 Liye Chong1,2†
Liye Chong1,2† Yuxing Lou3,2†
Yuxing Lou3,2† Xue Chen4†
Xue Chen4† Wenji Zhao1,2
Wenji Zhao1,2 Wei Zhang1,2
Wei Zhang1,2 Ziwei Zhang1,2
Ziwei Zhang1,2 Fan Yang1,2
Fan Yang1,2 Ping Li3,1,2*
Ping Li3,1,2*- 1Department of Endocrinology, Endocrine and Metabolic Disease Medical Center, Nanjing Drum Tower Hospital, Affiliated Hospital of Medical School, Nanjing University, Nanjing, China
- 2Nanjing Drum Tower Hospital, Branch of National Clinical Research Center for Metabolic Diseases, Nanjing, China
- 3Department of Endocrinology, Endocrine and Metabolic Disease Medical Center, Nanjing Drum Tower Hospital Clinical College of Nanjing University of Chinese Medicine, Nanjing, China
- 4Department of Pathology, Drum Tower Hospital, Medical School of Nanjing University, Nanjing, China
Context: Acromegaly is caused by somatotroph tumors. Recently, the WHO recommended the use of transcription factors (TFs) together with pituitary hormones to accurately classify the subtypes.
Objective: This study aims to evaluate differences in the clinical and prognostic characteristics of acromegaly patients with different pathological types.
Methods: A retrospective study was conducted on 94 acromegaly patients who underwent surgical treatment. Patients were classified into two groups on the basis of TFs expression by IHC. PIT1 tumors were positive only for PIT1, and PIT1/SF1 tumors were positive for both PIT1 and SF1. Additionally, on the basis of the expression of GH and PRL by IHC, PIT1 tumors were further subdivided into GH positive tumors (those positive for only GH) and GH/PRL positive tumors (those positive for both GH and PRL). Differences in clinical and prognostic features among the pathological groups were evaluated.
Results: PIT1/SF1 tumors represented 30.9% (n = 29) of the acromegaly patients in this cohort. PIT1/SF1 tumors had a higher baseline IGF-1 index (2.77 ± 0.73 vs. 2.39 ± 0.74, P = 0.024) than PIT1 tumors. Despite the higher proportion of postoperative GH < 1 μg/L, the biochemical remission rate of PIT1/SF1 tumors (30.8% vs. 27.6%, P = 0.812) was similar to that of PIT1 tumors. Compared with those with GH positive tumors, patients with GH/PRL positive tumors were younger at diagnosis (42.50 ± 13.36 vs. 49.05 ± 11.69, P = 0.046), and the proportion of male patients was higher (50.0% vs. 23.3%, P = 0.048). Furthermore, patients with GH/PRL positive tumors had a significantly higher postoperative GH level [7.30 (3.18–11.08) vs. 2.49 (1.57–6.84), P = 0.011] and IGF-1 index (1.82 ± 0.94 vs. 1.31 ± 0.63, P = 0.011) during follow-up. The biochemical remission rate in GH/PRL positive tumors was lower, but the difference was not statistically significant (18.2% vs. 37.2%, P = 0.159).
Conclusion: PIT1/SF1 tumors represent approximately 30.0% of acromegaly patients. Despite higher baseline IGF-1 levels, the clinical and prognostic features of patients with PIT1/SF1 tumors are similar to those of patients with PIT1 tumors. GH/PRL positive tumors, characterized by their earlier age at diagnosis and male predominance, tend to exhibit a lower biochemical remission rate compared to GH positive tumors.
Introduction
Acromegaly is a chronic systemic disease characterized by elevated growth hormone (GH) and insulin-like growth factor 1 (IGF-1) levels (1). Chronic excess GH and IGF-1 lead to multisystemic complications, including cardiovascular disease, osteoarthropathy, and metabolic disorders (2). More than 99% of acromegaly cases are attributed to pituitary somatotroph adenomas (3). According to the WHO classification of pituitary adenoma (PA), which is based primarily on the expression of pituitary hormones detected by immunohistochemistry (IHC), somatotroph adenomas are categorized as densely granulated somatotroph adenoma, sparsely granulated somatotroph adenoma, mammosomatotroph adenoma, or mixed somatotroph and lactotroph adenoma (4). With further studies on the pathogenesis and biological behavior of PAs, the proposal to change the nomenclature from PA to pituitary neuroendocrine tumor (PitNET) was adopted (5). Recently, the WHO updated the classification and recommended the use of pituitary transcription factors (TFs) and pituitary hormones to determine cell lineages for the subclassification of PitNETs (6). PIT1 (pituitary transcription factor 1), encoded by the POU1F1 gene, determines the differentiation of somatotrophs, lactotrophs, and thyrotrophs. SF1 (steroidogenic factor 1), encoded by the NR5A1 gene, regulates gonadotroph cell differentiation. TPIT (T-box pituitary transcription factor), encoded by the TBX19 gene, is responsible for the development of corticotroph cells.
Pituitary TFs play crucial roles in determining the differentiation of adenohypophyseal stem cells and accurately classifying the subtypes of PitNETs (6). With the routine application of TFs, studies have reported few acromegaly patients harboring atypical tumors that stain positive for both PIT1 and SF1 (PIT1/SF1 tumors) (7–9). Few studies on PIT1/SF1 tumors have been conducted and the clinical and prognostic characteristics of these tumors remain uncertain. Additionally, approximately 16–27% of acromegaly patients present elevated GH and prolactin (PRL) levels (10). The GH and PRL cosecreting tumors have garnered increased attention in recent years. However, the definition and prognosis of these tumors are still controversial.
To address these knowledge gaps, we retrospectively reviewed medical records and conducted a comprehensive comparative analysis between PIT1/SF1 and PIT1 tumors in a large cohort of operated acromegaly patients. Furthermore, according to the 2022 WHO classification of PitNETs, acromegaly patients harboring PIT1 tumors were further categorized on the basis of the expression of GH and PRL by IHC. In this study, we aimed to assess the differences in the demographic, imaging, pathological, metabolic, and prognostic features of acromegaly patients with different pathological types.
Materials and methods
Patients and inclusion criteria
We retrospectively reviewed the clinical data of acromegaly patients who underwent surgical treatment at Nanjing Drum Tower Hospital between January 2017 and May 2024. The diagnostic criteria for acromegaly were as follows (11): (1) characteristic clinical signs and symptoms related to excessive GH and IGF-1; (2) nadir GH > 1.0 μg/L during an oral glucose tolerance test (OGTT); and (3) IGF-1 levels above the upper limit of normal (ULN) adjusted for age. Patients who met the following criteria were included: (1) newly diagnosed with acromegaly who underwent surgery for somatotroph tumors; (2) sufficient preoperative and postoperative demographic, imaging, pathological, metabolic, hormonal, and prognostic data; and (3) regular follow-up at least three months after surgery. Patients who had undergone surgery, medical therapy (somatostatin receptor ligand, dopamine agonist, or GH receptor antagonist) or radiotherapy for tumors prior to hospital admission were excluded from the study.
Endocrinological evaluation and biochemical remission after the operation
All patients underwent an OGTT after an overnight fast for the diagnosis of acromegaly and assessment of glucose metabolism status. Blood samples were simultaneously collected to measure plasma glucose, insulin, and serum GH levels at 0, 30, 60, 90, and 120 minutes after the oral administration of 75 g of glucose. The nadir GH level was defined as the lowest GH level measured during the OGTT. Abnormal glucose metabolism includes impaired fasting glucose, impaired glucose tolerance, and diabetes mellitus (12). The homeostasis model assessment (HOMA) was utilized to evaluate insulin resistance and pancreatic β-cell function (13, 14).
The samples were also submitted to the same laboratory for the measurement of biochemical parameters and hormone levels. Serum GH levels were determined with a two-site chemiluminescent immunometric assay (Immulite 2000, Siemens Healthcare Diagnostics Products Limited) with a detection range of 0.05–40 μg/L and an analytical sensitivity of 0.01 μg/L. Serum IGF-1 levels were measured with a solid-phase chemiluminescent immunometric assay (Immulite 2000, Siemens Healthcare Diagnostics Products Limited) with a detection range of up to 1000 ng/mL. To ensure that the IGF-1 levels were comparable between patients, the IGF-1 index was used to standardize the IGF-1 levels. IGF-1 index= IGF-1/ULN adjusted for age. In particular, the normal range of PRL varies by sex, being 2.1–17.7 μg/L for men, 2.8–29.2 μg/L for premenopausal and nonpregnant women, 9.7–208.5 μg/L for pregnant women, and 1.8–20.3 μg/L for postmenopausal women.
Postoperative GH was defined as random serum GH within one day after surgery. Biochemical remission was defined as a random serum GH < 1.0 μg/L with normalization of serum IGF-1 adjusted for age at three months after surgery (11).
Imaging evaluation
To evaluate the imaging characteristics of tumors preoperatively and postoperatively, each patient underwent enhanced 3.0 T magnetic resonance imaging (MRI). Data on the maximum diameter of the tumor, compression of the optic nerve, invasion of the cavernous sinus, and internal carotid artery were obtained and recorded. Based on the maximum diameter, pituitary adenomas were categorized as microadenomas (< 1 cm) or macroadenomas (≥ 1 cm). The Knosp grade (grade 0–4) was determined from coronal sections of T1-weighted contrast-enhanced MRI images, and tumors were considered invasive if the Knosp grade was 3 or 4.
Pathology
The resected tissue was immediately fixed in 10% buffered formalin and subsequently embedded in paraffin. Blocks were cut into 3–4 µm paraffin sections and then stained with hematoxylin and eosin. IHC staining of the sections was uniformly performed on an automated IHC system (Ultra 60, ZSGB-Bio Co., Ltd., CHN). Immunohistochemical analysis was conducted for cytokeratin, the proliferation marker Ki-67 and hormonal markers, including GH, PRL, thyroid-stimulating hormone (TSH), luteinizing hormone (LH), follicle-stimulating hormone (FSH) and adrenocorticotropic hormone (ACTH) for each tumor. Antibodies against PIT, SF1, and TPIT (all monoclonal, prediluted, ZSGB-Bio Co., Ltd., CHN) were used to detect TF expression in the tumor samples. The sections were reviewed by a board-certified neuropathologist who was blinded to the patients’ medical records.
On the basis of the expression of TFs by IHC, acromegaly patients were classified into two groups: PIT1 tumors (defined as a positive stain only for PIT1) and PIT1/SF1 tumors (defined as a positive stain for both PIT1 and SF1). Additionally, on the basis of GH and PRL expression by IHC, PIT1 tumors were further subdivided into GH positive tumors (those positive for only GH) and GH/PRL positive tumors (those positive for both GH and PRL). Specifically, tumors in which PRL staining was positive in more than 10% of the cells were classified as PRL positive (15). GH positive tumors, also known as somatotroph tumors, include densely granulated somatotroph tumors and sparsely granulated somatotroph tumors. Multiple types of PIT1 lineage PitNETs can manifest as acromegaly and stain positive for both GH and PRL by IHC, including mammosomatotroph tumors (MSTs), mixed somatotroph and lactotroph tumors (MSLTs), and acidophil stem cell tumors. Given that precise differentiation between these tumor subtypes relies on electron microscopy, they were grouped together into GH/PRL positive tumor group.
Statistical analysis
Statistical analyses were conducted with SPSS (version 27.0, IBM, USA), and graphs were generated with GraphPad Prism (version 9.1.0.221, GraphPad, USA). Continuous variables are expressed as the mean ± standard deviation for normally distributed data or as median with interquartile range (IQR) for nonnormally distributed data. Student’s t test and the Mann–Whitney U test were performed for between-group comparisons. Categorical variables are presented as numbers and percentages and were compared by means of the chi-square test or Fisher’s exact test. A two-tailed P < 0.05 was considered to indicate statistically significant differences.
Results
Patient characteristics
In total, 109 acromegaly patients who underwent surgery were enrolled in the study, 12 patients were excluded because of insufficient medical records, and 3 patients were excluded due to a short follow-up (less than three months after surgery). Finally, 94 patients were included in the analysis. The characteristics of these patients at baseline are summarized in Table 1. The average age at diagnosis was 47.37 ± 12.25 years, with a median disease duration of 60 months. There were 64 female patients (68.1%), and the vast majority of patients (80.9%) harbored macroadenomas. At baseline, the median random GH level was 11.45 μg/L (IQR 7.05–29.23), and the nadir GH level during the OGTT was 9.34 μg/L (IQR 5.27–27.30). The mean IGF-1 index in these patients was 2.51 ± 0.75.
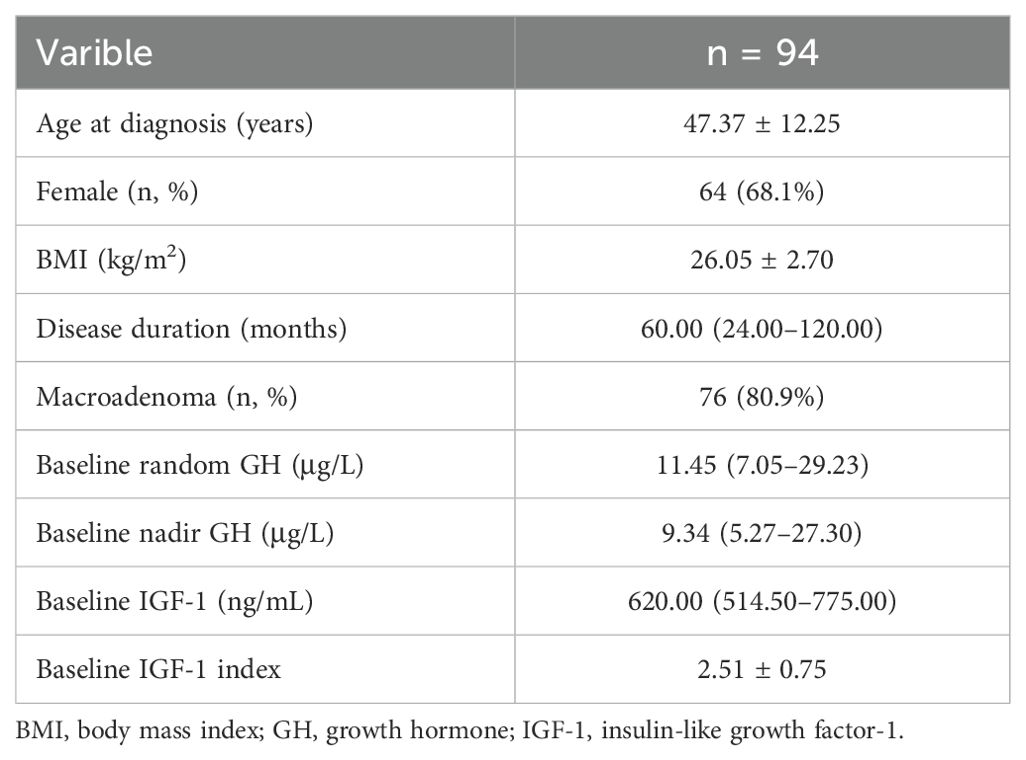
Table 1. General and clinical characteristics of acromegaly patients at baseline.
Clinical characteristics and treatment outcomes of patients with PIT1 and PIT1/SF1 tumors
All tumors were positive for PIT1 and negative for TPIT. Notably, 29 tumors stained positive for both PIT1 and SF1. Therefore, on the basis of the expression of TFs by IHC, patients were classified into PIT1 and PIT1/SF1 tumors. PIT1 tumors accounted for 69.1% (n = 65) of patients, whereas PIT1/SF1 tumors accounted for the other 30.9%. Figure 1 shows the histological appearance and immunostaining profile of a representative PIT1/SF1 tumor.
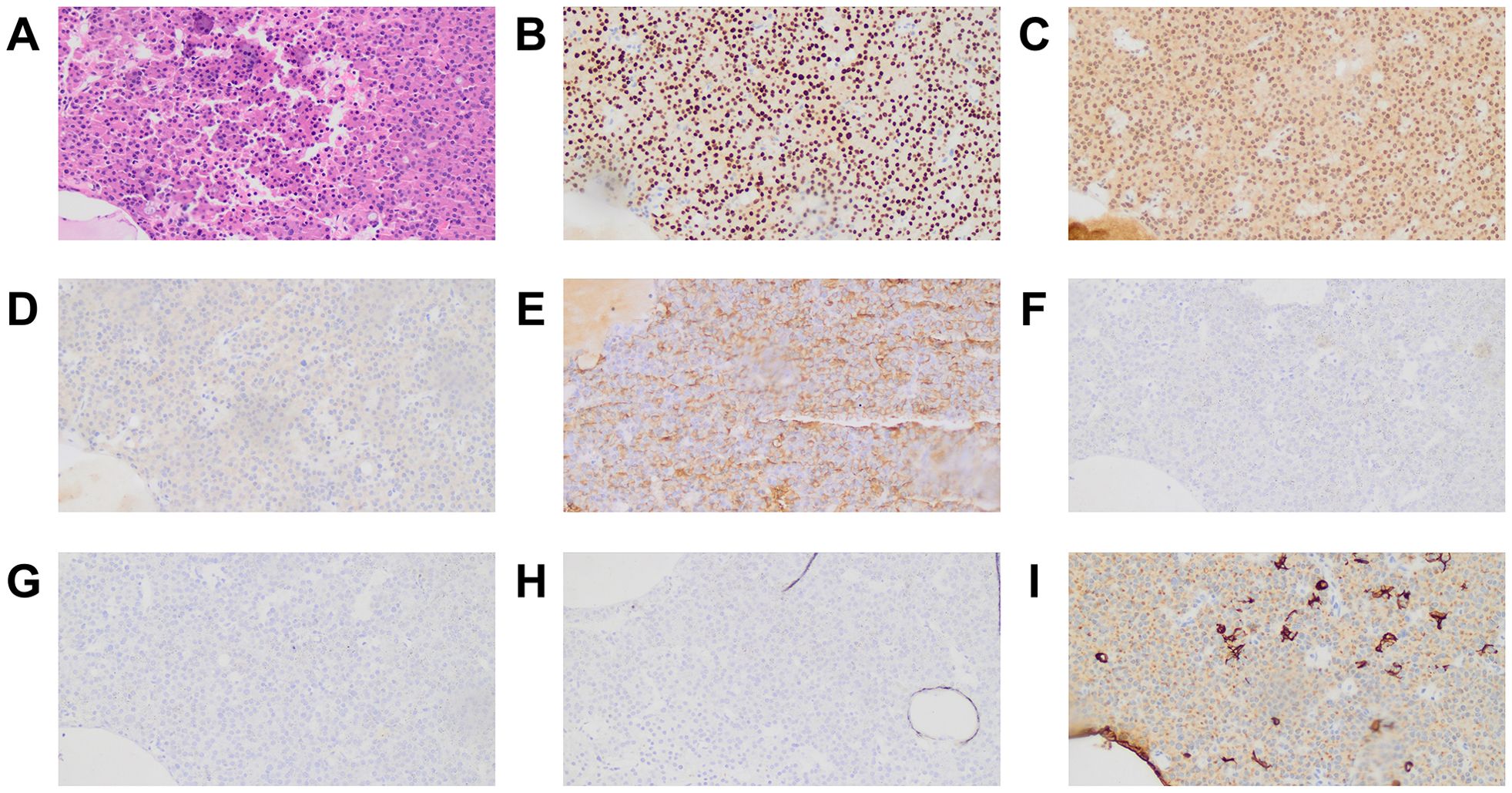
Figure 1. Histological appearance and immunostaining profile of a PIT1/SF1 tumor. The tumor was composed of a monomorphic cell population with abundant eosinophilic cytoplasm on hematoxylin and eosin stain (A). Tumor cells exhibited intense nuclear reactivity for PIT1 (B) and SF1 (C) but were negative for TPIT (D). Immunostaining for GH was obviously and diffusely positive (E), whereas immunostaining for PRL (F), FSH (G) and LH (H) was negative. Some tumor cells displayed a perinuclear positivity pattern on CK immunostaining, whereas others presented fibrous bodies in the cytoplasm (I). All microphotographs were performed on magnification ×200.
As shown in Table 2, there were no significant differences in demographic characteristics, including age at diagnosis, sex distribution, BMI and disease duration, between the two groups. The PIT1 tumors did not differ from the PIT1/SF1 tumors in terms of tumor diameter or the proportions of macroadenomas and invasive tumors (Knosp grade 3–4). Since SF1 regulates gonadotroph cell differentiation, we assessed the expression of FSH and LH in these tumors. Only 2 of 29 tumors in the PIT/SF1 group stained positive for FSH or LH, while the expression of these hormones was absent in the PIT1 tumors. Pathological studies revealed that PIT1/SF1 tumors more frequently displayed a perinuclear cytokeratin pattern than PIT1 tumors (86.2% vs. 52.3%, P = 0.002), whereas few PIT1/SF1 tumors presented fibrous bodies in the cytoplasm. The glucose metabolism status and pancreatic β-cell function, as evaluated by HOMA-β, were similar in the two cohorts. Moreover, no differences in the incidence of abnormal glucose metabolism or hypertension were found between PIT1 and PIT1/SF1 tumors.
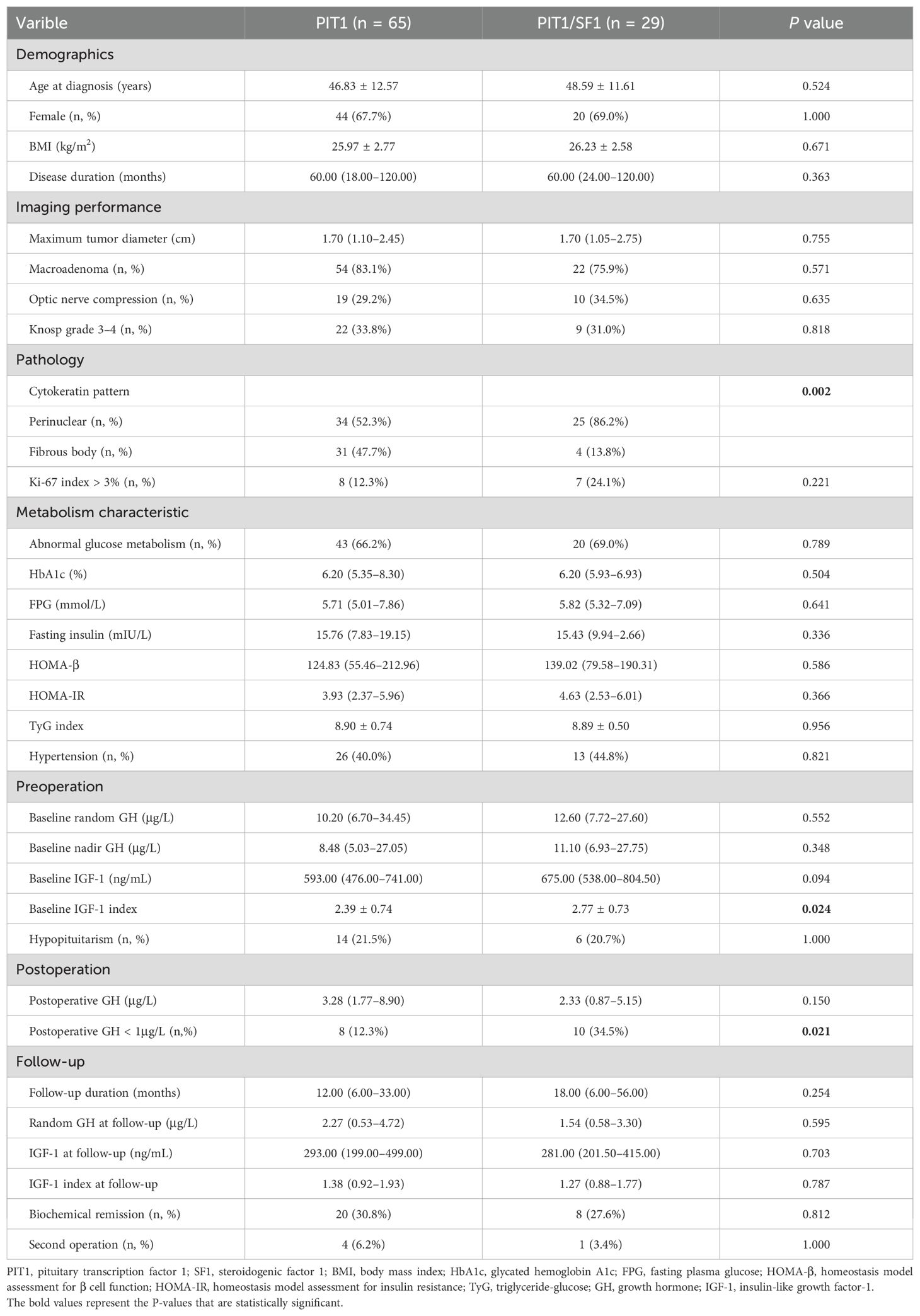
Table 2. Clinical characteristics and treatment outcomes of patients with PIT1 and PIT1/SF1 tumors.
Before surgery, although baseline random GH and nadir GH levels during the OGTT did not significantly differ among the groups, we observed that PIT1/SF1 tumors had a higher IGF-1 level [675.00 (538.00–804.50) vs. 593.00 (476.00–741.00), P = 0.094] and mean IGF-1 index (2.77 ± 0.73 vs. 2.39 ± 0.74, P = 0.024) than PIT1 tumors at baseline. After surgery, the serum GH levels were significantly decreased, and the proportion of postoperative GH < 1 μg/L among the PIT1/SF1 tumors was higher than that among the PIT1 tumors (34.5% vs. 12.3%, P = 0.021). However, the differences in the biochemical remission rates did not reach significance (30.8% vs. 27.6%, P = 0.812). More details regarding treatment outcomes are listed in Table 2.
Clinical characteristics and treatment outcomes of patients with GH positive and GH/PRL positive tumors
PIT1 tumors were further subdivided into GH positive and GH/PRL positive tumors on the basis of the expression of GH and PRL by IHC. Forty-three patients (66.2%) had GH positive tumors, whereas 22 patients (33.8%) had GH/PRL positive tumors. Figure 2 illustrates the patterns and incidences of these subgroups. Moreover, we further characterized the GH and PRL staining profiles of PIT1/SF1 tumors. Among 29 PIT1/SF1 tumors, 62.1% (n = 18) stained positive for only GH and 37.9% (n = 11) stained positive for both GH and PRL, which was similar to the staining profiles observed in the PIT1 tumors (62.1% vs. 66.2% P = 0.816).
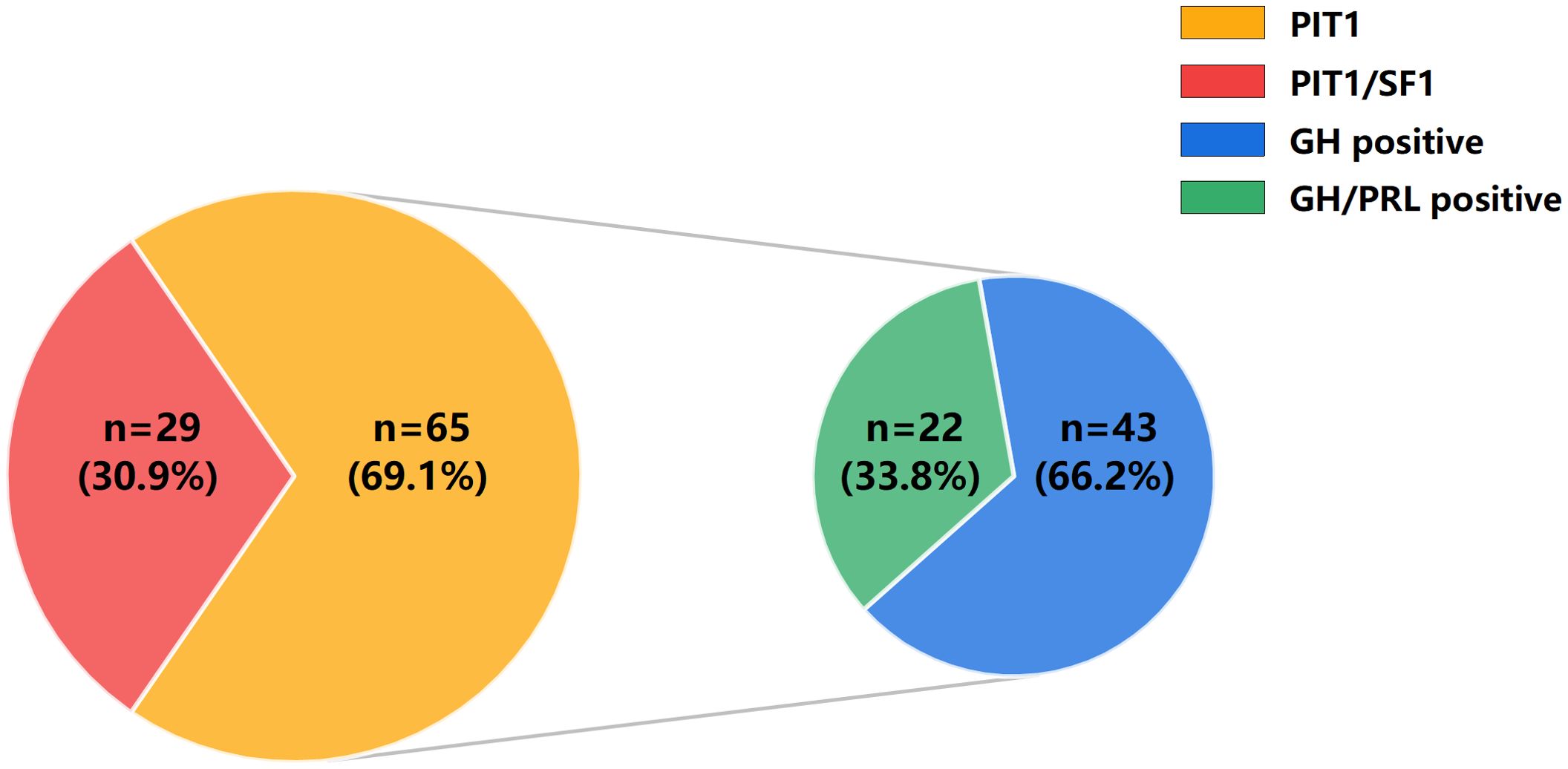
Figure 2. Distribution of acromegaly patients with different pathological types. On the basis of the expression of TFs by IHC, 94 acromegaly patients were classified into PIT1 tumors (n = 65) and PIT1/SF1 tumors (n = 29). Additionally, on the basis of the expression of GH and PRL by IHC, PIT1 tumors were further subdivided into GH positive tumors (n = 43) and GH/PRL positive tumors (n = 22).
The demographic, imaging, pathological, metabolic characteristics, and treatment outcomes of these patients are presented in Table 3. The age at diagnosis was lower in the GH/PRL positive tumors than in the GH positive tumors (42.50 ± 13.36 vs. 49.05 ± 11.69, P = 0.046). Moreover, the proportion of male patients in the GH/PRL positive tumors was higher than that in the GH positive tumors (50.0% vs. 23.3%, P = 0.048). In terms of imaging characteristics, no differences were found among the groups regarding tumor diameter or the prevalence of macroadenomas or invasive tumors. IHC staining of resected samples revealed that GH/PRL positive tumors more frequently exhibited a perinuclear cytokeratin pattern than GH positive tumors (86.4% vs. 34.9%, P < 0.001). With respect to metabolic comorbidities, we observed no significant difference in glucose metabolism parameters or the incidence of hypertension between the two groups.
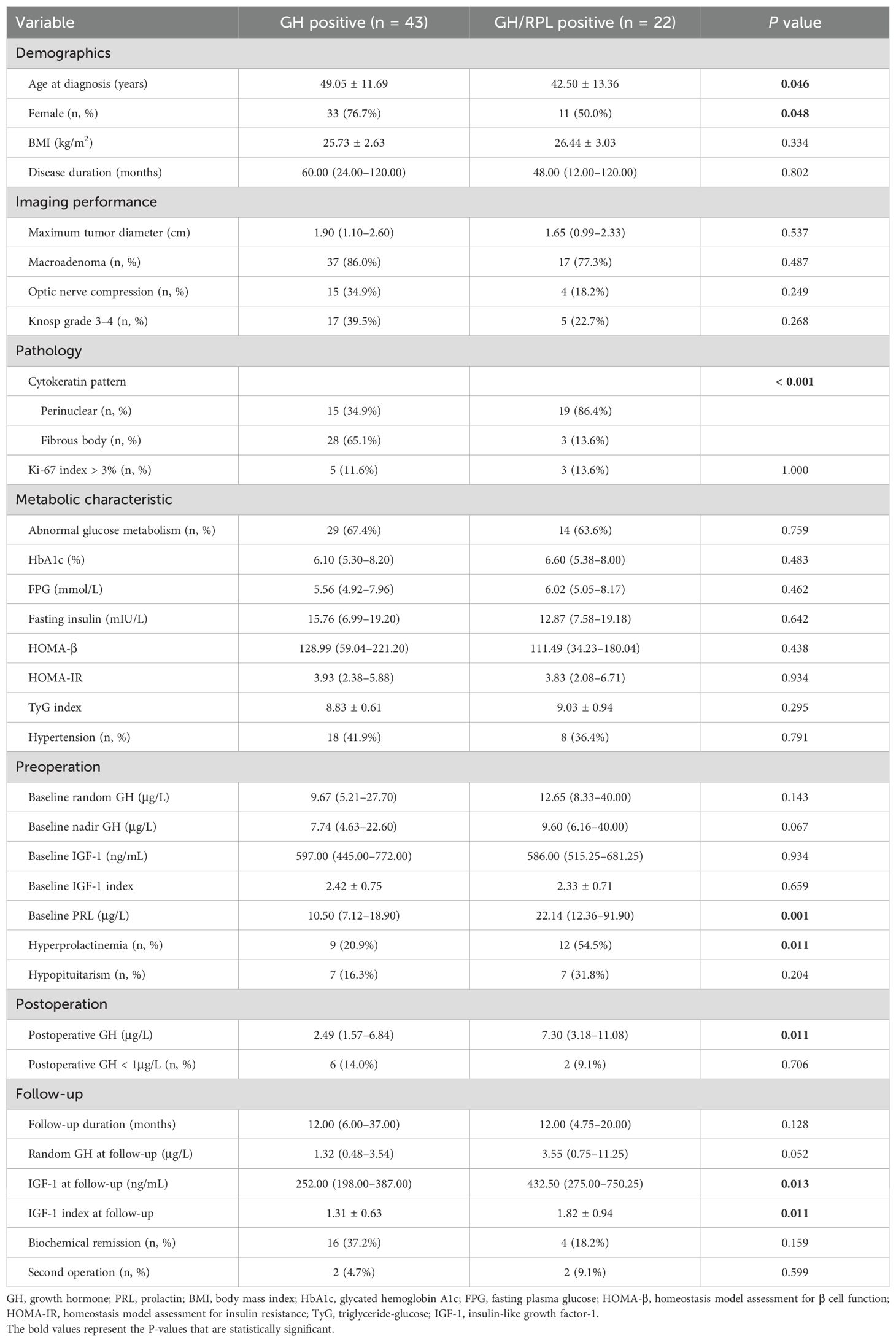
Table 3. Clinical characteristics and treatment outcomes of patients with GH positive and GH/PRL positive tumors.
Preoperative endocrine assessments revealed that the two groups had similar random GH levels, nadir GH levels, and IGF-1 index at baseline. As expected, hyperprolactinemia was more common in patients with GH/PRL positive tumors (54.5% vs. 20.9%, P = 0.011), and the median PRL level was significantly higher in the GH/PRL positive tumors than in the GH positive tumors [22.14 (12.36–91.90) vs. 10.50 (7.12–18.90), P = 0.001]. After surgery, the patients with GH/PRL positive tumors had significantly higher postoperative GH levels [7.30 (3.18–11.08) vs. 2.49 (1.57–6.84), P = 0.011] and IGF-1 index during follow-up (1.82 ± 0.94 vs. 1.31 ± 0.63, P = 0.011). Although there was no significant difference, patients with GH/PRL positive tumors tended to have a lower biochemical remission rate (18.2% vs. 37.2%, P = 0.159) than patients with GH positive tumors.
Discussion
To our knowledge, this is the first study to systematically investigate the clinicopathological characteristics and treatment outcomes of acromegaly patients with PIT1/SF1 tumors. Additionally, PIT1 tumors were further subdivided into GH positive and GH/PRL positive tumors on the basis of the expression of GH and PRL by IHC, and the clinical and prognostic characteristics of the groups were compared. The results indicated that 1) PIT1/SF1 tumors, as a specific subtype of plurihormonal tumors, were not rare and represented approximately 30.0% of the acromegaly patients in this cohort. Furthermore, despite higher baseline IGF-1 levels, the demographic, imaging, metabolic, and prognostic features of patients with PIT1/SF1 tumors were similar to those of patients with PIT1 tumors in the context of acromegaly. 2) Compared with those with GH positive tumors, patients with GH/PRL positive tumors, characterized by their earlier age at diagnosis and male predominance, tended to exhibit a lower biochemical remission rate.
Acromegaly is a heterogeneous disease with complex pathological subtypes. The WHO classification categorizes tumors composed of a single cell population expressing multiple TFs as plurihormonal tumors (6). PIT1/SF1 tumors, the main subtype of plurihormonal tumors in acromegaly, were previously considered rare (8). Based on pangenomic analysis of 134 PitNETs, Neou et al. first reported that the gonadotroph marker NR5A1, which encodes SF1, was also expressed in GNAS wild-type somatotroph tumors (16). Rymuza et al. subsequently analyzed the RNA sequencing and genome-wide DNA methylation results of 48 somatotroph tumors and confirmed that there was a group of tumors co-expressing NR5A1 (17, 18). Recently, Dottermusch et al. summarized previous research and proposed the term “somatogonadotroph PitNET” for tumors with PIT1/SF1 co−expression (19).
The incidence of PIT1/SF1 tumors varies across studies and ranges from 0.5% to 8% in studies conducted on all types of pituitary adenomas with or without function (20, 21). Dottermusch et al. first reported that 52% of somatotroph tumors stained positive for PIT1 and SF1 (19). In our cohort, PIT1/SF1 tumors accounted for approximately 30.0% of acromegaly patients, which is lower than the prevalence reported in prior study. Geographic and ethnic disparities and the divergence of the study subjects may have contributed to this difference (22). The Dottermusch et al. study solely focused on somatotroph tumors and excluded MSTs and MSLTs, whereas in our study, we included all of the above. Therefore, strict inclusion criteria may inflate the prevalence of PIT1/SF1 tumors, and further studies are necessary to validate our findings.
To date, there have been few studies in which the clinicopathological features of acromegaly patients with PIT1/SF1 tumors were investigated. One study by a Chinese neurosurgical team revealed that PIT1/SF1 tumors exhibited more aggressive behavior and had a poorer prognosis (more postoperative complications but similar biochemical remission) than PIT1 tumors in a cohort of 215 PitNETs (21). Furthermore, the team retrospectively reviewed the clinical data of 193 patients with growth hormone-secreting pituitary adenoma (GHPA) and reported that the tumor sizes of GHPAs with multiple TF positivity were smaller and that these tumors were less invasiveness; however, the rate of hormonal remission was lower than that for GHPAs with only PIT1 positivity (23). In contrast to earlier findings, our results revealed that the clinical and prognostic features of patients with PIT1/SF1 tumors were similar to those of patients with PIT1 tumors. The discrepancy in the subjects significantly contributed to the differences between our study and other studies. In contrast to our study which focused on acromegaly, Wang et al. included nonfunctional adenomas, and the proportion of nonfunctional adenomas among PIT1/SF1 tumors was significantly higher than that among PIT1 tumors (40.4% vs. 18.2%, P = 0.003) (21). The study has shown that nonfunctional macroadenomas in surgically treated patients tended to have larger tumor diameters and be more invasive than GH macroadenomas (24). On the other hand, in the study by Zhang et al., GHPAs with multiple TF positivity included not only PIT1/SF1 tumors (63.5%) but also PIT1/TPIT tumors (28.8%) and PIT1/SF1/TPIT tumors (7.7%). As mentioned in the discussion, the expression of ACTH in GHPAs with multiple TF positivity may lead to a poorer prognosis (23). Furthermore, SF1 tumors frequently manifest as nonfunctional tumors with normal serum FSH and LH levels (6). Consequently, PIT1/SF1 tumors typically present with clinical manifestations related only to excess GH in acromegaly (25, 26). The above evidence also supports our findings.
For the first time, we found that acromegaly patients with PIT1/SF1 tumors had significantly higher baseline IGF-1 levels than patients with PIT1 tumors. We suspect that the gastric inhibitory polypeptide receptor (GIPR) plays a role in it. Upon binding GIP, GIPR activates the signaling pathway and ultimately promotes GH synthesis and secretion (27). Therefore, GIPR+ somatotroph adenomas had higher baseline serum IGF-1 levels than GIPR- adenomas (28). Recent research has demonstrated that SF1 is closely correlated with GIPR (29). Consistent with prior research, Rymuza et al. reported that GIPR-high somatotroph tumors also highly expressed NR5A1 and concluded that activated GIPR related signaling pathways triggered the expression of NR5A1 (17). Collectively, the high expression of GIPR in PIT1/SF1 tumors may account for higher IGF-1 levels than in PIT1 tumors in acromegaly.
GH and PRL cosecreting tumors have emerged as a focus in recent years, but the precise definition of these tumors is still lacking in the current literature. Some previous studies classified somatotroph tumors with hyperprolactinemia as GH and PRL cosecreting tumors (10, 30), whereas other authors considered cosecreting tumors when tumors stained positive for both GH and PRL by IHC (31–33). In the latest multicenter retrospective study conducted on 604 acromegaly patients, the authors suggested that cosecreting tumors should be considered when the tumors stained positive for GH and PRL with elevated PRL levels or serum PRL levels above 100 ng/dL, regardless of the IHC result of PRL (34). The limitation of the above inclusion criteria, as noted by Wildemberg et al., is that some somatotroph tumors with intratumoral PRL expression in up to 5% of cells have elevated PRL levels due to the stalk effect, and some patients with GH/PRL positive tumors may not present with hyperprolactinemia (35). Given that the 2022 WHO classification recommends categorizing PitNETs on the basis of pituitary TFs combined with classical pituitary hormones and the cutoff of 10% for PRL positive cells favors the diagnosis of somatolactotroph tumors (36), we consider the PIT1 tumor to be a GH/PRL positive tumor when more than 10% of cells are positive for PRL by IHC.
This study is the first to evaluate the clinical and prognostic characteristics of GH/PRL positive tumors in the context of PIT1 positivity. In accordance with previous studies (10, 30, 34), we found that patients with GH/PRL positive tumors were younger at diagnosis than those with GH-positive tumors and predominantly male. Moreover, there is a debate regarding the tumor size and invasiveness of GH/PRL positive tumors. Some studies reported that these tumors were larger and more invasive (30, 34), while others did not find a difference (31, 32). In our series, no differences were found in tumor diameter or the prevalence of invasive tumors between GH/PRL positive and GH positive tumors. The possible reason for this discrepancy is the varying proportions of MSLTs among the subjects in different studies. Lv et al. demonstrated that MSLTs were aggressive tumors characterized by a larger tumor size, greater invasiveness, and worse biochemical remission (33). We hypothesize that the proportion of MSLTs is lower in our series.
Whether somatotroph tumors cosecreting PRL affect biochemical remission remains controversial (37). Araujo and Wang (10, 34) detected no difference in biochemical remission rates between GH/PRL positive and GH positive tumors. In contrast, a worse prognosis in patients with GH/PRL positive tumors has been reported in other studies (38). Our results revealed that patients with GH/PRL positive tumors had significantly higher IGF-1 levels during follow-up and tended to exhibit a lower biochemical remission rate than those with GH positive tumors, although the difference was not significant. Differences across studies probably resulted from the divergence of study subjects, remission criteria, and follow-up durations. Therefore, patients with GH and PRL stained tumors seem to require more time to achieve long-term biochemical remission (31), and we suggest that patients with these tumors require a close follow-up.
The current study has some limitations. As a single-center retrospective study, it was subject to the inherent limitations of retrospective analysis and possibly led to selection bias compared with multicenter research. Additionally, the relatively small sample size was a major limitation of our study. A considerable number of patients were excluded from the study because of insufficient medical records. Furthermore, the short follow-up duration also limits our ability to assess long-term prognosis. Multicenter prospective studies with long follow-up are needed to evaluate the clinical and prognostic characteristics of patients with different pathological types in acromegaly.
Conclusion
PIT1/SF1 tumors, as a specific subtype of plurihormonal tumors, are not rare among acromegaly patients. Additional studies are necessary to clarify whether PIT1/SF1 tumors have distinctive clinical and prognostic features in acromegaly. Patients with GH/PRL positive tumors differ from those with GH positive tumors in terms of clinical manifestations and prognosis, and these patients may require more attention during long-term follow-up.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving humans were approved by Ethics committee of Nanjing Drum Tower Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
LC: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Software, Visualization, Writing – original draft, Writing – review & editing. YL: Conceptualization, Data curation, Formal analysis, Methodology, Writing – original draft. XC: Investigation, Methodology, Writing – original draft, Data curation, Software. WJZ: Investigation, Software, Writing – original draft. WZ: Investigation, Software, Writing – original draft. ZZ: Project administration, Resources, Writing – review & editing. FY: Supervision, Validation, Writing – review & editing. PL: Conceptualization, Funding acquisition, Resources, Supervision, Validation, Writing – review & editing, Writing – original draft.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the National Key Research and Development Program of China (grant number 2022YFC2505306) and the Nanjing Health Technology Development Project (grant number ZKX23021).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Fleseriu M, Langlois F, Lim DST, Varlamov EV, Melmed S. Acromegaly: pathogenesis, diagnosis, and management. Lancet Diabetes Endocrinol. (2022) 10:804–26. doi: 10.1016/s2213-8587(22)00244-3
2. Gadelha MR, Kasuki L, Lim DST, Fleseriu M. Systemic complications of acromegaly and the impact of the current treatment landscape: an update. Endocr Rev. (2019) 40:268–332. doi: 10.1210/er.2018-00115
3. Zendran I, Gut G, Kaluzny M, Zawadzka K, Bolanowski M. Acromegaly caused by ectopic growth hormone releasing hormone secretion: A review. Front Endocrinol (Lausanne). (2022) 13:867965. doi: 10.3389/fendo.2022.867965
4. Lopes MBS. The 2017 World Health Organization classification of tumors of the pituitary gland: a summary. Acta Neuropathol. (2017) 134:521–35. doi: 10.1007/s00401-017-1769-8
5. Asa SL, Casar-Borota O, Chanson P, Delgrange E, Earls P, Ezzat S, et al. From pituitary adenoma to pituitary neuroendocrine tumor (PitNET): an International Pituitary Pathology Club proposal. Endocr Relat Cancer. (2017) 24:C5–8. doi: 10.1530/ERC-17-0004
6. Asa SL, Mete O, Perry A, Osamura RY. Overview of the 2022 WHO classification of pituitary tumors. Endocr Pathol. (2022) 33:6–26. doi: 10.1007/s12022-022-09703-7
7. Asa SL, Mete O, Riddle ND, Perry A. Multilineage pituitary neuroendocrine tumors (PitNETs) expressing PIT1 and SF1. Endocr Pathol. (2023) 34:273–8. doi: 10.1007/s12022-023-09777-x
8. Tang H, Wang X, Bie Z, Yang Z, Wang B, Li S, et al. Pituitary adenomas with multiple cell lineage combinations: clinicopathological features and short-term prognosis. J Neurosurg. (2023) 139:810–21. doi: 10.3171/2022.12.JNS222118
9. Fookeerah P, Varikatt W, Shingde M, Dexter MAJ, McLean M. Somatostatin receptor expression and clinical outcome of multilineage pituitary tumours expressing PIT1 and SF1. Endocr Connect. (2023) 12:e230328. doi: 10.1530/EC-23-0328
10. Wang M, Mou C, Jiang M, Han L, Fan S, Huan C, et al. The characteristics of acromegalic patients with hyperprolactinemia and the differences in patients with merely GH-secreting adenomas: clinical analysis of 279 cases. Eur J Endocrinol. (2012) 166:797–802. doi: 10.1530/eje-11-1119
11. Katznelson L, Laws ER Jr, Melmed S, Molitch ME, Murad MH, Utz A, et al. Acromegaly: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. (2014) 99:3933–51. doi: 10.1210/jc.2014-2700
12. Alberti KG, Zimmet PZ. Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1: diagnosis and classification of diabetes mellitus provisional report of a WHO consultation. Diabetes Med. (1998) 15:539–53. doi: 10.1002/(sici)1096-9136(199807)15:7<539::Aid-dia668>3.0.Co;2-s
13. Guerrero-Romero F, Simental-Mendia LE, Gonzalez-Ortiz M, Martinez-Abundis E, Ramos-Zavala MG, Hernandez-Gonzalez SO, et al. The product of triglycerides and glucose, a simple measure of insulin sensitivity. Comparison with the euglycemic-hyperinsulinemic clamp. J Clin Endocrinol Metab. (2010) 95:3347–51. doi: 10.1210/jc.2010-0288
14. Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. (1985) 28:412–9. doi: 10.1007/bf00280883
15. Villa C, Vasiljevic A, Jaffrain-Rea ML, Ansorge O, Asioli S, Barresi V, et al. A standardised diagnostic approach to pituitary neuroendocrine tumours (PitNETs): a European Pituitary Pathology Group (EPPG) proposal. Virchows Arch. (2019) 475:687–92. doi: 10.1007/s00428-019-02655-0
16. Neou M, Villa C, Armignacco R, Jouinot A, Raffin-Sanson ML, Septier A, et al. Pangenomic classification of pituitary neuroendocrine tumors. Cancer Cell. (2020) 37:123–34 e5. doi: 10.1016/j.ccell.2019.11.002
17. Rymuza J, Kober P, Rusetska N, Mossakowska BJ, Maksymowicz M, Nyc A, et al. Transcriptomic classification of pituitary neuroendocrine tumors causing acromegaly. Cells. (2022) 11:3846. doi: 10.3390/cells11233846
18. Kober P, Rymuza J, Baluszek S, Maksymowicz M, Nyc A, Mossakowska BJ, et al. DNA methylation pattern in somatotroph pituitary neuroendocrine tumors. Neuroendocrinology. (2024) 114:51–63. doi: 10.1159/000533692
19. Dottermusch M, Ryba A, Ricklefs FL, Flitsch J, Schmid S, Glatzel M, et al. Pituitary neuroendocrine tumors with PIT1/SF1 co-expression show distinct clinicopathological and molecular features. Acta Neuropathol. (2024) 147:16. doi: 10.1007/s00401-024-02686-1
20. Mete O, Cintosun A, Pressman I, Asa SL. Epidemiology and biomarker profile of pituitary adenohypophysial tumors. Modern Pathol. (2018) 31:900–9. doi: 10.1038/s41379-018-0016-8
21. Wang X, Tang H, Bie Z, Wang Y, Yuan R, Zhang Z, et al. Clinical and pathological features of pit1/SF1 multilineage pituitary neuroendocrine tumor. Neurosurgery. (2024) 95:94–102. doi: 10.1227/neu.0000000000002846
22. Lavrentaki A, Paluzzi A, Wass JA, Karavitaki N. Epidemiology of acromegaly: review of population studies. Pituitary. (2017) 20:4–9. doi: 10.1007/s11102-016-0754-x
23. Zhang Y, Tang H, Li S, Bie Z, Ma X, Wu H, et al. Co-expression of multiple transcription factors is associated with clinical features and endocrine prognosis in growth hormone-secreting pituitary adenomas. Endocrine. (2024) 87:788–99. doi: 10.1007/s12020-024-04082-x
24. Zada G, Lin N, Laws ER Jr. Patterns of extrasellar extension in growth hormone-secreting and nonfunctional pituitary macroadenomas. Neurosurg Focus. (2010) 29:E4. doi: 10.3171/2010.7.FOCUS10155
25. Mihajlovic M, Pekic S, Doknic M, Stojanovic M, Rasic D, Miljic D, et al. Plurihormonal pituitary neuroendocrine tumours - A single centre experience. Int J Surg Pathol. (2024) 32:470–7. doi: 10.1177/10668969231183712
26. Dogukan FM, Karatay H, Yuzkan S, Burhan S, Erkan B, Yilmaz-Ozguven B. Clinicopathological correlates of PIT1 and SF1-multilineage pituitary neuroendocrine tumors and the diagnostic utility of NKX2.2 immunohistochemistry in pituitary pathology. Arch Pathol Lab Med. (2024) 32:470–7. doi: 10.5858/arpa.2023-0543-OA
27. Hage M, Janot C, Salenave S, Chanson P, Kamenický P. MANAGEMENT OF ENDOCRINE DISEASE: Etiology and outcome of acromegaly in patients with a paradoxical GH response to glucose. Eur J Endocrinol. (2021) 184:R261–r8. doi: 10.1530/eje-20-1448
28. Hage M, Chaligné R, Viengchareun S, Villa C, Salenave S, Bouligand J, et al. Hypermethylator phenotype and ectopic GIP receptor in GNAS mutation-negative somatotropinomas. J Clin Endocrinol Metab. (2019) 104:1777–87. doi: 10.1210/jc.2018-01504
29. Bates HE, Campbell JE, Ussher JR, Baggio LL, Maida A, Seino Y, et al. Gipr is essential for adrenocortical steroidogenesis; however, corticosterone deficiency does not mediate the favorable metabolic phenotype of Gipr(-/-) mice. Diabetes. (2012) 61:40–8. doi: 10.2337/db11-1060
30. Guo X, Zhang R, Zhang D, Wang Z, Gao L, Yao Y, et al. Hyperprolactinemia and hypopituitarism in acromegaly and effect of pituitary surgery: long-term follow-up on 529 patients. Front Endocrinol (Lausanne). (2021) 12:807054. doi: 10.3389/fendo.2021.807054
31. Rick J, Jahangiri A, Flanigan PM, Chandra A, Kunwar S, Blevins L, et al. Growth hormone and prolactin-staining tumors causing acromegaly: a retrospective review of clinical presentations and surgical outcomes. J Neurosurg. (2019) 131:147–53. doi: 10.3171/2018.4.Jns18230
32. Varlamov EV, Wood MD, Netto JP, Thiessen J, Kim J, Lim DST, et al. Cystic appearance on magnetic resonance imaging in bihormonal growth hormone and prolactin tumors in acromegaly. Pituitary. (2020) 23:672–80. doi: 10.1007/s11102-020-01075-7
33. Lv L, Jiang Y, Yin S, Hu Y, Chen C, Ma W, et al. Mammosomatotroph and mixed somatotroph-lactotroph adenoma in acromegaly: a retrospective study with long-term follow-up. Endocrine. (2019) 66:310–8. doi: 10.1007/s12020-019-02029-1
34. Araujo-Castro M, Biagetti B, Menéndez Torre E, Novoa-Testa I, Cordido F, Pascual Corrales E, et al. Differences between GH- and PRL-cosecreting and GH-secreting pituitary adenomas: a series of 604 cases. J Clin Endocrinol Metab. (2024) 109:e2178–e87. doi: 10.1210/clinem/dgae126
35. Wildemberg LE, Gadelha MR. GH and prolactin co-secreting adenomas: it is time for a definition. J Clin Endocrinol Metab. (2024) 110:e192–3. doi: 10.1210/clinem/dgae262
36. Van Laethem D, Michotte A, Cools W, Velkeniers B, Unuane D, Andreescu CE, et al. Hyperprolactinemia in acromegaly is related to prolactin secretion by somatolactotroph tumours. Horm Metab Res. (2020) 52:647–53. doi: 10.1055/a-1207-1132
37. Agrawal N, Ioachimescu AG. Prognostic factors of biochemical remission after transsphenoidal surgery for acromegaly: a structured review. Pituitary. (2020) 23:582–94. doi: 10.1007/s11102-020-01063-x
Keywords: acromegaly, PIT1/SF1 tumor, GH/PRL positive tumor, clinical characteristics, prognosis
Citation: Chong L, Lou Y, Chen X, Zhao W, Zhang W, Zhang Z, Yang F and Li P (2025) Comparison of the clinical and prognostic characteristics of patients with different pathological types in acromegaly. Front. Endocrinol. 16:1571598. doi: 10.3389/fendo.2025.1571598
Received: 05 February 2025; Accepted: 15 April 2025;
Published: 12 May 2025.
Edited by:
Sadishkumar Kamalanathan, Jawaharlal Institute of Postgraduate Medical Education and Research (JIPMER), IndiaReviewed by:
Somdatta Giri, Kalyani (AIIMS Kalyani), IndiaAnupriya Kaliyappan, All India Institute of Medical Sciences Jodhpur, India
Copyright © 2025 Chong, Lou, Chen, Zhao, Zhang, Zhang, Yang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ping Li, bGk3ODMyMUB5ZWFoLm5ldA==
†These authors have contributed equally to this work and share first authorship