 Afaf I. Alsagheir1,2*
Afaf I. Alsagheir1,2* Bassam Bin-Abbas1,2
Bassam Bin-Abbas1,2 Nujud M. Alghamdi3
Nujud M. Alghamdi3 Raghad T. Alhuthil1
Raghad T. Alhuthil1 Sarah A. Murad4
Sarah A. Murad4 Tala H. Husein2
Tala H. Husein2 M. Zulf Mughal5Zehour E. Alsabban4
M. Zulf Mughal5Zehour E. Alsabban4 Layla M. Almarzoug6
Layla M. Almarzoug6 Khushnooda Ramzan7
Khushnooda Ramzan7- 1Department of Pediatrics, King Faisal Specialist Hospital & Research Centre, Riyadh, Saudi Arabia
- 2College of Medicine, Alfaisal University, Riyadh, Saudi Arabia
- 3Department of Nursing Development and Saudization, King Faisal Specialist Hospital & Research Centre, Riyadh, Saudi Arabia
- 4Department of Radiology, King Faisal Specialist Hospital & Research Centre, Jeddah, Saudi Arabia
- 5Department of Pediatric Endocrinology, Al Jalila Children’s Specialty Hospital, Dubai, United Arab Emirates
- 6College of Medicine, Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia
- 7Department of Clinical Genomics, King Faisal Specialist Hospital & Research Centre, Riyadh, Saudi Arabia
Introduction: X-linked hypophosphatemia (XLH) is a lifelong, progressive genetic condition affecting patients’ physical health and quality of life.
Methods: This cross-sectional study aimed to understand the burden of XLH on four generations of family members with XLH. 26 family members with XLH from Saudi Arabia were assessed via a home visit and clinical assessment in hospital. Patient demographics, biochemical parameters, and radiological and skeletal ndings were collected. Quality of life was assessed using the 36-Item Short Form Survey (SF-36) and Pediatric Quality of Life Inventory (PedsQL 4.0). Further assessment involved the 6-minute walk test (6MWT) and Western Ontario and McMaster Universities Arthritis Index (WOMAC) pain assessment.
Results: Our results showed low quality of life for the adults and children, with mean SF-36 and PedsQL (8–18 years) scores of 34.12 (standard deviation [SD] 25.02) and 55.04 (SD 29.47), respectively. High levels of complications of XLH and surgical interventions were common, including dental abscesses (92%), tooth loss (73.07%), osteotomies (76.92%) and craniosynostosis (76.90%). In 15 adult patients, aged 35–55 years, moderate WOMAC scores for pain, stiffness, and function of hip and knee joints and low 6MWT scores were reported. Skeletal deformities in the hip (53.85%) and skull (76.90%), and fractures and pseudofractures (38.40%), were common among older patients.
Discussion: These ndings demonstrate that the burden of XLH in these family members who had delayed diagnosis and were non-compliant to medical treatment and supportive care was high. Greater awareness and early diagnosis are essential for identi cation of cases and early initiation of treatment.
1 Introduction
X-linked hypophosphatemia (XLH) is a hereditary condition caused by mutations in the phosphate-regulating endopeptidase homolog X-linked (PHEX) gene (1). The loss of function of PHEX causes increased levels of fibroblast growth factor 23 (FGF23), leading to renal phosphate wasting as well as reduced serum 1,25-dihydroxyvitamin D levels, which lead to decreased absorption of phosphate and calcium from the intestines (2). Chronically reduced serum phosphate leads to impaired mineralization of the growth plate and osteoid, which in turn result in skeletal deformities, dental issues, and other systemic disorders (3). The incidence of the disease is approximately 1 in 20,000 to 1 in 70,000, according to the International XLH Registry (4).
In addition to the physical impacts of the disease, health-related quality of life is also deeply impacted in patients with XLH (1). Several studies have shown that XLH significantly impacts patients’ quality of life. Studies utilizing standardized questionnaires like the 36-Item Short Form Survey (SF-36) and EuroQol-5D have revealed that patients with XLH experience substantial limitations in physical and mental health domains, including pain, mobility, and daily activities (5, 6). These findings underscore the urgent need for targeted interventions to address the multifaceted challenges faced by individuals with XLH. Using the Oral Health Impact Profile questionnaire, Hanisch et al. examined the impact of XLH on oral health-related quality of life (HRQoL) (7). The study demonstrated that the majority of patients with XLH suffered from oral symptoms, including dental abscesses and fistulas, which negatively impacted oral HRQoL. The patients with XLH from the study had worse oral HRQoL, with a score of 10.30, compared with the general population, with a score of 4.09 (7).
Additionally, quality-of-life evaluations in XLH have gone beyond the evaluation of oral and physical health. The psychosocial effects of XLH have been investigated by Lo et al., who reported that patients experienced low self-esteem and well-being, depression, and frustration resulting from negative social encounters such as bullying (8). Furthermore, Yanes et al. reported that 65% of adult patients with XLH in their study experienced varying degrees of anxiety or depression (9). A Clinical Practice Research Datalink study in the UK demonstrated that patients with XLH were three times more likely to have depression versus a matched population of patients without XLH (10).
Conventional treatment for patients with XLH involves supplementation with phosphate and active vitamin D (5). Treatment in children aims to correct skeletal and radiographic abnormalities and lessen the effects of rickets and osteomalacia (5). In adults, treatment can help to minimize pain, improve healing of fractures or following surgery, and lessen the effects of osteomalacia. However, treatment is often discontinued once the bones are matured (5, 6). Despite treatment with conventional therapy, some patients continue to experience symptoms as well as side effects such as hyperparathyroidism, diarrhea, nephrocalcinosis, nausea, and abdominal pain (11). Dose adjustments and regular monitoring are recommended to overcome these limitations (5). Burosumab, an anti-FGF23 monoclonal antibody that increases levels of serum phosphate, has been shown to improve the symptoms of rickets and improve growth rates in children (12). In adults, burosumab therapy has been shown to lead to improved healing of fractures or pseudofractures and improvements in physical function, pain, and stiffness (13). Early diagnosis and treatment initiation is essential to prevent complications in patients with XLH. Such complications can include osteoarthritis, deafness, enthesopathies, or spinal stenosis (14).
This aims of this observational cohort study were:
● To describe patient background and demographics and to assess the quality of life in four generations of patients with XLH from the same family residing in a semi-remote area of Mecca, Saudi Arabia
● To assess the impact of XLH on joint stiffness and mobility across the life course
● To describe the current biochemical parameters and radiological and skeletal findings in these patients.
2 Materials and methods
This cross-sectional study was conducted at King Faisal Specialist Hospital & Research Centre (KFSHRC), Riyadh, Saudi Arabia. Following the establishment of an FGF23-related hypophosphatemic rickets and osteomalacia registry at KFSHRC and loss of follow-up of 12 family members who previously attended the hospital due to poor socioeconomic status and the COVID pandemic, the medical team from KFSHRC visited the entire family residing in a semi-remote area of Mecca, Saudi Arabia to conduct a clinical assessment. The visit was conducted on 25 June 2022 by a team of three pediatric endocrine consultants and two nurse practitioners.
Ethics approval for this study was granted by the institutional review board at KFSHRC (ethics approval number: 2211051).
2.1 Patient cohort
The patient cohort consisted of 30 individuals across four generations of the same family who manifested with XLH (Figure 1). Of these family members, 26 were assessed during the medical teams’ visit and had genetic confirmation of pathogenic mutations in the PHEX gene. The remaining four family members did not undergo assessment or genetic testing but had skeletal manifestations and had previously been diagnosed with hypophosphatemic rickets at KFSHRC based on clinical and biochemical data. Three of these four family members were deceased at the time of study. Clinical data was available for the final family member, who was not assessed at the medical teams’ visit.
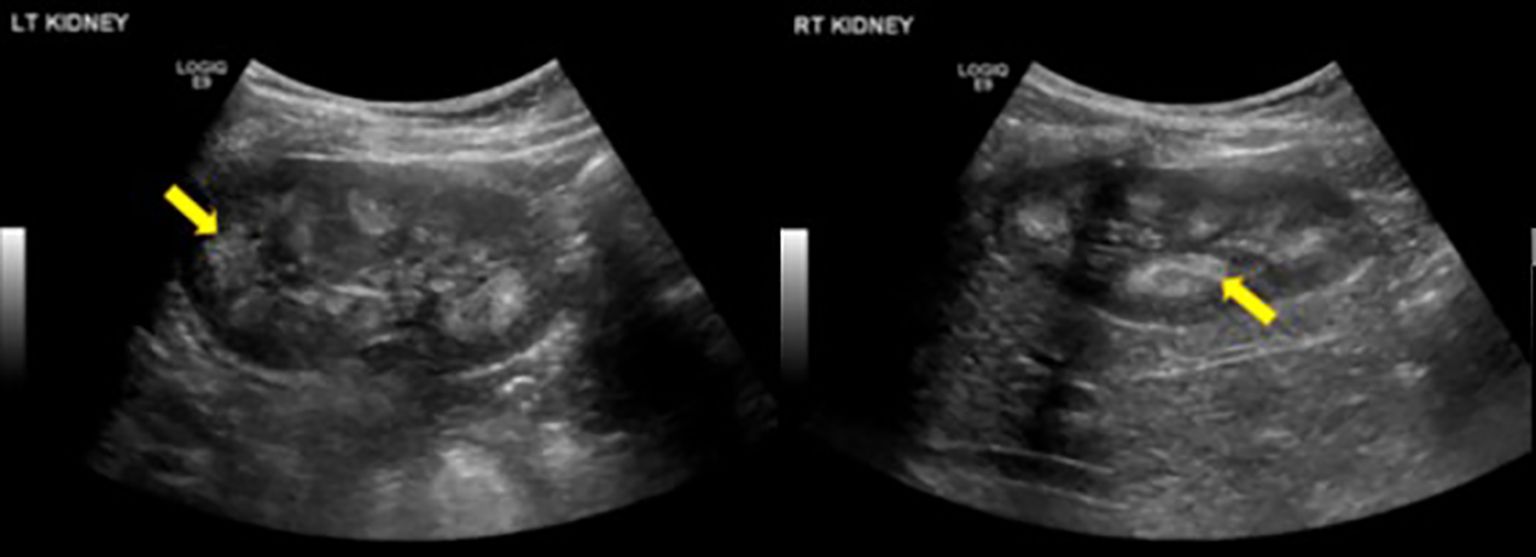
Figure 1. Renal Ultrasound of bilateral kidneys in a 9-year-old patient showed diffusely echogenic pyramids (yellow arrows), suggestive of bilateral medullary nephrocalcinosis.
2.2 Clinical evaluation
During the medical teams’ visit, patients were interviewed. Information was collected using a standardized case report form regarding symptoms related to their condition (pain, muscle weakness, ability to walk and do their daily activity, fractures, skeletal deformity), medical treatment, compliance to treatment, and surgical history. Visits to the physiotherapist at KFSHRC were arranged for further clinical assessment, which included the 6-minute walk test (6MWT) and Western Ontario and McMaster Universities Arthritis Index (WOMAC) pain assessment. Growth status was assessed by comparing the latest documented weight and height or length measurements to age–gender standard deviation (SD) scores. Laboratory tests were performed in the laboratory at KFSHRC, following the clinical team visit to the family, which included fasting levels of serum calcium, serum phosphate, parathyroid hormone (PTH), serum alkaline phosphatase (ALP), tubular reabsorption of phosphate, ratio of tubular maximum reabsorption rate of phosphate to glomerular filtration rate (TmP/GFR), spot urine, creatinine, calcium, and phosphate (from second urine sample passed in the morning).
Radiological assessments included X-rays of the left hand and knee, renal ultrasound to evaluate for nephrocalcinosis, and brain and spine magnetic resonance imaging (MRI). Bone densitometry was assessed by DEXA scan for adult patients only using the GE Lunar iDXA (GE Healthcare Lunar, Madison, WI), scanning both proximal femurs and lumbar spine.
2.3 Genetic analysis
The proband, who continues to be followed up at KFSHRC, was a 48-year-old male. He presented with abnormal skeletal morphology, elevated circulating ALP levels, hypophosphatemia, and short stature. He was genetically screened for the PHEX gene due to these clinical manifestations. The pathogenic PHEX variant identified in the proband was c.2239C>Tp.(Arg747*) using CentoXome® Solo at the CENTOGENE laboratory. Targeted screening testing for the same variant was undertaken in the rest of the family members at the CENTOGENE laboratory and supported by Kyowa Kirin Pharma.
2.4 Quality-of-life surveys
Family members completed validated quality-of-life surveys during the medical teams’ visit to Mecca. Two main validated Arabic-translated instruments were administered in this study to assess the quality of life of the patients with XLH: the SF-36 and the Pediatric Quality of Life Inventory (PedsQL 4.0).
The SF-36 questionnaire includes multiple-item subscales that assesses physical function, social functioning, role constraints brought on by physical difficulties, role restrictions brought on by emotional problems, mental health, vitality, pain, and overall perception of health (15). The questionnaire was self-administrated for adult patients (age >18 years). Each SF-36 subscale has a total score between 0 and 100. A higher score indicates better HRQoL.
The PedsQL 4.0 questionnaire evaluates the generic HRQoL in children and adolescents. It comprises 23 items that measure physical, emotional, social, and school functioning and has been demonstrated to be psychometrically valid and reliable. Developmentally appropriate versions were used for children and adolescents: child self-report for ages 8–12 years and 13–18 years, and interview-based parent-proxy report for children ages 5–7 years (administered by a nurse practitioner). Higher scores indicate a greater HRQoL. The score is not given if more than 50% of items are missing from a scale (16, 17).
Both questionnaires have been linguistically validated in the literature (English to Arabic) (18, 19). Nevertheless, internal consistency was measured using the Cronbach’s alpha reliability test.
2.5 Statistical analysis
Data analysis was performed using STATA (version 17). Continuous data were presented as mean, SD, range, median, or interquartile range, as appropriate. Categorical data were presented as frequencies and percentages.
3 Results
3.1 Patient demographics
Demographic information of the family members is summarized in Table 1. All adult patients (n = 19) were reported to have presented with genu varum (bowed legs) since early childhood, but only 12 patients (3 females and 9 males) sought medical advice at KFSHRC. The proband first visited KFSHRC in 1982 at the age of 8 years. The remaining 18 patients had a mean age at diagnosis of 7 years (SD 4.5) and were diagnosed based on clinical and radiological signs of rickets, as well as low mean serum phosphate (0.55 mmol/L, SD 0.32; reference range adults 0.80–1.50, children 0.90–1.80), high mean ALP (565 IU/L, SD 115.90; reference range adults 30–130, children [3–10 years] 130–260, children [10–14 years] 130–340, children [14–18 years] 30–180), and low tubular reabsorption of phosphate (reference range <75%). The patients were started on calcitriol, the active form of vitamin D, and oral phosphate supplements. However, the patients were not compliant with their treatment, and they did not follow up regularly. During their teenage years, they underwent surgical correction of their lower limbs by osteotomy.
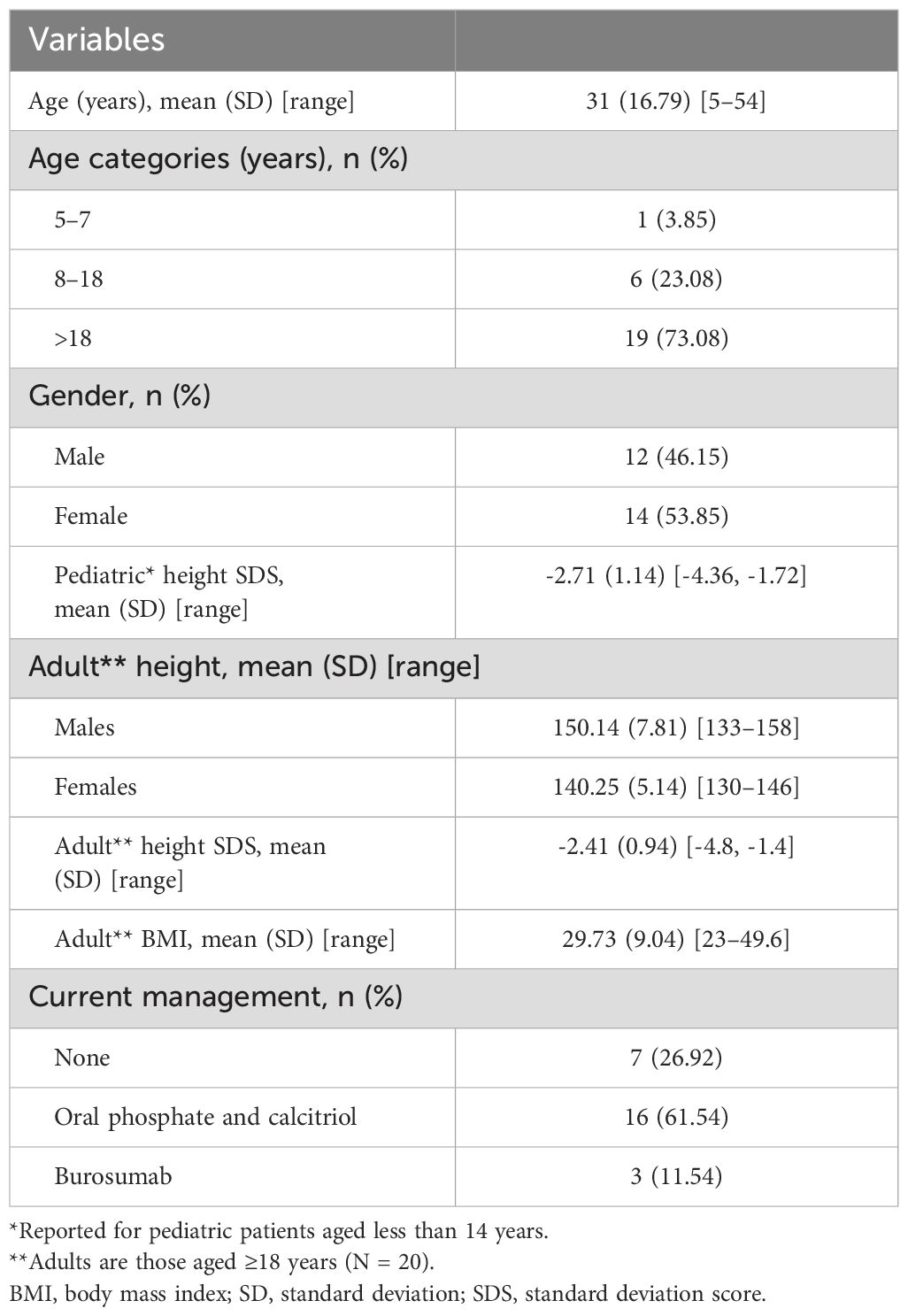
Table 1. Demographic information (N = 26).
3.2 Clinical evaluation
Laboratory findings revealed high mean PTH and ALP and low mean TmP/GFR in these family members (Table 2). Radiological and skeletal findings are shown in Table 3. Renal ultrasound revealed nephrocalcinosis in two patients. Common skeletal abnormalities in the family members included skull shape abnormality (N = 20), coxa vara or valga (N = 14), and genu varum or valgum (N = 11). Figures 2–4 show examples of renal ultrasound, X-ray, and MRI results.
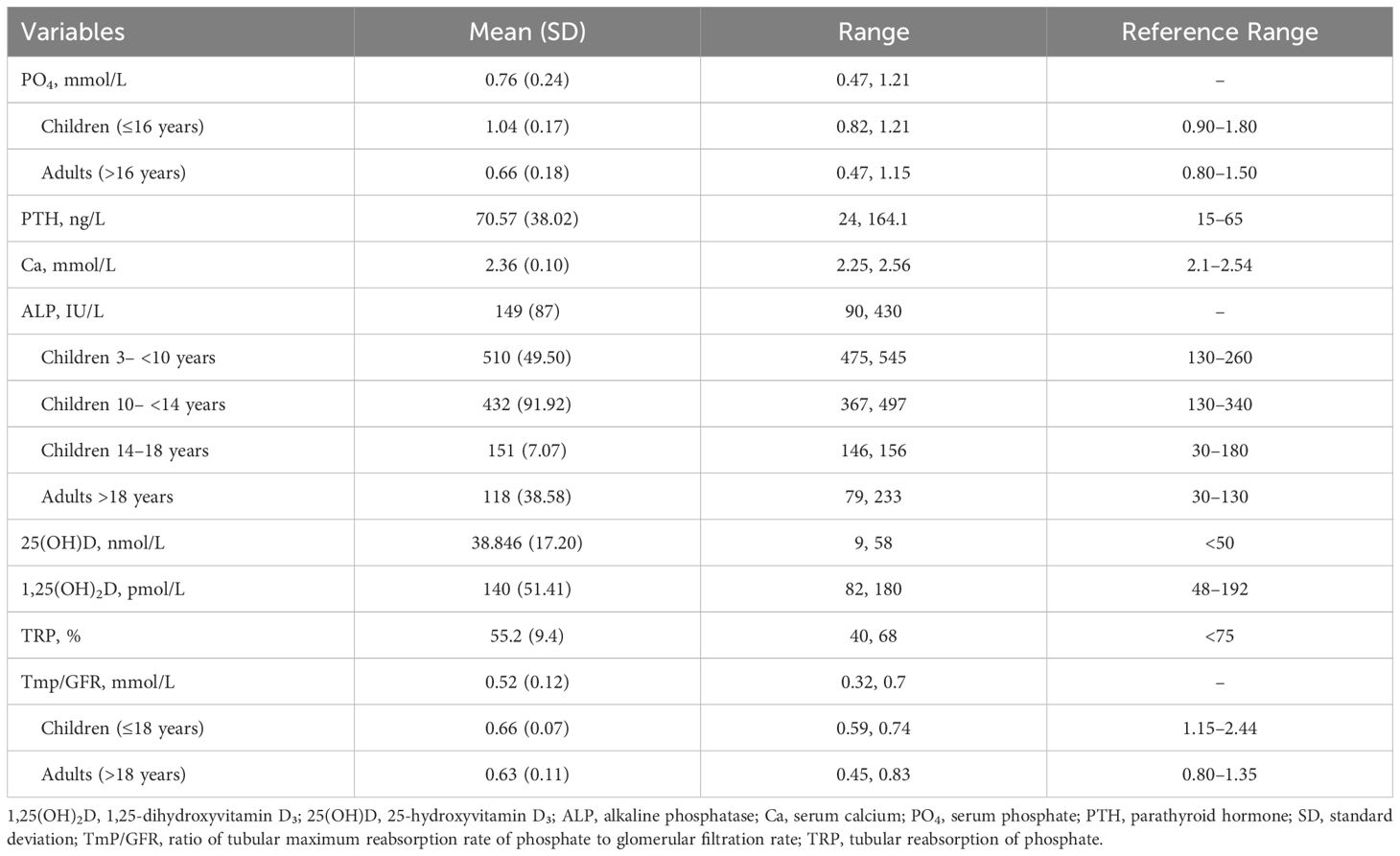
Table 2. Laboratory findings at presentation (n=26).
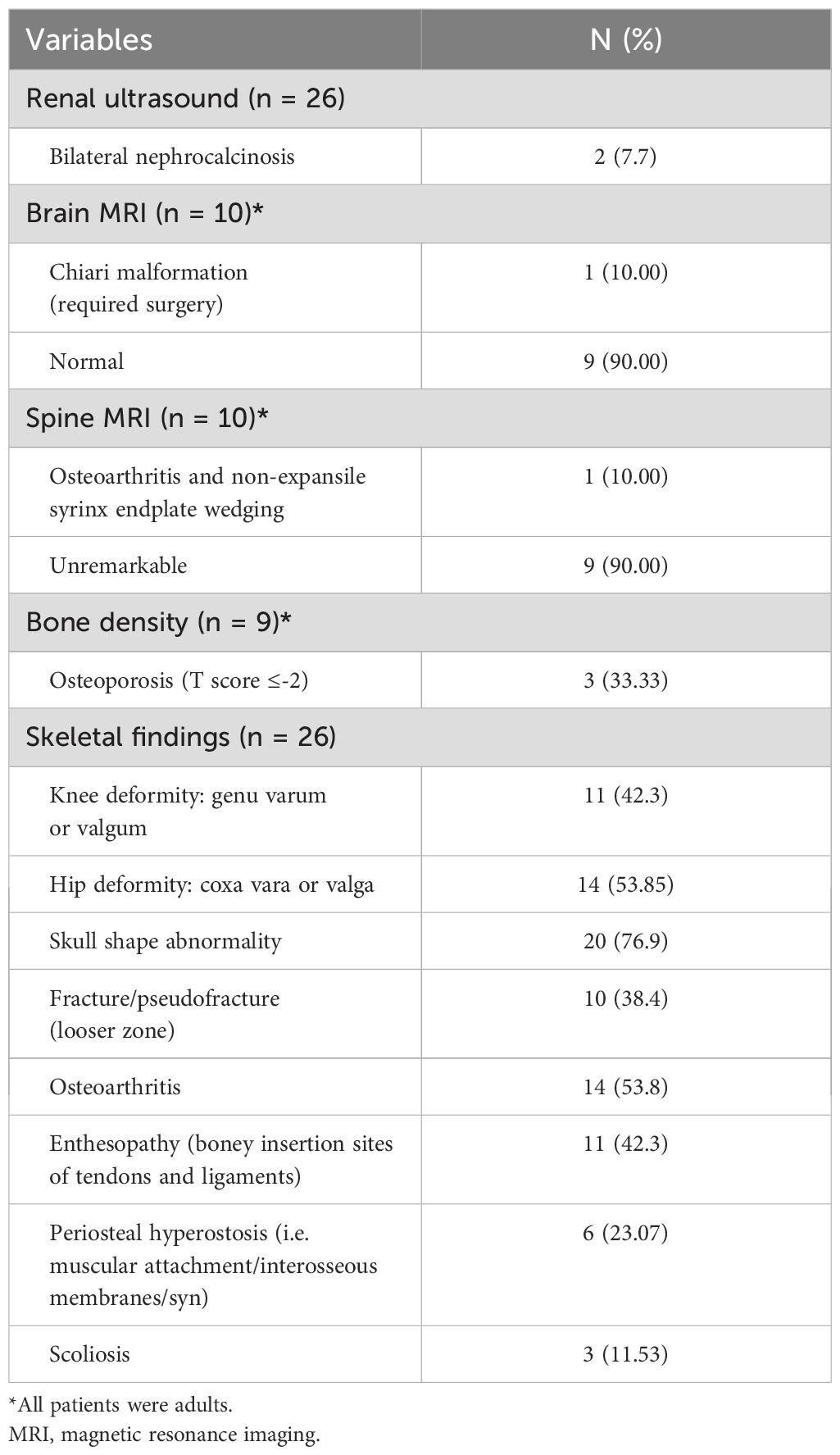
Table 3. Radiological and skeletal findings.
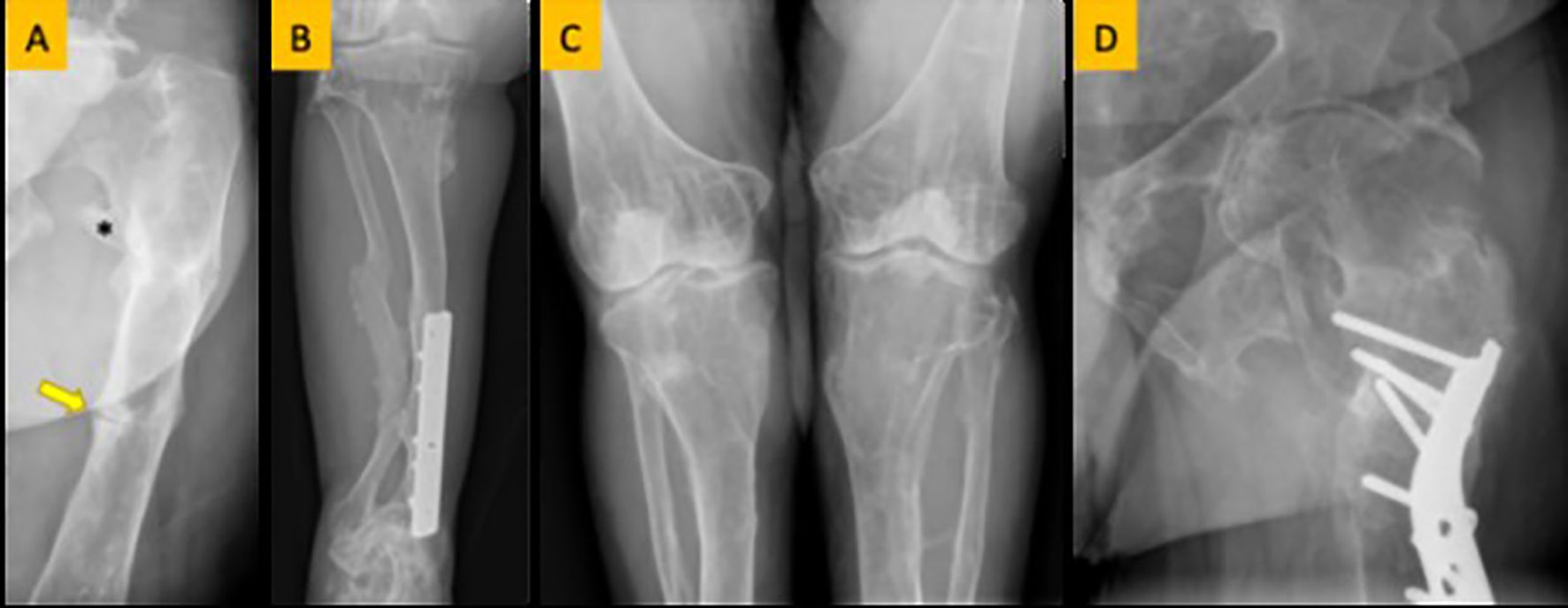
Figure 2. (A) Femoral looser zone (yellow arrow) and enthesopathy (asterisk). (B) Bowing deformity and remodeling of the tibia and fibula. (C) Genu valgus (knock knee) deformity. (D) Coxa valgus deformity.
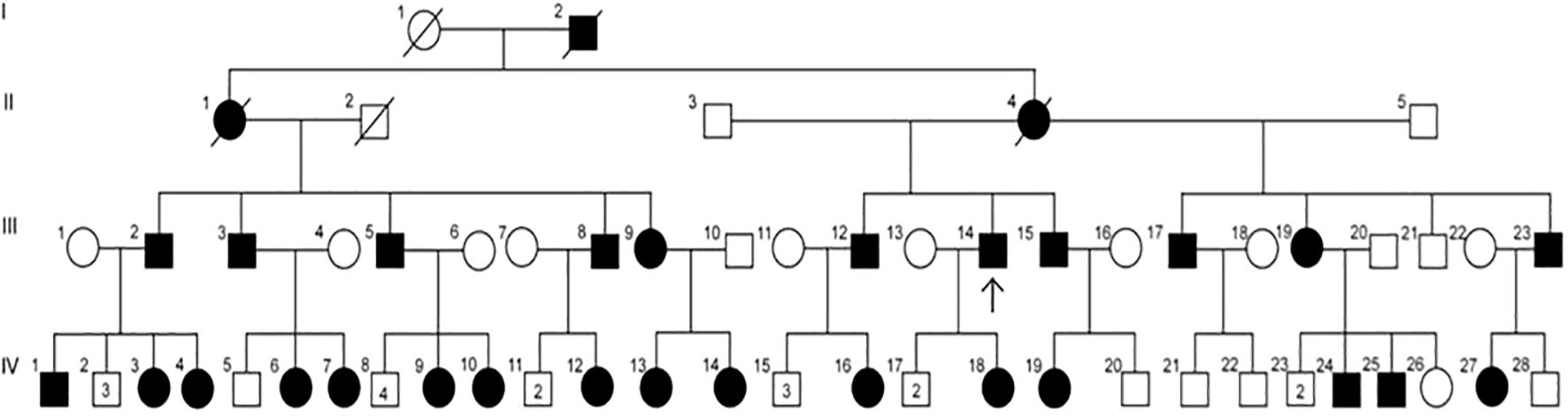
Figure 3. Pedigree of a family with XLH, suggesting X-linked dominant inheritance. Circles = females; squares = males; solid symbols = affected individuals; open symbols = unaffected individuals; strike-through symbols = deceased; arrow = proband; numbers within symbols = number of male or female sibling (not affected).
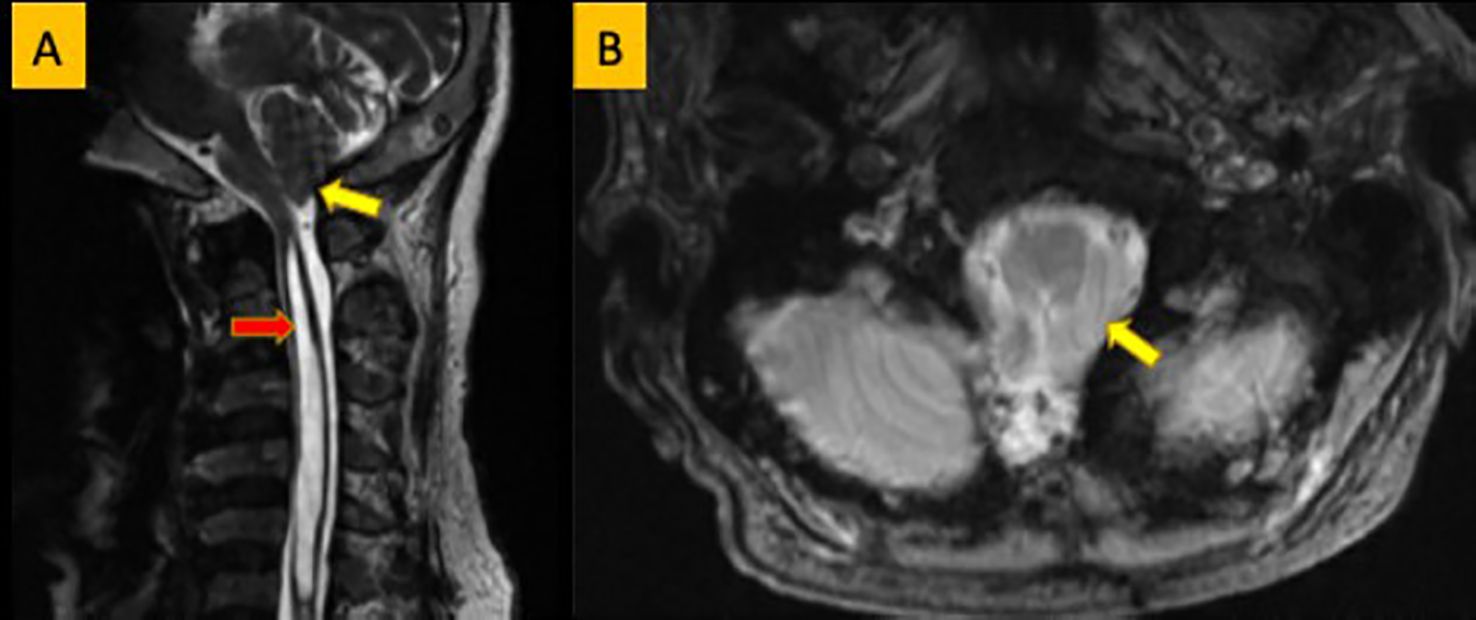
Figure 4. (A) Sagittal T2WI showing low-laying tonsil (yellow arrow). Large syrinx is seen in the cervical spinal cord (red arrow). (B) Axial T2WI showing crowding of the foramen magnum by the low laying tonsil (yellow arrow).
Physiotherapy assessment results (Table 4) showed low mobility, assessed using the 6MWT, and moderate levels of pain, stiffness, and function, measured using WOMAC. Patient procedures and complications are shown in Table 5. Dental abscesses and tooth loss were the most common complications, as well as osteoarthritis and craniosynostosis. Of the 20 adult patients, 1 (5%) is currently bedridden and 2 (10%) are using a wheelchair.
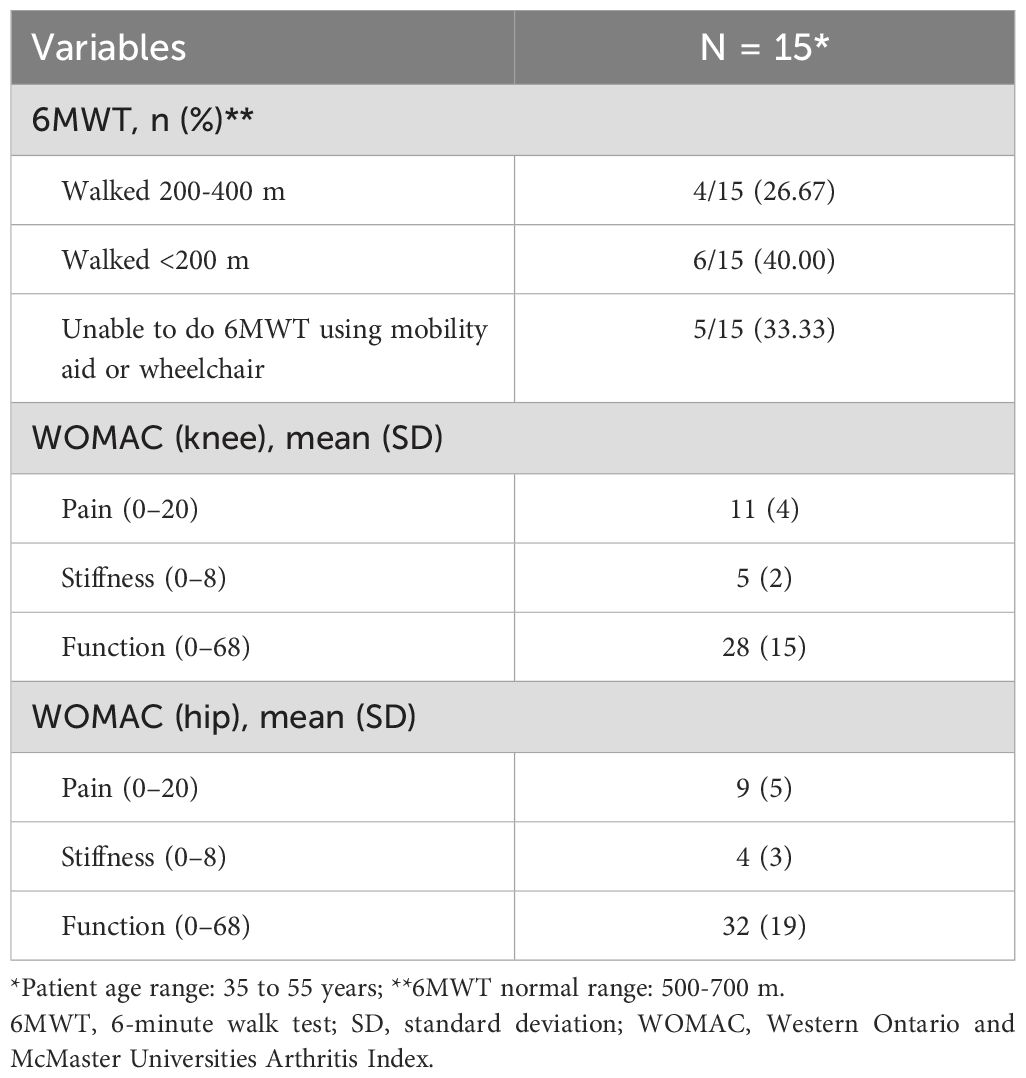
Table 4. Physiotherapy assessment results.
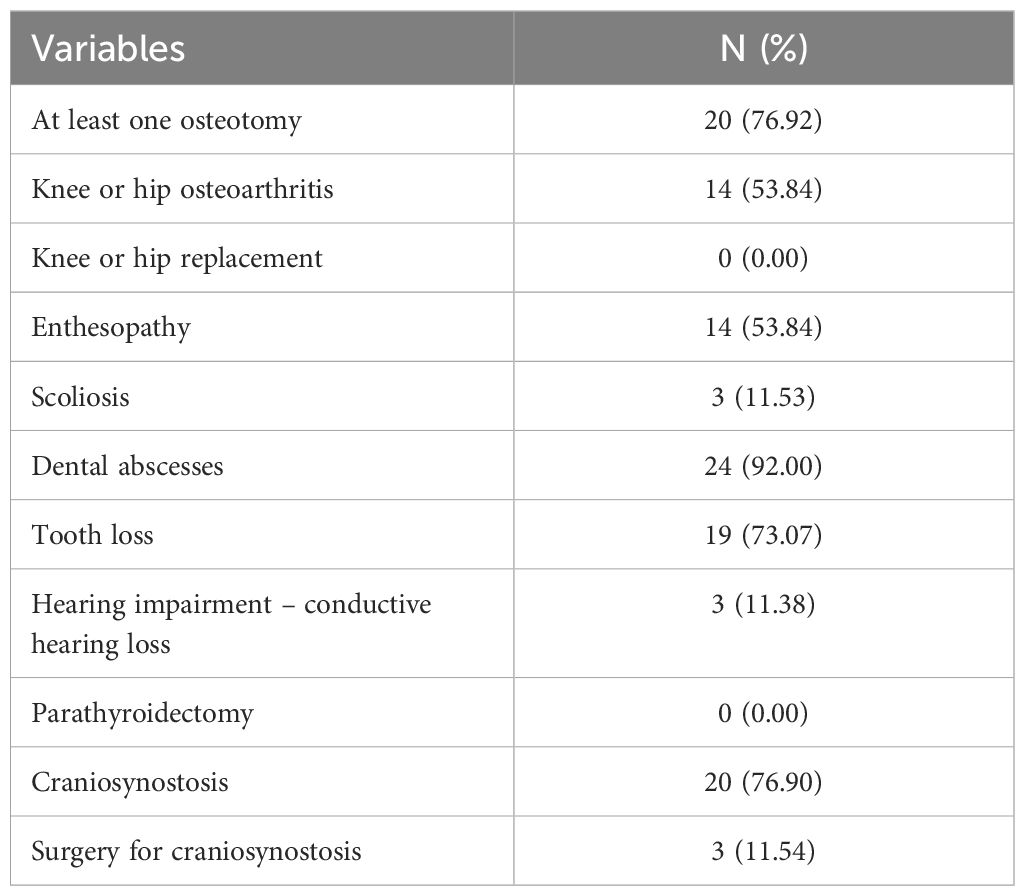
Table 5. Procedure/complications.
3.3 Genetic analysis
Family members were confirmed to be hemizygous for a C>T nucleotide substitution in exon 22 of the PHEX gene, resulting in the replacement of an arginine codon (CGA) with a stop codon (TGA) at amino acid position 747. This mutation is denoted as c.2239 C>T at the cDNA level or p.Arg747 top (R747X) at the protein level. According to Human Gene Mutation Database Professional release 2022.1, this variant has previously been associated with disease causing hypophosphataemic rickets by Francis et al. (20), Dixon et al. (21), and Hernandez-Frias et al. (22). In addition, well-established in vitro or in vivo functional studies are supportive of a damaging effect on the gene or gene product (23). ClinVar (National Library of Medicine) lists this variant as “pathogenic/likely pathogenic” under variation ID279873. The variant is also classified as “pathogenic” (class 1) according to the recommendations of CENTOGENE and the American College of Medical Genetics.
3.4 Quality of life
Results of the SF-36 and PedsQL surveys and Cronbach’s alpha reliability test are shown in Table 6. The SF−36 questionnaire was applied to adult participants (>18 years, n = 19) with a mean score of 34.12. The lowest scores were in the role limitations due to physical and emotional health domains (mean 23.75 and 26.67, respectively). For PedsQL (ages 8–18 years, n = 6), the overall mean score was 55.04. School functioning and emotional functioning scored the lowest (means of 48.96 and 52.5, respectively). For the PedsQL (ages 5–7 years), only one patient was tested, with an overall score of 34.8. They scored the lowest in the social functioning domain, with a score of 20. Both SF-36 and PedsQL (8–18 years) achieved excellent internal reliability scores (0.9682 and 0.9689, respectively).
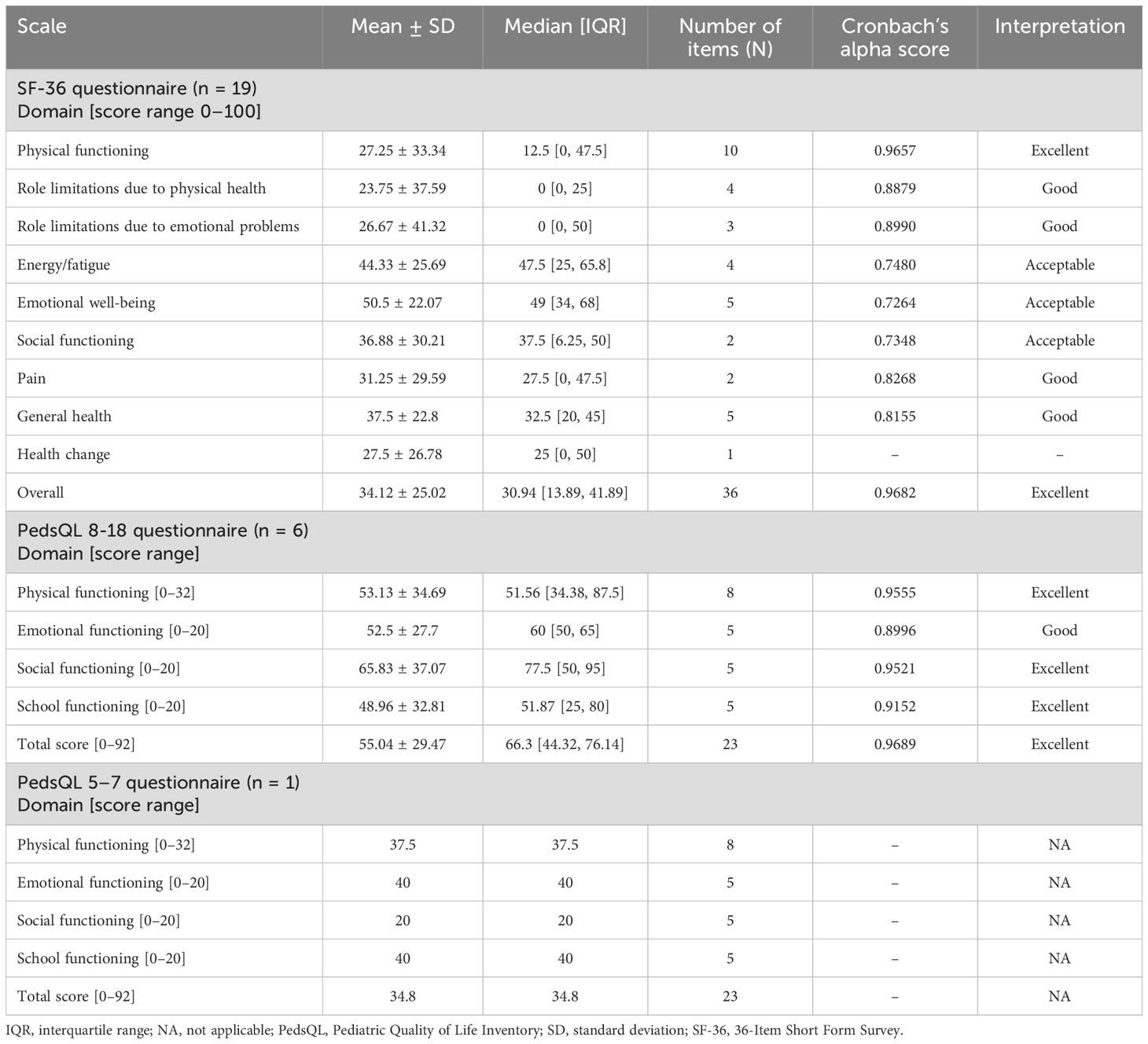
Table 6. SF-36 and PedsQL scores and reliability test results.
4 Discussion
This cross-sectional study of 26 patients described the impact of XLH on four generations of patients from the same family who resided in a semi-remote area of Mecca, Saudi Arabia, and who have had delayed diagnosis and poor access to medical treatment and supportive care over the past 50 years. All family members who underwent genetic testing were confirmed to have the c.2239C>Tp.(Arg747*) PHEX variant. Quality of life was low in this set of patients, with adults reporting low scores in role limitations due to physical and emotional health domains and children having low scores in school functioning and emotional functioning domains. High levels of complications and procedures were also reported in this group of patients, including dental abscesses, tooth loss, osteotomies, craniosynostosis, enthesopathy, and hip or knee arthritis. Moderate levels of pain, stiffness, and function were recorded for hip and knee joints, and low mobility was also reported. Skeletal deformities in the knee, hip, and skull, as well as fractures and pseudofractures, were common among the patients. Biochemical testing revealed high mean PTH and ALP and low mean TmP/GFR in these family members. Raised PTH in this set of patients is potentially a result of concomitant vitamin D and/or dietary calcium deficiency, or an inadequate dose of calcitriol in comparison to oral phosphate supplements.
Low quality of life in adults with XLH, as reported using the SF-36, has been described in other studies. In a French prospective cohort study of 52 adults with XLH, mean SF-36 physical component score (PCS) was 49.5 (SD 20.5) and mean SF-36 mental component score (MCS) was 57.9 (SD 21.3) (24). This French study reported similarly high levels of complications and procedures, with 95.6% having deformities in the lower limbs, 62.5% with dental defects, 61.5% with structural lesions, 64.0% with enthesopathies, and 85.4% having radiological arthritis. Furthermore, the majority of patients were on conventional therapy, with 64.6% taking oral phosphate, 59.2% vitamin D, and 66.7% vitamin D analogs (24). A further prospective cohort study conducted in the UK reported overall mean SF-36 PCS of 32.67 (SD 14.19) and SF-36 MCS of 48.38 (10.41) for 48 adult patients with XLH (25). In the XLH Burden of Disease Survey conducted in 232 adult patients with XLH, SF-36 PCS was 37.0 and MCS was 45.9, demonstrating low quality of life in this set of patients despite the use of conventional therapy (1). These patients also reported bowing of the legs (77%), surgical procedures (94%), history of fractures and pseudofractures (40%), and dental abscesses (82%), showing similar levels of complications and procedures to our study participants (1).
Quality of life in pediatric patients with XLH, as reported using PedsQL, has also been reported in other studies. One study reported a mean PedsQL score of 75.62 (SD 0.31) in 59 pediatric patients with XLH prior to initiation of a patient support program, with emotional functioning being the lowest domain with a mean of 65.75 (SD 0.31) (26). In the same study, the PedsQL score was even lower in the subset of patients who were not receiving treatment at enrollment, with a mean of 71.28 (SD 0.32). Further studies, using different measures of HRQoL, such as the EuroQol-5D, have also shown reduction in HRQoL in adults and children with XLH (9). Quality-of-life scores were much lower in our study, potentially due to delayed diagnosis and poor access to medical treatment and supportive care experienced by the patients.
Ambulatory function (6MWT) and patient-reported joint pain, stiffness, and physical function (WOMAC) have been assessed in clinical trials of patients with XLH. In a phase 3 extension trial, adult patients with XLH had reduced ambulatory function at baseline with a mean 6MWT of 362.0 meters (SD 106.3) (27). Furthermore, according to WOMAC, patients had reduced physical function as well as stiffness and pain at baseline, with a mean of 47.4 (SD 20.0) for physical function, 63.1 (SD 20.5) for stiffness, and 49.3 (16.8) for pain (27). WOMAC was also assessed in the XLH Burden of Disease Survey, where adult patients with XLH had mean scores of 40.8 for physical function, 39.5 for pain, and 50.3 for stiffness, which were much higher than the United States normative values (1). In a prospective cohort study, Orlando et al. reported a mean 6MWT of 335 meters (SD 102) in 26 adult patients XLH, which is 38.6% less than the distance expected based on the age and gender of patients (28). This group of patients had similar levels of skeletal deformities to our study participants, including 56% with genu varum, 32% with genu valgus, and osteoarthritis of the hip or knee in 44% and 28%, respectively. Furthermore, 23% were receiving no treatment and 57.7% were being treated with conventional therapy (28). The WOMAC scores in the subset of patients from our study were lower than previous study findings mentioned above, indicating less pain and stiffness and less impaired physical function in our cohort of patients.
Patients in this study were largely untreated, resulting from poor compliance to medical treatment and supportive care, or were undiagnosed because they did not seek medical advice, which might explain the high levels of complications and procedures in these family members. Clinical complications, such as pain, stiffness, dental problems, and skeletal abnormalities, as well as the effects of these clinical symptoms on daily life, social life, mental health, and functional capabilities, are commonly reported in patients with XLH and worsened by limited access to treatment (29). In a Japanese study of 25 adult patients, ossification of spinal ligaments (80%), nephrocalcinosis (72%), hearing impairment (32%), and osteophytes in the hip and knee joints (96% and 68%, respectively) were commonly reported complications (30). In addition to these, fractures and pseudofractures, deformities of the lower extremities, and gait abnormalities are further complications reported in adults (31).
Burosumab has shown promising results in improving the quality of life of patients with XLH. A phase 3 extension study reported improvements in pain, fatigue, and ambulatory function after 48 weeks of burosumab use (32). An observational study of children at a London hospital demonstrated an improvement in PedsQL at 9 months (mean 80.9, SD 15.1) compared with baseline (mean 66.1, SD 16.6) in patients with XLH starting burosumab (33). Results from a further phase 3 extension trial by Briot et al. showed improvements at 24 weeks compared with baseline in WOMAC (least square mean -3.94, SD 2.19) and 6MWT (least square mean +2.56, SD 1.11) (27). Furthermore, a prospective case series in adolescent patients with XLH demonstrated an improvement in patient-reported outcomes of WOMAC and the revised Graded Chronic Pain Scale (GCPS-R) following 12 to 48 months of burosumab use (34). Additionally, serum phosphate, TmP/GFR and 1,25-dihydroxyvitamin D levels significantly increased following burosumab initiation while ALP levels significantly decreased (34). Critically, this case series demonstrated that these positive outcomes were not sustained following burosumab discontinuation, suggesting that on-going burosumab treatment throughout adolescence is essential (34). Early treatment with burosumab for symptomatic individuals aged 1 to 17 years with XLH is recommended in international guidelines (35). For symptomatic adults, treatment with oral phosphate and active vitamin D remains the standard of care, except for patients with pseudofractures or those who experience a lack of response or adverse events with oral phosphate and active vitamin D for whom burosumab treatment is recommended (35).
Although this study provides valuable insights into the impact of XLH in this cohort of 26 family members, there are some limitations to the study. The observational nature of the study and use of patient-reported outcomes can lead to some level of bias. Nevertheless, patient-reported outcomes are essential to understand quality of life in patients with XLH. Missing data for some patients is a further limitation; however, there were difficulties faced by some family members in attending appointments at KFSHRC due to the semi-remote location and poor socioeconomic status. While it is known that some the family members are from low socio-economic backgrounds, additional information on social engagement of the patients, level of instruction, and profession was not available. It would be useful to assess the impact of these factors on patient attendance at follow-up appointments as well as to understand how XLH impacts social engagement, education and work productivity. Future studies will aim to further assess HRQoL in patients with XLH in Saudi Arabia, especially in patients initiating burosumab treatment, to further understand how treatment can impact quality of life. Future work might also focus on the economic impact of the disease on the Saudi Arabia health system.
4.1 Conclusions
This study demonstrates the huge impact of XLH in 26 family members from Saudi Arabia who had delayed diagnosis and poor access to medical treatment and supportive care. Poor quality of life, skeletal deformities, and multiple complications and procedures are likely exacerbated by a lack of treatment in this set of patients. Greater awareness and early diagnosis of XLH are essential for identification of cases and early initiation of treatment. We propose that healthcare professionals should perform full family mapping for every patient diagnosed with XLH.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving humans were approved by the institutional review board at KFSHRC. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants and their legal guardians/next of kin.
Author contributions
AA: Conceptualization, Investigation, Supervision, Writing – original draft, Writing – review & editing. BB-A: Conceptualization, Investigation, Supervision, Writing – original draft, Writing – review & editing. NA: Data curation, Investigation, Writing – original draft, Writing – review & editing. RA: Formal Analysis, Methodology, Writing – original draft, Writing – review & editing. SM: Data curation, Formal Analysis, Investigation, Validation, Writing – original draft, Writing – review & editing. TH: Data curation, Writing – original draft, Writing – review & editing. MM: Conceptualization, Investigation, Supervision, Writing – original draft, Writing – review & editing. ZA: Data curation, Investigation, Validation, Writing – original draft, Writing – review & editing. LA: Data curation, Writing – original draft, Writing – review & editing. KR: Formal Analysis, Methodology, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. Medical writing support was funded by Kyowa Kirin Pharma as an unrestricted educational grant, Kyowa Kirin Pharma has had no influence in the writing of this manuscript.
Acknowledgments
Medical writing support was provided by Abigail Holland of Connect Communications, Dubai, United Arab Emirates.
Conflict of interest
AA and BB-A received personal honoraria for lectures and for participation in advisory boards from Kyowa Kirin. ZM has received personal honoraria for lectures and for participation in advisory boards from Kyowa Kirin.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Skrinar A, Dvorak-Ewell M, Evins A, Macica C, Linglart A, Imel EA, et al. The lifelong impact of X-linked hypophosphatemia: results from a burden of disease survey. J Endocr Soc. (2019) 3:1321–34. doi: 10.1210/js.2018-00365
2. Mughal MZ, Baroncelli GI, de-Lucas-Collantes C, Linglart A, Magnolato A, Raimann A, et al. Burosumab for X-linked hypophosphatemia in children and adolescents: opinion based on early experience in seven European countries. Front Endocrinol (Lausanne). (2022) 13:1034580. doi: 10.3389/fendo.2022.1034580
3. Baroncelli GI, Mora S. X-linked hypophosphatemic rickets: multisystemic disorder in children requiring multidisciplinary management. Front Endocrinol (Lausanne). (2021) 12:688309. doi: 10.3389/fendo.2021.688309
4. Ariceta G, Beck-Nielsen SS, Boot AM, Brandi ML, Briot K, de Lucas Collantes C, et al. The international X-linked hypophosphatemia (Xlh) registry: first interim analysis of baseline demographic, genetic and clinical data. Orphanet J Rare Dis. (2023) 18:304. doi: 10.1186/s13023-023-02882-4
5. Carpenter TO, Imel EA, Holm IA, Jan de Beur SM, Insogna KL. A clinician’s guide to X-linked hypophosphatemia. J Bone Miner Res. (2011) 26:1381–8. doi: 10.1002/jbmr.340
6. Dahir K, Dhaliwal R, Simmons J, Imel EA, Gottesman GS, Mahan JD, et al. Health care transition from pediatric- to adult-focused care in X-linked hypophosphatemia: expert consensus. J Clin Endocrinol Metab. (2022) 107:599–613. doi: 10.1210/clinem/dgab796
7. Hanisch M, Bohner L, Sabandal MMI, Kleinheinz J, Jung S. Oral symptoms and oral health-related quality of life of individuals with X-linked hypophosphatemia. Head Face Med. (2019) 15:8. doi: 10.1186/s13005-019-0192-x
8. Lo SH, Lachmann R, Williams A, Piglowska N, Lloyd AJ. Exploring the burden of X-linked hypophosphatemia: A european multi-country qualitative study. Qual Life Res. (2020) 29:1883–93. doi: 10.1007/s11136-020-02465-x
9. Yanes MIL, Diaz-Curiel M, Peris P, Vicente C, Marin S, Ramon-Krauel M, et al. Health-related quality of life of X-linked hypophosphatemia in Spain. Orphanet J Rare Dis. (2022) 17:298. doi: 10.1186/s13023-022-02452-0
10. Hawley S, Shaw NJ, Delmestri A, Prieto-Alhambra D, Cooper C, Pinedo-Villanueva R, et al. Higher prevalence of non-skeletal comorbidity related to X-linked hypophosphataemia: A uk parallel cohort study using cprd. Rheumatol (Oxford). (2021) 60:4055–62. doi: 10.1093/rheumatology/keaa859
11. Al-Juraibah F, Al Shaikh A, Al-Sagheir A, Babiker A, Al Nuaimi A, Al Enezi A, et al. Experience of X-linked hypophosphatemic rickets in the gulf cooperation council countries: case series. Endocrinol Diabetes Metab Case Rep. (2024) 2024:23–0098. doi: 10.1530/EDM-23-0098
12. Whyte MP, Carpenter TO, Gottesman GS, Mao M, Skrinar A, San Martin J, et al. Efficacy and safety of burosumab in children aged 1-4 years with X-linked hypophosphataemia: A multicentre, open-label, phase 2 trial. Lancet Diabetes Endocrinol. (2019) 7:189–99. doi: 10.1016/S2213-8587(18)30338-3
13. Portale AA, Carpenter TO, Brandi ML, Briot K, Cheong HI, Cohen-Solal M, et al. Continued beneficial effects of burosumab in adults with X-linked hypophosphatemia: results from a 24-week treatment continuation period after a 24-week double-blind placebo-controlled period. Calcif Tissue Int. (2019) 105:271–84. doi: 10.1007/s00223-019-00568-3
14. Al Juraibah F, Al Amiri E, Al Dubayee M, Al Jubeh J, Al Kandari H, Al Sagheir A, et al. Diagnosis and management of X-linked hypophosphatemia in children and adolescent in the gulf cooperation council countries. Arch Osteoporos. (2021) 16:52. doi: 10.1007/s11657-021-00879-9
15. Ware JE Jr., Sherbourne CD. The mos 36-item short-form health survey (Sf-36). I. Conceptual framework and item selection. Med Care. (1992) 30:473–83. doi: 10.1097/00005650-199206000-00002
16. Varni JW, Seid M, Kurtin PS. Pedsql 4.0: reliability and validity of the pediatric quality of life inventory version 4.0 generic core scales in healthy and patient populations. Med Care. (2001) 39:800–12. doi: 10.1097/00005650-200108000-00006
17. Varni JW, Seid M, Knight TS, Uzark K, Szer IS. The pedsql 4.0 generic core scales: sensitivity, responsiveness, and impact on clinical decision-making. J Behav Med. (2002) 25:175–93. doi: 10.1023/a:1014836921812
18. Coons SJ, Alabdulmohsin SA, Draugalis JR, Hays RD. Reliability of an arabic version of the rand-36 health survey and its equivalence to the us-english version. Med Care. (1998) 36:428–32. doi: 10.1097/00005650-199803000-00018
19. Arabiat D, Elliott B, Draper P, Al Jabery M. Cross-cultural validation of the pediatric quality of life inventory 4.0 (Pedsql) generic core scale into arabic language. Scand J Caring Sci. (2011) 25:828–33. doi: 10.1111/j.1471-6712.2011.00889.x
20. Francis F, Strom TM, Hennig S, Boddrich A, Lorenz B, Brandau O, et al. Genomic organization of the human pex gene mutated in X-linked dominant hypophosphatemic rickets. Genome Res. (1997) 7:573–85. doi: 10.1101/gr.7.6.573
21. Dixon PH, Christie PT, Wooding C, Trump D, Grieff M, Holm I, et al. Mutational analysis of phex gene in X-linked hypophosphatemia. J Clin Endocrinol Metab. (1998) 83:3615–23. doi: 10.1210/jcem.83.10.5180
22. Hernandez-Frias O, Gil-Pena H, Perez-Roldan JM, Gonzalez-Sanchez S, Ariceta G, Chocron S, et al. Risk of cardiovascular involvement in pediatric patients with X-linked hypophosphatemia. Pediatr Nephrol. (2019) 34:1077–86. doi: 10.1007/s00467-018-4180-3
23. Zheng B, Wang C, Chen Q, Che R, Sha Y, Zhao F, et al. Functional characterization of phex gene variants in children with X-linked hypophosphatemic rickets shows no evidence of genotype-phenotype correlation. J Bone Miner Res. (2020) 35:1718–25. doi: 10.1002/jbmr.4035
24. Che H, Roux C, Etcheto A, Rothenbuhler A, Kamenicky P, Linglart A, et al. Impaired quality of life in adults with X-linked hypophosphatemia and skeletal symptoms. Eur J Endocrinol. (2016) 174:325–33. doi: 10.1530/EJE-15-0661
25. Cole S, Sanchez-Santos MT, Kolovos S, Javaid MK, Pinedo-Villanueva R. Patient-reported outcomes measures of X-linked hypophosphataemia participants: findings from a prospective cohort study in the uk. Orphanet J Rare Dis. (2023) 18:26. doi: 10.1186/s13023-023-02620-w
26. Rothenbuhler A, Gueorguieva I, Lichtenberger-Geslin L, Audrain C, Soskin S, Bensignor C, et al. Young xlh patients-reported experience with a supportive care program. Patient Prefer Adherence. (2023) 17:1393–405. doi: 10.2147/PPA.S391025
27. Briot K, Portale AA, Brandi ML, Carpenter TO, Cheong HI, Cohen-Solal M, et al. Burosumab treatment in adults with X-linked hypophosphataemia: 96-week patient-reported outcomes and ambulatory function from a randomised phase 3 trial and open-label extension. RMD Open. (2021) 7:e001714. doi: 10.1136/rmdopen-2021-001714
28. Orlando G, Bubbear J, Clarke S, Keen R, Roy M, Anilkumar A, et al. Physical function and physical activity in adults with X-linked hypophosphatemia. Osteoporos Int. (2022) 33:1485–91. doi: 10.1007/s00198-022-06318-w
29. Cheung M, Rylands AJ, Williams A, Bailey K, Bubbear J. Patient-reported complications, symptoms, and experiences of living with X-linked hypophosphatemia across the life-course. J Endocr Soc. (2021) 5:bvab070. doi: 10.1210/jendso/bvab070
30. Kato H, Koga M, Kinoshita Y, Taniguchi Y, Kobayashi H, Fukumoto S, et al. Incidence of complications in 25 adult patients with X-linked hypophosphatemia. J Clin Endocrinol Metab. (2021) 106:e3682–e92. doi: 10.1210/clinem/dgab282
31. Imanishi Y, Shoji T, Emoto M. Complications and treatments in adult X-linked hypophosphatemia. Endocrines. (2022) 3:560–9. doi: 10.3390/endocrines3030047
32. Kamenicky P, Briot K, Brandi ML, Cohen-Solal M, Crowley RK, Keen R, et al. Benefit of burosumab in adults with X-linked hypophosphataemia (Xlh) is maintained with long-term treatment. RMD Open. (2023) 9:e002676. doi: 10.1136/rmdopen-2022-002676
33. Gilbey-Cross R, Sandy JL, Morris M, Cocca A, Sakka SD, Massey J, et al. (2019). Burosumab can improve pain and quality of life for children with X-linked hypophosphataemia and their families: A london centre’s experience, in: 9th International Conference on Children’s Bone Health, Salzburg, Austria. p. P74, Bone Abstracts.
34. Baroncelli GI, Grandone A, Aversa A, Sessa MR, Pelosini C, Michelucci A, et al. Safety and efficacy of burosumab in improving phosphate metabolism, bone health, and quality of life in adolescents with X-linked hypophosphatemic rickets. Eur J Med Genet. (2024) 70:104958. doi: 10.1016/j.ejmg.2024.104958
Keywords: X-linked hypophosphatemia, quality of life, complications, pain, physical function
Citation: Alsagheir AI, Bin-Abbas B, Alghamdi NM, Alhuthil RT, Murad SA, Husein TH, Mughal MZ, Alsabban ZE, Almarzoug LM and Ramzan K (2025) Quality of life of 26 family members from four generations with X-linked hypophosphatemia: a cross-sectional study. Front. Endocrinol. 16:1544016. doi: 10.3389/fendo.2025.1544016
Received: 12 December 2024; Accepted: 21 March 2025;
Published: 14 April 2025.
Edited by:
Tommaso Aversa, University of Messina, ItalyReviewed by:
Anna Grandone, University of Campania Luigi Vanvitelli, ItalyGustavo Roberto Cointry, National University of Rosario, Argentina
Copyright © 2025 Alsagheir, Bin-Abbas, Alghamdi, Alhuthil, Murad, Husein, Mughal, Alsabban, Almarzoug and Ramzan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Afaf I. Alsagheir, YXNhZ2hlaXJAa2ZzaHJjLmVkdS5zYQ==