 Mingxiao Li
Mingxiao Li Yili Xiao1†
Yili Xiao1†- 1Medical School, Hunan University of Chinese Medicine, Changsha, China
- 2Department of Cardiology, The First Affiliated Hospital of Hunan University of Chinese Medicine, Changsha, China
- 3The Domestic First-class Discipline Construction Project of Chinese Medicine of Hunan University of Chinese Medicine, Changsha, China
Atherosclerosis (AS) is a global public health concern and involves a complex pathogenesis characterized by lipid abnormalities, oxidative stress, and inflammatory responses at the cellular and molecular levels. The crosstalk between the endoplasmic reticulum (ER) and mitochondria, mediated by mitochondria-associated membranes (MAMs), plays a critical role in the pathogenesis of atherosclerosis. As two key cellular organelles, the ER and mitochondria interact physically and functionally through MAMs, which serve as bridges between their close contact and interdependence. MAMs maintain lipid homeostasis, promote calcium ion transport, the oxidative stress response, apoptosis, and autophagy. Recent studies have highlighted the significance of ER-mitochondria crosstalk in the progression of AS, as indicated by mitochondrial and ER structural and functional integrity, redox homeostasis, and calcium homeostasis. This review comprehensively explores the novel mechanisms of ER-mitochondria crosstalk in AS and emphasizes the potential of MAMs as therapeutic targets, aiming to provide new perspectives and strategies for the treatment of cardiovascular diseases.
1 Introduction
Atherosclerosis (AS), characterized by lipid accumulation, chronic inflammation, and vascular dysfunction, is a complex pathological process and a core contributor to global cardiovascular disease-related disability and mortality (1). With improvements in living standards and dietary changes, its incidence has increased annually, severely threatening human quality of life (2). According to the 2019 Global Burden of Disease (GBD) statistics, approximately one-third of annual deaths are attributed to cardiovascular diseases, with AS-related complications being the dominant factor (3). Although statins, anti-inflammatory therapies, and vascular intervention technologies have significantly improved clinical outcomes, acute cardiovascular events (e.g., myocardial infarction and stroke) caused by plaque instability remain a major challenge (4). Therefore, there is an urgent need to explore the molecular mechanisms underlying AS.
Traditionally, AS research has focused on singular pathological processes such as the oxidation of low-density lipoprotein (LDL), macrophage foam cell transformation, and smooth muscle cell proliferation; however, the dynamic interactions between organelles that systemically facilitate disease progression have been overlooked. Recent breakthroughs in super-resolution microscopy, spatial transcriptomics, and protein interactionomics have propelled the study of organelle communication networks to the forefront of life sciences (5). Notably, the physical and functional coupling between the endoplasmic reticulum (ER) and mitochondria via mitochondria-associated membranes (MAMs) has been identified as a core hub for coordinating lipid metabolism, calcium signaling, oxidative stress, and cell fate decisions (6–9), offering a novel perspective for AS research.
The ER, the largest membrane system in the cell, performs fundamental functions such as protein synthesis and modification, lipid generation, and calcium storage, while also serving as a platform for stress sensing and signal integration (10). Mitochondria drive energy metabolism and inflammatory responses via oxidative phosphorylation and reactive oxygen species (ROS) generation (11). Over the past decade, research has revealed that the ER and mitochondria are not independent but are functionally coupled through MAMs. MAMs are dynamic structures formed by specific protein anchors between the ER and mitochondrial membranes, enabling the efficient transport of lipids, calcium ions, and signaling molecules (12). For example, calcium flux mediated by the IP3R-GRP75-voltage-dependent anion channel (VDAC) complex (13), mitochondrial autophagy promoted by FUN14 domain-containing protein 1 (FUNDC1) (6), and membrane fusion events involving Mitofusin 2 (Mfn2) (7) depend on the precise spatiotemporal regulation of MAMs.
AS is a vascular pathology driven by metabolic and inflammatory synergies. Pathological conditions, such as endothelial cell damage induced by oxidized LDL (ox-LDL), macrophage inflammatory polarization, and smooth muscle cell phenotype switching, are accompanied by ER stress and mitochondrial dysfunction (14, 15). Recent studies have identified multiple mechanisms by which MAMs contribute to these processes: (1) lipid metabolism, where MAM-enriched enzymes such as ACAT1 and phosphatidylethanolamine N-methyltransferase 2 (PEMT2) facilitate cholesterol esterification and phospholipid remodeling, promoting lipid droplet formation and foam cell transformation (16); (2) inflammation regulation, where MAMs serve as a platform for NLRP3 inflammasome assembly, amplifying inflammatory signals through calcium overload and mitochondrial reactive oxygen species (mtROS) bursts (17); (3) apoptosis, where MAMs promote interactions between Bcl-2 family proteins and mitochondrial membrane permeability transitions, determining cellular survival within plaques (18).
Despite significant progress, many gaps remain in our understanding of the molecular mechanisms of MAMs in AS. For instance, how do MAMs dynamically assemble in response to mechanical stimuli such as blood flow shear forces? Do specific cell types (e.g., endothelial cells and macrophages) exhibit spatial heterogeneity in MAM function? Are there temporal heterogeneities in the MAMs function across different disease phases? Existing drugs (e.g., statins (19) and metformin (20)) have been shown to modulate ER-mitochondrial crosstalk by targeting key MAM proteins (e.g., VDAC1 and Mfn2); however, their multi-target characteristics may lead to off-target effects, highlighting the need for developing specific MAM-targeted therapies. Addressing these questions requires the integration of multi-omics analysis, organoid models, and gene editing technologies to further dissect the regulatory network of MAMs across spatiotemporal dimensions and provide a comprehensive strategy for AS treatment.
This review aims to systematically summarize the latest research progress on ER-mitochondria crosstalk in AS, focusing on the structural and functional characteristics of MAMs and their regulatory roles in lipid metabolism, inflammatory responses, and cell death. By organizing key molecular mechanisms and evaluating their clinical translation potential, we aim to reveal innovative therapeutic strategies targeting MAMs and provide a theoretical framework for the development of precision medicine based on organelle interaction regulation.
2 The structural basis of ER-mitochondria crosstalk
The signaling network between the ER and mitochondria constitutes a complex and precise system within the cell. Research on physical and functional interactions between these organelles has primarily focused on physical connections (e.g., MAMs and Endoplasmic Reticulum-Mitochondria Encounter Structure [ERMES]), which provide direct channels for material exchange (21–24). ERMES also contributes to maintaining the mitochondrial function (25–29). Additionally, proteins on MAMs and ERMES participate in various signaling pathways, thereby promoting calcium signaling (22–24), cellular stress and apoptosis (30–35), and other processes. These interactions not only facilitate material exchange and energy metabolism coordination but also provide essential protective mechanisms for cells to respond to environmental changes.
2.1 mitochondria-associated membranes
MAMs, which are the membrane structures between mitochondria and the ER, were first described by Copeland et al. (12) and confirmed in subsequent studies. Despite the narrow membrane gap of MAMs (10–80 nm), these structures efficiently support multiple critical biological processes including calcium homeostasis, lipid metabolism, autophagy, inflammatory responses, ER stress, and mitochondrial dynamics (36).
MAMs enable a more direct material exchange and information communication between the ER and mitochondria, a process that relies on diverse proteins within MAMs. Proteins in MAMs are categorized into three types: (1) Proteins exclusively localized to MAMs (MAMs-resident proteins), (2) Proteins localized to MAMs but also present in other cellular regions (MAMs-enriched proteins), (3) Proteins transiently associated with MAMs (MAMs-associated proteins) (37). Owing to the dynamic nature of MAMs, their exact composition remains unclear.
MAMs contain numerous proteins that perform diverse functions and regulate various cellular and biological processes. Enriched proteins in MAMs, such as glucose-regulated protein 75 (GRP75) and inositol 1,4,5-trisphosphate receptor (IP3R), contribute to maintaining the structure and function of MAMs. IP3R participates in ER calcium release. FUNDC1 typically acts as a mitochondrial autophagy receptor, while Sigma-1 receptor (Sig-1R) regulates ER stress, mitochondrial function, and oxidative stress (6). Mfn2, an important MAM-enriched protein, maintains calcium homeostasis, and mitochondrial dynamics. Mfn2 is believed to protect mitochondria and inhibit apoptosis by suppressing activation of the PERK pathway (7). Increased Mfn2 expression can also ameliorate mitochondrial calcium overload (8). MAM-resident proteins, such as phosphatidylserine synthase 1/2 (PSS1/2), are highly enriched in MAMs and participate in the transport of phospholipids between the mitochondria and ER, thereby promoting lipid synthesis. Mitofusin 1 (Mfn1) collaborates with other MAM-enriched proteins to maintain the structure of MAMs (6, 9).
As a key site for ER-mitochondrial crosstalk, MAMs, with their intricate structure, participate in and regulate various physiological and pathological processes, and play a crucial role in maintaining cellular homeostasis and responding to diverse stress reactions. Related studies are ongoing.
2.2 Endoplasmic reticulum-mitochondria encounter structure
The endoplasmic reticulum-mitochondrial encounter structure (ERMES) acts as a bridge and regulatory center within the cell. ERMES is a multi-subunit complex composed of transmembrane anchoring components (ER membrane protein Mmm1 and mitochondrial outer membrane proteins Mdm10 and Mdm34), soluble connecting components (Mdm12 interacts with the synaptic binding protein-like mitochondrial lipid-binding protein [SMP] domain of Mmm1 and Mdm34 to stabilize ERMES conformation (11)), and dynamic regulatory components (RhoGTPase Gem1 hydrolyzes GTP to assemble and disassemble ERMES, though its presence is condition-dependent and not all ERMES complexes contain this subunit (10)). The SMP domains of Mdm34, Mdm12, and Mmm1 specifically interact to form a stable complex that bridges the ER and mitochondrial outer membranes. Two Mdm12 and two Mmm1 SMP domains interact in a head-to-tail manner to form a tetrameric hydrophobic channel (12), providing structural support for transmembrane transport (e.g., Ca2+, phospholipids) and signal communication.
Gem1, Mdm10, and Mmm1 play key roles in maintaining mitochondrial morphology. Gem1 has two Ca2+-binding EF-hand motifs, and Ca2+ from the ER can bind to these motifs to activate Gem1, promoting Ca2+ transfer to mitochondria and thereby regulating mitochondrial movement (13). Therefore, the ER can influence mitochondrial morphology through the ERMES (38). Rasul et al. (14) demonstrated that Mdm12 interacts with the MAMs regulatory protein Emr1, and that in the absence of Emr1, the number of ERMES structures decreases, leading to abnormalities in mitochondrial morphological. This study further confirms that ERMES mediates mitochondrial dynamics.
Various cellular processes mediated by ERMES are influenced by ERMES regulatory proteins. In addition to Emr1, Arf1, Lam6, and Gem1 help to maintain the number of ERMES foci. Overexpression of Lam6 causes ERMES expansion, Tom7 increases the specificity of Mdm10 binding to ERMES, preventing excessive leakage of Mdm10 from ERMES and binding to the SAM complex, and Sar1 regulates the area of the ERMES complex (15).
3 Biological processes of endoplasmic reticulum-mitochondria crosstalk
3.1 Lipid synthesis
Lipid synthesis is not confined to the ER; multiple enzymes in MAMs participate in this process. For example, in the synthesis of phosphatidylcholine (PC) and phosphatidylethanolamine (PE), phosphatidylserine synthase 1 (PSS1) in the ER catalyzes the formation of phosphatidylserine (PS) from phosphatidic acid (PA). PS enters the mitochondria via MAMs and is converted to PE by related enzymes. PE is then transported out of the mitochondria and converted to PC by PEMT2 in the ER (17). PEMT2, a key enzyme in PC synthesis, has only been identified in MAMs (18). Studies have explored the mechanism of PS entry into the mitochondria. In the liver tissue of mice with non-alcoholic steatohepatitis (NASH), Mfn2 expression is significantly reduced. Mfn2 knockout in mouse hepatocytes causes triglyceride accumulation and inflammatory responses (19). Hernández et al. demonstrated that Mfn2 binds to PS and transports it to the mitochondria for subsequent reactions (19). Enzymes involved in phospholipid biosynthesis are present in both ER and mitochondrial membranes, and the intermediates generated during this process are translocated to ER-mitochondria contact sites (20).
The rate-limiting enzyme in cholesterol synthesis, 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR), is inhibited by statins, which are commonly used to treat AS. Godoy et al. (39) found that mice treated with atorvastatin lacked ER-mitochondria connections, but this was not proven to be based on HMGCR inhibition. Zhong et al. (40) experimentally confirmed that in aldehyde dehydrogenase 2 (ALDH2)-deficient mice, serum total cholesterol and HMGCR levels increased due to ALDH2 promotion of HMGCR ubiquitination and degradation. This process relies on ALDH2 escaping mitochondria to promote the binding of Insig1 to HMGCR in the ER and recruit GP78, indicating that ER-mitochondrial crosstalk is involved in HMGCR-regulated cholesterol synthesis.
Acyl-CoA:diacylglycerol acyltransferase 2 (DGAT2) in MAMs catalyzes the synthesis, and promotes the formation, of lipid droplets. Acyl-CoA:cholesterol acyltransferase 1 (ACAT1/SOAT1), a key rate-limiting enzyme in MAMs, catalyzes the esterification of free cholesterol with long-chain fatty acids to form cholesterol esters (CE), thereby driving CE storage within lipid droplets and directly regulating the early stages of lipid droplet biogenesis (21). Presenilin 2 (PS2), a protein highly enriched in MAMs, corrects excessive lipid droplet formation in Alzheimer’s disease (AD) cells (22). Zhao et al. (23) found that the ATAD3A protein is enriched in MAMs of AD mouse brain tissue and that ATAD3A oligomerization inhibits CYP46A1-mediated brain cholesterol metabolism in AD mice. Szabo et al. (24) demonstrated that abnormal TAU disrupts ER-mitochondria coupling, reduces cholesterol transfer from the ER to the mitochondria, and decreases pregnenolone production, highlighting the importance of ER-mitochondria crosstalk in lipid synthesis. Phosphofurin acidic cluster sorting protein 2 (PACS2), an early MAM protein, regulates lipid homeostasis. Arruda et al. (25) found that PACS2 and IP3R1/2 levels are significantly increased in tissues from overweight mice. Reducing the expression of PACS2 and IP3R1/2 improves mitochondrial antioxidant capacity and insulin sensitivity in obese animals. In summary, collaboration between the ER and mitochondria ensures the smooth progression of lipid synthesis, thereby maintaining cellular membrane integrity and normal cellular function.
3.2 Inflammasome formation
Inflammasomes are signaling complexes that play crucial roles in immune and inflammatory responses. Among the various family members, the most well characterized are NLRP3, NLRP1, NLRC4, and AIM2 (26). ER-mitochondria crosstalk (MAMs) regulates activation of the NLRP3 inflammasome. ER stress induces mitochondrial damage, leading to the release of mtROS and mitochondrial damage-associated molecular patterns (mtDAMPs) such as mitochondrial DNA (mtDNA) and cardiolipin. These are transmitted via MAMs to NLRP3 molecules on the ER membrane, triggering oligomerization and inflammasome activation (27, 28). Moreover, overexpression of RTN1A, a key MAMs protein, can disrupt the binding of HK1 to VDAC1, leading to free VDAC1 driving inflammasome assembly (29). extracellular ATP, through the P2X7R-mediated pathway, disrupts the functional balance of MAMs and induces the assembly of the NLRP3 inflammasome. GRP75 can inhibit eATP-P2X7R pathway-induced NLRP3 inflammasome aggregation (30).
The NLRP3 inflammasome is the only inflammasome currently confirmed to be associated with MAMs and exerts unique functions by sensing damage-associated molecular patterns (DAMPs) in the MAMs microenvironment. DAMPs (e.g., Ca2+ signaling, mtROS) can be produced by damaged cells, with Ca2+ signaling activating the NLRP3 inflammasome through calcium flux between the ER and mitochondria (31). Ye et al. (32) further investigated the relationship between Ca2+ and inflammation and finding that IP3R-mediated excessive Ca2+ release could induce mitochondrial dysfunction and promote NLRP3 inflammasome activation. Based on the above studies, it is evident that ER-mitochondrial crosstalk can activate NLRP3. However, inhibition of NLRP3 is also related to MAMs. Missiroli et al. found that PML, located in MAMs, inhibits NLRP3 activation by tightly binding to NLRP3 and P2X7R (33).
3.3 Calcium flux and signal transduction
One of the key roles of MAMs is to maintain calcium homeostasis between the ER and mitochondria. Arruda et al. (25) demonstrated that an increase in MAMs under obese conditions exacerbates mitochondrial calcium accumulation and disrupts mitochondrial function. Moreover, when subjected to external stimuli, changes in various components of MAMs can lead to MAMs dysfunction, thereby disrupting Ca2+ transport between the ER and mitochondria (36).
MAMs regulates calcium ion flux through several key proteins. FUNDC1, by binding to IP3R2 and localizing to MAMs, increases mitochondrial and cytosolic Ca2+ levels while decreasing ER Ca2+ levels when overexpressed. Conversely, in the absence of FUNDC1, mitochondrial and cytosolic Ca2+ levels decreased, while ER Ca2+ levels increase (37). In addition, the IP3R-GRP75-VDAC complex is a classic structure in MAMs involved in calcium homeostasis, facilitating Ca2+ transfer from the ER to the mitochondria (41). IP3R mediates Ca2+ release from the ER, whereas VDAC is responsible for Ca2+ transfer into the mitochondrial intermembrane space. PTEN and Akt, which are localized in MAMs, promote ER calcium release by phosphorylating IP3R. PTEN knockout inhibits ER Ca2+ release, thereby maintaining mitochondrial Ca2+ homeostasis and negatively regulating apoptosis (42, 43). The Sig-1R-BiP complex in MAMs can rapidly respond to decreases in Ca2+ concentration, thereby regulating ER-mitochondria Ca2+ signaling (44). Additionally, SERCA2b, enriched in MAMs, exhibits high Ca2+ affinity. Its activity is enhanced through interaction with CANX (calnexin) and inhibited via interaction with TMX1 (thioredoxin-related transmembrane protein 1), thereby regulating Ca2+ influx into either the endoplasmic reticulum (ER) or mitochondria. (45). Beyond ER Ca2+ release and ER-mitochondrial Ca2+ transfer, MAM-associated proteins also inhibit mitochondrial Ca2+ release. For example, Bcl-2 can reduce mitochondrial calcium release, thereby inhibiting apoptosis (38).
3.4 Mitochondrial dynamics and homeostasis
Mitochondria undergo dynamic changes in shape, size, and number via fusion and fission. Previous studies have hypothesized that ER plays a significant role in important mitochondrial functions, including mitochondrial dynamics (46). Under the action of certain mitochondrial dynamic proteins, including MFF, dynamin‐related protein 1(Drp1) accumulates on the mitochondrial outer membrane and is arranged in a helical pattern, causing mitochondrial fission (47). However, in the experiments by Friedman et al. (48), MAMs exhibited abnormal mitochondrial contractions in the absence of MFF. Arasaki et al. (49) further elaborated on this view, indicating that the SNARE protein Syn17 on MAMs activates Drp1 and determines its localization, thereby promoting mitochondrial fission. Under conditions of energy stress, AMPK accumulates extensively and interacts with Mfn2 in MAMs, promoting mitochondrial fission (50). Under hypoxic conditions, FUNDC1 accumulates in MAMs and binds to Drp1, promoting Drp1-mediated mitochondrial fission. Inhibition of FUNDC1 disrupts MAMs integrity, leading to decreased cytoplasmic and mitochondrial Ca2+ concentrations and the suppression of mitochondrial fission (24, 51).
The ERMES is often associated with mitochondrial dynamics. During cell division, Mmm1, Mdm10, and Mdm12 connect mitochondria to the actin cytoskeleton and participate in polarized mitochondrial movement (38). ERMES plays a widespread role in the regulation of mitochondrial homeostasis in fungi. Garrido-Bazán et al. (39) found that downregulation of MdmB expression in yeast ERMES leads to mitochondrial fission defects. Experiments also showed that ER-mitochondrial crosstalk is indispensable for H2O2-induced mitochondrial contraction. In an earlier study, Sogo et al. (40) observed changes in mitochondrial networks and aggregation in yeast Mdm10 mutants. Similarly, giant spherical mitochondria have been discovered after Mdm12 knockout in cells (52). Regarding the specific mechanism, Esposito et al. (53) demonstrated that the absence of Mdm10 or Mdm12 in ERMES leads to an increase in peroxisomes, causing mitochondrial dysfunction. Their experiments provide further evidence for the role of ERMES in maintaining mitochondrial homeostasis.
3.5 Autophagy and apoptosis
Autophagy, a universal cellular metabolic process, is characterized by the formation of autophagosomes. Hamasaki et al. found that the autophagy-related protein ATG14 is located in MAMs, where autophagosome formation begins (54). During mitophagy induction, PINK1 and Beclin 1 are expressed in MAMs, promoting the formation of autophagosomes and MAMs (55). The autophagy-related proteins Atg8 and Atg11 co-localize with Mdm12 and Mdm34 in ERMES, facilitating mitophagy (56).
ER-mitochondrial crosstalk provides the structural basis for autophagy. Kohle et al. (28) suggested that the contact between the ER and mitochondria is central to autophagy. In cases of myocardial ischemia-reperfusion injury, Mfn2 can promote the transfer of phospholipids from the ER to the mitochondria, facilitating autophagosome membrane formation, and thereby activating protective mitophagy processes (25). Ikeda et al. (26) experimentally demonstrated that downregulation of Drp1 expression in mouse cells leads to mitochondrial dysfunction, indicating a positive correlation between Drp1 expression and the intensity of Mitophagy. Additionally, the STX17-Fis axis is involved in inducing mitochondrial autophagy; Fis1 inhibits the transfer of STX17 from MAMs to the mitochondria, thereby suppressing mitochondrial phagocytosis during autophagy (27). In summary, ER-mitochondria contact determines mitochondrial fission points, which are a prerequisite for subsequent engulfment by autophagosomes or lysosomes. Böckler et al. (29) further confirmed that ERMES co-localizes with phagophore sites and promotes phagophore membrane formation, possibly by facilitating lipid transfer to phagophores. Böckler et al. suggested that ERMES functions only in mitochondrial autophagy and not in the broader autophagy processes (57). Garofalo et al. indicated that GD3, a core protein that initiates autophagy, is strongly associated with MAMs. They speculated that GD3 may be enriched in MAMs lipid rafts under autophagy stimulation, promoting autophagosome formation, and playing a role in early autophagy (58).
During acute nutrient deficiency, ERMCS can alter its structure to dynamically enhance the transfer of calcium ions (Ca2+) and lipids from the ER to the mitochondria, stimulate apoptosis, and promote oxidative phosphorylation (30, 31). Mitochondrial Ca2+ overload is a key trigger of apoptosis, with MAMs protein complexes (e.g., IP3Rs, VDAC1) directly involved by promoting Ca2+ transmembrane transport. Studies also indicate that Bcl-2 family protein Bok amplifies Ca2+ signaling by binding to IP3Rs (33, 34), while PACS-2 deficiency reduces Ca2+ flux and inhibits apoptosis by disrupting ER-mitochondria connections (35). Notably, neuronal apoptosis is closely linked to ER-mitochondria crosstalk dysfunction, marked by disorders of proteins, such as Grp75 and Sigma1R, and excessive release of ROS and pro-inflammatory factors. Interventions in calcium homeostasis or the suppression of inflammatory responses can significantly restore neuronal function (59). In summary, ER-mitochondrial crosstalk, which integrates calcium homeostasis, lipid metabolism, and inflammatory responses, serves as a central regulator of cell autophagy and apoptosis (Figure 1; Table 1).
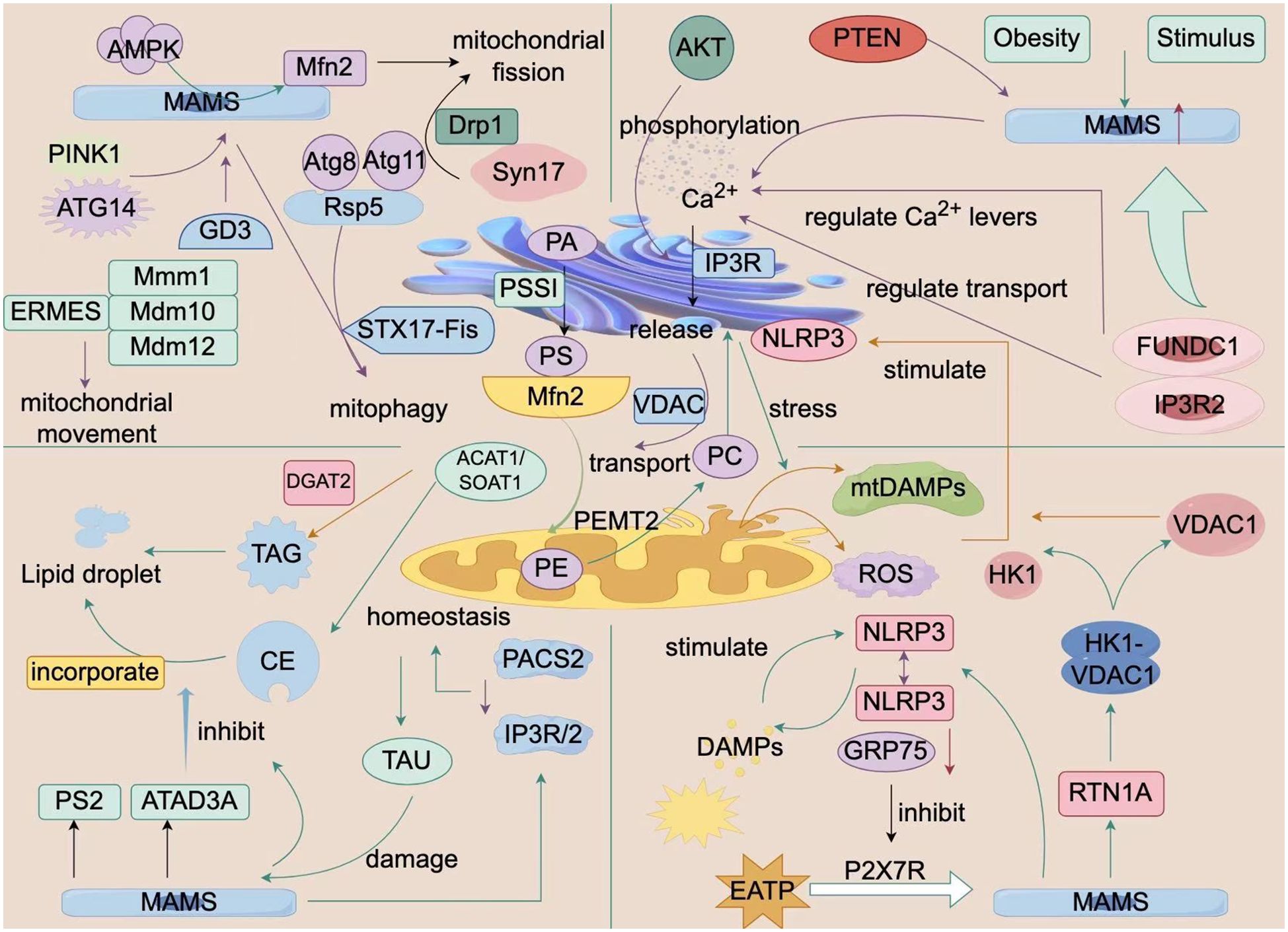
Figure 1. Biological Processes of ER-Mitochondria Crosstalk. This figure is a schematic diagram of the biological processes of interaction between the endoplasmic reticulum and mitochondria within a cell. It shows a variety of molecules and signaling pathways, including mitochondrial fission and fusion-related proteins (e.g., Mfn2, Drp1), autophagy-related proteins (e.g., PINK1, PARKIN), calcium ion signaling, and ER-related proteins (e.g., ERMES, VDAC1). These molecules and signaling pathways interact to regulate cellular metabolism, energy balance, and autophagy processes. Additionally, the figure illustrates the impact of external factors such as obesity and stimulation on these processes. The structures and molecules in the figure represent the biological processes of ER-mitochondria crosstalk (MAMs). The main structural molecules are described as follows: AMPK: AMP-activated protein kinase; MAMs: Mitochondria-associated membranes; PINK1: PTEN-induced putative kinase 1; ATG14: Autophagy-related protein 14; GD3: Disialyl ganglioside; ERMES: Endoplasmic reticulum-mitochondria contact site complex; STX17-Fis: Synaptophysin 17-mitochondrial fission protein 1 complex; PACS2: Phosphorylated protein sorting and transport protein 2; TAU: Microtubule-associated protein Tau; CE: Cholesterol ester; PS2: Presenilin 2; ATAD3A: ATPase family AAA domain-containing protein 3A; NLRP3: NOD-like receptor pyrin domain-containing protein 3; RTN1A: Reticulon 1A; FUNDC1: FUN14 domain-containing protein 1; VDAC1: Voltage-dependent anion channel 1; Mfn2: Mitofusin 2. The image is created with Figdraw.
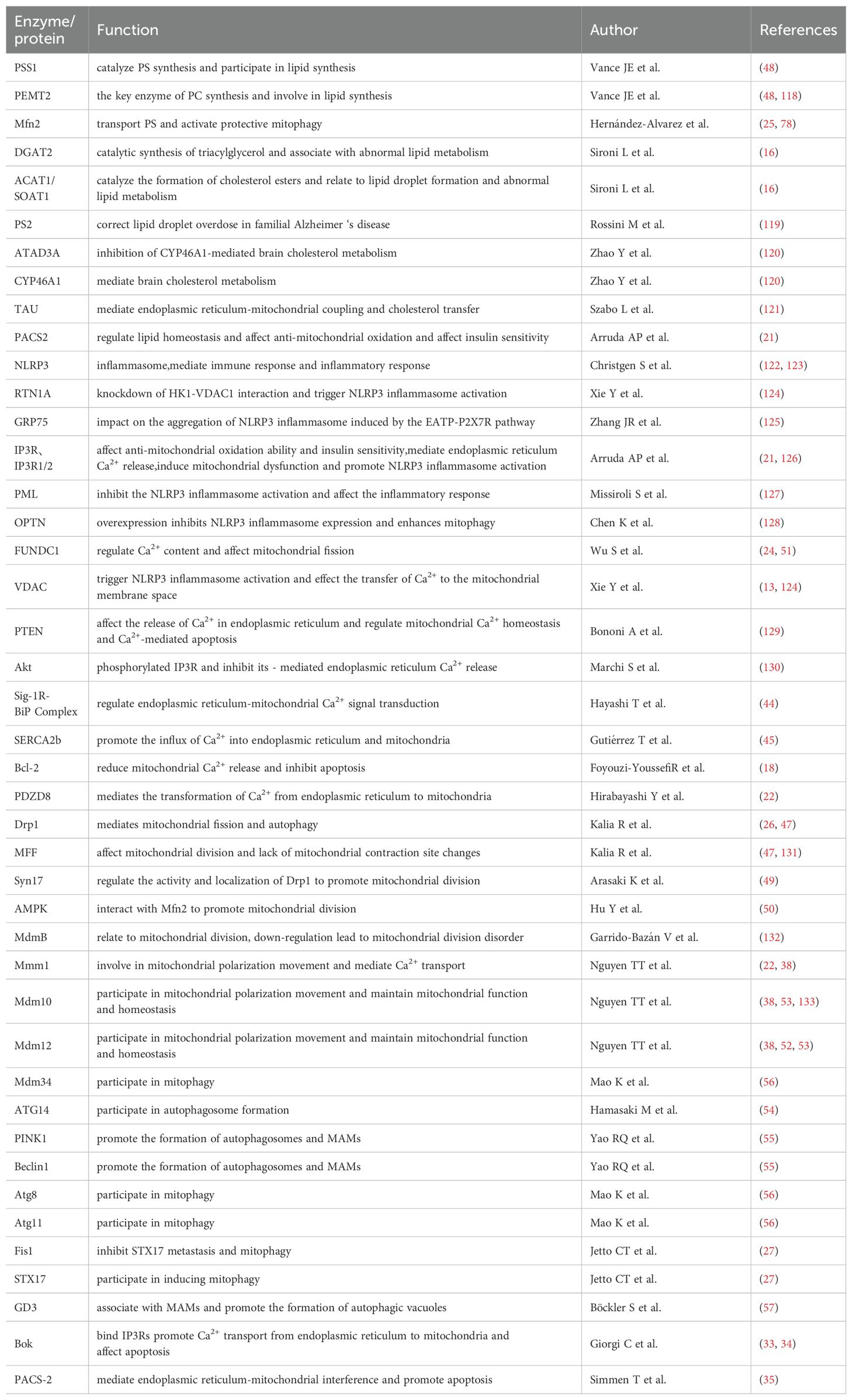
Table 1. Biological Processes of ER-Mitochondria Crosstalk.
4 ER-mitochondria crosstalk in atherosclerosis
In the pathological environment of AS, the ER and mitochondria form a dynamic signaling network through multiple mechanisms. This network involves various biological processes, including calcium homeostasis, lipid transport homeostasis, and enrichment and activation of key MAMs proteins, thereby accelerating the progression of AS. Elucidating these mechanisms is crucial for understanding AS pathogenesis and developing new diagnostic and therapeutic targets. Herein, we summarize and critically analyze the basic forms of ER-mitochondrial crosstalk in the context of AS (Figure 2; Table 2), highlighting the potential key role of the Mfn2 family of proteins in this process. However, the specific underlying mechanisms remain to be explored.
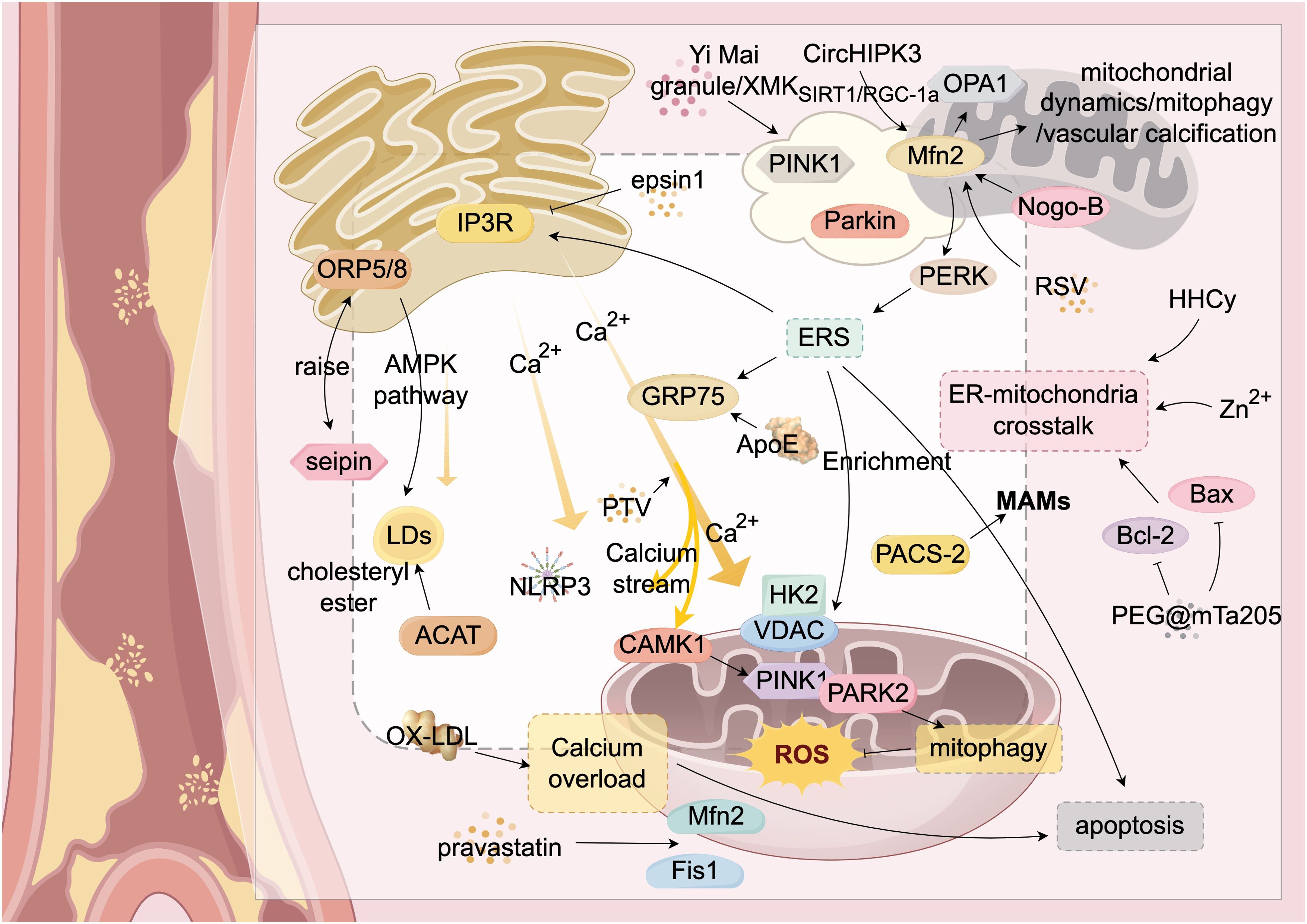
Figure 2. ER-Mitochondria Crosstalk in Atherosclerosis. This figure provides a detailed illustration of the complex mechanisms by which the endoplasmic reticulum (ER) and mitochondria interact through mitochondria-associated membranes (MAMs) and their associated proteins during atherosclerosis. It highlights key molecules and signaling pathways, including calcium ion flux and lipid transfer. Mfn2 serves as a central hub for communication between the ER and mitochondria. The figure also shows the roles of various proteins (such as ORP5/8, IP3R, GRP75, PERK, PAC5-2, VDAC, and Mfn2) in these interactions. Additionally, it illustrates abnormal biological processes such as ER stress (ERS), mitochondrial autophagy (mitophagy), and apoptosis, which collectively impact the progression of atherosclerosis. Through the interplay of these molecules and signaling pathways, ER-mitochondria crosstalk plays a critical role in the development of atherosclerosis. Structures and Molecules in the Figure: White rectangular areas: Mitochondria-associated membranes (MAMs); Yellow structure (upper left): Endoplasmic reticulum (ER); Yellow structures (upper right and lower right): Mitochondria; Yellow arrow (center): Calcium ion flow between the ER and mitochondria. The main structural molecules are described as follows: PINK1: PTEN-induced putative kinase 1; Parkin: Parkin protein; Mfn2: Mitofusin 2; Nogo-B: Reticulon 4B; Bax: Bcl-2-associated X protein; Bcl-2: B-cell lymphoma-2; Fis1: Mitochondrial fission protein 1; OX-LDL: Oxidized low-density lipoprotein; Seipin: Fat differentiation-related protein; ACAT: Acyl-CoA:cholesterol acyltransferase; ORP5/8: Oxysterol-binding protein-related proteins 5/8; GRP75: Glucose-regulated protein 75; CAMK1: Calcium/calmodulin-dependent protein kinase 1; NLRP3: NOD-like receptor pyrin domain-containing protein 3; LDs: Lipid droplets; PERK: RNA-dependent protein kinase-like ER kinase. The image is created with Figdraw.
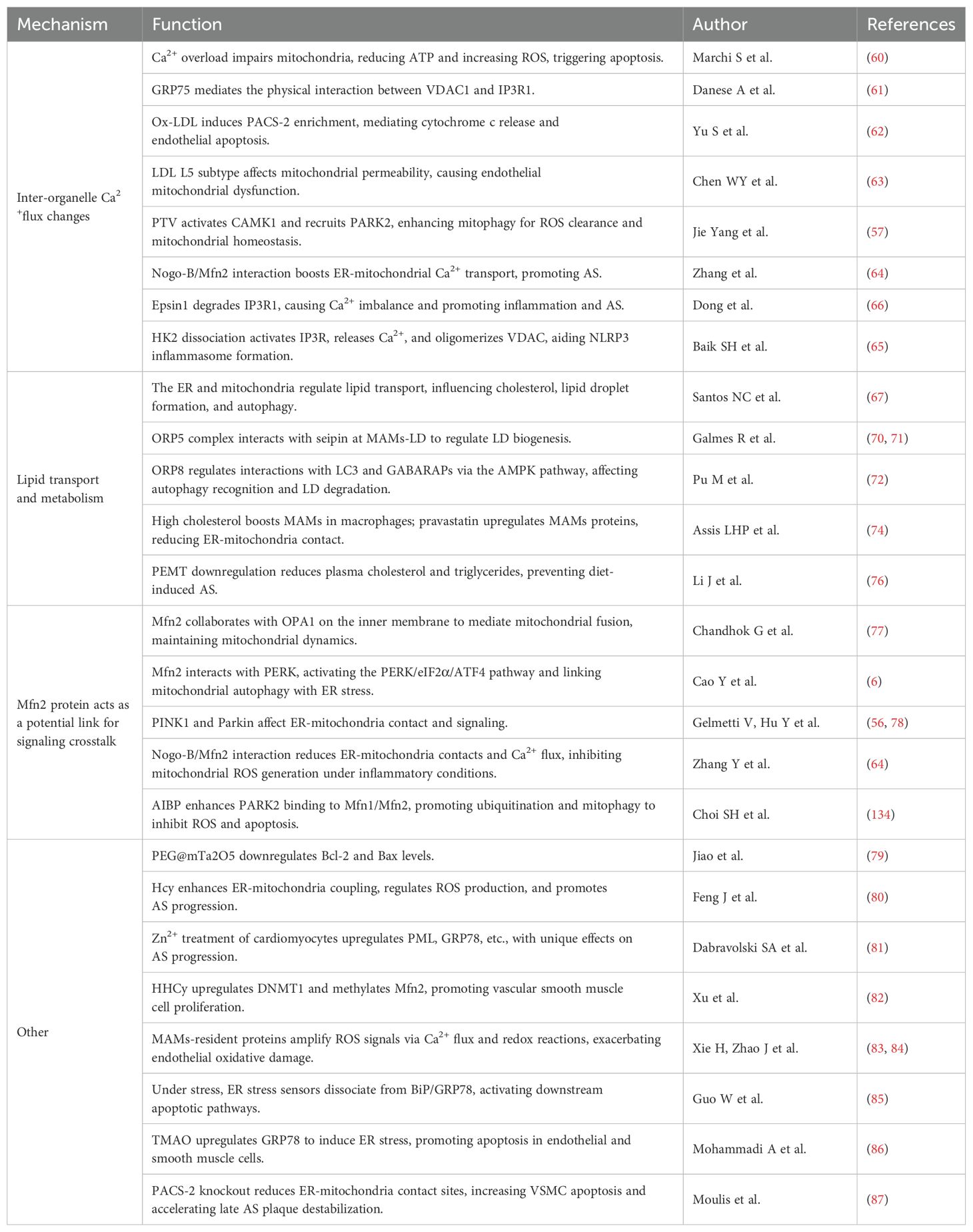
Table 2. Mechanisms of ER-Mitochondria Signaling Crosstalk in Atherosclerosis.
4.1 Crosstalk between organelles based on calcium ion flux
Calcium ion flux between the endoplasmic reticulum (ER) and mitochondria is a key signaling pathway mediating organelle communication. The ER, acting as a cellular calcium reservoir, releases Ca2+ into the cytosol upon cellular demand, thereby dynamically regulating mitochondrial Ca2+ homeostasis. However, mitochondrial calcium overload directly inhibits basic mitochondrial function, leading to reduced ATP production and increased ROS generation, and can even trigger mitochondrial apoptosis (60). Within the cell, Ca2+ is released from the ER through IP3R and is transferred to the mitochondria via voltage-dependent anion channels (VDAC) on the outer mitochondrial membrane (OMM). VDAC1 physically connects to IP3R1 through GRP75, a MAM-associated protein, allowing GRP75 to directly promote calcium transfer from the ER to the mitochondria (61). During early ER stress, MAM proteins accumulate, increasing the ER-mitochondrial contact sites and accelerating mitochondrial calcium uptake to mitigate ER stress. This highlights the close collaborative relationship between the ER and mitochondria, with Ca2+ flux serving as the fundamental pathway for communication.
ER-mitochondrial interactions based on Ca2+ flux also play a crucial role in the progression of AS. Early AS lesions involve ox-LDL-induced endothelial cell apoptosis, a process linked to mitochondrial Ca2+ overload. PACS-2, enriched in MAM regions, promotes the formation of more ER-mitochondria contact sites under ox-LDL induction. This accelerates Ca2+ flux between the two organelles, leading to mitochondrial membrane potential loss and increased ROS production. These changes promote cytochrome c release and endothelial cell apoptosis (62). Additionally, the L5 subtype of LDL containing Apolipoprotein E (ApoE) interacts with VDAC, increasing mitochondrial permeability and causing mitochondrial dysfunction in endothelial cells (63). Yang et al. (57) demonstrated that pitavastatin (PTV) promotes calcium release from mitochondria to activate CAMK1, increasing the phosphorylation of PINK1. PINK1, in turn, recruits and phosphorylates PARK2 on the mitochondrial membrane, thereby activating mitophagy. This process is beneficial for ROS clearance and mitochondrial homeostasis in endothelial cells. Additionally, Zhang et al. (64) found that in ApoE−/− mice lacking Reticulon 4B(Nogo-B), Mfn2 protein levels decrease, and the development of AS lesions is inhibited. Further research indicated that Nogo-B interacts with Mfn2 in endothelial cells, increasing ER-mitochondria Ca2+ transport via ER-mitochondrial crosstalk. This process promotes ROS generation and activates the ROS-p38-p65 signaling pathway, enhancing inflammatory responses and promoting AS development. Collectively, these studies show that the Ca2+ flux between the ER and mitochondria in AS regulates mitochondrial homeostasis, ROS clearance, and apoptosis in endothelial cells, representing a specific mechanism by which ER-mitochondria crosstalk promotes AS progression.
Activation of the NLRP3 inflammasome in macrophages within plaques has also been linked to Ca2+ flux homeostasis in MAMs. A previous study revealed that NLRP3 inflammasome formation occurs as follows: hexokinase 2 (HK2) dissociates from VDAC on the outer mitochondrial membrane, activating IP3R signaling and causing Ca2+ release from the ER into the mitochondria. Increased mitochondrial calcium concentrations induce VDAC oligomerization, setting the stage for NLRP3 inflammasome formation (65). Furthermore, Dong et al. (66) found that epsin1 accelerates the ubiquitin-dependent degradation of IP3R1, leading to abnormal ER-mitochondrial calcium signaling and cytosolic free calcium imbalance, thereby promoting inflammation and AS. Thus, Ca2+ flux between the ER and MAMs also regulates inflammatory responses within AS plaques.
4.2 Lipid transport and metabolism as a crosstalk pathway
AS, a lipid metabolism-related disease, involves ER mitochondrial lipid transport via MAMs contact sites, including cholesterol esterification, lipid droplet formation, and autophagy (67). ORP5/8 proteins, key members of the oxysterol-binding protein-related protein (Osh/ORP) family, are the only proteins of the ORP family that are anchored to the ER membrane via their C-terminal transmembrane domains (68, 69). Studies have revealed that ORP5/8 proteins are critically involved in ER-mitochondria lipid trafficking (70). Specifically, the ORP5 complex localizes to MAMs subdomains and interacts with seipin to recruit it to MAMs-lipid droplet (LD) contact sites, thereby promoting LD biogenesis (71). In contrast, ORP8 regulates the interactions between LC3 and GABARAPs through the AMP-activated protein kinase (AMPK) pathway, thereby facilitating autophagic recognition and LD degradation (72). Although the homeostatic mechanisms of lipid transport homeostasis have been well studied, their molecular-level realization remains unclear. Scholars have determined the ORP8 domain crystal structure when bound to “cargo” lipids, using computer simulations to identify PS and PI4P binding to ORP8 (73), highlighting the unique importance of the “lid” structure for ORP8 function.
Hypercholesterolemia, a key risk factor of atherosclerosis, increases the number of MAMs contact sites in macrophages. Statins such as pravastatin upregulate MAM-related proteins such as Mfn2 and Fis1, also reducing ER-mitochondrial interactions and hinting at the link between ER-mitochondrial crosstalk and lipid metabolism/transport (74). Moreover, MAMs host key enzymes for cholesterol metabolism balance, such as acyl-CoA cholesterol acyltransferase (ACAT), which esterifies free cholesterol for storage in lipid droplets. ACAT dysfunction can also facilitate the development of AS (75). Downregulation of the MAMs protein PEMT also lowers plasma cholesterol and triglyceride levels, thereby preventing diet-induced AS (76). In summary, ER-mitochondrial lipid transport plays multiple roles in AS development, from cholesterol esterification and transport to triglyceride synthesis and lipid metabolism-protein regulation. The dysfunction of these processes may promote the formation of AS. Understanding these molecular mechanisms can help to clarify AS’s pathophysiology of AS and underpin new therapeutic approaches.
4.3 Mfn2 protein: a potential link for ER-mitochondria crosstalk
From the existing literature, we found that Mfn2 is a key link in the ER–mitochondrial crosstalk in AS pathology. The process of mitochondrial fission and fusion necessitates the recruitment of Drp1, Optic atrophy 1 (OPA1) and Mfn2 (77).Mfn2 on the mitochondrial outer membrane interacts with OPA1 on the inner membrane to promote mitochondrial fusion and maintain mitochondrial dynamics (77). Moreover, Mfn2 interacts with PERK protein, activating the PERK/eIF2α/ATF4 pathway upstream of UPR, thus linking mitochondrial autophagy and ER stress (6). Recent studies have expanded our understanding of the role of the PINK1/Parkin pathway, which mediates mitophagy. PINK1 and Parkin proteins, found in MAMs (56), interact with ER-mitochondrial communication proteins such as Mfn2 (78), promoting ER-mitochondrial contact and crosstalk. Furthermore, Nogo-B protein expression is upregulated in carotid/coronary atherosclerotic plaques. The Nogo-B protein stabilize Mfn2 to underpin ER-mitochondrial connections and Ca2+ flux homeostasis, thereby suppressing mitochondrial ROS generation during inflammatory responses (64). Thus, Mfn2, which intersects with multiple pathways, is a central hub protein in the ER-mitochondrial interaction network in AS.
4.4 Other factors
Advances in nanotechnology have enabled the use of various nanoparticles in disease diagnosis, bioimaging, and drug delivery. However, long-term exposure to nanoparticles can damage endothelial cells and exacerbate AS. Jiao et al. (79) found that the mesoporous tantalum oxide nanomaterial PEG@mTa2O5 downregulates Bcl-2 and Bax expression. While Bcl-2 family proteins are linked to ER-mitochondria crosstalk in apoptosis, whether PEG@mTa2O5 worsens AS via this crosstalk remains unclear.
Hyperhomocysteinemia (HHCy) contributes to AS progression. Homocysteine (Hcy) increases ER-mitochondrial coupling and promotes mitochondrial ROS production, which can disrupt Ca2+ homeostasis and alter membrane potential. This reprograms mitochondrial metabolism and activates T cells, driving AS progression through cytokine/chemokine release, immune responses, and Treg regulation (80). Zn2+ homeostasis also promotes AS progression via ER-mitochondria crosstalk. Treating cardiomyocytes with Zn2+ upregulates ER-mitochondria contact proteins like PML, ER stress proteins such as GRP78, and calmodulin (81). Epigenetic regulation studies further suggest that HHCy promotes AS via ER-mitochondrial crosstalk. Xu et al. (82) found that HHCy upregulates DNMT1 and increases Mfn2 methylation. Downregulation of Mfn2 in AS plaques drives abnormal vascular smooth muscle cell proliferation (82).
Excessive ROS generation is associated with several cardiovascular diseases, including AS. MAMs serve as key hubs for ROS production. Mitochondrial cytochrome b5, a target of cytochrome c, activates the CYP-dependent monooxygenase system, thereby increasing ROS production. MAM-resident proteins (e.g., GRP75, ERO1, SIG-1R, and VDAC) amplify ROS signals by promoting Ca2+ flux and redox reactions, and exacerbating endothelial cell oxidative damage (83, 84).
ER stress integrates with mitochondrial apoptotic signals via MAMs and is directly involved in AS pathology. Under stress, ER stress sensors (PERK and IRE1) dissociate from the chaperone BiP/GRP78, activating downstream apoptotic pathways (85). Trimethylamine N-oxide (TMAO), an AS risk factor, induces ER stress by upregulating GRP78 and promoting the apoptosis of endothelial and smooth muscle cells (86). MAM dysfunction has also been linked to the loss of AS plaque stability. Moulis et al. found that PACS-2 knockout reduces ER-mitochondrial contact sites, which increased vascular smooth muscle cell (VSMC) apoptosis, and accelerated late-stage AS plaque instability (87).
Research indicates that age-related cardiovascular diseases, including AS, are closely related to mitochondrial dysfunction and abnormal ER-mitochondrial interactions. Specifically, the MAM protein PACS-2 inhibits apoptosis in vascular endothelial and smooth muscle cells, promoting age-related AS pathology (87, 88). Additionally, mtDNA mutations in Mfn2 can impair MAMs functions such as phospholipid synthesis/transport and calcium homeostasis, thereby reducing ER-mitochondria contact sites (89). Similarly, Granatiero et al. (90) observed that the 13514A>G mtDNA mutation decreased ER-mitochondrial contact sites in MELAS cells (primary skin fibroblasts derived from MELAS or Leigh syndrome patients), accompanied by blocked calcium ion flow and reduced mitochondrial calcium uptake. However, these mtDNA mutations have not been confirmed in AS. Given that mitochondrial dysfunction and mitochondrial DNA mutations are characteristics of mitochondrial aging, combined with the aforementioned research findings, does mitochondrial aging promote age-related atherosclerotic (AS) progression by disrupting ER-mitochondria crosstalk? This represents a promising avenue for future research.
5 Targeting ER-mitochondria crosstalk in AS: promising drugs
A number of pharmaceutical reagents target ER-mitochondrial crosstalk through distinct mechanisms (Figure 3; Table 3), demonstrating their potential for treating cardiovascular diseases, including AS.
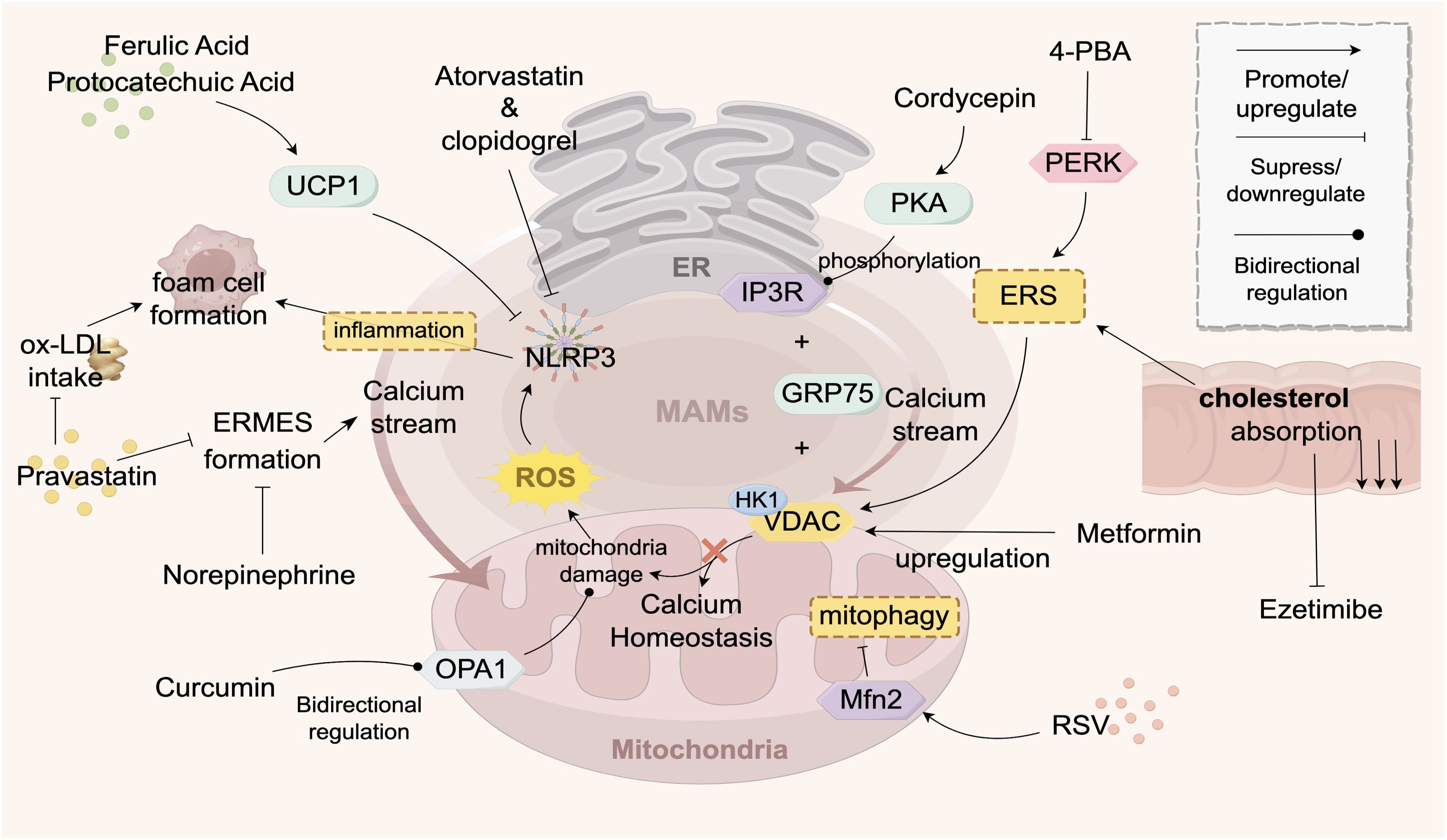
Figure 3. Drugs Targeting ER-Mitochondria Crosstalk in AS. This figure illustrates the signaling exchange between the ER and mitochondria via MAMs contact sites in an atherosclerotic environment. The ER transfers Ca2+ to mitochondria to maintain their function, but Ca2+ overload can inhibit mitochondrial function, reducing ATP and increasing ROS, which triggers apoptosis. Statins stabilize endothelial function by inhibiting a key enzyme in cholesterol synthesis. Various drugs (e.g., pravastatin, atorvastatin, 4-PBA) target ER-mitochondria crosstalk to improve calcium ion flow, maintain mitochondrial function, reduce inflammation, and decrease foam cell formation, thereby improving AS. Traditional Chinese medicine components, such as curcumin and resveratrol, modulate key proteins (e.g., OPA1, MFN2) to influence mitochondrial function and show therapeutic potential. The structures and molecules in the figure represent the targets of AS drugs that act on ER-mitochondria crosstalk. The gray structure at the top represents the ER, the pink structure at the bottom represents the mitochondrion, and the gray-pink area between them represents the MAMs. The meanings of the various arrows are labeled in the legend in the top right corner of the figure. The main structural molecules are described as follows: UCP1: Uncoupling protein 1; ROS: Reactive oxygen species; OPA1: Optic atrophy protein 1; NLRP3: NOD-like receptor pyrin domain-containing protein 3; VDAC: Voltage-dependent anion channel; HK1: Hexokinase 1; PERK: Protein kinase R-like ER kinase; PKA: Protein kinase A; RSV: Resveratrol; ERS: Endoplasmic reticulum stress; GRP75: Glucose-regulated protein 75; IP3R: Inositol 1,4,5-trisphosphate receptor; 4-PBA: 4-phenylbutyric acid. The image is created with Figdraw.
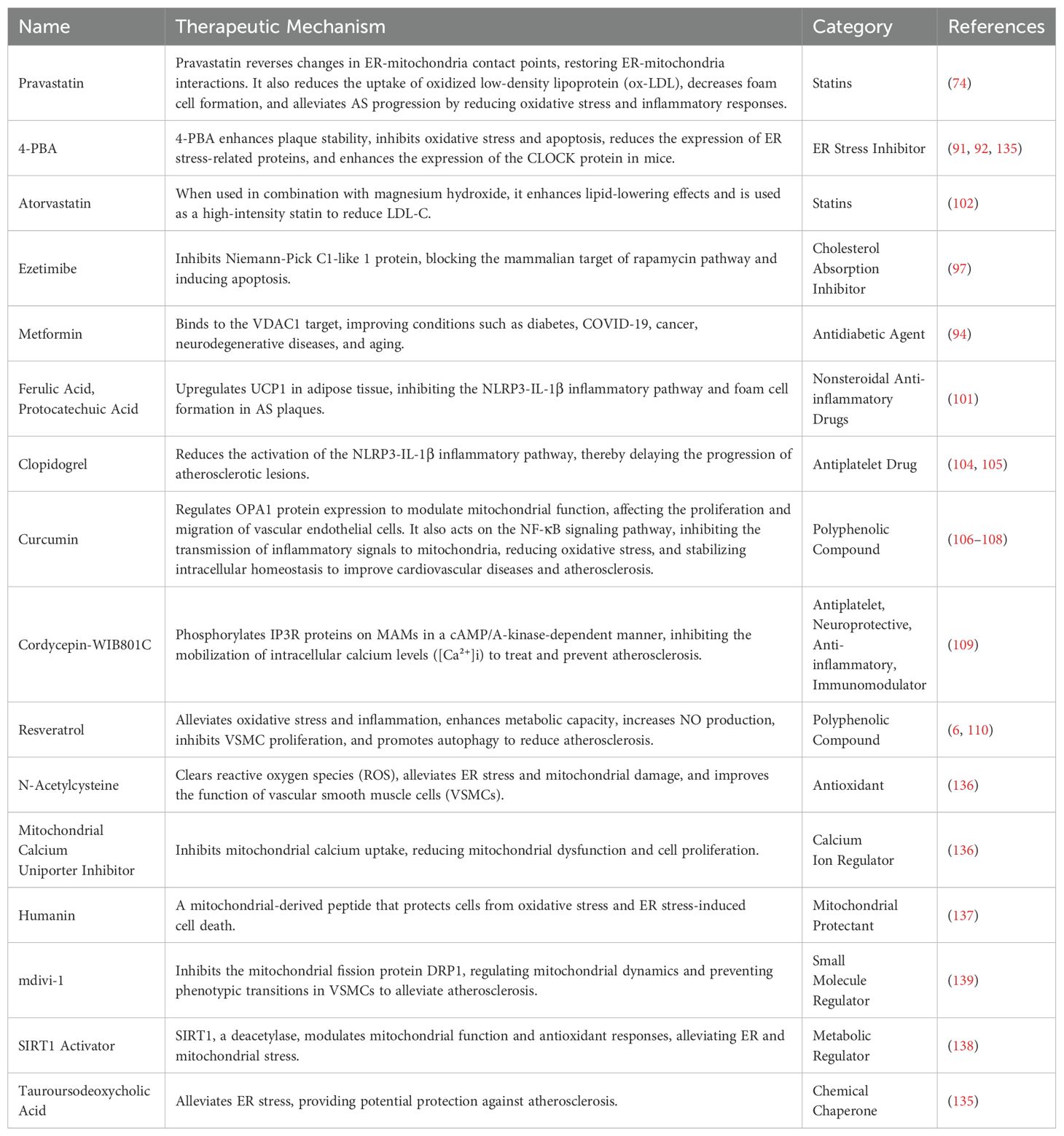
Table 3. Drugs Targeting ER-Mitochondria Crosstalk in AS.
Some drugs improve AS by inhibiting the Ca2+ flow between the ER and mitochondria. Pravastatin reduces ER-mitochondria interaction sites, inhibiting Ca2+ transfer and decreasing foam cell formation by reducing macrophage uptake of ox-LDL (81). Noradrenaline downregulates mitochondrial Ca2+ uptake and ER-mitochondrial coupling sites in cardiomyocytes (74). The chemical 4-Phenylbutyric acid (4-PBA) reduces ER stress levels, suppresses PERK activation to maintain Ca2+ homeostasis (91), and prevents adverse effects caused by excessive Ca2+ influx, such as mitochondrial membrane potential depolarization and increased ROS production (91–93). Metformin upregulates the expression of VDAC1 protein in MAMs (94), thereby preserving mitochondrial Ca2+ homeostasis and reducing ROS generation (95).
Several agents target lipid synthesis to ameliorate AS. Statins exert their therapeutic effects by inhibiting key enzymes involved in cholesterol synthesis, such as HMG-CoA reductase, thereby reducing cholesterol biosynthesis and indirectly suppressing lipid transport and signaling crosstalk between the ER and mitochondria (74). This mechanism stabilizes vascular endothelial cell function, attenuates foam cell formation, and ultimately slows AS progression (96). Ezetimibe reduces systemic cholesterol levels by inhibiting intestinal cholesterol absorption, thereby mitigating excessive cholesterol-induced ER stress and its detrimental effects on mitochondrial function (97, 98). Yimai granules improve AS by activating the Pink1-Mfn2-Parkin pathway via miRNA-125a-5p, which enhances mitophagy, suppresses proinflammatory factor release, inhibits vasoconstrictor production (99).
Moreover, emerging data indicate that certain agents can directly target MAMs to suppress inflammatory responses and are a platform for NLRP3 inflammasome activation, since reducing ER stress or MAM formation can inhibit NLRP3 (100). Ferulic acid and protocatechuic acid upregulate UCP1 in adipose tissue to inhibit the NLRP3-IL-1β pathway and foam cell formation (101). Atorvastatin inhibits the activation of the NLRP3 inflammasome, reduces the release of inflammatory factors, mitigates vascular inflammatory responses, and thereby suppresses the progression of atherosclerosis (102, 103). Clopidogrel delays the progression of atherosclerotic lesions by inhibiting the activation of the NLRP3-IL-1β inflammatory pathway (104, 105).
Specific herbal formulas and active components of traditional Chinese medicine (TCM) can also ameliorate AS by targeting the ER-mitochondrial crosstalk. Curcumin improves AS by modulating the expression OPA1, bidirectionally regulating mitochondrial function, thereby influencing the proliferation and migration of vascular endothelial cells (106). Additionally, curcumin acts on the NF-κB signaling pathway (107) by inhibiting the propagation of inflammatory signals to the mitochondria, reducing oxidative stress damage, stabilizing intracellular homeostasis, and lowering the risk of cardiovascular diseases and AS (108). Cordycepin (CE)-WIB801C suppresses AS progression via cAMP-dependent protein kinase (PKA)-mediated phosphorylation of the inositol trisphosphate receptor (IP3R) on MAMs, thereby inhibiting intracellular calcium mobilization ([Ca2+]i) (109). Resveratrol (RSV) upregulates the expression of Mfn1 and Mfn2 in MAMs (110), stabilizing mitochondrial dynamics and improving mitochondrial function. Mfn2, a key protein promoting ER-mitochondrial communication, modulates inter-organelle signaling crosstalk through its expression levels (6). Taken together, it appears that these TCM components target key MAM proteins such as OPA1, IP3R, Mfn1, and Mfn2.
6 Discussion
The ER and mitochondria exert profound effects on various physiological processes within cells through complex molecular mechanisms. However, crosstalk between the two organelles plays an even more critical role in maintaining lipid and calcium homeostasis, promoting inflammatory responses, and apoptosis. Exploring the ER-mitochondria crosstalk in the context of AS not only deepens our understanding of the pathophysiology of the disease, but also shifts the research focus from isolated organelles to their dynamic interactions, providing new insights for future studies.
Although research on ERMES-related proteins as biomarkers for AS remains limited, mtDNA abnormalities have been established as effective molecular markers for AS (111). Studies indicate that dysfunctional ERMES can induce excessive mitochondrial fission (e.g., Drp1-dependent fission), compromising mitochondrial membrane integrity and significantly increasing the risk of mtDNA leakage into the cytoplasm (112). Moreover, mitochondrial dynamic disorders reduce oxidative phosphorylation efficiency, leading to decreased ATP synthesis and reactive oxygen species (ROS) accumulation, further increasing the risk of mtDNA leakage (113). In clinical translation, the peripheral blood cell mitochondrial DNA copy number, owing to its high sensitivity and accessibility, has emerged as a promising indicator for early screening and risk assessment of AS (114). Whole-genome sequencing technology can precisely quantify fluctuations in mtDNA-CN, and its abnormal reduction has been linked to plaque instability and increased risk of cardiovascular events (111). Thus, molecules downstream of MAMs (e.g., mtDNA) show promise for transitioning from basic research to clinical applications. Future studies should investigate the potential of MAM-related proteins as biomarkers and explore the dynamic regulatory strategies targeting MAMs to disrupt the AS cycle.
The current clinical management of AS primarily relies on pharmacological and interventional therapies. Statins are commonly used lipid-lowering drugs that reduce blood cholesterol levels, stabilize plaques, and reduce the risk of cardiovascular events. Other medications, such as metformin and ezetimibe, also act through distinct mechanisms. Interventional therapies such as coronary artery stenting (115) and carotid endarterectomy (116), directly address vascular stenosis or occlusion and restore blood flow. However, these approaches carry risks including drug resistance, intolerance, side effects, and post-procedural complications such as restenosis and thrombosis (117).
Given these challenges, there is an urgent need to address the limitations of the existing therapies and explore novel approaches. The precise regulation of the ER-mitochondrial crosstalk in AS offers a new perspective. Key questions include ensuring therapeutic efficacy, enhancing safety, and identifying more effective and safer treatment strategies. To achieve this, further elucidation of the molecular mechanisms underlying the ER-mitochondrial crosstalk in AS, as well as the functions and interactions of specific MAMs proteins, is required. Identifying potential drug targets within MAMs and understanding the precise regulation of calcium signaling and lipid transport-related molecules will provide a robust foundation for drug design. Additionally, exploring the unique advantages and targets of TCM and its active components in modulating ER-mitochondrial crosstalk, combined with modern biotechnology, may unlock novel therapeutic potential. This may lead to the development of multi-target and highly efficacious anti-AS drugs. Moreover, the development of nanotechnology-based drug delivery systems could further enhance the targeted drug delivery to AS lesions. In summary, in-depth research on ER-mitochondria crosstalk in AS holds promise for revolutionizing cardiovascular disease prevention and treatment, addressing current diagnostic and therapeutic gaps, and offering safer and more effective strategies for patients with AS.
Author contributions
ML: Conceptualization, Writing – original draft. YX: Writing – review & editing. LD: Writing – review & editing, Validation. SC: Writing – review & editing, Visualization. WP: Writing – review & editing, Visualization. CT: Writing – review & editing, Funding acquisition, Supervision.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. The present study was supported by the Hunan Provincial natural Science Foundation (grant no.2022JJ70112), the second batch of key talent project in traditional Chinese medicine of Hunan during the‘14th Five-Year Plan' period [grant no. Xiang Zhong Yi Yao (2024)3] and the undergraduate Scientific research and innovation Fund Project of Hunan university of Chinese Medicine (grant no.S202410541114).
Acknowledgments
We would like to express our gratitude to Tao Jing, Jiang Chenyu, and Zhang Guomin for their assistance during the manuscript preparation process. And we would like to thank Editage (www.editage.cn) for English language editing.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Perrotta I. Atherosclerosis: from molecular biology to therapeutic perspective 2.0. Int J Mol Sci. (2022) 23:15158. doi: 10.3390/ijms232315158
2. Copeland DE and Dalton AJ. An association between mitochondria and the endoplasmic reticulum in cells of the pseudobranch gland of a teleost. J Biophys Biochem Cytol. (1959) 5:393–6. doi: 10.1083/jcb.5.3.393
3. Luan Y, Luan Y, Yuan RX, Feng Q, Chen X, Yang Y, et al. Structure and function of mitochondria-associated endoplasmic reticulum membranes (MAMs) and their role in cardiovascular diseases. Oxid Med Cell Longev. (2021) 2021:4578809. doi: 10.1155/2021/4578809
4. Janikiewicz J, Szymański J, Malinska D, Patalas-Krawczyk P, Michalska B, Duszyński J, et al. Mitochondria-associated membranes in aging and senescence: structure, function, and dynamics. Cell Death Dis. (2018) 9:332. doi: 10.1038/s41419-017-0105-5
5. Wang N, Wang C, Zhao H, He Y, Lan B, Sun L, et al. The MAMs structure and its role in cell death. Cells. (2021) 10(3):657. doi: 10.3390/cells10030657
6. Cao Y, Chen Z, Hu J, Feng J, Zhu Z, Fan Y, et al. Mfn2 regulates high glucose-induced MAMs dysfunction and apoptosis in podocytes via PERK pathway. Front Cell Dev Biol. (2021) 9:769213. doi: 10.3389/fcell.2021.769213
7. Song Z, Song H, Liu D, Yan B, Wang D, Zhang Y, et al. Overexpression of MFN2 alleviates sorafenib-induced cardiomyocyte necroptosis via the MAM-CaMKIIdelta pathway in vitro and in vivo. Theranostics. (2022) 12:1267–85. doi: 10.7150/thno.65716
8. Stone SJ and Vance JE. Phosphatidylserine synthase-1 and -2 are localized to mitochondria-associated membranes. J Biol Chem. (2000) 275:34534–40. doi: 10.1074/jbc.m002865200
9. Kornmann B, Currie E, Collins SR, Schuldiner M, Nunnari J, Weissman JS, et al. An ER-mitochondria tethering complex revealed by a synthetic biology screen. Science. (2009) 325(5939):477–81. doi: 10.1126/science.1175088
10. Okamoto M, Nakano K, Takahashi-Nakaguchi A, Sasamoto K, Yamaguchi M, Teixeira MC, et al. In ERMES component controls mitochondrial morphology, mtROS, and drug efflux pump expression, resulting in azole susceptibility. J Fungi. (2023) 9:240. doi: 10.3390/jof9020240
11. Wozny MR, Di Luca A, Morado DR, Picco A, Khaddaj R, Campomanes P, et al. In situ architecture of the ER-mitochondria encounter structure. Nature. (2023) 618:188–92. doi: 10.1038/s41586-023-06050-3
12. Jeong H, Park J, Jun Y, and Lee C. Crystal structures of Mmm1 and Mdm12-Mmm1 reveal mechanistic insight into phospholipid trafficking at ER-mitochondria contact sites. Proc Natl Acad Sci United States America. (2017) 114:E9502–11. doi: 10.1073/pnas.1715592114
13. Kornmann B and Walter P. ERMES-mediated ER-mitochondria contacts: molecular hubs for the regulation of mitochondrial biology. J Cell Sci. (2010) 123:1389–93. doi: 10.1242/jcs.058636
14. Nguyen TT, Lewandowska A, Choi JY, Markgraf DF, Junker M, Bilgin M, et al. Gem1 and ERMES do not directly affect phosphatidylserine transport from ER to mitochondria or mitochondrial inheritance. Traffic. (2012) 13:880–90. doi: 10.1111/j.1600-0854.2012.01352.x
15. Rasul F, Zheng F, Dong F, He J, Liu L, Liu W, et al. Emr1 regulates the number of foci of the endoplasmic reticulum-mitochondria encounter structure complex. Nat Commun. (2021) 12:521. doi: 10.1038/s41467-020-20866-x
16. Cheema JY, He J, Wei W, and Fu C. The endoplasmic reticulum-mitochondria encounter structure and its regulatory proteins. Contact (Thousand Oaks). (2021) 4:25152564211064491. doi: 10.1177/25152564211064491
17. Vance JE. Inter-organelle membrane contact sites: implications for lipid metabolism. Biol Direct. (2020) 15:24. doi: 10.1186/s13062-020-00279-y
18. Cui Z, Vance JE, Chen MH, Voelker DR, and Vance DE. Cloning and expression of a novel phosphatidylethanolamine N-methyltransferase. A specific biochemical and cytological marker for a unique membrane fraction in rat liver. J Biol Chem. (1993) 268:16655–63. doi: 10.1016/s0021-9258(19)85468-6
19. Hernandez-Alvarez MI, Sebastián D, Vives S, Ivanova S, Bartoccioni P, Kakimoto P, et al. Deficient endoplasmic reticulum-mitochondrial phosphatidylserine transfer causes liver disease. Cell. (2019) 177:881–895.e17. doi: 10.1016/j.cell.2019.04.010
20. Ganji R, Paulo JA, Xi Y, Kline I, Zhu J, Clemen CS, et al. The p97-UBXD8 complex regulates ER-Mitochondria contact sites by altering membrane lipid saturation and composition. Nat Commun. (2023) 14:638. doi: 10.1038/s41467-023-36298-2
21. Sironi L, Restelli LM, Tolnay M, Neutzner A, and Frank S. Dysregulated interorganellar crosstalk of mitochondria in the pathogenesis of parkinson’s disease. Cells. (2020) 9:233. doi: 10.3390/cells9010233
22. Rossini M, García-Casas P, Filadi R, and Pizzo P. Loosening ER-mitochondria coupling by the expression of the presenilin 2 loop domain. Cells. (2021) 10:1968. doi: 10.3390/cells10081968
23. Zhao Y, Hu D, Wang R, Sun X, Ropelewski P, Hubler Z, et al. ATAD3A oligomerization promotes neuropathology and cognitive deficits in Alzheimer’s disease models. Nat Commun. (2022) 13:1121. doi: 10.1038/s41467-022-28769-9
24. Szabo L, Cummins N, Paganetti P, Odermatt A, Papassotiropoulos A, Karch C, et al. ER-mitochondria contacts and cholesterol metabolism are disrupted by disease-associated tau protein. EMBO Rep. (2023) 24:e57499. doi: 10.15252/embr.202357499
25. Arruda AP, Pers BM, Parlakgül G, Güney E, Inouye K, and Hotamisligil GS. Chronic enrichment of hepatic endoplasmic reticulum-mitochondria contact leads to mitochondrial dysfunction in obesity. Nat Med. (2014) 20:1427–35. doi: 10.1038/nm.3735
26. Christgen S and Kanneganti TD. Inflammasomes and the fine line between defense and disease. Curr Opin Immunol. (2020) 62:39–44. doi: 10.1016/j.coi.2019.11.007
27. Missiroli S, Patergnani S, Caroccia N, Pedriali G, Perrone M, Previati M, et al. Mitochondria-associated membranes (MAMs) and inflammation. Cell Death Dis. (2018) 9:329. doi: 10.1038/s41419-017-0027-2
28. Bronner DN, Abuaita BH, Chen X, Fitzgerald KA, Nuñez G, He Y, et al. Endoplasmic reticulum stress activates the inflammasome via NLRP3- and caspase-2-driven mitochondrial damage. Immunity. (2015) 43:451–62. doi: 10.1016/j.immuni.2015.08.008
29. Xie YF, Cai H, Zhong F, Xiao W, Gordon RE, et al. Reticulon-1A mediates diabetic kidney disease progression through endoplasmic reticulum-mitochondrial contacts in tubular epithelial cells. Kidney Int. (2022) 102:293–306. doi: 10.1016/j.kint.2022.02.038
30. Zhang JR, Shen SY, Zhai MY, Shen ZQ, Li W, Liang LF, et al. Augmented microglial endoplasmic reticulum-mitochondria contacts mediate depression-like behavior in mice induced by chronic social defeat stress. Nat Commun. (2024) 15:5199. doi: 10.1038/s41467-024-49597-z
31. Gao P, Yang WX, and Sun L. Mitochondria-associated endoplasmic reticulum membranes (MAMs) and their prospective roles in kidney disease. Oxid Med Cell Longevity. (2020) 2020:3120539. doi: 10.1155/2020/3120539
32. Ye L, Zeng Q, Ling M, Ma R, Chen H, Lin F, et al. Inhibition of IP3R/ca2+Dysregulation protects mice from ventilator-induced lung injury endoplasmic reticulum and mitochondrial pathways. Front Immunol. (2021) 12:729094. doi: 10.3389/fimmu.2021.729094
33. Missiroli S, Perrone M, Gafà R, Nicoli F, Bonora M, Morciano G, et al. PML at mitochondria-associated membranes governs a trimeric complex with NLRP3 and P2X7R that modulates the tumor immune microenvironment. Cell Death Differentiation. (2023) 30:429–41. doi: 10.1038/s41418-022-01095-9
34. Chen KH, Feng L, Hu W, Chen J, Wang X, Wang L, et al. Optineurin inhibits NLRP3 inflammasome activation by enhancing mitophagy of renal tubular cells in diabetic nephropathy. FASEB J. (2019) 33:4571–85. doi: 10.1096/fj.201801749rrr
35. Hirabayahi Y, Kwon SK, Paek H, Pernice WM, Paul MA, Lee J, et al. ER-mitochondria tethering by PDZD8 regulates Ca dynamics in mammalian neurons. Science. (2017) 358:623–9. doi: 10.1126/science.aan6009
36. López-Otín C, Pietrocola F, Roiz-Valle D, Galluzzi L, and Kroemer G. Meta-hallmarks of aging and cancer. Cell Metab. (2023) 35:12–35. doi: 10.1016/j.cmet.2022.11.001
37. Wu SN, Lu Q, Wang Q, Ding Y, Ma Z, Mao X, et al. Binding of FUN14 domain containing 1 with inositol 1,4,5-trisphosphate receptor in mitochondria-associated endoplasmic reticulum membranes maintains mitochondrial dynamics and function in hearts in vivo. Circulation. (2017) 136:2248–66. doi: 10.1161/circulationaha.117.030235
38. Foyouzi-Youssefi R, et al. Bcl-2 decreases the free Ca2+ concentration within the endoplasmic reticulum. Proc Natl Acad Sci U S A. (2000) 97:5723–8. doi: 10.1073/pnas.97.11.5723
39. Garrido-Bazán V and Aguirre J. H2O2 induces calcium and ERMES complex-dependent mitochondrial constriction and division as well as mitochondrial outer membrane remodeling in. J Fungi. (2022) 8:829. doi: 10.3390/jof8080829
40. Sogo LF and Yaffe MP. Regulation of mitochondrial morphology and inheritance by Mdm10p, a protein of the mitochondrial outer membrane. J Cell Biol. (1994) 126:1361–73. doi: 10.1083/jcb.126.6.1361
41. Yuan M, Gong M, He J, Xie B, Zhang Z, Meng L, et al. IP3R1/GRP75/VDAC1 complex mediates endoplasmic reticulum stress-mitochondrial oxidative stress in diabetic atrial remodeling. Redox Biol. (2022) 52:102289. doi: 10.1016/j.redox.2022.102289
42. Marchi S, Marinello M, Bononi A, Bonora M, Giorgi C, Rimessi A, et al. Selective modulation of subtype III IP(3)R by Akt regulates ER Ca(2)(+) release and apoptosis. Cell Death Dis. (2012) 3:e304. doi: 10.1038/cddis.2012.45
43. Bononi A, Bonora M, Marchi S, Missiroli S, Poletti F, Giorgi C, et al. Identification of PTEN at the ER and MAMs and its regulation of Ca(2+) signaling and apoptosis in a protein phosphatase-dependent manner. Cell Death Differ. (2013) 20:1631–43. doi: 10.1038/cdd.2013.77
44. Belgareh-Touzé N, Cavellini L, and Cohen MM. Ubiquitination of ERMES components by the E3 ligase Rsp5 is involved in mitophagy. Autophagy. (2017) 13:114–32. doi: 10.1080/15548627.2016.1252889
45. Zhou H, Wang S, Hu S, Chen Y, and Ren J. ER-mitochondria microdomains in cardiac ischemia-reperfusion injury: A fresh perspective. Front Physiol. (2018) 9:755. doi: 10.3389/fphys.2018.00755
46. Ikeda Y, Shirakabe A, Maejima Y, Zhai P, Sciarretta S, Toli J, et al. Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ Res. (2015) 116:264–78. doi: 10.1161/circresaha.116.303356
47. Jetto CT, Nambiar A, and Manjithaya R. Mitophagy and neurodegeneration: between the knowns and the unknowns. Front Cell Dev Biol. (2022) 10. doi: 10.3389/fcell.2022.837337
48. Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, and Voeltz GK. ER tubules mark sites of mitochondrial division. Science. (2011) 334:358–62. doi: 10.1126/science.1207385
49. Böckler S and Westermann B. Mitochondrial ER contacts are crucial for mitophagy in yeast. Dev Cell. (2014) 28:450–8. doi: 10.1016/j.devcel.2014.01.012
50. Böckler S and Westermann B. ER-mitochondria contacts as sites of mitophagosome formation. Autophagy. (2014) 10:1346–7. doi: 10.4161/auto.28981
51. Garofalo T, Matarrese P, Manganelli V, Marconi M, Tinari A, Gambardella L, et al. Evidence for the involvement of lipid rafts localized at the ER-mitochondria associated membranes in autophagosome formation. Autophagy. (2016) 12:917–35. doi: 10.1080/15548627.2016.1160971
52. Ham SJ, Yoo H, Woo D, Lee DH, Park KS, and Chung J. PINK1 and Parkin regulate IP R-mediated ER calcium release. Nat Commun. (2023) 14(1):5202. doi: 10.1038/s41467-023-40929-z
53. Kuroda Y, Mitsui T, Kunishige M, Shono M, Akaike M, Azuma H, et al. Parkin enhances mitochondrial biogenesis in proliferating cells. Hum Mol Genet. (2006) 15:883–95. doi: 10.1093/hmg/ddl006
54. Vincow ES, Merrihew G, Thomas RE, Shulman NJ, Beyer RP, MacCoss MJ, et al. The PINK1-Parkin pathway promotes both mitophagy and selective respiratory chain turnover in vivo. Proc Natl Acad Sci USA. (2013) 110:6400–5. doi: 10.1073/pnas.1221132110
55. Docherty CK, Bresciani J, Carswell A, Chanderseka A, Friel E, Stasi M, et al. An inducible and vascular smooth muscle cell-specific pink1 knockout induces mitochondrial energetic dysfunction during atherogenesis. Int J Mol Sci. (2021) 22:9993. doi: 10.3390/ijms22189993
56. Gelmetti V, De Rosa P, Torosantucci L, Marini ES, Romagnoli A, Di Rienzo M, et al. PINK1 and BECN1 relocalize at mitochondria-associated membranes during mitophagy and promote ER-mitochondria tethering and autophagosome formation. Autophagy. (2017) 13:654–69. doi: 10.1080/15548627.2016.1277309
57. Yang J, Sun M, Cheng R, Tan H, Liu C, Chen R, et al. Pitavastatin activates mitophagy to protect EPC proliferation through a calcium-dependent CAMK1-PINK1 pathway in atherosclerotic mice. Commun Biol. (2022) 5:124. doi: 10.1038/s42003-022-03081-w
58. Zhang YK, Wang S, Chen X, Wang Z, Wang X, Zhou Q, et al. Liraglutide prevents high glucose induced HUVECs dysfunction via inhibition of PINK1/Parkin-dependent mitophagy. Mol Cell Endocrinol. (2022) 545:111560. doi: 10.1016/j.mce.2022.111560
59. Chen X, Mi L, Gu G, Gao X, Gao X, Shi M, et al. Dysfunctional endoplasmic reticulum-mitochondrion coupling is associated with endoplasmic reticulum stress-induced apoptosis and neurological deficits in a rodent model of severe head injury. J Neurotrauma. (2022) 39:560–76. doi: 10.1089/neu.2021.0347
60. Marchi S, Patergnani S, Missiroli S, Morciano G, Rimessi A, Wieckowski MR, et al. Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell Calcium. (2018) 69:62–72. doi: 10.1016/j.ceca.2017.05.003
61. Danese A, Patergnani S, Bonora M, Wieckowski MR, Previati M, Giorgi C, et al. Calcium regulates cell death in cancer: Roles of the mitochondria and mitochondria-associated membranes (MAMs). Biochim Et Biophys Acta-Bioenergetics. (2017) 1858:615–27. doi: 10.1016/j.bbabio.2017.01.003
62. Yu SJ, Zhang L, Liu C, Yang J, Zhang J, and Huang L. PACS2 is required for ox-LDL-induced endothelial cell apoptosis by regulating mitochondria-associated ER membrane formation and mitochondrial Ca elevation. Exp Cell Res. (2019) 379:191–202. doi: 10.1016/j.yexcr.2019.04.002
63. Chen WY, Chen YF, Chan HC, Chung CH, Peng HY, Ho YC, et al. Role of apolipoprotein E in electronegative low -density lipoprotein-induced mitochondrial dysfunction in cardiomyocytes. Metabolism-Clinical Exp. (2020) 107:154227. doi: 10.1016/j.metabol.2020.154227
64. Zhang Y, Li JJ, Xu R, Wang XP, Zhao XY, Fang Y, et al. Nogo-B mediates endothelial oxidative stress and inflammation to promote coronary atherosclerosis in pressure-overloaded mouse hearts. Redox Biol. (2023) 68:102944. doi: 10.1016/j.redox.2023.102944
65. Baik SH, Ramanujan VK, Becker C, Fett S, Underhill DM, and Wolf AJ. Hexokinase dissociation from mitochondria promotes oligomerization of VDAC that facilitates NLRP3 inflammasome assembly and activation. Sci Immunol. (2023) 8:eade7652. doi: 10.1126/sciimmunol.ade7652
66. Dong YZ, Lee Y, Cui K, He M, Wang B, Bhattacharjee S, et al. Epsin-mediated degradation of IP3R1 fuels atherosclerosis. Nat Commun. (2020) 11:928. doi: 10.1038/s41467-020-17848-4
67. Santos NC, Girik V, and Nunes-Hasler P. ORP5 and ORP8: sterol sensors and phospholipid transfer proteins at membrane contact sites? Biomolecules. (2020) 10. doi: 10.3390/biom10060928
68. Jaworski CJ, Moreira E, Li A, Lee R, and Rodriguez IR. A family of 12 human genes containing oxysterol-binding domains. Genomics. (2001) 78:185–96. doi: 10.1006/geno.2001.6663
69. Du X, Kumar J, Ferguson C, Schulz TA, Ong YS, Hong W, et al. A role for oxysterol-binding protein-related protein 5 in endosomal cholesterol trafficking. J Cell Biol. (2011) 192:121–35. doi: 10.1083/jcb.201004142
70. Galmes R, Houcine A, van Vliet AR, Agostinis P, Jackson CL, and Giordano F. ORP5/ORP8 localize to endoplasmic reticulum-mitochondria contacts and are involved in mitochondrial function. EMBO Rep. (2016) 17:800–10. doi: 10.15252/embr.201541108
71. Guyard V, Monteiro-Cardoso VF, Omrane M, Sauvanet C, Houcine A, Boulogne C, et al. ORP5 and ORP8 orchestrate lipid droplet biogenesis and maintenance at ER-mitochondria contact sites. J Cell Biol. (2022) 221:e202112107. doi: 10.1083/jcb.202112107
72. Pu MM, Zheng W, Zhang H, Wan W, Peng C, Chen X, et al. ORP8 acts as a lipophagy receptor to mediate lipid droplet turnover. Protein Cell. (2023) 14:653–67. doi: 10.1093/procel/pwac063
73. Eisenreichova A, Klima M, Anila MM, Koukalova A, Humpolickova J, Różycki B, et al. Crystal structure of the ORP8 lipid transport ORD domain: model of lipid transport. Cells. (2023) 12:1974. doi: 10.3390/cells12151974
74. Assis LHP, Dorighello GdG, Rentz T, de Souza JC, Vercesi AE, de Oliveira HCF, et al. In vivo pravastatin treatment reverses hypercholesterolemia induced mitochondria-associated membranes contact sites, foam cell formation, and phagocytosis in macrophages. Front Mol Biosci. (2022) 9:839428. doi: 10.3389/fmolb.2022.839428
75. Ding YQ, Liu N, Zhang D, Guo L, Shang Q, Liu Y, et al. Mitochondria-associated endoplasmic reticulum membranes as a therapeutic target for cardiovascular diseases. Front Pharmacol. (2024) 15:1398381. doi: 10.3389/fphar.2024.1398381
76. Li JY, Xin Y, Li J, Chen H, and Li H. Phosphatidylethanolamine N-methyltransferase: from functions to diseases. Aging Dis. (2023) 14:879–91. doi: 10.14336/ad.2022.1025
77. Chandhok G, Lazarou M, and Neumann B. Structure, function, and regulation of mitofusin-2 in health and disease. Biol Rev. (2018) 93:933–49. doi: 10.1111/brv.12378
78. Hu Y, Chen H, Zhang L, Lin X, Li X, Zhuang H, et al. The AMPK-MFN2 axis regulates MAM dynamics and autophagy induced by energy stresses. Autophagy. (2021) 17:1142–56. doi: 10.1080/15548627.2020.1749490
79. Jiao YY, Zhang X, Yang H, Ma H, and Zou J. Mesoporous tantalum oxide nanomaterials induced cardiovascular endothelial cell apoptosis via mitochondrial-endoplasmic reticulum stress apoptotic pathway. Drug Delivery. (2023) 30:108–20. doi: 10.1080/10717544.2022.2147251
80. Feng J, Lü S, Ding Y, Zheng M, and Wang X. Homocysteine activates T cells by enhancing endoplasmic reticulum-mitochondria coupling and increasing mitochondrial respiration. Protein Cell. (2016) 7:391–402. doi: 10.1007/s13238-016-0245-x
81. Dabravolski SA, Sadykhov NK, Kartuesov AG, Borisov EE, Sukhorukov VN, and Orekhov AN. Interplay between zn homeostasis and mitochondrial functions in cardiovascular diseases and heart ageing. Int J Mol Sci. (2022) 23:6890. doi: 10.3390/ijms23136890
82. Xu L, Hao H, Hao Y, Wei G, Li G, Ma P, et al. Aberrant MFN2 transcription facilitates homocysteine-induced VSMCs proliferation via the increased binding of c-Myc to DNMT1 in atherosclerosis. J Cell Mol Med. (2019) 23:4611–26. doi: 10.1111/jcmm.14341
83. Zhao JH, Li J, Li G, and Chen M. The role of mitochondria-associated membranes mediated ROS on NLRP3 inflammasome in cardiovascular diseases. Front Cardiovasc Med. (2022) 9:1059576. doi: 10.3389/fcvm.2022.1059576
84. Xie H, Tang J, Song L, Xu G, Li W, Zhu J, et al. Mitochondria-endoplasmic reticulum crosstalk in apoptosis: The interactions of cytochrome c with monooxygenase and its reductase. Int J Biol Macromolecules. (2024) 279:135160. doi: 10.1016/j.ijbiomac.2024.135160
85. Guo W, Diao Z, and Liu W. Asymmetric dimethylarginine downregulates sarco/endoplasmic reticulum calcium−ATPase 3 and induces endoplasmic reticulum stress in human umbilical vein endothelial cells. Mol Med Rep. (2017) 16:7541–7. doi: 10.3892/mmr.2017.7529
86. Mohammadi A, Gholamhoseyniannajar A, Yaghoobi MM, Jahani Y, and Vahabzadeh Z. Expression levels of heat shock protein 60 and glucose-regulated protein 78 in response to trimethylamine-N-oxide treatment in murine macrophage J774A.1 cell line. Cell Mol Biol. (2015) 61:94–100.
87. Moulis M, Grousset E, Faccini J, Richetin K, Thomas G, and Vindis C. The multifunctional sorting protein PACS-2 controls mitophagosome formation in human vascular smooth muscle cells through mitochondria-ER contact sites. Cells. (2019) 8:638. doi: 10.3390/cells8060638
88. Lin W, Chen S, Wang Y, Wang M, Lee WY, Jiang X, et al. Dynamic regulation of mitochondrial-endoplasmic reticulum crosstalk during stem cell homeostasis and aging. Cell Death Dis. (2021) 12:794. doi: 10.1038/s41419-021-03912-4
89. Larrea D, Pera M, Gonnelli A, Quintana-Cabrera R, Akman HO, Guardia-Laguarta C, et al. MFN2 mutations in Charcot-Marie-Tooth disease alter mitochondria-associated ER membrane function but do not impair bioenergetics. Hum Mol Genet. (2019) 28:1782–800. doi: 10.1093/hmg/ddz008
90. Granatiero V, Giorgio V, Calì T, Patron M, Brini M, Bernardi P, et al. Reduced mitochondrial Ca(2+) transients stimulate autophagy in human fibroblasts carrying the 13514A>G mutation of the ND5 subunit of NADH dehydrogenase. Cell Death Differ. (2016) 23:231–41. doi: 10.1038/cdd.2015.84
91. Ma Z, Du X, Sun Y, Jia Y, Liang X, and Gao Y. Attenuation of PM2.5-induced lung injury by 4-phenylbutyric acid: maintenance of [Ca(2+)]i stability between endoplasmic reticulum and mitochondria. Biomolecules. (2024) 14:1135. doi: 10.3390/biom14091135
92. Zhu GL, Gao H, Li Y, Li X, Yang X, Wang C, et al. Suppression of endoplasmic reticulum stress by 4-PBA enhanced atherosclerotic plaque stability via up-regulating CLOCK expression. Pathol Res Pract. (2024) 253:154969. doi: 10.1016/j.prp.2023.154969
93. Laksono RM, Kalim H, Rohman MS, Widodo N, Ahmad MR, and Halim W. Pulsed radiofrequency decreases pERK and affects intracellular Ca2+ influx, cytosolic ATP level, and mitochondrial membrane potential in the sensitized dorsal root ganglion neuron induced by N-methyl D-aspartate. J Pain Res. (2023) 16:1697–711. doi: 10.2147/JPR.S425900
94. Shoshan-Barmatz V, Anand U, Nahon-Crystal E, Di Carlo M, and Shteinfer-Kuzmine A. Adverse effects of metformin from diabetes to COVID-19, cancer, neurodegenerative diseases, and aging: is VDAC1 a common target? Front Physiol. (2021) 12:730048. doi: 10.3389/fphys.2021.730048
95. Jenkins AJ, Welsh P, and Petrie JR. Metformin, lipids and atherosclerosis prevention. Curr Opin Lipidology. (2018) 29:346–53. doi: 10.1097/mol.0000000000000532
96. Zhou WP, Fan XR, Li SH, Zeng ZL, and Wei YM. Statins combined with AAV8-TBG-LOX-1 reduce the vascular lipid-driven inflammatory response and inhibit atherosclerosis. Curr Med Sci. (2024) 44:1097–102. doi: 10.1007/s11596-024-2954-3
97. Yin Y, Wu C, Zhou Y, Zhang M, Mai S, Chen M, et al. Ezetimibe induces paraptosis through niemann-pick C1-like 1 inhibition of mammalian-target-of-rapamycin signaling in hepatocellular carcinoma cells. Genes (Basel). (2023) 15:4. doi: 10.3390/genes15010004
98. Ali AH, Younis N, Abdallah R, Shaer F, Dakroub A, Ayoub MA, et al. Lipid-lowering therapies for atherosclerosis: statins, fibrates, ezetimibe and PCSK9 monoclonal antibodies. Curr Med Chem. (2021) 28:7427–45. doi: 10.2174/1875533xmte03ndeo0
99. Kong DZ, Sun P, Lu Y, Yang Y, Min DY, Zheng SC, et al. Yi Mai granule improve energy supply of endothelial cells in atherosclerosis via miRNA-125a-5p regulating mitochondrial autophagy through Pink1-Mfn2-Parkin pathway. J Ethnopharmacology. (2024) 319:117114. doi: 10.1016/j.jep.2023.117114
100. Li ZY, Wang B, Tian L, Zheng B, Zhao X, and Liu R. Methane-rich saline suppresses ER-mitochondria contact and activation of the NLRP3 inflammasome by regulating the PERK signaling pathway to ameliorate intestinal ischemia-reperfusion injury. Inflammation. (2024) 47:376–89. doi: 10.1007/s10753-023-01916-0
101. Hong KX, Wang J, Kang XXue H, Gao Y, Liang H, et al. Ferulic acid and protocatechuic acid alleviate atherosclerosis by promoting UCP1 expression to inhibit the NLRP3-IL-1β signaling pathway. Food Funct. (2025) 16:40–53. doi: 10.1039/d4fo02955k
102. Chen W, Zhang Y, Miao G, Ying Y, Ren Z, Sun X, et al. The augment effects of magnesium hydride on the lipid lowering effect of atorvastatin: an in vivo and in vitro investigation. Med Gas Res. (2025) 15:148–55. doi: 10.4103/mgr.medgasres-d-23-00047
103. Aldossary KM, Ali LS, Abdallah MSBahaa MM, Elmasry TA, Elberri EI, et al. Effect of a high dose atorvastatin as added-on therapy on symptoms and serum AMPK/NLRP3 inflammasome and IL-6/STAT3 axes in patients with major depressive disorder: randomized controlled clinical study. Front Pharmacol. (2024) 15:1464358. doi: 10.3389/fphar.2024.1381523
104. Li FY, Xu D, Hou K, Gou X, Lv N, Fang W, et al. Pretreatment of indobufen and aspirin and their combinations with clopidogrel or ticagrelor alleviates inflammasome mediated pyroptosis via inhibiting NF-κB/NLRP3 pathway in ischemic stroke. J Neuroimmune Pharmacol. (2021) 16:835–53. doi: 10.1007/s11481-020-09978-9
105. Golledge J. Update on the pathophysiology and medical treatment of peripheral artery disease. Nat Rev Cardiol. (2022) 19:456–74. doi: 10.1038/s41569-021-00663-9
106. Gutierrez T, Parra V, Troncoso R, Pennanen C, Contreras-Ferrat A, Vasquez-Trincado C, et al. Alteration in mitochondrial Ca(2+) uptake disrupts insulin signaling in hypertrophic cardiomyocytes. Cell Commun Signal. (2014) 12:68. doi: 10.1186/s12964-014-0068-4
107. Ruan H, Huang Q, Wan B, and Yang M. Curcumin alleviates lipopolysaccharides-induced inflammation and apoptosis in vascular smooth muscle cells via inhibition of the NF-kappaB and JNK signaling pathways. Inflammopharmacology. (2022) 30:517–25. doi: 10.1007/s10787-021-00912-w
108. Li X, Zhu J, Lin Q, Yu M, Lu J, Feng J, et al. Effects of curcumin on mitochondrial function, endoplasmic reticulum stress, and mitochondria-associated endoplasmic reticulum membranes in the jejunum of oxidative stress piglets. J Agric Food Chem. (2022) 70:8974–85. doi: 10.1021/acs.jafc.2c02824
109. Lee DH, Kim HH, Cho HJ, Yu YB, Kang HC, Kim JL, et al. Cordycepin-Enriched WIB801C from Cordyceps militaris Inhibits Collagen-Induced [Ca(2+)]i Mobilization via cAMP-Dependent Phosphorylation of Inositol 1, 4, 5-Trisphosphate Receptor in Human Platelets. Biomol Ther (Seoul). (2014) 22:223–31. doi: 10.4062/biomolther.2014.025
110. Gao J, Wang H, Li Y, and Li W. Resveratrol attenuates cerebral ischaemia reperfusion injury via modulating mitochondrial dynamics homeostasis and activating AMPK-Mfn1 pathway. Int J Exp Pathol. (2019) 100:337–49. doi: 10.1111/iep.12336
111. Zhang RF, Jiang Y, Zhang G, Zeng W, Suo Y, Zhang F, et al. Mitochondrial DNA in atherosclerosis: Mechanisms, biomarker potential, and therapeutic perspectives. Int Immunopharmacol. (2025) 152:114449. doi: 10.1016/j.intimp.2025.114449
112. Kalia R, Wang RY, Yusuf A, Thomas PV, Agard DA, Shaw JM, et al. Structural basis of mitochondrial receptor binding and constriction by DRP1. Nature. (2018) 558:401–5. doi: 10.1038/s41586-018-0211-2
113. Liu Y, Huang Y, Xu C, An P, Luo Y, Jiao L, et al. Mitochondrial dysfunction and therapeutic perspectives in cardiovascular diseases. Int J Mol Sci. (2022) 23:16053. doi: 10.3390/ijms232416053
114. Longchamps RJ, Castellani CA, Yang SY, Newcomb CE, Sumpter JA, Lane J, et al. Evaluation of mitochondrial DNA copy number estimation techniques. PLoS One. (2020) 15:e0228166. doi: 10.1371/journal.pone.0228166
115. Cutlip DE, Chhabra AG, Baim DS, Chauhan MS, Marulkar S, Massaro J, et al. Beyond restenosis: five-year clinical outcomes from second-generation coronary stent trials. Circulation. (2004) 110:1226–30. doi: 10.1161/01.CIR.0000140721.27004.4B
116. Hara T and Rai Y. Carotid endarterectomy. Adv Tech Stand Neurosurg. (2022) 44:187–207. doi: 10.1007/978-3-030-87649-4_10
117. Chen PP, Patel PB, Ding J, Krimbill J, Siracuse JJ, O'Donnell TFX, et al. Asian race is associated with peripheral arterial disease severity and postoperative outcomes. J Vasc Surg. (2023) 78:175–83.e3. doi: 10.1016/j.jvs.2023.02.015
118. Arasaki K, Shimizu H, Mogari H, Nishida N, Hirota N, Furuno A, et al. A role for the ancient SNARE syntaxin 17 in regulating mitochondrial division. Dev Cell. (2015) 32:304–17. doi: 10.1016/j.devcel.2014.12.011
119. Berger KH, Sogo LF, and Yaffe MP. Mdm12p, a component required for mitochondrial inheritance that is conserved between budding and fission yeast. J Cell Biol. (1997) 136:545–53. doi: 10.1083/jcb.136.3.545
120. Esposito M, Hermann-Le Denmat S, and Delahodde A. Contribution of ERMES subunits to mature peroxisome abundance. PLoS One. (2019) 14:e0214287. doi: 10.1371/journal.pone.0214287
121. Obara CJ, Nixon-Abell J, Moore AS, Riccio F, Hoffman DP, Shtengel G, et al. Motion of VAPB molecules reveals ER-mitochondria contact site subdomains. Nature. (2024) 626:169–76. doi: 10.1038/s41586-023-06956-y
122. Simmen T, Aslan JE, Blagoveshchenskaya AD, Thomas L, Wa , et al. PACS-2 controls endoplasmic reticulum-mitochondria communication and Bid-mediated apoptosis. EMBO J. (2005) 24:717–29. doi: 10.1038/sj.emboj.7600637
123. Çoku J, Booth DM, Skoda JPedrotty MC, Vogel J, Liu K, et al. Reduced ER-mitochondria connectivity promotes neuroblastoma multidrug resistance. EMBO J. (2022) 41:e108272. doi: 10.15252/embj.2021108272
124. Giorgi C, Baldassari F, Bononi ABonora M, De Marchi E, Marchi S, et al. Mitochondrial ca(2+) and apoptosis. Cell Calcium. (2012) 52:36–43. doi: 10.1016/j.ceca.2012.02.008
125. Carpio MA, Means RE, Brill AL, Sainz A, Ehrlich BE, and Katz SG. BOK controls apoptosis by Ca transfer through ER-mitochondrial contact sites. Cell Rep. (2021) 34:108827. doi: 10.1016/j.celrep.2021.108827
126. Liu WK, Song H, Xu J, Guo Y, Zhang C, Yao Y, et al. Low shear stress inhibits endothelial mitophagy via caveolin-1/miR-7-5p/SQSTM1 signaling pathway. Atherosclerosis. (2022) 356:9–17. doi: 10.1016/j.atherosclerosis.2022.07.014
127. Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, et al. Autophagosomes form at ER-mitochondria contact sites. Nature. (2013) 495:389–93. doi: 10.1038/nature11910
128. Cao YH, Chen X, Pan FWang M, Zhuang H, Chen J, et al. Xinmaikang-mediated mitophagy attenuates atherosclerosis via the PINK1/Parkin signaling pathway. Phytomedicine. (2023) 119:154955. doi: 10.1016/j.phymed.2023.154955
129. Yao RQ, Ren C, Xia ZF, and Yao YM. Organelle-specific autophagy in inflammatory diseases: a potential therapeutic target underlying the quality control of multiple organelles. Autophagy. (2021) 17:385–401. doi: 10.1080/15548627.2020.1725377
130. Mao K and Klionsky DJ. Mitochondrial fission facilitates mitophagy in. Autophagy. (2013) 9:1900–1. doi: 10.4161/auto.25804
131. Kohler V, Aufschnaiter A, and Büttner S. Closing the gap: membrane contact sites in the regulation of autophagy. Cells. (2020) 9:1184. doi: 10.3390/cells9051184
132. Geisler S, Holmström KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. (2010) 12:119–31. doi: 10.1038/ncb2012
133. El Kodsi DN, Tokarew JM, Sengupta R, Lengacher NA, Chatterji A, Nguyen AP, et al. Parkin coregulates glutathione metabolism in adult mammalian brain. Acta Neuropathol Commun. (2023) 11:19. doi: 10.1186/s40478-022-01488-4
134. Choi SH, Agatisa-Boyle C, Gonen A, Kim A, Kim J, Alekseeva E, et al. Intracellular AIBP (Apolipoprotein A-I binding protein) regulates oxidized LDL (Low-density lipoprotein)-induced mitophagy in macrophages. Arterioscler Thromb Vasc Biol. (2021) 41:E82–96. doi: 10.1161/atvbaha.120.315485
135. Wang ZX, Sun W, Zhang K, Ke X, and Wang Z. New insights into the relationship of mitochondrial metabolism and atherosclerosis. Cell Signalling. (2025) 127:111580. doi: 10.1016/j.cellsig.2024.111580
136. Zhng ZB, Valero RA, Quintana A, Hoth M, Núñez L, and Villalobos C. Research progress on the role of endoplasmic reticulum-mitochondrial interaction in cardiovascular diseases. Heart J. (2020) 32:522–7. doi: 10.1074/jbc.M110.198952
137. Sreekumar PG, Hinton DR, and Kannan R. Endoplasmic reticulum-mitochondrial crosstalk: a novel role for the mitochondrial peptide humanin. Neural Regeneration Res. (2017) 12:35–8. doi: 10.4103/1673-5374.198970
138. Yang YS, Liu Y, Wang Y, Chao Y, Zhang J, Jia Y, et al. Regulation of SIRT1 and its roles in inflammation. Front Immunol. (2022) 13:831168. doi: 10.3389/fimmu.2022.831168
Keywords: atherosclerosis, endoplasmic reticulum-mitochondrial crosstalk, mitochondria-associated membranes (MAMs), endoplasmic reticulum contact complex, endoplasmic reticulum, mitochondria
Citation: Li M, Xiao Y, Dai L, Chen S, Pei W and Tan C (2025) Endoplasmic reticulum-mitochondria crosstalk: new mechanisms in the development of atherosclerosis. Front. Endocrinol. 16:1573499. doi: 10.3389/fendo.2025.1573499
Received: 09 February 2025; Accepted: 06 May 2025;
Published: 05 June 2025.
Edited by:
Ki Ho Park, University of Virginia, United StatesReviewed by:
Fernanda Marques da Cunha, Universidade Federal de São Paulo, BrazilSubhoshree Ghose, Oxford Nanopore Technologies, United Kingdom
Copyright © 2025 Li, Xiao, Dai, Chen, Pei and Tan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chao Tan, dGFuY2hhbzMxMDE3OUBobnVjbS5lZHUuY24=
†These authors have contributed equally to this work and share first authorship