 Xinru Jiang
Xinru Jiang Wei Zhao1†
Wei Zhao1† Botao Shen
Botao Shen Kexin Chen
Kexin Chen- 1Department of Cardiology, The First Hospital of Jilin University, Changchun, China
- 2Core Facility of the First Hospital of Jilin University, Changchun, China
Metabolic dysfunction-associated steatotic liver disease (MASLD), previously known as non-alcoholic fatty liver disease (NAFLD), has gradually become a leading cause of end-stage liver disease as a heterogeneous group of diseases. While the underlying mechanisms of MASLD remain incompletely understood, it is clear that glycolipid metabolism, coupled with subsequent disruptions in hepatic sinusoidal homeostasis and cellular senescence play significant roles in its onset and progression. In recent years, CD47 has been recognized not only as a critical target in cancer therapy but also as a participant in the development of metabolic diseases through complex signaling pathways. Increasing evidence suggests that CD47 is closely associated with the development of MASLD; however, its role in MASLD has not yet been widely explored. Therefore, this review aims to summarize current research on the potential role of CD47 in the pathogenesis of MASLD, particularly in relation to disturbances in glucose and lipid metabolism.
1 Introduction
In recent years, with the increasing prevalence of obesity and insulin resistance (IR) in the population, the incidence of metabolic dysfunction-associated steatotic liver disease (MASLD) has been rising. Recent data reveal that the global prevalence of MASLD has grown from 25.3% between 1990 and 2006 to 38.0% during the period 2016–2019. At present, MASLD is considered the most prevalent chronic liver disease worldwide (1). MASLD was previously known as non-alcoholic fatty liver disease (NAFLD). In 2023, three leading liver associations proposed replacing the NAFLD with MASLD, and renaming non-alcoholic steatohepatitis (NASH) to metabolic dysfunction-associated steatohepatitis (MASH), highlighting the importance of metabolic dysfunction (2). Despite the differences in definitions between MASLD and NAFLD, it is widely believed that data from NAFLD studies are also applicable to MASLD (3).
MASLD is defined by the accumulation of lipid droplets in more than 5% of hepatocytes, along with at least one cardiometabolic risk factor, such as obesity, diabetes, dyslipidemia, or hypertension, while ruling other causes of fatty liver disease. Additionally, affected individuals are generally non-drinkers, with alcohol intake below 20 g/day for women and 30 g/day for men (4). MASLD encompasses a range of liver disease states, ranging from simple hepatic steatosis to more severe forms involving hepatic inflammation and hepatocyte ballooning, referred to as metabolic dysfunction-associated steatohepatitis (MASH). It can progress to varying degrees of fibrosis, potentially culminating in cirrhosis and significantly increasing potentially culminating in cirrhosis and significantly increasing the risk of hepatocellular carcinoma (5).
CD47 is a transmembrane protein of the immunoglobulin superfamily that is almost universally expressed on the surface of human cells. It has a crucial role in regulating cellular processes such as renewal, adhesion, apoptosis, and phagocytosis. With a molecular weight of approximately 50 kDa, CD47 belongs to the immunoglobulin superfamily and exists in four isoforms. Its molecular structure includes an N-terminal extracellular immunoglobulin variable (IgV) domain, five transmembrane domains, and a C-terminal cytoplasmic tail that undergoes alternative splicing (6). In 1987, this protein was initially identified as a complex of Rh antigens on red blood cells and was later found to copurify with integrin αVβ3. Molecular cloning subsequently revealed that the protein is identical to the cancer antigen OV-3, leading to its alternative names, integrin-associated protein (IAP) and ovarian tumor marker (OA3) (7). In 2000, CD47 was identified as a marker that enables red blood cells to evade phagocytosis by binding to the signal regulatory protein alpha (SIRPα) on phagocytes. This interaction transmits a “don’t eat me” signal, preventing the clearance of red blood cells by the immune system (8). This role has been extensively studied in the context of cancer, However, in recent years, increasing research has shown that CD47 is closely associated with metabolic diseases such as diabetes, coronary atherosclerosis, and MASLD (9, 10). Given the critical role of metabolic dysfunction in MASLD pathogenesis, we conducted this review to offer an in-depth analysis and synthesis of the available literature. The following sections will review the role of CD47 in MASLD, with particular emphasis on the mechanisms underlying disturbances in glucose and lipid metabolism.
2 The roles of CD47
CD47 is a ubiquitously expressed integral membrane protein that functions as a high-affinity receptor for secreted matricellular protein thrombospondin-1 (TSP1), while serve as a ligand for the inhibitory phagocyte receptor signal-regulatory protein-α (SIRPα). In addition, other molecules such as SIRPγ and integrins have also been implicated in CD47 interactions. Although SIRPγ binds CD47 with lower affinity and lacks classical signaling motifs, it can promote T cell adhesion and activation (11–13). CD47 also interacts in cis with integrins such as β1 and β3, influencing cell adhesion and migration through indirect regulation rather than classical ligand-receptor signaling (6). Therefore, the dual roles of CD47 exerts various cellular processes such as proliferation, angiogenesis, migration, apoptosis, differentiation, stress responses, and metabolism.
2.1 Signal inhibitory receptor protein- alpha
Signal regulatory protein alpha (SIRPα), the receptor for CD47, shares structural similarities with CD47. It consists of three extracellular immunoglobulin-like domains, a transmembrane domain, and a C-terminal intracellular domain, which includes two immunoreceptor tyrosine-based inhibitory motifs (ITIMs) (14). The single IgV domain of CD47 binds to the N-terminal domain of SIRPα, leading to tyrosine phosphorylation of the ITIMs on SIRPα. This phosphorylation then recruits and activates protein tyrosine phosphatases, inhibiting macrophage phagocytic activity. As a result, a “don’t eat me” signal is transmitted, preventing the cell from being engulfed and eliminated by phagocytes (15).
2.2 Thrombospondin-1
Thrombospondin-1 (TSP1) is an endogenous ligand of CD47 and a homotrimeric glycoprotein with multiple functional domains (16). With a molecular weight of approximately 420–450 kDa, each subunit of TSP1 includes an N-terminal heparin-binding domain, a central repeat region, and a C-terminal (COOH) domain that promotes cell adhesion (17). TSP1 is a major component of the α-granules released by platelets upon activation (18). In addition to its presence in platelets, TSP1 is expressed at low levels in nearly all human tissues, including the liver (19). Due to its multiple specialized domains, TSP1 can bind to various cell receptors and perform diverse functions (19). The interaction between the C-terminal domain of TSP1 and CD47 has a critical role in numerous pathways, including cell renewal, inflammatory responses, redox regulation, adipocyte function, and vascular endothelial function (16). This interaction is essential in both physiological and pathological contexts.
3 CD47 in MASLD
The primary characteristic of MASLD is the accumulation of triglycerides (TG) in the liver. While the mechanisms underlying the onset and progression of MASLD are not fully understood, the most widely accepted model to explain the development of MASLD and the progression from simple steatosis to MASH is the “two-hit” hypothesis, which suggests that the first hit is an imbalance in fatty acid metabolism, eventually causing hepatic TG accumulation. The “second hit” leads to inflammation, hepatocyte injury, and fibrosis. Factors initiating the second hit are proinflammatory cytokines, oxidative stress and subsequent lipid peroxidation, adipokines, and mitochondrial dysfunction (20). However, subsequent research has revealed that the pathogenesis of MASLD is far more complex than the two-hit hypothesis suggests. Studies have found that factors, including oxidative stress, immunometabolism, disorders of glucose and lipid metabolism, intrahepatic cellular interactions, alterations in gut microbiota, and genetic susceptibility collectively contribute to the development and progression ofMASLD/MASH (21). Accumulating evidence indicates that metabolic disorders, including obesity, diabetes, insulin resistance, dyslipidemia, metabolic syndrome, and hyperuricemia, play critical roles in the development and progression of MASLD/MASH. Disturbances in glucose and lipid metabolism affect mitochondrial function, hepatic sinusoidal homeostasis, oxidative stress, and cellular senescence, with CD47 playing a crucial role in these processes. This review will primarily focus on glucose and lipid metabolism and provide an overview of the role of CD47 in the associated mechanisms. Table 1 summarizes selected representative studies.
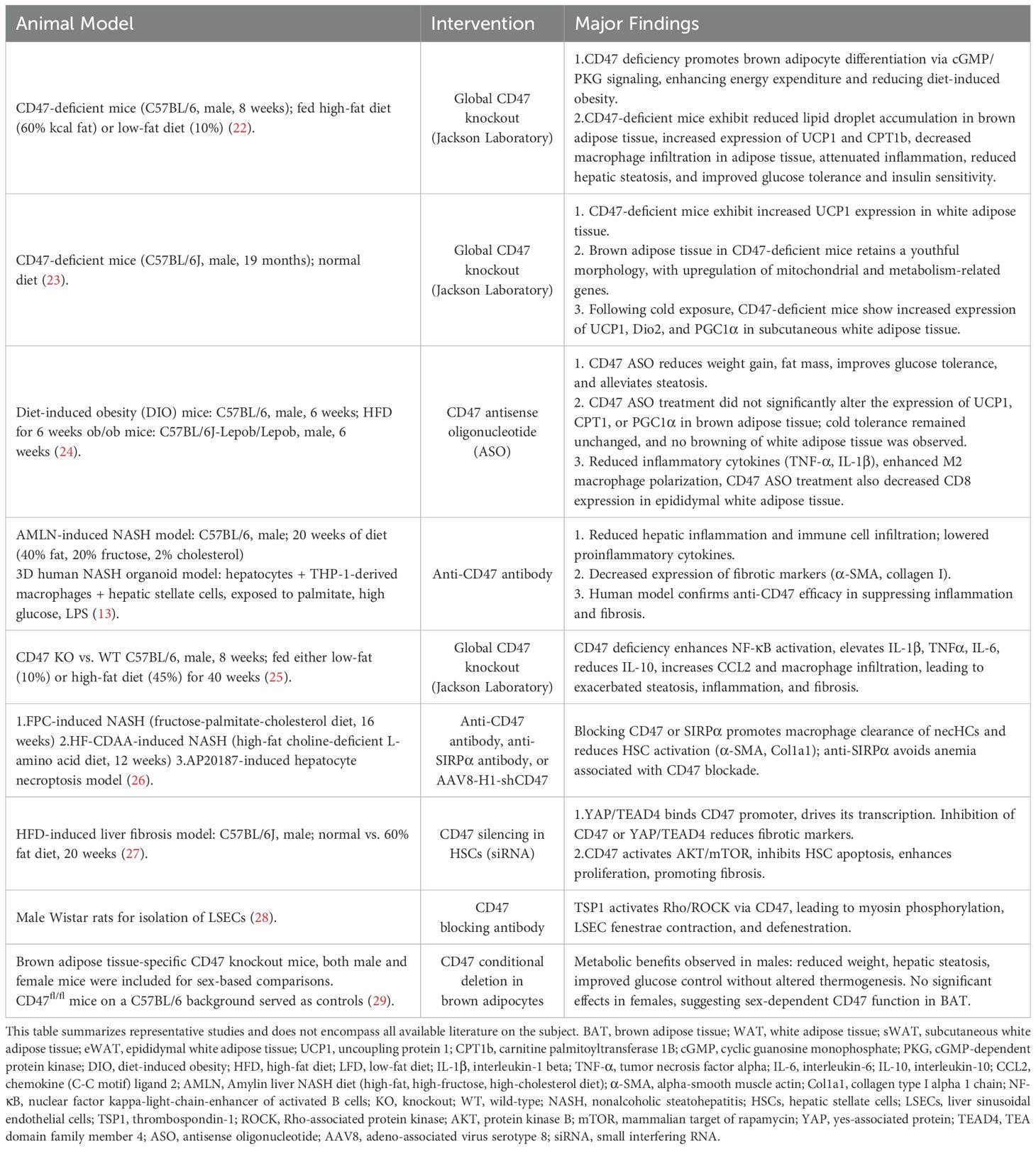
Table 1. Preclinical evidence of CD47 function in MASLD: insights from animal and organoid models.
3.1 Disorder of glycolipid metabolism
MASLD is closely associated with metabolic dysfunction and is considered a manifestation of insulin resistance in the liver. Up to 95% of obese patients and 75% of diabetic patients may suffer from MASLD (30). The key feature of MASLD is the accumulation of triglycerides (TG) in the liver. The sources of TG include dietary TG, TG synthesized from free fatty acids (FFA) through de novo lipogenesis (DNL) in the liver, and TG formed from FFA released by adipose tissue breakdown and transported to the liver. TG can be transported out of the liver as very low-density lipoprotein (VLDL) or metabolized via β-oxidation. In the context of insulin resistance, increased breakdown of white adipose tissue (WAT) leads to substantial release of FFA, which activates the DNL pathway. At the same time, impaired β-oxidation, decreased adiponectin secretion, and leptin resistance contribute to excessive accumulation of TG in the liver (31, 32). TSP1 has been shown to play an important role in IR (33), and CD47, as its ligand, has gained increasing attention for its involvement in IR (Figure 1). CD47 deficiency protects mice from glucose intolerance and insulin resistance by diet and aging (22, 23). CD47 antisense oligonucleotide (ASO) treatment in two obesity mouse models (diet-induced obesity or genetically obese models) improved glucose homeostasis and hepatic steatosis (24). Moreover, recent research (34) has revealed that CD47 can inhibit insulin secretion through a new mechanism, by suppressing the activation of cell division cycle 42(Cdc42), a small GTPase in the Rho family involved in actin remodeling and vesicle exocytosis. Inhibiting CD47 expression enhances β-cell function by increasing pancreatic islet size and β-cell proliferation through the upregulation of c-myc expression, thereby maintaining glucose homeostasis and insulin sensitivity. These findings highlight the multifaceted role of CD47 in modulating insulin secretion, glucose homeostasis, and hepatic lipid metabolism.
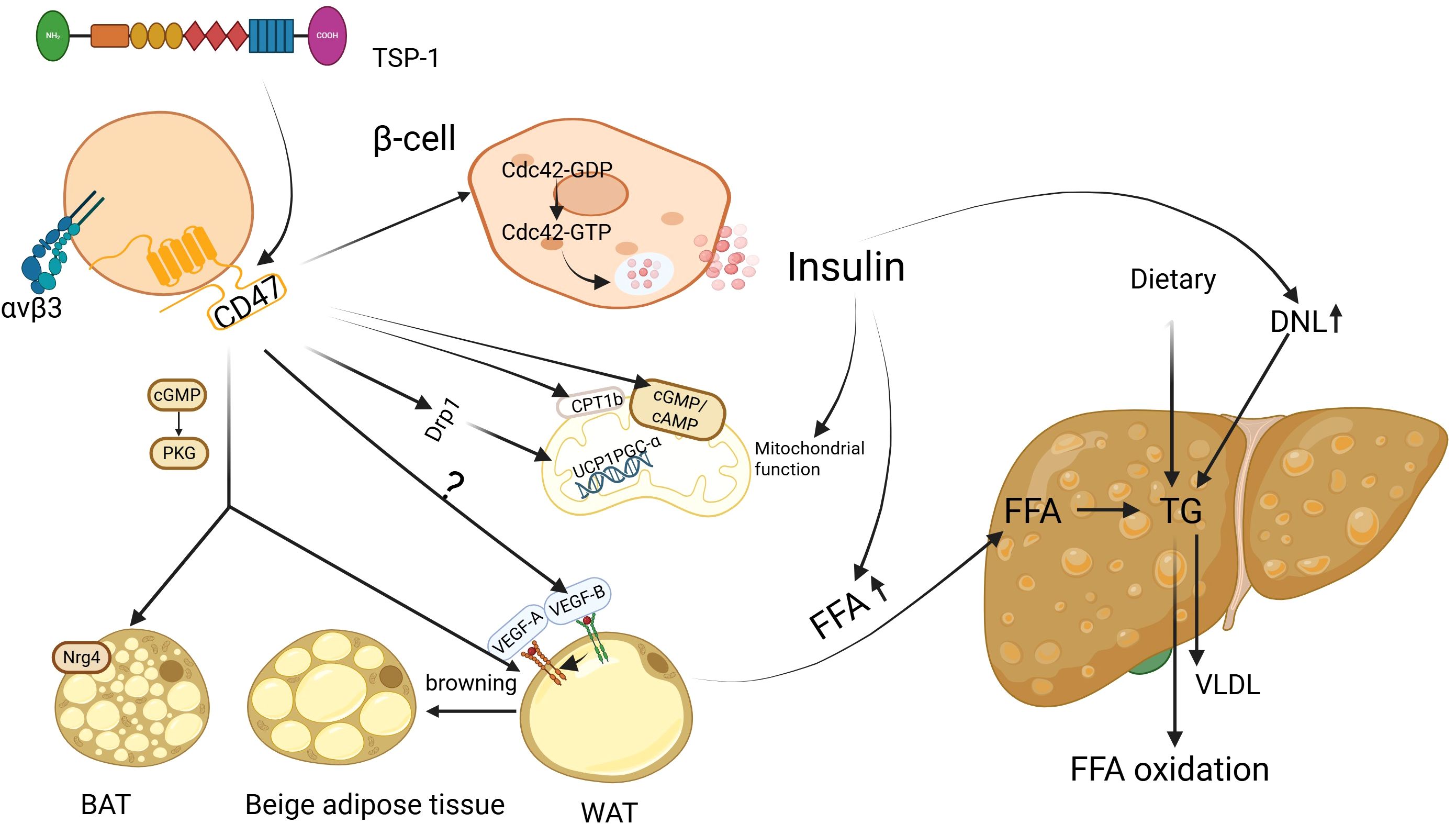
Figure 1. CD47 regulation of glucose and lipid metabolism in MASLD. Insulin resistance (IR) is recognized as a critical factor in the development of metabolic dysfunction-associated steatotic liver disease (MASLD). In the presence of IR, fat accumulation and mitochondrial dysfunction are observed. Suppressing CD47 expression has the potential to improve both lipid and mitochondrial function via multiple mechanisms, thereby reducing hepatic lipid accumulation. Additionally, this inhibition may help mitigate insulin resistance and the cascade of metabolic disturbances associated with it. BAT, brown adipose tissue; WAT, white adipose tissue; FFA, free fatty acids; TG, triglycerides; DNL, de novo lipogenesis; VLDL, very-low-density lipoprotein; Cdc42, cell division cycle 42; GDP, guanosine diphosphate; GTP, guanosine triphosphate; cGMP, cyclic guanosine monophosphate; AMP, cyclic adenosine mono-phosphate; PKG, protein kinase G; VEGF-A, vascular endothelial growth factor-A; VEGF-B, vascular endothelial growth factor-B; CPT1b, carnitine palmitoyl l transferase 1B; UCP1, Uncoupling protein 1;PGC-α, PGC-1 alpha; Drp1, Dynamin-related protein 1; Nrg4, neuregulin 4; Image created using the www.biorender.com.
The human body primarily contains three types of adipose tissue: WAT, brown adipose tissue (BAT), and beige adipose tissue. WAT, particularly visceral adipose tissue, functions as an endocrine organ, influencing liver metabolism by secretion of regulatory factors such as adiponectin, leptin, and interleukin -6(IL-6), On the other hand, BAT and beige adipose tissue generate heat through uncoupled oxidative phosphorylation. Increased BAT activity can potentially improve metabolic disturbances and reduce hepatic steatosis (35). Under certain conditions, WAT can convert into beige adipose tissue, a process known as browning. Thus, the balance between WAT and BAT function plays a crucial role in regulating liver lipid homeostasis and MASLD progression.
Under conditions of IR, the levels of FFA increase, leading to heightened energy expenditure by brown adipocytes in an attempt to lipid metabolic balance. However, when FFA levels continue to rise and the oxidative capacity of brown fat cells fails to match it, excessive lipid accumulation occurs (36). Also, adipose tissue dysfunction is considered a key mechanism in developing MASLD. For example, studies have shown that patients with MASLD exhibit reduced BAT activity, and the degree of hepatic lipid accumulation is influenced by BAT activity (35). This is thought to be primarily related to the thermogenic capacity of BAT and its secretion of regulatory factors, such as neuregulin 4 (Nrg4), which exerts its effects by activating epidermal growth factor receptor 3 (ErbB3) and epidermal growth factor receptor (ErbB4) signaling pathways in hepatocytes, helping counteract diet-induced hepatic steatosis (37). These observations suggest that targeting CD47 may improve systemic metabolism by enhancing BAT function and WAT browning.
During adipocyte browning, mitochondrial biogenesis increases alongside upregulation of uncoupling protein 1 (UCP1). Cold exposure or norepinephrine can induce PGC-1 alpha (PGC-1α), expression via activation of the β3-adrenergic receptor and cAMP/PKA pathway. Similarly, AMPK activation in response to energy stress, physical exercise, or pharmacological agents also promotes this process (38–40). CD47 is significantly upregulated in the brown adipose tissue of mice with high-fat diet-induced obesity. CD47 deficiency enhances energy expenditure by promoting brown adipocyte differentiation through upregulation of the cGMP/PKG signaling pathway, thereby alleviating obesity induced by a high-fat diet in these mice (22). In aged CD47-deficient mice, although there was no significant change in fat mass compared to wild-type mice, smaller epididymal white adipocytes were observed. In addition, there was an enhancement in the expression of (UCP1, type 2 deiodinase (Dio2), and PGC-1α, indicating enhanced browning of WAT. This adjustment protected the mice from obesity and glucose intolerance associated with aging (23). Notably, these effects displayed sexual dimorphism, with male mice showing more pronounced improvements in body weight regulation and lipid metabolism compared to females (29). Thus, CD47 deficiency appears to improve systemic metabolism by promoting BAT activation and WAT browning, offering protection against diet- and age-related metabolic disorders. Functionally, CD47 signaling intersects with β-adrenergic and AMPK pathways, converging on PGC-1α as a common downstream target. These findings suggest that CD47 may act as a regulatory node within classical browning networks, contributing to adipose tissue remodeling and the maintenance of energy balance.
Additionally, a recent study demonstrated that inhibiting vascular endothelial growth factor-B (VEGF-B) signaling can prevent the development of MASLD by blocking lipolysis in WAT (41). VEGF-B, a member of the VEGF family, is not only involved in angiogenesis but is also thought to be a crucial regulator of lipid metabolic disorders and glucose dysregulation (42–44). The expression level of VEGF-B is significantly elevated in patients with MASLD compared to those without the disease (45). VEGF-B exerts a lipid-lowering effect by binding to vascular endothelial growth factor receptor 1 (VEGFR1), activating adenosine monophosphate-activated protein kinase (AMPK), and partially through the indirect activation of the VEGF-A/VEGFR2 pathway (46, 47). It is well established that the binding of TSP1 to CD47 inhibits VEGFR2 activation and its downstream signaling, thereby suppressing nitric oxide (NO) production and cyclic guanosine monophosphate (cGMP) signal transduction (48). Both CD47 and VEGF-B regulate lipid metabolism by modulating lipolysis in white adipose tissue, with a partial overlap in their signaling pathways. However, whether CD47 can influence lipid metabolism via VEGF-B remains to be further explored. Also, a recent study (49) found that the Nrg4 expression level was remarkably elevated in brown fat of TSP1 knockout mice, The absence of Nrg4 has been shown to accelerate liver damage, fibrosis, inflammation, and cell death in a NASH mouse model. These findings suggest that the TSP1-CD47 axis may serve as a key regulator of adipose-liver crosstalk, influencing both lipid metabolism and hepatic steatosis in MASLD.
3.2 Mitochondrial dysfunction
Mitochondria are well-known as the primary sites of aerobic respiration and are fundamental to cellular energy metabolism and apoptosis. The liver, which contains an abundance of mitochondria, relies on these organelles to metabolize energy efficiently. When the body ingests excess lipids, the enhanced capacity of the mitochondrial tricarboxylic acid (TCA) cycle and β-oxidation help prevent the progression from obesity to MASLD (50). However, as hepatic steatosis progresses, the persistent excess of free fatty acids (FFAs) leads to mitochondrial dysfunction, characterized by impaired β-oxidation, reduced respiratory chain activity, alterations in mitochondrial morphology and membrane permeability, and compromised mitophagy (51, 52). These changes result in increased reactive oxygen species (ROS) production, contributing to lipid accumulation, inflammation, necrosis, and fibrosis in the liver, which in turn leads to a vicious loop of MASLD and mitochondrial dysfunction. Current evidence suggests that the signaling strength of the TSP1-CD47 pathway is inversely correlated with mitochondrial quantity and function within cells (53). The TSP1-CD47 pathway downregulates cGMP and cyclic adenosine monophosphate (cAMP), affecting mitochondrial biogenesis. Frazier et al. found that the expression levels of mitochondrial-related genes, such as PGC-1α, cytochrome b and c(cytb/c), and nuclear respiratory factor 1(NRF-1), are significantly elevated in the skeletal muscle of the CD47-null C57Bl/6J mouse model (54). This suggests that CD47 plays a crucial role in regulating mitochondrial function, and its absence may promote mitochondrial biogenesis, potentially offering therapeutic avenues for MASLD.
CD47 may also lead to the loss of mitochondrial membrane potential, increased ROS production, and promotion of cell death (55–57). Dynamin-related protein 1 (Drp1) has been shown to regulate mitochondrial-dependent cell death signaling pathways, and the binding of CD47 with TSP1 can promote Drp1 translocation from the cytoplasm to the mitochondria (13, 58). Another study (22) found an increased rate of mitochondrial uncoupling by upregulating the mRNA levels of UCP1 and carnitine palmitoyltransferase 1B (CPT1b) in BAT of CD47-null mice compared to wild-type mice. This enhanced mitochondrial uncoupling improved diet-induced hepatic steatosis. In summary, CD47 regulates mitochondrial dynamics and function, and its deficiency appears to have protective effects on mitochondrial activity, potentially mitigating liver steatosis.
3.3 Hepatic sinusoidal homeostasis
Liver sinusoidal endothelial cells (LSECs), hepatic stellate cells (HSCs), and hepatocyte macrophages, primarily Kupffer cells (KCs), are key components of the liver sinusoids. These cells work in concert to maintain the integrity and function of the unique hepatic microcirculatory system. In conditions of IR or lipid metabolism disorders, the accumulation of lipids and their metabolites in the liver disrupts the hepatic sinusoidal microenvironment, leading to damage and dysfunction of liver sinusoidal endothelial cells. This disruption of hepatic sinusoidal homeostasis further promotes hepatic fat deposition, thereby creating a vicious cycle that plays critical roles in the pathogenesis of MASLD and MASH (Figure 2).
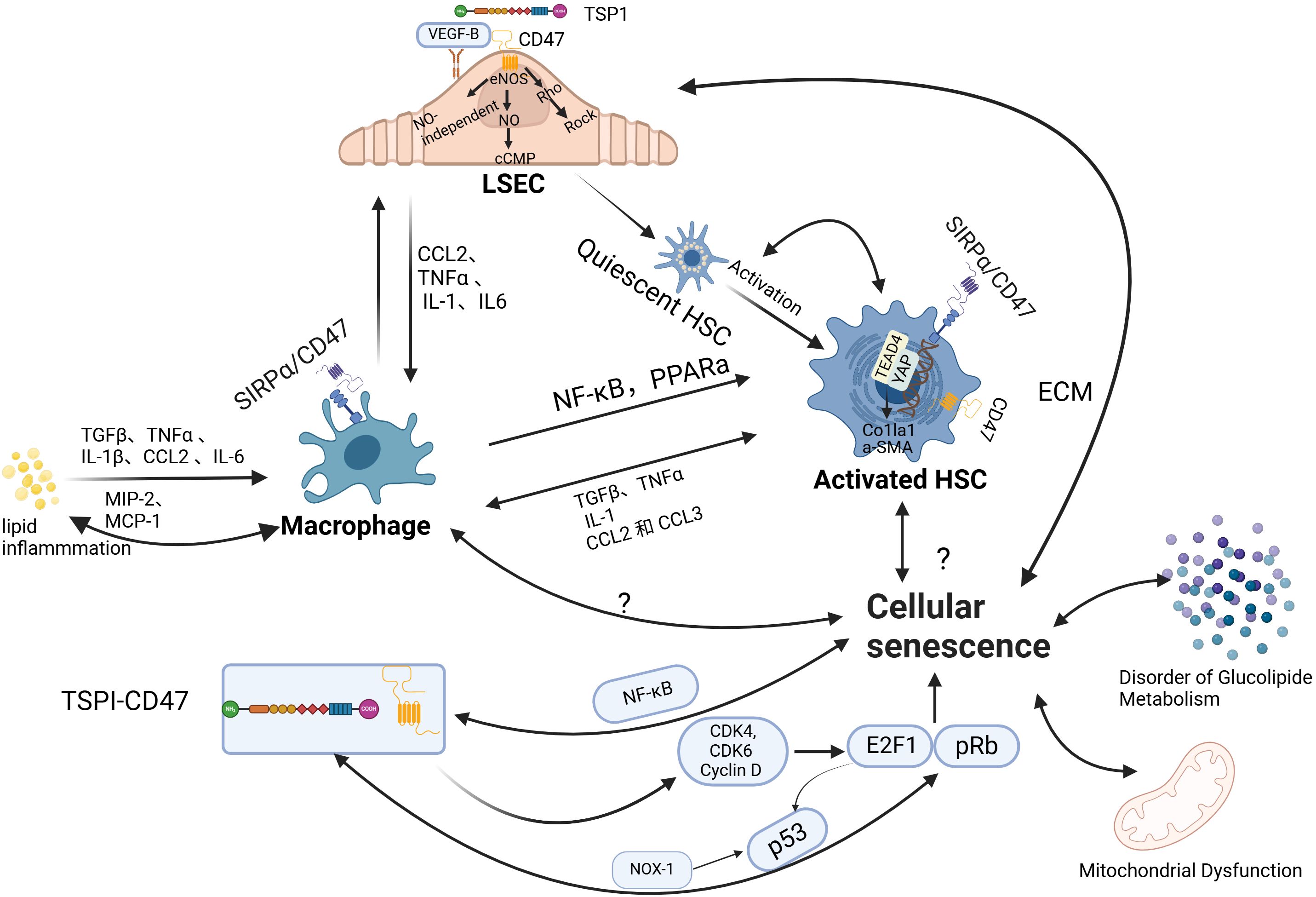
Figure 2. The role of CD47 in regulating hepatic sinusoidal function and cellular senescence. (1) liver sinusoidal endothelial cells (LSECs) play a pivotal role in maintaining sinusoidal homeostasis, supporting the normal function of hepatic stellate cells (HSCs) and macrophages. Under physiological conditions, LSECs exhibit anti-inflammatory and anti-fibrotic properties. However, phenotypic alterations in LSECs can lead to HSC activation and exacerbate macrophage infiltration. The maintenance of LSEC phenotype stability is dependent on nitric oxide (NO). The binding of TSP1 to CD47 inhibits vascular endothelial growth factor receptor 2 (VEGFR2) activation and its downstream signaling, reducing NO production. This, in turn, induces LSEC defenestration through myosin activation via the Rho-ROCK pathway. Additionally, studies suggest that targeting CD47 can modulate macrophage and HSC function via pathways such as Signal regulatory protein alpha (SIRPα) and YAP/TEAD4, thereby reducing hepatic inflammatory infiltration and fibrosis. (2) Aging contributes to the progression of metabolic dysfunction-associated steatotic liver disease (MASLD)/metabolic dysfunction-associated steatohepatitis (MASH) through various mechanisms, including its effects on hepatic glycolipid metabolism, mitochondrial function, and intrahepatic cellular processes. The interaction between TSP1 and CD47 influences the pRb-E2F1 and p53 pathways, thereby regulating the cell cycle. Nuclear factor-κB (NF-κB), a central regulator of inflammatory responses, is also modulated by CD47, with CD47 levels positively correlated with NF-κB expression. LSEC, liver sinusoidal endothelial cell;TSP1, thrombospondin-1; eNOS, endothelial nitric oxide synthase;NO, nitric oxide;cGMP, cyclic guanosine monophosphate;Rho, Ras homolog (GTPase);Rock, Rho-associated, coiled-coil-containing protein kinase;CCL2, C-C motif chemokine ligand 2;TNFα, tumor necrosis factor alpha;IL-1, interleukin-1;IL6, interleukin-6;NF-κB, nuclear factor-κB;PPARα, peroxisome proliferator-activated receptor alpha;HSC, hepatic stellate cell;TGFβ, transforming growth factor beta;MIP-2, macrophage inflammatory protein 2;MCP-1, monocyte chemoattractant protein 1;CCL3, C-C motif chemokine ligand 3;YAP1, Yes-associated protein 1;TEAD1, TEA domain transcription factor 1;Col1a1, collagen I chain;α-SMA, alpha-smooth muscle actin;SIRPα, signal regulatory protein alpha; ECM, extracellular matrix; CDK4, cyclin-dependent kinase 4; CDK6, cyclin-dependent kinase 6; E2F1, E2F transcription factor 1; pRb, phosphorylated Rb; NOX-1, NADPH oxidase 1; p53, tumor protein p53; Image created using the www.biorender.com.
3.3.1 Macrophage infiltration
Liver macrophages are primarily composed of resident Kupffer cells and monocyte-derived macrophages, which can be classified into pro-inflammatory M1 and anti-inflammatory M2 phenotypes. Monocyte recruitment is mainly regulated by CC chemokine receptor 2 (CCR2), while Kupffer cells are regulated by C-C motif chemokine ligand 2 (CCL2). Studies have shown that monocyte-derived macrophages exhibit more pronounced pro-inflammatory properties compared to resident Kupffer cells (59). Inflammatory cytokines can activate macrophages, leading to the increased release of macrophage inflammatory protein 2 (MIP-2) and monocyte chemoattractant protein 1(MCP-1), which further exacerbate macrophage infiltration in the liver (60). This process mainly involves nuclear factor-κB (NF-κB) and peroxisome proliferator-activated receptor alpha (PPARα) (61, 62). Macrophages can be activated by fatty acids, excess cholesterol, and their metabolites (such as leptin and adiponectin), resulting in the release of tumor necrosis factor (TNF-α) and interleukin-1 (IL-1), which affect hepatocyte function and activity (63). In MASLD, a distinct subset of liver lipid-associated macrophages (LAMs) expressing TREM2 is predominantly localized within steatotic regions and closely correlates with disease severity. TREM2 plays a critical role in the clearance of apoptotic cells and lipid metabolism regulation, and its deficiency exacerbates hepatic inflammation. Bariatric surgery has been shown to improve MASH progression by enhancing the reparative functions of TREM2+ macrophages (64). In addition to macrophages, adaptive immune cells such as CD8+ T cells and Th1/Th17-polarized CD4+ T cells contribute to liver inflammation by secreting IFN-γ and TNF-α. B cells also promote disease progression via antibody production and pro-inflammatory cytokine release. Moreover, natural killer (NK) cells and neutrophils participate in hepatocyte injury and inflammatory responses (65–67).
CD47 deficiency has been associated with decreased levels of the pro-inflammatory cytokines TNF-α and interleukin-6(IL-6), and higher levels of the anti-inflammatory cytokine interleukin-10 (IL-10) in mice fed with a high-fat diet. This reduction in inflammation correlates with lower levels of MCP-1 and CCR2, leading to decreased macrophage infiltration in adipose tissue (22). Similar findings have been corroborated by in vitro studies (22) Moreover, anti-CD47 therapy has been demonstrated to inhibit MCP-1 expression and secretion, resulting in diminished liver infiltration of monocytes/macrophages. Additionally, this therapy reduces hepatic stellate cell activation via transforming growth factor-beta (TGF-β) signaling, thereby alleviating hepatic inflammation and fibrosis (13). A recent study (58) reported that the expression levels of CD47 on necrotic hepatic apoptotic cells (necHCs) and SIRPα on liver macrophages are elevated, impairing the macrophages’ ability to clear necHCs. This impairment contributes to the exacerbation of liver fibrosis and inflammatory infiltration. Blocking the CD47-SIRPα axis has been demonstrated to promote the phagocytic clearance of necHCs by liver macrophages, thus inhibiting the progression of liver fibrosis. Besides, Anti-CD47 therapy has been shown to reduce neutrophil infiltration in both circulation and liver tissue in NASH models, and to inhibit neutrophil migration (13). Additionally, CD47 blockade suppresses dendritic cell maturation and activation, modulates T cell responses, and promotes the differentiation of naïve T cells into regulatory T cells. In B cells, CD47 enhances maturation and activation, while CD47 expression in NK cells contributes to their recruitment and activation (68, 69). Overall, CD47 orchestrates the migration, activation, and apoptosis of various immune cell populations, working in concert with TREM2+ macrophages and other immune subsets to shape the hepatic inflammatory microenvironment and drive MASLD progression. These findings suggest that targeting CD47 has therapeutic potential for reducing liver inflammation and fibrosis by modulating immune responses and enhancing apoptotic cell clearance.
However, contradictory findings have emerged. In another study, CD47 knockout mice raised on a high-fat diet exhibited decreased expression of PPARα and sirtuin1(SIRT1)—a NAD+-dependent deacetylase that enhances PPARα activity, upregulates fatty acid oxidation (FAO), and downregulates lipogenic gene expression (70). These mice also showed increased phosphorylation, nuclear translocation of the NF-κB p65 subunit, and elevated hepatic CCL2 levels, leading to increased monocyte/macrophage infiltration (25). CD47 gene knockout and anti-CD47 antibody treatment exhibit distinct effects in liver disease models, likely due to differences in mechanisms of action and duration of intervention. Global and sustained CD47 deficiency resulting from genetic knockout may elicit compensatory responses that disrupt lipid metabolic homeostasis, potentially impairing lipid export via downregulation of apolipoproteins or undermining innate hepatic defense mechanisms against steatosis. In contrast, anti-CD47 antibody therapy primarily functions by blocking the CD47–SIRPα interaction, thereby enhancing macrophage-mediated clearance of apoptotic cells, reducing pro-inflammatory cytokine production, and inhibiting monocyte/macrophage infiltration and HSC activation. This form of therapy typically involves short-term and localized interventions, thereby avoiding long-term disturbances in lipid metabolism. This conflicting evidence underscores the complexity of CD47’s role in metabolic and inflammatory diseases, suggesting that CD47 may exert both beneficial and detrimental effects depending on the context. Further research is necessary to reconcile these findings and better understand the full spectrum of CD47’s involvement in liver pathology.
3.3.2 Hepatic stellate cells
Hepatic stellate cells (HSCs) are non-parenchymal perisinusoidal cells that remain quiescent under normal conditions. However, upon stimulation by lipotoxic metabolites, inflammation, and oxidative stress, they become activated and transform into myofibroblasts. Activated HSCs secrete procollagen, a component of the extracellular matrix (ECM), pro-inflammatory, as well as pro-fibrotic cytokines, influencing surrounding cells via paracrine or autocrine signaling to promote liver fibrosis. It is currently believed that the persistent overactivation of HSCs is a key factor in liver fibrosis and a critical step in the progression to MASH (71). CD47 is significantly upregulated in activated HSCs. Previous research (26) by Shi et al. showed that blocking the CD47-SIRPα axis could alleviate diet-induced NASH-related liver fibrosis. Recently, Li et al. reported that CD47 knockdown reduced the expression of alpha-smooth muscle actin (α-SMA) and collagen I(COL1A1)by inhibiting the AKT/mTOR signaling pathway in a high-fat diet-induced mouse model of MASLD, thus preventing HSC activation and reducing liver fibrosis, similar to previous findings. The study further suggested that this process may be mediated by the YAP/TEAD4/CD47 signaling axis (27). Yes-associated protein (YAP), a key transcriptional regulator in the Hippo pathway, plays a pivotal role in liver regeneration and fibrogenesis. Notably, upregulation and nuclear translocation of YAP have been shown to activate HSCs both in vitro and in vivo (72). One of YAP’s canonical target genes, connective tissue growth factor (CTGF), is significantly overexpressed in fibrotic liver tissue and is capable of activating HSCs while promoting the synthesis and secretion of ECM proteins. CTGF also contributes to cell proliferation, migration, and phenotypic transformation, positioning the YAP–CTGF axis as a central driver of hepatic fibrogenesis (73, 74). Importantly, extensive crosstalk exists between the YAP/TAZ and TGF-β/Smad signaling pathways under various physiological and pathological conditions. Although direct interaction between YAP and Smad3 is relatively weak, YAP can form functional complexes through the transcriptional coactivator p300 and the transcription factor TEAD4, synergistically regulating the expression of pro-fibrotic genes such as CTGF and CYR61 (75–77). This interaction may represent a critical node in the integration of TGF-β and YAP/TEAD signaling.
Collectively, these findings indicate that the AKT/mTOR and YAP/TEAD4 pathways converge in mediating CD47’s role in HSC activation, and that their interplay with TGF-β/Smad signaling amplifies fibrogenic responses. but the interactions between these pathways remain unclear. Further research is needed to elucidate the specific regulatory mechanisms of CD47 in this process.
3.3.3 Liver sinusoidal endothelial cells
LSECs are specialized endothelial cells that lack a basement membrane and possess small pores known as fenestrae. Under physiological conditions, LSECs are involved in lipid exchange between the blood and liver, keeping Kupffer cells and hepatic stellate cells inactive, modulating intrahepatic vascular resistance and portal vein pressure, all while exhibiting anti-inflammatory and anti-fibrotic properties (78). However, harmful stimuli such as FFA and microbial endotoxins can induce LSEC capillarization—a phenomenon characterized by the loss of fenestrae and the formation of a basement membrane—through the generation of reactive ROS and inflammation (79). This phenotypic change in LSECs impairs hepatic lipid uptake and metabolism, thereby promoting liver injury, inflammatory cell infiltration, and fibrosis (80).
The stability of the LSEC phenotype is closely associated with NO production. Also, LSECs can secrete NO to maintain the quiescence of hepatic stellate cells (81) VEGF is thought to be a crucial regulator of the LSEC phenotype, exerting its effects through both NO-dependent (eNOS-NO-cGMP) and NO-independent pathways (82). It is now clear that TSP1 binds to CD47 to inhibit VEGFR2 activation and downstream signaling, leading to reduced NO production and cGMP signal transduction (48). However, there is currently no direct evidence that blocking CD47 in NAFLD leads to increased NO secretion by LSECs, thereby improving fibrosis. Moreover, a study has demonstrated that the CD47-TSP1 pathway can induce LSEC defenestration through the activation of myosin by the Rho-ROCK pathway, and targeting CD47 has been shown to reduce LSEC defenestration (28). All these findings suggest that CD47 may influence the development and progression of MASLD by modulating both the function and morphology of LSECs.
3.4 Senescence
Aging is characterized by the gradual loss of physiological integrity and is a major risk factor for many diseases. Cellular senescence, considered one of the hallmarks of aging, is defined as a permanent cell cycle arrest state (83). While senescent cells lose their proliferative capacity, they gain an increased ability to secrete pro-inflammatory factors, a phenomenon known as the senescence-associated secretory phenotype (SASP), which affects surrounding cells through paracrine signaling and contributes to chronic inflammation (84). Although the causal relationship between MASLD and senescence remains unclear, it is undeniable that aging is closely linked to the occurrence and progression of MASLD/MASH. Recent research has gradually revealed that both aging and CD47 contribute to the development of MASLD/NASH by influencing glucolipid metabolic dysfunction, mitochondrial function, and hepatic sinusoidal homeostasis through several shared pathways (13, 22, 26).
Recently, a study (85) showed that aging WAT cells release increased levels of FFA, exacerbating hepatic steatosis. After treatment with senolytics (drugs that selectively eliminate senescent cells), the liver steatosis was alleviated by clearing the senescent adipocytes. Previous studies (86) have shown that CD47 deficiency can prevent aging-induced glucolipid metabolic dysfunction, thereby alleviating hepatic steatosis. The glucolipid metabolism disorders caused by senescent cells are currently thought to be related to mitochondrial metabolic dysregulation (87). As cells age, mitochondria may exhibit reduced energy production, weakened antioxidant capacity, and impaired autophagy, leading to abnormal lipid accumulation within cells (88, 89). In the aging liver, multiple cellular dysfunctions can be observed: 1) A reduction in the number of LSECs, with decreased expression of markers maintaining their normal morphology (VEGFR2, CD32b) and reduced vasodilation capacity (90). 2) Considering the heterogeneity of hepatic macrophages, the conclusions on the effect of aging on the phagocytic clearance capacity of hepatic macrophages are not uniform, and most studies now suggest that aging leads to an enhanced pro-inflammatory response in macrophages thereby leading to further deterioration of cellular function (91–93). 3) The specific role of senescent HSC on fibrosis is unclear; senescent HSC can ameliorate hepatic fibrosis, but a recent study showed that the number of senescent HSC increased in MASH and showed pro-fibrotic properties (94–96). Collectively, these findings highlight that aging-driven cellular senescence, particularly through dysregulated adipose tissue metabolism, mitochondrial dysfunction, and hepatic non-parenchymal cell impairment, plays a central role in the progression of hepatic steatosis and fibrosis.
Recent research has revealed that aging not only elevates the expression levels of TSP1 (54, 97–99) but also enhances the clustering of CD47 on the cell surface, thereby intensifying TSP1-CD47 signaling. Furthermore, several studies (9, 86, 99) have demonstrated that TSP1 can induce endothelial cell senescence via CD47. The phosphorylated Rb- E2F transcription factor 1 (pRb-E2F1) pathway is recognized as a crucial mechanism for cell cycle regulation, TSP1, acting as a downstream effector of E2F1, binds to CD47 on endothelial cells, inhibiting the activity of cyclin D1 and cyclin-dependent kinase 4/6(CDK4/6), activating the NADPH oxidase 1(Nox1) complex and playing an important role in the pRb pathway through p53-induced DNA damage response. In addition, E2F1 regulates the cell cycle by inducing p53 phosphorylation (99–102). Research (103, 104) has indicated that CD47 deficiency results in the reactivation of several stem cell markers, such as c-myc and SRY-Box transcription factor 2 (Sox2), in fully differentiated adult cells, enabling them to recapture some stem cell-like characteristics. As aging progresses, senescent cells accumulate in many tissues, but these cells are typically cleared by macrophages. When this clearance function is impaired, senescent cells accumulate, leading to a series of pathological processes. Macrophages play a critical role in reducing the number of senescent cells through phagocytosis (105). However, senescent cells increase CD47 expression, which binds to the SIRP-α molecule on macrophages, enhancing the “don’t eat me” signal. This not only inhibits macrophages from engulfing and phagocytosing these senescent cells, but also reduces their ability to clear surrounding cells (97). Additionally, NF-κB, known as the master regulator of inflammatory cytokines, has been identified as an activator of age-related transcriptional changes. Its expression is elevated in MASLD patients, and various stimuli can exacerbate liver steatosis via NF-kB signaling, Inhibition of its activity ameliorates hepatic steatosis (106–108). CD47 is positively correlated with NF-κB levels (109), and CD47 deficiency suppresses NF-κB activity, resulting in the downregulation of IL-1β, IL-6, and TNFα in the livers of aging mice (102). However, in CD47 knockout (CD47KO) mice, the phosphorylation and nuclear translocation of the p65 subunit of NF-κB were significantly enhanced, promoting the progression of MASLD and liver fibrosis (25). This mechanism parallels the upregulation of advanced glycation end-products (AGEs) with age, which accelerates triglyceride accumulation and, in turn, the development of MASLD (110). This seems to contradict previous research findings, CD47 regulates the development of MASLD/MASH through various pathways, and multiple stimuli can aggravate hepatic steatosis via the NF-kB signaling pathway. Experimental variables, including feeding duration and methods used to generate CD47-deficient mouse models, can impact these outcomes. In summary, accumulating evidence suggests that aging is an emerging risk factor for MASLD, and further research is needed to elucidate the specific mechanisms by which CD47 influences this process. Collectively, these findings suggest that CD47, by integrating signals from TSP1, NF-κB, and immune evasion pathways, plays a complex and context-dependent role in aging-related hepatic steatosis and fibrosis. Further clarification of these mechanisms will help resolve existing contradictions and guide targeted interventions.
4 Conclusions
The increasing prevalence of Metabolic dysfunction-associated steatotic liver disease (MASLD) will lead to a substantial global disease burden and public health costs, and its pathogenesis remains unclear. In this review, we summarize the regulatory role of CD47 in various pathogenic mechanisms of MASLD/MASH, with a focus on glucose and lipid metabolism. Although significant progress has been made in understanding the role of CD47 in metabolic dysfunction-associated steatotic liver disease (MASLD), key questions remain. The specific functions of CD47 in hepatocytes, liver sinusoidal endothelial cells, and hepatic stellate cells require further clarification, particularly regarding its context-dependent effects on inflammation and fibrosis. Its interactions with metabolic pathways—such as PI3K/AKT/mTOR, cGMP/PKG, NF-κB, and YAP/TEAD4—may uncover synergistic targets but remain insufficiently explored. Evidence also suggests a sex-dependent role of CD47 in lipid metabolism and aging-related disorders, highlighting the importance of sex-stratified research. To date, most findings linking CD47 to MASLD come from animal or in vitro studies, with limited validation in human tissues. While anti-CD47 antibodies show promise in oncology, their metabolic effects are unclear and raise safety concerns. Given CD47’s ubiquitous expression on erythrocytes, its blockade may disrupt “self” recognition and lead to hemolytic anemia—a major adverse event in clinical trials. Addressing these issues may help advance our understanding of the link between CD47 and MASLD and promote its clinical application in metabolic diseases.
Author contributions
XJ: Writing – original draft. WZ: Writing – original draft. BS: Writing – original draft. YH: Writing – original draft. KC: Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by grants from Jilin Provincial Department of Finance (JLSWSRCZX2023-101).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Wong VW-S, Ekstedt M, Wong GL-H, and Hagström H. Changing epidemiology, global trends and implications for outcomes of nafld. J Hepatol. (2023) 79:842–52. doi: 10.1016/j.jhep.2023.04.036
2. Rinella ME, Lazarus JV, Ratziu V, Francque SM, Sanyal AJ, Kanwal F, et al. A multisociety delphi consensus statement on new fatty liver disease nomenclature. J Hepatol. (2023) 79:1542–56. doi: 10.1016/j.jhep.2023.06.003
3. Younossi ZM, Paik JM, Stepanova M, Ong J, Alqahtani S, and Henry L. Clinical profiles and mortality rates are similar for metabolic dysfunction-associated steatotic liver disease and non-alcoholic fatty liver disease. J Hepatol. (2024) 80:694–701. doi: 10.1016/j.jhep.2024.01.014
4. Eslam M, Newsome PN, Sarin SK, Anstee QM, Targher G, Romero-Gomez M, et al. A new definition for metabolic dysfunction-associated fatty liver disease: an international expert consensus statement. J Hepatol. (2020) 73:202–9. doi: 10.1016/j.jhep.2020.03.039
5. Powell EE, Wong VW-S, and Rinella M. Non-alcoholic fatty liver disease. Lancet. (2021) 397:2212–24. doi: 10.1016/s0140-6736(20)32511-3
6. Brown EJ and Frazier WA. Integrin-associated protein (Cd47) and its ligands. Trends Cell Biol. (2001) 11:130–5. doi: 10.1016/s0962-8924(00)01906-1
7. Mawby WJ, Holmes CH, Anstee DJ, Spring FA, and Tanner MJ. Isolation and characterization of cd47 glycoprotein: A multispanning membrane protein which is the same as integrin-associated protein (Iap) and the ovarian tumour marker oa3. Biochem J. (1994) 304:525–30. doi: 10.1042/bj3040525
8. Oldenborg PA, Gresham HD, and Lindberg FP. Cd47-signal regulatory protein alpha (Sirpα) regulates fcγ and complement receptor-mediated phagocytosis. J Exp Med. (2001) 193:855–62. doi: 10.1084/jem.193.7.855
9. Bitar M. Diabetes impairs angiogenesis and induces endothelial cell senescence by up-regulating thrombospondin-cd47-dependent signaling. Int J Mol Sci. (2019) 20:673. doi: 10.3390/ijms20030673
10. Kojima Y, Volkmer JP, McKenna K, Civelek M, Lusis AJ, Miller CL, et al. Cd47 blocking antibodies restore phagocytosis and prevent atherosclerosis. Nature. (2016) 536:86–90. doi: 10.1038/nature18935
11. Stefanidakis M, Newton G, Lee WY, Parkos CA, and Luscinskas FW. Endothelial cd47 interaction with sirpgamma is required for human T-cell transendothelial migration under shear flow conditions in vitro. Blood. (2008) 112:1280–9. doi: 10.1182/blood-2008-01-134429
12. Brooke G, Holbrook JD, Brown MH, and Barclay AN. Human lymphocytes interact directly with cd47 through a novel member of the signal regulatory protein (Sirp) family. J Immunol. (2004) 173:2562–70. doi: 10.4049/jimmunol.173.4.2562
13. Gwag T, Ma E, Zhou C, and Wang S. Anti-cd47 antibody treatment attenuates liver inflammation and fibrosis in experimental non-alcoholic steatohepatitis models. Liver Int. (2022) 42:829–41. doi: 10.1111/liv.15182
14. Barclay AN. Signal regulatory protein alpha (Sirpalpha)/cd47 interaction and function. Curr Opin Immunol. (2009) 21:47–52. doi: 10.1016/j.coi.2009.01.008
15. Barclay AN and van den Berg TK. The interaction between signal regulatory protein alpha (Sirpα) and cd47: structure, function, and therapeutic target. Annu Rev Immunol. (2014) 32:25–50. doi: 10.1146/annurev-immunol-032713-120142
16. Kale A, Rogers NM, and Ghimire K. Thrombospondin-1 cd47 signalling: from mechanisms to medicine. Int J Mol Sci. (2021) 22:4062. doi: 10.3390/ijms22084062
17. Roberts DD. Thrombospondins: from structure to therapeutics. Cell Mol Life Sci. (2008) 65:669–71. doi: 10.1007/s00018-007-7483-2
18. Gwag T, Lee S, Li Z, Newcomb A, Otuagomah J, Weinman SA, et al. Platelet-derived thrombospondin 1 promotes immune cell liver infiltration and exacerbates diet-induced steatohepatitis. JHEP Rep. (2024) 6:101019. doi: 10.1016/j.jhepr.2024.101019
19. Stenina-Adognravi O. Invoking the power of thrombospondins: regulation of thrombospondins expression. Matrix Biol. (2014) 37:69–82. doi: 10.1016/j.matbio.2014.02.001
20. Day CP and James OF. Steatohepatitis: A tale of two “Hits”? Gastroenterology. (1998) 114:842–5. doi: 10.1016/s0016-5085(98)70599-2
21. Buzzetti E, Pinzani M, and Tsochatzis EA. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (Nafld). Metabolism. (2016) 65:1038–48. doi: 10.1016/j.metabol.2015.12.012
22. Maimaitiyiming H, Norman H, Zhou Q, and Wang S. Cd47 deficiency protects mice from diet-Induced obesity and improves whole body glucose tolerance and insulin sensitivity. Sci RepInt J Mol Sci. (2015) 5:8846. doi: 10.1038/srep08846
23. Li D, Gwag T, and Wang S. Absence of cd47 maintains brown fat thermogenic capacity and protects mice from aging-related obesity and metabolic disorder. Biochem Biophys Res Commun. (2021) 575:14–9. doi: 10.1016/j.bbrc.2021.08.062
24. Gwag T, Li D, Ma E, Guo Z, Liang Y, and Wang S. Cd47 antisense oligonucleotide treatment attenuates obesity and its-associated metabolic dysfunction. Sci Rep. (2023) 13:2748. doi: 10.1038/s41598-023-30006-2
25. Tao H-C, Chen K-X, Wang X, Chen B, Zhao W-O, Zheng Y, et al. Cd47 deficiency in mice exacerbates chronic fatty diet-induced steatohepatitis through its role in regulating hepatic inflammation and lipid metabolism. Front Immunol. (2020) 11:148. doi: 10.3389/fimmu.2020.00148
26. Shi H, Wang X, Li F, Gerlach BD, Yurdagul A, Moore MP, et al. Cd47-sirpα Axis blockade in nash promotes necroptotic hepatocyte clearance by liver macrophages and decreases hepatic fibrosis. Sci Transl Med. (2022) 14:eabp8309. doi: 10.1126/scitranslmed.abp8309
27. Li Y, Dong L, Yin X, Wang X, Zhu X, Zheng P, et al. Cd47, a novel yap target gene, contributes to hepatic stellate cell activation and liver fibrosis induced by high-fat diet. Heliyon. (2024) 10(10):e31621. doi: 10.1016/j.heliyon.2024.e31621
28. Venkatraman L and Tucker-Kellogg L. The cd47-binding peptide of thrombospondin-1 induces defenestration of liver sinusoidal endothelial cells. Liver Int. (2013) 33:1386–97. doi: 10.1111/liv.12231
29. Li D, Gwag T, and Wang S. Sex differences in the effects of brown adipocyte cd47 deficiency on age-related weight change and glucose homeostasis. Biochem Biophys Res Commun. (2023) 676:78–83. doi: 10.1016/j.bbrc.2023.07.042
30. Byrne CD, Olufadi R, Bruce KD, Cagampang FR, and Ahmed MH. Metabolic disturbances in non-alcoholic fatty liver disease. Clin Sci (Lond). (2009) 116:539–64. doi: 10.1042/cs20080253
31. Smith GI, Shankaran M, Yoshino M, Schweitzer GG, Chondronikola M, Beals JW, et al. Insulin resistance drives hepatic de novo lipogenesis in nonalcoholic fatty liver disease. J Clin Invest. (2020) 130:1453–60. doi: 10.1172/jci134165
32. Bo T, Gao L, Yao Z, Shao S, Wang X, Proud CG, et al. Hepatic selective insulin resistance at the intersection of insulin signaling and metabolic dysfunction-associated steatotic liver disease. Cell Metab. (2024) 36:947–68. doi: 10.1016/j.cmet.2024.04.006
33. Matsuo Y, Tanaka M, Yamakage H, Sasaki Y, Muranaka K, Hata H, et al. Thrombospondin 1 as a novel biological marker of obesity and metabolic syndrome. Metabolism. (2015) 64:1490–9. doi: 10.1016/j.metabol.2015.07.016
34. Ghimire K, Kale A, Li J, Julovi SM, O’Connell P, Grey ST, et al. A metabolic role for cd47 in pancreatic B Cell insulin secretion and islet transplant outcomes. Sci Transl Med. (2023) 55:eadd2387. doi: 10.1126/scitranslmed.add2387
35. Ahmed BA, Ong FJ, Barra NG, Blondin DP, Gunn E, Oreskovich SM, et al. Lower brown adipose tissue activity is associated with non-alcoholic fatty liver disease but not changes in the gut microbiota. Cell Rep Med. (2021) 2:100397. doi: 10.1016/j.xcrm.2021.100397
36. Koliaki C, Szendroedi J, Kaul K, Jelenik T, Nowotny P, Jankowiak F, et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. (2015) 21:739–46. doi: 10.1016/j.cmet.2015.04.004
37. Wang G-X, Zhao X-Y, Meng Z-X, Kern M, Dietrich A, Chen Z, et al. The brown fat–enriched secreted factor nrg4 preserves metabolic homeostasis through attenuation of hepatic lipogenesis. Nat Med. (2014) 20:1436–43. doi: 10.1038/nm.3713
38. Wu L, Zhang L, Li B, Jiang H, Duan Y, Xie Z, et al. Amp-activated protein kinase (Ampk) regulates energy metabolism through modulating thermogenesis in adipose tissue. Front Physiol. (2018) 9:122. doi: 10.3389/fphys.2018.00122
39. MaChado SA, Pasquarelli-do-Nascimento G, da Silva DS, Farias GR, de Oliveira Santos I, Baptista LB, et al. Browning of the white adipose tissue regulation: new insights into nutritional and metabolic relevance in health and diseases. Nutr Metab (Lond). (2022) 19:61. doi: 10.1186/s12986-022-00694-0
40. Desjardins EM and Steinberg GR. Emerging role of ampk in brown and beige adipose tissue (Bat): implications for obesity, insulin resistance, and type 2 diabetes. Curr Diabetes Rep. (2018) 18:80. doi: 10.1007/s11892-018-1049-6
41. Falkevall A, Mehlem A, Folestad E, Ning FC, Osorio-Conles Ó, Radmann R, et al. Inhibition of vegf-B signaling prevents non-alcoholic fatty liver disease development by targeting lipolysis in the white adipose tissue. J Hepatol. (2023) 78:901–13. doi: 10.1016/j.jhep.2023.01.014
42. Luo X, Li R-R, Li Y-Q, Yu H-P, Yu H-N, Jiang W-G, et al. Reducing vegfb expression regulates the balance of glucose and lipid metabolism in mice via vegfr1. Mol Med Rep. (2022) 26:1–15. doi: 10.3892/mmr.2022.12801
43. Shen Y, Chen W, Han L, Bian Q, Fan J, Cao Z, et al. Vegf-B antibody and interleukin-22 fusion protein ameliorates diabetic nephropathy through inhibiting lipid accumulation and inflammatory responses. Acta Pharm Sin B. (2021) 11:127–42. doi: 10.1016/j.apsb.2020.07.002
44. Hagberg CE, Mehlem A, Falkevall A, Muhl L, Fam BC, Ortsäter H, et al. Targeting vegf-B as a novel treatment for insulin resistance and type 2 diabetes. Nature. (2012) 490:426–30. doi: 10.1038/nature11464
45. Ye X, Kong W, Zafar MI, Zeng J, Yang R, and Chen LL. Plasma vascular endothelial growth factor B is elevated in non-alcoholic fatty liver disease patients and associated with blood pressure and renal dysfunction. EXCLI J. (2020) 19:1186–95. doi: 10.17179/excli2020-2647
46. Hu L, Shan Z, Wang F, Gao X, and Tong Y. Vascular endothelial growth factor B exerts lipid-lowering effect by activating ampk via vegfr1. Life Sci. (2021) 276:119401. doi: 10.1016/j.lfs.2021.119401
47. Robciuc Marius R, Kivelä R, Williams Ian M, de Boer Jan F, van Dijk Theo H, Elamaa H, et al. Vegfb/vegfr1-induced expansion of adipose vasculature counteracts obesity and related metabolic complications. Cell Metab. (2016) 23:712–24. doi: 10.1016/j.cmet.2016.03.004
48. Kaur S, Martin-Manso G, Pendrak ML, Garfield SH, Isenberg JS, and Roberts DD. Thrombospondin-1 inhibits vegf receptor-2 signaling by disrupting its association with cd47. J Biol Chem. (2010) 285:38923–32. doi: 10.1074/jbc.M110.172304
49. Guo L, Zhang P, Chen Z, Xia H, Li S, Zhang Y, et al. Hepatic neuregulin 4 signaling defines an endocrine checkpoint for steatosis-to-nash progression. J Clin Invest. (2017) 127:4449–61. doi: 10.1172/jci96324
50. Ramanathan R, Ali AH, and Ibdah JA. Mitochondrial dysfunction plays central role in nonalcoholic fatty liver disease. Int J Mol Sci. (2022) 23:7280. doi: 10.3390/ijms23137280
51. Moore MP, Cunningham RP, Meers GM, Johnson SA, Wheeler AA, Ganga RR, et al. Compromised hepatic mitochondrial fatty acid oxidation and reduced markers of mitochondrial turnover in human nafld. Hepatology. (2022) 76:1452–65. doi: 10.1002/hep.32324
52. Pérez-Carreras M, Del Hoyo P, Martín MA, Rubio JC, Martín A, Castellano G, et al. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology. (2003) 38:999–1007. doi: 10.1002/hep.1840380426
53. Miller TW, Soto-Pantoja DR, Schwartz AL, Sipes JM, DeGraff WG, Ridnour LA, et al. Cd47 receptor globally regulates metabolic pathways that control resistance to ionizing radiation. J Biol Chem. (2015) 290:24858–74. doi: 10.1074/jbc.M115.665752
54. Frazier EP, Isenberg JS, Shiva S, Zhao L, Schlesinger P, Dimitry J, et al. Age-dependent regulation of skeletal muscle mitochondria by the thrombospondin-1 receptor cd47. Matrix Biol. (2011) 30:154–61. doi: 10.1016/j.matbio.2010.12.004
55. Manna PP, Dimitry J, Oldenborg P-A, and Frazier WA. Cd47 augments fas/cd95-mediated apoptosis. J Biol Chem. (2005) 280:29637–44. doi: 10.1074/jbc.M500922200
56. Roue G, Bitton N, Yuste VJ, Montange T, Rubio M, Dessauge F, et al. Mitochondrial dysfunction in cd47-mediated caspase-independent cell death: ros production in the absence of cytochrome C and aif release. Biochimie. (2003) 85:741–6. doi: 10.1016/s0300-9084(03)00129-9
57. Isenberg JS, Annis DS, Pendrak ML, Ptaszynska M, Frazier WA, Mosher DF, et al. Differential interactions of thrombospondin-1, -2, and -4 with cd47 and effects on cgmp signaling and ischemic injury responses. J Biol Chem. (2009) 284:1116–25. doi: 10.1074/jbc.M804860200
58. Bouwstra R, van Meerten T, and Bremer E. Cd47-sirpα Blocking-based immunotherapy: current and prospective therapeutic strategies. Clin Transl Med. (2022) 12:e943. doi: 10.1002/ctm2.943
59. Morinaga H, Mayoral R, Heinrichsdorff J, Osborn O, Franck N, Hah N, et al. Characterization of distinct subpopulations of hepatic macrophages in hfd/obese mice. Diabetes. (2015) 64:1120–30. doi: 10.2337/db14-1238
60. Zhang W and Lang R. Macrophage metabolism in nonalcoholic fatty liver disease. Front Immunol. (2023) 14:1257596. doi: 10.3389/fimmu.2023.1257596
61. Stienstra R, Saudale F, Duval C, Keshtkar S, Groener JEM, van Rooijen N, et al. Kupffer cells promote hepatic steatosis via interleukin-1β-dependent suppression of peroxisome proliferator-activated receptor α Activity. Hepatology. (2010) 51:511–22. doi: 10.1002/hep.23337
62. Locatelli I, Sutti S, Vacchiano M, Bozzola C, and Albano E. Nf-κb1 deficiency stimulates the progression of non-alcoholic steatohepatitis (Nash) in mice by promoting nkt-cell-mediated responses. Clin Sci. (2012) 124:279–87. doi: 10.1042/cs20120289
63. Kazankov K, Jørgensen SMD, Thomsen KL, Møller HJ, Vilstrup H, George J, et al. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat Rev Gastroenterol Hepatol. (2018) 16:145–59. doi: 10.1038/s41575-018-0082-x
64. Fredrickson G, Florczak K, Barrow F, Mahmud S, Dietsche K, Wang H, et al. Trem2 macrophages mediate the beneficial effects of bariatric surgery against mash. Hepatology. (2025) 81:1776–91. doi: 10.1097/HEP.0000000000001098
65. Hirsova P, Bamidele AO, Wang H, Povero D, and Revelo XS. Emerging roles of T cells in the pathogenesis of nonalcoholic steatohepatitis and hepatocellular carcinoma. Front Endocrinol (Lausanne). (2021) 12:760860. doi: 10.3389/fendo.2021.760860
66. Barrow F, Khan S, Wang H, and Revelo XS. The emerging role of B cells in the pathogenesis of nafld. Hepatology. (2021) 74:2277–86. doi: 10.1002/hep.31889
67. Barrow F, Khan S, Fredrickson G, Wang H, Dietsche K, Parthiban P, et al. Microbiota-driven activation of intrahepatic B cells aggravates nash through innate and adaptive signaling. Hepatology. (2021) 74:704–22. doi: 10.1002/hep.31755
68. Liu Y, Weng L, Wang Y, Zhang J, Wu Q, Zhao P, et al. Deciphering the role of cd47 in cancer immunotherapy. J Adv Res. (2024) 63:129–58. doi: 10.1016/j.jare.2023.10.009
69. Chen Q, Guo X, and Ma W. Opportunities and challenges of cd47-targeted therapy in cancer immunotherapy. Oncol Res. (2023) 32:49–60. doi: 10.32604/or.2023.042383
70. Wan J, Wu X, Chen H, Xia X, Song X, Chen S, et al. Aging-induced aberrant rage/pparα Axis promotes hepatic steatosis via dysfunctional mitochondrial β Oxidation. Aging Cell. (2020) 19:e13238. doi: 10.1111/acel.13238
71. Higashi T, Friedman SL, and Hoshida Y. Hepatic stellate cells as key target in liver fibrosis. Adv Drug Del Rev. (2017) 121:27–42. doi: 10.1016/j.addr.2017.05.007
72. Mannaerts I, Leite SB, Verhulst S, Claerhout S, Eysackers N, Thoen LF, et al. The hippo pathway effector yap controls mouse hepatic stellate cell activation. J Hepatol. (2015) 63:679–88. doi: 10.1016/j.jhep.2015.04.011
73. Ye L, Ziesch A, Schneider JS, Ofner A, Nieß H, Denk G, et al. The inhibition of yap signaling prevents chronic biliary fibrosis in the abcb4(-/-) model by modulation of hepatic stellate cell and bile duct epithelium cell pathophysiology. Aging Dis. (2024) 15:338–56. doi: 10.14336/ad.2023.0602
74. Ramazani Y, Knops N, Elmonem MA, Nguyen TQ, Arcolino FO, van den Heuvel L, et al. Connective tissue growth factor (Ctgf) from basics to clinics. Matrix Biol. (2018) 68-69:44–66. doi: 10.1016/j.matbio.2018.03.007
75. Fujii M, Toyoda T, Nakanishi H, Yatabe Y, Sato A, Matsudaira Y, et al. Tgf-β Synergizes with defects in the hippo pathway to stimulate human Malignant mesothelioma growth. J Exp Med. (2012) 209:479–94. doi: 10.1084/jem.20111653
76. Zhang C, Wei W, Tu S, Liang B, Li C, Li Y, et al. Upregulation of cyr61 by tgf-β and yap signaling exerts a counter-suppression of hepatocellular carcinoma. J Biol Chem. (2024) 300:107208. doi: 10.1016/j.jbc.2024.107208
77. Fujii M, Nakanishi H, Toyoda T, Tanaka I, Kondo Y, Osada H, et al. Convergent signaling in the regulation of connective tissue growth factor in Malignant mesothelioma: tgfβ Signaling and defects in the hippo signaling cascade. Cell Cycle. (2012) 11:3373–9. doi: 10.4161/cc.21397
78. Shetty S, Lalor PF, and Adams DH. Liver sinusoidal endothelial cells - gatekeepers of hepatic immunity. Nat Rev Gastroenterol Hepatol. (2018) 15:555–67. doi: 10.1038/s41575-018-0020-y
79. Gracia-Sancho J, Caparrós E, Fernández-Iglesias A, and Francés R. Role of liver sinusoidal endothelial cells in liver diseases. Nat Rev Gastroenterol Hepatol. (2021) 18:411–31. doi: 10.1038/s41575-020-00411-3
80. Herrnberger L, Hennig R, Kremer W, Hellerbrand C, Goepferich A, Kalbitzer HR, et al. Formation of fenestrae in murine liver sinusoids depends on plasmalemma vesicle-associated protein and is required for lipoprotein passage. PloS One. (2014) 9:e115005. doi: 10.1371/journal.pone.0115005
81. DeLeve LD, Wang X, and Guo Y. Sinusoidal endothelial cells prevent rat stellate cell activation and promote reversion to quiescence. Hepatology. (2008) 48:920–30. doi: 10.1002/hep.22351
82. Xie G, Wang X, Wang L, Wang L, Atkinson RD, Kanel GC, et al. Role of differentiation of liver sinusoidal endothelial cells in progression and regression of hepatic fibrosis in rats. Gastroenterology. (2012) 142:918–27. e6. doi: 10.1053/j.gastro.2011.12.017
83. He S and Sharpless NE. Senescence in health and disease. Cell. (2017) 169:1000–11. doi: 10.1016/j.cell.2017.05.015
84. Wang B, Han J, Elisseeff JH, and Demaria M. The senescence-associated secretory phenotype and its physiological and pathological implications. Nat Rev Mol Cell Biol. (2024) 25(12):958–78. doi: 10.1038/s41580-024-00727-x
85. Tang Q, Xing X, Huang H, Yang J, Li M, Xu X, et al. Eliminating senescent cells by white adipose tissue–targeted senotherapy alleviates age-related hepatic steatosis through decreasing lipolysis. GeroScience. (2024) 46:3149–67. doi: 10.1007/s11357-024-01068-5
86. Ghimire K, Li Y, Chiba T, Julovi SM, Li J, Ross MA, et al. Cd47 promotes age-associated deterioration in angiogenesis, blood flow and glucose homeostasis. Cells. (2020) 9:1695. doi: 10.3390/cells9071695
87. Ogrodnik M, Miwa S, Tchkonia T, Tiniakos D, Wilson CL, Lahat A, et al. Cellular senescence drives age-dependent hepatic steatosis. Nat Commun. (2017) 8:15691. doi: 10.1038/ncomms15691
88. Lee YH, Kuk MU, So MK, Song ES, Lee H, Ahn SK, et al. Targeting mitochondrial oxidative stress as a strategy to treat aging and age-related diseases. Antioxidants (Basel). (2023) 12:934. doi: 10.3390/antiox12040934
89. Miwa S, Kashyap S, Chini E, and von Zglinicki T. Mitochondrial dysfunction in cell senescence and aging. J Clin Invest. (2022) 132:e158447. doi: 10.1172/jci158447
90. Maeso-Díaz R, Ortega-Ribera M, Fernández-Iglesias A, Hide D, Muñoz L, Hessheimer AJ, et al. Effects of aging on liver microcirculatory function and sinusoidal phenotype. Aging Cell. (2018) 17:e12829. doi: 10.1111/acel.12829
91. van Beek AA, Van den Bossche J, Mastroberardino PG, de Winther MPJ, and Leenen PJM. Metabolic alterations in aging macrophages: ingredients for inflammaging? Trends Immunol. (2019) 40:113–27. doi: 10.1016/j.it.2018.12.007
92. Wang L, Hong W, Zhu H, He Q, Yang B, Wang J, et al. Macrophage senescence in health and diseases. Acta Pharm Sin B. (2024) 14:1508–24. doi: 10.1016/j.apsb.2024.01.008
93. Stranks AJ, Hansen AL, Panse I, Mortensen M, Ferguson DJP, Puleston DJ, et al. Autophagy controls acquisition of aging features in macrophages. J Innate Immun. (2015) 7:375–91. doi: 10.1159/000370112
94. Yashaswini CN, Qin T, Bhattacharya D, Amor C, Lowe S, Lujambio A, et al. Phenotypes and ontogeny of senescent hepatic stellate cells in metabolic dysfunction-associated steatohepatitis. J Hepatol. (2024) 81:207–17. doi: 10.1016/j.jhep.2024.03.014
95. Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, et al. Senescence of activated stellate cells limits liver fibrosis. Cell. (2008) 134:657–67. doi: 10.1016/j.cell.2008.06.049
96. Maeso-Díaz R, Ortega-Ribera M, Lafoz E, Lozano JJ, Baiges A, Francés R, et al. Aging influences hepatic microvascular biology and liver fibrosis in advanced chronic liver disease. Aging Dis. (2019) 10:684–98. doi: 10.14336/ad.2019.0127
97. Schloesser D, Lindenthal L, Sauer J, Chung K-J, Chavakis T, Griesser E, et al. Senescent cells suppress macrophage-mediated corpse removal via upregulation of the cd47-qpct/L axis. J Cell Biol. (2023) 222:e202207097. doi: 10.1083/jcb.202207097
98. Tanaka M, Kanazashi M, Matsumoto T, Kondo H, Ishihara A, and Fujino H. Mild hyperbaric oxygen exposure attenuates rarefaction of capillary vessels in streptozotocin-induced diabetic soleus muscle in rats. BioMed Res. (2021) 42(1):1–11. doi: 10.2220/biomedres.42.1
99. Meijles DN, Sahoo S, Al Ghouleh I, Amaral JH, Bienes-Martinez R, Knupp HE, et al. The matricellular protein tsp1 promotes human and mouse endothelial cell senescence through cd47 and nox1. Sci Signal. (2017) 10:eaaj1784. doi: 10.1126/scisignal.aaj1784
100. Harbour JW and Dean DC. The rb/E2f pathway: expanding roles and emerging paradigms. Genes Dev. (2000) 14:2393–409. doi: 10.1101/gad.813200
101. Ji W, Zhang W, and Xiao W. E2f-1 directly regulates thrombospondin 1 expression. PloS One. (2010) 5:e13442. doi: 10.1371/journal.pone.0013442
102. Zhao W, Shen B, Cheng Q, Zhou Y, and Chen K. Roles of tsp1-cd47 signaling pathway in senescence of endothelial cells: cell cycle, inflammation and metabolism. Mol Biol Rep. (2023) 50:4579–85. doi: 10.1007/s11033-023-08357-w
103. Gao Q, Chen K, Gao L, Zheng Y, and Yang Y-G. Thrombospondin-1 signaling through cd47 inhibits cell cycle progression and induces senescence in endothelial cells. Cell Death Dis. (2016) 7:e2368–e. doi: 10.1038/cddis.2016.155
104. Kaur S, Soto-Pantoja DR, Stein EV, Liu C, Elkahloun AG, Pendrak ML, et al. Thrombospondin-1 signaling through cd47 inhibits self-renewal by regulating C-myc and other stem cell transcription factors. Sci Rep. (2013) 3:1673. doi: 10.1038/srep01673
105. Stout RD and Suttles J. Immunosenescence and macrophage functional plasticity: dysregulation of macrophage function by age-associated microenvironmental changes. Immunol Rev. (2005) 205:60–71. doi: 10.1111/j.0105-2896.2005.00260.x
106. Ding Y, Xu X, Meng B, Wang L, Zhu B, Guo B, et al. Myeloid-derived growth factor alleviates non-alcoholic fatty liver disease alleviates in a manner involving ikkbeta/nf-kappab signaling. Cell Death Dis. (2023) 14:376. doi: 10.1038/s41419-023-05904-y
107. Fan C, Ling-Hu A, Sun D, Gao W, Zhang C, Duan X, et al. Nobiletin ameliorates hepatic lipid deposition, oxidative stress, and inflammation by mechanisms that involve the nrf2/nf-κb axis in nonalcoholic fatty liver disease. J Agric Food Chem. (2023) 71:20105–17. doi: 10.1021/acs.jafc.3c06498
108. Yang S, Xu B, Han Y, Jiang M, Luo T, Wu N, et al. Taf15 exacerbates nonalcoholic steatohepatitis progression by regulating lipid metabolism and inflammation via fasn and P65 nf-kappab. Liver Int. (2023) 43:1920–36. doi: 10.1111/liv.15607
109. Lo J, Lau EY, Ching RH, Cheng BY, Ma MK, Ng IO, et al. Nuclear factor kappa B-mediated cd47 up-regulation promotes sorafenib resistance and its blockade synergizes the effect of sorafenib in hepatocellular carcinoma in mice. Hepatology. (2015) 62:534–45. doi: 10.1002/hep.27859
Keywords: CD47, MASLD, glucose metabolism, lipid metabolism, mitochondrial, hepatic sinusoidal, senescence
Citation: Jiang X, Zhao W, Shen B, Han Y and Chen K (2025) CD47-mediated regulation of glucose and lipid metabolism: implications for the pathogenesis of MASLD. Front. Endocrinol. 16:1535382. doi: 10.3389/fendo.2025.1535382
Received: 27 November 2024; Accepted: 06 June 2025;
Published: 24 June 2025.
Edited by:
Alexandre Gabarra Oliveira, São Paulo State University, BrazilReviewed by:
Santosh Panda, Washington University in St. Louis, United StatesHitarthi Vyas, University of Michigan, United States
Copyright © 2025 Jiang, Zhao, Shen, Han and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kexin Chen, Y2hlbmtleGluQGpsdS5lZHUuY24=
†These authors have contributed equally to this work