 Dandan Dai
Dandan Dai Jing Xie*
Jing Xie*- Department of Pathology, Ruijin Hospital Affiliated to Shanghai Jiaotong University School of Medicine, Shanghai, China
Pheochromocytoma is a neuroendocrine neoplasm that originates from chromaffin cells of the adrenal medulla. Langerhans cell histiocytosis (LCH) is a proliferative disease of histiocyte-like cells, often associated with activating mutations of the mitogen-activated protein kinase (MAPK) pathway. We present a case of a 49-year-old male with a history of pheochromocytoma, which metastasized to the inferior vena cava eight years after left adrenalectomy. At the same time, it was found that the pheochromocytoma in the metastasis was complicated with LCH, a combination that has not been previously reported. Genetic analysis was carried out by next-generation sequencing (NGS) technology. Somatic mutations of BRAF and RAD54B were detected in Langerhans cells and EPAS1 in pheochromocytoma.
1 Introduction
Pheochromocytomas are rare tumors originating in the adrenal medulla (1) and usually secrete catecholamines leading to hypertension and myocardial degenerative effects (2). Metastatic Pheochromocytoma is most commonly reported in the local lymph nodes, bone, liver and lung (3). Since it is impossible to differentiate non-metastatic and metastatic Pheochromocytoma based upon clinical or even histopathological findings, all Pheochromocytoma are currently considered potentially metastatic tumours (WHO 2022 classification) (4). As a result, all patients with Pheochromocytoma require long and intensive follow up. Pheochromocytoma mostly results from pathogenic variants of predisposing genes, with a genetic contribution that now stands at around 70%. Germline variants account for approximately 40%, while the remaining 30% is attributable to somatic variants (5). Langerhans cell histiocytosis (LCH), the most common histiocytic disorder, encompasses conditions characterized by aberrant function and differentiation or proliferation of cells of the mononuclear phagocyte system (6). LCH is a histiocytic neoplasm characterized by a mass of CD1a + CD207+ histiocytes, exhibiting a diverse range of clinical manifestations from a self-healing rash or single bone destruction to multi-organ disease with potentially fatal consequences (7). In this article, we present a unique case of metastatic pheochromocytoma complicated with LCH. A 49-year-old male had undergone a left adrenalectomy in 2016 and pathology confirmed pheochromocytoma. In January 2024, the patient presented with intermittent low-grade fever, prompting a comprehensive examination that revealed a mass within the inferior vena cava. Elevated catecholamine levels were detected in both blood and urine. While high resolution CT showed multiple nodules in both lungs. The patient was considered to have multiple metastases of pheochromocytoma and a biopsy of the mass in the inferior vena cava was performed. The pathology confirmed metastatic pheochromocytoma complicated with LCH. Molecular pathology showed EPAS1 mutation in pheochromocytoma. BRAF insertion mutation and RAD54B frameshift mutation was detected in in Langerhans cells. This report is, to our knowledge, the first case of pheochromocytoma coexisting with LCH, and also the primary report of detecting RAD54B mutation in LCH. It highlights the possibility of intravascular metastasis occurring simultaneously with LCH in patients with a previous history of pheochromocytoma even years after adrenalectomy and emphasizes the need to adopt a comprehensive next-generation sequencing (NGS) panel. According to the latest guidelines, it is mandatory to perform genetic analysis in all pheochromocytoma cases regardless of phenotype. Besides, We propose testing for RAD54B gene variants. A possible correlation between RAD54B pathogenic variants and LCH clinical course should be considered. For patients diagnosed with pheochromocytoma following surgical intervention, long-term blood pressure monitoring and regular follow-up are recommended to find recurrence or metastasis in time.
2 Clinical presentation
A 49-year-old man without specific family history presented with nocturnal episodic headaches in 2016. The headache lasted about 2 hours, accompanied by sweating, no obvious palpitation or dizziness. The patient presented at the local hospital with elevated blood pressure (specific value unknown). Urinary measurements demonstrated elevated level of vanillylmandelic acid at 15.28 mg/24h (normal range, 2-6 mg/24 h), while aldosterone and serum calcium remained within normal limits. Abdominal computed tomography revealed a left adrenal nodule approximately 6.1x4.5cm in size. On the basis of these findings, pheochromocytoma was suspected. In the same year, the patient underwent a left adrenalectomy. The headache and sweating recovered soon after operation. The pathology examination revealed the presence of pheochromocytoma with vascular invasion and capsular invasion(Figures 1A–C). Tumour cells were strongly immunopositive for synaptophysin (Figure 1D), chromogranin A (Figure 1E), SDHB (Figure 1F). Sustentacular cells were immunopositive for S100 protein(Figure 1G). Moreover, immunostains for AE1/AE3, CD56, CD34, Her2, Inhibin and HMB45 were negative (data not shown). Pheochromocytoma of the Adrenal Gland Scaled Score (PASS) of 4 was assigned and the Ki67 proliferation index was 1%. According to the guidelines for genetic screening of Pheochromocytomas and paragangliomas (PPGLs), our patient was proposed for next-generation sequencing (NGS) targeting. EPAS1 mutation was detected in pheochromocytoma. No germline mutations were found. The patient was treated with antihypertensive medication resulting in blood pressure control within the normal range. After discharge, the patient has not been regularly followed up.
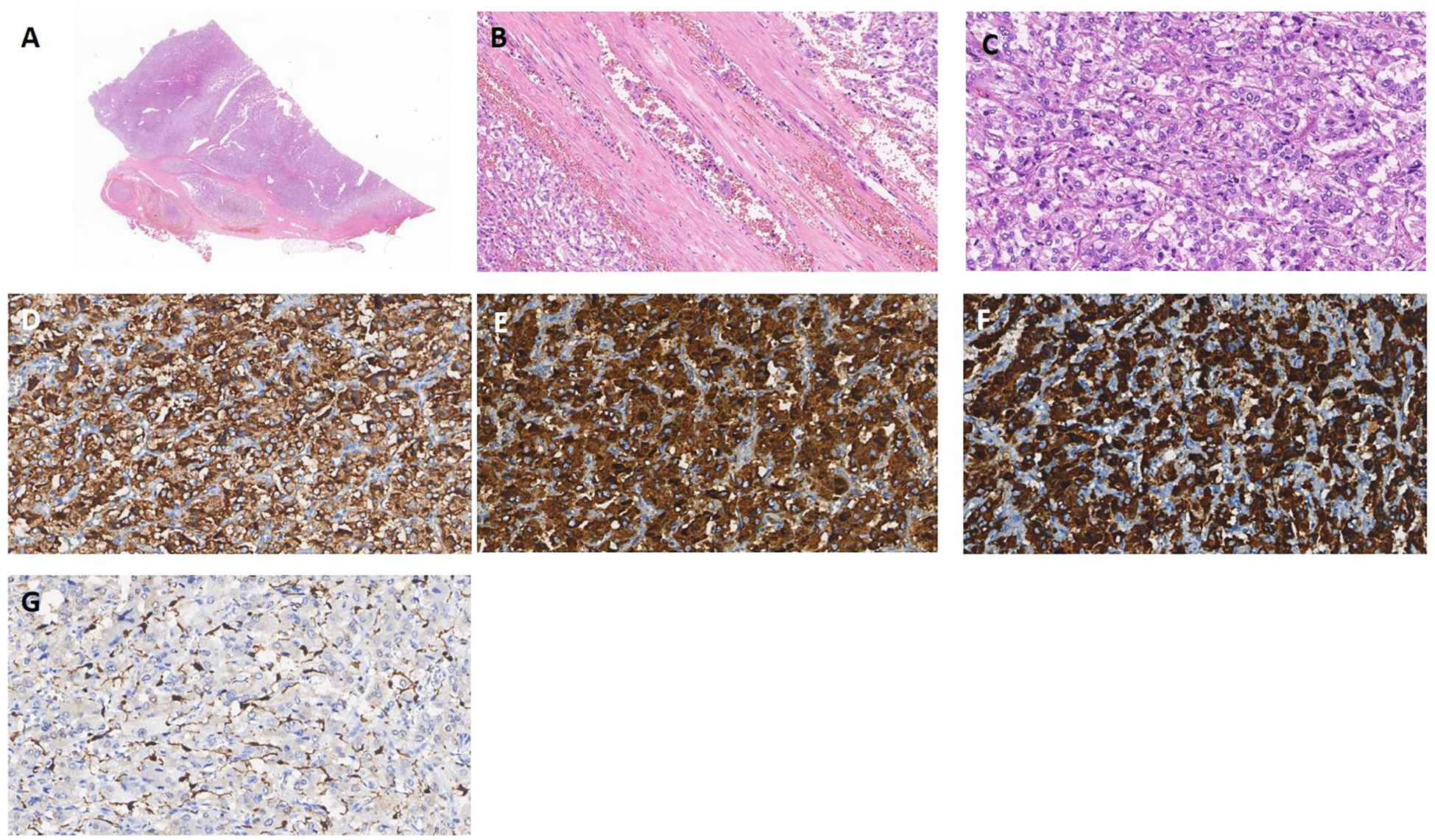
Figure 1. Pheochromocytoma (A) Pheochromocytoma with vascular invasion (x5 HE). (B) Intravascular tumor embolus (x100 HE). (C) Classic histoarchitecture with uniform cell nests(Zallballen)(x200 HE). (D-F) Syn, CgA and SDHB expression is positive, supporting the diagnosis of pheochromocytoma(x200). (G) Immunohistochemical stain for S100 showing classic distribution of sustentacular cells, mostly at the periphery of cell nests(x200).
In January 2024, the patient developed intermittent low-grade fever. He received treatment at the local hospital for 2 days without improvement (specific details unknown). Abdominal color Doppler ultrasound showed foreign bodies in the inferior vena cava, while high resolution CT showed multiple nodules in both lungs. 68Ga-DOTATATE PET/CT scan(0.59mCi) was performed showing a soft tissue density mass in the left renal vein-inferior vena cava with abnormally high metabolism and multiple solid nodules in both lungs with slightly high metabolism. Combined with the medical history, the patient was considered to have tumor recurrence invading the inferior vena cava, along with multiple metastases in both lungs. The patient’s low-grade fever resolved spontaneously after hospitalization and did not recur.
In March 2024, the patient came to the hospital for further examination. Laboratory tests revealed an elevated level of plasma normetanephrine at 5279.1 pg/ml (normal range, 19–121 pg/ml), while metanephrine remained within normal limits. Urinary measurements also revealed elevation levels of epinephrine at 22.57 ug/24h (normal range, <22ug/24h) and norepinephrine at 2290.15 ug/24h (normal range, 7-65ug/24h). Computed tomographic angiography of the abdominal aorta revealed the presence of a mass extending from the left renal vein to the inferior vena cava (Figures 2A–C). No obvious abnormality was found in the right adrenal gland. After two weeks of prophylactic treatment with alpha-blockers, a biopsy of the tumor in the inferior vena cava was conducted under local anesthesia. Pathology confirmed metastatic pheochromocytoma accompanied by LCH. Immunophenotyping of chromaffin cells was consistent with the primary lesion (Figures 2E, F). The pheochromocytoma area was accompanied by an adjacent region comprising of large monocytic cells with reniform-to-oval nuclei and a central horizontal groove, and plentiful of eosinophils (Figures 2D, G). The mononuclear cells revealed positivity for CD1a (Figure 2H), Langerin (Figure 2I) S100 and the Ki67 proliferation index was 10%. Molecular pathology identified the c.1457_1471del; p.N486_P490del mutation in the 12th exon of the BRAF gene, and the c.528dupT; p.V177Cfs*9 mutation in the 5th exon of the RAD54B gene. The patient underwent a 68Ga-DOTATATE PET/CT scan(0.59mCi). Multiple solid pulmonary nodules were found in both lungs, some of which had slightly higher metabolism. It may suggest the potential pulmonary metastasis from pheochromocytoma. The patient was recommended to undergo further diagnostic tests in order to confirm the diagnosis. However, the patient expressed no intention to pursue additional investigations. Considering the difficulty of the operation, the patient postponed the operation and took antihypertensive treatment including doxazosin mesylate extended release tablets and arotinolol hydrochloride tablets. The 24-hour ambulatory blood pressure showed that systolic blood pressure and diastolic blood pressure were normal during the day and night after drug use. The circadian rhythm of systolic blood pressure and diastolic blood pressure disappeared. The 24-hour systolic blood pressure and diastolic blood pressure increased (120/77mmHg). After discharge, the patient’s blood pressure was monitored and stabilized, and regular follow-up was requested. The clinical course is shown in Table 1.
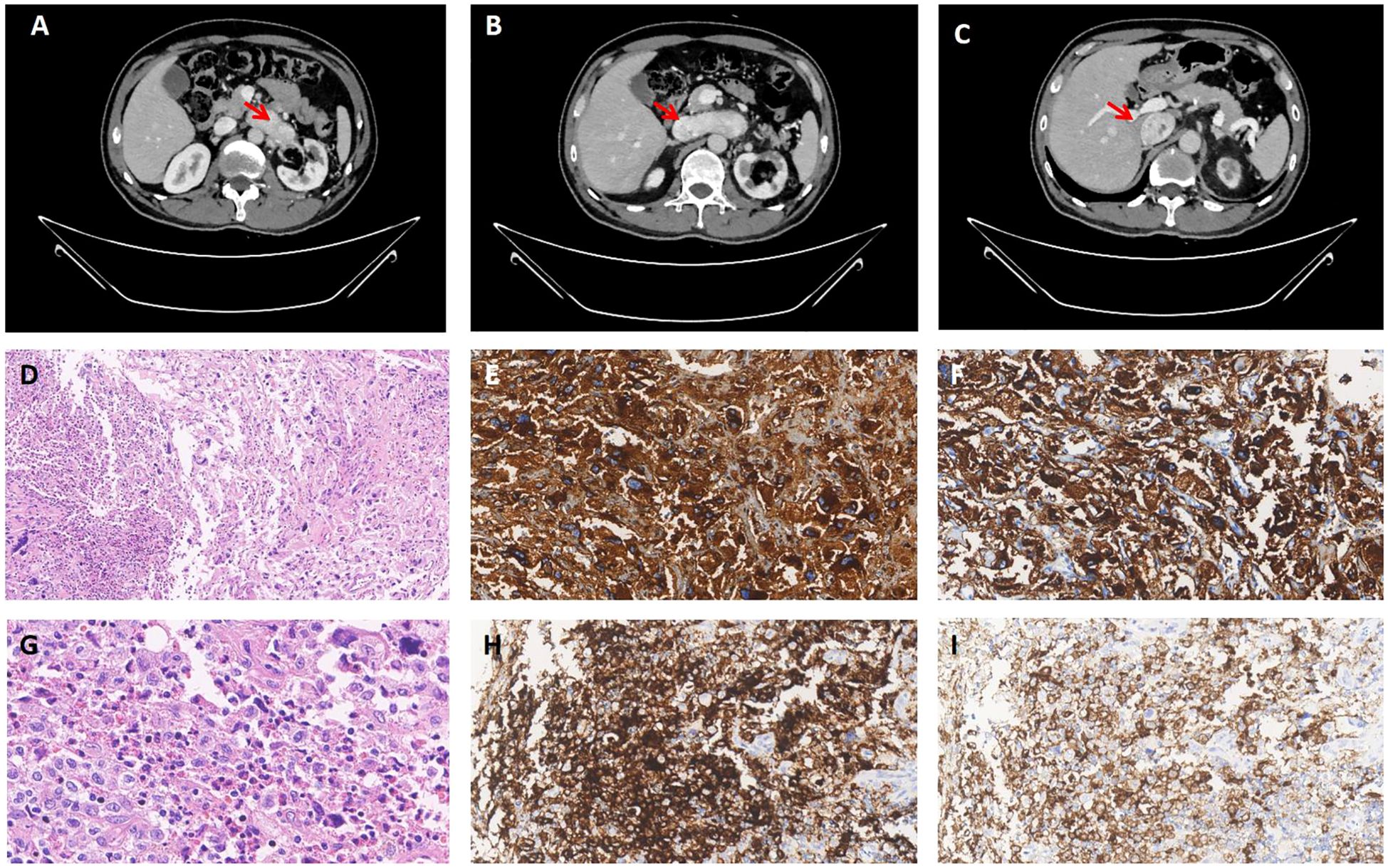
Figure 2. Computed tomographic angiography and pathological images. (A-C) The lumen of the left renal vein showed a soft tissue mass extending from its junction with the inferior vena cava, along the course of the inferior vena cava. The enhanced scan exhibited significant enhancement. Pheochromocytoma in metastasis. (D) The mononuclear cell area on the left side exhibits a significant presence of numerous eosinophils(x100 HE). (E, F) Pheochromocytoma expressing CgA and syn (x200). (G) The monocytic cells have reniform-to-oval nuclei and a central horizontal groove(x400 HE). (H, I) The mononuclear cells are positive for CD1a and Langerin (x200).
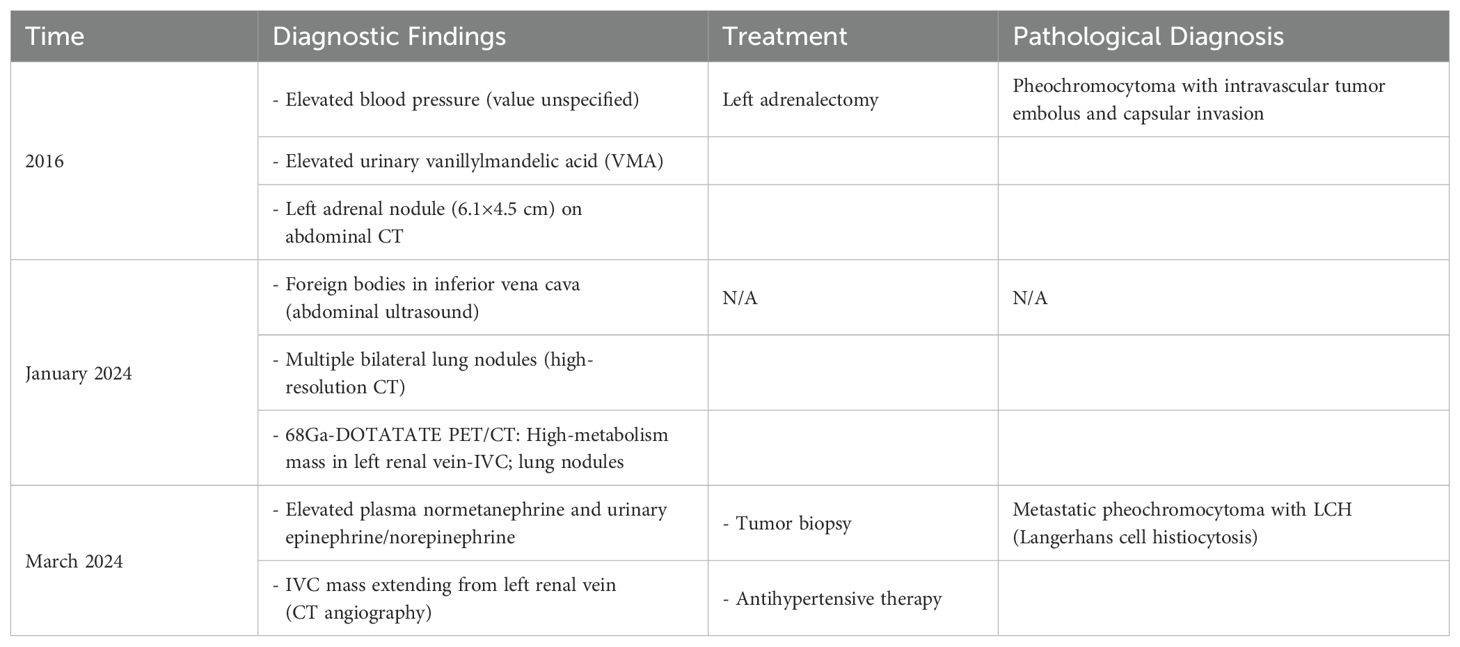
Table 1. Clinical timeline.
3 Discussion
This case represents a rare complication of pheochromocytoma metastasizing to the inferior vena cava with concomitant LCH. The metastasis of pheochromocytoma to the inferior vena cava has not been reported. In this case EPAS1(HIF2α) mutation was detected. EPAS1has been identified as one of the susceptibility genes associated with Pheochromocytomas (8).
PPGLs are rare neuroendocrine tumors (NETs) derived from adrenomedullary chromaffin cells and from the autonomic paraganglia, respectively. Pheochromocytomas (PCCs) represent about 80–85% of chromaffin-cell neoplasms, whereas paragangliomas (PGLs) account for the remaining 15–20%. Based on transcriptional profile, PPGLs are classified into three clusters. The tree clusters are: 1) hypoxia/pseudohypoxia, 2) kinase signaling group, 3) Wnt signaling pathway (9). Cluster 1 includes PPGLs with variants in genes encoding the hypoxia-inducible factor (HIF) 2α, the Von Hippel–Lindau tumor suppressor (VHL), the prolyl hydroxylase domain (PHD), fumarate hydratase (FH), and succinate dehydrogenase subunits (SDHx) (10). All these mutations promote HIFa stabilization and accumulation resulting in increased angiogenesis via changes in vascular endothelial growth factor-1 and -2 receptors (VEGFR1/2) and platelet-derived growth factor-b receptor (PDGFR) transcription (11). Cluster 2 consists of germline or somatic mutations in RET, NF1, TMEM127, MAX, HRAS and KIF1Bβ which associated with PI3 kinase pathways the “PI3K/AKT/mTOR and MAPK/ERK”. Cluster 2 PPGLs are mostly benign exhibiting a mature catecholamine phenotype (12). Cluster 3 PPGLs are due to somatic mutations of the CSDE1 gene or somatic gene fusions of the MAML3 gene (13). Cluster 3 tumors have a more aggressive behavior (5).
LCH is caused by clonal expansion of myeloid precursors that differentiate into CD1a+/CD207+ cells in lesions that leads to a spectrum of organ involvement and dysfunction (14). LCH is often characterized by activating mutations of the mitogen-activated protein kinase (MAPK) pathway with BRAFV600E being the most recurrent mutation. The remaining cases of LCH that do not bear the BRAFV600E mutation are often characterized by other mutations in the BRAF gene (15). BRAF mutations have been reported in >50% of patients with LCH (16). BRAF is a core component of the MAPK/ERK1/2 signaling cascade and involves the sequential phosphorylation and activation of RAS-RAF-MEK-ERK (17). In this case, molecular pathology identified the c.1457_1471del; p.N486_P490del mutation in the 12th exon of the BRAF gene, which is atypical.
Notably, for the first time, a somatic mutation in the oncogene RAD54B was identified in Langerhans cells. RAD54B belongs to the SNF2/SWI2 superfamily, involving in cell cycle regulation after DNA damage and participating in homologous recombinational repair, which ensures the precise repair of the most deleterious DNA lesions, double-stranded breaks (18). RAD54B displays oncogene-like characteristics and is amplified or overexpressed in a diverse array of cancer types, including colorectal, lung, prostate and breast (19). It has not been proven yet that RAD54B mutation is associated with LCH.
Tumorigenesis is a multiphase process dependent on several modifications at cellular and tissue levels, leading to sustain proliferative signalling, evasion from growth suppressors and from cell death, replicative immortality, and induction of angiogenesis, invasion, and metastasis (20). Beyond genetic alterations, the interplay among cancer cells and tumour microenvironment (TME) components has a central role in tumour initiation and progression (21). Angiogenesis, the development of new blood vessels from established vasculature, provides growth and hematogenous dissemination of the cancer cells. In this case EPAS1 mutation was detected in pheochromocytoma. EPAS1 mutation promotes HIFa stabilization and accumulation resulting in increased angiogenesis, which might enhance the likelihood of the formation and metastasis of other tumors, resulting in the coexistence of the two types of tumors.
More conclusive evidence is still required to ascertain whether there is a definitive correlation between pheochromocytoma and LCH concerning their pathogenesis. Currently, it remains uncertain whether this case represents a simple collision tumor or if it is an instance of LCH triggered by a pheochromocytoma. Due to the presence of multiple nodules in lungs, we were more inclined to suspect that the patient may have systemic LCH and recommended further examination. However, the patient did not cooperate. We recommend regular monitoring of blood pressure and periodic reviews for the patient.
Presently, clinical and hystopatological scoring systems have been studied and validated to assist in predicting the risk of disease recurrence. Parasiliti-Caprino et al. (22) conducted a retrospective multicenter study on 177 PCC patients who underwent radical surgery and proposed a multivariable continuous model for post-surgical PCC recurrence prediction. The model was named the SGAP-model (size, genetic, age, and PASS). It was created on an 8-point scale, by assigning 1 point for tumor size > 50 mm, 3 points for positive genetic testing, 1 point for age ≤ 35 years, and 3 points for PASS ≥ 3. Patients with a SGAP-score of 0-2 showed a virtually absent risk of recurrence; patients with a SGAP-score of 3-4 showed an intermediate risk profile; patients with a SGAP-score of 5-8 showed a markedly elevated risk of recurrence that exceeded 60% after 10 years. An accurate estimation of recurrence risk would be of fundamental importance in clinical practice, as it may allow clinicians to suggest a higher-intensity monitoring when the estimated recurrence risk is high.
4 Conclusion
In conclusion, the current case reminds us that pheochromocytoma can metastasize to the inferior vena cava. Due to its rare occurrence and non-specific clinical manifestations, imaging may still be the most valuable method for discovering metastasis. Pheochromocytoma accompanied by LCH has not been reported yet. This rare complication may indicate a potential relationship in the pathogenesis between the two. Whether RAD54B gene mutation is a pathogenic mutation of LCH needs to be further explored. Importantly, no pheochromocytoma can be considered fully benign and all patients should be followed for life for recurrence, new primary pheochromocytoma, and metastatic disease.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Ethics statement
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
DD: Writing – original draft. JX: Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
We are deeply grateful to Dr. Yinchun Jin for his expertise in histological section preparation. Special thanks to Dr. Zhongyu Wang for his insightful interpretation of molecular mechanisms. We also wish to acknowledge Dr. Xu Zhong for his invaluable assistance in clinical data curation.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Farrugia FA, Charalampopoulos A. Pheochromocytoma. Endocr Regul. (2019) 53:191–212. doi: 10.2478/enr-2019-0020
2. Tischler AS, Kimura N, Mcnicol AM. Pathology of pheochromocytoma and extra-adrenal paraganglioma. Ann New York Acad Sci. (2006) 1073:557–70. doi: 10.1196/annals.1353.059
3. Angelousi A, Kassi E, Zografos G, Kaltsas G. Metastatic pheochromocytoma and paraganglioma. Eur J Clin Invest. (2015) 45:986–97. doi: 10.1111/eci.12495
4. Raleigh DR, Solomon DA, Lloyd SA, Lazar A, Garcia MA, Sneed PK, et al. Histopathologic review of pineal parenchymal tumors identifies novel morphologic subtypes and prognostic factors for outcome. Neuro Oncol. (2017) 19:78–88. doi: 10.1093/neuonc/now105
5. Provenzano A, Chetta M, De Filpo G, Cantini G, La Barbera A, Nesi G, et al. Novel germline PHD2 variant in a metastatic pheochromocytoma and chronic myeloid leukemia, but in the absence of polycythemia. Med (Kaunas). (2022) 58:1113. doi: 10.3390/medicina58081113
6. Allen CE, Merad M, McClain KL. Langerhans-cell histiocytosis. N Engl J Med. (2018) 379:856–68. doi: 10.1056/NEJMra1607548
7. Li Q. Successful treatment of Langerhans cell histiocytosis in an infant with vemurafenib: a case report and literature review. J Dermatol Treat. (2023) 34:2279901. doi: 10.1080/09546634.2023.2279901
8. Buffet A, Burnichon N, Favier J, Gimenez-Roqueplo AP. An overview of 20 years of genetic studies in pheochromocytoma and paraganglioma. Best Pract Res Clin Endocrinol Metab. (2020) 34:101416. doi: 10.1016/j.beem.2020.101416
9. Crona J, Taïeb D, Pacak K. New perspectives on pheochromocytoma and paraganglioma: toward a molecular classification. Endocr Rev. (2017) 38:489–515. doi: 10.1210/er.2017-00062
10. Pacak K, Wimalawansa SJ. Pheochromocytoma and paraganglioma. Endocr Pract. (2015) 21:406–12. doi: 10.4158/EP14481.RA
11. Martinelli S, Amore F, Canu L, Maggi M, Rapizzi E. Tumour microenvironment in pheochromocytoma and paraganglioma. Front Endocrinol (Lausanne). (2023) 14:1137456. doi: 10.3389/fendo.2023.1137456
12. Martinelli S, Maggi M, Rapizzi E. Pheochromocytoma/paraganglioma preclinical models: which to use and why? Endocr Connect. (2020) 9:R251–60. doi: 10.1530/EC-20-0472
13. Majewska A, Budny B, Ziemnicka K, Ruchała M, Wierzbicka M. Head and neck paragangliomas-A genetic overview. Int J Mol Sci. (2020) 21:7669. doi: 10.3390/ijms21207669
14. Rodriguez-Galindo C, Allen CE. Langerhans cell histiocytosis. Blood. (2020) 135:1319–31. doi: 10.1182/blood.2019000934
15. Sconocchia T, Foßelteder J, Sconocchia G, Reinisch A. Langerhans cell histiocytosis: current advances in molecular pathogenesis. Front Immunol. (2023) 14:1275085. doi: 10.3389/fimmu.2023.1275085
16. Whitlock JA, Geoerger B, Dunkel IJ, Roughton M, Choi J, Osterloh L. alone or in combination with trametinib, in BRAF V600-mutated pediatric Langerhans cell histiocytosis. Blood Adv. (2023) 7:3806–15. doi: 10.1182/bloodadvances.2022008414
17. Zhang W, Liu HT. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. (2002) 12:9–18. doi: 10.1038/sj.cr.7290105
18. Mai Y, Lin T, Zhang L, Yang W, Liu S, Wang M, et al. RAD54B inhibits vascular endothelial senescence via suppression of CHK1/p53/p21 pathway. Can J Physiol Pharmacol. (2024) 102:137–49. doi: 10.1139/cjpp-2023-0192
19. McAndrew EN, McManus KJ. The enigmatic oncogene and tumor suppressor-like properties of RAD54B: Insights into genome instability and cancer. Genes Chromosomes Cancer. (2017) 56:513–23. doi: 10.1002/gcc.22458
20. Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
21. Shoucair I, Weber Mello F, Jabalee J, Maleki S, Garnis C. The role of cancerassociated fibroblasts and extracellular vesicles in tumorigenesis. Int J Mol Sci. (2020) 21:6837. doi: 10.3390/ijms21186837
Keywords: Langerhans cell histiocytosis, pheochromocytoma, metastasis, case report, EPAS1 gene
Citation: Dai D and Xie J (2025) Metastatic pheochromocytoma complicated with Langerhans cell histiocytosis: a case report. Front. Endocrinol. 16:1494783. doi: 10.3389/fendo.2025.1494783
Received: 12 September 2024; Accepted: 24 March 2025;
Published: 11 April 2025.
Edited by:
Suja Pillai, The University of Queensland, AustraliaReviewed by:
Serena Martinelli, University of Florence, ItalyChiara Lopez, University of Turin, Italy
Copyright © 2025 Dai and Xie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jing Xie, eGoxMTU1OUByamguY29tLmNu