 Ke Qin*
Ke Qin* Tingyuan Zhang
Tingyuan Zhang- Department of Critical Care Medicine, Henan Provincial People’s Hospital, Zhengzhou, China
Objective: This study aimed to investigate the linear association between lipoprotein(a) [Lp(a)] levels and all-cause and cardiovascular mortality in patients with acute coronary syndrome (ACS).
Methods: This retrospective cohort study included 578 patients with ACS who were hospitalized at Henan Provincial People’s Hospital between January 2020 and January 2024. Patients were categorized into two groups: lower Lp(a) group (≤ 300 mg/L) and higher Lp(a) group (> 300 mg/L). Kaplan-Meier survival analysis, Cox regression models, subgroup and sensitivity analyses were used to evaluate the association between Lp(a) and all-cause and cardiovascular mortality. Restricted cubic spline (RCS) analysis was conducted to explore nonlinear associations.
Results: During a median follow-up of 27.5 months, a total of 124 all-cause deaths occurred (21.5%), of which 79 cases (13.7%) were classified as cardiovascular deaths. Compared to the lower Lp(a) group, the higher Lp(a) group exhibited a significantly increased risk of all-cause and cardiovascular mortality across all models. In the fully adjusted model (Model 3), the hazard ratio (HR) for all-cause mortality was 1.719 (95% confidence interval [CI]: 1.197–2.470, P = 0.003), while the HR for cardiovascular mortality was 2.505 (95% CI: 1.529-4.102, P < 0.001). In an additional analysis using a 500 mg/L cut-off, patients with Lp(a) > 500 mg/L had a significantly higher risk of cardiovascular mortality (HR = 2.209, P = 0.001), while the association with all-cause mortality (P = 0.284) was not statistically significant in the fully adjusted model. When Lp(a) was analyzed as a continuous variable, each 90 mg/L increase in Lp(a) was associated with a 5% higher risk of all-cause mortality (HR = 1.052, 95% CI: 1.003-1.104, P = 0.038), and each 45 mg/L increase was associated with a 5% higher risk of cardiovascular mortality (HR = 1.054, 95% CI: 1.026-1.084, P < 0.001). For log10-transformed Lp(a), the HR was 1.954 (95% CI: 1.252-3.050, P = 0.003) for all-cause mortality and 3.913 (95% CI: 2.108-7.265, P < 0.001) for cardiovascular mortality. Similarly, for standardized Lp(a) (Z-score), the HR was 1.178 (95% CI: 1.009-1.375, P = 0.038) for all-cause mortality and 1.408 (95% CI: 1.179-1.681, P < 0.001) for cardiovascular mortality. Most subgroup analyses showed that elevated Lp(a) levels were significantly associated with an increased risk of all-cause and cardiovascular mortality (P < 0.05). Sensitivity analyses confirmed the robustness of the findings, with significant associations persisting after excluding patients with early mortality or without stent implantation. Kaplan-Meier analysis showed that both all-cause and cardiovascular survival rates were significantly lower in the high Lp(a) group compared to the low Lp(a) group (P < 0.001 for both). RCS analyses revealed a linear positive association between Lp(a) levels and both all-cause and cardiovascular mortality.
Conclusions: Higher Lp(a) levels were independently and linearly associated with an increased risk of all-cause and cardiovascular mortality in ACS patients.
1 Introduction
Acute coronary syndrome (ACS) is a group of acute cardiovascular events caused by the rupture or erosion of atherosclerotic plaques in the coronary arteries, representing a major contributor to cardiovascular-related mortality and disease burden worldwide (1). Epidemiological data indicate that millions of ACS patients are hospitalized annually, and the long-term mortality rate remains high, particularly among high-risk individuals with heart failure, chronic kidney disease (CKD), or diabetes (2–4). Despite significant improvements in ACS prognosis due to percutaneous coronary intervention (PCI), pharmacological treatment (such as antiplatelet agents, lipid-lowering drugs, and β-blockers), a substantial proportion of patients still experience major adverse cardiovascular events (MACE) or death during follow-up (5–7). Current evidence has confirmed that certain biomarkers, such as those related to inflammation, can independently predict the risk of cardiovascular events in patients with ACS (8–10). However, even after controlling for these known risk factors, residual cardiovascular risk remains, suggesting the need to identify additional risk indicators. Therefore, identifying other residual cardiovascular risk factors that can accurately predict long-term mortality risk in ACS patients is crucial for optimizing individualized risk assessment and developing more effective treatment strategies.
Lipoprotein(a) [Lp(a)] is a cholesterol-rich low-density lipoprotein (LDL)-like particle that is structurally unique due to its apolipoprotein B-100 (ApoB-100), which is covalently linked via disulfide bonds to apolipoprotein(a) [Apo(a)] (11). Lp(a) is primarily synthesized and secreted by the liver into the bloodstream, with serum levels varying widely among individuals, mainly determined by genetic factors (LPA gene polymorphisms) (12). Unlike other lipid parameters, Lp(a) levels are relatively unaffected by lifestyle factors or commonly used lipid-lowering medications, such as statins (13). Lp(a) has significant pro-atherogenic, pro-inflammatory, and pro-thrombotic properties, which may accelerate the development of coronary artery disease (CAD) and its complications through multiple mechanisms (14). Given these mechanisms, elevated Lp(a) levels in ACS patients may significantly impact their prognosis and increase the risk of all-cause mortality.
Lp(a) has been extensively studied in relation to cardiovascular disease (CVD) onset and progression, with substantial evidence supporting its role as an independent risk factor (15–17). However, the specific relationship between Lp(a) and all-cause mortality remains controversial, particularly in the ACS patient population. Some studies have reported a significant positive correlation between elevated Lp(a) levels and higher all-cause mortality, while others have not observed such an association (18–21). These discrepancies may be attributed to heterogeneity in study populations, differences in follow-up durations, variations in Lp(a) measurement methods, and inconsistencies in statistical approaches. Additionally, most prior studies have relied on categorical analysis of Lp(a) (e.g., using thresholds of 30 mg/dL or 50 mg/dL) rather than exploring its linear association with all-cause mortality. Therefore, one of the primary objectives of this study was to use continuous variable analysis to clarify the dose-response relationship between Lp(a) and all-cause and cardiovascular mortality and to investigate its clinical significance in ACS patients.
Based on this background, we hypothesize that Lp(a) levels are linearly and positively associated with all-cause and cardiovascular mortality in ACS patients. The objectives of this study are as follows: (1) to evaluate the association between Lp(a) levels and all-cause and cardiovascular mortality and determine whether Lp(a) serves as an independent predictor; (2) to validate the robustness of this association by using different continuous variable analysis strategies for Lp(a) (raw values, log-transformed values, and standardized values); (3) to examine this relationship across different clinical subgroups (such as age, sex, hypertension, diabetes, CKD); and (4) to explore the potential nonlinearity of this association to determine whether the relationship follows a linear pattern. The findings of this study will contribute to a better understanding of the role of Lp(a) in predicting all-cause and cardiovascular mortality risk in ACS patients and provide valuable insights for optimizing individualized treatment and follow-up strategies.
2 Methods
2.1 Study population
This retrospective cohort study was conducted at Henan Provincial People’s Hospital, including patients enrolled between January 2020 and January 2024. A total of 578 participants were included and categorized into two groups based on their Lp(a) levels: the lower Lp(a) group (n = 350) and the higher Lp(a) group (n = 228). Patients were included in the study if they met the following criteria: (a) Aged ≥ 18 years at the time of enrollment; (2) Underwent serum Lp(a) measurement at the hospital; (3) Had complete baseline clinical data, including demographics, laboratory results, and medical history; (4) Had follow-up data available for mortality outcomes. Patients were excluded from the study based on the following criteria: (1) Missing key data, including Lp(a) levels or mortality status; (2) History of malignancy, autoimmune diseases, or severe infections that could affect Lp(a) levels; (3) Severe hepatic or renal dysfunction at baseline; (4) Loss to follow-up or withdrawal from the study. This study was approved by the Ethics Committee of Henan Provincial People’s Hospital and was conducted in accordance with the Declaration of Helsinki. Informed consent was obtained from all participants or their legal representatives before data collection.
2.2 Measurement of lipoprotein(a)
Serum Lp(a) levels were measured using an immunoturbidimetric assay, a widely used method for the quantitative determination of Lp(a), performed on an automated biochemical analyzer following the manufacturer’s standardized protocol. The reference range for Lp(a) was 0–300 mg/L. Based on this threshold, participants were categorized into two groups: those with Lp(a) ≤ 300 mg/L were classified as the lower Lp(a) group, while those with Lp(a) > 300 mg/L were assigned to the higher Lp(a) group. In addition, an analysis was conducted using a cut-off value of Lp(a) = 500 mg/L, according to which patients were further divided into two groups: Lp(a) ≤ 500 mg/L and Lp(a) > 500 mg/L.
In statistical analyses, Lp(a) was treated as both a categorical and a continuous variable. For continuous analysis, three approaches were used: (1) raw Lp(a) values in mg/L, (2) log10-transformed Lp(a) values to normalize skewed distributions, and (3) standardized Lp(a) values (Z-scores) to facilitate effect size comparisons across different models.
2.3 Outcome assessment
The primary outcome of this study was all-cause mortality, defined as death from any cause occurring during the follow-up period. In addition, cardiovascular mortality was analyzed as a secondary outcome, defined as death resulting from cardiovascular causes, including ACS, sudden cardiac death, heart failure, stroke, and other fatal cardiovascular events. All patients were followed from the time of hospital discharge until death or the end of the study on January 2025. Mortality data were obtained from hospital records and verified through follow-up assessments, including phone interviews and electronic medical records.
2.4 Collection and definition of covariates
Baseline demographic and clinical characteristics were collected from medical records, including age, gender, types of ACS (including ST-segment elevation myocardial infarction (STEMI), non-ST-segment elevation myocardial infarction, and unstable angina pectoris), smoking status, and body mass index (BMI). Hypertension was defined as a self-reported history of hypertension, systolic blood pressure (SBP) ≥ 140 mmHg, diastolic blood pressure (DBP) ≥ 90 mmHg, or the use of antihypertensive medications (22). Diabetes was defined as a self-reported history of diabetes, fasting glucose ≥ 7.0 mmol/L, glycated hemoglobin (HbA1c) ≥ 6.5%, or the use of insulin or oral hypoglycemic drugs (23). Hyperlipidemia was defined as a self-reported history of hyperlipidemia, total cholesterol (TC) ≥ 5.2 mmol/L, low-density lipoprotein cholesterol (LDL-C) ≥ 3.4 mmol/L, triglycerides ≥ 1.7 mmol/L, or the use of lipid-lowering agents (24). CKD was defined as an estimated glomerular filtration rate (eGFR) < 60 mL/min/1.73m2 or a history of CKD (25).
Coronary severity was assessed using the Gensini score, which quantifies the extent of coronary artery stenosis, and the number of diseased vessels. Left ventricular ejection fraction (LVEF) was measured via echocardiography to assess cardiac function. Stent implantation status was recorded to identify patients who had undergone PCI. Laboratory parameters were obtained from fasting blood samples, including TC, LDL-C, high-density lipoprotein cholesterol (HDL-C), triglycerides, apolipoprotein A1 (ApoA1), apolipoprotein B (ApoB), eGFR, uric acid, fibrinogen, albumin, and fasting glucose. Medication use was documented, including antiplatelet agents (aspirin, ticagrelor), lipid-lowering drugs (statins), antihypertensive drugs [beta-blockers, angiotensin-converting enzyme inhibitors (ACEI), angiotensin receptor blockers (ARB), calcium channel blockers (CCB)], diuretics (furosemide, spironolactone), and antidiabetic medications (insulin, oral hypoglycemic drugs).
2.5 Statistical methods
Baseline characteristics were compared between the lower and higher Lp(a) groups using the chi-square test for categorical variables and the independent t-test for normally distributed continuous variables or the Wilcoxon rank-sum test for non-normally distributed continuous variables. The normality of continuous variables was assessed using the Shapiro-Wilk test. Kaplan-Meier survival analysis was conducted to compare cumulative all-cause and cardiovascular mortality between the two Lp(a) groups, and differences in survival curves were assessed using the log-rank test. Cox proportional hazards regression models were used to assess the association between Lp(a) and all-cause and cardiovascular mortality, with hazard ratios (HRs) and 95% confidence intervals (CIs) reported. Three models were constructed: Model 1 adjusted for age and gender; Model 2 further adjusted for smoking, hypertension, diabetes, and CKD; and Model 3 further adjusted for ApoA1, ApoB, albumin, uric acid, eGFR, fibrinogen, LVEF, and Gensini score. Subgroup analyses were performed to evaluate potential effect modifications by age, gender, hypertension, diabetes, CKD, and STEMI. Sensitivity analyses were conducted by excluding patients who died within two years of follow-up or had a follow-up duration of less than two years or those without stent implantation. Restricted cubic spline (RCS) models were used to explore potential nonlinear associations between Lp(a) and all-cause and cardiovascular mortality. All statistical analyses were performed using SPSS version 26.0 and R version 4.3.4, with a two-tailed P-value < 0.05 considered statistically significant.
3 Results
3.1 Clinical characteristics
As shown in Table 1, the total population consisted of 578 individuals with a mean age of 65.0 years, including 459 males (79.4%). Compared to the lower Lp(a) group (≤ 300 mg/L), the higher Lp(a) group (> 300 mg/L) was older (P < 0.001), exhibited a higher prevalence of CKD (P = 0.001), showed a lower eGFR (P < 0.001), presented with a higher Gensini score (P < 0.001) and a greater number of diseased vessels (P < 0.001), demonstrated a lower LVEF (P = 0.042), recorded higher SBP (P = 0.017), displayed elevated TC (P = 0.007), LDL-C (P < 0.001), and ApoB levels (P = 0.002), showed increased fibrinogen levels (P < 0.001), and experienced a higher all-cause and cardiovascular mortality rate (P < 0.001). During a median follow-up of 27.5 months, a total of 124 all-cause deaths occurred (21.5%), of which 79 cases (13.7%) were classified as cardiovascular deaths. The number of events per variable (EPV) in the fully adjusted Cox model was approximately 9, indicating an acceptable level of model stability. Figure 1 presented the results of Kaplan-Meier survival analysis evaluating the association between serum Lp(a) levels and all-cause mortality (Figure 1A) as well as cardiovascular mortality (Figure 1B). Figure 1A showed that the all-cause survival rate during the follow-up period was significantly lower in the high Lp(a) group compared to the low Lp(a) group, with a statistically significant difference between the survival curves (Log-rank test, P < 0.001). Figure 1B further indicated that cardiovascular survival was also markedly lower in the high Lp(a) group, showing a continuously declining trend throughout the follow-up period, with a significant difference compared to the low Lp(a) group (Log-rank P < 0.001).
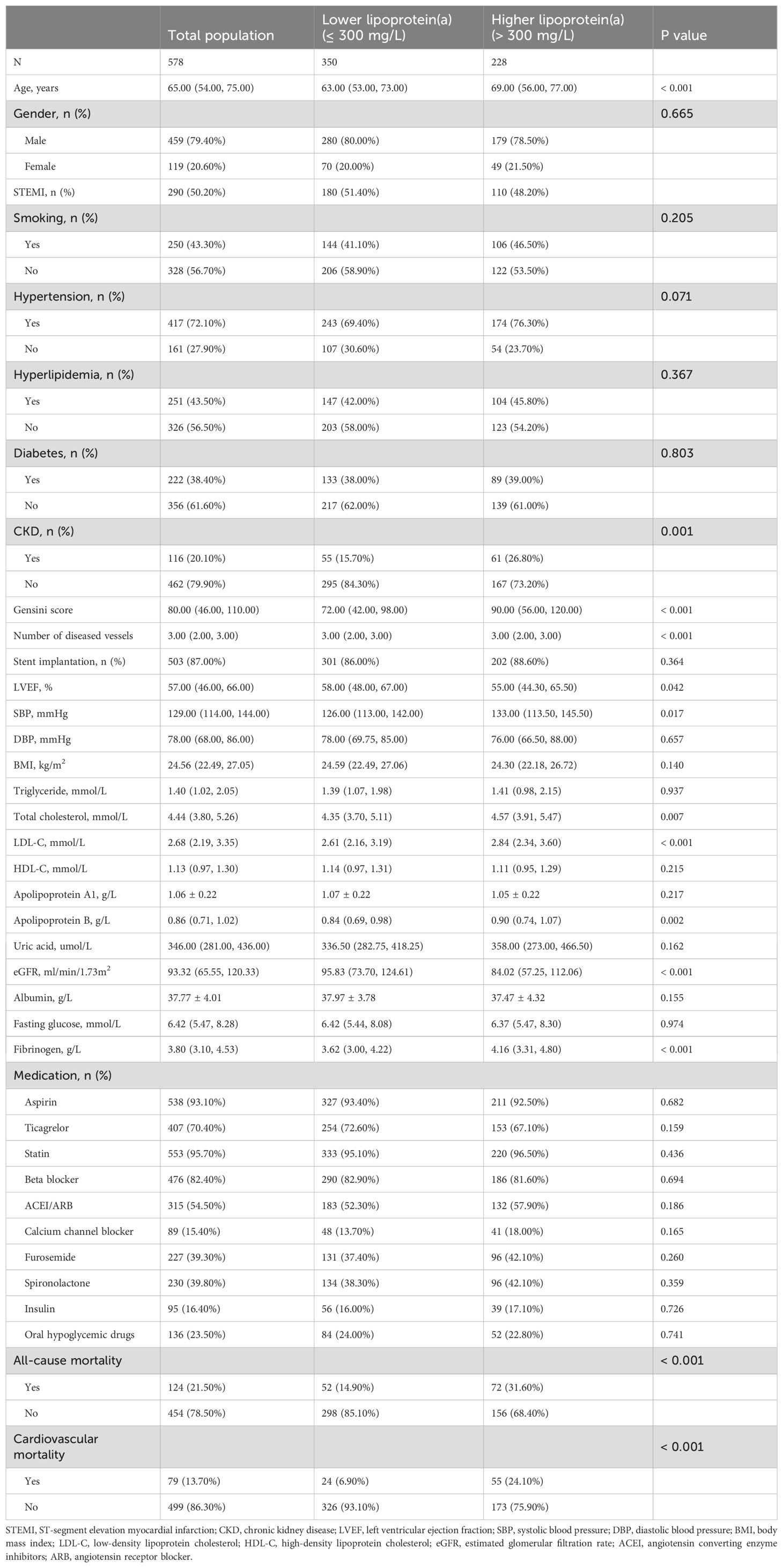
Table 1. Baseline characteristics grouped by lipoprotein(a).
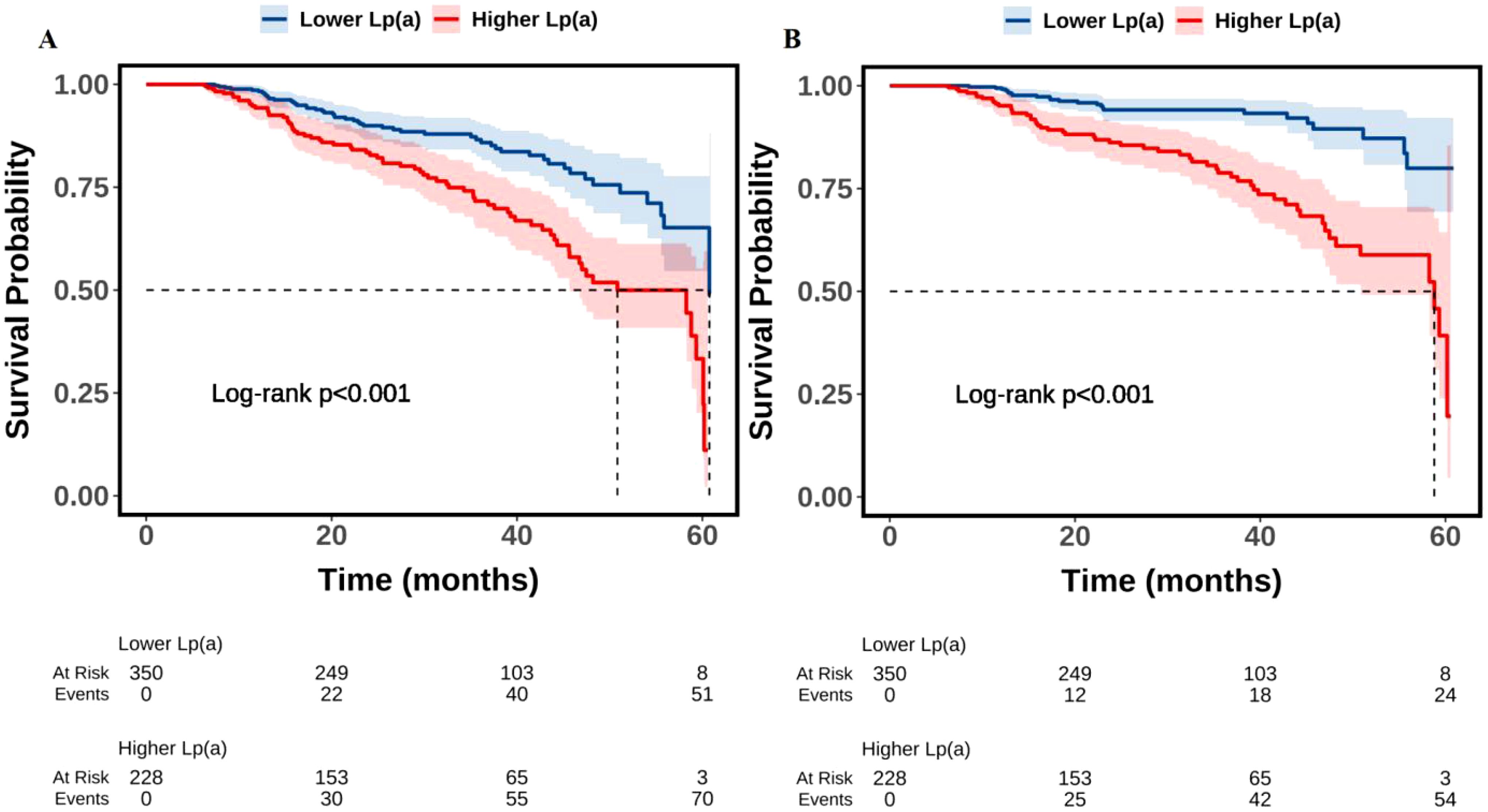
Figure 1. Kaplan-Meier survival analysis of the association between Lp(a) and all-cause (A) and cardiovascular (B) mortality. Lp(a), lipoprotein(a).
3.2 Multivariable association between lipoprotein(a) and all-cause mortality
As shown in Table 2, compared to the lower Lp(a) group (≤ 300 mg/L), the higher Lp(a) group (> 300 mg/L) exhibited a significantly increased risk of all-cause and cardiovascular mortality across all models. For all-cause mortality, the HRs were 1.862 (95% CI: 1.299-2.669, P = 0.001) in Model 1 (adjusted for age and gender), 1.848 (95% CI: 1.290–2.648, P = 0.001) in Model 2 (further adjusted for smoking, hypertension, diabetes, and CKD), and 1.719 (95% CI: 1.197-2.470, P = 0.003) in Model 3 (fully adjusted for clinical and laboratory variables). Correspondingly, for cardiovascular mortality, the HRs were 3.065 (95% CI: 1.895-4.960, P < 0.001), 3.027 (95% CI: 1.871-4.897, P < 0.001), and 2.505 (95% CI: 1.529-4.102, P < 0.001) across the three models, respectively.
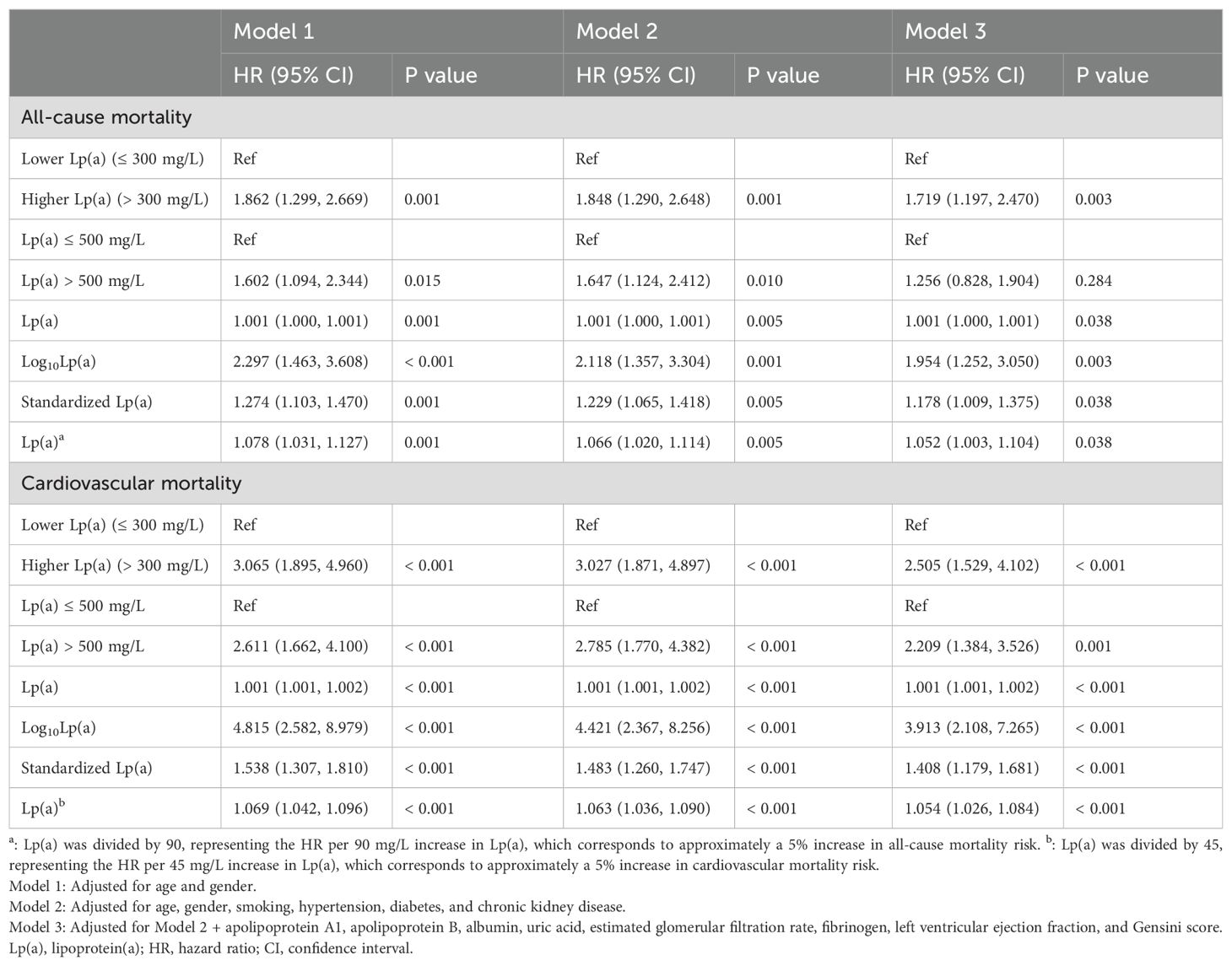
Table 2. Multivariable association between lipoprotein(a) and all-cause and cardiovascular mortality.
To further explore the prognostic value of more widely recognized clinical thresholds, an additional analysis was conducted using 500 mg/L as the cut-off point. Compared to patients with Lp(a) ≤ 500 mg/L, those with Lp(a) > 500 mg/L exhibited a significantly higher risk of all-cause mortality in Models 1 and 2, with HRs of 1.602 (95% CI: 1.094-2.344, P = 0.015) and 1.647 (95% CI: 1.124-2.412, P = 0.010), respectively. However, in the fully adjusted Model 3, the association was attenuated and did not reach statistical significance (HR = 1.256, 95% CI: 0.828-1.904, P = 0.284). In contrast, for cardiovascular mortality, the elevated risk associated with Lp(a) > 500 mg/L remained statistically significant across all three models: HRs were 2.611 (95% CI: 1.662-4.100, P < 0.001) in Model 1, 2.785 (95% CI: 1.770-4.382, P < 0.001) in Model 2, and 2.209 (95% CI: 1.384-3.526, P = 0.001) in Model 3.
When Lp(a) was analyzed as a continuous variable, elevated levels were significantly associated with increased risks of both all-cause and cardiovascular mortality across all models. For all-cause mortality, the HRs in Model 1 were 1.001 (95% CI: 1.000-1.001, P = 0.001) for Lp(a), 2.297 (95% CI: 1.463-3.608, P < 0.001) for log10Lp(a), and 1.274 (95% CI: 1.103-1.470, P = 0.001) for standardized Lp(a); in Model 2, the HRs were 1.001 (95% CI: 1.000-1.001, P = 0.005), 2.118 (95% CI: 1.357-3.304, P = 0.001), and 1.229 (95% CI: 1.065-1.418, P = 0.005), respectively; and in Model 3, the HRs were 1.001 (95% CI: 1.000-1.001, P = 0.038), 1.954 (95% CI: 1.252-3.050, P = 0.003), and 1.178 (95% CI: 1.009-1.375, P = 0.038). Similarly, for cardiovascular mortality, the HRs in Model 1 were 1.001 (95% CI: 1.001-1.002, P < 0.001), 4.815 (95% CI: 2.582-8.979, P < 0.001), and 1.538 (95% CI: 1.307-1.810, P < 0.001); in Model 2, they were 1.001 (95% CI: 1.001-1.002, P < 0.001), 4.421 (95% CI: 2.367-8.256, P < 0.001), and 1.483 (95% CI: 1.260-1.747, P < 0.001); and in Model 3, the HRs were 1.001 (95% CI: 1.001-1.002, P < 0.001), 3.913 (95% CI: 2.108-7.265, P < 0.001), and 1.408 (95% CI: 1.179-1.681, P < 0.001), respectively.
When Lp(a)/90 and Lp(a)/45 were used as standardized variables in the analysis, the results showed that every 90 mg/L increase in Lp(a) was associated with approximately a 5% higher risk of all-cause mortality (HR = 1.052, 95% CI: 1.003-1.104, P = 0.038), and every 45 mg/L increase in Lp(a) was associated with approximately a 5% higher risk of cardiovascular mortality (HR = 1.054, 95% CI: 1.026-1.084, P < 0.001).
3.3 Subgroup analysis
Subgroup analyses of Table 3 demonstrated that elevated Lp(a) levels were significantly associated with increased all-cause mortality in patients aged ≥ 60 years (higher Lp(a) vs lower Lp(a): HR = 1.785, P = 0.003; log10Lp(a): HR = 1.917, P = 0.006; standardized Lp(a): HR = 1.223, P = 0.009), males (HR = 1.892, P = 0.005; standardized Lp(a): HR = 1.240, P = 0.043), patients with hypertension (HR = 1.502, P = 0.045; log10Lp(a): HR = 1.860, P = 0.011), without hypertension (HR = 4.001, P = 0.009; Lp(a): HR = 1.002, P = 0.003; log10Lp(a): HR = 6.546, P = 0.002; standardized Lp(a): HR = 1.671, P = 0.003), without diabetes (HR = 1.967, P = 0.009; Lp(a): HR = 1.001, P = 0.014; log10Lp(a): HR = 2.422, P = 0.012; standardized Lp(a): HR = 1.318, P = 0.014), and without CKD (log10Lp(a): HR = 2.509, P = 0.006), as well as in STEMI patients (HR = 2.408, P = 0.003; log10Lp(a): HR = 2.896, P = 0.005).
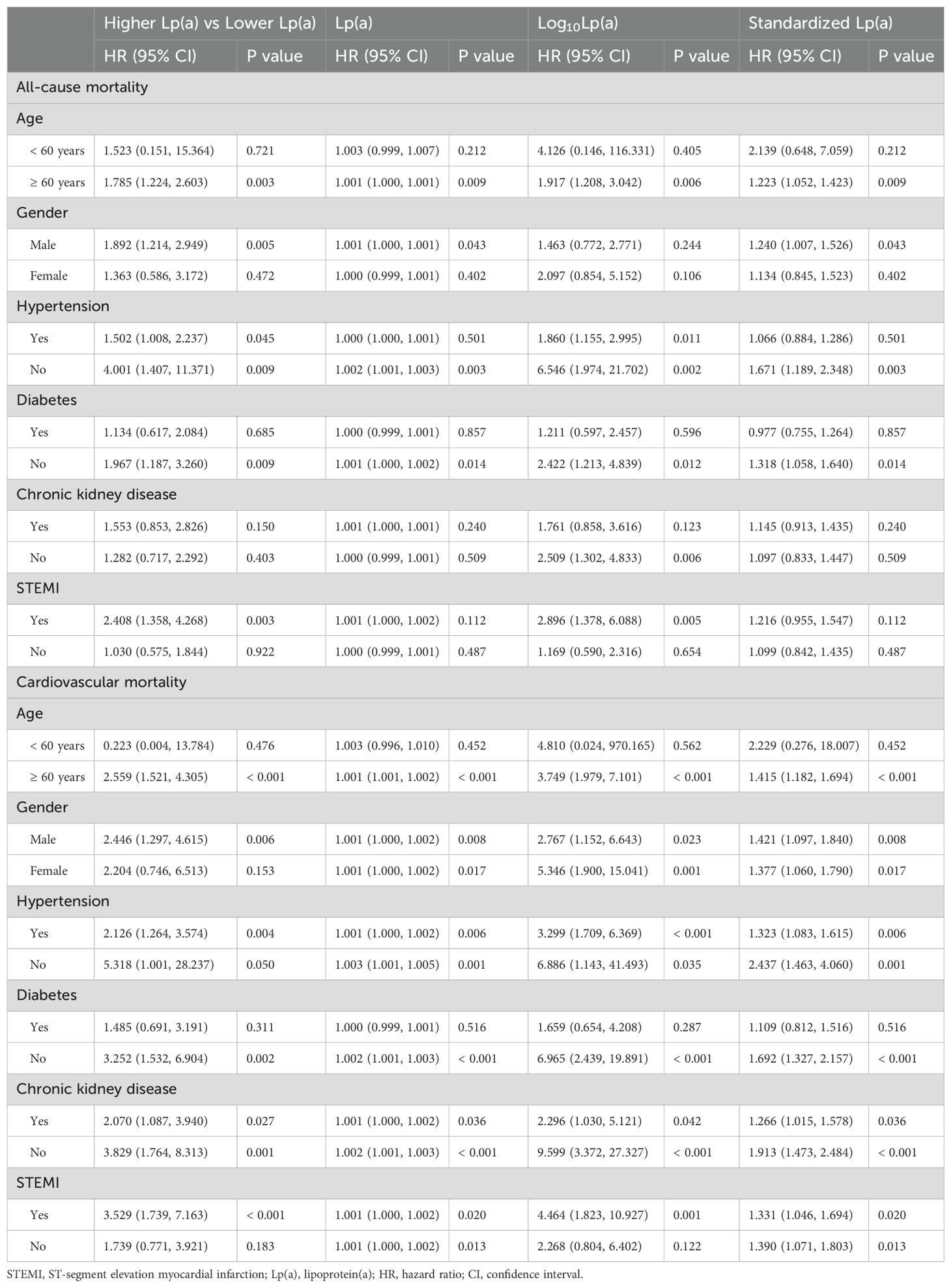
Table 3. Subgroup analysis of the association between lipoprotein(a) and all-cause and cardiovascular mortality.
For cardiovascular mortality, significant associations were observed in patients aged ≥ 60 years (HR = 2.559, P < 0.001; Lp(a): HR = 1.001, P < 0.001; log10Lp(a): HR = 3.749, P < 0.001; standardized Lp(a): HR = 1.415, P < 0.001), males (HR = 2.446, P = 0.006; Lp(a): HR = 1.001, P = 0.008; log10Lp(a): HR = 2.767, P = 0.023; standardized Lp(a): HR = 1.421, P = 0.008), and females (Lp(a): HR = 1.001, P = 0.017; log10Lp(a): HR = 5.346, P = 0.001; standardized Lp(a): HR = 1.377, P = 0.017). Similar associations were found in patients with hypertension (HR = 2.126, P = 0.004; Lp(a): HR = 1.001, P = 0.006; log10Lp(a): HR = 3.299, P < 0.001; standardized Lp(a): HR = 1.323, P = 0.006), without hypertension (Lp(a): HR = 1.003, P = 0.001; log10Lp(a): HR = 6.886, P = 0.035; standardized Lp(a): HR = 2.437, P = 0.001), without diabetes (HR = 3.252, P = 0.002; Lp(a): HR = 1.002, P < 0.001; log10Lp(a): HR = 6.965, P < 0.001; standardized Lp(a): HR = 1.692, P < 0.001), with CKD (HR = 2.070, P = 0.027; Lp(a): HR = 1.001, P = 0.036; log10Lp(a): HR = 2.296, P = 0.042; standardized Lp(a): HR = 1.266, P = 0.036), and without CKD (HR = 3.829, P = 0.001; Lp(a): HR = 1.002, P < 0.001; log10Lp(a): HR = 9.599, P < 0.001; standardized Lp(a): HR = 1.913, P < 0.001). In terms of ACS type, both STEMI (HR = 3.529, P < 0.001; Lp(a): HR = 1.001, P = 0.020; log10Lp(a): HR = 4.464, P = 0.001; standardized Lp(a): HR = 1.331, P = 0.020) and non-STEMI patients (Lp(a): HR = 1.001, P = 0.013; standardized Lp(a): HR = 1.390, P = 0.013) demonstrated significant associations.
3.4 Sensitivity analysis
In the sensitivity analysis excluding patients with mortality or follow-up duration less than two years (Table 4), the associations between elevated Lp(a) levels and mortality remained robust in the fully adjusted model (Model 3). For all-cause mortality, the higher Lp(a) group showed a significantly increased risk compared to the lower Lp(a) group (HR = 2.424, 95% CI: 1.420-4.138, P = 0.001). Continuous variable analyses also demonstrated significant associations: Lp(a) (HR = 1.001, P = 0.002), log10-transformed Lp(a) (HR = 3.646, P < 0.001), and standardized Lp(a) (HR = 1.389, P = 0.002). For cardiovascular mortality, the higher Lp(a) group had a markedly increased risk compared to the lower group (HR = 5.083, 95% CI: 2.199-11.752, P < 0.001). Similarly, significant associations were observed for Lp(a) as a continuous variable (HR = 1.002, P < 0.001), log10-transformed Lp(a) (HR = 13.424, P < 0.001), and standardized Lp(a) (HR = 1.855, P < 0.001).
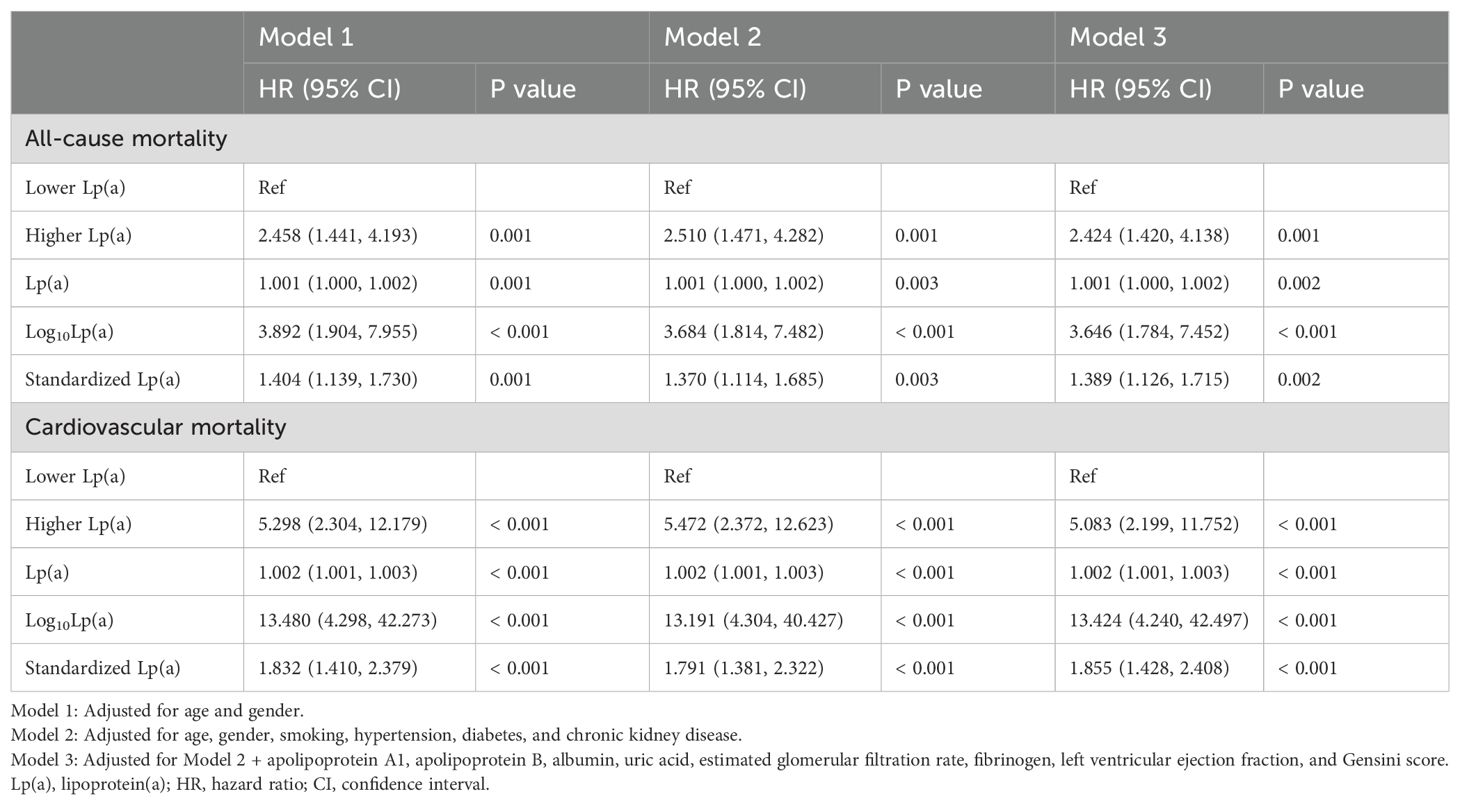
Table 4. Sensitivity analysis: exclusion of patients with mortality or follow-up duration less than two years.
In the sensitivity analysis excluding patients without stent implantation (Table 5), for all-cause mortality, the higher Lp(a) group showed a significantly elevated risk compared to the lower group (HR = 1.922, 95% CI: 1.281-2.885, P = 0.002). Significant associations were also observed for Lp(a) as a continuous variable (HR = 1.001, P = 0.006), log10-transformed Lp(a) (HR = 2.267, P = 0.002), and standardized Lp(a) (HR = 1.268, P = 0.006). For cardiovascular mortality, the higher Lp(a) group had a significantly increased risk (HR = 2.924, 95% CI: 1.690-5.061, P < 0.001), with consistent associations across continuous forms: Lp(a) (HR = 1.001, P < 0.001), log10Lp(a) (HR = 4.015, P < 0.001), and standardized Lp(a) (HR = 1.505, P < 0.001).
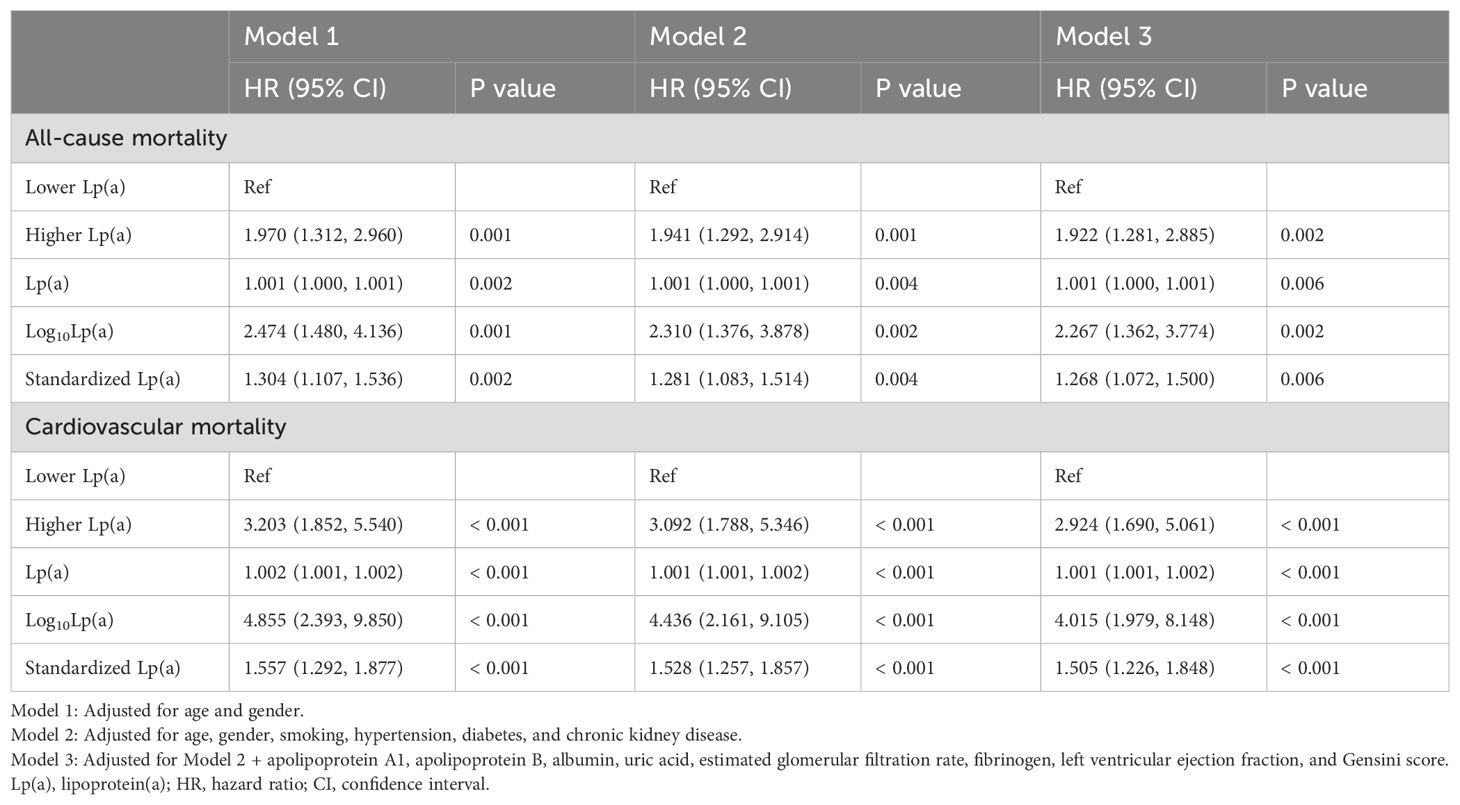
Table 5. Sensitivity analysis: excluding patients without stent implantation.
3.5 Restricted cubic spline analysis
Figure 2 presented the RCS analysis of the association between Lp(a) and all-cause and cardiovascular mortality. In the fully adjusted model, which included age, gender, smoking, hypertension, diabetes, CKD, ApoA1, ApoB, albumin, uric acid, eGFR, fibrinogen, LVEF, and Gensini score, Lp(a) remained linearly and positively associated with all-cause (Figure 2A) and cardiovascular (Figure 2B) mortality (P-nonlinear = 0.528 and 0.859).
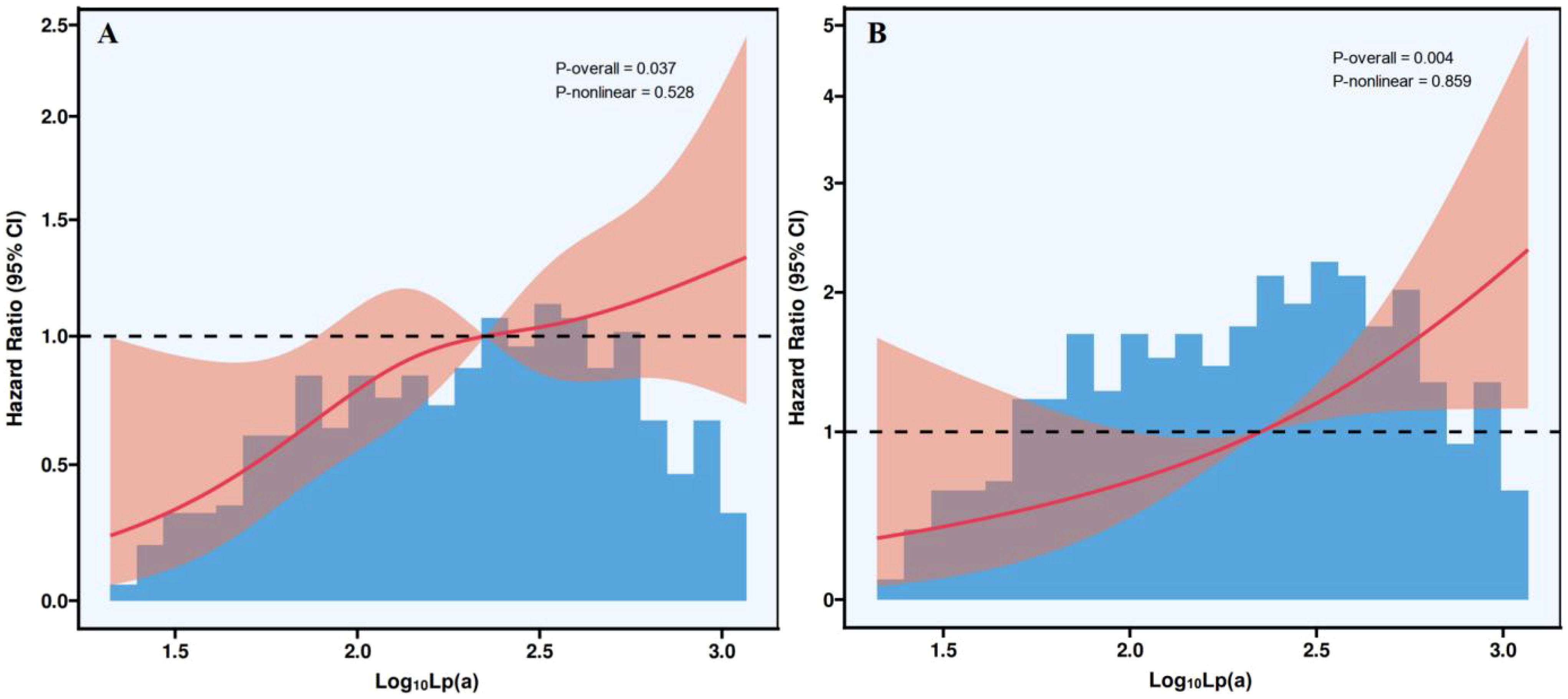
Figure 2. Restricted cubic spline plots of the association between Lp(a) and all-cause (A) and cardiovascular (B) mortality. Lp(a), lipoprotein(a); CI, confidence interval.
4 Discussion
This study systematically analyzed the linear association between Lp(a) levels and all-cause and cardiovascular mortality in patients with ACS. The results indicate that elevated Lp(a) levels are an independent risk factor for increased all-cause and cardiovascular mortality. Compared to the Lp(a) ≤ 300 mg/L group, the Lp(a) > 300 mg/L group exhibited a significantly higher risk of all-cause and cardiovascular mortality. In the additional analysis using the 500 mg/L threshold, Lp(a) > 500 mg/L was also significantly associated with increased cardiovascular mortality, although the association with all-cause mortality was not statistically significant after full adjustment. When Lp(a) was analyzed as a continuous variable, each 90 mg/L increase in Lp(a) was associated with a 5% higher risk of all-cause mortality, and each 45 mg/L increase was associated with a 5% higher risk of cardiovascular mortality. Additionally, analyses based on log10-transformed Lp(a) and standardized Z-score further supported this trend. Subgroup analyses revealed that elevated Lp(a) levels were significantly associated with increased all-cause mortality in patients aged ≥ 60 years, males, those with or without hypertension, those without diabetes or CKD, and in STEMI patients. For cardiovascular mortality, significant associations were observed across a broader range of subgroups, including both males and females, patients aged ≥ 60 years, individuals with or without hypertension, diabetes, or CKD, as well as both STEMI and non-STEMI populations. Sensitivity analyses confirmed the robustness of these findings. Notably, RCS analysis did not reveal a nonlinear trend, further supporting the stable linear relationship between Lp(a) levels and all-cause and cardiovascular mortality. Overall, this study underscores the potential clinical value of Lp(a) as a prognostic biomarker in ACS patients and provides critical evidence for improving cardiovascular risk management.
Lp(a) is a genetically determined lipoprotein, and its association with CVD has been confirmed in multiple international studies (26–29). Large-scale prospective studies, such as the UK Biobank and the Copenhagen General Population Study, have demonstrated that elevated Lp(a) levels are closely related to an increased risk of all-cause mortality and cardiovascular events (30–32). Besides, based on a prospective study of the general Danish population, Langsted et al. analyzed 69,764 individuals with Lp(a) measurements and found that Lp(a) levels >93 mg/dL (199 nmol/L, 96th-100th percentiles) were associated with an increased risk of cardiovascular and all-cause mortality compared to Lp(a) < 10 mg/dL (18 nmol/L, 1st-50th percentiles), with a median survival reduction of 1.2 years (33). For every 50 mg/dL (105 nmol/L) increase in Lp(a), the observational HR for cardiovascular mortality was 1.16, while the genetic risk was 1.23 (based on LPA KIV-2) and 0.98 (based on LPA rs10455872). This suggests that elevated Lp(a) levels increase mortality risk primarily through a lower number of LPA KIV-2 repeats rather than cholesterol content. Furthermore, in a systematic review and meta-analysis conducted by Amiri et al., which included 75 studies with a total of 957,253 participants, the association between Lp(a) and mortality risk was investigated (19). The results showed that, in both the general population and CVD patients, the highest Lp(a) level group had all-cause mortality risk ratios of 1.09 and 1.18, respectively. Elevated Lp(a) levels were also associated with an increased risk of CVD-related mortality, a trend that was evident in the general population, CVD patients, and individuals with diabetes. For every 50 mg/dL increase in Lp(a), the risk of CVD-related mortality increased by 31% in the general population and by 15% in CVD patients. These findings support the European Society of Cardiology (ESC) and European Atherosclerosis Society (EAS) guidelines, which recommend that all adults measure Lp(a) at least once to assess mortality risk. Additionally, Wohlfahrt et al. examined 851 patients with acute myocardial infarction (AMI) and found a U-shaped relationship between Lp(a) levels and all-cause mortality (16). Compared to those with Lp(a) levels of 10–30 nmol/L, individuals with Lp(a) < 7 nmol/L and those with Lp(a) ≥ 125 nmol/L had an increased risk of mortality. Moreover, both low and high Lp(a) levels were associated with an increased risk of recurrent cardiovascular events, and this association was partially attenuated by heart failure-related factors. Another study not only identified an association between Lp(a) and all-cause and cardiovascular mortality in AMI patients but also demonstrated that the combination of Lp(a) and high-sensitivity C-reactive protein (Hs-CRP) provided a better prediction of mortality risk (34). This predictive value of Lp(a) in combination with other biomarkers has also been validated in the general population, where Lp(a) was found to be independently associated with both all-cause and cardiovascular mortality. Additionally, when combined with fibrinogen, Lp(a) provided an even stronger predictive value for higher mortality risk compared to either marker alone (35). Furthermore, Kim et al. conducted a cohort study involving 275,430 Korean adults and found that high Lp(a) levels were associated with an increased risk of all-cause and cardiovascular mortality (36). After a follow-up period of 6.6 years, individuals with Lp(a) ≥ 50 mg/dL had a significantly higher risk of cardiovascular and all-cause mortality. Those with Lp(a) ≥ 100 mg/dL had a 2.45-fold increased risk of cardiovascular mortality compared to those with Lp(a) < 10 mg/dL. This association was independent of LDL-C and was only influenced by HDL-C levels, suggesting that Lp(a) is an independent risk factor for cardiovascular mortality in the Korean population. Beyond baseline Lp(a) levels, persistently high Lp(a) levels over long-term follow-up have also been strongly associated with mortality. For example, in a cohort study of 1,131 AMI patients, participants were categorized into four groups based on Lp(a) levels at admission and after one year: persistently low, increased, decreased, and persistently high Lp(a) levels (37). Over a median follow-up period of 50 months, the results indicated that, compared to the persistently low Lp(a) group, the persistently high Lp(a) group had significantly increased risks of major adverse cardiovascular and cerebrovascular events (MACCE), nonfatal stroke, unplanned revascularization, all-cause mortality, and cardiovascular mortality. This study demonstrated that persistently high Lp(a) levels in AMI patients were closely associated with an increased risk of MACCE, stroke, revascularization, and mortality.
Despite these compelling findings highlighting the strong association between Lp(a) and mortality risk, our study offers several distinct advantages compared to previous research. First, we utilized RCS analysis to examine the dose-response relationship between Lp(a) levels and mortality risk, confirming the linear nature of this association. This approach provides a clearer and more quantitative basis for the clinical application of Lp(a). Second, we selected 300 mg/L as the threshold for Lp(a) group division based on the upper reference limit commonly used in Chinese clinical laboratories (0-300 mg/L), which has also been widely adopted in domestic studies (38–40). In addition, we conducted a supplementary analysis using 500 mg/L, a widely recognized international cut-off value, and found that Lp(a) > 500 mg/L was significantly associated with increased cardiovascular mortality, further supporting the clinical relevance of this threshold. However, due to the retrospective nature of this study and incomplete clinical records, we were unable to accurately identify specific subgroups at extremely high cardiovascular risk, such as those with multivessel disease, peripheral artery disease, or familial hypercholesterolemia, and therefore could not perform further stratified analyses in these populations. Considering the greater vulnerability of these individuals to adverse outcomes, future prospective studies incorporating more detailed baseline data are warranted to clarify the prognostic significance of Lp(a) in this particularly high-risk group. In addition, current guidelines emphasize the need for intensified lipid-lowering therapy in extremely high-risk individuals (41–43). Although specific Lp(a)-targeted treatments are not yet widely available, such patients may benefit from aggressive LDL-C control through high-intensity statins, or ezetimibe. As Lp(a) contributes residual risk beyond traditional lipids, incorporating Lp(a) assessment into risk stratification could help identify those who may require more comprehensive therapeutic strategies. Third, our study was conducted in a cohort of Chinese ACS patients, filling a critical gap in the literature regarding the association between Lp(a) and all-cause and cardiovascular mortality in this population. This enhances the generalizability and relevance of our findings to Chinese patients. Fourth, we performed detailed subgroup and sensitivity analyses, further validating the robustness of our results and identifying specific high-risk populations, such as elderly males and individuals without comorbidities, who may be at an even greater risk of Lp(a)-related mortality. Moreover, due to the wide numerical range of Lp(a) concentrations, the risk increment associated with each 1 mg/dL increase is relatively small. Thus, although Lp(a) as a continuous variable shows statistically significant associations with mortality, the effect size per unit appears modest. To better reflect its clinical relevance, we additionally performed a standardized analysis, and the results indicated that a one standard deviation increase in Lp(a) was significantly associated with increased risks of both all-cause and cardiovascular mortality. This suggests that Lp(a) may have greater value in risk stratification when evaluated using standardized values or categorized by clinically meaningful thresholds to help identify individuals at higher risk. In summary, our study builds upon existing literature and further refines the clinical significance of Lp(a). These findings provide valuable insights for future precision medicine strategies aimed at optimizing risk assessment and management in ACS patients. And these results also support current international guideline recommendations advocating for at least one lifetime measurement of Lp(a), and suggest that such recommendations may also be applicable and beneficial in the Chinese ACS population (44). In addition, recent studies have further emphasized the importance of Lp(a) in CVD prevention and management (45, 46). For example, a review by Sosnowska et al. published in 2025 highlighted that Lp(a) is one of the current focal points in cardiovascular research (46). It not only contributes to the process of atherosclerosis but is also closely associated with aortic valve stenosis, inflammation, and thrombosis. The review emphasized that Lp(a) measurement should be incorporated into routine cardiovascular risk assessment and that the development of targeted Lp(a)-lowering therapies should be actively pursued. Future updates to local or regional clinical guidelines may consider integrating Lp(a) assessment as part of standard care for ACS patients.
Lp(a) may influence mortality risk in patients with ACS through multiple biological mechanisms. First, Lp(a) promotes atherosclerosis and thrombosis by facilitating atherosclerotic plaque formation through its cholesterol-rich LDL-like structure, while its Apo(a) component, which resembles plasminogen, competitively inhibits the fibrinolytic system, thereby enhancing thrombotic potential and increasing the risk of ACS recurrence (47–49). Second, Lp(a)-mediated inflammatory responses exacerbate cardiovascular damage by activating the monocyte-macrophage system, which stimulates the release of inflammatory cytokines such as interleukin-6 (IL-6) and tumor necrosis factor-alpha (TNF-α), thereby accelerating vascular inflammation and atherosclerosis progression (50, 51). Additionally, Lp(a) is closely associated with aortic valve and coronary artery calcification, microcirculatory dysfunction, and left ventricular hypertrophy, with elevated levels contributing to increased coronary plaque instability, impaired ventricular contractile function, reduced ejection capacity, and valvular regurgitation, ultimately heightening the susceptibility of ACS patients to sudden cardiac death and increasing long-term mortality risk (52–55). Overall, Lp(a) plays a crucial role in coagulation, inflammation, vascular injury, calcification, and myocardial hypertrophy, which may explain its strong association with poor prognosis in ACS patients.
Although this study provides important findings, certain limitations should be acknowledged. First, as a single-center retrospective study, there is a potential for selection bias, and the generalizability of the results needs further validation through large-scale, multicenter studies. Second, this study only measured baseline Lp(a) levels and did not assess the impact of temporal variations in Lp(a) on ACS prognosis. Although Lp(a) levels are relatively stable, they may still be influenced by acute inflammation or liver function changes in the short term. Future studies should consider dynamic monitoring of Lp(a) to more accurately evaluate its long-term effects. Third, despite adjusting for multiple confounders, residual confounding may still exist, as factors such as genetic polymorphisms and Lp(a) particle size were not included in the analysis and may have influenced the results. In addition, although comorbidities such as diabetes, hypertension, and CKD were included as covariates, they were defined only as binary variables. Detailed information on severity, duration, and control (such as blood pressure, blood glucose, and dynamic renal function) was not available. However, due to the retrospective nature of the study and reliance on historical electronic medical records, these data were largely missing. As a result, we were unable to incorporate more nuanced indicators of disease status, which may have limited the granularity of the multivariate models. Fourth, information on post-discharge treatment and medication adherence was not available due to limitations in retrospective data collection. Since long-term outcomes such as mortality are significantly affected by ongoing pharmacologic management—especially in ACS patients—this may introduce a potential source of bias. We recommend that future prospective studies include systematic follow-up of medication use to improve the accuracy and clinical relevance of outcome assessment. Fifth, although we performed comprehensive cardiovascular mortality analyses, the study did not incorporate competing risk models (such as accounting for non-cardiovascular death), which may be particularly relevant in older populations. Future research using competing risk approaches, such as Fine-Gray models, may help improve the accuracy and robustness of risk estimation. Lastly, given the lack of specific therapeutic interventions aimed at lowering Lp(a) levels, the findings of this study are primarily applicable to risk prediction rather than guiding treatment strategies. Further research is needed to explore whether lowering Lp(a) can improve clinical outcomes in ACS patients.
5 Conclusions
This study confirms a significant linear positive correlation between higher Lp(a) levels and all-cause and cardiovascular mortality in patients with ACS, with this association being more pronounced in specific subgroups, such as elderly males and those without comorbidities. The findings not only support Lp(a) as an independent prognostic predictor for ACS but also highlight the need for enhanced monitoring and management of patients with elevated Lp(a) levels in clinical practice. Future research should further explore the potential clinical benefits of Lp(a)-lowering therapies and integrate genetic studies to elucidate its biological mechanisms.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by Ethics Committee of Henan Provincial People’s Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
KQ: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. TZ: Conceptualization, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Bhatt DL, Lopes RD, and Harrington RA. Diagnosis and Treatment of Acute Coronary Syndromes: A Review [published correction appears in JAMA. 2022 May 03;327(17):1710. doi: 10.1001/jama.2022.6185. JAMA. (2022) 327:662–75. doi: 10.1001/jama.2022.0358
2. Breen K, Finnegan L, Vuckovic K, Fink A, Rosamond W, and DeVon HA. Multimorbidity in patients with acute coronary syndrome is associated with greater mortality, higher readmission rates, and increased length of stay: A systematic review. J Cardiovasc Nurs. (2020) 35:E99–E110. doi: 10.1097/JCN.0000000000000748
3. Bueno H, Rossello X, Pocock SJ, Van de Werf F, Chin CT, Danchin N, et al. In-hospital coronary revascularization rates and post-discharge mortality risk in non-ST-segment elevation acute coronary syndrome. J Am Coll Cardiol. (2019) 74:1454–61. doi: 10.1016/j.jacc.2019.06.068
4. Gong W, Yan Y, Liu J, Wang X, Zheng W, Que B, et al. In-hospital mortality and treatment in patients with acute coronary syndrome with and without standard modifiable cardiovascular risk factors: findings from the CCC-ACS project. J Am Heart Assoc. (2024) 13:e029252. doi: 10.1161/JAHA.122.029252
5. Wei C, Zhu ZW, Hu XQ, and Zhou SH. Incidence, predictors and associations between in-hospital bleeding and adverse events in patients with acute coronary syndrome above 75 years of age – the real-world scenario. Cardiovasc Innov Appl. (2023) 8:1–10. doi: 10.15212/CVIA.2023.0029
6. Xie JX, Gunzburger EC, Kaun L, Plomondon ME, Barón AE, Waldo SW, et al. Medical therapy utilization and long-term outcomes following percutaneous coronary intervention: five-year results from the veterans affairs clinical assessment, reporting, and tracking system program. Circ Cardiovasc Qual Outcomes. (2019) 12:e005455. doi: 10.1161/CIRCOUTCOMES.118.005455
7. Bainey KR, Alemayehu W, Armstrong PW, Westerhout CM, Kaul P, and Welsh RC. Long-term outcomes of complete revascularization with percutaneous coronary intervention in acute coronary syndromes. JACC Cardiovasc Interv. (2020) 13:1557–67. doi: 10.1016/j.jcin.2020.04.034
8. Pruc M, Kubica J, Banach M, Świeczkowski D, Rafique Z, Peacock WF, et al. Prognostic value of the monocyte-to-high-density lipoprotein-cholesterol ratio in acute coronary syndrome patients: A systematic review and meta-analysis. Kardiol Pol. (2025) 83:52–61. doi: 10.33963/v.phj.102773
9. Zhen L, Chen XH, Fan JY, Wang X, Ai H, Que B, et al. Prognostic value of neutrophil-to-lymphocyte ratio for patients with acute coronary syndrome and obstructive sleep apnea. Cardiovasc Innov Appl. (2024) 9:1–11. doi: 10.15212/CVIA.2024.0016
10. van der Stouwe JG, Godly K, Kraler S, Godly J, Matter CM, Wenzl FA, et al. Body temperature, systemic inflammation and risk of adverse events in patients with acute coronary syndromes. Eur J Clin Invest. (2024) 54:e14314. doi: 10.1111/eci.14314
11. Tsimikas S. A test in context: lipoprotein(a): diagnosis, prognosis, controversies, and emerging therapies. J Am Coll Cardiol. (2017) 69:692–711. doi: 10.1016/j.jacc.2016.11.042
12. Koschinsky ML and Marcovina SM. Structure-function relationships in apolipoprotein(a): insights into lipoprotein(a) assembly and pathogenicity. Curr Opin Lipidol. (2004) 15:167–74. doi: 10.1097/00041433-200404000-00009
13. de Boer LM, Oorthuys AOJ, Wiegman A, Langendam MW, Kroon J, Spijker R, et al. Statin therapy and lipoprotein(a) levels: a systematic review and meta-analysis. Eur J Prev Cardiol. (2022) 29:779–92. doi: 10.1093/eurjpc/zwab171
14. Volgman AS, Koschinsky ML, Mehta A, and Rosenson RS. Genetics and pathophysiological mechanisms of lipoprotein(a)-associated cardiovascular risk. J Am Heart Assoc. (2024) 13:e033654. doi: 10.1161/JAHA.123.033654
15. Duarte Lau F and Giugliano RP. Lipoprotein(a) and its Significance in Cardiovascular Disease: A Review [published correction appears in JAMA Cardiol. 2022 Jul 1;7(7):776. doi: 10.1001/jamacardio.2022.2074. JAMA Cardiol. (2022) 7:760–9. doi: 10.1001/jamacardio.2022.0987
16. Leistner DM, Laguna-Fernandez A, Haghikia A, Abdelwahed YS, Schatz AS, Erbay A, et al. Impact of elevated lipoprotein(a) on coronary artery disease phenotype and severity [published correction appears in Eur J Prev Cardiol. 2024 May 11;31(7):915. doi: 10.1093/eurjpc/zwae104. Eur J Prev Cardiol. (2024) 31:856–65. doi: 10.1093/eurjpc/zwae007
17. Arsenault BJ and Kamstrup PR. Lipoprotein(a) and cardiovascular and valvular diseases: A genetic epidemiological perspective. Atherosclerosis. (2022) 349:7–16. doi: 10.1016/j.atherosclerosis.2022.04.015
18. Emerging Risk Factors Collaboration, Erqou S, Kaptoge S, Perry PL, Di Angelantonio E, Thompson A, et al. Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. JAMA. (2009) 302:412–23. doi: 10.1001/jama.2009.1063
19. Amiri M, Raeisi-Dehkordi H, Verkaar AJCF, Wu Y, van Westing AC, Berk KA, et al. Circulating lipoprotein (a) and all-cause and cause-specific mortality: a systematic review and dose-response meta-analysis. Eur J Epidemiol. (2023) 38:485–99. doi: 10.1007/s10654-022-00956-4
20. Wohlfahrt P, Jenča D, Melenovský V, Franeková J, Jabor A, Šramko M, et al. Very low lipoprotein(a) and increased mortality risk after myocardial infarction. Eur J Intern Med. (2021) 91:33–9. doi: 10.1016/j.ejim.2021.04.012
21. Wang ZW, Li M, Li JJ, and Liu NF. Association of lipoprotein(a) with all-cause and cause-specific mortality: A prospective cohort study. Eur J Intern Med. (2022) 106:63–70. doi: 10.1016/j.ejim.2022.09.010
22. Whelton PK, Carey RM, Aronow WS, Casey DE Jr, Collins KJ, Dennison Himmelfarb C, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines [published correction appears in Hypertension. 2018 Jun;71(6):e136-e139. doi: 10.1161/HYP.0000000000000075.] [published correction appears in Hypertension. 2018 Sep;72(3):e33. doi: 10.1161/HYP.0000000000000080. Hypertension. (2018) 71:1269–324. doi: 10.1161/HYP.0000000000000066
23. American Diabetes Association. 2. Classification and diagnosis of diabetes: standards of medical care in diabetes-2019. Diabetes Care. (2019) 42:S13–28. doi: 10.2337/dc19-S002
24. Fang Y, Mei W, Wang C, Ren X, Hu J, Su F, et al. Dyslipidemia and hyperuricemia: a cross-sectional study of residents in Wuhu, China. BMC Endocr Disord. (2024) 24:2. doi: 10.1186/s12902-023-01528-7
25. Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2024 clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney Int. (2024) 105:S117–314. doi: 10.1016/j.kint.2023.10.018
26. Afzal Z, Cao H, Chaudhary M, Chigurupati HD, Neppala S, Alruwaili W, et al. Elevated lipoprotein(a) levels: A crucial determinant of cardiovascular disease risk and target for emerging therapies. Curr Probl Cardiol. (2024) 49:102586. doi: 10.1016/j.cpcardiol.2024.102586
27. Kamstrup PR. Lipoprotein(a) and cardiovascular disease. Clin Chem. (2021) 67:154–66. doi: 10.1093/clinchem/hvaa247
28. Marcovina SM. Lipoprotein(a): a genetically determined risk factor for Cardiovascular disease. Crit Rev Clin Lab Sci. (2023) 60:560–72. doi: 10.1080/10408363.2023.2229915
29. Tsioulos G, Kounatidis D, Vallianou NG, Poulaki A, Kotsi E, Christodoulatos GS, et al. Lipoprotein(a) and atherosclerotic cardiovascular disease: where do we stand? Int J Mol Sci. (2024) 25:3537. doi: 10.3390/ijms25063537
30. Lu J, Wang Z, Zhang J, Jiao F, Zou C, Han L, et al. Causal association of blood lipids with all-cause and cause-specific mortality risk: a Mendelian randomization study. J Lipid Res. (2024) 65:100528. doi: 10.1016/j.jlr.2024.100528
31. Arsenault BJ, Pelletier W, Kaiser Y, Perrot N, Couture C, Khaw KT, et al. Association of long-term exposure to elevated lipoprotein(a) levels with parental life span, chronic disease-free survival, and mortality risk: A Mendelian randomization analysis. JAMA Netw Open. (2020) 3:e200129. doi: 10.1001/jamanetworkopen.2020.0129
32. Simony SB, Mortensen MB, Langsted A, Afzal S, Kamstrup PR, and Nordestgaard BG. Sex differences of lipoprotein(a) levels and associated risk of morbidity and mortality by age: The Copenhagen General Population Study. Atherosclerosis. (2022) 355:76–82. doi: 10.1016/j.atherosclerosis.2022.06.1023
33. Langsted A, Kamstrup PR, and Nordestgaard BG. High lipoprotein(a) and high risk of mortality. Eur Heart J. (2019) 40:2760–70. doi: 10.1093/eurheartj/ehy902
34. Wang Z, Tang J, Shi Q, Fang L, Liu N, and Zhang J. Synergistic effect of lipoprotein(a) and high-sensitivity C-reactive protein on the risk of all-cause and cardiovascular death in patients with acute myocardial infarction: a large prospective cohort study. Front Endocrinol (Lausanne). (2024) 15:1392859. doi: 10.3389/fendo.2024.1392859
35. Wang Z, Yan X, Fang L, Tang J, and Zhang J. Association between lipoprotein(a), fibrinogen and their combination with all-cause, cardiovascular disease and cancer-related mortality: findings from the NHANES. BMC Public Health. (2024) 24:1927. doi: 10.1186/s12889-024-19443-4
36. Kim BJ, Lee MY, Choi HI, Kwon MJ, and Kang JG. Lipoprotein(a)-related cardiovascular and all-cause mortalities in Korean adults. Eur J Prev Cardiol. (2023) 30:308–17. doi: 10.1093/eurjpc/zwac271
37. Wang Z, Tang J, Shi Q, Fang L, Liu N, and Zhang J. Persistent lipoprotein(a) exposure and its association with clinical outcomes after acute myocardial infarction: a longitudinal cohort study. Ann Med. (2025) 57:2454975. doi: 10.1080/07853890.2025.2454975
38. Di Muro FM, Crociani MF, Nardi G, Ciardetti N, Biagiotti L, Bigi E, et al. Coronary plaque characteristics assessed by optical coherence tomography and plasma lipoprotein(a) levels in patients with acute coronary syndrome. Catheter Cardiovasc Interv. (2024). doi: 10.1002/ccd.31363
39. Calmarza P, Trejo JM, Lapresta C, and Lopez P. Relationship between lipoprotein(a) concentrations and intima-media thickness: a healthy population study. Eur J Prev Cardiol. (2012) 19:1290–5. doi: 10.1177/1741826711423216
40. Ma KL, Gong TK, Hu ZB, Zhang Y, Wang GH, Liu L, et al. Lipoprotein(a) accelerated the progression of atherosclerosis in patients with end-stage renal disease. BMC Nephrol. (2018) 19:192. doi: 10.1186/s12882-018-0986-2
41. Banach M, Reiner Ž, Surma S, Bajraktari G, Bielecka-Dabrowa A, Bunc M, et al. 2024 recommendations on the optimal use of lipid-lowering therapy in established atherosclerotic cardiovascular disease and following acute coronary syndromes: A position paper of the international lipid expert panel (ILEP). Drugs. (2024) 84:1541–77. doi: 10.1007/s40265-024-02105-5
42. Dyrbuś K, Gąsior M, Penson PE, and Banach M. Extreme cardiovascular risk-do we need a new risk category? Eur Heart J. (2022) 43:1784–6. doi: 10.1093/eurheartj/ehab771
43. Ray KK, Reeskamp LF, Laufs U, Banach M, Mach F, Tokgözoğlu LS, et al. Combination lipid-lowering therapy as first-line strategy in very high-risk patients. Eur Heart J. (2022) 43:830–3. doi: 10.1093/eurheartj/ehab718
44. Kronenberg F, Mora S, and Stroes ESG. Consensus and guidelines on lipoprotein(a) - seeing the forest through the trees. Curr Opin Lipidol. (2022) 33:342–52. doi: 10.1097/MOL.0000000000000855
45. Dyrbuś K, Mędrala Z, Konsek K, Nowowiejska-Wiewióra A, Trzeciak P, Skrzypek M, et al. Lipoprotein(a) and its impact on cardiovascular disease - the Polish perspective: design and first results of the Zabrze-Lipoprotein(a) Registry. Arch Med Sci. (2024) 20:1069–76. doi: 10.5114/aoms/188294
46. Sosnowska B, Toth PP, Razavi AC, Remaley AT, Blumenthal RS, and Banach M. 2024: the year in cardiovascular disease - the year of lipoprotein(a) – research advances and new findings. Arch Med Sci. (2025) 21:355–73. doi: 10.5114/aoms/202213
47. Boffa MB and Koschinsky ML. Lipoprotein (a): truly a direct prothrombotic factor in cardiovascular disease? J Lipid Res. (2016) 57:745–57. doi: 10.1194/jlr.R060582
48. Ugovšek S and Šebeštjen M. Lipoprotein(a)-the crossroads of atherosclerosis, atherothrombosis and inflammation. Biomolecules. (2021) 12:26. doi: 10.3390/biom12010026
49. Rehberger Likozar A, Zavrtanik M, and Šebeštjen M. Lipoprotein(a) in atherosclerosis: from pathophysiology to clinical relevance and treatment options. Ann Med. (2020) 52:162–77. doi: 10.1080/07853890.2020.1775287
50. Reyes-Soffer G and Westerterp M. Beyond Lipoprotein(a) plasma measurements: Lipoprotein(a) and inflammation. Pharmacol Res. (2021) 169:105689. doi: 10.1016/j.phrs.2021.105689
51. Simantiris S, Antonopoulos AS, Papastamos C, Benetos G, Koumallos N, Tsioufis K, et al. Lipoprotein(a) and inflammation- pathophysiological links and clinical implications for cardiovascular disease. J Clin Lipidol. (2023) 17:55–63. doi: 10.1016/j.jacl.2022.10.004
52. Arsenault BJ, Loganath K, Girard A, Botezatu S, Zheng KH, Tzolos E, et al. Lipoprotein(a) and calcific aortic valve stenosis progression: A systematic review and meta-analysis. JAMA Cardiol. (2024) 9:835–42. doi: 10.1001/jamacardio.2024.1882
53. Qiu Y, Hao W, Guo Y, Guo Q, Zhang Y, Liu X, et al. The association of lipoprotein (a) with coronary artery calcification: A systematic review and meta-analysis. Atherosclerosis. (2024) 388:117405. doi: 10.1016/j.atherosclerosis.2023.117405
54. Sbrana F, Dal Pino B, Aquaro GD, Bigazzi F, Vergaro G, and Sampietro T. Lipoprotein(a) apheresis restores coronary microcirculation in refractory angina. Rev Port Cardiol. (2022) 41:437–9. doi: 10.1016/j.repc.2021.06.018
Keywords: lipoprotein(a), acute coronary syndrome, all-cause mortality, cardiovascular mortality, restricted cubic spline
Citation: Qin K and Zhang T (2025) Lipoprotein(a) and its linear association with all-cause and cardiovascular mortality in patients with acute coronary syndrome. Front. Endocrinol. 16:1580298. doi: 10.3389/fendo.2025.1580298
Received: 20 February 2025; Accepted: 12 May 2025;
Published: 27 May 2025.
Edited by:
Maciej Banach, Medical University of Lodz, PolandReviewed by:
Weichieh Lee, Chi Mei Medical Center, TaiwanArianna Toscano, University Hospital of Policlinico G. Martino, Italy
Copyright © 2025 Qin and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ke Qin, c2p0dXFrZUAxNjMuY29t