 Yanan Bai
Yanan Bai Jianglan Wu1
Jianglan Wu1- 1College of Traditional Chinese Medicine, Hunan University of Traditional Chinese Medicine, Changsha, Hunan, China
- 2Diagnostics of Traditional Chinese Medicine, National Key Discipline, Hunan University of Traditional Chinese Medicine, Changsha, Hunan, China
Diabetes is a metabolic disorder primarily characterized by persistent hyperglycemia. Diabetes-induced inflammation significantly compromises cardiovascular health, greatly increasing the risk of atherosclerosis. The increasing prevalence of harmful lifestyle habits and overconsumption has contributed substantially to the global rise in diabetes-related cardiovascular diseases, creating a significant economic and healthcare burden. Although current therapeutic strategies focus on blood glucose control and metabolic regulation, clinical observations show that diabetic patients still face persistent residual risk of AS even after achieving metabolic stability. Recent studies suggest that this phenomenon is linked to diabetes-induced trained immunity. Diabetes can induce trained immunity in bone marrow progenitor cells and myeloid cells, thus promoting the long-term development of AS. This article first introduces the concept and molecular mechanisms of trained immunity, with particular emphasis on metabolic and epigenetic reprogramming, which plays a crucial role in sustaining chronic inflammation during trained immunity. Next, it summarizes the involvement of trained immunity in diabetes and its contribution to AS, outlining the cell types that can be trained in AS. Finally, it discusses the connection between diabetes-induced trained immunity and AS, as well as the potential of targeting trained immunity as an intervention strategy. Understanding the molecular mechanisms of trained immunity and their impact on disease progression may provide innovative strategies to address the persistent clinical challenges in managing diabetes and its complications.
1 Introduction
Diabetes represents a persistent metabolic condition marked by increased blood glucose concentrations, which arise from inadequate insulin production, insulin resistance, or a combination of these mechanisms. The swift rise in its global occurrence has rendered it a notable issue for public health (1). A recent forecast from the International Diabetes Federation indicates that around 536.6 million adults between the ages of 20 and 79 were diagnosed with diabetes in 2021, accounting for roughly 10.5% of the worldwide population. By the year 2045, projections indicate that the population of individuals diagnosed with diabetes will increase to 783.2 million, reflecting a prevalence rate of around 12.2% (2). In China, the prevalence of diabetes exhibited a significant rise, escalating from 9.7% in 2007 to 11.2% in 2017. This trend positions the nation as having the highest number of diabetes cases and the most substantial healthcare burden on a global scale (3). Diabetes is categorized into two main forms: type 1 diabetes (T1D) and T2D. Both forms are linked to serious complications and pose a considerable challenge to public health (4).
Atherosclerosis(AS) is one of the most common complications of diabetes, and the atherosclerotic cardiovascular diseases (ASCVDs) it triggers are the leading cause of death among individuals with diabetes (5). Epidemiological studies suggest that individuals diagnosed with T1D or T2D exhibit a two to fourfold increased likelihood of developing ASCVDs (6). Prospective studies have shown that hyperglycemia, a defining characteristic of diabetes, serves as an independent and significant risk factor for cardiovascular diseases (CVDs) in individuals with diabetes (7, 8). The low-grade systemic inflammation triggered by hyperglycemia notably accelerates AS and results in significant vascular lesions (9). Furthermore, this heightened risk is intricately associated with various significant pathological mechanisms observed in individuals with diabetes, such as dyslipidemia, genetic predisposition, insulin resistance, and obesity (5, 10). The presence of these metabolic comorbidities not only heightens the likelihood of cardiovascular incidents but also intensifies the progression of diabetes, creating a detrimental feedback loop. Individuals with diabetes exhibit elevated residual risks in the management of CVDs, rendering conventional treatment approaches frequently inadequate for the comprehensive control of cardiovascular events. Research indicates that previous conditions such as dyslipidemia, hyperglycemia, or being overweight can result in enduring “legacy effects,” where the likelihood of vascular complications continues even after achieving normalized blood glucose and metabolic (11, 12). Recent findings indicate that this phenomenon could be associated with the trained immunity observed in individuals diagnosed with diabetes.
Trained immunity describes the phenomenon where innate immune cells develop memory-like responses to previous stimuli, allowing them to respond more robustly to future identical or varied challenges (13). Trained immunity presents a complex duality. On one hand, it functions as a defensive strategy against recurring infectious agents and neoplasms; conversely, when innate immune cells play a role in the pathophysiology of chronic inflammatory conditions, such as AS, trained immunity fosters enduring disease-associated characteristics and hastens disease advancement (14, 15). Recent studies have indicated the possible causal involvement of high glucose-induced activation of the innate immune response in AS, substantiating that trained immunity may serve as a mechanism connecting diabetes and AS (16). Thus, a thorough exploration of the mechanisms underlying trained immunity could yield essential new perspectives on the prevention and management of cardiovascular complications associated with diabetes.
2 Trained immunity
2.1 Brief introduction to trained immunity
The human immune system can be categorized into two primary components: innate immunity and adaptive immunity. The innate immune system identifies pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs) via pattern recognition receptors (PRRs). This recognition triggers a swift activation of immune cells and the release of various substances, resulting in a rapid yet non-specific defense response (17). In contrast, the adaptive immune response operates at a slower pace yet produces highly specific reactions to pathogens, establishing long-term memory that enables the organism to eradicate pathogens more efficiently and effectively during subsequent exposures (18). Historically, immune memory has been regarded as a characteristic solely of adaptive immunity, whereas innate immunity was perceived as offering only non-specific defense mechanisms. In 2011, Netea et al. (13) presented the concept of “trained immunity,” demonstrating that innate immune cells possess the ability to establish enduring, non-specific memory following their initial encounter with pathogens. This leads to a more robust reaction when faced with either identical or varied stimuli, thereby questioning the conventional perspective that innate immunity is devoid of memory. The mechanisms, however, exhibit distinct characteristics compared to those of adaptive immune memory (Figure 1).
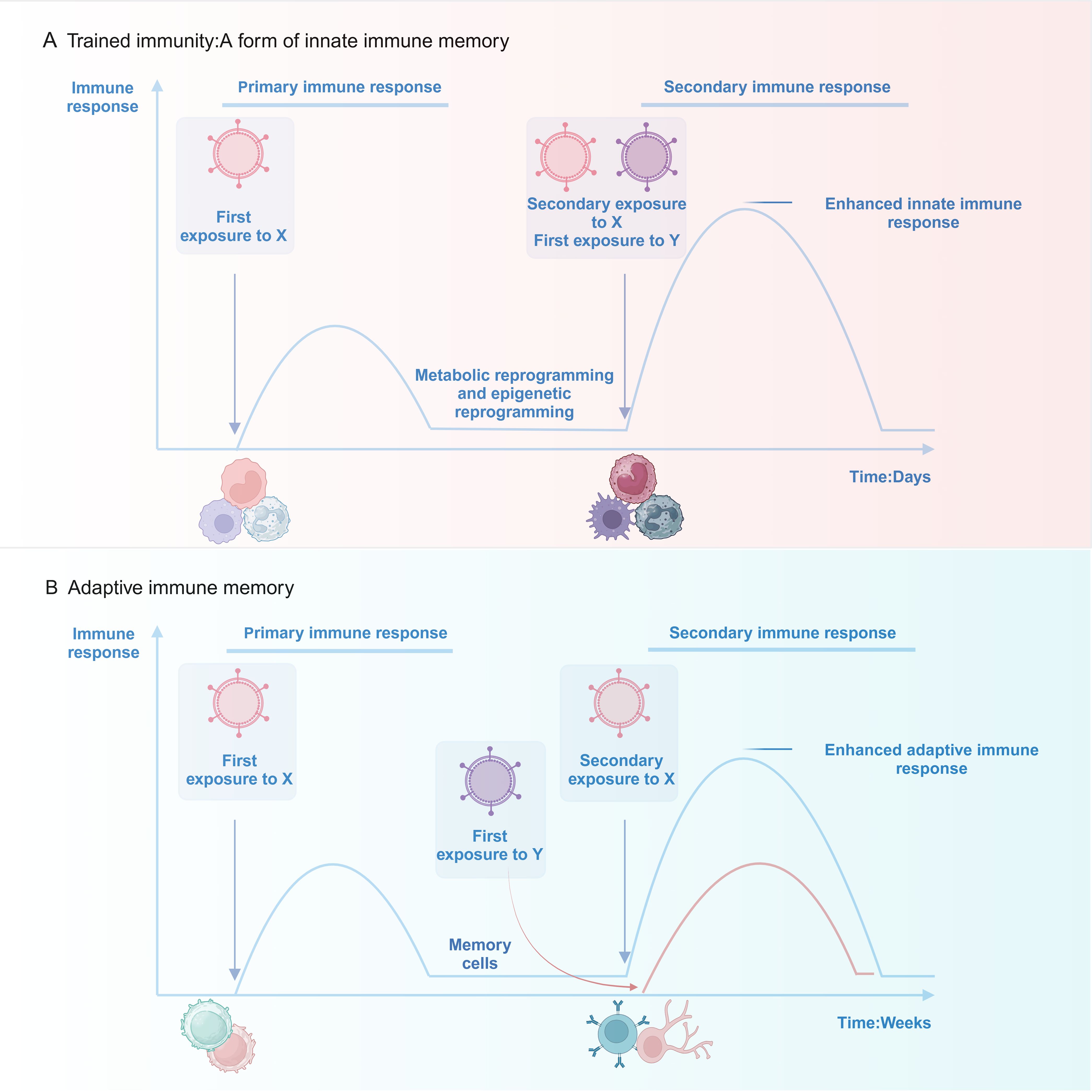
Figure 1. Upon exposure to stimulus X, innate immune cells (e.g., monocytes/macrophages and neutrophils) activate rapidly and develop memory via metabolic and epigenetic reprogramming. This improves their response to subsequent exposures of stimulus X or an unrelated stimulus Y. Innate immune memory can last up to a year. In contrast, adaptive immune cells (T and B lymphocytes) require weeks to form memory after activation by stimulus X, resulting in memory lasting decades. Adaptive immune memory is highly specific: re-exposure to stimulus X triggers a strong recall response, while a new stimulus Y initiates a fresh immune response.
Trained immunity was originally believed to be triggered by external factors, including microbial infections or BCG vaccination, resulting in the enduring functional reprogramming of immune cells (19). Following the cessation of stimuli, immune cells persist in a “trained” condition, facilitating a more rapid and robust response upon subsequent exposure (20). Recent investigations have demonstrated that endogenous molecules, including oxidized low-density lipoprotein (oxLDL) (21), aldosterone (22), high glucose (16),and catecholamines (23), can also induce trained immunity. This internal stimulation could transition the innate immune system from a protective to a more hostile function, which may result in overactivation of the immune response, fostering persistent sterile inflammation, and playing a role in the emergence of chronic inflammatory conditions (24).
Trained immunity was initially recognized in myeloid cells, commonly referred to as peripheral trained immunity (25). Nonetheless, its duration (extending over months or even years) stands in stark contrast to the brief lifespan of mature myeloid cells (merely a few days). Further investigations have validated that trained immunity influences hematopoietic stem and progenitor cells (HSPCs) within the bone marrow via mechanisms of epigenetic reprogramming. (termed central trained immunity). This enables newly differentiated myeloid cells (such as monocytes, macrophages, and neutrophils) to maintain their “trained” state, thereby preserving long-term immune memory (26). Furthermore, other innate immune cells, including natural killer(NK) cells (27), innate lymphoid cells (28) and dendritic cells(DCs) (29), display comparable traits. It is important to note that trained immunity extends beyond myeloid cells; certain non-immune cells, including vascular endothelial cells (VECs) and epithelial cells (ECs), have demonstrated characteristics akin to trained immunity. This observation implies that the occurrence of this phenomenon may be more prevalent than earlier assumptions suggested (30, 31).
2.2 Molecular mechanisms mediating trained immunity
Research has confirmed that trained immunity is primarily mediated through two interconnected mechanisms: metabolic reprogramming and epigenetic modification (Figure 2). Different stimuli may activate specific pathways and influence the expression of distinct genes (32). Additionally, post-transcriptional regulation, such as N6-methyladenosine (m6A) modification, may also play a role in trained immunity, although its exact role remains to be verified (32). Notably, trained immunity exhibits transgenerational effects: newborns whose parents received BCG vaccination show stronger non-specific protective effects and higher survival rates following BCG vaccination, suggesting that BCG confers a synergistic transgenerational immune-enhancing effect (33).
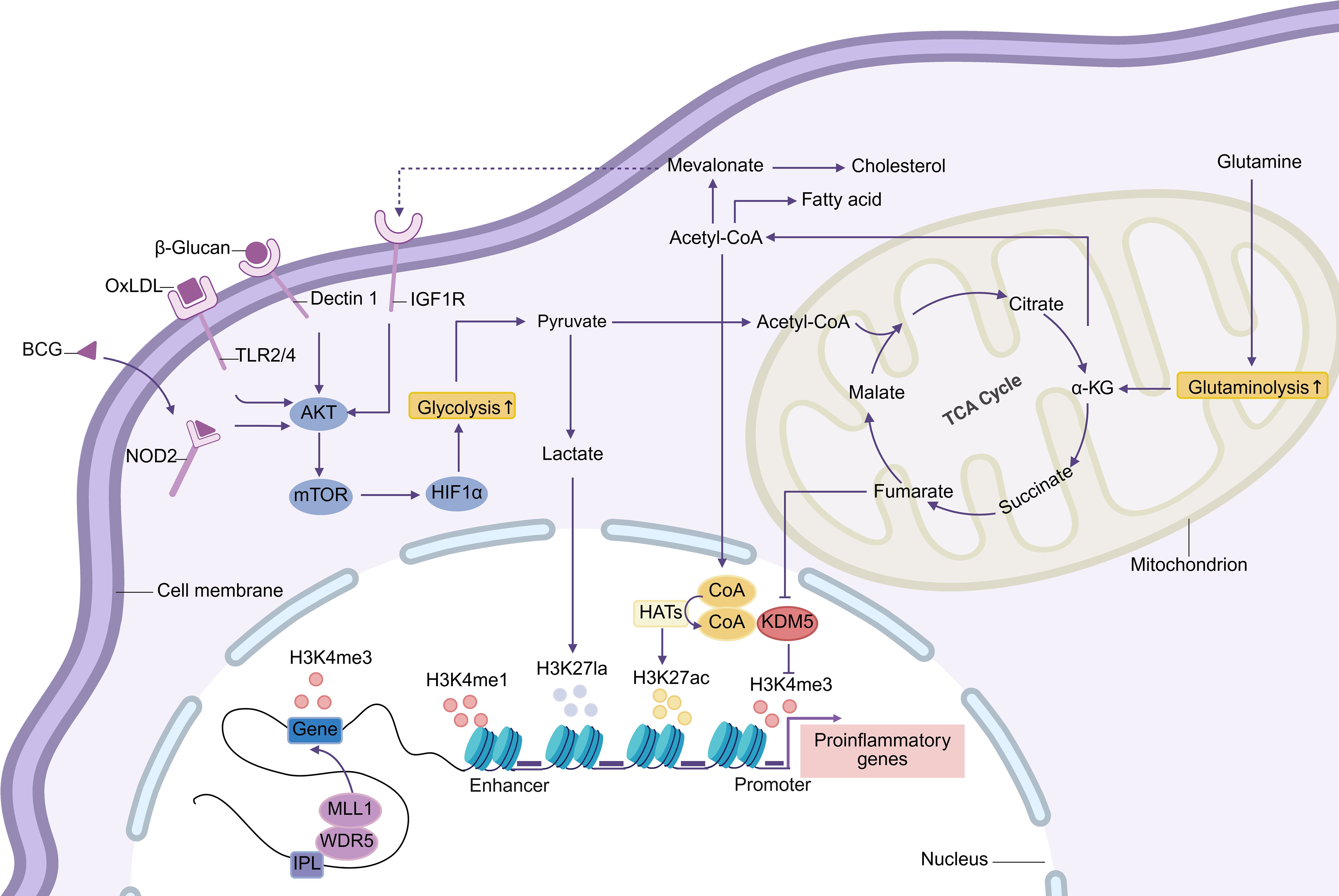
Figure 2. The Akt-mTOR-HIF1α pathway initiates trained immunity by triggering glycolytic reprogramming. This enhances glucose uptake and breakdown into pyruvate, which can convert to lactate or oxidize to acetyl-CoA for tricarboxylic acid (TCA) cycle citrate production. Lactate modifies histones via lactylation, while cytoplasmic citrate regenerates acetyl-CoA. Acetyl-CoA supports fatty acid/cholesterol synthesis and drives epigenetic remodeling as an HAT acetyl donor. Enhanced glutamine catabolism accumulates fumarate, inhibiting KDM5 activity. Immune gene-priming long noncoding RNAs (IPLs) gene promoters, mediating histone methylation and enhancing transcription. These metabolic-epigenetic networks form the basis of trained immunity. Abbreviations: HAT: histone acetyltransferases; oxLDL: oxidized low-density lipoprotein; IGF1R: insulin-like growth factor 1 receptor; NOD2: nucleotide-binding oligomerization domain protein 2; TLR: Toll-like receptor; α-KG: α-ketoglutarate.
2.2.1 Metabolic reprogramming
Metabolic reprogramming serves as the fundamental process that facilitates the development of trained immunity. Upon stimulation, innate immune cells experience notable metabolic reconfiguration, which entails modifications across various pathways, such as oxidative phosphorylation (OXPHOS), fatty acids metabolism, and glutaminolysis (34). The metabolic alterations observed serve to fulfill the energy requirements necessary for the establishment of immune memory, while simultaneously modulating epigenetic modifications via intermediate metabolites. This process ultimately improves the cells’ ability to respond to future stimuli (25).
Glycolysis serves as the pivotal metabolic center in the context of trained immunity. Glucose is metabolized into pyruvate, which can subsequently be transformed into lactic acid or proceed into the tricarboxylic acid (TCA) cycle (35). The hallmark “aerobic glycolysis” (Warburg effect) in immune cells enables them to preferentially produce lactic acid under normoxic conditions, a process that is not confined to hypoxic environments (36). In their resting state, immune cells rely on OXPHOS for energy, a more efficient but slower process (37). Upon activation, they rapidly switch to aerobic glycolysis to meet increased energy needs, a metabolic shift crucial for the establishment of trained immunity (38). From a mechanistic perspective, β-glucan enhances aerobic glycolysis in monocytes through the Akt-mTOR-HIF1α signaling pathway, and the inhibition of this pathway abolishes the trained immunity phenotype (39). In a comparable manner, the activation of glycolysis is crucial in the trained immunity elicited by BCG and oxLDL, underscoring the fundamental role of glycolysis in innate immune memory (21, 40). Beyond its role in energy provision, glycolysis may also contribute to immune regulation through the following mechanisms: 1) The upregulation of glycolysis increases the NAD+/NADH ratio, thereby reducing Sirtuin-1 histone deacetylase activity. In vitro addition of the Sirtuin-1 activator resveratrol prevents β-glucan-induced trained immunity in monocytes, highlighting the negative regulatory function of Sirtuin-1 in trained immunity (39). 2) Glycolysis supplies substrates for the pentose phosphate pathway (PPP), which generates nucleotides and NADPH. However, inhibiting the oxidative branch of PPP does not impair the induction of trained immunity, suggesting its dispensability in this context (41, 42).
The conventional view of lactic acid as merely the end product of glycolysis has been fundamentally revised. Growing evidence underscores its multifaceted roles in immune regulation. LPS stimulation induces lactic acid accumulation in bone marrow-derived macrophages (BMDMs), triggering lactylation through binding to histone lysine residues (43). This novel post-translational modification does not directly suppress early inflammatory responses but functions as a “lactate clock”, initiating after histone acetylation. It subsequently activates the homeostatic genes of M2 anti-inflammatory macrophages during the later stages of acute inflammation (44). Through epigenetic regulation, lactylation orchestrates the gene expression network during the resolution of inflammation. Although this mechanism primarily contributes to the resolution of inflammation, its role in “trained immunity” macrophages, which require sustained activation, is limited. Recent breakthroughs, however, have unveiled a dual-pathway regulatory mechanism of lactic acid in trained immunity: metabolically, lactic acid is catalyzed by lactate dehydrogenase A (LDHA) to generate acetyl-CoA, which exhibits greater mitochondrial oxidation efficiency than glucose, supporting the energy needs of innate immune cells via enhanced ATP synthesis; epigenetically, lactylation enhances chromatin accessibility, directly activating the transcription of inflammatory cytokines such as IL-6 and TNF-α (44). This process depends on LDHA and monocarboxylate transporter 1 (MCT1). Blocking either component disrupts cytokine secretion and impaired anti-infection capabilities, underscoring lactic acid’s bridging role between metabolic reprogramming and epigenetic regulation. Notably, current research faces three critical limitations: first, LDHA is involved in both lactic acid metabolism and glycolysis (the latter being a hallmark of trained immunity), complicating the ability to separate the independent contribution of lactylation (44); second, the regulatory differences between lactylation in acute inflammatory resolution and the chronic inflammatory environment of trained immunity remain unclear; third, the complex interactions between lactylation and other histone modifications, such as acetylation, are yet to be fully understood. Future studies should aim to develop conditional lactylation-deficient models and incorporate multi-dimensional analyses of metabolomics and epigenomics to systematically investigate the spatiotemporal regulatory foundation of the lactic acid metabolic network. This approach will provide a solid theoretical foundation for the development of targeted immune intervention strategies focused on the lactic acid pathway.
Glutamine catabolism is another critical feature of trained immunity. In monocytes activated by BCG or β-glucan, glutamine metabolism is significantly enhanced, leading to the production of glutamate and α-ketoglutarate (α-KG). The latter enters the TCA cycle, contributing to the accumulation of fumarate, succinate, and malate (42). Notably, fumarate specifically inhibits histone lysine demethylase 5 (KDM5), increasing the level of H3K4me3 at the promoters of pro-inflammatory genes and facilitating epigenetic reprogramming (42). Moreover, fumarate stabilizes HIF1α protein, thereby maintaining glycolytic flux (45). Additionally, α-KG serves as a cofactor for TET demethylases and JMJD3 histone demethylases (KDM6), contributing indirectly to epigenetic regulation (46, 47). Collectively, these findings outline a critical mechanism by which metabolic intermediates regulate epigenetic modifications to establish trained immunity.
Acetyl-CoA functions as a central node in the metabolic regulation of trained immunity. Within mitochondria, pyruvate is converted into acetyl-CoA, which enters the TCA cycle. Subsequently, citrate is transported to the cytoplasm, where it is re-synthesized into acetyl-CoA via the ATP-citrate lyase (ACLY) (48, 49). This crucial metabolite regulates trained immunity through three mechanisms: 1) it participates in fatty acid synthesis (FAS), which is essential for aldosterone-induced trained immunity (22); 2) it is converted into mevalonate, entering the cholesterol synthesis pathway. This process critical for oxLDL and β-glucan-induced trained immunity, as statins can block it by inhibiting HMG-CoA reductase and thereby suppress trained immunity (50). Studies have shown that mevalonate promotes glycolysis by activating the IGF-1R-mTOR pathway, thereby forming a positive feedback loop during the induction of trained immunity. Its accumulation leads to an endogenous trained immunity phenotype (50); 3) as an acetyl donor for histone acetyltransferases (HATs), it contributes to the epigenetic regulation of trained immunity (51).
OXPHOS plays a stimulus-dependent role in trained immunity. BCG, oxLDL, catecholamines, and low-dose β-glucan (1 μg/ml) all enhance OXPHOS activity in monocytes (21, 23, 40, 52). Low-dose β-glucan achieves this by upregulating H3K4me1 levels at enhancer regions of TCA cycle enzyme genes via Set7 methyltransferase (52). Conversely, high-dose β-glucan (10 μg/ml) inhibits OXPHOS (39), and the underlying dose-dependent mechanism requires further investigation. Notably, the OXPHOS inhibitor oligomycin blocks oxLDL and β-glucan-induced trained immunity, but does not affect the BCG model (21, 41, 52), suggesting that different stimuli may activate divergent metabolic pathways.
2.2.2 Epigenetic reprogramming
Epigenetic reprogramming dynamically alters chromatin structure and accessibility, facilitating gene expression remodeling, which is a central mechanism in the establishment of trained immunity. During initial stimulation, innate immune cells undergo significant epigenetic modifications (53). Even after the stimulus diminishes, certain epigenetic changes remain, enhancing gene transcription upon subsequent stimulation, thereby bolstering the non-specific immune response (20), This process involves the regulation of histone modifications, DNA methylation, and non-coding RNA s(ncRNAs) (54).
In trained immunity, key histone modifications include the enrichment of H3K4me3 at promoter regions, the accumulation of H3K4me1 at distal enhancer regions, and enrichment of histone 3 lysine 27 acetylation (H3K27ac) in both promoters and enhancers (55, 56). Among these, H3K27ac gradually diminishes after the stimulus ceases, while H3K4me1/3 persists, maintaining long-term cellular hyper-reactivity (57). Furthermore, H3K4me3 is found in the promoter regions of glycolysis-related genes (e.g., hexokinase) in trained monocytes, linking epigenetic modifications directly to metabolic reprogramming (39). The reduction of repressive marks further enhances chromatin accessibility, thereby facilitating trained immunity (40, 42). Recently, histone lactylation has been identified as a metabolic modification that plays a critical role in regulating trained immunity (44).
Epigenetic enzymes also play pivotal roles in trained immunity; Set7 and lysine methyltransferase G9A mediate the “writing” of H3K4me1 and H3K9me2, respectively (52, 57), while KDM4, KDM5, and Sirtuin-1 deacetylases form the “erasure system”, removing H3K4me3, H3K9me3, and H3K27ac marks (42, 58, 59). While the core function of histone modifications are well understood, the precise targeting of specific genomic regions remains unclear.
Recent studies have also emphasized the significant role of ncRNAs in integrating metabolic and epigenetic regulation within trained immunity. In the β-glucan-trained monocyte model, microRNAs (miRNAs), such as miR-9-5p, suppress the expression of the metabolic enzyme isocitrate dehydrogenase 3α (IDH3α), reducing α-KG levels and promoting fumarate accumulation. This metabolic shift enhances inflammation through two mechanisms: First, fumarate stabilizes HIF-1α, driving glycolysis and pro-inflammatory cytokine secretion. Second, fumarate inhibits histone demethylase KDM5, increasing H3K4me3 modification at the promoters of pro-inflammatory genes, enhancing chromatin accessibility and transcription activation. Monocytes deficient in miR-9-5p show a metabolic shift toward OXPHOS, alongside reduced H3K4me3 levels and impaired anti-infection capacity. These results indicate that miR-9-5p is a critical link between metabolic reprogramming and epigenetic remodeling (60).
Additionally, long non-coding RNAs (lncRNAs) that regulate immune gene promoters(IPLs) achieve precise epigenetic reprogramming by altering the chromatin’s three-dimensional structure. For instance, the upstream master lncRNA of inflammatory chemokine loci (UMLILO) recruits the histone methyltransferase MLL1 to the CXCL gene promoter within a topologically associated domain (TAD), enriching H3K4me3 and promoting transcription (61). Mouse models have shown that deleting UMLILO prevents the trained immunity phenotype induced by β-glucan, whereas its insertion into the chemokine TAD restores immune memory function, underscoring its necessity (61). In the β-glucan model, UMLILO activation is dependent on the nuclear factor of activated T cells, and many IPL promoters contain multiple transcription factor binding sites, which could explain how different stimuli activate common pathways. Although the specific deposition of IPLs may contribute to the specificity of epigenetic modifications, studies so far have focused solely on the β-glucan stimulation model, and their applicability to other immune stimulations remains uncertain. Notably, miRNAs and IPLs may miRNAs and IPLs may work together within a regulatory network, where miR-9-5p indirectly influences chromatin accessibility through the metabolic-epigenetic axis, while IPLs such as UMLILO directly reconfigure chromatin structure, jointly maintaining the long-term hyper-reactivity of pro-inflammatory genes. Future studies should explore the interactions among ncRNAs, metabolic intermediates, and chromatin modification enzymes to elucidate the dynamic mechanisms that govern epigenetic memory in trained immunity.
The role of DNA methylation in trained immunity requires further investigation. Preliminary studies suggest that, in the β-glucan-induced monocyte trained immunity model, DNA methylation pattern remain largely unchanged, while LPS-induced immune tolerance is associated with stable, long-term DNA methylation modifications, indicating that DNA methylation may primarily regulate gene expression inhibition (56). Clinical research has shown that the DNA methylation changes induced by BCG vaccination in neonatal monocytes can persist up to 14 months post-vaccination (62). In adult vaccine recipients, responders show widespread loss of DNA methylation at inflammatory gene promoters. Genome-wide analysis has identified significant methylation differences at 43 loci, which may serve as biomarkers for predicting individual responses to trained immunity (63, 64). These findings suggest that DNA methylation may be crucial in the establishment and maintenance of immune memory through dynamic modifications.
In conclusion, during trained immunity, the metabolic reprogramming of innate immune cells connects various metabolic pathways. Metabolic intermediates act as substrates or cofactors for epigenetic enzymes, linking metabolic alterations to the expression of inflammatory genes via epigenetic reprogramming.
3 Trained immunity in diabetes
Chronic low-grade inflammation associated with diabetes is a critical driver of vascular lesions (65). Even when metabolic parameters, such as blood glucose, are controlled, prior metabolic disturbances can perpetuate increased cardiovascular risk through the mechanism of “metabolic memory” (11). Recent studies show that diabetes induces “trained immunity” in innate immune cells via epigenetic and metabolic reprogramming, thereby sustaining chronic inflammation (66). Specifically, pathological factors such as hyperglycemia, dyslipidemia, obesity, and unhealthy lifestyle habits remodel the epigenetic landscape of HSPCs, leading to the persistent activation of pro-inflammatory pathways in innate immune cells (12). This immune memory enables these cells to maintain elevated glycolytic activity and increased secretion of inflammatory mediators, even after removal from a metabolically imbalanced environment, ultimately accelerating the progression of vascular diseases (67).
3.1 Hyperglycemia
In diabetic patients, the mechanism underlying trained immunity is complex. Hyperglycemia can induce monocytes and macrophages to establish innate immune memory. Specifically, human primary monocytes pretreated with 25 mmol/L high glucose in vitro exhibit increased secretion of IL-6 and TNF-α, along with enhanced glycolysis upon subsequent LPS re-stimulation, even after being cultured under normal glucose conditions for 5 days. This process is mediated by MLL family-dependent H3K4me3 epigenetic modification (68). Clinical evidence reveals that H3K4me3 and H3K9ac modifications at pro-inflammatory gene loci in monocytes from diabetic patients are abnormally enriched, confirming that metabolic memory sustains the pro-inflammatory phenotype via epigenetic modifications over an extended period (69). Similarly, neutrophils are influenced by hyperglycemia, where metabolic reprogramming leads to coenzymeA accumulation, triggering histone acetylation and promoting excessive formation of neutrophil extracellular traps (NETs). This phenomenon not only intensifies inflammatory responses but also delays wound healing in diabetic patients (70).
The long-term influence of hyperglycemia on the immune system is largely attributed to its effect on HSPCs. In streptozotocin-induced diabetic mouse models, hyperglycemia drives bone marrow hematopoiesis by upregulating S100A8/A9, leading to an increase in circulating neutrophils and Ly6-C(hi) monocytes, thereby accelerating AS progression (71). Further studies have revealed that hyperglycemic exposure can induce the accumulation of Set7-mediated H3K4me1 marks in the NF-κB-p65 promoter region of HSPCs. The monocytes derived from these cells display significantly elevated IL-6 and TNF-α secretion upon LPS stimulation, and this pro-inflammatory phenotype persists even after blood glucose normalization (72). HSPCs in the bone marrow of patients with T2D also exhibit upregulated expression of pro-inflammatory genes, and the monocytes derived from these cells have accumulated H3K4me1 marks at the NF-κB-p65 promoter, validating that hyperglycemia can convey immune memory across generations through epigenetic “imprinting” (72).
Non-immune cells also exhibit hyperglycemic memory. Vascular smooth muscle cells(VSMCs) in diabetic mice manifest a pro-atherosclerotic phenotype, characterized by enhanced migratory capacity and sustained high expression of pro-inflammatory genes (73). VECs are highly responsive to blood glucose fluctuations: short-term hyperglycemia induces sustained inflammatory responses by increasing H3K4me1 modification in the promoter regions of pro-inflammatory genes, with this effect remaining irreversible even after subsequent blood glucose normalization (74). This epigenetic memory closely resembles the mechanism of trained innate immunity, suggesting that vascular cells and the immune system form a synergistic pro-inflammatory network under metabolic stress.
Short-term blood glucose fluctuations, such as transient intermittent hyperglycemia (TIH), also exert significant pathological effects. In Apolipoprotein E knockout mouse models (ApoE-/-), TIH accelerates AS progression by increasing circulating monocytes and neutrophils via activation of bone marrow hematopoiesis. This process depends on the binding of S100A8/A9 to the receptor for advanced glycation end products (AGEs) on myeloid progenitor cells (75). Additionally, the interaction between AGEs and their receptors can activate macrophage inflammatory responses, creating a pro-inflammatory positive feedback loop (76). Notably, TIH does not affect HbA1c levels, suggesting that traditional blood glucose monitoring may underestimate the risk of metabolic memory. Thus, an integrated assessment system combining continuous glucose monitoring with epigenetic markers is urgently required.
Hyperinsulinemia associated with insulin resistance might enhance the secretion and migratory capacity of inflammatory factors in monocytes via the PI3K/Akt/mTOR pathway (77), though insulin itself may inhibit NF-κB activation in a hyperglycemic environment (78), illustrating the complexity of its immunomodulatory actions. While insulin may play a role in regulating trained immunity, its precise mechanism remains to be fully elucidated. Furthermore, hyperglycemia-induced lactate accumulation in diabetic patients may mediate the activation of trained immunity via histone lactylation modifications.
The above findings demonstrate that hyperglycemia remodels chromatin accessibility through metabolic reprogramming and histone modification, keeping pro-inflammatory genes in a pre-activated state and establishing trained immunity. This metabolic and epigenetic reprogramming mechanism offers a novel and insightful perspective on the pathological progression of chronic complications in diabetes.
3.2 Lipid metabolism disorders
In metabolic disorders like diabetes, lipid metabolism abnormalities are not only a byproduct of abnormal glucose metabolism but also a significant pathological foundation that drives chronic inflammation and AS (10). Recent studies show that aberrant lipid metabolites, such as oxLDL and Lp(a), can induce “trained immunity” in innate immune cells, establishing a persistent low-grade inflammatory state and accelerating vascular lesion progression (30, 79).
In vitro experiments have demonstrated that monocytes exposed to oxLDL exhibit sustained glycolytic activity and enhanced secretion of pro-inflammatory cytokines even after the stimulus is removed, alongside enrichment of H3K4me3 marks at pro-inflammatory gene promoters (21). This process involves activation of the mTOR/HIF-1α axis via Toll-like receptors (TLR4/2), upregulation of key glycolytic enzymes, disruption of cholesterol homeostasis through enhanced expression of scavenger receptors (CD36/SR-A), and downregulation of efflux transporters like ABCA1 and ABCG1, which ultimately promotes foam cell formation (80, 81). Moreover, mitochondrial reprogramming induced by oxLDL—characterized by accumulation of TCA cycle intermediates, reactive oxygen species (ROS) production, and enhanced OXPHOS—amplifies the inflammatory response (81).
Clinical evidence confirms that monocytes from patients with familial hypercholesterolemia exhibit a trained immunity phenotype, with elevated H3K4me3 levels, enhanced glycolysis-related gene expression, and persistence of cytokine hyper-reactivity despite 3 months of statin therapy reducing LDL-C levels (82). Analysis of bone marrow progenitor cells reveals that a high-lipid microenvironment induces myeloid-biased differentiation of hematopoietic stem cells (HSCs), with progenitors retaining pro-inflammatory transcriptional signatures even after lipid normalization (83).
Lp(a), the main carrier of oxidized phospholipids (OxPL), contributes to trained immunity through its pro-inflammatory effects (84). In vitro studies show that co-incubating healthy human monocytes with plasma from patients with elevated Lp(a) for 24 hours followed by restimulation with Pam3Cys significantly enhances IL-6 and TNF-α secretion. This effect can be blocked by anti-OxPL antibodies, confirming OxPL as the key mediator of Lp(a)-induced trained immunity (85). Clinical observations further reveal that monocytes from patients with elevated Lp(a) exhibit exaggerated pro-inflammatory cytokine secretion and maintain a persistently activated state upon in vitro stimulation (85), suggesting that Lp(a) may establishes immune memory through epigenetic or metabolic reprogramming mechanisms.
3.3 Obesity
In diabetes, metabolic memory is not only linked to obesity-related metabolic disorders but is also directly induced by obesity itself. Obesity can perpetuate metabolic memory, thereby exacerbating disease progression through the continuous reprogramming of the immune system. Insulin resistance, in conjunction with an unhealthy lifestyle characterized by a high-sugar, high-fat diet (HFD) and physical inactivity, plays a pivotal role in promoting obesity in diabetic patients. Despite successful weight loss, around 80% of patients experience weight regain, which leads to a worsening of metabolic complications (86). Research has demonstrated that chronic inflammation in adipose tissue during obesity is marked by the infiltration of pro-inflammatory macrophages. Although weight loss partially alleviates this inflammation, key immune markers remain elevated, creating an “obesity memory” (87). This long-term alteration in the phenotype and function of immune cells triggered by obesity may underlie the recurrence of metabolic complications upon weight regain.
The formation of obesity-related metabolic memory involves a synergistic interaction between metabolic reprogramming and epigenetic remodeling. Metabolic disturbances in fatty acid metabolism among obese individuals may lead to the accumulation of palmitic acid, a saturated fatty acid associated with various metabolic dysfunctions. Studies have shown that macrophages treated with palmitic acid or secretions from obese adipose tissue exhibit increased glycolysis and OXPHOS activity, along with enhanced secretion of TNF-α and IL-6 upon LPS re-stimulation (88). This process is mediated by the activation of the TLR4/mTOR pathway and epigenetic modifications such as H3K4me3, catalyzed by histone methyltransferases. Inhibition of mTOR or methyltransferases can reverse this “trained” effect, confirming that the interaction between metabolic and epigenetic networks represents the core regulatory mechanism (88). Further investigations have shown that, in comparison to persistent obesity, weight cycling (repeated cycles of weight loss and regain) in animal models induces more severe metabolic dysregulation. The underlying mechanisms include an enhanced lipid-handling capacity of adipose tissue macrophages, hyperactivation of antigen-presenting cells, and T cell exhaustion (89). Under these conditions, metabolic memory amplifies inflammation through a positive feedback loop, leading to sustained chemokine release from adipose tissue and recruitment of pro-inflammatory monocytes, further disrupting metabolic homeostasis.
A growing body of evidence supports the existence of obesity-related immune memory. For example, upon in vitro stimulation, monocytes isolated from obese patients exhibit significantly greater cytokine secretion capacity compared to those from normal-weight controls, indicative of a baseline activated state (90). Specifically, in vitro LPS stimulation triggers higher levels of IL-1β, IL-6, IL-8, and TNF production by monocytes derived from obese patients (90). Moreover, elevated serum leptin levels in obese patients correlate with higher IL-1β and IL-6 concentrations. In vitro experiments further confirm that monocytes pretreated with leptin demonstrate an enhanced inflammatory cytokine secretion capacity upon subsequent LPS re-stimulation (91). Collectively, these findings suggest that monocytes from obese patients possess an intrinsic pro-inflammatory tendency, potentially associated with a memory mechanism akin to trained immunity.
Obesity also drives myeloid-biased differentiation via the fat-bone marrow axis. Similar to the effects of hyperglycemia on bone marrow, obese adipose tissue releases S100A8/S100A9 and stimulates IL-1β secretion from bone marrow progenitor cells via activation of TLR4/MyD88 signaling and NLRP3 inflammasomes, promoting the expansion of monocytes and neutrophils (92). Notably, even after repeated bone marrow transplants, HSCs in obese mice still preferentially differentiate into inflammatory adipose tissue macrophages, an effect that depends on MyD88 signaling (93). This finding reveals the underlying mechanism of persistent immune memory following weight normalization.
3.4 Dietary stimulation
Unhealthy dietary habits, such as those associated with the Western diet (WD), are commonly observed during the progression of diabetes. WD is characterized by high levels of sugar, calories, fat, and salt, as well as low fiber and mineral content. It not only contributes directly to obesity, metabolic syndrome, and disorders in glucose/lipid metabolism but also remodels immune homeostasis via multiple mechanisms (94).
Studies have shown that WD and its metabolites can exacerbate chronic inflammation-related diseases by inducing trained immunity. WD can alter the composition of the gut microbiota, decrease protective metabolites, and increase intestinal permeability, allowing LPS to translocate into the circulatory system and trigger metabolic endotoxemia. In ApoE-/- mice, LPS-simulated metabolic endotoxemia upregulates miR-24, enhances monocyte inflammatory reprogramming, and accelerates AS progression (95). Notably, low-dose LPS (≤10 pg/mL) induces trained immunity in monocytes, characterized by sustained high expression of pro-inflammatory factors such as TNF-α and IL-6, while high-concentration LPS induces immune tolerance, revealing a dose-dependent regulatory mechanism (96).
In an AS-susceptible model (Ldlr -/-mice), WD activates the NLRP3 inflammasome and downstream IL-1β signaling, driving transcriptional and epigenetic reprogramming iv granulocyte-monocyte progenitor cells (GMPs) (97). Even after reverting to a normal diet for 4 weeks, myeloid cells retain a hyper-inflammatory phenotype, accompanied by DNA hypomethylation in genes and the expansion of circulating monocyte (97, 98).
A comparative study of urban and rural populations in Tanzania further supports the immunomodulatory effects of WD. Urban residents, whose diet is deficient in flavonoids like apigenin, produce more pro-inflammatory factors in monocytes upon LPS stimulation. In contrast, the high concentration of apigenin in traditional rural diets partially inhibits trained immunity and reduces the inflammatory response (99). Additionally, apigenin blocks oxLDL-induced monocyte trained immunity in vitro, suggesting its potential anti-inflammatory properties. In a HFD-induced C57BL/6J mouse obesity model, even when body weight normalizes, bone marrow monocytes and adipose tissue macrophages persist with a pro-inflammatory phenotype (100). Mechanistically, long-term alterations in chromatin accessibility at activator protein 1 (AP-1) binding sites are critical, and this phenomenon can be recapitulated in mouse macrophages through short-term exposure to stearic acid, indicating that some nutrients can directly drive epigenetic reprogramming (99). Moreover, irregular diets are also associated with trained immunity. For example, intermittent HFD increases the number of neutrophils in peripheral blood by reprogramming neutrophil progenitor cells, eliciting a stronger inflammatory response compared to continuous HFD (101).
3.5 Gut microbiota dysbiosis
In diabetic patients, the GM typically exhibits reduced diversity, an increase in pro-inflammatory bacteria, and a significant decline in commensal bacteria that produce (SCFAs) (102). This reduction in SCFAs contributes to disturbances in glucose and lipid metabolism, impaired intestinal barrier function, enhanced endotoxin translocation into the bloodstream, and the induction of chronic low-grade inflammation, all of which promote insulin resistance (103). Metabolic abnormalities, such as hyperglycemia and lipid metabolism disorders, and endotoxemia have been shown to trigger trained immunity via epigenetic reprogramming, as discussed previously.
One key metabolite of gut microbiota dysbiosis—trimethylamine N-oxide (TMAO)—plays a crucial role in trained immunity. TMAO is generated by intestinal microbiota through the metabolism of dietary choline and carnitine. Diabetic individuals, due to their tendency to consume high-fat and high-red meat diets (which are rich in choline and carnitine) (104), provide an abundant substrate for TMAO production. TMAO induces endothelial trained immunity by activating endoplasmic reticulum (ER) stress (105). Research has shown that TMAO utilizes PERK (protein kinase R-like ER kinase) as a conditional DAMP receptor (106), triggering ER stress in human aortic endothelial cells (HAECs). This leads to PERK phosphorylation and the activation of the downstream transcription factor cAMP response element-binding protein (CREB), linking ER stress signals with mitochondrial dysfunction and enhanced glycolytic metabolism via the PERK-CREB signaling axis (106). As a result, HAECs adopt a pro-inflammatory phenotype, similar to innate immune cells, resulting in the upregulation of adhesion molecules such as ICAM-1 and inflammatory factors, thereby maintaining a chronic pro-inflammatory state (107, 108). In summary, TMAO induces trained immunity in HAECs through the PERK-CREB axis, involving multiple regulatory mechanisms, including ER stress and metabolic reprogramming. This highlights TMAO as a potential therapeutic target for preventing and treating AS.
Chronic low-grade inflammation, common in diabetes, accelerates vascular lesions through trained immunity. Pathological processes such as hyperglycemia, lipid metabolism disorders, gut microbiota dysbiosis, and obesity induce a persistent pro-inflammatory phenotype in immune cells—especially innate immune cells—through epigenetic and metabolic reprogramming.
4 Trained immunity in AS
AS is characterized by chronic inflammation of the arterial intima, which is central to its pathology. The onset, progression, and thrombotic complications of AS are closely linked to the abnormal activation of the innate immune system (109). Trained immunity has a dual effect: while it enhances the host’s resistance to infections via broad-spectrum immune responses, excessive activation leads innate immune cells to form persistent pro-inflammatory phenotypes, exacerbating chronic inflammatory diseases such as AS (14, 110).
Monocytes and macrophages, as the core myeloid cells in AS lesions, can adopt pro-inflammatory and pro-AS phenotypes upon exposure to microbial stimuli or endogenous metabolites (80). Microbial stimulation activates trained immunity by upregulating the transcription of pro-inflammatory genes (111), while endogenous stimuli like oxLDL promote foam cell formation and sustained inflammation through metabolic reprogramming and epigenetic modifications (21). Clinical studies indicate that monocytes in AS patients exhibit trained immunity traits, including enhanced secretion of pro-inflammatory cytokines, increased glycolytic activity, and altered epigenetic/metabolic markers, maintaining heightened reactivity even after in vitro differentiation into macrophages (112, 113). Notably, myocardial infarction induces trained immunity in monocytes, thereby increasing the risk of AS-related cardiovascular events (114). Recent studies have demonstrated that type I interferons (IFNs) in sicca syndrome induce monocytes to secrete pro-AS cytokines and enhance cholesterol uptake. This interaction with trained immunity is bidirectional: trained immunity modulates type I IFN production and the transcriptional response to receptor restimulation (115).
In addition to monocytes/macrophages, other myeloid cells such as neutrophils (116) and DCs (117), have been implicated in AS-related immune memory. BCG vaccination induces long-lasting pro-inflammatory phenotypes in neutrophils, upregulating activation markers and enriching H3K4me3 modifications across the genome, which boosts their antibacterial functions (118). Neutrophils from β-glucan-pretreated mice exhibit significantly increased IL-6 secretion, myeloperoxidase production, and resistance to Listeria monocytogenes upon LPS stimulation (119), demonstrating the reprogramming of an anti-tumor phenotype via type I IFN signaling and ROS-mediated mechanisms. This reprogramming effect can be transferred to naïve mice via neutrophil adoptive transfer or bone marrow transplantation, independent of the adaptive immune system (120). Periodontitis, through trained immunity, may elevate neutrophil numbers and pro-inflammatory responses, increasing AS risk (121). However, the role of trained immunity in DCs dendritic cells in AS remains unclear (117).
The persistence of trained immunity depends on the epigenetic and functional reprogramming of bone marrow progenitor cells (122). BCG vaccination triggers transcriptional, epigenetic, and functional reprogramming of HSPCs for up to 3 months, which is then transmitted to peripheral monocytes (123). In AS models, GMPs in Ldlr-/- mice undergo continuous reprogramming after 4 weeks of WD feeding, and the differentiated monocytes exhibit a hyper-responsive state under TLR ligand stimulation, which persists even 4 weeks after returning to a conventional diet (97). Clinical studies have shown that HSPC transcriptional profiles in patients with severe coronary artery disease are skewed towards myeloid differentiation, with enrichment of monocyte/neutrophil pathways, although insufficient epigenetic evidence exists to confirm its association with trained immunity (124).
Moreover, non-immune vascular cells may also contribute to AS via trained immunity mechanisms. In VECs, short-term hyperglycemia induces epigenetic remodeling via H3K4me1 enrichment, mediated by Set7 methyltransferase, resulting in sustained high expression of pro-inflammatory genes and maintenance of inflammatory responses even after blood glucose normalization (74, 125). Lysophosphatidylcholine (LPC) activates key trained immunity pathways, inducing metabolic and epigenetic reprogramming in VECs and promoting a persistent pro-atherosclerotic phenotype (126). OxLDL induces prolonged inflammatory factor secretion in HAECs through metabolic reprogramming and histone modification (30).VSMCs can transdifferentiate into macrophage-like cells under certain conditions, promoting AS development. Furthermore, oxLDL or BCG vaccination induces sustained pro-inflammatory responses in VSMCs via metabolic and epigenetic mechanisms, contributing to plaque instability (127). Non-immune cells acquire memory akin to innate immune cells through epigenetic and metabolic reprogramming, collaborating with myeloid cells to drive chronic inflammation in AS. This finding expands the cellular scope of trained immunity and presents new perspectives for targeted intervention.
In conclusion, the pro-atherosclerotic role of trained immunity is increasingly recognized. In AS, effector cells of trained immunity include not only myeloid cells or HSPCs but also vascular non-immune cells. The interactions among these diverse cell types in vivo contribute to the onset and progression of AS.
5 Trained immunity in diabetes and its role in accelerating AS
Diabetes-induced pathological changes can significantly contribute to complications such as AS. Despite therapeutic interventions aimed at regulating blood glucose and correcting other metabolic disturbances, patients’ cardiovascular risks often persist—this phenomenon is referred to as “metabolic memory” (11). Studies indicate that the long-term impact of diabetes on innate immune cells remains largely irreversible even after metabolic intervention, suggesting that the mechanisms involved extend beyond mere metabolic regulation. Recent research has revealed that diabetes-induced epigenetic reprogramming in the bone marrow niche locks myeloid cells into a pro-inflammatory state via trained immunity, which continues to drive AS progression even after metabolic abnormalities are corrected (128), thus offering a critical molecular explanation for “metabolic memory” (Figure 3).
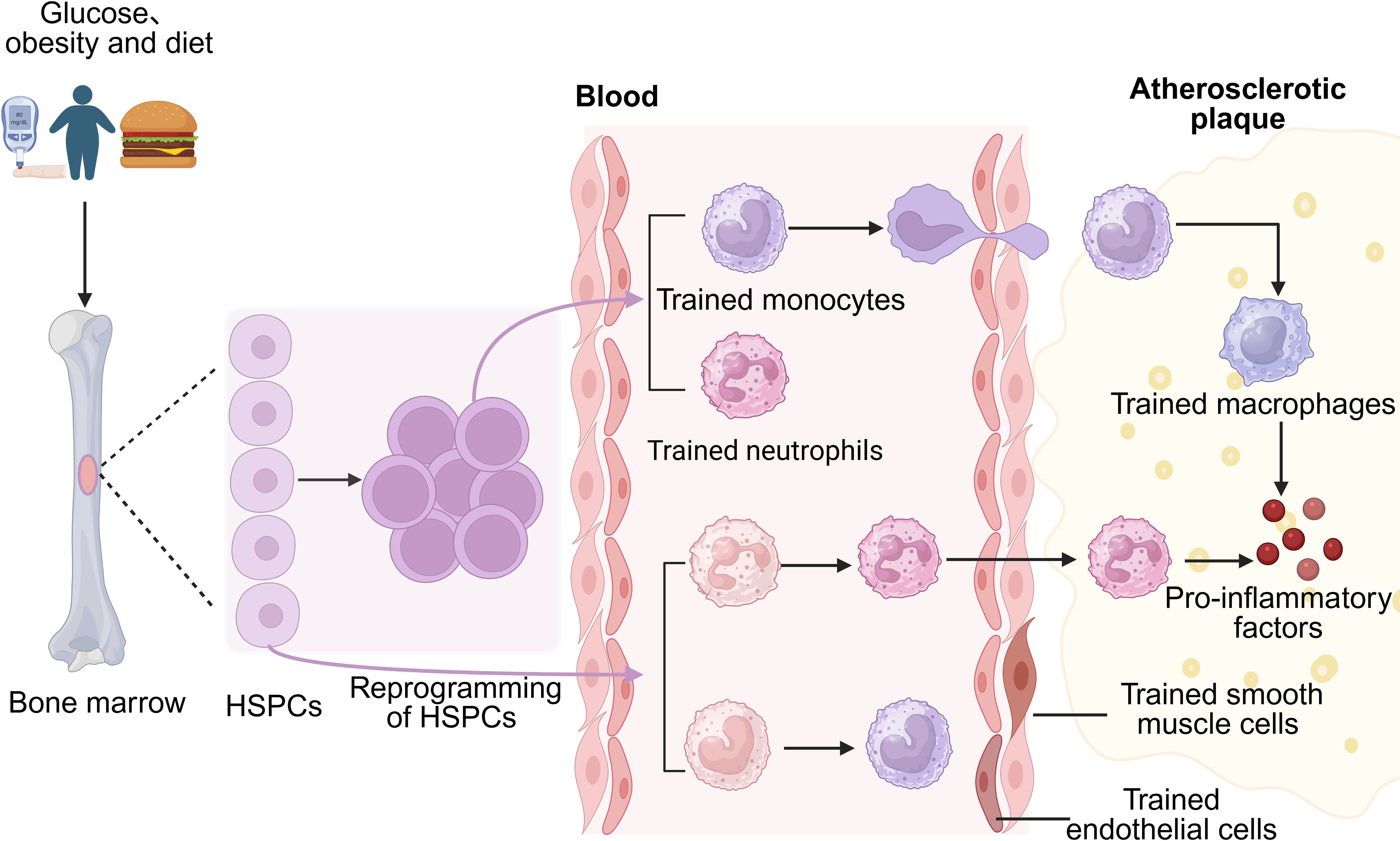
Figure 3. Diabetes reprograms hematopoietic stem and progenitor cells (HSPCs) to induce trained immunity, differentiating into trained monocytes and neutrophils. Normal HSPC-derived monocytes and neutrophils can also be trained. Trained neutrophils infiltrate atherosclerotic plaques directly, while trained monocytes migrate to plaques and transform into macrophages. Both cell types secrete pro-inflammatory factors, promoting plaque formation. Additionally, trained immunity can occur in non-immune vascular cells, such as endothelial cells.
To explore the connection between hyperglycemia, trained immunity, and AS, a study using streptozotocin to induce diabetes in mice conducted a comprehensive investigation (16). The findings showed that bone marrow cells from diabetic mice, when differentiated into BMDMs under normal blood glucose conditions and stimulated, demonstrated significantly increased IL-6 expression, alongside enhanced uptake of modified LDL and foam cell formation—hallmarks of a pro-AS phenotype. When bone marrow from diabetic mice was transplanted into Ldlr-/- mice with normal blood glucose levels and placed on a WD, AS progression accelerated dramatically. Mechanistically, high-glucose culture of HSCs led to the upregulation of H3K4me3 and H3K27ac modifications, which are associated with glycolytic metabolic reprogramming. Macrophages within the aortic plaques of these mice displayed enriched H3K4me3 modification, indicating that hyperglycemia promotes trained immunity via epigenetic memory in progenitor cells, a process that is transmittable across generations. Additional studies demonstrated that this mechanism depends on the regulation of the transcription factor Runx1 (16). Importantly, similar epigenetic and transcriptional changes were observed in peripheral blood mononuclear cells and AS plaque macrophages from diabetic patients, confirming the presence of a hyperglycemia-induced trained immunity mechanism in human diabetic AS (16).
Another study focused on the epigenetic regulator histone deacetylase 3 (HDAC3), revealing that its expression in plaque macrophages of diabetic AS patients was significantly elevated and positively correlated with serum LDL, triglycerides, and blood glucose levels (129). In vitro experiments demonstrated that in a high-glucose environment, LPS stimulation markedly increased glucose uptake and lactate/succinate production in BMDMs, promoting polarization toward the pro-inflammatory M1 phenotype and enhancing secretion of IL-6, IL-1β, and TNF-α, which facilitated foam cell transformation and AS plaque progression. Animal studies further showed that specific knockout of HDAC3 in macrophages mitigated AS lesions in high-fat-diet (HFD)-fed ApoE-/- mice, indicating that HDAC3 exacerbates AS progression by mediating metabolic dysregulation and epigenetic modifications in macrophages (129). While these observations suggest a role for trained immunity, the exact regulatory patterns of epigenetic changes on specific target genes and their temporal effects remain unclear. Future studies should investigate the persistent association between pro-inflammatory phenotypes and epigenetic modifications to elucidate the molecular mechanisms of HDAC3 in diabetes-associated AS.
Diet-induced trained immunity is another significant factor driving AS progression. A HFD enhances the pro-inflammatory activity and abundance of circulating neutrophils in Ldlr-/- mice by reprogramming neutrophil progenitor cells, thereby accelerating AS development (101). In AS model mice fed a WD for 4 weeks, granulocyte-monocyte progenitors (GMPs) and differentiating monocytes/macrophages exhibited persistent functional and epigenetic reprogramming. Notably, even after reverting to a normal diet, these myeloid cells retained a highly inflammatory phenotype (97). Further experiments demonstrated that bone marrow transplantation from WD-fed mice into Ldlr-/- mice on a regular diet significantly increased AS plaque size, independent of serum cholesterol levels. This effect was closely linked to abnormal DNA methylation in myeloid cells and the expansion of circulating monocytes (98). Another study demonstrated that HDAC9 expression was significantly upregulated in plaque macrophages of WD-fed Ldlr-/- mice. HDAC9 reduced histone 3 acetylation, inhibiting the expression of ABCA1, ABCG1, and the nuclear receptor PPARγ, thus impairing cholesterol efflux and promoting macrophage polarization toward the M1 phenotype. In contrast, HDAC9 deficiency enhanced cholesterol efflux and reduced inflammatory mediators like IL-1β and MCP-1, thereby suppressing AS progression (130). Together with the findings on HDAC3, both regulators affect innate immunity by modulating TLR and IFN signaling pathways, suggesting that HDAC inhibitors could suppress trained immunity activation by targeting these pathways (131).
In obesity-related chronic low-grade inflammation, the adipokine leptin secreted by adipocytes plays a crucial regulatory role through trained immunity. Studies show that brief exposure to physiological leptin concentrations significantly enhances the response of monocytes differentiated in vitro to LPS stimulation, increasing TNF-α secretion, phagocytic function, and foam cell formation—processes directly linked to AS pathogenesis (91). Clinical data further support that serum leptin levels in obese individuals correlate positively with inflammatory markers such as IL-6 and IL-1β. Leptin gene polymorphisms also demonstrate gender-specific regulation of IL-6, with more pronounced effects observed in men (91). These findings suggest that leptin continuously activates monocytes/macrophages via trained immunity, driving inflammation and lipid metabolism disorders that accelerate AS progression. Gender differences may influence the molecular regulatory pathways involved. In studies of lipid metabolism disorders, pre-stimulation of monocytes with oxLDL for 6 days followed by re-stimulation with TLR agonists induces sustained enhancement of AS-related pro-inflammatory factor secretion. This phenotype is maintained through glycolysis upregulation and metabolic-epigenetic reprogramming mediated by histone H3K4me3 modifications (21). Similar mechanisms are observed in metabolic endotoxemia induced by gut microbiota dysbiosis and in TMAO-induced ER stress via the PERK pathway, both of which promote AS progression through trained immunity (95, 108).
Recent studies have highlighted that miRNAs play a pivotal role in diabetic AS by targeting metabolic and immune pathways. Specifically, miRNA-425-5p exacerbates endothelial dysfunction by suppressing monocarboxylate transporter MCT4 expression (132). In high-glucose or IL-1β-stimulated human umbilical vein endothelial cells (HUVECs) derived from diabetic patients, the significant upregulation of miRNA-425-5p leads to reduced MCT4 expression, resulting in impaired lactate efflux and intracellular accumulation. This ultimately triggers metabolic disturbances and apoptosis in endothelial cells. These findings suggest that the miRNA-425-5p-mediated imbalance of the glycolysis-lactate axis is a critical factor in diabetic vascular injury. Silencing this miRNA may offer a potential strategy for restoring MCT4 expression and improving endothelial cell function, providing an intervention for diabetic vascular complications (132). Further research has demonstrated that miRNAs contribute to AS progression by regulating immune responses and metabolic reprogramming. For instance, high glucose induces macrophage polarization toward the pro-inflammatory M1 phenotype via miR-33-mediated glycolytic reprogramming, thereby intensifying vascular wall inflammation and plaque instability (133). Conversely, miR-638 inhibits the proliferation, migration, and glycolysis of VSMCs by targeting lactate dehydrogenase A, exhibiting anti-AS potential (134). These two miRNAs regulate AS pathogenesis from both pro-inflammatory and anti-proliferative perspectives. More importantly, miRNAs may serve as a crucial nexus between metabolic abnormalities and epigenetic memory. As previously described, miR-9-5p reshapes the epigenetic landscape of pro-inflammatory and glycolysis-related genes in β-glucan-trained monocytes by suppressing the expression of the metabolic enzyme IDH3α. This indicates that miR-9-5p mediates metabolic reprogramming to regulate epigenetic modifications, thus modulating trained immunity. However, the precise regulatory mechanisms underlying miR-9-5p’s role in diabetes-associated trained immunity remain to be further investigated. Future studies should integrate multi-omics analyses with functional validation to explore how miRNAs coordinate metabolic reprogramming and epigenetic modifications, revealing their global regulatory networks in diabetic AS. The development of specific inhibitors or agonists targeting key miRNAs holds promise for advancing precision therapies for diabetic vascular complications.
In conclusion, even with targeted interventions, trained immunity remains a critical factor in sustaining chronic low-grade inflammation in diabetes and driving AS. This may explain why interventions focusing solely on metabolic disturbances, without addressing inflammation, are limited in reducing cardiovascular risk.
6 Preventive and therapeutic modulation of trained immunity
Targeting the chronic inflammation mediated by trained immunity presents a promising strategy for the prevention and treatment of AS and CVDs. Current anti-inflammatory approaches have been shown to suppress trained immunity in AS patients and reduce inflammatory responses (135). Additionally, colchicine, an NLRP3 inflammasome inhibitor, significantly lowers the risk of cardiovascular events in patients with coronary heart disease (136). However, broad-spectrum anti-inflammatory treatments come with increased risks of infections and limited therapeutic effectiveness (137), highlighting the need for more precise interventions that specifically target the mechanisms underlying trained immunity.
The establishment of innate immune memory is driven by metabolic reprogramming and epigenetic remodeling, both of which present potential therapeutic targets. In terms of metabolic regulation, metformin inhibits monocyte immune training by suppressing the mTOR pathway, effectively eliminating the trained immune phenotype induced by oxLDL (138). Statins inhibit monocyte immune training by blocking cholesterol synthesis, although they are ineffective in familial hypercholesterolemia patients who have already developed trained immunity (82). Concerning epigenetic regulation, the histone methyltransferase inhibitor 5’-methylthioadenosine reverses oxLDL-induced histone methylation, abolishing trained immune characteristics such as foam cell formation (139). Inhibition of histone deacetylases (HDACs), such as HDAC3 and HDAC9, can reduce atherosclerotic lesions in AS mouse models, with the class I HDAC inhibitor sodium valproate showing promising effects in alleviating AS in diabetic ApoE-/- mice (140).
Since epigenetic and metabolic mechanisms are critical to cellular function and essential for maintaining normal physiological processes, enhancing therapeutic specificity is crucial to minimize side effects in AS treatments. One strategy for achieving precise intervention involves nanotechnology-based delivery systems (141). For example, statin-loaded recombinant high-density lipoprotein (HDL) nanoparticles can specifically target plaque macrophages in AS mice, effectively reducing local inflammation. Similarly, rapamycin (an mTOR inhibitor)-loaded HDL nanoparticles not only block monocyte trained immunity in vitro but also continuously suppress it in mouse heart transplant models by targeting HSCs, significantly improving transplant survival rates (142–144). These delivery systems help reduce systemic toxicity by concentrating the drug locally, offering valuable insights for clinical application. Moreover, identifying specific molecular targets is crucial for increasing intervention accuracy. Rather than broadly inhibiting epigenetic enzymes, targeting activation marks in the promoter regions of inflammatory genes (e.g., IPLs) could more precisely suppress pro-inflammatory phenotypes. On the metabolic side, the small molecule 3PO partially inhibits the inducible glycolytic enzyme PFKFB3, blocking oxLDL-induced trained immunity while preserving basal glycolytic function and avoiding the adverse effects of complete metabolic inhibition (143). Additionally, the timing of intervention is pivotal in determining therapeutic efficacy and safety. Short-term interventions during the critical window of trained immunity initiation can help prevent AS complications while maintaining the plasticity of the innate immune system. Dynamic monitoring of immune activation markers and precise regulation during key disease progression stages can minimize long-term interference with normal immune function, thus maximizing therapeutic benefits.
Aberrant activation of trained immunity is a critical factor in the progression of AS, involving pathways such as type IFN signaling and the PI3K-AKT pathway. These key regulatory nodes provide potential therapeutic targets for AS treatment. Studies have shown that the E3 ligase TRIM29 regulates innate immune responses through multiple mechanisms (145). In respiratory viral infections, TRIM29 suppresses type IFN production, diminishing the antiviral activity of alveolar macrophages and airway ECs. Its deficiency significantly enhances IFN secretion and protects mice from influenza infection (146, 147). Under metabolic stress, TRIM29 stabilizes the ER stress pathway by interacting with the PERK protein, promoting PERK-mediated inflammation and oxidative stress; inhibiting PERK disrupts this interaction and alleviates its pathological effects (148). Moreover, TRIM29 inhibits inflammasome activation by ubiquitinating and degrading NLRP6/NLRP9b, reducing IL-18 production, a key pro-inflammatory cytokine (149). These findings suggest that targeting TRIM29 may suppress AS progression through various pathways, including blocking IFN activation, mitigating ER stress, and inhibiting inflammasome activation.
The PI3K-AKT pathway has bidirectional regulatory effects in AS: moderate activation stabilizes plaques by inhibiting NF-κB-mediated inflammatory cytokine production and promoting collagen synthesis, while excessive activation contributes to insulin resistance and abnormal proliferation of VSMCs, increasing the risk of plaque rupture (150). Further research has identified poly(ADP-ribose) polymerase 9 (PARP9), an upstream regulator of this pathway, which induces type I IFN production in DCsdendritic cells and macrophages via the PI3K/AKT3 pathway (151). In the AS microenvironment, sustained activation of immune cells within plaques leads to excessive IFN production, which may induce trained immunity and create a positive feedback loop of chronic pro-inflammatory cytokine release. Targeting PARP9 may inhibit plaque deterioration by modulating PI3K/AKT3 activity and disrupting the IFN-driven inflammatory cascade.
In conclusion, targeting TRIM29 or PARP9 signaling could offer novel therapeutic strategies for the prevention and treatment of AS by inhibiting the formation of trained immunity. However, the specific roles of these targets, such as the dynamic expression patterns of TRIM29 in AS plaques, and the clinical feasibility of translating these findings, including the tolerability and efficacy of PARP9 inhibitors, require thorough validation. Future research should incorporate single-cell multi-omics and dynamic pathological models to clarify their precise regulatory networks in AS, paving the way for the clinical translation of these discoveries into targeted interventions.
7 Conclusion and future perspectives
It is projected that in the near future, the incidence of diabetes and its associated AS will continue to increase, particularly in regions with a high prevalence of metabolic syndrome, such as China. While current clinical guidelines emphasize the management of blood glucose and lipid levels, large-scale studies have demonstrated that even diabetic patients with well-controlled metabolic parameters still face significant residual cardiovascular risks. This highlights the need for a shift in treatment paradigms, with the “trained immunity” mechanism driven by diabetes offering a novel approach to address this challenge.
Recent research has revealed that diabetes can substantially accelerate the pathological progression of AS by inducing prolonged activation of trained immunity. This process is characterized by persistent metabolic reprogramming in myeloid cells, dysregulated epigenetic changes, and excessive production of pro-inflammatory mediators. These findings explain why interventions targeting only metabolic disturbances, such as hyperglycemia, are insufficient to effectively mitigate cardiovascular risk, while also emphasizing the potential of precision medicine through the modulation of trained immunity. Possible therapeutic targets include the inhibition of inflammatory signaling pathways, the reversal of aberrant epigenetic modifications, or the modulation of critical metabolic processes. However, current therapeutic strategies face two main challenges: first, the molecular mechanisms underlying trained immunity, such as metabolic reprogramming, are intricately linked to various physiological functions; second, much of the evidence to date relies on in vitro monocyte models and animal studies, with limited validation in human clinical data.
To address these challenges, a stratified intervention approach could be employed. This might include the use of nanocarriers for targeted drug delivery to myeloid cells, the development of epigenetic editing technologies for targeting inflammation-related gene loci, and the implementation of short-term interventions during the critical initiation phase of trained immunity. Additionally, a more detailed examination of specific molecular markers (e.g., IPLs) could pave the way for the development of more selective and effective therapeutic strategies. Despite the current translational challenges, targeting trained immunity represents a promising new paradigm for overcoming the “metabolic memory” of diabetic AS and may drive a strategic shift in cardiovascular treatments from focusing solely on metabolic regulation to incorporating immune remodeling.
Author contributions
YB: Conceptualization, Investigation, Writing – original draft, Writing – review & editing. JW: Conceptualization, Visualization, Writing – review & editing. WJ: Funding acquisition, Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was supported by The National Natural Science Foundation of China (82374334). And supported by Hunan Provincial Natural Science Foundation Project (2022JJ30433 and 2024JJ9466).
Acknowledgments
We sincerely thank every member of the team for their work. The figures created by biorender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Antar SA, Ashour NA, Sharaky M, Khattab M, Ashour NA, Zaid RT, et al. Diabetes mellitus: Classification, mediators, and complications; A gate to identify potential targets for the development of new effective treatments. BioMed Pharmacother. (2023) 168:115734. doi: 10.1016/j.biopha.2023.115734
2. Sun H, Saeedi P, Karuranga S, Pinkepank M, Ogurtsova K, Duncan BB, et al. Erratum to “IDF Diabetes Atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045” [Diabetes Res. Clin. Pract. 183 (2022) 109119. Diabetes Res Clin Pract. (2023) 204:110945. doi: 10.1016/j.diabres.2023.110945
3. Ji L. Current challenges of diabetes and metabolic disorders in China. Diabet Obes Metab. (2023) 25:3–4. doi: 10.1111/dom.15048
4. Yu MG, Gordin D, Fu J, Park K, Li Q, King GL. Protective factors and the pathogenesis of complications in diabetes. Endocr Rev. (2024) 45:227–52. doi: 10.1210/endrev/bnad030
5. Chait A, Eckel RH, Vrablik M, Zambon A. Lipid-lowering in diabetes: An update. Atherosclerosis. (2024) 394:117313. doi: 10.1016/j.atherosclerosis.2023.117313
6. Jia G, Bai H, Mather B, Hill MA, Jia G, Sowers JR. Diabetic vasculopathy: molecular mechanisms and clinical insights. Int J Mol Sci. (2024) 25:804. doi: 10.3390/ijms25020804
7. Stratton IM, Adler AI, Neil HA, Matthews DR, Manley SE, Cull CA, et al. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): prospective observational study. BMJ. (2000) 321:405–12. doi: 10.1136/bmj.321.7258.405
8. Nathan DM, Cleary PA, Backlund J-YC, Genuth SM, Lachin JM, Orchard TJ, et al. Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications (DCCT/EDIC) Study Research Group. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med. (2005) 353:2643–53. doi: 10.1056/NEJMoa052187
9. Ye J, Li L, Wang M, Ma Q, Tian Y, Zhang Q, et al. Diabetes mellitus promotes the development of atherosclerosis: the role of NLRP3. Front Immunol. (2022) 13:900254. doi: 10.3389/fimmu.2022.900254
10. Goldberg RB. Dyslipidemia in diabetes: when and how to treat? Endocrinol Metab Clin North Am. (2022) 51:603–24. doi: 10.1016/j.ecl.2022.02.011
11. Chen Z, Natarajan R. Epigenetic modifications in metabolic memory: What are the memories, and can we erase them? Am J Physiol Cell Physiol. (2022) 323:C570–82. doi: 10.1152/ajpcell.00201.2022
12. Bekkering S, Saner C, Riksen NP, Netea MG, Sabin MA, Saffery R, et al. Trained immunity: linking obesity and cardiovascular disease across the life-course? Trends Endocrinol Metab. (2020) 31:378–89. doi: 10.1016/j.tem.2020.01.008
13. Netea MG, Quintin J, van der Meer JWM. Trained immunity: a memory for innate host defense. Cell Host Microbe. (2011) 9:355–61. doi: 10.1016/j.chom.2011.04.006
14. Domínguez-Andrés J, Dos Santos JC, Bekkering S, Mulder WJM, van der Meer JWM, Riksen NP, et al. Trained immunity: adaptation within innate immune mechanisms. Physiol Rev. (2023) 103:313–46. doi: 10.1152/physrev.00031.2021
15. Ziogas A, Bruno M, van der Meel R, Mulder WJM, Netea MG. Trained immunity: Target for prophylaxis and therapy. Cell Host Microbe. (2023) 31:1776–91. doi: 10.1016/j.chom.2023.10.015
16. Edgar L, Akbar N, Braithwaite AT, Krausgruber T, Gallart-Ayala H, Bailey J, et al. Hyperglycemia induces trained immunity in macrophages and their precursors and promotes atherosclerosis. Circulation. (2021) 144:961–82. doi: 10.1161/CIRCULATIONAHA.120.046464
17. Turvey SE, Broide DH. Chapter 2: innate immunity. J Allergy Clin Immunol. (2010) 125:S24–32. doi: 10.1016/j.jaci.2009.07.016
18. Chi H, Pepper M, Thomas PG. Principles and therapeutic applications of adaptive immunity. Cell. (2024) 187:2052–78. doi: 10.1016/j.cell.2024.03.037
19. Kleimann P, Irschfeld L-M, Grandoch M, Flögel U, Temme S. Trained innate immunity in animal models of cardiovascular diseases. Int J Mol Sci. (2024) 25:2312. doi: 10.3390/ijms25042312
20. Netea MG, Domínguez-Andrés J, Barreiro LB, Chavakis T, Divangahi M, Fuchs E, et al. Defining trained immunity and its role in health and disease. Nat Rev Immunol. (2020) 20:375–88. doi: 10.1038/s41577-020-0285-6
21. Keating ST, Groh L, Thiem K, Bekkering S, Li Y, Matzaraki V, et al. Rewiring of glucose metabolism defines trained immunity induced by oxidized low-density lipoprotein. J Mol Med (Berl). (2020) 98:819–31. doi: 10.1007/s00109-020-01915-w
22. van der Heijden CDCC, Keating ST, Groh L, Joosten LAB, Netea MG, Riksen NP. Aldosterone induces trained immunity: the role of fatty acid synthesis. Cardiovasc Res. (2020) 116:317–28. doi: 10.1093/cvr/cvz137
23. van der Heijden CDCC, Groh L, Keating ST, Kaffa C, Noz MP, Kersten S, et al. Catecholamines induce trained immunity in monocytes. In Vitro In Vivo Circ Res. (2020) 127:269–83. doi: 10.1161/CIRCRESAHA.119.315800
24. Bekkering S, Domínguez-Andrés J, Joosten LAB, Riksen NP, Netea MG. Trained immunity: reprogramming innate immunity in health and disease. Annu Rev Immunol. (2021) 39:667–93. doi: 10.1146/annurev-immunol-102119-073855
25. Fanucchi S, Domínguez-Andrés J, Joosten LAB, Netea MG, Mhlanga MM. The intersection of epigenetics and metabolism in trained immunity. Immunity. (2021) 54:32–43. doi: 10.1016/j.immuni.2020.10.011
26. Mitroulis I, Ruppova K, Wang B, Chen L-S, Grzybek M, Grinenko T, et al. Modulation of myelopoiesis progenitors is an integral component of trained immunity. Cell. (2018) 172:147–161.e12. doi: 10.1016/j.cell.2017.11.034
27. Terrén I, Orrantia A, Astarloa-Pando G, Amarilla-Irusta A, Zenarruzabeitia O, Borrego F. Cytokine-induced memory-like NK cells: from the basics to clinical applications. Front Immunol. (2022) 13:884648. doi: 10.3389/fimmu.2022.884648
28. Acevedo OA, Berrios RV, Rodríguez-Guilarte L, Lillo-Dapremont B, Kalergis AM. Molecular and cellular mechanisms modulating trained immunity by various cell types in response to pathogen encounter. Front Immunol. (2021) 12:745332. doi: 10.3389/fimmu.2021.745332
29. Li Z, Zhao M, Yang Y, Zou Z, Zhang L, Jiang F, et al. Treatment of a MyD88 inhibitor alleviates rejection and inflammation in xenotransplantation by inhibiting dendritic cells activation and trained immunity in macrophages. Int Immunopharmacol. (2024) 130:111664. doi: 10.1016/j.intimp.2024.111664
30. Sohrabi Y, Lagache SMM, Voges VC, Semo D, Sonntag G, Hanemann I, et al. OxLDL-mediated immunologic memory in endothelial cells. J Mol Cell Cardiol. (2020) 146:121–32. doi: 10.1016/j.yjmcc.2020.07.006
31. Naik S, Larsen SB, Gomez NC, Alaverdyan K, Sendoel A, Yuan S, et al. Inflammatory memory sensitizes skin epithelial stem cells to tissue damage. Nature. (2017) 550:475–80. doi: 10.1038/nature24271
32. Sadeghi M, Divangahi M. Discovering adaptive features of innate immune memory. Immunol Rev. (2024) 323:186–196. doi: 10.1111/imr.13328
33. Berendsen M, Schaltz-Buchholzer F, Bles P, Biering-Sørensen S, Jensen KJ, Monteiro I, et al. Parental Bacillus Calmette-Guérin vaccine scars decrease infant mortality in the first six weeks of life: A retrospective cohort study. EClinicalMedicine. (2021) 39:101049. doi: 10.1016/j.eclinm.2021.101049
34. Domínguez-Andrés J, Joosten LA, Netea MG. Induction of innate immune memory: the role of cellular metabolism. Curr Opin Immunol. (2019) 56:10–6. doi: 10.1016/j.coi.2018.09.001
35. Chandel NS. Glycolysis. Cold Spring Harbor Perspect Biol. (2021) 13:a040535. doi: 10.1101/cshperspect.a040535
36. Li L, Wang M, Ma Q, Ye J, Sun G. Role of glycolysis in the development of atherosclerosis. Am J Physiol Cell Physiol. (2022) 323:C617–29. doi: 10.1152/ajpcell.00218.2022
37. Leng S, Zhang X, Wang S, Qin J, Liu Q, Liu A, et al. Ion channel Piezo1 activation promotes aerobic glycolysis in macrophages. Front Immunol. (2022) 13:976482. doi: 10.3389/fimmu.2022.976482
38. Liu Q-J, Yuan W, Yang P, Shao C. Role of glycolysis in diabetic atherosclerosis. World J Diabetes. (2023) 14:1478. doi: 10.4239/wjd.v14.i10.1478
39. Cheng S-C, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, et al. mTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science. (2014) 345:1250684. doi: 10.1126/science.1250684
40. Arts RJW, Carvalho A, La Rocca C, Palma C, Rodrigues F, Silvestre R, et al. Immunometabolic pathways in BCG-induced trained immunity. Cell Rep. (2016) 17:2562–71. doi: 10.1016/j.celrep.2016.11.011
41. TeSlaa T, Ralser M, Fan J, Rabinowitz JD. The pentose phosphate pathway in health and disease. Nat Metab. (2023) 5:1275–89. doi: 10.1038/s42255-023-00863-2
42. Arts RJW, Novakovic B, Ter Horst R, Carvalho A, Bekkering S, Lachmandas E, et al. Glutaminolysis and fumarate accumulation integrate immunometabolic and epigenetic programs in trained immunity. Cell Metab. (2016) 24:807–19. doi: 10.1016/j.cmet.2016.10.008
43. Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y, et al. Metabolic regulation of gene expression by histone lactylation. Nature. (2019) 574:575–80. doi: 10.1038/s41586-019-1678-1
44. Cai H, Chen X, Liu Y, Chen Y, Zhong G, Chen X, et al. Lactate activates trained immunity by fueling the tricarboxylic acid cycle and regulating histone lactylation. Nat Commun. (2025) 16:3230. doi: 10.1038/s41467-025-58563-2
45. Koivunen P, Hirsilä M, Remes AM, Hassinen IE, Kivirikko KI, Myllyharju J. Inhibition of hypoxia-inducible factor (HIF) hydroxylases by citric acid cycle intermediates: POSSIBLE LINKS BETWEEN CELL METABOLISM AND STABILIZATION OF HIF *. J Biol Chem. (2007) 282:4524–32. doi: 10.1074/jbc.M610415200
46. Dhat R, Mongad D, Raji S, Arkat S, Mahapatra NR, Singhal N, et al. Epigenetic modifier alpha-ketoglutarate modulates aberrant gene body methylation and hydroxymethylation marks in diabetic heart. Epigenet Chromatin. (2023) 16:12. doi: 10.1186/s13072-023-00489-4
47. Montano EN, Bose M, Huo L, Tumurkhuu G, De Los Santos G, Simental B, et al. α-ketoglutarate-dependent KDM6 histone demethylases and interferon-stimulated gene expression in lupus. Arthritis Rheumatol. (2024) 76:396–410. doi: 10.1002/art.42724
48. Spinelli JB, Haigis MC. The multifaceted contributions of mitochondria to cellular metabolism. Nat Cell Biol. (2018) 20:745–54. doi: 10.1038/s41556-018-0124-1
49. Fernandez-Fuente G, Rigby MJ, Puglielli L. Intracellular Citrate/acetyl-CoA flux and endoplasmic reticulum acetylation: Connectivity is the answer. Mol Metab. (2022) 67:101653. doi: 10.1016/j.molmet.2022.101653
50. Bekkering S, Arts RJW, Novakovic B, Kourtzelis I, van der Heijden CDCC, Li Y, et al. Metabolic induction of trained immunity through the mevalonate pathway. Cell. (2018) 172:135–146.e9. doi: 10.1016/j.cell.2017.11.025
51. Christ A, Latz E. The Western lifestyle has lasting effects on metaflammation. Nat Rev Immunol. (2019) 19:267–8. doi: 10.1038/s41577-019-0156-1
52. Keating ST, Groh L, van der Heijden CDCC, Rodriguez H, dos Santos JC, Fanucchi S, et al. The set7 lysine methyltransferase regulates plasticity in oxidative phosphorylation necessary for trained immunity induced by β-glucan. Cell Rep. (2020) 31:107548. doi: 10.1016/j.celrep.2020.107548
53. Liotti A, Ferrara AL, Loffredo S, Galdiero MR, Varricchi G, Di Rella F, et al. Epigenetics: An opportunity to shape innate and adaptive immune responses. Immunology. (2022) 167:451–70. doi: 10.1111/imm.13571
54. Riksen NP, Netea MG. Immunometabolic control of trained immunity. Mol Aspects Med. (2021) 77:100897. doi: 10.1016/j.mam.2020.100897
55. Novakovic B, Habibi E, Wang S-Y, Arts RJW, Davar R, Megchelenbrink W, et al. β-glucan reverses the epigenetic state of LPS-induced immunological tolerance. Cell. (2016) 167:1354–1368.e14. doi: 10.1016/j.cell.2016.09.034
56. Saeed S, Quintin J, Kerstens HHD, Rao NA, Aghajanirefah A, Matarese F, et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science. (2014) 345:1251086. doi: 10.1126/science.1251086
57. Mourits VP, van Puffelen JH, Novakovic B, Bruno M, Ferreira AV, Arts RJ, et al. Lysine methyltransferase G9a is an important modulator of trained immunity. Clin Transl Immunol. (2021) 10:e1253. doi: 10.1002/cti2.1253
58. Moorlag SJCFM, Matzaraki V, van Puffelen JH, van der Heijden C, Keating S, Groh L, et al. An integrative genomics approach identifies KDM4 as a modulator of trained immunity. Eur J Immunol. (2022) 52:431–46. doi: 10.1002/eji.202149577
59. Stelmaszyk A, Mikołajczak P, Dworacka M. Sirtuin 1 as the mechanism of action of agents used in the diabetes mellitus pharmacotherapy. Eur J Pharmacol. (2021) 907:174289. doi: 10.1016/j.ejphar.2021.174289
60. Su H, Liang Z, Weng S, Sun C, Huang J, Zhang T, et al. miR-9-5p regulates immunometabolic and epigenetic pathways in β-glucan-trained immunity via IDH3α. JCI Insight. (2022) 7:e159640. doi: 10.1172/jci.insight.159640
61. Fanucchi S, Fok ET, Dalla E, Shibayama Y, Börner K, Chang EY, et al. Immune genes are primed for robust transcription by proximal long noncoding RNAs located in nuclear compartments. Nat Genet. (2019) 51:138–50. doi: 10.1038/s41588-018-0298-2
62. Bannister S, Kim B, Domínguez-Andrés J, Kilic G, Ansell BRE, Neeland MR, et al. Neonatal BCG vaccination is associated with a long-term DNA methylation signature in circulating monocytes. Sci Adv. (2022) 8:eabn4002. doi: 10.1126/sciadv.abn4002
63. Verma D, Parasa VR, Raffetseder J, Martis M, Mehta RB, Netea M, et al. Anti-mycobacterial activity correlates with altered DNA methylation pattern in immune cells from BCG-vaccinated subjects. Sci Rep. (2017) 7:12305. doi: 10.1038/s41598-017-12110-2
64. Das J, Verma D, Gustafsson M, Lerm M. Identification of DNA methylation patterns predisposing for an efficient response to BCG vaccination in healthy BCG-naïve subjects. Epigenetics. (2019) 14:589–601. doi: 10.1080/15592294.2019.1603963
65. Okdahl T, Wegeberg A-M, Pociot F, Brock B, Størling J, Brock C. Low-grade inflammation in type 2 diabetes: a cross-sectional study from a Danish diabetes outpatient clinic. BMJ Open. (2022) 12:e062188. doi: 10.1136/bmjopen-2022-062188
66. Charles-Messance H, Sheedy FJ. Train to lose: innate immune memory in metaflammation. Mol Nutr Food Res. (2021) 65:e1900480. doi: 10.1002/mnfr.201900480
67. Thiem K, Stienstra R, Riksen NP, Keating ST. Trained immunity and diabetic vascular disease. Clin Sci (Lond). (2019) 133:195–203. doi: 10.1042/CS20180905
68. Thiem K, Keating ST, Netea MG, Riksen NP, Tack CJ, van Diepen J, et al. Hyperglycemic memory of innate immune cells promotes in vitro proinflammatory responses of human monocytes and murine macrophages. J Immunol. (2021) 206:807–13. doi: 10.4049/jimmunol.1901348
69. Miao F, Chen Z, Genuth S, Paterson A, Zhang L, Wu X, et al. Evaluating the role of epigenetic histone modifications in the metabolic memory of type 1 diabetes. Diabetes. (2014) 63:1748–62. doi: 10.2337/db13-1251
70. Shrestha S, Lee Y-B, Lee H, Choi Y-K, Park B-Y, Kim M-J, et al. Diabetes primes neutrophils for neutrophil extracellular trap formation through trained immunity. Res (Wash D C). (2024) 7:365. doi: 10.34133/research.0365
71. Nagareddy PR, Murphy AJ, Stirzaker RA, Hu Y, Yu S, Miller RG, et al. Hyperglycemia promotes myelopoiesis and impairs the resolution of atherosclerosis. Cell Metab. (2013) 17:695–708. doi: 10.1016/j.cmet.2013.04.001
72. Vinci MC, Costantino S, Damiano G, Rurali E, Rinaldi R, Vigorelli V, et al. Persistent epigenetic signals propel a senescence-associated secretory phenotype and trained innate immunity in CD34+ hematopoietic stem cells from diabetic patients. Cardiovasc Diabetol. (2024) 23:107. doi: 10.1186/s12933-024-02195-1
73. Li S, Reddy MA, Cai Q, Meng L, Yuan H, Lanting L, et al. Enhanced proatherogenic responses in macrophages and vascular smooth muscle cells derived from diabetic db/db mice. Diabetes. (2006) 55:2611–9. doi: 10.2337/db06-0164
74. El-Osta A, Brasacchio D, Yao D, Pocai A, Jones PL, Roeder RG, et al. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J Exp Med. (2008) 205:2409–17. doi: 10.1084/jem.20081188
75. Flynn MC, Kraakman MJ, Tikellis C, Lee MK, Hanssen NM, Kammoun HL, et al. Transient intermittent hyperglycemia accelerates atherosclerosis by promoting myelopoiesis. Circ Res. (2020) 127:877–92. doi: 10.1161/CIRCRESAHA.120.316653
76. Twarda-Clapa A, Olczak A, Białkowska AM, Koziołkiewicz M. Advanced glycation end-products (AGEs): formation, chemistry, classification, receptors, and diseases related to AGEs. Cells. (2022) 11:1312. doi: 10.3390/cells11081312
77. Ratter JM, van Heck JIP, Rooijackers HMM, Jansen HJ, van Poppel PCM, Tack CJ, et al. Insulin acutely activates metabolism of primary human monocytes and promotes a proinflammatory phenotype. J Leukoc Biol. (2021) 110:885–91. doi: 10.1002/JLB.3AB0120-019RR
78. Dandona P, Aljada A, Mohanty P, Ghanim H, Hamouda W, Assian E, et al. Insulin inhibits intranuclear nuclear factor kappaB and stimulates IkappaB in mononuclear cells in obese subjects: evidence for an anti-inflammatory effect? J Clin Endocrinol Metab. (2001) 86:3257–65. doi: 10.1210/jcem.86.7.7623
79. Bekkering S, Quintin J, Joosten LAB, van der Meer JWM, Netea MG, Riksen NP. Oxidized low-density lipoprotein induces long-term proinflammatory cytokine production and foam cell formation via epigenetic reprogramming of monocytes. Arterioscler Thromb Vasc Biol. (2014) 34:1731–8. doi: 10.1161/ATVBAHA.114.303887
80. Wang J, Liu Y-M, Hu J, Chen C. Trained immunity in monocyte/macrophage: Novel mechanism of phytochemicals in the treatment of atherosclerotic cardiovascular disease. Front Pharmacol. (2023) 14:1109576. doi: 10.3389/fphar.2023.1109576
81. Groh LA, Ferreira AV, Helder L, van der Heijden CDCC, Novakovic B, van de Westerlo E, et al. oxLDL-induced trained immunity is dependent on mitochondrial metabolic reprogramming. Immunometabolism. (2021) 3:e210025. doi: 10.20900/immunometab20210025
82. Bekkering S, Stiekema LCA, Bernelot Moens S, Verweij SL, Novakovic B, Prange K, et al. Treatment with statins does not revert trained immunity in patients with familial hypercholesterolemia. Cell Metab. (2019) 30:1–2. doi: 10.1016/j.cmet.2019.05.014
83. Bahrar H, Bekkering S, Stienstra R, Netea MG, Riksen NP. Innate immune memory in cardiometabolic disease. Cardiovasc Res. (2023) 119:2774–86. doi: 10.1093/cvr/cvad030
84. Guo Z, Wang L, Liu H, Xie Y. Innate immune memory in monocytes and macrophages: the potential therapeutic strategies for atherosclerosis. Cells. (2022) 11:4072. doi: 10.3390/cells11244072
85. van der Valk FM, Bekkering S, Kroon J, Yeang C, van den Bossche J, van Buul JD, et al. Oxidized phospholipids on lipoprotein(a) elicit arterial wall inflammation and an inflammatory monocyte response in humans. Circulation. (2016) 134:611–24. doi: 10.1161/CIRCULATIONAHA.116.020838
86. Rhee E-J. Weight cycling and its cardiometabolic impact. J Obes Metab Syndr. (2017) 26:237–42. doi: 10.7570/jomes.2017.26.4.237
87. Blaszczak AM, Bernier M, Wright VP, Gebhardt G, Anandani K, Liu J, et al. Obesogenic memory maintains adipose tissue inflammation and insulin resistance. Immunometabolism. (2020) 2:e200023. doi: 10.20900/immunometab20200023
88. Caslin HL, Cottam MA, Piñon JM, Boney LY, Hasty AH. Weight cycling induces innate immune memory in adipose tissue macrophages. Front Immunol. (2022) 13:984859. doi: 10.3389/fimmu.2022.984859
89. Thaiss CA, Itav S, Rothschild D, Meijer MT, Levy M, Moresi C, et al. Persistent microbiome alterations modulate the rate of post-dieting weight regain. Nature. (2016) 540:544–51. doi: 10.1038/nature20796
90. Dorneles GP, Boeira MCR, Schipper LL, Silva IRV, Elsner VR, Dal Lago P, et al. Acute strenuous exercise induces an imbalance on histone H4 acetylation/histone deacetylase 2 and increases the proinflammatory profile of PBMC of obese individuals. Oxid Med Cell Longev. (2017) 2017:1530230. doi: 10.1155/2017/1530230
91. Flores Gomez D, Bekkering S, Ter Horst R, Cossins B, van den Munckhof ICL, Rutten JHW, et al. The effect of leptin on trained innate immunity and on systemic inflammation in subjects with obesity. J Leukoc Biol. (2024) 115:374–84. doi: 10.1093/jleuko/qiad118
92. Nagareddy PR, Kraakman M, Masters SL, Stirzaker RA, Gorman DJ, Grant RW, et al. Adipose tissue macrophages promote myelopoiesis and monocytosis in obesity. Cell Metab. (2014) 19:821–35. doi: 10.1016/j.cmet.2014.03.029
93. Singer K, DelProposto J, Lee Morris D, Zamarron B, Mergian T, Maley N, et al. Diet-induced obesity promotes myelopoiesis in hematopoietic stem cells. Mol Metab. (2014) 3:664–75. doi: 10.1016/j.molmet.2014.06.005
94. Christ A, Lauterbach M, Latz E. Western diet and the immune system: an inflammatory connection. Immunity. (2019) 51:794–811. doi: 10.1016/j.immuni.2019.09.020
95. Geng S, Chen K, Yuan R, Peng L, Maitra U, Diao N, et al. The persistence of low-grade inflammatory monocytes contributes to aggravated atherosclerosis. Nat Commun. (2016) 7:13436. doi: 10.1038/ncomms13436
96. Ifrim DC, Quintin J, Joosten LAB, Jacobs C, Jansen T, Jacobs L, et al. Trained immunity or tolerance: opposing functional programs induced in human monocytes after engagement of various pattern recognition receptors. Clin Vaccine Immunol. (2014) 21:534–45. doi: 10.1128/CVI.00688-13
97. Christ A, Günther P, Lauterbach MAR, Duewell P, Biswas D, Pelka K, et al. Western diet triggers NLRP3-dependent innate immune reprogramming. Cell. (2018) 172:162–175.e14. doi: 10.1016/j.cell.2017.12.013
98. van Kampen E, Jaminon A, van Berkel TJC, Van Eck M. Diet-induced (epigenetic) changes in bone marrow augment atherosclerosis. J Leukoc Biol. (2014) 96:833–41. doi: 10.1189/jlb.1A0114-017R
99. Temba GS, Kullaya V, Pecht T, Mmbaga BT, Aschenbrenner AC, Ulas T, et al. Urban living in healthy Tanzanians is associated with an inflammatory status driven by dietary and metabolic changes. Nat Immunol. (2021) 22:287–300. doi: 10.1038/s41590-021-00867-8
100. Hata M, Andriessen EMMA, Hata M, Diaz-Marin R, Fournier F, Crespo-Garcia S, et al. Past history of obesity triggers persistent epigenetic changes in innate immunity and exacerbates neuroinflammation. Science. (2023) 379:45–62. doi: 10.1126/science.abj8894
101. Lavillegrand J-R, Al-Rifai R, Thietart S, Guyon T, Vandestienne M, Cohen R, et al. Alternating high-fat diet enhances atherosclerosis by neutrophil reprogramming. Nature. (2024) 634:447–56. doi: 10.1038/s41586-024-07693-6
102. Yi C, Huang S, Zhang W, Guo L, Xia T, Huang F, et al. Synergistic interactions between gut microbiota and short chain fatty acids: Pioneering therapeutic frontiers in chronic disease management. Microb Pathog. (2025) 199:107231. doi: 10.1016/j.micpath.2024.107231
103. Wang S, Cui Z, Yang H. Interactions between host and gut microbiota in gestational diabetes mellitus and their impacts on offspring. BMC Microbiol. (2024) 24:161. doi: 10.1186/s12866-024-03255-y
104. Wang F, Glenn AJ, Tessier A-J, Mei Z, Haslam DE, Guasch-Ferré M, et al. Integration of epidemiological and blood biomarker analysis links haem iron intake to increased type 2 diabetes risk. Nat Metab. (2024) 6:1807–18. doi: 10.1038/s42255-024-01109-5
105. Lu Y, Sun Y, Saaoud F, Shao Y, Xu K, Jiang X, et al. ER stress mediates Angiotensin II-augmented innate immunity memory and facilitates distinct susceptibilities of thoracic from abdominal aorta to aneurysm development. Front Immunol. (2023) 14:1268916. doi: 10.3389/fimmu.2023.1268916
106. Benson TW, Conrad KA, Li XS, Wang Z, Helsley RN, Schugar RC, et al. Gut microbiota-derived trimethylamine N-oxide contributes to abdominal aortic aneurysm through inflammatory and apoptotic mechanisms. Circulation. (2023) 147:1079–96. doi: 10.1161/CIRCULATIONAHA.122.060573
107. Saaoud F, Liu L, Xu K, Cueto R, Shao Y, Lu Y, et al. Aorta- and liver-generated TMAO enhances trained immunity for increased inflammation via ER stress/mitochondrial ROS/glycolysis pathways. JCI Insight. (2023) 8:e158183. doi: 10.1172/jci.insight.158183
108. Bingyu W, Jun Q, Bingyang L, Xi Y, Jianqing Z, Jiangfang L. Trimethylamine N-oxide promotes PERK-mediated endothelial-mesenchymal transition and apoptosis thereby aggravates atherosclerosis. Int Immunopharmacol. (2024) 142:113209. doi: 10.1016/j.intimp.2024.113209
109. Kong P, Cui Z-Y, Huang X-F, Zhang D-D, Guo R-J, Han M. Inflammation and atherosclerosis: signaling pathways and therapeutic intervention. Signal Transduct Target Ther. (2022) 7:131. doi: 10.1038/s41392-022-00955-7
110. Pan Y, Li J, Xia X, Wang J, Jiang Q, Yang J, et al. β-glucan-coupled superparamagnetic iron oxide nanoparticles induce trained immunity to protect mice against sepsis. Theranostics. (2022) 12:675–88. doi: 10.7150/thno.64874
111. Quintin J, Saeed S, Martens JHA, Giamarellos-Bourboulis EJ, Ifrim DC, Logie C, et al. Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe. (2012) 12:223–32. doi: 10.1016/j.chom.2012.06.006
112. Shirai T, Nazarewicz RR, Wallis BB, Yanes RE, Watanabe R, Hilhorst M, et al. The glycolytic enzyme PKM2 bridges metabolic and inflammatory dysfunction in coronary artery disease. J Exp Med. (2016) 213:337–54. doi: 10.1084/jem.20150900
113. Bekkering S, van den Munckhof I, Nielen T, Lamfers E, Dinarello C, Rutten J, et al. Innate immune cell activation and epigenetic remodeling in symptomatic and asymptomatic atherosclerosis in humans. vivo Atheroscl. (2016) 254:228–36. doi: 10.1016/j.atherosclerosis.2016.10.019
114. Dong Z, Hou L, Luo W, Pan L-H, Li X, Tan H-P, et al. Myocardial infarction drives trained immunity of monocytes, accelerating atherosclerosis. Eur Heart J. (2024) 45:669–84. doi: 10.1093/eurheartj/ehad787
115. Huijser E, van Helden-Meeuwsen CG, Grashof DGB, Tarn JR, Brkic Z, Huisman JMA, et al. Trained immunity in primary sjögren’s syndrome: linking type I interferons to a pro-atherogenic phenotype. Front Immunol. (2022) 13:840751. doi: 10.3389/fimmu.2022.840751
116. Kalafati L, Hatzioannou A, Hajishengallis G, Chavakis T. The role of neutrophils in trained immunity. Immunol Rev. (2023) 314:142–57. doi: 10.1111/imr.13142
117. Hole CR, Wager CML, Castro-Lopez N, Campuzano A, Cai H, Wozniak KL, et al. Induction of memory-like dendritic cell responses. vivo Nat Commun. (2019) 10:2955. doi: 10.1038/s41467-019-10486-5
118. Moorlag SJCFM, Rodriguez-Rosales YA, Gillard J, Fanucchi S, Theunissen K, Novakovic B, et al. BCG vaccination induces long-term functional reprogramming of human neutrophils. Cell Rep. (2020) 33:108387. doi: 10.1016/j.celrep.2020.108387
119. Théroude C, Reverte M, Heinonen T, Ciarlo E, Schrijver IT, Antonakos N, et al. Trained immunity confers prolonged protection from listeriosis. Front Immunol. (2021) 12:723393. doi: 10.3389/fimmu.2021.723393
120. Kalafati L, Kourtzelis I, Schulte-Schrepping J, Li X, Hatzioannou A, Grinenko T, et al. Innate immune training of granulopoiesis promotes anti-tumor activity. Cell. (2020) 183:771–785.e12. doi: 10.1016/j.cell.2020.09.058
121. Irwandi RA, Chiesa ST, Hajishengallis G, Papayannopoulos V, Deanfield JE, D’Aiuto F. The roles of neutrophils linking periodontitis and atherosclerotic cardiovascular diseases. Front Immunol. (2022) 13:915081. doi: 10.3389/fimmu.2022.915081
122. Mitroulis I, Hajishengallis G, Chavakis T. Bone marrow inflammatory memory in cardiometabolic disease and inflammatory comorbidities. Cardiovasc Res. (2023) 119:2801–12. doi: 10.1093/cvr/cvad003
123. Cirovic B, de Bree LCJ, Groh L, Blok BA, Chan J, van der Velden WJFM, et al. BCG vaccination in humans elicits trained immunity via the hematopoietic progenitor compartment. Cell Host Microbe. (2020) 28:322–334.e5. doi: 10.1016/j.chom.2020.05.014
124. Noz MP, Bekkering S, Groh L, Nielen TM, Lamfers EJ, Schlitzer A, et al. Reprogramming of bone marrow myeloid progenitor cells in patients with severe coronary artery disease. eLife. (2020) 9:e60939. doi: 10.7554/eLife.60939
125. Okabe J, Orlowski C, Balcerczyk A, Tikellis C, Thomas MC, Cooper ME, et al. Distinguishing hyperglycemic changes by Set7 in vascular endothelial cells. Circ Res. (2012) 110:1067–76. doi: 10.1161/CIRCRESAHA.112.266171
126. Lu Y, Sun Y, Drummer C IV, Nanayakkara GK, Shao Y, Saaoud F, et al. Increased acetylation of H3K14 in the genomic regions that encode trained immunity enzymes in lysophosphatidylcholine-activated human aortic endothelial cells - Novel qualification markers for chronic disease risk factors and conditional DAMPs. Redox Biol. (2019) 24:101221. doi: 10.1016/j.redox.2019.101221
127. Schnack L, Sohrabi Y, Lagache SMM, Kahles F, Bruemmer D, Waltenberger J, et al. Mechanisms of trained innate immunity in oxLDL primed human coronary smooth muscle cells. Front Immunol. (2019) 10:13. doi: 10.3389/fimmu.2019.00013
128. Choudhury RP, Edgar L, Rydén M, Fisher EA. Diabetes and metabolic drivers of trained immunity: new therapeutic targets beyond glucose. Arterioscler Thromb Vasc Biol. (2021) 41:1284–90. doi: 10.1161/ATVBAHA.120.314211
129. Huang D, Gao W, Zhong X, Wu H, Zhou Y, Ma Y, et al. Epigenetically altered macrophages promote development of diabetes-associated atherosclerosis. Front Immunol. (2023) 14:1196704. doi: 10.3389/fimmu.2023.1196704
130. Cao Q, Rong S, Repa JJ, Clair RST, Parks JS, Mishra N. HDAC9 represses cholesterol efflux and generation of alternatively activated macrophages in atherosclerosis development. Arterioscler Thromb Vasc Biol. (2014) 34:1871–9. doi: 10.1161/ATVBAHA.114.303393
131. El Omari N, Khalid A, Makeen HA, Alhazmi HA, Albratty M, Mohan S, et al. Stochasticity of anticancer mechanisms underlying clinical effectiveness of vorinostat. Heliyon. (2024) 10:e33052. doi: 10.1016/j.heliyon.2024.e33052
132. Luo E, Wang D, Yan G, Qiao Y, Zhu B, Liu B, et al. The NF-κB/miR-425-5p/MCT4 axis: A novel insight into diabetes-induced endothelial dysfunction. Mol Cell Endocrinol. (2020) 500:110641. doi: 10.1016/j.mce.2019.110641
133. Ouimet M, Ediriweera HN, Gundra UM, Sheedy FJ, Ramkhelawon B, Hutchison SB, et al. MicroRNA-33-dependent regulation of macrophage metabolism directs immune cell polarization in atherosclerosis. J Clin Invest. (2015) 125:4334–48. doi: 10.1172/JCI81676
134. Chen S, Chen H, Yu C, Lu R, Song T, Wang X, et al. MiR-638 repressed vascular smooth muscle cell glycolysis by targeting LDHA. Open Med (Wars). (2019) 14:663–72. doi: 10.1515/med-2019-0077
135. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. (2017) 377:1119–31. doi: 10.1056/NEJMoa1707914
136. Nidorf SM, Fiolet ATL, Mosterd A, Eikelboom JW, Schut A, Opstal TSJ, et al. Colchicine in patients with chronic coronary disease. N Engl J Med. (2020) 383:1838–47. doi: 10.1056/NEJMoa2021372
137. Flores-Gomez D, Bekkering S, Netea MG, Riksen NP. Trained immunity in atherosclerotic cardiovascular disease. Arteriosclerosis Thromb Vasc Biol. (2021) 41:62–9. doi: 10.1161/ATVBAHA.120.314216
138. Elliehausen CJ, Anderson RM, Diffee GM, Rhoads TW, Lamming DW, Hornberger TA, et al. Geroprotector drugs and exercise: friends or foes on healthy longevity? BMC Biol. (2023) 21:287. doi: 10.1186/s12915-023-01779-9
139. Leentjens J, Bekkering S, Joosten LAB, Netea MG, Burgner DP, Riksen NP. Trained innate immunity as a novel mechanism linking infection and the development of atherosclerosis. Circ Res. (2018) 122:664–9. doi: 10.1161/CIRCRESAHA.117.312465
140. Bowes AJ, Khan MI, Shi Y, Robertson L, Werstuck GH. Valproate attenuates accelerated atherosclerosis in hyperglycemic apoE-deficient mice: evidence in support of a role for endoplasmic reticulum stress and glycogen synthase kinase-3 in lesion development and hepatic steatosis. Am J Pathol. (2009) 174:330–42. doi: 10.2353/ajpath.2009.080385
141. Teunissen AJP, van Leent MMT, Prévot G, Brechbühl EES, Pérez-Medina C, Duivenvoorden R, et al. Targeting trained innate immunity with nanobiologics to treat cardiovascular disease. Arterioscler Thromb Vasc Biol. (2021) 41:1839–50. doi: 10.1161/ATVBAHA.120.315448
142. Duivenvoorden R, Tang J, Cormode DP, Mieszawska AJ, Izquierdo-Garcia D, Ozcan C, et al. A statin-loaded reconstituted high-density lipoprotein nanoparticle inhibits atherosclerotic plaque inflammation. Nat Commun. (2014) 5:3065. doi: 10.1038/ncomms4065
143. Braza MS, van Leent MMT, Lameijer M, Sanchez-Gaytan BL, Arts RJW, Pérez-Medina C, et al. Inhibiting inflammation with myeloid cell-specific nanobiologics promotes organ transplant acceptance. Immunity. (2018) 49:819–828.e6. doi: 10.1016/j.immuni.2018.09.008
144. van Leent MMT, Meerwaldt AE, Berchouchi A, Toner YC, Burnett ME, Klein ED, et al. A modular approach toward producing nanotherapeutics targeting the innate immune system. Sci Adv. (2021) 7:eabe7853. doi: 10.1126/sciadv.abe7853
145. Xing J, Zhang A, Minze LJ, Li XC, Zhang Z. TRIM29 negatively regulates the type I interferon production in response to RNA virus. J Immunol. (2018) 201:183–92. doi: 10.4049/jimmunol.1701569
146. Xing J, Zhang A, Zhang H, Wang J, Li XC, Zeng M-S, et al. TRIM29 promotes DNA virus infections by inhibiting innate immune response. Nat Commun. (2017) 8:945. doi: 10.1038/s41467-017-00101-w
147. Xing J, Weng L, Yuan B, Wang Z, Jia L, Jin R, et al. Identification of a role for TRIM29 in the control of innate immunity in the respiratory tract. Nat Immunol. (2016) 17:1373–80. doi: 10.1038/ni.3580
148. Wang J, Lu W, Zhang J, Du Y, Fang M, Zhang A, et al. Loss of TRIM29 mitigates viral myocarditis by attenuating PERK-driven ER stress response in male mice. Nat Commun. (2024) 15:3481. doi: 10.1038/s41467-024-44745-x
149. Wang J, Wang L, Lu W, Farhataziz N, Gonzalez A, Xing J, et al. TRIM29 controls enteric RNA virus-induced intestinal inflammation by targeting NLRP6 and NLRP9b signaling pathways. Mucosal Immunol. (2025) 18:135–50. doi: 10.1016/j.mucimm.2024.10.004
150. Zhao Y, Qian Y, Sun Z, Shen X, Cai Y, Li L, et al. Role of PI3K in the progression and regression of atherosclerosis. Front Pharmacol. (2021) 12:632378. doi: 10.3389/fphar.2021.632378
Keywords: diabetes, atherosclerosis, trained immunity, metabolism, inflammation, epigenetics
Citation: Bai Y, Wu J and Jian W (2025) Trained immunity in diabetes: emerging targets for cardiovascular complications. Front. Endocrinol. 16:1533620. doi: 10.3389/fendo.2025.1533620
Received: 24 November 2024; Accepted: 24 April 2025;
Published: 14 May 2025.
Edited by:
Ricardo Pujol Borrell, Autonomous University of Barcelona, SpainReviewed by:
Junji Xing, Houston Methodist Research Institute, United StatesShanel Raghubeer, Cape Peninsula University of Technology, South Africa
Copyright © 2025 Bai, Wu and Jian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Weixiong Jian, ZGF4aW9uZzEwMjBAMTI2LmNvbQ==