur as a hereditary tumor and, like other head and neck paragangliomas, is most frequently associated with mutations in the SDHx genes. However, data regarding the genetics of VPGL are limited. Herein, we report a rare case of a 41-year-old woman with VPGL carrying a germline variant in the FH gene. Using whole-exome sequencing, a variant, FH p.S249R, was identified; no variants were found in other PPGL susceptibility and candidate genes. Loss of heterozygosity analysis revealed the loss of the wild-type allele of the FH gene in the tumor. The pathogenic effect of the p.S249R variant on FH activity was confirmed by immunohistochemistry for S-(2-succino)cysteine (2SC). Potentially deleterious somatic variants were found in three genes, SLC7A7, ZNF225, and MED23. The latter two encode transcriptional regulators that can impact gene expression deregulation and are involved in tumor development and progression. Moreover, FH-mutated VPGL was characterized by a molecular phenotype different from SDHx-mutated PPGLs. In conclusion, the association of genetic changes in the FH gene with the development of VPGL was demonstrated. The germline variant FH: p.S249R and somatic deletion of the second allele can lead to biallelic gene damage that promotes tumor initiation. These results expand the clinical and mutation spectra of FH-related disorders and improve our understanding of the molecular genetic mechanisms underlying the pathogenesis of VPGL.
1 Introduction
Vagal paraganglioma (VPGL) is a neuroendocrine tumor with an extremely low annual incidence rate (1 per 100,000) (1). VPGL arises from a parasympathetic paraganglion within or adjacent to the vagus nerve accounting for approximately 13% of head and neck paragangliomas (HNPGLs) (2). The clinical signs and symptoms of VPGL vary; the most frequent symptoms include neck mass, pulsatile tinnitus, pharyngeal mass, hoarseness, and hearing loss. Metastasis occurs in 16–19% of cases (3). Up to 40% of VPGLs are characterized by multicentricity (multifocal cases) and predominantly occur together with HNPGLs in other locations (4). VPGL present challenges in terms of diagnostics and treatment. Despite the use of various instrumental diagnostic methods (computed tomography [CT], magnetic resonance imaging, and angiography), the diagnosis of VPGL is often made at the time of surgery. Tumor resection carries the risk of cranial nerve damage but remains the only treatment option for individuals diagnosed with VPGL.
VPGL, a part of PPGL, is highly heritable (5). However, the genetics of VPGL have been poorly investigated owing to its rarity. Data on VPGL are mostly limited to reports on cases and case series, usually devoted to the clinical description of tumors, treatment experience, and patient management. Recently, we presented a series of genetic analyses of HNPGLs covering VPGLs. Germline mutations have been observed in PPGL susceptibility genes, such as SDHB, SDHD, NF1, FH, and IDH2, as well as in candidate PPGL-associated genes, ACLY, OGDH, and PDHA2 (6). Most SDHx-mutated VPGLs were multifocal tumors that manifest simultaneously as carotid paragangliomas. Somatic variants of ACO1, PIK3CA, and TP53 have been identified (6). Ding et al. reported germline mutations in SDHB and MAP3K13 in malignant VPGLs (7). A few other studies have also found germline variants in the SDHB and SDHD genes (8–11). Familial VPGLs are associated with earlier disease onset (mean age, 45 vs. 60 years) and a higher risk of multifocality (78% vs. 23%) than sporadic tumors (4, 12). Due to the rarity of VPGLs, their etiology remains unclear. However, several conditions such as familial inheritance, genetic alterations in susceptibility genes, sex (female predominance), young age, Carney’s triad, and possibly chronic hypoxia may increase the risk of VPGL development (12, 13).
This study presents an analysis of the clinical phenotype and molecular genetics of VPGL with germline missense variant in the FH gene, which have been previously revealed in a comprehensive mutation profile analysis of HNPGLs (6). FH is considered a susceptibility gene for PPGLs; however, the frequency of FH-related cases is low (~ 1%) (14). VPGLs are predominantly caused by mutations in SDHx genes. To date, there have been no reports of FH-mutated VPGLs in the literature. Therefore, this case can be regarded as extremely rare and interesting.
2 Materials and methods
2.1 Patient
The patient was admitted to the Vishnevsky Institute of Surgery, Ministry of Health of the Russian Federation. Informed consent was obtained from the patient for molecular genetic studies and use of the data for scientific purposes and publication. The study was approved by the ethics committee of the Vishnevsky Institute of Surgery (ethics committee approval no. 007/18, October 2, 2018) and was performed in accordance with the Declaration of Helsinki (1964).
2.2 Immunohistochemistry
Pathomorphological studies were performed by the chief pathologist at the Department of Pathology, of Vishnevsky Institute of Surgery. Immunohistochemistry (IHC) was done on 3–5 μm thick sections made from the formalin-fixed, paraffin-embedded (FFPE) blocks with tumor and lymph node tissues of the patient as previously described (15). Immunoreactions were performed using the following primary antibodies: Chromogranin A (DAK-A3, DAKO, USA), Synaptophysin (MRQ-40, Cell Marque, USA), CD56 (123C3, DAKO), S100 protein (polyclonal, DAKO), Pancytokeratin (AE1/AE3, Biocare, USA), Ki67 (MIB-1, DAKO), SDHB (21A11AE7, Abcam, UK), FH (monoclonal, clone J-13, from Santa Cruz Biotechnology, USA), 2SC (polyclonal, Cambridge Research Biochemicals, UK), and 5-hydroxymethylcytosine (5-hmC) (polyclonal, Active Motif, USA). Secondary antibodies were conjugated to horseradish peroxidase (HRP) and detected using a Histofine DAB-2V system (Nichirei Biosciences, Japan). Automated staining was performed using a Lab Vision Autostainer 360-2D (Thermo Fisher Scientific). The slides were visualized using an Axio Imager 2 microscope (Carl Zeiss Microscopy, Germany). Stromal cells were used as internal positive controls for FH and 5-hmC, and as internal negative controls for 2SC. FH staining was considered “negative” in the absence of FH expression in tumor cells compared to a positive internal control and “positive” in other cases. 2SC staining was defined as “positive” if tumor cells displayed strong/diffuse staining in cytoplasm, nuclear, or both compared to negative internal control and “negative” in other cases. Slides were classified as “low 5-hmC” or “high 5-hmC” if tumor cells showed absent/low staining or slightly inferior/equivalent staining compared to markedly high staining of endothelial cells, respectively. Sporadic VPGL (without mutations in any susceptibility genes) and normal lymph node tissues were used as external negative controls for 2SC IHC (Supplementary Figure S1). Negative reagent controls for primary antibodies and the detection system were used to ensure the specificity of the IHC tests. Uterine leiomyomatous tissue from patients with hereditary leiomyomatosis was used as an external positive control for 2SC staining, and as an external negative control for FH immunoreactivity (Supplementary Figure S1). This control was kindly provided by Alexandra Asaturova, head of the Pathology Department, Gynecology and Perinatology Named after Academician V.I. Kulakov of Ministry of Healthcare of Russian Federation.
2.3 Whole-exome sequencing
Whole-exome sequencing was performed on tumor and normal (lymph node) tissues of the patient. Genomic DNA was extracted using a High Pure FFPET DNA Isolation Kit (Roche, Basel, Switzerland). Exome libraries were prepared with a TruSeq Exome Library Prep Kit (Illumina, USA) and sequenced using an Illumina NextSeq 500 system under 76 × 2 bp paired-end mode. The raw reads were aligned to the reference human genome (GRCh37/hg19) using BWA-MEM (16). Default parameters were used to start the alignment. Secondary (supplementary) alignments in the BWA output were removed with samtools [samtools view -F 2048] (17). Mapping statistics were obtained with samtools flagstat. The BAM files were preprocessed using Picard-tools (18) [picard.jar FixMateInformation ADD_MATE_CIGAR=true]. Then, duplicated reads were identified and marked [picard.jar MarkDuplicatesWithMateCigar MINIMUM_DISTANCE=600]. Germline variant calling was performed using GATK4 HaplotypeCaller, including the options -A StrandBiasBySample, -A StrandOddsRatio, -A BaseQualityRankSumTest, -A MappingQualityRankSumTest, -A RMSMappingQuality, -A ReadPosRankSumTest, and -A FisherStrand (19). Other parameters were set to default, except for –max-reads-per-alignment-start 0. GATK VariantFiltration was used for germline variant filtering [SNVs - QD < 2.0, QUAL < 35.0, MQ < 40, MQRankSum < -12.5, FS > 60.0, SOR > 3.0, ReadPosRankSum < -8.0; indels - QD < 2.0, QUAL < 33.0, FS > 200.0, ReadPosRankSum < -20.0]. Somatic variants were discovered using GATK Mutect2 (20). First, we created a list of variants observed in normal tissues, which were obtained from patients with HNPGLs (PoN, panel of norms) (6), with the GATK Mutect2, GenomicsDBImport and CreateSomaticPanelOfNormals tools. Then we supplied it to the final GATK Mutect2 search for tumor-paired sample which was started with the default parameters except for max-reads-per-alignment-start 0, –f1r2-tar-gz <filename>, –germline-resource <gnomad_vcf>, -A StrandBiasBySample, -A StrandOddsRatio, -A AS_StrandOddsRatio. Somatic variant filtration was performed with GATK FilterMutectCalls. However, because FFPE samples were examined, preliminary steps were performed to eliminate FFPE artifacts and potential cross-sample contamination. Orientation bias artifacts were evaluated using GATK LearnReadOrientationModel. Read counts supporting reference, alternate, and other alleles for GnomAD known SNP sites were calculated using the GATK GetPileupSummaries tool (with the –min-mapping-quality 27 argument). Cross-sample contamination and tumor segmentation were evaluated using GATK CalculateContamination. Finally, GATK FilterMutectCalls was run with the derived information on tumor segmentation, cross-sample contamination, and orientation bias. All variants were annotated using ANNOVAR (21). The identified variants were filtered based on objective criteria, including mutation region (exonic and splicing), mutation type (missense, nonsense, insertion, deletion, stop-loss, start-loss), population allele frequency (<0.01, gnomAD), genomic region conservation score (>0.5, phastCons), clinical significance (not benign, ClinVar), and predicted as deleterious by more than 3 pathogenicity prediction algorithms (SIFT, PolyPhen2, LRT, and others). InterVar (22) and Varsome (23) tools were used for clinical interpretation of genetic variants. To verify all variants of interest, the IGV browser was utilized (24). The mutational load (ML) was calculated using an algorithm previously developed for FFPE PPGL samples (25). Copy number variations were analyzed using the beta allele frequency (BAF) method (6).
A total of 34.4 and 93.9 million reads were identified in normal and tumor samples, respectively; of these, 97.8% and 96.52% were mapped. 91.5% and 87.9% of reads were properly paired; 1.63% and 2.27% of reads were singletons; 16.1% and 42.9% of reads were duplicates.
2.4 Transcriptome sequencing
Total RNA was isolated from FFPE tumor tissues using a High Pure FFPET RNA Isolation Kit (Roche) and subjected to cDNA library preparation with a TruSeq Stranded Total RNA Ribo-Zero H/M/R Gold Kit (Illumina) following the manufacturer’s protocols. Sample sequencing was performed along with the collection of 104 HNPGLs on an Illumina NextSeq 500 system in a single-end run with a sequencing read length of 76 bp. Totally, 51.3 million reads (read quality > 30) were obtained for the sample. Primary bioinformatics analysis of raw sequencing data included quality control using FastQC (26) and read trimming, filtering, and adapter removal using Trimmomatic (27). Further sequencing reads were aligned using STAR (28) with the GRCh38.p12. The parameters that differed from the default were: outFilterMismatchNmax – 6, outFilterMultimapNmax – 1, quantMode – TranscriptomeSAM, outSAMstrandField – intronMotif, outFilterIntronMotifs – RemoveNoncanonicalUnannotated. The mapped reads were counted at the transcriptional level using featureCounts (Subread package) (29). Gene Set Variation Analysis (GSVA) was performed using the GSVA 1.46.0 (30) and clusterProfiler 4.6.2 (31) packages from Bioconductor, using the KEGG database for SDHx-mutated and non-mutated tumors. A total of 340 KEGG pathways were identified. The pathways identified with a p-value < 0.05 were sorted based on the [(1-adj. p-value)*(1-p-value)*absolute LogFC] score to identify the most significant and striking changes. A hierarchical clustering analysis was performed using the top 50 biological pathways based on this score. The clinical and pathological characteristics of the patient cohort with HNPGLs are shown in Supplementary Table S1.
2.5 Loss of heterozygosity analysis
LOH analysis was performed in tumor and normal tissues of the patient using three dinucleotide microsatellite repeats flanking the FH locus (1q43, GRCh37/hg19): D1S304, D1S204, and D1S321 (1q43), D1S235 (1q42.3) and D1S423 (1q44), as well as seven microsatellite markers located at different regions on chromosome 11: D11S1984, D11S1313, D11S907, D11S4088, D11S969, D11S1339, and D11S5030) as described in (32). Polymerase chain reaction (PCR) was performed using primers for microsatellites described previously (33). The obtained fluorescence-labeled PCR products were processed on a NANOPHORE-05 (Syntol, Russia) and analyzed using GeneMarker software (SoftGenetics, USA). A score < 0.7 or > 1.3 indicates LOH.
3 Results
3.1 Case description
A 42-year-old woman was referred to the Vishnevsky Institute of Surgery, Ministry of Health of the Russian Federation, with a mass on the right side of the neck, which had been growing for a long time. The patient had experienced frequent episodes of throat pain since the age of 26 years. She underwent tonsillectomy, which had no positive effects. She also complained of hoarseness.
A computed tomography (CT) revealed a solid tumor in the area of the right carotid artery bifurcation, with an oval shape and clear smooth contours, measuring 47 × 30 × 70 mm. An early arterial phase study revealed bright heterogeneous accumulation of the contrast agent and lobulations with hypodense central areas. Laterally, it deformed the surface of the neck and pushed the larynx. The lower pole of the lesion was located medial to the common carotid artery (CCA) bifurcation, in the area of the fork of the internal carotid artery (ICA) and external carotid artery (ECA), pushing the vessels anteriorly. Superiorly the lesion spread to the base of the skull (Figure 1).
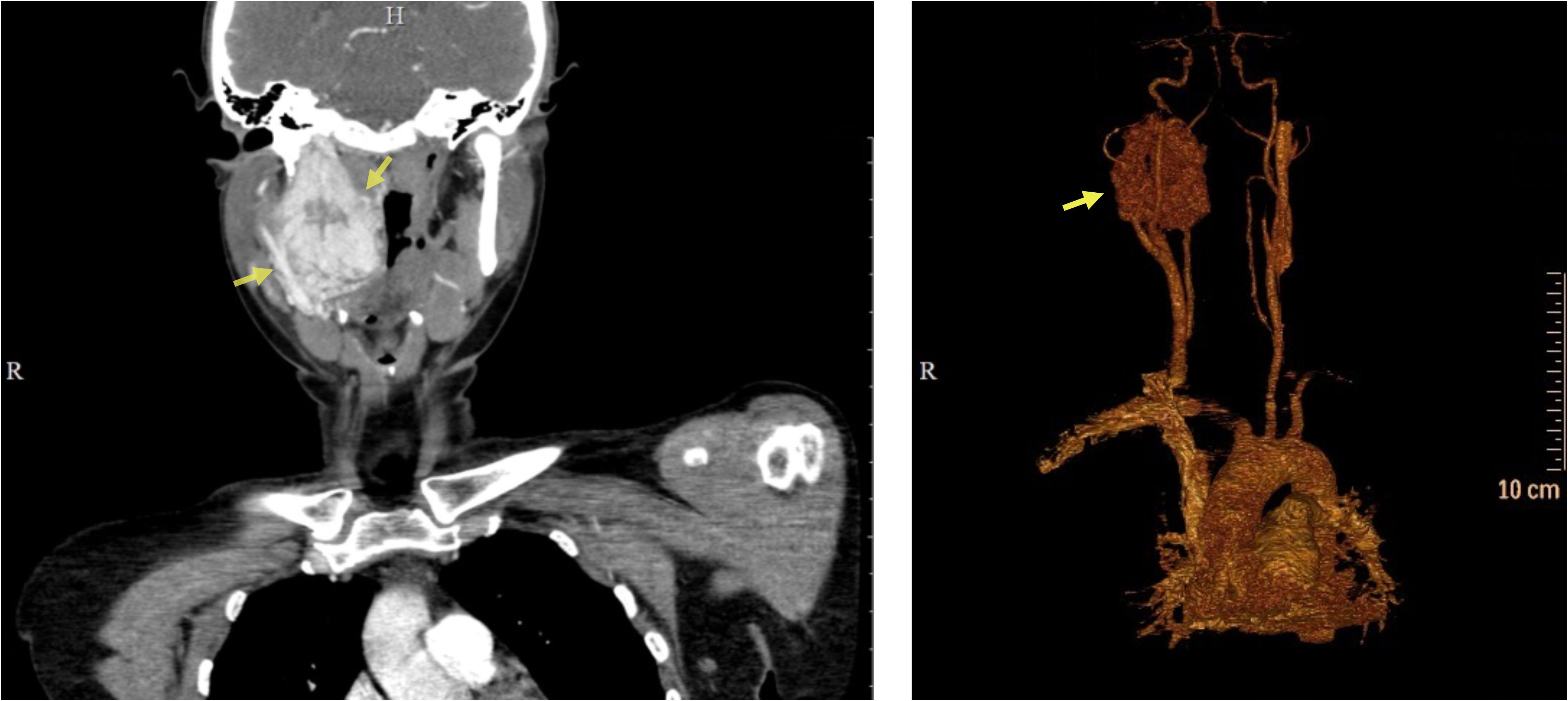
Figure 1 Preoperative computed tomography of the patient’s head and neck. CT scan (left), 3D reconstruction (right). Yellow arrows indicate the tumor.
In 2018, the patient underwent surgical tumor removal. During the procedure, the CCA, ICA, and ECA were isolated. The ICA was surrounded by the tumor up to its entry into the skull. When isolating the carotid arteries, three enlarged lymph nodes had to be removed. The tumor originated from the vagus nerve and partly from the sympathetic trunk. The vagus nerve was carefully dissected, ligated and sutured. During isolation of the ICA from the tumor, hypervascularization of the mass was noted due to active bleeding. The ICA was isolated up to the cranial entrance and requiring expansion of access, crossing of the digastric muscle, and removal of the styloid process. Following isolation of the ICA, the paraganglioma was excised from the base of the skull. It is noteworthy that the vagus nerve expanded to approximately 1 cm at the entrance to the skull. Careful hemostasis, wound drainage, and layer-by-layer suturing of the wound were performed after tumor extraction. The patient was transferred to the intensive care unit without experiencing any neurological symptoms.
Histological examination of the resected tumor confirmed paraganglioma (Figure 2). Hematoxylin and eosin staining revealed a Zellballen structure typical of PPGLs. The chief tumor cells were positive for chromogranin A, synaptophysin, and CD56 antibodies, indicating a neuroendocrine tumor. S100 protein was expressed in sustentacular cells. The tumor cells tested negative for pancytokeratin. Ki67 stained about 2% of the cells. SDHB was positively expressed in tumor cells, suggesting no deleterious mutation in the SDHx genes and the presence of a stable succinate dehydrogenase complex (15, 34).
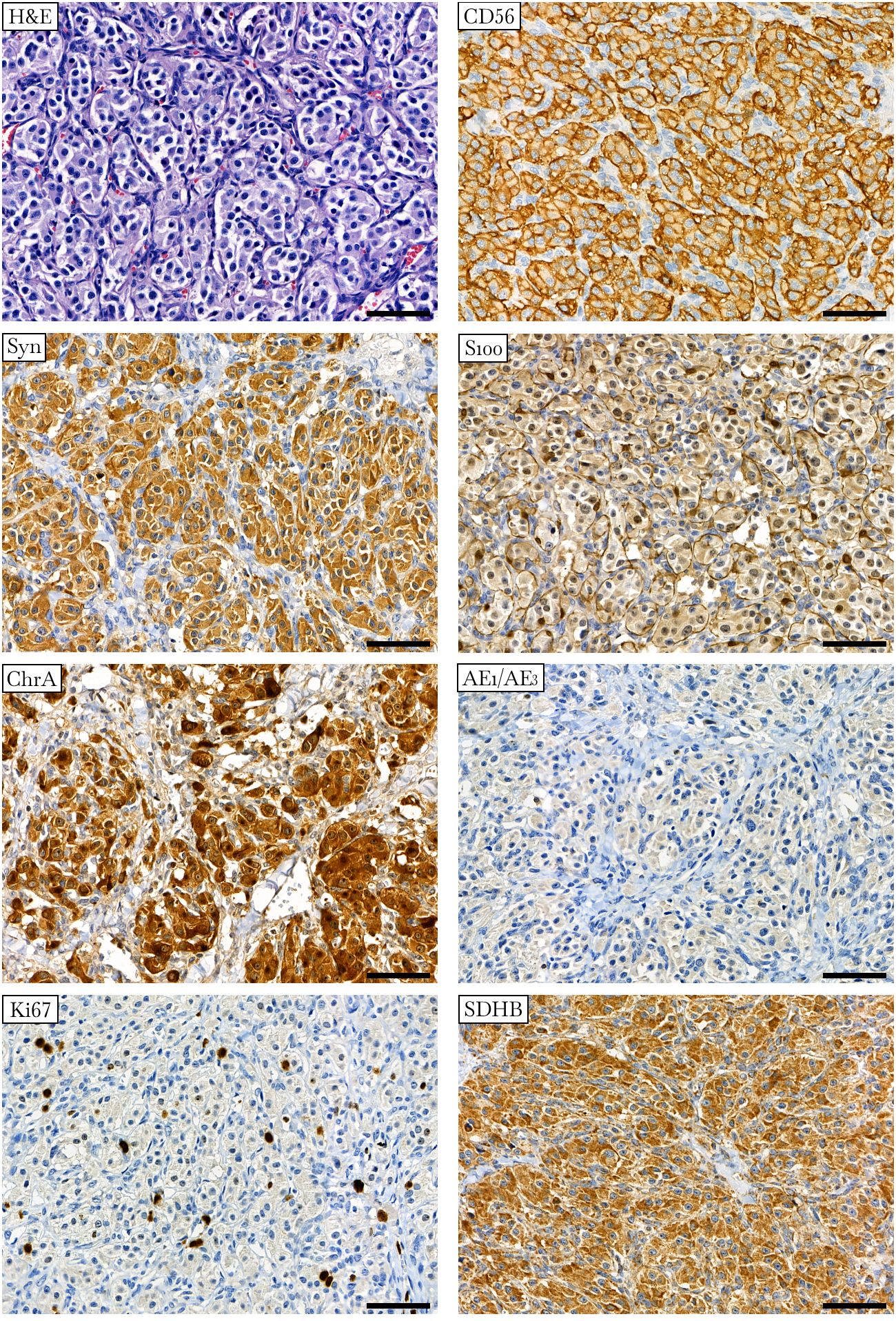
Figure 2 Histologic and immunohistochemical definition of paraganglioma. Hematoxylin-eosin (H&E) staining displays a specific “Zellballen” growth pattern with small nests of chief cells surrounded by supporting cells and further separated by fibrovascular stroma. Chief tumor cells stain positive for CD56, synaptophysin (Syn), chromogranin A (ChrA), and SDHB antibodies and negative for pancytokeratin (AE1/AE3). S100 staining occurs predominantly in sustentacular cells. Ki67 stains approximately 2% of the nuclei. ×400 magnification, scale bar 50 μm.
3.2 Mutation profiling and biological pathways enrichment
To reveal tumor-associated genetic changes, whole-exome sequencing was performed on the tumor and normal (lymph node) tissues of the patient as described previously (6). FFPE lymph node tissue slides containing normal cells were examined by a pathologist. Based on these results, a heterozygous germline variant, NM_000143: c.747T>A, p.S249R (chr1:241669460), was identified in the FH gene, with a variant allele frequency (VAF) of 0.57 in tumor and 0.45 in lymph node. This variant was recently submitted to the ClinVar database and was classified as a variant of uncertain significance. The FH: p.S249R variant was characterized by low population frequency (close to 0), high genomic region conservation score (0.89, set of 20 placental mammals), and “strong deleterious” predicted by all used in silico prediction algorithms (Table 1). According to the ACMG/AMP 2015 guideline (InterVar), this variant was interpreted as “uncertain significance” in the same way as in the GeneBe platform, whereas Varsome classified this variant as “likely pathogenic.” In the protein crystal structure of FH (35), serine 249 is located in the alpha chain in the central domain and forms a hydrogen bond with asparagine 310, another monomer in the functional homotetrameric enzyme (Supplementary Figure S2) (35–37). The substitution of serine 249 for arginine can lead to the destabilization of the tetrameric assembly and dynamics, leading to reduced FH activity. No mutations were found in other PPGL susceptibility genes (EGLN1, EGLN2, MDH2, SDHA, SDHB, SDHC, SDHD, SDHAF2, MAX, RET, TMEM127, VHL, EPAS1, NF1, H3F3A, IDH1, IDH2, ATRX, and HRAS) and 60 candidate genes in the reported panel (38). The identified FH: p.S249R variant was visualized using IGV (24) and was verified using Sanger sequencing (Supplementary Figure S3).

Table 1 Germline and somatic variants identified in the patient.
A total of 21 somatic variants that affected 18 genes were identified. Potential deleterious somatic variants were found in only three genes (SLC7A7, ZNF225, and MED23) with variant allele frequency (VAF) ranging from 0.06 to 0.13 (Table 1). The SLC7A7 gene carried two frameshift deletions. Mutational load (ML) was calculated based on exome sequencing data. VPGL was characterized by a low ML, 0.01 at VAF 0.15 (or 0.06 at VAF 0.2). An analysis of copy number variations was performed for all chromosomes using the BAF method. The tumor showed a potential loss of chromosomes 1, 11, and 20 in some cells (Supplementary Figure S4).
RNA sequencing was performed on the tumor tissue of the patient, along with the collection of 104 HNPGLs. Only the mutational profile has been previously studied for a subset of these tumors (6). Hierarchical clustering analysis of the top-50 enriched KEGG biological pathways between SDHx-mutated and non-mutated tumors (GSVA, P < 0.05) was conducted. This revealed that FH-mutated VPGL were related to a cluster consisting predominantly of non-SDHx-mutated HNPGLs. Genes upregulated in SDHx-mutated tumors were mainly downregulated in FH-mutated VPGL. Sample clustering did not depend on carotid or vagal tumor localization (Supplementary Figure S5).
3.3 Biallelic inactivation of the FH gene
Based on BAF analysis (6), probable FH wild-type allele loss was observed. To validate these data, LOH analysis was performed on the tumor and normal tissues of the patient using three dinucleotide microsatellite repeats located near the FH locus (1q43, GRCh37/hg19), D1S304, D1S204, and D1S321 (1q43), as well as two more distant markers, D1S235 (1q42.3) and D1S423 (1q44) (Supplementary Figure S6). Two microsatellite markers, one close to the FH gene (D1S321) and the left distant marker (D1S235) demonstrated LOH with allelic imbalance ratios of < 0.7 or > 1.3 (LOH = 1.36 and 1.41, respectively); the right distant marker (D1S423) showed retention of heterozygosity. The other microsatellite repeats, D1S304 and D1S204, were homozygous (noninformative). Microsatellite analysis confirmed allelic loss in regions near the FH locus, indicating inactivation of the normal copy of the gene (Supplementary Figure S6).
BAF analysis revealed a potential loss of chromosome 11 in FH-mutated tumors. The loss of chromosome 11 has previously been shown in PPGLs with germline mutations in SDHAF2, SDHD, VHL, and SDHB genes (39). To examine this result, LOH was analyzed for seven microsatellite markers located in different regions of chromosome 11 (D11S1984, D11S1313, D11S907, D11S4088, D11S969, D11S1339, and D11S5030) (Supplementary Figure S6). Three of these seven markers (D11S5030, D11S1984, and D11S1339) were not informative. Two microsatellite repeats showed LOH at 11p13 (D11S907) and 11q25 (D11S969) regions, whereas loci 11q.12.1 (D11S1313) and 11p15.5 (D11S4088) were characterized by the retention of both alleles in tumor tissue (Supplementary Figure S6). Thus, the summarized results from the BAF analysis and microsatellite examination indicated deletions of particular regions on chromosome 11 rather than the loss of the whole chromosome in the studied patient.
3.4 Fumarate hydratase deficiency
The presence of FH deficiency in the tumor was estimated by IHC staining of two markers: FH and 2SC. The latter is a marker of loss or reduction in FH enzymatic activity associated with elevated fumarate levels, resulting in increased protein succination and production of 2SC (40). Strong positive staining for FH was observed in the studied tumor, indicating that the mutant protein was stable and detectable using the anti-FH antibody (Figure 3). Simultaneously, the tumor showed positive diffuse cytoplasmic staining for 2SC, a marker of deficient FH activity (41).
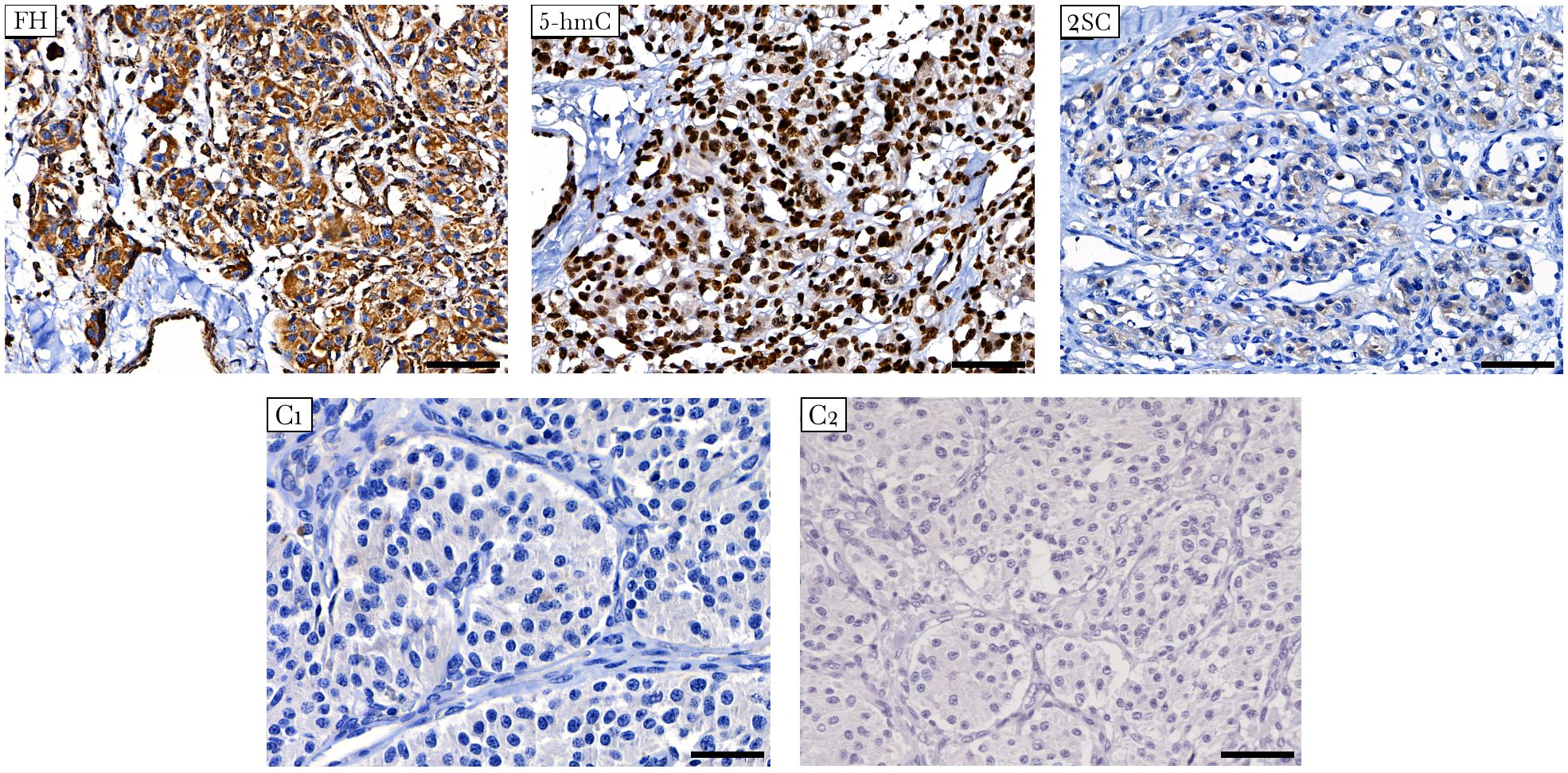
Figure 3 Immunostaining for FH, 5-hmC, and 2SC in FH-mutated VPGL. The tumor shows strong positive staining for FH (top left), diffuse cytoplasmic staining for 2SC (top right), and high nuclear labeling for 5-hmC (top middle). (C1-C2) Negative control reactions were performed for the antibodies used in the study. Negative immunostaining of 2SC in non-FH-mutated vagal paraganglioma (C1, bottom left). Isotype control image for 5-hmC and 2SC antibodies (C2, bottom right). Other control images can be found in Supplementary Figure S1. ×400 magnification, scale bar 50 μm.
In addition, the impact of the FH variant on methylation was studied using 5-hmC IHC. Accumulation of succinate and fumarate blocks the hydroxylation of 5-mC to 5-hmC catalyzed by ten-eleven translocation (TET) enzymes, leading to the loss of 5-hmC, which is an evidence of DNA hypermethylation (42). The 5-hmC level was assessed by comparison with that of adjacent endothelial cells as a positive internal control. In tumor cells, 5-hmC staining was characterized by nuclear labeling with an intensity equivalent to that in endothelial cells (Figure 3).
4 Discussion
The FH gene encodes for the fumarate hydratase enzyme, which is a component of the tricarboxylic acid (TCA) cycle and catalyzes the conversion of fumarate to malate. FH is a housekeeping and tumor suppressor gene, and its germline inactivating mutations cause severe pathologies such as uterine and cutaneous leiomyomata and renal cell carcinoma (43). Recently, several studies revealed FH as a new susceptibility gene for PPGLs, with a mutation frequency varying from 0.83% to 2.8% (Table 2) (14, 44–46). We detected an FH mutation frequency in HNPGLs of 0.66%, which is lower than that reported for PPGLs predominantly enriched with pheochromocytomas (PCCs). Only four cases of FH-mutated HNPGLs (three carotid body tumors and one case of VPGL - the index case) have been reported to date (14, 45, 47). The age of the identified PPGL patients with germline FH variants ranged from 6–70 years, and somatic FH variants were found in patients aged 27–77 years. Importantly, FH mutations were associated with metastasis and multifocal development of PPGLs in approximately one-fourth of the cases (Table 2). The index patient with the germline FH: p.S249R variant was diagnosed with VPGL at 41 years of age; however, symptoms of the disease had been a concern for her for more than 16 years. No regional or distant metastases were detected at the time of diagnosis by CT or histopathological analyses. However, the patient lacked long-term follow-up; therefore, possible metastasis cannot be excluded. Thus, FH-related PPGLs appear to manifest at a young age and are likely to predispose to malignant tumors. Therefore, the identification of germline FH mutations and subsequent monitoring of carriers are important for early diagnosis and prevention of disease progression.
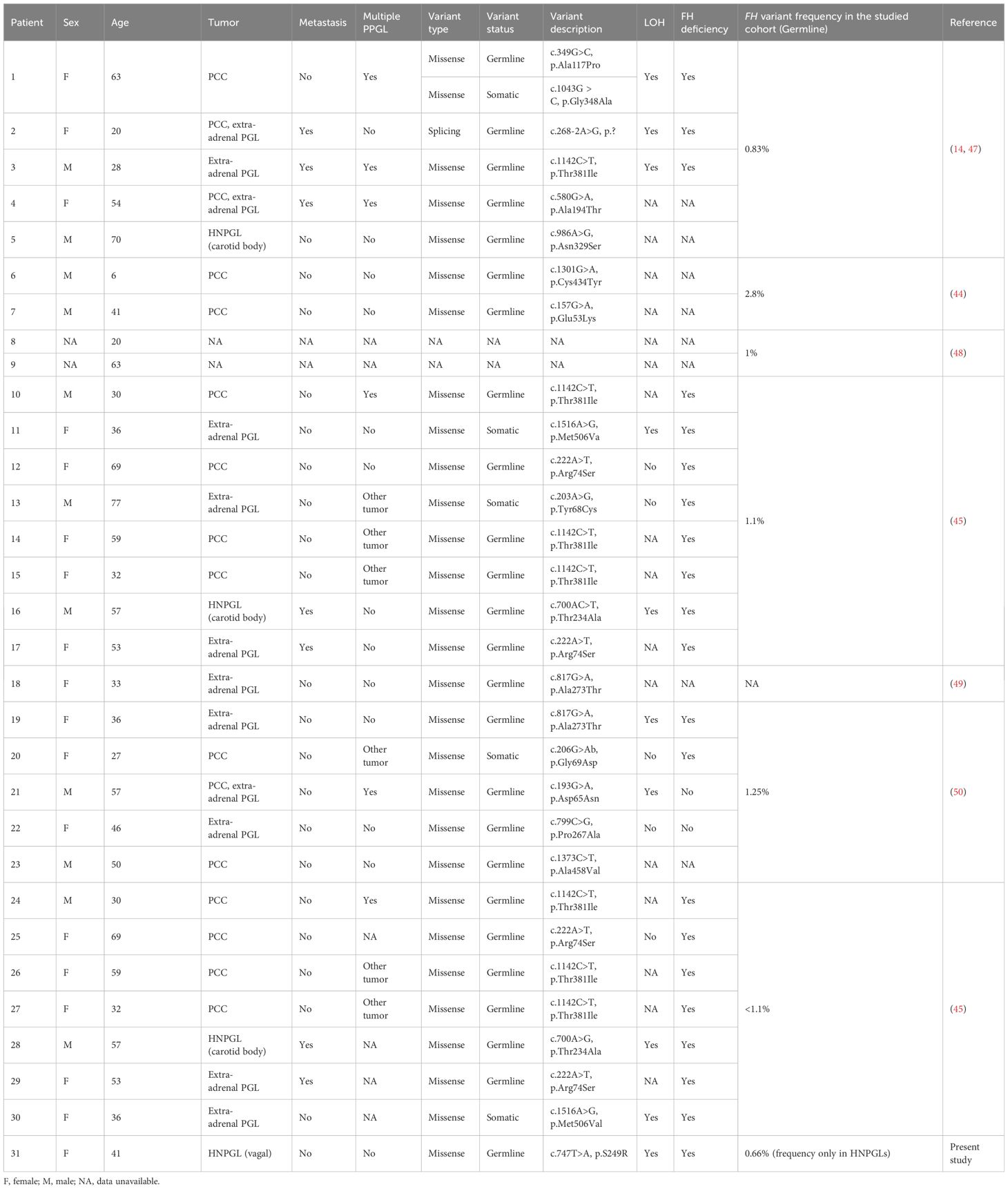
Table 2 Characteristics of reported PPGL cases with FH variants.
The identified FH variant p.S249R was previously poorly described in the clinical databases. Its effect on protein was interpreted as “likely pathogenic” (Varsome) and “uncertain significance” (ACMG/AMP 2015). According to the protein crystal structure, the replaced amino acid (serine 249) is located in the central domain and is involved in the interactions of monomers in homotetrameric mitochondrial FH proteins. FH acts as a tumor suppressor, and the biallelic loss of this gene can cause cancer (51). LOH analysis revealed the loss of the wild-type FH allele in the studied tumor. Moreover, the potential loss of chromosome 11 predicted by BAF analysis was examined, but only specific chromosomal deletions were confirmed. Similar deletions are typical along chromosomes 1 and 20, which have also been detected with potential loss using BAF analysis.
The FH damage was confirmed by IHC staining of the 2SC. Tumor cells displayed 2SC positivity with simultaneous positive immunostaining for the FH protein. Immunostaining for FH and 2SC is widely used to detect FH deficiency caused by germline FH mutations in hereditary leiomyomatosis and renal cell carcinoma syndrome (52). A recent study demonstrated the rational utility of this approach for identifying FH variants in PPGL (45). Fuchs et al. screened 589 PPGLs using FH and 2SC staining and identified eight FH-deficient tumors, the majority of which had germline FH mutations. They also observed that FH-deficient tumors did not exhibit SDH complex deficiencies. This is consistent with our results, as FH-mutated VPGL were shown to have a positive SDHB expression pattern, indicating a stable SDH complex. The study of Fuchs et al. also demonstrated the high specificity of 2SC IHC for detecting FH deficiency in PPGLs associated with germline FH mutations. Interestingly, they found only one case of FH-mutated HNPGLs, a carotid body tumor. Similar to our case, the tumor showed positive FH and 2SC staining and carried a pathogenic/likely pathogenic FH: p.Thr234Ala variant. Buelow et al. reported several cases of positive FH and 2SC IHC results associated with pathogenic FH: p.R233H mutation in multiple cutaneous leiomyomas (53). Thus, our finding of positive FH expression and positive diffuse cytoplasmic staining in 2SC is consistent with the presence of a pathogenic/likely pathogenic missense mutation. Together with the results of LOH analysis, a positive FH pattern indicated the expression of a stable mutant protein. Simultaneously, the FH: p.S249R variant impaired FH activity and caused increased levels of protein succination, as reflected by positive diffuse 2SC IHC. This demonstrates the potential pathogenic effect of the FH p.S249R variant and the presence of FH deficiency in the tumor. We believe that the results obtained may provide evidence to classify the variant as “likely pathogenic” based on the following ACMG/AMP 2015 criteria: PS3, PM2, PP2 and PP3.
A deficiency of SDH or FH in successive reactions in the TCA cycle leads to the accumulation of the oncometabolites succinate and fumarate. Both succinate and fumarate can act as antagonists of α-ketoglutarate (α-KG) inhibiting α-KG-dependent dioxygenases, including JmjC domain-containing histone demethylases (KDMs) and the TET family of DNA hydroxylases that lead to alterations in histone and DNA methylation (42). FH- and SDH-deficient PPGLs are likely characterized by similar molecular phenotypes, which were confirmed by the common development of a hypermethylation phenotype and related gene expression profiles (47). Interestingly, the opposite results were observed. Cluster analysis of the biological pathways revealed that FH-mutated tumors did not display an “SDH-like” phenotype. This might be explained by the specificity of the molecular profiles of HNPGLs, which are not characterized by clear SDHx-mutated and non-mutated transcriptomic clusters compared with PCCs. However, an additional IHC test for a specific epigenetic marker, 5-hmC, showed the absence of DNA hypermethylation in the tumor and confirmed a different molecular phenotype of the studied FH-mutated tumor. Different DNA methylation has previously been shown in FH-deficient renal cell carcinoma (RCC) (54). Approximately 20% of FH-deficient RCC did not exhibited the CpG island methylator phenotype (CIMP), which was associated with lower incidence of metastasis. Thus, our result is consistent with the heterogeneity of DNA methylation in FH-deficient tumors and may also be related to benign tumor behavior. Notably, the clustering analysis of FH-related tumors was limited to one sample. Moreover, the sampling and assay designs were different from those in a previously reported study, which can also explain the specific sample clustering.
Somatic mutation profile analysis revealed three genes, SLC7A7, ZNF225, and MED23, with potentially deleterious variants. The SLC7A7 gene encodes for the light subunit of a cationic amino acid transmembrane transporter that is involved in the activity of the y+L amino acid transport system (55, 56). No variants in SLC7A7 have been previously reported in PPGLs; the role of short SLC7A7 deletions in FH-mutated VPGL remains unclear. ZNF225 and MED23 encode proteins involved in the transcriptional activation of genes. ZNF225 is predicted to be a DNA-binding transcriptional activator specific for RNA polymerase II; however, little is known about this. MED23 is a subunit of the mediator complex that plays a key role in regulating gene expression via RNA polymerase II and other post-transcriptional steps (57). Interestingly, decreased MED23 expression was correlated with malignant PCCs with a high degree of sensitivity but suboptimal specificity (58). Thus, somatic genetic alterations in transcriptional activators may be involved in the development of FH-mutated VPGL. These mutations could lead to the downregulation of many genes, as observed in the pathway enrichment analysis of the tumors (Figure 3).
5 Conclusion
The fourth case of FH-mutated HNPGLs has been reported; notably, this is the first reported case of VPGL. In this patient, the germline FH variant p.S249R co-occurred with LOH at the gene locus and FH deficiency. This strongly suggests that FH acts as a tumor suppressor gene in paragangliomas and may serve as evidence of variant pathogenicity. Despite the clear common change in cell metabolism driven by FH and SDH mutations, FH-mutated VPGL did not display an “SDH-like” molecular phenotype, indicating a different mechanism of tumor development. The identified somatic variants in the transcriptional activators ZNF225 and MED23 seem to be associated with the deregulation of gene expression and play a role in tumor pathogenesis and potential progression (particularly MED23). This case highlights the association between HNPGLs and hereditary FH mutations and firmly supports its consideration as a tumor susceptibility gene. Despite the rarity of FH mutations in PPGLs, carriers seem to be inclined toward early onset tumors with potential for metastasis. Genetic testing for germline mutations in FH is recommended for patients with HNPGL without variants in the SDHx genes, with subsequent screening of family members.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA993587.
Ethics statement
The studies involving humans were approved by The ethics committee of the Vishnevsky Institute of Surgery, Ministry of Health of the Russian Federation. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
AS: Conceptualization, Data curation, Funding acquisition, Investigation, Project administration, Supervision, Writing – original draft, Writing – review & editing. VP: Data curation, Formal analysis, Methodology, Visualization, Writing – review & editing, Writing – original draft. DK: Formal analysis, Methodology, Resources, Writing – original draft. EP: Methodology, Writing – original draft. GK: Methodology, Writing – original draft. AA: Formal analysis, Methodology, Writing – original draft. AK: Methodology, Writing – original draft. AD: Investigation, Writing – review & editing. DA: Formal analysis, Methodology, Writing – review & editing. MF: Investigation, Methodology, Writing – original draft. AK: Funding acquisition, Project administration, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was funded by the Russian Science Foundation, grant numbers 19-15-00419 and 21-14-00353. Clinical and pathomorphological case study, whole-exome analysis, and transcriptome profiling were supported by grant 19-15-00419. Analyses of LOH and IHC markers were performed with the financial assistance of grant 21-14-00353. The funder played no role in study design, data collection, analysis and interpretation of data, or the writing of this manuscript.
Acknowledgments
We thank the Vishnevsky Institute of Surgery for sample collection and the Center for Precision Genome Editing and Genetic Technologies for Biomedicine, EIMB RAS for providing computing power. The study was performed using the equipment of EIMB RAS “Genome” center (http://www.eimb.ru/rus/ckp/ccu_genome_c.php).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2024.1381093/full#supplementary-material
References
1. Zanoletti E, Mazzoni A. Vagal paraganglioma. Skull Base. (2006) 16:161–7. doi: 10.1055/s-2006-949519
2. Lloyd R, Osamura R, Klöppel G, Rosai J. WHO classification of tumours of endocrine organs. 4th edition. Lyon: International Agency for Research on Cancer (2017).
3. Snezhkina A, Pavlov V, Dmitriev A, Melnikova N, Kudryavtseva A. Potential biomarkers of metastasizing paragangliomas and pheochromocytomas. Life (Basel). (2021) 11. doi: 10.3390/life11111179
4. Netterville JL, Jackson CG, Miller FR, Wanamaker JR, Glasscock ME. Vagal paraganglioma: a review of 46 patients treated during a 20-year period. Arch Otolaryngol Head Neck Surg. (1998) 124:1133–40. doi: 10.1001/archotol.124.10.1133
5. El-Naggar AK, Chan JKC, Rubin Grandis J, Takata T, Slootweg PJ, International Agency for Research on C. WHO classification of head and neck tumours. Lyon: IARC Press. (2017).
6. Savvateeva M, Kudryavtseva A, Lukyanova E, Kobelyatskaya A, Pavlov V, Fedorova M, et al. Somatic mutation profiling in head and neck paragangliomas. J Clin Endocrinol Metab. (2022) 107:1833–42. doi: 10.1210/clinem/dgac250
7. Ding Y, Li L, Han D, Wang S, Chen X. Head and neck Malignant paragangliomas: experience from a single institution. Ear Nose Throat J. (2021), 1455613211052338. doi: 10.1177/01455613211052338
8. Michalowska I, Lewczuk A, Cwikla J, Prejbisz A, Swoboda-Rydz U, Furmanek MI, et al. Evaluation of head and neck paragangliomas by computed tomography in patients with pheochromocytoma-paraganglioma syndromes. Pol J Radiol. (2016) 81:510–8. doi: 10.12659/PJR.897490
9. Bugalho MJ, Montalvao P, Domingues R, Duarte H. Clinical usefulness of (6)(8)Ga-DOTA-NOC PET/CT in staging a vagal paraganglioma associated with a novel SDHB mutation. BMJ Case Rep. (2016) 2016. doi: 10.1136/bcr-2015-213438
10. Ding Y, Feng Y, Wells M, Huang Z, Chen X. SDHx gene detection and clinical Phenotypic analysis of multiple paraganglioma in the head and neck. Laryngoscope. (2019) 129:E67–71. doi: 10.1002/lary.27509
11. van Nederveen FH, Dannenberg H, Sleddens HF, de Krijger RR, Dinjens WN. p53 alterations and their relationship to SDHD mutations in parasympathetic paragangliomas. Mod Pathol. (2003) 16:849–56. doi: 10.1097/01.MP.0000084111.03922.4D
12. Prasad SC, Paties CT, Pantalone MR, Mariani-Costantini R, Sanna M. Carotid body and vagal paragangliomas: epidemiology, genetics, clinicopathological features, imaging, and surgical management. In: Mariani-Costantini R, editor. Paraganglioma: A Multidisciplinary Approach. Brisbane (AU), Codon Publications. (2019).
13. Carney JA. Carney triad: a syndrome featuring paraganglionic, adrenocortical, and possibly other endocrine tumors. J Clin Endocrinol Metab. (2009) 94:3656–62. doi: 10.1210/jc.2009-1156
14. Castro-Vega LJ, Buffet A, De Cubas AA, Cascon A, Menara M, Khalifa E, et al. Germline mutations in FH confer predisposition to Malignant pheochromocytomas and paragangliomas. Hum Mol Genet. (2014) 23:2440–6. doi: 10.1093/hmg/ddt639
15. Snezhkina AV, Kalinin DV, Pavlov VS, Lukyanova EN, Golovyuk AL, Fedorova MS, et al. Immunohistochemistry and mutation analysis of SDHx genes in carotid paragangliomas. Int J Mol Sci. (2020) 21. doi: 10.3390/ijms21186950
16. Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv: Genomics. (2013). doi: 10.48550/arXiv.1303.3997
17. Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, et al. Twelve years of SAMtools and BCFtools. Gigascience. (2021) 10. doi: 10.1093/gigascience/giab008
18. Broad I. Picard Tools. (2018). Available at: https://broadinstitute.github.io/picard/.
19. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. (2010) 20:1297–303. doi: 10.1101/gr.107524.110
20. DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. (2011) 43:491–8. doi: 10.1038/ng.806
21. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. (2010) 38:e164. doi: 10.1093/nar/gkq603
22. Li Q, Wang K. InterVar: clinical interpretation of genetic variants by the 2015 ACMG-AMP guidelines. Am J Hum Genet. (2017) 100:267–80. doi: 10.1016/j.ajhg.2017.01.004
23. Kopanos C, Tsiolkas V, Kouris A, Chapple CE, Albarca Aguilera M, Meyer R, et al. VarSome: the human genomic variant search engine. Bioinformatics. (2019) 35:1978–80. doi: 10.1093/bioinformatics/bty897
24. Robinson JT, Thorvaldsdottir H, Wenger AM, Zehir A, Mesirov JP. Variant review with the integrative genomics viewer. Cancer Res. (2017) 77:e31–e4. doi: 10.1158/0008-5472.CAN-17-0337
25. Kudryavtseva AV, Lukyanova EN, Kalinin DV, Zaretsky AR, Pokrovsky AV, Golovyuk AL, et al. Mutational load in carotid body tumor. BMC Med Genomics. (2019) 12:39. doi: 10.1186/s12920-019-0483-x
26. Andrews S, FASTQC. A quality control tool for high throughput sequence data. (2010). Available at: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
27. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. (2014) 30:2114–20. doi: 10.1093/bioinformatics/btu170
28. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. (2013) 29:15–21. doi: 10.1093/bioinformatics/bts635
29. Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. (2014) 30:923–30. doi: 10.1093/bioinformatics/btt656
30. Hanzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinf. (2013) 14:7. doi: 10.1186/1471-2105-14-7
31. Wu T, Hu E, Xu S, Chen M, Guo P, Dai Z, et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation (Camb). (2021) 2:100141. doi: 10.1016/j.xinn.2021.100141
32. Snezhkina A, Fedorova M, Kobelyatskaya A, Markova D, Lantsova M, Ikonnikova A, et al. The SDHD:p.H102R variant is frequent in Russian patients with head and neck paragangliomas and associated with loss of 11p15.5 region and hypermethylation of H19-DMR. Int J Mol Sci. (2022) 24. doi: 10.3390/ijms24010628
33. Gyapay G, Morissette J, Vignal A, Dib C, Fizames C, Millasseau P, et al. The 1993-94 Genethon human genetic linkage map. Nat Genet. (1994) 7:246–339. doi: 10.1038/ng0694supp-246
34. Gill AJ. Succinate dehydrogenase (SDH) and mitochondrial driven neoplasia. Pathology. (2012) 44:285–92. doi: 10.1097/PAT.0b013e3283539932
35. Kavanagh KL, Oppermann U, (SGC) SGC. Crystal structure of human fumarate hydratase. (2008). doi: 10.2210/pdb3e04/pdb
36. Sehnal D, Bittrich S, Deshpande M, Svobodova R, Berka K, Bazgier V, et al. Mol* Viewer: modern web app for 3D visualization and analysis of large biomolecular structures. Nucleic Acids Res. (2021) 49:W431–W7. doi: 10.1093/nar/gkab314
37. Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. The protein data bank. Nucleic Acids Res. (2000) 28:235–42. doi: 10.1093/nar/28.1.235
38. Gieldon L, William D, Hackmann K, Jahn W, Jahn A, Wagner J, et al. Optimizing genetic workup in pheochromocytoma and paraganglioma by integrating diagnostic and research approaches. Cancers (Basel). (2019) 11. doi: 10.3390/cancers11060809
39. Hoekstra AS, Hensen EF, Jordanova ES, Korpershoek E, van der Horst-Schrivers AN, Cornelisse C, et al. Loss of maternal chromosome 11 is a signature event in SDHAF2, SDHD, and VHL-related paragangliomas, but less significant in SDHB-related paragangliomas. Oncotarget. (2017) 8:14525–36. doi: 10.18632/oncotarget.v8i9
40. Trpkov K, Hes O, Agaimy A, Bonert M, Martinek P, Magi-Galluzzi C, et al. Fumarate hydratase-deficient renal cell carcinoma is strongly correlated with fumarate hydratase mutation and hereditary leiomyomatosis and renal cell carcinoma syndrome. Am J Surg Pathol. (2016) 40:865–75. doi: 10.1097/PAS.0000000000000617
41. Bardella C, El-Bahrawy M, Frizzell N, Adam J, Ternette N, Hatipoglu E, et al. Aberrant succination of proteins in fumarate hydratase-deficient mice and HLRCC patients is a robust biomarker of mutation status. J Pathol. (2011) 225:4–11. doi: 10.1002/path.2932
42. Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu H, et al. Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. (2012) 26:1326–38. doi: 10.1101/gad.191056.112
43. Tomlinson IP, Alam NA, Rowan AJ, Barclay E, Jaeger EE, Kelsell D, et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet. (2002) 30:406–10. doi: 10.1038/ng849
44. Clark GR, Sciacovelli M, Gaude E, Walsh DM, Kirby G, Simpson MA, et al. Germline FH mutations presenting with pheochromocytoma. J Clin Endocrinol Metab. (2014) 99:E2046–50. doi: 10.1210/jc.2014-1659
45. Fuchs TL, Luxford C, Clarkson A, Sheen A, Sioson L, Elston M, et al. A clinicopathologic and molecular analysis of fumarate hydratase-deficient pheochromocytoma and paraganglioma. Am J Surg Pathol. (2022) 47:25–36. doi: 10.1097/PAS.0000000000001945
46. Muller M, Guillaud-Bataille M, Richard S, Benusiglio PR. Correction to: Reassessing the clinical spectrum associated with Hereditary Leiomyomatosis and Renal Cell Carcinoma syndrome in French FH mutation carriers. Clin Genet. (2018) 93:1118. doi: 10.1111/cge.13222
47. Letouze E, Martinelli C, Loriot C, Burnichon N, Abermil N, Ottolenghi C, et al. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell. (2013) 23:739–52. doi: 10.1016/j.ccr.2013.04.018
48. Muller M, Ferlicot S, Guillaud-Bataille M, Le Teuff G, Genestie C, Deveaux S, et al. Reassessing the clinical spectrum associated with hereditary leiomyomatosis and renal cell carcinoma syndrome in French FH mutation carriers. Clin Genet. (2017) 92:606–15. doi: 10.1111/cge.13014
49. Snabboon T, Houngngam N, Tangjittrong S, Sasiwimonphan K, Iampenkhae K, Prichayudhrichayudh S. Paraganglioma of the organ of Zuckerkandl and FH gene mutation. Endokrynol Pol. (2020) 71:583–4. doi: 10.5603/EP.a2020.0068
50. Ma X, Cui Y, Gao Y, Zhang X, Nie M, Tong A. Fumarate hydratase gene germline variants and mosaicism associated with pheochromocytoma and paraganglioma. Ann N Y Acad Sci. (2022) 1516:262–270. doi: 10.1111/nyas.14866
51. Schmidt C, Sciacovelli M, Frezza C. Fumarate hydratase in cancer: A multifaceted tumour suppressor. Semin Cell Dev Biol. (2020) 98:15–25. doi: 10.1016/j.semcdb.2019.05.002
52. Trpkov K, Hes O, Williamson SR, Adeniran AJ, Agaimy A, Alaghehbandan R, et al. New developments in existing WHO entities and evolving molecular concepts: The Genitourinary Pathology Society (GUPS) update on renal neoplasia. Mod Pathol. (2021) 34:1392–424. doi: 10.1038/s41379-021-00779-w
53. Buelow B, Cohen J, Nagymanyoki Z, Frizzell N, Joseph NM, McCalmont T, et al. Immunohistochemistry for 2-succinocysteine (2SC) and fumarate hydratase (FH) in cutaneous leiomyomas may aid in identification of patients with HLRCC (Hereditary leiomyomatosis and renal cell carcinoma syndrome). Am J Surg Pathol. (2016) 40:982–8. doi: 10.1097/PAS.0000000000000626
54. Sun G, Zhang X, Liang J, Pan X, Zhu S, Liu Z, et al. Integrated molecular characterization of fumarate hydratase-deficient renal cell carcinoma. Clin Cancer Res. (2021) 27:1734–43. doi: 10.1158/1078-0432.CCR-20-3788
55. Torrents D, Estevez R, Pineda M, Fernandez E, Lloberas J, Shi YB, et al. Identification and characterization of a membrane protein (y+L amino acid transporter-1) that associates with 4F2hc to encode the amino acid transport activity y+L. A candidate gene for lysinuric protein intolerance. J Biol Chem. (1998) 273:32437–45. doi: 10.1074/jbc.273.49.32437
56. Noguchi A, Takahashi T. Overview of symptoms and treatment for lysinuric protein intolerance. J Hum Genet. (2019) 64:849–58. doi: 10.1038/s10038-019-0620-6
57. Borggrefe T, Yue X. Interactions between subunits of the Mediator complex with gene-specific transcription factors. Semin Cell Dev Biol. (2011) 22:759–68. doi: 10.1016/j.semcdb.2011.07.022
Keywords: head and neck paraganglioma, vagal paraganglioma, mutation profile, LOH, biallelic inactivation, FH
Citation: Snezhkina AV, Pavlov VS, Kalinin DV, Pudova EA, Krasnov GS, Ayupova AF, Kobelyatskaya AA, Dmitriev AA, Atiakshin DA, Fedorova MS and Kudryavtseva AV (2024) Genetic changes in the FH gene cause vagal paraganglioma. Front. Endocrinol. 15:1381093. doi: 10.3389/fendo.2024.1381093
Received: 02 February 2024; Accepted: 12 April 2024;
Published: 24 April 2024.
Edited by:
Anna Angelousi, National and Kapodistrian University of Athens, GreeceReviewed by:
Lawrence S. Kirschner, The Ohio State University, United StatesAlberto Cascon, Spanish National Cancer Research Center, Spain
Rafael Torres Perez, National Center for Biotechnology, Spanish National Research Council (CSIC) Madrid, Spain, in collaboration with reviewer AC
Copyright © 2024 Snezhkina, Pavlov, Kalinin, Pudova, Krasnov, Ayupova, Kobelyatskaya, Dmitriev, Atiakshin, Fedorova and Kudryavtseva. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anastasiya V. Snezhkina, bGVmdGdlckByYW1ibGVyLnJ1