Abstract
Osteosarcopenia (OS), a recently recognized syndrome characterized by the simultaneous occurrence of osteopenia/osteoporosis and sarcopenia, has emerged as an important concept in clinical practice. This integrated framework provides a comprehensive view of the musculoskeletal system, addressing a previously underappreciated aspect of muscle health. OS notably increases the risk of falls, fractures, hospitalization, and mortality in elderly patients with chronic diseases. Despite its growing clinical relevance, OS remains underdiagnosed, and its classification as a distinct syndrome is not universally accepted. The persistently high global prevalence of chronic diseases, along with their substantial medical, economic, and social burdens, underscores the urgent need for updated prevention and management strategies. This review advocates for greater awareness and improved management of OS in patients with chronic diseases. It examines the relationship between OS and chronic conditions, emphasizing its epidemiology, adverse outcomes, diagnostic approaches, pathophysiology, and potential management strategies.
1 Background
Osteosarcopenia (OS) is defined as the concurrent presence of osteopenia/osteoporosis and sarcopenia (1), a concept first introduced by Duque et al. (1). It is a prevalent condition, particularly among the elderly. Prior to recognizing OS as a distinct syndrome, muscle-related disorders were often overlooked. However, acknowledging OS is crucial, as it significantly increases the risk of falls, fractures, hospitalization, and mortality in older individuals (2). Recent studies have also emphasized that OS contributes to the onset and progression of chronic diseases, thereby worsening outcomes for patients with these conditions (3, 4). Despite its growing significance, OS remains underrecognized in the management of chronic diseases, with much of the focus still placed solely on bone health. Given the global aging population, the prevalence of OS is expected to rise sharply in the coming years (5), underscoring the need for a more comprehensive approach to its role in chronic disease management.
Chronic diseases are defined as conditions lasting one year or more that require ongoing medical care, limit daily activities, or both (6). Often referred to as the “plague” of the 21st century, these conditions pose a significant threat to global health, second only to infectious diseases (7, 8) Cardiovascular diseases, chronic obstructive pulmonary disease (COPD), diabetes, chronic kidney disease, and chronic liver disease have long been major burdens on healthcare systems, collectively contributing to more deaths worldwide than all other causes combined (7). Furthermore, the limited treatment options available have resulted in recurrent hospitalizations, driving up medical costs and creating substantial challenges for healthcare economies, particularly in certain Asian countries (9). In response to these pressing issues, there is an urgent need to update and improve prevention and management strategies for chronic diseases.
The management of chronic diseases is centered on preserving physiological reserves and maintaining long-term homeostasis. The musculoskeletal (MSK) system not only facilitates locomotion but also plays a critical role in functional metabolism and nutrient storage (10). The onset of OS significantly undermines the health and stability of patients with chronic diseases. As such, there is an urgent need to reassess and expand our understanding of OS to develop more effective and up-to-date management strategies. This review aims to explore the impact of OS on chronic diseases, focusing particularly on its diagnosis, pathophysiology, and potential management approaches.
2 Osteosarcopenia: epidemiology and clinical diagnosis
2.1 Clinical diagnosis
Currently, there are no universally accepted diagnostic criteria for OS. Its clinical diagnosis is typically based on the concurrent presence of osteopenia/osteoporosis and sarcopenia, as identified through ongoing research and expert consensus. In clinical practice, OS is diagnosed when both of these conditions are present simultaneously.
2.1.1 Diagnosis of osteopenia/osteoporosis
The World Health Organization (WHO) defines osteoporosis using bone mineral density (BMD) measurements obtained through Dual-Energy X-ray Absorptiometry (DXA). Osteoporosis is diagnosed when BMD is 2.5 standard deviations (SD) or more below the young adult reference mean (11). The T-score, which reflects BMD values at key sites such as the lumbar spine and femoral neck, is used to assess both osteopenia and osteoporosis (12). A T-score of ≥ -1.0 SD is considered normal; a T-score between -2.5 SD and -1.0 SD indicates osteopenia; and a T-score of ≤ -2.5 SD signifies osteoporosis (13). This classification is widely accepted and utilized globally.
2.1.2 Diagnosis of sarcopenia
In 2018, the European Working Group on Sarcopenia in Older People (EWGSOP) established diagnostic criteria for sarcopenia. The key indicators for muscle strength include: grip strength <27 kg for men and <16 kg for women, or a chair stand test time >15 seconds for more than five repetitions. For muscle mass, the threshold is defined as appendicular skeletal muscle mass <20 kg for men and <15 kg for women. Regarding muscle function, a gait speed ≤0.8 m/s is considered critical. The most reliable methods for assessing muscle mass in clinical settings are bioelectrical impedance analysis (BIA) and Dual-Energy X-ray Absorptiometry (DXA), with DXA regarded as the gold standard for diagnosis (14). The recommended cutoff values for muscle mass measurement are 7.0 kg/m² for men and 5.4 kg/m² for women using DXA, and 7.0 kg/m² for men and 5.7 kg/m² for women using BIA (15).
2.2 Epidemiology
The prevalence of OS increases with age among community-dwelling individuals. In men, it ranges from 14.3% in those aged 60–64 to 59.4% in those aged ≥75 years; in women, it ranges from 20.3% in those aged 60–64 to 48.3% in those aged ≥75 years. Overall, the prevalence tends to be higher in women than in men (5). These findings align with the aging process, suggesting that as the global population ages, the prevalence of OS is expected to rise, although it may vary across regions or ethnic groups.
3 The effect of osteosarcopenia on chronic diseases
3.1 Chronic liver disease
OS significantly increases the risk and worsens the prognostic outcomes of chronic liver disease. Recent studies have shown that OS is associated with higher mortality rates and a reduced quality of life (QOL) in patients with cirrhosis (LC) (16). Additionally, OS exacerbates severe late-stage liver disease complications, including hepatic encephalopathy and infections (17). A retrospective study by Chisato Saeki et al. found that OS significantly elevated the risk of mortality in cirrhosis patients compared to controls (hazard ratio (HR): 4.798; 95% Confidence Intervals (CI): 1.885–12.212; p = 0.001), making it a critical independent prognostic factor for these individuals (18). Furthermore, Saeki’s study highlighted that in patients with primary biliary cholangitis and OS, the incidence of vertebral fractures was as high as 55.6%, which was significantly greater than in other groups (19). Additionally, a prospective cohort study revealed that the prevalence of OS increases with the progression of liver fibrosis. Notably, 7.0% of chronic hepatitis C patients without cirrhosis developed OS before the onset of cirrhosis (20). This suggests a potential association between OS and the progression of liver fibrosis, although the underlying pathophysiological mechanisms warrant further exploration.
3.2 Cardiovascular diseases
OS and cardiovascular diseases are both age-related conditions that share numerous common risk factors (21). A study involving 70,697 participants found that OS was independently associated with a 17% increased risk of heart failure (HF) events (22). Additionally, OS was strongly and independently linked to major electrocardiographic abnormalities in the elderly and significantly correlated with the onset and progression of coronary heart disease (23). Previous studies have also demonstrated that OS increases the risk of falls and fractures (24). Clinically, elderly individuals with chronic heart disease are at heightened risk of falls and fractures. Among adults with chronic heart conditions like heart failure (HF), the fall rate is 43%, compared to approximately 30% in those with other chronic diseases (25). This increased risk cannot be solely attributed to low cardiac output and polypharmacy. The significant role of OS as a common condition in elderly individuals with chronic heart disease may often be underrecognized. However, research exploring the relationship between these two conditions remains limited, and the mechanisms through which OS contributes to the heightened cardiovascular risk warrant further investigation.
3.3 Chronic kidney disease
OS is strongly associated with poor health outcomes and declining renal function in patients with chronic kidney disease (CKD) (26). A study by Yuta Nakano et al. demonstrated that patients with OS experienced significantly worse primary adverse outcomes compared to those with a single condition (HR: 3.28; 95% CI: 1.52-7.08) and had an elevated risk of renal composite adverse outcomes (HR: 2.07; 95% CI: 1.10-3.89) (26). Another study reported that in CKD patients, those with OS had a 33% higher risk of mortality compared to those without OS (HR: 1.33; 95% CI: 1.07–1.66; P = 0.011), and the likelihood of progression to end-stage renal disease (ESRD) was more than twice as high as in the control group (HR: 2.08; 95% CI: 1.53–2.82; P < 0.001) (27). Furthermore, patients receiving renal replacement therapy are often in a more weakened and energy-deprived state (28). Since MSK provides essential energy and functional support for these patients, OS poses a potentially “fatal” threat to individuals with chronic kidney disease.
3.4 Chronic respiratory diseases
OS significantly increases the risk of hospitalization and mortality in patients with chronic obstructive pulmonary disease (COPD) (29). COPD patients often experience long-term overinflation and heightened dyspnea due to worsening airflow obstruction. The weakening of the musculoskeletal system (MSK) in these patients can lead to a reduction in type I muscle fibers, decreased oxidative enzyme activity, and reduced muscle capillaries (30). all of which exacerbate breathing difficulties. COPD is particularly prevalent among the elderly, and in its advanced stages, systemic cachexia is frequently observed. This condition is marked by mitochondrial damage and oxidative stress resulting from prolonged hypoxia, contributing to a negative nitrogen balance and systemic depletion. OS-induced frailty, combined with the decline in respiratory muscle function, may worsen the severity of dyspnea in COPD patients, leading to more frequent hospitalizations.
In summary, OS plays a crucial role in mediating the occurrence of various adverse outcomes in chronic diseases, underlining its critical importance in the long-term management of these conditions. Despite its significance, OS remains insufficiently recognized and addressed in the management of chronic diseases, particularly in conditions such as diabetes and cancer. It is strongly recommended that OS be actively identified (Table 1). Furthermore, prior studies suggest that chronic diseases may contribute to the development of OS through mechanisms such as systemic inflammation, metabolic disorders, long-term polypharmacy, and frailty. Recent research highlights the potential interactions between these factors (Figure 1). However, research on how OS influences the onset of chronic diseases is still in its early stages and remains a challenging area of investigation.
Table 1
| Author/year | Article type | Chronic disease | Results | Cite |
|---|---|---|---|---|
| Chisato Saeki et al.; 2023. | Retrospective Study | Liver Cirrhosis | Osteosarcopenia is significantly associated with mortality in patients with liver cirrhosis. | (18) |
| Tatiana Bering et al.;2018 | Retrospective Study | Chronic Hepatitis C | The prevalence of osteosarcopenia increases concurrently with the progression of liver fibrosis. | (20) |
| Chisato Saeki et al.;2021 | Retrospective Study | Primary Biliary Cholangitis | The prevalence of vertebral fractures in patients with primary biliary cholangitis and osteosarcopenia is as high as 55.6%, significantly higher compared to other groups. | (19) |
| Zhenjie Gu et al.;2020 | Meta-Analysis | Heart Failure | Osteoporosis is significantly associated with an increased risk of heart failure. | (22) |
| Ramin Heshmat et al.;2021 | Cross-Sectional Study | Cardiovascular Disease | Sarcopenia and its components are associated with electrocardiographic abnormalities in elderly Iranians. Sarcopenia may be identified as a specific risk factor for future cardiovascular events. | (23) |
| Yuta Nakano et al.;2024 | Prospective Cohort Study | Chronic Kidney Disease | Osteosarcopenia is associated with poor survival rates and declined renal function in elderly CKD patients. | (26) |
| Thomas J Wilkinson et al.;2021 | Cross-Sectional Study | Chronic Kidney Disease | The presence of sarcopenia increases the risk of death and end-stage renal disease (ESRD) in patients with chronic kidney disease. | (27) |
| Kai Ma et al.;2022 | Review. | chronic obstructive pulmonary disease | ostosarcopenia is widely observed as a common feature of several chronic diseases, including aging and chronic obstructive pulmonary disease (COPD). | (30) |
Research on the impact of osteosarcopenia on chronic diseases.
Figure 1

Interaction between osteosarcopenia and chronic diseases. Sarcopenia is a significant risk factor for increased prevalence, hospitalization rates, mortality, fractures, and falls, particularly in elderly populations. Chronic diseases serve as key modulators in the intricate interplay between osteoporosis and sarcopenia. These conditions accelerate the progression of Osteosarcopenia by promoting systemic inflammation, hormonal imbalances, and metabolic dysfunction, which in turn exacerbate the detrimental effects of Osteosarcopenia on overall health. The mutual interaction between Osteosarcopenia and chronic diseases worsens both skeletal and muscular integrity, creating a cycle of deterioration that significantly impacts patient prognosis.
4 The pathogenesis of osteosarcopenia associated with chronic disease
4.1 NF-κB signaling pathway
The NF-κB signaling pathway plays a pivotal role in the development and metabolic processes of both bone and muscle cells, making it a key pathway in the pathogenesis of OS. Chronic systemic inflammation (SCI) is a central factor in the progression of this condition (31). As a well-established inflammatory signaling pathway, the activation of NF-κB leads to the production of pro-inflammatory cytokines that promote muscle degradation and bone resorption. Consequently, the progression of OS is not only driven by the NF-κB pathway itself, but is also significantly aggravated by chronic inflammatory diseases.
4.1.1 RANK/RANKL-OPG—NF-κB
The receptor activator of NF-κB (RANK)-RANK ligand (RANKL)-osteoprotegerin (OPG) system is central to the regulation of bone remodeling (32). RANK, RANKL, and OPG are all components of the tumor necrosis factor (TNF) receptor superfamily. RANKL, a soluble ligand primarily produced by osteoblasts and T cells, inhibits osteogenesis while promoting osteoclast differentiation. In this system, RANKL binds to its receptor RANK, triggering downstream signaling molecules, such as colony-stimulating factor 1 receptor (CSF-1) and NF-κB receptors, which initiate osteoclast differentiation (32). OPG, another receptor for RANKL, inhibits osteoclastogenesis and bone loss by binding to RANKL, thus blocking the RANK-RANKL signaling cascade (33).
Additionally, RANKL is expressed in skeletal muscles, where its activation suppresses myogenic differentiation and induces muscle atrophy through the regulation of the NF-κB signaling pathway (34). Hamoudi et al. found that anti-RANKL treatment could shift macrophages to the M2 phenotype and inhibit NF-κB activation in Duchenne muscular dystrophy, thereby protecting muscles from chronic inflammation and improving their mechanical properties (35). They also observed that OPG knockout mice exhibited selective atrophy of fast-twitch type IIb muscle fibers, and C2C12 myotubes exposed to RANKL showed a reduction in cross-sectional area (34). These findings suggest that the OPG/RANKL/RANK-NF-κB axis plays a pivotal role in the crosstalk between bone and muscle, further linking bone loss and muscle atrophy in OS (36).
Systemic chronic inflammation leads to elevated TNFα levels, which directly promote osteoclast formation in the early stages by stimulating the expression of the colony-stimulating factor-1 receptor gene. This process increases osteoclast precursors and enhances osteoclastogenesis (37) (38) (39). Additionally, IL-6 and IL-1 can directly enhance osteoclast activity or indirectly increase RANKL production in osteocytes, further facilitating osteoclast activation and supporting bone resorption mediated by osteoblasts (40) (41) (42) (4).Recent studies have identified a novel mechanism linked to mutations in the GPNMB gene in COPD, which is associated with osteoporosis. The GPNMB gene encodes a type I transmembrane protein that acts as a positive regulator of osteoblast function. Research has shown that mutations in the GPNMB gene disrupt bone homeostasis by mediating abnormal RANKL levels. Furthermore, these mutations exacerbate pulmonary inflammation in COPD patients by modulating MYC expression, adding another layer of complexity to the relationship between bone metabolism, chronic inflammation, and pulmonary disease (43). In summary, the RANK/RANKL-OPG-NF-κB pathway plays a critical role in both bone loss and muscle atrophy. Osteoblasts, as a major source of RANKL, are central to this pathway, underscoring their significant role in the complex “bone-muscle crosstalk” that contributes to the development of OS. This highlights the potential of osteoblast function as an important area of research, especially in the context of chronic diseases.
4.1.2 NF-κB—UPS
The ubiquitin-proteasome system (UPS) plays a central role in regulating protein degradation, selectively eliminating damaged or misfolded proteins, and maintaining cellular homeostasis (44). In bone remodeling, the UPS significantly impacts osteoblast function, with several E3 ubiquitin ligases involved in negatively regulating bone metabolism (45). For example, WWP1, a key negative regulator of osteoblast activity, promotes the degradation of the osteogenic gene Runx2, inhibiting the differentiation of osteoprogenitors into mature osteoblasts (46). This process is tightly regulated by the TNF-α-mediated NF-κB signaling pathway (46). However, NF-κB may not be the only pathway contributing to the suppression of osteogenic potential in bone marrow stromal cells (BMSCs).
At the same time, the UPS is also the primary for myofibril degradation. The UPS operates by attaching ubiquitin to substrate proteins, marking them for degradation by the 26S proteasome. E3 ubiquitin ligases, such as Atrogin-1 and MuRF-1, serve as rate-limiting factors in this pathway, and their upregulation is associated with muscle wasting in various chronic conditions (47). Chronic inflammatory factors, such as TNF-α, contribute to muscle atrophy by activating the NF-κB signaling pathway, which leads to the translocation of NF-κB from the cytoplasm to the nucleus (30) (48).This activation enhances the expression of E3 ligases like Atrogin-1 and MuRF-1, thereby increasing the degradation of myofibrillar proteins in skeletal muscle.
In summary, inflammatory factors such as TNF-α, IL-6, and IL-1 play a crucial role in the NF-κB-mediated development of OS (49). Since NF-κB is central to linking chronic diseases with OS, targeting this pathway could provide promising strategies for rethinking and treating the condition.
4.2 GH-IGF-1 axis
Insulin-like growth factor 1 (IGF-1) is synthesized by nearly all tissues and plays a pivotal role in mediating cell growth, differentiation, and transformation (50). Its secretion is primarily regulated by the growth hormone (GH) axis, with the liver serving as the primary source of circulating IGF-1 production and secretion. IGF-1 primarily by enhancing protein synthesis via the PI3K/Akt/mTOR signaling pathway (51). While current studies indicate that IGF-1 exhibits genetic polymorphisms, both systemic and locally produced skeletal IGF-1 are crucial for regulating bone mass formation and maintenance by promoting bone matrix synthesis and inhibiting bone resorption (52–54). Moreover, IGF-1 is increasingly recognized as an anabolic hormone that governs skeletal modeling and remodeling throughout the lifespan.
IGF-1 is involved in several anabolic pathways in skeletal muscle (55). In an animal study, localized expression of the IGF-1Ea or IGF-1Eb isoforms within muscle tissue was shown to counteract sarcopenia without causing significant side effects in other tissues or organs, nor did it affect the animals’ lifespan (56). This suggests that local expression of IGF-1 may play a role in preserving the youthful state of muscle tissue. However, it is important to recognize that OS frequently coexists with aging. The decline in IGF-1 levels associated with both aging and chronic diseases is a key factor contributing to the pathogenesis of OS.
In aging or end-stage liver disease, hepatic dysfunction, along with a reduction in growth hormone (GH) receptors, leads to decreased serum levels of insulin-like growth factor 1 (IGF-1) (57). Concurrently, impaired synthesis of branched-chain amino acids (BCAAs) disrupts BCAA-activated mTOR signaling, consequently interfering with glycogen and protein synthesis (58). mTOR, an atypical protein kinase, plays a central role in regulating growth and metabolism in response to nutrients, growth factors (such as IGF), and cellular energy status (59). Decreased BCAA levels further exacerbate the suppression of IGF-1 secretion. Moreover, prior studies have demonstrated that compensatory increases in angiotensin II in patients with chronic heart failure (CHF) contribute to sarcopenia and suppress osteoblast function by disrupting IGF-1 signaling (60). Zhang et al. identified a novel mechanism in a CKD mouse model, in which impaired IGF-1 receptor signaling compromised satellite cell function during muscle regeneration, leading to muscle fibrosis (61). Furthermore, glucocorticoids (GCs), frequently used in the long-term management of various chronic conditions, influence the regulation of the GH-IGF-1 axis. On one hand, GCs induce osteoblast apoptosis by inhibiting Wnt signaling and negatively regulating IGF-1 signaling, thereby hindering osteoblast precursor differentiation and maturation (62). On the other hand, GCs enhance osteoclast activity by modulating the RANKL/RANK/OPG pathway, which promotes bone resorption (63). With regard to muscle tissue, GCs not only inhibit IGF-1 production but also increase myostatin expression, thereby attenuating muscle protein synthesis (30). In conclusion, the GH-IGF-1 axis plays a predominantly anabolic role in the regulation of both bone and muscle health. However, factors that impair IGF-1 synthesis are significant contributors to the pathogenesis of OS. Consequently, IGF-1 replacement therapy may offer a promising therapeutic strategy for managing chronic diseases associated with OS.
4.3 Vitamin D
Vitamin D is widely recognized as a critical regulator of bone formation and has long been a central focus in the prevention and treatment of osteoporosis. Factors such as poor nutrition and insufficient sunlight exposure are commonly associated with impaired vitamin D synthesis and metabolism, often leading to vitamin D deficiency (64). This deficiency directly affects bone mineralization and matrix formation, resulting in abnormal bone growth and development. A recent study by Michael Booth and colleagues identified a high prevalence of vitamin D deficiency among young orthopedic trauma patients, suggesting an inverse relationship between vitamin D levels above 30 ng/mL and BMD (65).
Furthermore, recent studies have confirmed that decreased vitamin D levels are associated with an increased risk of sarcopenia. A study by Mori K et al. demonstrated that calcitriol could alleviate skeletal muscle changes induced by CKD (66, 67) (68). First, Vitamin D plays a key role in muscle health by promoting myoblast self-renewal and maintaining the satellite stem cell pool, through the regulation of the Forkhead box O (FOXO)3 and Notch signaling pathways (69). Second, the active form of vitamin D, 1,25(OH)2D, binds to the vitamin D receptor (VDR) to exert its biological effects and promote myogenic differentiation. Finally, 1,25(OH)2D enhances the expression of MyoD, which, in turn, inhibits myostatin production in a time-dependent manner (70). The active form of vitamin D, 1,25(OH)2D, is primarily synthesized in the kidneys via the mitochondrial enzyme 1α-hydroxylase, which is encoded by the Cyp27b1 gene. Vitamin D, widely recognized as a key regulator of bone metabolism, has long been a focal point in clinical interventions. However, its role in muscle tissue regulation has only recently garnered increased attention. Vitamin D replacement therapy has become well-established and may represent a promising therapeutic strategy for the treatment of OS.
4.4 Sex hormones
Disruption of sex hormone metabolism is considered one of the primary contributors to bone loss (71). Testosterone directly promotes osteoblast differentiation and indirectly facilitates it through its effects on various cytokines (72). Estrogen also exerts osteogenic effects, although to a lesser extent than testosterone. Numerous studies have demonstrated that the incidence of bone loss is significantly higher in elderly women compared to men. Postmenopausal women are particularly vulnerable to bone loss due to a decrease in the secretion of estrogen and progesterone from the ovaries. This results in estrogen deficiency, which accelerates bone turnover (73, 74). The increased activity of the basic multicellular unit (BMU) during bone remodeling leads to enhanced osteoblast apoptosis, shortened bone formation time, and reduced osteoclast apoptosis (75).
Additionally, sex hormones play a critical role in the metabolic processes of muscle tissue. In the cytoplasm, testosterone binds to androgen receptors and promotes protein synthesis through the mitogen-activated protein kinase (MAPK) pathway, thereby increasing muscle protein synthesis and muscle mass (76). Estradiol, on the other hand, facilitates muscle tissue repair through specific estrogen receptors (77). In chronic conditions such as cirrhosis, patients often exhibit reduced testosterone levels and an imbalance in the estrogen-to-androgen ratio. This imbalance leads to decreased osteoblast and osteocyte formation (78), reduced muscle protein turnover, and inhibition of myoblast differentiation into skeletal muscle cells (59), ultimately contributing to an imbalance in bone remodeling and a decline in muscle mass. The development of OS is closely associated with sex hormone deficiency, which is influenced by both aging and chronic diseases (60). It is important to note that while hormone replacement therapy is often associated with adverse effects, targeted therapies acting on specific receptors may offer a more promising therapeutic approach.
Current research on the pathogenesis of OS remains a subject of ongoing debate. Traditionally, osteoporosis and sarcopenia have been considered separate entities. However, recent studies have highlighted the significant interactions between bone and muscle tissues. A comprehensive understanding of the pathogenesis of both osteoporosis and sarcopenia is essential for advancing clinical knowledge. Furthermore, exploring the potential crosstalk between bone and muscle could offer valuable insights and novel therapeutic strategies for the prevention and treatment of OS.
4.5 Endocrine role of osteocytes and myocytes
4.5.1 FGF-23
Fibroblast growth factor 23 (FGF-23) was the first bone-derived factor to be identified (79), primarily synthesized by osteoblasts. Elevated levels of FGF-23 were later discovered in the circulation of patients with rickets, leading to the recognition of its negative regulatory role in bone metabolism (80). The osteomalacia induced by FGF-23 is closely linked to the bone-kidney axis. FGF-23 primarily targets the proximal renal tubules, where it reduces the expression of co-transporters involved in phosphate absorption and reabsorption, thereby impairing bone deposition (81). Additionally, FGF-23 regulates vitamin D metabolism by inhibiting 1-α-hydroxylase, the enzyme responsible for converting 25-hydroxyvitamin D (25(OH)D) to its active form 1,25(OH)2D). This inhibition reduces the synthesis of 1,25(OH)2D, ultimately affecting bone mineralization (82). The Klotho protein acts as a downstream activator of the FGF-23 receptor pathway. Reduced expression of Klotho leads to elevated levels of FGF-23, which increases phosphate excretion in the proximal renal tubules (83). In CKD, Klotho levels gradually decline as the glomerular filtration rate decreases, often serving as one of the earliest indicators of disturbances in bone metabolism (84).
Moreover, Kido et al. found that FGF-23 is associated with muscle atrophy in CKD (85). The effects of FGF-23 on muscle may occur independently of s-Klotho and could directly interact with FGF receptors in skeletal muscle to exert inhibitory effects (86). Additionally, Chisato Sato et al. conducted an in vitro study to investigate the effects of FGF-23 on isolated human BMSCs. Their findings revealed that FGF-23 promotes the p53/p21/oxidative stress pathway, inducing premature senescence of human BMSCs in a Klotho-independent manner (87). In muscle tissue, BMSCs, which serve as the primary source of satellite cell differentiation, play a crucial role in maintaining skeletal muscle mass and repairing muscle fibers. In conclusion, osteocytes play a pivotal role in bone and muscle homeostasis. Unlike other members of the fibroblast growth factor (FGF) family, FGF-23 is almost exclusively produced in the skeletal system (79). BMSCs, which serve as precursors to osteoblasts, osteoclasts, and satellite cells, hold significant potential for repairing both bone and muscle damage. Further investigation into the effects of bone-derived, hormone-like factors such as FGF-23 on BMSCs may provide valuable insights and therapeutic strategies for addressing OS (88).
4.5.2 Sclerostin
Sclerostin (28 kDa) is a small secretory glycoprotein synthesized by osteocytes and encoded by the SOST gene. It belongs to the Dan/Cerberus family of bone morphogenetic protein (BMP) antagonists (33). In a study by Graciolli FG et al. on CKD, serum sclerostin levels were found to increase as the disease progressed, leading to the inhibition of osteoblastogenesis by suppressing the WNT/β-catenin signaling pathway. Sclerostin also promotes bone resorption by inducing the synthesis of RANK-L, thereby enhancing osteoclastogenesis (89). Additionally, sclerostin interacts with BMP to inhibit BMP-induced Smad phosphorylation and disrupts the canonical Wnt pathway by competitively binding to the co-receptor of the Wnt pathway, LDL receptor-related protein 5/6 (LRP5/6) (90). Furthermore, studies have shown that hypoxia/HIF-1α, in collaboration with Osterix, inhibits the Wnt pathway, thereby suppressing osteoblast proliferation. Activation of HIF-1α induces sclerostin (Sost gene) expression, representing a novel mechanism by which HIF-1α impedes osteoblast Wnt signaling (91).
Recent studies have also emphasized the critical role of sclerostin in regulating muscle mass. Research by von Maltzahn et al. demonstrated that sclerostin inhibits muscle stem cell differentiation by suppressing the Wnt signaling pathway (92). Additionally, a study by Soohyun P. Kim et al. found that SOST gene knockout mice exhibited a significant increase in lean body mass compared to control groups (93). Moreover, it was observed that sclerostin suppresses the crosstalk between MLO-Y4 osteocytes and muscle cells (C2C12) mediated by WNT3a through the regulation of the Wnt/β-catenin pathway (94). These findings suggest that the effects of sclerostin on both bone and muscle are not merely independent signaling pathways. More importantly, osteocytes, as the primary source of sclerostin, may mediate the interactive signaling mechanisms between bone and muscle cells.
4.5.3 Myostatin
Myostatin (Mstn), the first identified myokine, is primarily secreted by muscle fibers and functions as a negative regulator of skeletal homeostasis. In vitro studies have demonstrated that Mstn inhibits the expression of critical osteogenic transcription factors, such as Osterix and Runx2, thereby suppressing osteoblast differentiation (33). In vivo, Chen et al. observed that recombinant Mstn reduced the number of osteoblasts present on bone surfaces (95). Additionally, Qin et al. showed that Mstn upregulates the expression of Wnt pathway inhibitors, including SOST and Dickkopf Wnt signaling pathway inhibitor 1 (DKK1), which impedes osteocyte differentiation (96). Mstn also enhances the expression of RANKL, a pivotal gene involved in osteoclastogenesis, within osteocytes. However, in contrast to its effects on osteocytes, Mstn does not directly affect osteoclast activity. Instead, it promotes the expression of genes that mediate RANKL-induced osteoclastogenesis by facilitating SMAD2-dependent nuclear translocation of NFATc1, thus stimulating osteoclast proliferation independently of other signaling pathways (96).
As the name suggests, Mstn is inversely correlated with muscle mass. The regulation of muscle development by Mstn is complex, and although the precise mechanisms remain incompletely understood, it is generally accepted that Mstn mediates its effects through two primary signaling pathways: the MAPK pathway and the phosphoinositide 3-kinase (PI3K) pathway. Mstn downregulates the MEK/ERK1/2 MAPK pathway and/or the AKT/mTORC1 signaling cascade, leading to decreased expression of muscle-specific genes, such as Pax3, Myod1 (myogenic differentiation 1), and Myf5, thereby inhibiting myocyte differentiation (97). In addition, Mstn suppresses the persistent activation of eukaryotic translation initiation factor 4E (eIF4E) and eukaryotic translation initiation factor 4E-binding protein 1 (4E-BP1) within the PI3K/Akt/mTOR pathway, disrupting the balance between muscle protein synthesis and degradation, ultimately resulting in a reduction in myocyte cytoplasmic volume. Furthermore, Mstn promotes the expression of genes involved in the UPS, such as Murf-1, which accelerates muscle protein degradation (98).
Moreover, Mstn may act as a potential molecular mediator of bone-muscle loss in chronic diseases. For instance, uremic toxins, such as indoxyl sulfate, have been shown to elevate Mstn levels in CKD patients (99). Increased tumor necrosis factor-alpha (TNF-α) levels in chronic inflammatory diseases can also mediate the upregulation of Mstn via the NF-κB pathway, suppressing MyoD-induced Mstn expression (100). Additionally, IL-6 has been found to directly enhance Mstn expression in muscle fibers (30). It is noteworthy that Mstn also inhibits the differentiation of BMSCs into osteoblasts. Hamrick et al. demonstrated that Mstn may alter the mechanical responsiveness of BMSCs by suppressing the expression of osteogenic factors during mechanical loading (101). Additionally, Rebbapragada et al. showed that Mstn antagonizes bone morphogenetic protein (BMP) 7-induced adipogenesis, thereby inhibiting BMSC differentiation into adipocytes (102). BMPs play a pivotal role in regulating muscle growth and spatial patterning during embryonic development. The imbalance in BMSC differentiation is a key factor in the pathogenesis of bone density disorders, and genomic studies exploring adipogenic differentiation of BMSCs remain an active area of investigation. Recent research has identified DAAM2, TIMP2, and TMEM241 as potential therapeutic targets for bone degeneration and osteoporosis-related conditions (103). Furthermore, BMSCs, as precursors to satellite cells, are essential for muscle repair following injury. Although it remains uncertain whether the adipogenic imbalance of BMSCs directly influences satellite cell differentiation, this imbalance could represent a potential mechanism by which mesenchymal stem cells (MSCs) contribute to the treatment of bone-muscle loss syndromes.
4.5.4 Irisin
Irisin is another myokine, but its effects differ significantly from those of Mstn. As a positive regulator of both the skeletal and muscular systems, irisin plays a crucial role in maintaining metabolic homeostasis (33). It can promote osteoblast proliferation and their differentiation into osteocytes via the MAPK signaling pathway (104). Furthermore, Storlino et al. demonstrated that intermittent administration of irisin downregulated the expression of sost and inhibited osteocyte apoptosis under conditions of oxidative stress and/or microgravity (105). Additionally, irisin stimulates the proliferation of osteoclast precursors and suppresses their differentiation through the p38, c-Jun N-terminal kinase (JNK) signaling pathways, as well as the RANKL-induced NF-κB pathway (106).
Irisin has also been shown to promote myogenesis through autocrine mechanisms. Specifically, it increases the mRNA levels of muscle growth-related genes, such as insulin-like growth factor 1 (IGF-1) and peroxisome proliferator-activated receptor gamma coactivator 1-alpha (Pgc1α4), primarily via the ERK signaling pathway. Moreover, irisin inhibits the expression of Mstn, thereby synergistically enhancing muscle protein synthesis (107).
In the endocrine interaction pathways between osteocytes and muscle cells, it is evident that several shared pathways contribute to conditions resembling OS. Osteocytes play a pivotal role in the ‘bone-muscle crosstalk.’ Regardless of their origin—whether from bone or muscle—most factors activate downstream signaling pathways through osteocyte-mediated secretion or regulation, such as SOST and RANKL, which, in turn, influence the metabolic processes of both bone and muscle. A comprehensive understanding of the combined actions of these pathways could improve the management of conditions like OS. In conclusion, the development of OS is not solely driven by the mechanisms underlying osteopenia/osteoporosis or sarcopenia, but is more significantly influenced by the pathological and physiological interactions that perpetuate a vicious cycle between these two conditions (Figure 2).
Figure 2

Interaction between the skeletal and muscular systems. The skeletal and muscular systems both derive from a shared mesenchymal stem cell lineage, with their proliferation and differentiation regulated by overlapping signaling pathways and cytokines. These systems function as “endocrine” organs, secreting factors that influence one another and contribute to the overall homeostasis of the musculoskeletal system. Positive interactions between bone and muscle are primarily mediated by pathways such as WNT, MAPK/ERK, PI3K/AKT, and the vitamin D receptor. In contrast, the NF-κB pathway serves as a negative regulator. Exogenous cytokines, including growth hormone/IGF-1, vitamin D, estrogen, and testosterone, promote the proliferation and differentiation of both bone and muscle cells, enhancing anabolic processes in these tissues. Bone-derived factors such as sclerostin and FGF-23, and muscle-derived factors like myostatin and irisin, regulate muscle catabolism, protein synthesis, and bone remodeling. Specifically, irisin promotes the proliferation of both bone and muscle cells, while sclerostin and myostatin inhibit cell proliferation. FGF-23 inhibits bone remodeling but positively influences muscle cell proliferation.
Indeed, OS does not solely occur in healthy adults but is a complex condition influenced by various factors, including inflammation, malnutrition, prolonged polypharmacy, frailty, functional impairment, and aging. While research on the mechanisms linking chronic diseases to OS remains limited, the interplay of common risk factors for both conditions provides indirect insights into the impact of chronic diseases on the development of OS (Table 2). In the long-term management of chronic diseases, MSK plays a crucial role, as the pathological mechanisms associated with these conditions contribute to the onset of OS. Therefore, OS should not be overlooked in patients with chronic diseases. It is a syndrome that coexists with and interacts with chronic diseases (Figure 3).
Table 2
| Study objective | Author/tear | Study type | Disease & potential mechanisms | Results | Cite |
|---|---|---|---|---|---|
| The Relationship Between Osteosarcopenia in Asymptomatic Individuals and Coronary Artery Calcification. | Chul-Hyun Park et al.;2022 | Cross-Sectional Study | Coronary Artery Atherosclerosis & GH/IGF-1,RANK/RANKL‐OPG—NF-kB,NF-kB—UPS | The authors propose that the GH/IGF-1 axis plays a central role in the development of osteosarcopenia in coronary artery calcification. Another key mechanism is “inflammation,” where pro-inflammatory cytokines are highly associated with the development of osteosarcopenia through the ubiquitin-proteasome pathway, leading to accelerated bone resorption. | (38) |
| Factors Influencing Sarcopenia and Osteosarcopenia in Chronic Liver Disease and Their Molecular Pathogenesis. | Chisato Saeki et al.;2021 | Review | Chronic Liver Disease & NF-kB,IGF-1,Vitamin D,Sclerostin,FGF-23,Myostatin and Irisin | The authors believe that pro-inflammatory cytokines (such as IL-1β, IL-6, and TNF-α), IGF-1, vitamin D, sclerostin, FGF-23, myostatin, and irisinin play crucial roles in the pathogenesis of osteosarcopenia in chronic liver disease. | (57) |
| Sarcopenia as a Comorbidity of Cardiovascular Disease. | Ken-ichiro Sasaki et al.;2022 | Review | Heart Failure & RANK/RANKL/OPG—NF-kB,NF-kB—UPS,IGF-1 and BCAA | The authors suggest that inflammatory biomarkers in heart failure (HF), such as IL-6, TNF-α, and C-reactive protein, have a direct impact and influence the onset of osteosarcopenia by reducing IGF-1 levels and branched-chain amino acids. Oxidative stress activates the UPS system, promoting protein degradation. Additionally, low testosterone levels induced by HF also play a role. | (60) |
| An Overview of the Molecular Mechanisms Leading to Musculoskeletal Disorders in Chronic Liver Disease. | Young Joo Yang et al.;2021 | Review | Chronic Liver Disease & RANK/RANKL‐OPG—NF-kB,IGF-1,BCAA and Testosterone | The authors highlight the role of the inflammation-mediated RANK/RANKL-OPG-NF-kB pathway in chronic liver disease, as well as the impact of hepatocyte damage leading to decreased levels of IGF-1, BCAAs, and testosterone in the development of osteosarcopenia. | (58) |
| Osteosarcopenia and Long-COVID: a dangerous combination | Umberto Tarantino et al.;2022 | Review | COVID & RANK/RANKL‐OPG—NF-kB and NF-kB—UPS | The authors describe how chronic inflammation in long COVID mediates the RANK/RANKL-OPG-NF-kB pathway and the NF-kB-UPS system, influencing the development of osteosarcopenia. | (49) |
| Osteosarcopenia in Patients with Chronic Obstructive Pulmonary Disease (COPD). | Lorenzo Lippi et al.;2022 | Review | COPD & RANKL—NF-kB,NF-kB—UPS and IGF-1 | The authors state that long-term inflammation in COPD mediates the effects of NF-κB, RANKL (Receptor Activator of Nuclear Factor-kappa B Ligand), and M-CSF (Macrophage Colony-Stimulating Factor), while hypoxic damage activates the UPS system. Additionally, long-term hormone therapy leading to decreased IGF-1 levels plays a key role in the development of osteosarcopenia. | (39) |
| Osteosarcopenia in Chronic Pancreatitis. | I V Kozlova et al.;2021 | Case-control study | Chronic Pancreatitis & RANK/RANKL‐OPG—NF-kB and NF-kB—UPS | The correlation between lumbar spine T-score and IL-6 (r = -0.29; p = 0.03) and IL-8 (r = -0.29; p = 0.04) was revealed. A correlation was also established between osteosarcopenia and cytokine concentrations in the colonic mucosa in chronic pancreatitis (IL-2: r = 0.44; p < 0.001; IL-6: r = 0.48; p < 0.001; IL-8: r = 0.42; p < 0.001). | (42) |
| Aromatase Inhibitor-Induced Osteoporosis and Osteopenia in Elderly Breast Cancer Patients. | Andrea Casabella et al.;2024 | Case-control study | Breast Cancer & Estrogen and RANKL—NF-kB | The authors reveal that decreased estrogen secretion in elderly breast cancer patients leads to increased secretion of RANKL by osteoblasts, which in turn enhances osteoclast activity, resulting in accelerated bone remodeling. | (74) |
| Osteosarcopenia in Peritoneal Dialysis Patients. | Andrew Davenport et al.;2022 | Retrospective Study | Chronic Kidney Disease & Vitamin D | The authors suggest that chronic kidney disease, leading to abnormalities in calcium-phosphate metabolism and vitamin D synthesis, may be a contributing factor to the development of osteosarcopenia. | (67) |
| Recent Advances in Vitamin D and Osteosarcopenia. | Olivier Bruyère et al.;2017 | Review | Chronic Kidney Disease/Chronic Liver Disease & Vitamin D | The authors suggest that vitamin D metabolism is coordinated by the skin, liver, and kidneys. Chronic liver and kidney diseases may lead to vitamin D deficiency. Vitamin D deficiency enhances bone marrow adipogenesis and intramuscular fat deposition, thereby impairing skeletal function. In muscle tissue, vitamin D regulates calcium flux, mineral homeostasis, and signaling pathways that control protein synthesis and metabolism. | (68) |
| The Vicious Cycle of Osteosarcopenia in Inflammatory Bowel Disease. | Dorota Skrzypczak et al.;2021 | Review | Inflammatory Bowel Disease & RANKL/NF-kB and Hormone(GC) | The authors state that long-term inflammation in patients with inflammatory bowel disease mediates the RANKL/NF-kB pathway and the use of hormones, which may contribute to the development of osteosarcopenia. They also emphasize the endocrine interaction between osteocytes and myocytes. | (4) |
The potential molecular mechanisms by which chronic diseases affect osteosarcopenia.
Figure 3

Pathophysiological processes of chronic diseases and their direct effects on shared molecular mechanisms in bone-muscle interactions. This figure illustrates the shared molecular mechanisms underlying bone-muscle interactions, with “→” indicating a positive effect and “→”indicating a negative effect. The green dashed line highlights the regulation of bone resorption and muscle catabolism, while other pathways emphasize the regulation of bone formation and muscle anabolic processes. The pathophysiological processes associated with chronic diseases and sarcopenia are depicted above, with “→” indicating a promoting effect and “→” indicating an inhibitory effect. Chronic diseases converge on these pathways through elevated inflammatory factors (e.g., TNF-α, IL-6, IL-1) and reduced levels of insulin-like growth factor 1 (IGF-1) and active vitamin D, negatively impacting the musculoskeletal system by promoting bone resorption and muscle catabolism. Specifically, inflammatory cytokines increase bone resorption and muscle breakdown, while decreased IGF-1 and vitamin D levels hinder bone formation and muscle anabolism, resulting in an imbalance in bone and muscle mass. Furthermore, pathological interactions vary across chronic diseases: in chronic obstructive pulmonary disease (COPD), mutations in the GPNMB gene and upregulation of HIF-1α expression; in inflammatory bowel disease (IBD) and COPD, glucocorticoid (GC) use; and in chronic kidney disease (CKD) and liver cirrhosis (LC), the accumulation of toxic substances (such as hyperammonemia and indoxyl sulfate) further promote bone resorption and muscle catabolism. Reduced branched-chain amino acids (BCAAs) in LC patients and decreased synthesis of active vitamin D (1,25(OH)2D3) in CKD patients inhibit anabolic processes, exacerbating musculoskeletal deterioration.
5 Management
5.1 Non-pharmacological management
Exercise is widely regarded as one of the most cost-effective and beneficial strategies for managing OS, offering significant advantages for patients with chronic diseases. Research indicates that physical activity can effectively prevent the onset of OS and alleviate its associated negative outcomes. For OS linked to chronic conditions such as cardiovascular diseases, diabetes, cancer, and respiratory disorders, exercise is commonly recommended as a therapeutic intervention. A 2023 statement from the American Heart Association underscores that resistance training not only enhances the quality and strength of the MSK but also provides essential physiological and clinical benefits for individuals with cardiovascular diseases and related risk factors (108). In clinical practice, all patients with chronic diseases diagnosed with OS should be prescribed resistance training (RT). RT may include various forms of exercise, such as free weights (e.g., dumbbells), bodyweight exercises (e.g., push-ups, squats), machine weights, or resistance bands. Each session should consist of 1 to 3 sets at a moderate intensity, with 8 to 12 repetitions per set performed to the point of fatigue, at least twice a week (108). Furthermore, RT should be sustained as a preventive measure for patients with chronic diseases.
Additionally, studies have emphasized the critical role of proper nutritional support in managing OS. Patients are advised to maintain a diverse and balanced diet that includes essential trace elements, vitamins, proteins, and dairy products. Increasing the intake of foods rich in BCAAs, such as meat (e.g., beef, salmon, shrimp), legumes, and nuts (e.g., peanuts, walnuts, almonds), is particularly beneficial (109). Simultaneously, the consumption of soft drinks and alcohol should be minimized. Furthermore, it is recommended that patients with OS consistently consume 1000 to 1500 mg of calcium daily, along with 400–800 IU of vitamin D or 260 µg of 25-hydroxyvitamin D supplements (e.g., calcitriol) every two weeks as part of their daily nutritional regimen (110). This combination provides pharmacological support to complement basic nutritional intake.
5.2 Pharmacological management
5.2.1 Inflammation
In many chronic diseases, systemic inflammation persists throughout the disease course, playing a crucial role in long-term disease management (35). Celecoxib, a nonsteroidal anti-inflammatory drug (NSAID), has demonstrated effective anti-inflammatory properties in the MSK disorders and is commonly utilized in the clinical management of immune system diseases, osteoarthritis, cancer, cardiovascular diseases, and respiratory conditions. As a potential pharmacological approach for OS, celecoxib warrants further investigation. Additionally, the NF-κB pathway plays a pivotal role in the pathogenesis of OS. Furthermore, denosumab, a human monoclonal antibody that targets receptor activator of nuclear factor kappa-β ligand (RANKL), has been shown to effectively inhibit bone resorption and enhance bone mass. It is widely used in the treatment of osteoporosis (111). A study conducted over a three-year period, comparing women with OS, revealed that patients receiving denosumab treatment experienced significant improvements in both grip strength and lean body mass (112). These findings underscore the potential of denosumab as a promising therapeutic option for OS.
5.2.2 Hormone therapy
Currently, hormone replacement therapies, including selective androgen receptor modulators (SARMs), selective estrogen receptor modulators (SERMs), growth hormone, and insulin-like growth factor 1 (IGF-1), are under extensive investigation for the treatment of OS. Mechanistically, these therapies hold promising potential in promoting both muscle and bone health. However, the clinical benefits have not always been consistently observed (58). Moreover, the adverse effects associated with hormone replacement therapies remain a concern, limiting their widespread use. Nevertheless, targeted therapy for patients with chronic diseases may offer more favorable responses. For instance, in patients with chronic liver disease, treatment with SERMs and branched-chain amino acid supplements may prove effective. In individuals with chronic kidney disease, a regimen including calcium, phosphate supplements, and vitamin D could provide potential benefits. Further clinical validation is required to substantiate these approaches.
5.2.3 Endocrine targets in osteocytes and myocytes
Recent research on the management of OS has increasingly focused on the endocrine crosstalk between osteocytes and myocytes as a promising therapeutic target. Blosozumab, a human monoclonal antibody that targets sclerostin—an important regulator of bone metabolism—has shown considerable promise. A recent randomized, double-blind phase 2 clinical trial demonstrated that sclerostin inhibition with Blosozumab effectively treats osteoporosis in postmenopausal women, resulting in significant increases in bone mineral density (BMD) at the spine, femoral neck, and total hip (113). Additionally, ACE-031, a soluble form of activin receptor type IIB that functions as a Mstn inhibitor, can bind to and neutralize Mstn, thereby promoting muscle growth (114). A study demonstrated that ACE-031 was well tolerated, increased bone formation markers, and improved lean body mass in postmenopausal women (115). These findings suggest that both Blosozumab and ACE-031 may become viable treatment options for OS, particularly in patients with chronic diseases.
5.2.4 Mesenchymal stem cell therapy
Mesenchymal stem cells (MSCs) are undifferentiated progenitor cells with self-renewal capacity and the ability to differentiate into multiple cell types, including adipocytes, osteoblasts, myocytes, and satellite cells (116). Due to their regenerative properties, MSCs are increasingly utilized in clinical settings, particularly for the management of chronic and long-term diseases (116). MSCs hold significant potential for the treatment of OS, with demonstrated abilities to promote osteogenesis and myogenesis, as well as repair damaged bone and muscle tissues. First, MSC transplantation is feasible, and over 1,000 MSC transplantation clinical trials have been registered, with human umbilical cord-derived MSCs being applied in multiple fields (117). Additionally, preclinical studies consistently demonstrate that MSCs improve bone density in animal models of osteoporosis, providing strong evidence for their efficacy (118). Furthermore, MSCs have been shown to promote muscle regeneration. Recently, induced pluripotent stem cells (iPSCs) have emerged as a potential source of stem cells. When used as autologous transplants, iPSCs can differentiate into muscle tissue and eliminate the risk of immune rejection (119). Furthermore, genetic manipulation of MSCs presents a promising strategy to enhance their therapeutic potential. Identifying specific genes that promote MSC differentiation into osteoblasts and satellite cells could enable more precise and efficient treatment of bone and muscle damage associated with OS. Additionally, metabolomics evaluations may provide valuable insights for diagnosing OS, particularly in individuals with genetic predispositions (120). However, despite these advancements, the clinical application of MSCs in the treatment of OS remains a significant challenge.
Current research frequently addresses OS by treating osteoporosis and sarcopenia as distinct conditions. However, these approaches have demonstrated limited or no efficacy in slowing the progression of OS. Based on a comprehensive review of prior studies examining the “combinatory actions” between bone and muscle, we aim to identify promising strategies for the treatment of OS (Figure 4).
Figure 4

Prevention and treatment strategies for osteosarcopenia in patients with chronic diseases. This figure outlines key strategies for preventing and treating osteosarcopenia in patients with chronic diseases. Regular physical activity, including strength training and weight-bearing exercises, is essential for enhancing muscle mass and strength. Adequate nutrition, with sufficient daily intake of calcium, active vitamin D, dietary protein, and branched-chain amino acids (BCAAs), supports both muscle and bone health. Pharmacological treatments for osteosarcopenia include Denosumab (a monoclonal antibody targeting RANKL to prevent bone resorption), selective androgen receptor modulators (SARMs), selective estrogen receptor modulators (SERMs), growth hormone/IGF-1 therapy, myostatin inhibitors, and ACE-031 (an Activin receptor type IIB antagonist),Mesenchymal stem cell therapy.
6 Conclusion
This review underscores the significant impact of OS on chronic diseases. As discussed, OS exacerbates adverse outcomes in chronic disease. With the rising prevalence of chronic conditions requiring long-term medical management, the strain on healthcare systems continues to intensify (7). However, despite its profound impact, OS has not received adequate attention in the long-term management of these diseases, and much of the research remains underdeveloped. Moreover, the consequences of OS on chronic diseases extend beyond the scope of this review. The musculoskeletal system, essential for energy production and movement, plays a pivotal role in maintaining overall health (10). Traditionally, chronic disease management has focused on bone loss and osteoporosis, but it is only in recent years that a more nuanced understanding of OS has emerged (1). Despite these advancements, current research continues to treat bone and muscle systems separately. In reality, the integrity of the MSK must be considered holistically. Therefore, it is crucial to urge clinicians to reconsider the critical role of OS in managing chronic diseases.
Additionally, this review delves into the potential mechanisms driving OS in chronic diseases. Moving away from the traditional approach of addressing bone and muscle systems independently, it highlights the concept of “bone-muscle crosstalk” mediated by osteocytes. The onset of OS is not merely the disruption of skeletal or muscular systems in isolation but rather the interaction between skeletal and muscular dysfunctions that, through “bone-muscle crosstalk,” exacerbates both bone loss and muscle atrophy (33) (36). By examining these mechanisms, we have integrated current prevention and treatment strategies, which may pave the way for advancements in the long-term management of chronic diseases.
Additionally, future recommendations are provided. Current research suggests that chronic diseases mediate the onset of OS through specific pathophysiological pathways. However, the precise role of OS in influencing the pathogenesis of chronic diseases remains a significant area of debate and an important research gap. Despite this, existing studies still indicate that, on a macro level, sarcopenia contributes to the increased prevalence or progression of chronic diseases. Future research, however, faces a significant challenge in exploring the underlying molecular mechanisms at the microscopic level.
7 Methodology
This systematic review was conducted in strict accordance with the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) (121) guidelines.
A comprehensive literature search was performed in two electronic databases—PubMed and Google Scholar—covering studies published from January 2019 through January 2025. Notably, several of the included review articles incorporated data from high−quality studies conducted prior to 2010.The search strategy combined Medical Subject Headings (MeSH) terms with free−text keywords, including OS, osteoporosis, sarcopenia, bone mass loss, muscle mass loss, epidemiology, molecular mechanisms, management, chronic disease, and chronic inflammatory disease. Boolean operators (AND, OR) were applied to refine and expand the search results.
In addition, the reference lists of all retrieved articles were manually screened to identify any additional eligible studies. Two authors (YT−Y and HF−Z) independently screened titles and abstracts according to predefined inclusion criteria. Full texts were then reviewed to remove duplicates and exclude studies that did not meet the eligibility criteria. Any disagreements were resolved through discussion with a third reviewer (WZ−W). Eligible study designs included observational studies (cohort, case–control, and cross−sectional), systematic reviews, meta−analyses, and randomized double−blind controlled trials (RCTs). Animal studies and experimental model studies were excluded. Studies deemed irrelevant to the research focus of this review were also excluded. Additionally, case reports, editorials, letters to the editor, and conference proceedings were excluded. Ultimately, a total of 120 references were included in this review (Figure 5).
Figure 5
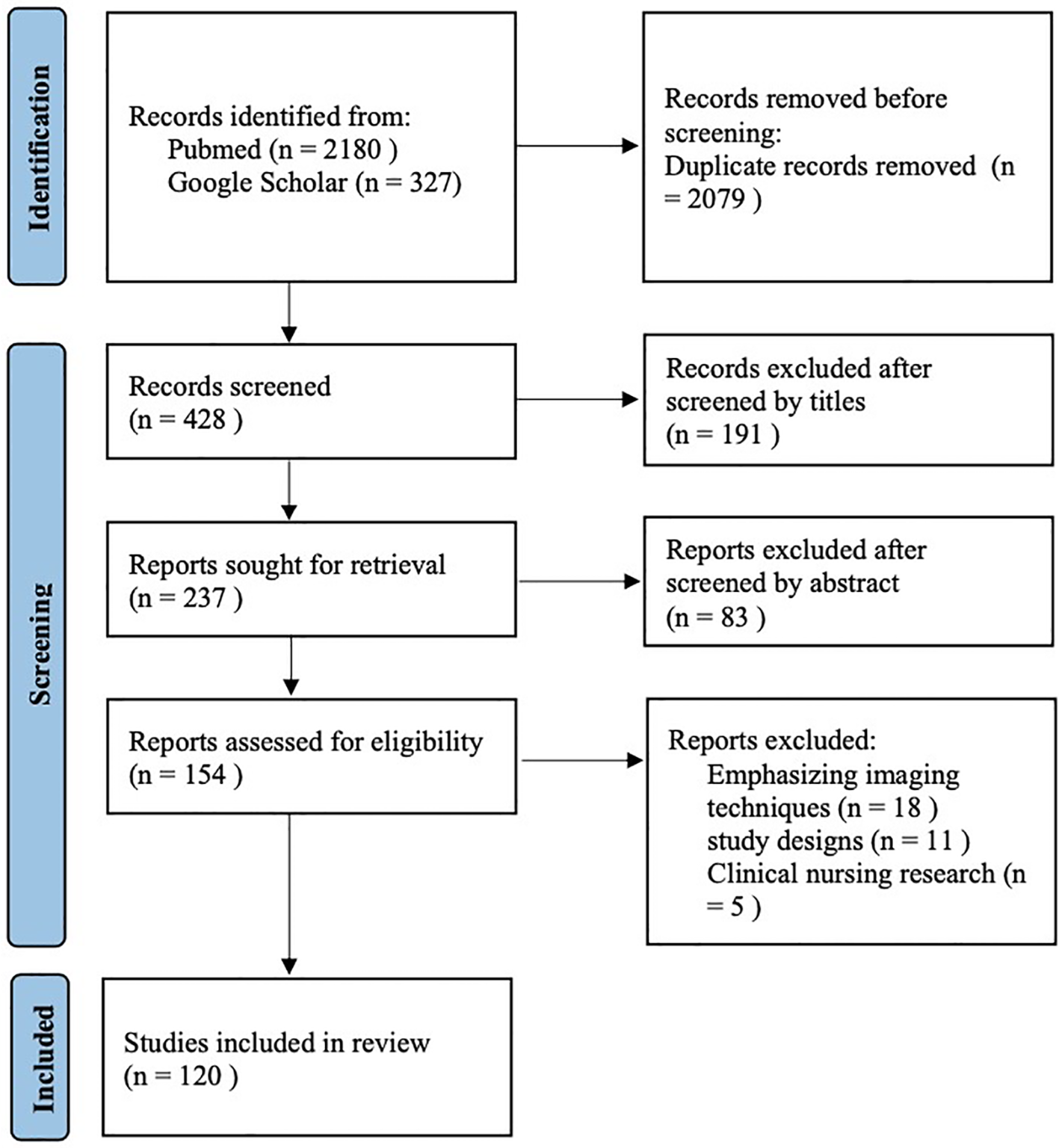
Flow diagram of the study selection process.
Statements
Author contributions
Y-TY: Data curation, Investigation, Methodology, Visualization, Writing – original draft, Writing – review & editing. H-FZ: Investigation, Writing – review & editing. W-ZW: Funding acquisition, Supervision, Validation, Writing – review & editing. XL: Funding acquisition, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This study was supported by The National Natural Science Foundation of China (82460428), Yunnan Provincial Department of Science and Technology- Kunming Medical University Joint Special Fund for Basic Research General Program (202501AY070001-166), the 76th Batch of the China Postdoctoral Science Foundation General Funding-Regional Special Support Program (2024MD763983), Yunnan Health Training Project of High Level talents (H-2024026), Youth Project of Yunnan Basic Research Programme of Yunnan Provincial Department of Science and Technology (202401AU070046), Teachers’ Project of Scientific Research Fund of Yunnan Provincial Department of Education - Special Project on Basic Research for Young Talents (2024J0180), The fifth batch of 535 young academic backbone training subjects of the First Affiliated Hospital of Kunming Medical University (2025535Q08), 2024 Yunnan Province Colorful Cloud Postdoctoral Program Innovation Project, Education and Teaching Research Project of the First Affiliated Hospital of Kunming Medical University (2024 JY-17), Golden Flower Enterprise Co., Ltd. Horizontal Project (Study on the Protective Effect of Nano-hydroxyapatite/Chitosan Scaffold Loaded with Artificial Tiger Bone Powder on Cartilage Injury in Osteoarthritis).
Acknowledgments
The authors of this manuscript sincerely thank Professor Bing Wang, Director of the Department of Orthopedics at the First Affiliated Hospital of Kunming Medical University, the faculty of the department, and Professor Xi Li for their guidance and assistance with this work.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1
Hirschfeld HP Kinsella R Duque G . Osteosarcopenia: where bone, muscle, and fat collide. Osteoporos Int. (2017) 28:2781–90. doi: 10.1007/s00198-017-4151-8
2
Sepúlveda-Loyola W Phu S Bani Hassan E Brennan-Olsen S L Zanker J Vogrin S et al . The joint occurrence of osteoporosis and sarcopenia (Osteosarcopenia): definitions and characteristics. J Am Med Dir Assoc. (2020) 21:220–5. doi: 10.1016/j.jamda.2019.09.005
3
Pugliese N Arcari I Aghemo A Lania A G Lleo A Mazziotti G . Osteosarcopenia in autoimmune cholestatic liver diseases: Causes, management, and challenges. World J Gastroenterol. (2022) 28:1430–43. doi: 10.3748/wjg.v28.i14.1430
4
Skrzypczak D Ratajczak AE Szymczak-Tomczak A Dobrowolska A Eder P Krela-Kaźmierczak I . A vicious cycle of osteosarcopeniain inflammatory bowel diseases-aetiology, clinical implications and therapeutic perspectives. Nutrients. (2021) 13:293–318. doi: 10.3390/nu13020293
5
Kirk B Zanker J Duque G . Osteosarcopenia: epidemiology, diagnosis, and treatment-facts and numbers. J Cachexia Sarcopenia Muscle. (2020) 11:609–18. doi: 10.1002/jcsm.12567
6
Airhihenbuwa CO Tseng TS Sutton VD Price L . Global perspectives on improving chronic disease prevention and management in diverse settings. Prev Chronic Dis. (2021) 18:E33. doi: 10.5888/pcd18.210055
7
Reardon S . A world of chronic disease. Science. (2011) 333:558–9. doi: 10.1126/science.333.6042.558
8
Sallis RE . Exercise in the treatment of chronic disease: an underfilled prescription. Curr Sports Med Rep. (2017) 16:225–6. doi: 10.1249/JSR.0000000000000378
9
Mi Y Xue Z Qu S Yin Y Huang J Kou R et al . The economic burden of coronary heart disease in mainland China. Public Health. (2023) 224:140–51. doi: 10.1016/j.puhe.2023.08.034
10
Daly RM Rosengren B E Alwis G Ahlborg HG Sernbo I Karlsson MK . Gender specific age-related changes in bone density, muscle strength and functional performance in the elderly: a-10 year prospective population-based study. BMC Geriatr. (2013) 13:71. doi: 10.1186/1471-2318-13-71
11
Kanis JA . Assessment of fracture risk and its application to screening for postmenopausal osteoporosis. Report of a WHO Study Group. World Health Organ Tech Rep Ser. (1994) 843:1–129. doi: 10.1007/BF01622200
12
Kanis JA Glüer CC . An update on the diagnosis and assessment of osteoporosis with densitometry. Committee of Scientific Advisors, International Osteoporosis Foundation. Osteoporos Int. (2000) 11:192–202. doi: 10.1007/s001980050281
13
Lamichhane AP . Osteoporosis-an update. JNMA J Nepal Med Assoc. (2005) 44:60–6.
14
Cruz-Jentoft AJ Bahat G Bauer J Boirie Y Bruyère O Cederholm T et al . Sarcopenia: revised European consensus on definition and diagnosis. Age Ageing. (2019) 48:601. doi: 10.1093/ageing/afz046
15
Cho MR Lee S Song SK . A review of sarcopenia pathophysiology, diagnosis, treatment and future direction. J Korean Med Sci. (2022) 37:e146. doi: 10.3346/jkms.2022.37.e146
16
Hanai T Shiraki M Nishimura K Ohnishi S Imai K Suetsugu A et al . Sarcopenia impairs prognosis of patients with liver cirrhosis. Nutrition. (2015) 31:193–9. doi: 10.1016/j.nut.2014.07.005
17
Silva LD Bering T Rocha GA . The impact of nutrition on quality of life of patients with hepatitis C. Curr Opin Clin Nutr Metab Care. (2017) 20:420–5. doi: 10.1097/MCO.0000000000000396
18
Saeki C Kanai T Ueda K Nakano M Oikawa T Torisu Y et al . Osteosarcopenia predicts poor survival in patients with cirrhosis: a retrospective study. BMC Gastroenterol. (2023) 23:196. doi: 10.1186/s12876-023-02835-y
19
Saeki C Oikawa T Kanai T Nakano M Torisu Y Sasaki N et al . Relationship between osteoporosis, sarcopenia, vertebral fracture, and osteosarcopenia in patients with primary biliary cholangitis. Eur J Gastroenterol Hepatol. (2021) 33:731–7. doi: 10.1097/MEG.0000000000001791
20
Bering T Diniz KGD Coelho MPP Vieira DA Soares MMS Kakehasi AM et al . Association between pre-sarcopenia, sarcopenia, and bone mineral density in patients with chronic hepatitis C. J Cachexia Sarcopenia Muscle. (2018) 9:255–68. doi: 10.1002/jcsm.12269
21
Shane E Mancini D Aaronson K Silverberg S J Seibel M J Addesso V et al . Bone mass, vitamin D deficiency, and hyperparathyroidism in congestive heart failure. Am J Med. (1997) 103:197–207. doi: 10.1016/S0002-9343(97)00142-3
22
Gu Z Yuanyuan Y Lingyu Z Cong C . Assessment of the risk of incident heart failure in patients with osteoporosis: a systematic review and meta-analysis of eligible cohort studies. Pol Arch Intern Med. (2020) 130:934–41. doi: 10.20452/pamw.15598
23
Heshmat R Shafiee G Ostovar A Fahimfar N Maleki Birjandi S Jabbari M et al . Relationship between sarcopenia and electrocardiographic abnormalities in older people: the bushehr elderly health program. Front Med (Lausanne). (2021) 8:656181. doi: 10.3389/fmed.2021.656181
24
Teng Z Zhu Y Teng Y Long Q Hao Q Yu X et al . The analysis of osteosarcopenia as a risk factor for fractures, mortality, and falls. Osteoporos Int. (2021) 32:2173–83. doi: 10.1007/s00198-021-05963-x
25
Denfeld QE Turrise S MacLaughlin EJ Chang PS Clair WK Lewis EF et al . Preventing and managing falls in adults with cardiovascular disease: A scientific statement from the american heart association. Circ Cardiovasc Qual Outcomes. (2022) 15:e000108. doi: 10.1161/HCQ.0000000000000108
26
Nakano Y Mandai S Naito S Fujiki T Mori Y Ando F et al . Effect of osteosarcopenia on longitudinal mortality risk and chronic kidney disease progression in older adults. Bone. (2024) 179:116975. doi: 10.1016/j.bone.2023.116975
27
Wilkinson TJ Miksza J Yates T Lightfoot CJ Baker LA Watson EL et al . Association of sarcopenia with mortality and end-stage renal disease in those with chronic kidney disease: a UK Biobank study. J Cachexia Sarcopenia Muscle. (2021) 12:586–98. doi: 10.1002/jcsm.12705
28
Hoshino J . Renal rehabilitation: exercise intervention and nutritional support in dialysis patients. Nutrients. (2021) 13:1444–65. doi: 10.3390/nu13051444
29
Pitta F Troosters T Probst VS Spruit M A Decramer M Gosselink R . Physical activity and hospitalization for exacerbation of COPD. Chest. (2006) 129:536–44. doi: 10.1378/chest.129.3.536
30
Ma K Huang F Qiao R Miao L . Pathogenesis of sarcopenia in chronic obstructive pulmonary disease. Front Physiol. (2022) 13:850964. doi: 10.3389/fphys.2022.850964
31
Furman D Campisi J Verdin E Carrera-Bastos P Targ S Franceschi C et al . Chronic inflammation in the etiology of disease across the life. span. Nat Med. (2019) 25:1822–32. doi: 10.1038/s41591-019-0675-0
32
Lacey DL Boyle WJ Simonet WS Kostenuik PJ Dougall WC Sullivan JK et al . Bench to bedside: elucidation of the OPG-RANK-RANKL pathway and the development of denosumab. Nat Rev Drug Discov. (2012) 11:401–19. doi: 10.1038/nrd3705
33
He C He W Hou J Chen K Huang M Yang M et al . Bone and muscle crosstalk in aging. Front Cell Dev Biol. (2020) 8:585644. doi: 10.3389/fcell.2020.585644
34
Hamoudi D Bouredji Z Marcadet L Yagita H Landry LB Argaw A et al . Muscle weakness and selective muscle atrophy in osteoprotegerin-deficient mice. Hum Mol Genet. (2020) 29:483–94. doi: 10.1093/hmg/ddz312
35
Hamoudi D Marcadet L Piette Boulanger A Yagita H Bouredji Z Argaw A et al . An anti-RANKL treatment reduces muscle inflammation and dysfunction and strengthens bone in dystrophic mice. Hum Mol Genet. (2019) 28:3101–12. doi: 10.1093/hmg/ddz124
36
Sheng R Cao M Song M Wang M Zhang Y Shi L et al . Muscle-bone crosstalk via endocrine signals and potential targets for osteosarcopenia-related fracture. J Orthop Translat. (2023) 43:36–46. doi: 10.1016/j.jot.2023.09.007
37
Zhao B Grimes SN Li S Hu X Ivashkiv LB . TNF-induced osteoclastogenesis and inflammatory bone resorption are inhibited by transcription factor RBP-J. J Exp Med. (2012) 209:319–34. doi: 10.1084/jem.20111566
38
Park CH Lee YT Yoon KJ . Association between osteosarcopenia and coronary artery calcification in asymptomatic individuals. Sci Rep. (2022) 12:2231. doi: 10.1038/s41598-021-02640-1
39
Lippi L Folli A Curci C D'Abrosca F Moalli S Mezian K et al . Osteosarcopenia in patients with chronic obstructive pulmonary diseases: which pathophysiologic implications for rehabilitation? Int J Environ Res Public Health. (2022) 19:14314–32. doi: 10.3390/ijerph192114314
40
Blaschke M Koepp R Cortis J Komrakova M Schieker M Hempel U et al . IL-6, IL-1β, and TNF-α only in combination influence the osteoporotic phenotype in Crohn’s patients via bone formation and bone resorption. Adv Clin Exp Med. (2018) 27:45–56. doi: 10.17219/acem/67561
41
Wu Q Zhou X Huang D Ji Y Kang F . IL-6 enhances osteocyte-mediated osteoclastogenesis by promoting JAK2 and RANKL activity in vitro. Cell Physiol Biochem. (2017) 41:1360–9. doi: 10.1159/000465455
42
Kozlova IV Bykova AP . Osteosarcopenia in chronic pancreatitis. Ter Arkh. (2021) 93:869–75. doi: 10.26442/00403660.2021.08.200971
43
Byun MK Cho EN Chang J Ahn CM Kim HJ . Sarcopenia correlates with systemic inflammation in COPD. Int J Chron Obstruct Pulmon Dis. (2017) 12:669–75. doi: 10.2147/COPD.S130790
44
Çetin G Klafack S Studencka-Turski M Krüger E Ebstein F . The ubiquitin-proteasome system in immune cells. Biomolecules. (2021) 11.
45
Zhang HR Wang YH Xiao ZP Yang G Xu YR Huang ZT et al . E3 ubiquitin ligases: key regulators of osteogenesis and potential therapeutic targets for bone disorders. Front Cell Dev Biol. (2024) 12:1447093. doi: 10.3389/fcell.2024.1447093
46
Zhao L Huang J Zhang H Wang Y Matesic L E Takahata M et al . Tumor necrosis factor inhibits mesenchymal stem cell differentiation into osteoblasts via the ubiquitin E3 ligase Wwp1. Stem Cells. (2011) 29:1601–10. doi: 10.1002/stem.703
47
Li H Malhotra S Kumar A . Nuclear factor-kappa B signaling in skeletal muscle atrophy. J Mol Med (Berl). (2008) 86:1113–26. doi: 10.1007/s00109-008-0373-8
48
Cai D Frantz JD Tawa NE Jr Melendez PA Oh BC Lidov HG et al . IKKbeta/NF-kappaB activation causes severe muscle wasting in mice. Cell. (2004) 119:285–98. doi: 10.1016/j.cell.2004.09.027
49
Tarantino U Visconti V V Bonanni R Gatti A Marcozzi M Calabrò D et al . Osteosarcopenia and Long-COVID: a dangerous combination. Ther Adv Musculoskelet Dis. (2022) 14:1759720x221130485. doi: 10.1177/1759720X221130485
50
Gow DJ Sester DP Hume DA . CSF-1, IGF-1, and the control of postnatal growth and development. J Leukoc Biol. (2010) 88:475–81. doi: 10.1189/jlb.0310158
51
Yoshida T Delafontaine P . Mechanisms of IGF-1-mediated regulation of skeletal muscle hypertrophy and atrophy. Cells. (2020) 9:1970–95. doi: 10.3390/cells9091970
52
Gao ST Lv ZT Zhou CK Mao C Sheng WB . Association between IGF-1 polymorphisms and risk of osteoporosis in Chinese population: a meta-analysis. BMC Musculoskelet Disord. (2018) 19:141. doi: 10.1186/s12891-018-2066-y
53
Lee DO Jee BC Ku SY Suh CS Kim SH Choi YM et al . Relationships between the insulin-like growth factor I (IGF-I) receptor gene G3174A polymorphism, serum IGF-I levels, and bone mineral density in postmenopausal Korean women. J Bone Miner Metab. (2008) 26:42–6. doi: 10.1007/s00774-007-0795-3
54
Giustina A Mazziotti G Canalis E . Growth hormone, insulin-like growth factors, and the skeleton. Endocr Rev. (2008) 29:535–59. doi: 10.1210/er.2007-0036
55
Shavlakadze T Winn N Rosenthal N Grounds M D . Reconciling data from transgenic mice that overexpress IGF-I specifically in skeletal muscle. Growth Horm IGF Res. (2005) 15:4–18. doi: 10.1016/j.ghir.2004.11.001
56
Ascenzi F Barberi L Dobrowolny G Villa Nova Bacurau A Nicoletti C Rizzuto E et al . Effects of IGF-1 isoforms on muscle growth and sarcopenia. Aging Cell. (2019) 18:e12954. doi: 10.1111/acel.12954
57
Saeki C Tsubota A . Influencing factors and molecular pathogenesis of sarcopenia and osteosarcopenia in chronic liver disease. Life (Basel). (2021) 11:899–920. doi: 10.3390/life11090899
58
Yang YJ Kim DJ . An overview of the molecular mechanisms contributing to musculoskeletal disorders in chronic liver disease: osteoporosis, sarcopenia, and osteoporotic sarcopenia. Int J Mol Sci. (2021) 22:2604–37. doi: 10.3390/ijms22052604
59
Kamimura H Sato T Natsui K Kobayashi T Yoshida T Kamimura K et al . Molecular mechanisms and treatment of sarcopenia in liver disease: A review of current knowledge. Int J Mol Sci. (2021) 22. doi: 10.3390/ijms22031425
60
Sasaki KI Fukumoto Y . Sarcopenia as a comorbidity of cardiovascular disease. J Cardiol. (2022) 79:596–604. doi: 10.1016/j.jjcc.2021.10.013
61
Zhang L Wang XH Wang H Du J Mitch WE . Satellite cell dysfunction and impaired IGF-1 signaling cause CKD-induced muscle atrophy. J Am Soc Nephrol. (2010) 21:419–27. doi: 10.1681/ASN.2009060571
62
Steell L Gray SR Russell RK MacDonald J Seenan JP Wong SC et al . Pathogenesis of musculoskeletal deficits in children and adults with inflammatory bowel disease. Nutrients. (2021) 13. doi: 10.3390/nu13082899
63
Watanabe H Enoki Y Maruyama T . Sarcopenia in chronic kidney disease: factors, mechanisms, and therapeutic interventions. Biol Pharm Bull. (2019) 42:1437–45. doi: 10.1248/bpb.b19-00513
64
Jean G Souberbielle JC Chazot C . Vitamin D in chronic kidney disease and dialysis patients. Nutrients. (2017) 9:328–43. doi: 10.3390/nu9040328
65
Booth M Sabacinski K Watkins C Butcho E Kramer E Meadows L et al . Vitamin D levels and bone mineral density: a prospective cross-sectional analysis of young orthopedic trauma patients at a rural United States trauma center. J Trauma Inj. (2024) 37:276–80. doi: 10.20408/jti.2024.0038
66
Mori K . Maintenance of skeletal muscle to counteract sarcopenia in patients with advanced chronic kidney disease and especially those undergoing hemodialysis. Nutrients. (2021) 13:1538–60. doi: 10.3390/nu13051538
67
Davenport A . Frailty, appendicular lean mass, osteoporosis and osteosarcopenia in peritoneal dialysis patients. J Nephrol. (2022) 35:2333–40. doi: 10.1007/s40620-022-01390-1
68
Bruyère O Cavalier E Reginster JY . Vitamin D and osteosarcopenia: an update from epidemiological studies. Curr Opin Clin Nutr Metab Care. (2017) 20:498–503. doi: 10.1097/MCO.0000000000000411
69
Olsson K Saini A Strömberg A Alam S Lilja M Rullman E et al . Evidence for vitamin D receptor expression and direct effects of 1α,25(OH)2D3 in human skeletal muscle precursor cells. Endocrinology. (2016) 157:98–111. doi: 10.1210/en.2015-1685
70
Garcia LA King KK Ferrini MG Norris KC Artaza JN . 1,25(OH)2vitamin D3 stimulates myogenic differentiation by inhibiting cell proliferation and modulating the expression of promyogenic growth factors and myostatin in C2C12 skeletal muscle cells. Endocrinology. (2011) 152:2976–86. doi: 10.1210/en.2011-0159
71
Du Y Xie B Wang M Zhong Y Lv Z Luo Y et al . Roles of sex hormones in mediating the causal effect of vitamin D on osteoporosis: A two-step Mendelian randomization study. Front Endocrinol (Lausanne). (2023) 14:1159241. doi: 10.3389/fendo.2023.1159241
72
Shigehara K Izumi K Kadono Y Mizokami A . Testosterone and bone health in men: A narrative review. J Clin Med. (2021) 10:530–42. doi: 10.3390/jcm10030530
73
Bolamperti S Villa I Rubinacci A . Bone remodeling: an operational process ensuring survival and bone mechanical competence. Bone Res. (2022) 10:48. doi: 10.1038/s41413-022-00219-8
74
Casabella A Paladin F Bighin C Ottaviani S Marelli C Ponzano M et al . Aromatase inhibitor-induced bone loss osteosarcopenia in older patients with breast cancer: effects of the RANK/RANKL system’s inhibitor denosumab vs. bisphosphonates. Intern Emerg Med. (2024) 19:2193–9. doi: 10.1007/s11739-024-03725-1
75
Riggs BL Khosla S Melton LJ 3rd . Sex steroids and the construction and conservation of the adult skeleton. Endocr Rev. (2002) 23:279–302. doi: 10.1210/edrv.23.3.0465
76
La Colla A Pronsato L Milanesi L Vasconsuelo A . 17β-Estradiol and testosterone in sarcopenia: Role of satellite cells. Ageing Res Rev. (2015) 24:166–77. doi: 10.1016/j.arr.2015.07.011
77
Geraci A Calvani R Ferri E Marzetti E Arosio B Cesari M . Sarcopenia and menopause: the role of estradiol. Front Endocrinol (Lausanne). (2021) 12:682012. doi: 10.3389/fendo.2021.682012
78
Guerra-Menéndez L Sádaba MC Puche JE Lavandera JL de Castro LF de Gortázar AR et al . IGF-I increases markers of osteoblastic activity and reduces bone resorption via osteoprotegerin and RANK-ligand. J Transl Med. (2013) 11:271. doi: 10.1186/1479-5876-11-271
79
Yamashita T Yoshioka M Itoh N . Identification of a novel fibroblast growth factor, FGF-23, preferentially expressed in the ventrolateral thalamic nucleus of the brain. Biochem Biophys Res Commun. (2000) 277:494–8. doi: 10.1006/bbrc.2000.3696
80
Feng JQ Ward LM Liu S Lu Y Xie Y Yuan B et al . Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat Genet. (2006) 38:1310–5. doi: 10.1038/ng1905
81
Gattineni J Bates C Twombley K Dwarakanath V Robinson ML Goetz R et al . FGF23 decreases renal NaPi-2a and NaPi-2c expression and induces hypophosphatemia in vivo predominantly via FGF receptor 1. Am J Physiol Renal Physiol. (2009) 297:F282–91. doi: 10.1152/ajprenal.90742.2008
82
Shimada T Kakitani M Yamazaki Y Hasegawa H Takeuchi Y Fujita T et al . Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J Clin Invest. (2004) 113:561–8. doi: 10.1172/JCI200419081
83
Khairallah P Nickolas TL . Updates in CKD-associated osteoporosis. Curr Osteoporos Rep. (2018) 16:712–23. doi: 10.1007/s11914-018-0491-3
84
Rotondi S Tartaglione L Muci M L Mandanici G Leonangeli C et al . Soluble α -klotho serum levels in chronic kidney disease. Int J Endocrinol 2015. (2015) p:872193. doi: 10.1155/2015/872193
85
Hanna RM Ghobry L Wassef O Rhee C M Kalantar-Zadeh K . A practical approach to nutrition, protein-energy wasting, sarcopenia, and cachexia in patients with chronic kidney disease. Blood Purif. (2020) 49:202–11. doi: 10.1159/000504240
86
Fukasawa H Ishigaki S Kinoshita-Katahashi N Niwa H Yasuda H Kumagai H et al . Plasma levels of fibroblast growth factor-23 are associated with muscle mass in haemodialysis patients. Nephrol (Carlton). (2014) 19:784–90. doi: 10.1111/nep.12333
87
Sato C Iso Y Mizukami T Otabe K Sasai M Kurata M et al . Fibroblast growth factor-23 induces cellular senescence in human mesenchymal stem cells from skeletal muscle. Biochem Biophys Res Commun. (2016) 470:657–62. doi: 10.1016/j.bbrc.2016.01.086
88
Lu W Xiao W Xie W Fu X Pan L Jin H et al . The role of osteokines in sarcopenia: therapeutic directions and application prospects. Front Cell Dev Biol. (2021) 9:735374. doi: 10.3389/fcell.2021.735374
89
Graciolli FG Neves KR Barreto F Barreto DV Dos Reis LM Canziani ME et al . The complexity of chronic kidney disease-mineral and bone disorder across stages of chronic kidney disease. Kidney Int. (2017) 91:1436–46. doi: 10.1016/j.kint.2016.12.029
90
Fijalkowski I Geets E Steenackers E Van Hoof V Ramos FJ Mortier G et al . A novel domain-specific mutation in a sclerosteosis patient suggests a role of LRP4 as an anchor for sclerostin in human bone. J Bone Miner Res. (2016) 31:874–81. doi: 10.1002/jbmr.2782
91
Chen D Li Y Zhou Z Wu C Xing Y Zou X et al . HIF-1α inhibits Wnt signaling pathway by activating Sost expression in osteoblasts. PloS One. (2013) 8:e65940. doi: 10.1371/journal.pone.0065940
92
von Maltzahn J Chang NC Bentzinger CF Rudnicki MA . Wnt signaling in myogenesis. Trends Cell Biol. (2012) 22:602–9. doi: 10.1016/j.tcb.2012.07.008
93
Kim SP Frey JL Li Z Kushwaha P Zoch ML Tomlinson RE et al . Sclerostin influences body composition by regulating catabolic and anabolic metabolism in adipocytes. Proc Natl Acad Sci U.S.A. (2017) 114:E11238–e11247. doi: 10.1073/pnas.1707876115
94
Huang J Romero-Suarez S Lara N Mo C Kaja S Brotto L et al . Crosstalk between MLO-Y4 osteocytes and C2C12 muscle cells is mediated by the Wnt/β-catenin pathway. JBMR Plus. (2017) 1:86–100. doi: 10.1002/jbm4.10015
95
Chen YS Guo Q Guo LJ Liu T Wu XP Lin ZY et al . GDF8 inhibits bone formation and promotes bone resorption in mice. Clin Exp Pharmacol Physiol. (2017) 44:500–8. doi: 10.1111/1440-1681.12728
96
Qin Y Peng Y Zhao W Pan J Ksiezak-Reding H Cardozo C et al . Myostatin inhibits osteoblastic differentiation by suppressing osteocyte-derived exosomal microRNA-218: A novel mechanism in muscle-bone communication. J Biol Chem. (2017) 292:11021–33. doi: 10.1074/jbc.M116.770941
97
Rodriguez J Vernus B Chelh I Cassar-Malek I Gabillard JC Hadj Sassi A et al . Myostatin and the skeletal muscle atrophy and hypertrophy signaling pathways. Cell Mol Life Sci. (2014) 71:4361–71. doi: 10.1007/s00018-014-1689-x
98
Wang DT Yang YJ Huang RH Zhang ZH Lin X . Myostatin activates the ubiquitin-proteasome and autophagy-lysosome systems contributing to muscle wasting in chronic kidney disease. Oxid Med Cell Longev. (2015) 2015:684965. doi: 10.1155/2015/684965
99
Enoki Y Watanabe H Arake R Sugimoto R Imafuku T Tominaga Y et al . Indoxyl sulfate potentiates skeletal muscle atrophy by inducing the oxidative stress-mediated expression of myostatin and atrogin-1. Sci Rep. (2016) 6:32084. doi: 10.1038/srep32084
100
Dhaliwal A Quinlan JI Overthrow K Greig C Lord JM Armstrong MJ et al . Sarcopenia in inflammatory bowel disease: A narrative overview. Nutrients. (2021) 13. doi: 10.3390/nu13020656
101
Hamrick MW Shi X Zhang W Pennington C Thakore H Haque M et al . Loss of myostatin (GDF8) function increases osteogenic differentiation of bone marrow-derived mesenchymal stem cells but the osteogenic effect is ablated with unloading. Bone. (2007) 40:1544–53. doi: 10.1016/j.bone.2007.02.012
102
Rebbapragada A Benchabane H Wrana JL Celeste AJ Attisano L . Myostatin signals through a transforming growth factor beta-like signaling pathway to block adipogenesis. Mol Cell Biol. (2003) 23:7230–42. doi: 10.1128/MCB.23.20.7230-7242.2003
103
Teng Z Zhu Y Lin D Hao Q Yue Q Yu X et al . Deciphering the chromatin spatial organization landscapes during BMMSC differentiation. J Genet Genomics. (2023) 50:264–75. doi: 10.1016/j.jgg.2023.01.009
104
Qiao X Nie Y Ma Y Chen Y Cheng R Yin W et al . Irisin promotes osteoblast proliferation and differentiation via activating the MAP kinase signaling pathways. Sci Rep. (2016) 6:18732. doi: 10.1038/srep18732
105
Storlino G Colaianni G Sanesi L Lippo L Brunetti G Errede M et al . Irisin prevents disuse-induced osteocyte apoptosis. J Bone Miner Res. (2020) 35:766–75. doi: 10.1002/jbmr.3944
106
Ma Y Qiao X Zeng R Cheng R Zhang J Luo Y et al . Irisin promotes proliferation but inhibits differentiation in osteoclast precursor cells. FASEB J. (2018) 9:fj201700983RR. doi: 10.1096/fj.201700983RR
107
Huh JY Dincer F Mesfum E Mantzoros CS . Irisin stimulates muscle growth-related genes and regulates adipocyte differentiation and metabolism in humans. Int J Obes (Lond). (2014) 38:1538–44. doi: 10.1038/ijo.2014.42
108
Paluch AE Boyer WR Franklin BA Laddu D Lobelo F Lee DC et al . Resistance exercise training in individuals with and without cardiovascular disease: 2023 update: A scientific statement from the american heart association. Circulation. (2024) 149:e217–31. doi: 10.1161/CIR.0000000000001189
109
Glass DJ . Molecular mechanisms modulating muscle mass. Trends Mol Med. (2003) 9:344–50. doi: 10.1016/S1471-4914(03)00138-2
110
Jeong HM Kim DJ . Bone diseases in patients with chronic liver disease. Int J Mol Sci. (2019) 20:4270–97. doi: 10.3390/ijms20174270
111
Aryana I Rini SS Setiati S . Retraction: denosumab’s therapeutic effect for future osteosarcopenia therapy: A systematic review and meta-analysis. Ann Geriatr Med Res. (2023) 27:361. doi: 10.4235/agmr.22.0139.r1
112
Bonnet N Bourgoin L Biver E Douni E Ferrari S . RANKL inhibition improves muscle strength and insulin sensitivity and restores bone mass. J Clin Invest. (2019) 129:3214–23. doi: 10.1172/JCI125915
113
Recker RR Benson CT Matsumoto T Bolognese MA Robins DA Alam J et al . A randomized, double-blind phase 2 clinical trial of blosozumab, a sclerostin antibody, in postmenopausal women with low bone mineral density. J Bone Miner Res. (2015) 30:216–24. doi: 10.1002/jbmr.2351
114
Becker C Lord SR Studenski SA Warden SJ Fielding RA Recknor CP et al . Myostatin antibody (LY2495655) in older weak fallers: a proof-of-concept, randomised, phase 2 trial. Lancet Diabetes Endocrinol. (2015) 3:948–57. doi: 10.1016/S2213-8587(15)00298-3
115
Attie KM Borgstein NG Yang Y Condon CH Wilson DM Pearsall AE et al . A single ascending-dose study of muscle regulator ACE-031 in healthy volunteers. Muscle Nerve. (2013) 47:416–23. doi: 10.1002/mus.23539
116
Krampera M Le Blanc K . Mesenchymal stromal cells: Putative microenvironmental modulators become cell therapy. Cell Stem Cell. (2021) 28:1708–25. doi: 10.1016/j.stem.2021.09.006
117
Noh JY Yang Y Jung H . Molecular mechanisms and emerging therapeutics for osteoporosis. Int J Mol Sci. (2020) 21:7623–45. doi: 10.3390/ijms21207623
118
Chen Y Huang Y Li J Jiao T Yang L . Enhancing osteoporosis treatment with engineered mesenchymal stem cell-derived extracellular vesicles: mechanisms and advances. Cell Death Dis. (2024) 15:119. doi: 10.1038/s41419-024-06508-w
119
Bodine SC Sinha I Sweeney HL . Mechanisms of skeletal muscle atrophy and molecular circuitry of stem cell fate in skeletal muscle regeneration and aging. J Gerontol A Biol Sci Med Sci. (2023) 78:14–8. doi: 10.1093/gerona/glad023
120
Arjmand B Sarvari M Alavi-Moghadam S Payab M Goodarzi P Gilany K et al . Prospect of stem cell therapy and regenerative medicine in osteoporosis. Front Endocrinol (Lausanne). (2020) 11:430. doi: 10.3389/fendo.2020.00430
121
Page MJ Moher D Bossuyt PM Boutron I Hoffmann TC Mulrow CD et al . PRISMA 2020 explanation and elaboration: updated guidance and exemplars for reporting systematic reviews. Bmj. (2021) 372:n160. doi: 10.1136/bmj.n160
Summary
Keywords
osteosarcopenia, osteoporosis, sarcopenia, chronic diseases, chronic inflammatory disease, mesenchymal stem cell therapy
Citation
Yi Y-T, Zhao H-F, Wang W-Z and Li X (2025) Osteosarcopenia: epidemiology, molecular mechanisms, and management. Front. Endocrinol. 16:1577758. doi: 10.3389/fendo.2025.1577758
Received
16 February 2025
Accepted
05 August 2025
Published
28 August 2025
Volume
16 - 2025
Edited by
Michela Rossi, Bambino Gesù Children’s Hospital (IRCCS), Italy
Reviewed by
Jian-xiong Wang, First Affiliated Hospital of Anhui Medical University, China
Tomislav Tosti, University of Belgrade, Serbia
Updates
Copyright
© 2025 Yi, Zhao, Wang and Li.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei-Zhou Wang, wangweizhou@kmmu.edu.cn; Xi Li, 970244480@qq.com
†ORCID: Xi Li, orcid.org/0009-0004-0333-0408
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.