 Xueqian Mao
Xueqian Mao Chaoming Mao
Chaoming Mao Jiameng Liu
Jiameng Liu Xi Wang1
Xi Wang1- 1Department of Nuclear Medicine, The Affiliated Hospital of Jiangsu University, Zhenjiang, China
- 2Department of Ultrasound Medicine, The Affiliated Hospital of Jiangsu University, Zhenjiang, China
The expanding clinical utilization of immune checkpoint inhibitors (ICIs) in oncology has brought increasing attention to thyroid dysfunction as a prominent immune-related adverse event (irAE). Elucidating the pathophysiological mechanisms underlying ICI-induced thyroiditis represents a critical step toward developing evidence-based diagnostic protocols and targeted therapeutic interventions for cancer patients undergoing immunotherapy. This comprehensive review systematically examines current advances in understanding the etiopathogenesis of ICI-induced thyroiditis. First, we described pharmacological characterization of ICIs, then discussed multifactorial analysis of cellular and molecular contributors to thyroid autoimmunity following ICI administration and finally analyzed critical evaluation of emerging hypotheses regarding primary pathogenic drivers. Through this review, we aim to establish mechanistic connections between ICI pharmacodynamics and thyroid tissue immunopathology.
Introduction
The therapeutic integration of immune checkpoint inhibitors (ICIs) in oncology has transformed clinical outcomes for diverse malignancies through enhanced T-cell-mediated antitumor immunity via blockade of inhibitory checkpoints including programmed cell death protein-1 (PD-1)/PD-1 ligand (PD-L1) and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4). Paradoxically, this immunomodulatory efficacy coincides with the breakdown of peripheral tolerance mechanisms, manifesting as multi-organ immune-related adverse events (irAEs) (1, 2). Current studies reveal differential irAE profiles demonstrating tumour-type specificity, temporal dependency on treatment duration, and immune microenvironmental modulation patterns (3). Clinically significant irAEs predominantly affect immune-privileged organs including the integumentary system, endocrine axes, gastrointestinal tract, pulmonary parenchyma, and hepatic tissue (4, 5). Particularly in patients treated with ICIs, the incidence of thyroiditis ranges from 10% to 20% (6). The clinical features of ICI-induced thyroiditis are predominantly characterized by destructive thyroiditis, which can lead to transient thyrotoxicosis, followed by the development of hypothyroidism (5, 7). The pathogenesis of thyroiditis may be related to the immune system’s attack on thyroid tissue, which is often associated with the loss of immune tolerance induced by ICIs. Thus, exploring the underlying mechanism of ICI-induced thyroiditis is of particular importance. Through analyzing the characteristics of the effects of ICIs on the immune system, the mechanisms of the autoimmune tendency of the thyroid gland, and the impacts of immune cells, immune molecules, and genetic factors on thyroiditis, we can provide a new theoretical basis and therapeutic guidance for clinical practice. This will enable better management of the side effects induced by ICIs. Clinically, ICI therapy should be discontinued only when symptomatic thyrotoxicosis is present, and long-term levothyroxine replacement therapy should be initiated for persistent hypothyroidism (8) (Figure 1).

Figure 1. Clinical features and incidence of immune checkpoint inhibitor (ICI)-induced thyroiditis. The clinical features of ICI-induced thyroiditis are thyrotoxicosis caused by destruction of thyroid follicular cells (TFC), followed by a decline in thyroid hormones and development of hypothyroidism.
Characteristics of the ICIs and irAEs
Immune checkpoints play a crucial role in the immune system by regulating antigen recognition by the T-cell receptor (TCR). These checkpoints encompass both inhibitory and stimulatory immune checkpoint molecules (9, 10). They modulate the intensity of the immune response and uphold immune tolerance, thereby safeguarding normal tissues from the detrimental effects of the immune response (11). During the process of tumor immune escape, inhibitory immune checkpoints have particularly emerged as the focal point of cancer immunotherapy research. Common inhibitory immune checkpoints include PD-1, PD-L1, and CTLA-4 targets (12).
PD-1 is a type I transmembrane protein that belongs to the CD28 immunoglobulin superfamily. It is mainly expressed on tumor-infiltrating activated T lymphocytes, B lymphocytes, natural killer cells, and other immune cells (13). PD-L1 is broadly expressed on tumor cells, antigen-presenting cells (APC), and stromal cells within the tumor microenvironment, with its expression regulated by Interferon-gamma (IFN-γ). In contrast, PD-L2 is predominantly expressed in dendritic cells (DCs) and pulmonary macrophages (14). The interaction between PD-1 and PD-L1 effectively inhibits T-cell activation and it can even induce apoptosis of T-cells and reduce cytokine production, thereby enabling tumor cells to evade immune surveillance (15). Through the use of PD-(L)1 blockers, the anti-tumor function of immune cells can be restored, and the immune escape of tumor cells can be circumvented (16).
CTLA-4 (CD152), a homologue of CD28, mainly exerts its function during T-cell activation (17, 18). CTLA-4 participates in the regulation of immune responses by downregulating the activity of CD4+ T effector cells and enhancing the function of regulatory T (Treg) cells (19). In the initial phase of T-cell activation, naive T cells bind to major histocompatibility complex (MHC) molecules on APC via the TCR. Meanwhile, CTLA-4 binds to CD80 and CD86 ligands on APC with a higher affinity than CD28 does, thereby inhibiting the activation signal of T cells (20). Blocking CTLA-4 in the tumor microenvironment potentiates T-cell activation, leading to stronger anti-tumor responses driven by CD8+ T cells (19, 21).
The blockade of PD-1 and CTLA-4 demonstrates significant differences in the autoimmune phenotype. CTLA-4 inhibitors primarily act in the early stage of the immune response, influencing the initiation phase of T cells in the lymph nodes. In contrast, PD-(L)1 inhibitors function in the effector phase (22). Inhibition of the PD-1 pathway typically leads to alterations in effector cell properties, presenting as a mild and chronic autoimmune phenotype. Conversely, inhibition of the CTLA-4 pathway triggers both intrinsic changes in effector cells and extrinsic changes in Foxp3+ T cells, resulting in a more severe non-specific autoimmune phenotype (23). This variability has been validated in irAEs generated by ICI treatment of tumors. Thyroiditis can be induced by both combination therapies (PD-1 inhibitors combined with CTLA-4 inhibitors) and monotherapies (PD-(L)1 inhibitors). The incidence rate is highest with combination therapy and lowest with CTLA-4 inhibitors (5, 6, 24), This suggests that differences in the mechanism of action of ICIs are associated with the varied incidence of ICI-induced thyroiditis. This raises an important question: why does the thyroid gland demonstrate particular vulnerability to immune checkpoint blockade? While the preceding discussion outlines the pharmacological basis of ICIs, the following analysis would elucidate the unique immunological characteristics of thyroid tissue that make it susceptible to autoimmune attack during checkpoint inhibition.
Common mechanisms of thyroid initiating autoimmune tendency
As a crucial endocrine organ, the thyroid gland exhibits a high incidence in irAEs and autoimmune diseases, including Hashimoto’s thyroiditis (HT) and Graves’ disease (GD). While there is no single definitive explanatory mechanism for its susceptibility, multiple factors interact to contribute to the development of thyroid autoimmunity. The following section examines three intrinsic characteristics of thyroid tissue that predispose it to autoimmune dysregulation during ICI therapy.
Thyroid-specific proteins as autoantigens
Functionally specific proteins in thyroid tissues, such as the thyroid-stimulating hormone receptor (TSHR), thyroid peroxidase (TPO), and thyroglobulin (Tg), possess unique biological characteristics, such as size, abundance, membrane-binding properties, glycosylation patterns, and polymorphisms, which may disrupt immunological tolerance (25). Clinical observations indicate that when the thyroid’s physiological structural barriers are disrupted by physicochemical factors or infection, function-specific proteins may be released in an “ectopic” manner. This abnormal release can then trigger the autoimmune response. Although the inherently limited central tolerance mechanisms typically eliminate most autoreactive cells, peripheral tolerance remains critical for maintaining immune protection in these organs, as evidenced by studies in animal models and humans with autoimmune disorders (26).
Immunological sensitivity and tertiary lymphoid structures
Thyroid tissue demonstrates a high level of immunological sensitivity. The observed effects may partly stem from antigen-driven tertiary lymphoid structures (TLSs) that emerge and sustain within pathological microenvironments (27, 28). TLSs are lymphoid aggregates that form postnatally in inflamed, infected, or neoplastic tissues (29). The formation of TLSs depends on the presence of antigens, and their development and persistence are driven by antigen exposure, with subsequent resolution upon antigen clearance (30). Currently, TLSs have garnered significant attention in tumor progression, immune therapy response (31) and irAEs (32). Studies have shown that ICIs can induce and enhance TLSs functionality (33). Although this intervention may improve tumor progression, it could simultaneously heighten immune sensitivity in thyroid tissue.
Role of thyroid cells in innate immunity
Thyroid follicular cells (TFC) function not only as key effectors of endocrine activity but also as active participants in innate immunity. TFC recognize and react to pathogen-associated molecular patterns (PAMPs) via pattern recognition receptors (PRRs) and also respond to damage-associated molecular patterns (DAMPs) resulting from cellular injury. In an autoimmune context, the response of thyroid cells to these patterns can trigger an up-regulation of inflammatory cytokines such as tumor necrosis factor-alpha (TNF-α), Interleukin(IL)-1, and IL-6, which promote specific immune responses (34). Moreover, TFC which are common targets of autoimmune attacks, also exhibit APC-like cell functionality (35, 36). In the study of autoimmune thyroid disease (AITD), cytokines such as IFN-γ induce TFC to express MHC-II molecules, which present thyroid autoantigens to CD4+ T cells, thereby breaking the immune tolerance of the host (37). Recent single-cell transcriptomic studies of GD have revealed that TFC abnormally express a full set of human leukocyte antigen (HLA) molecules, along with elevated expression of CD40 (36). CD40 expression is a characteristic feature of APC (38). However, TFC lack expression of the key costimulatory molecules CD80 and CD86, which are required for T cell activation (36).
Mechanisms of ICI-induced immune-associated thyroiditis
Although cancer immunotherapy has become a major focus of research, the underlying mechanisms responsible for ICI-induced thyroiditis remain inadequately elucidated (39). The cytopathology of ICI-induced thyroiditis exhibits distinct characteristics, such as large clusters of necrotic cells, lymphocytes, and CD163+ histiocytes (40). The rapid onset of ICI-induced thyroiditis, which differs markedly from typical cases like HT, suggests that the mechanism underlying ICI-induced thyroiditis is multifaceted and intricate. It involves multiple components of the immune system, including antigenic cross-reactivity, T-cell subsets and B cell activation, cytokine and chemokine activity and genetic susceptibility (Figure 2).
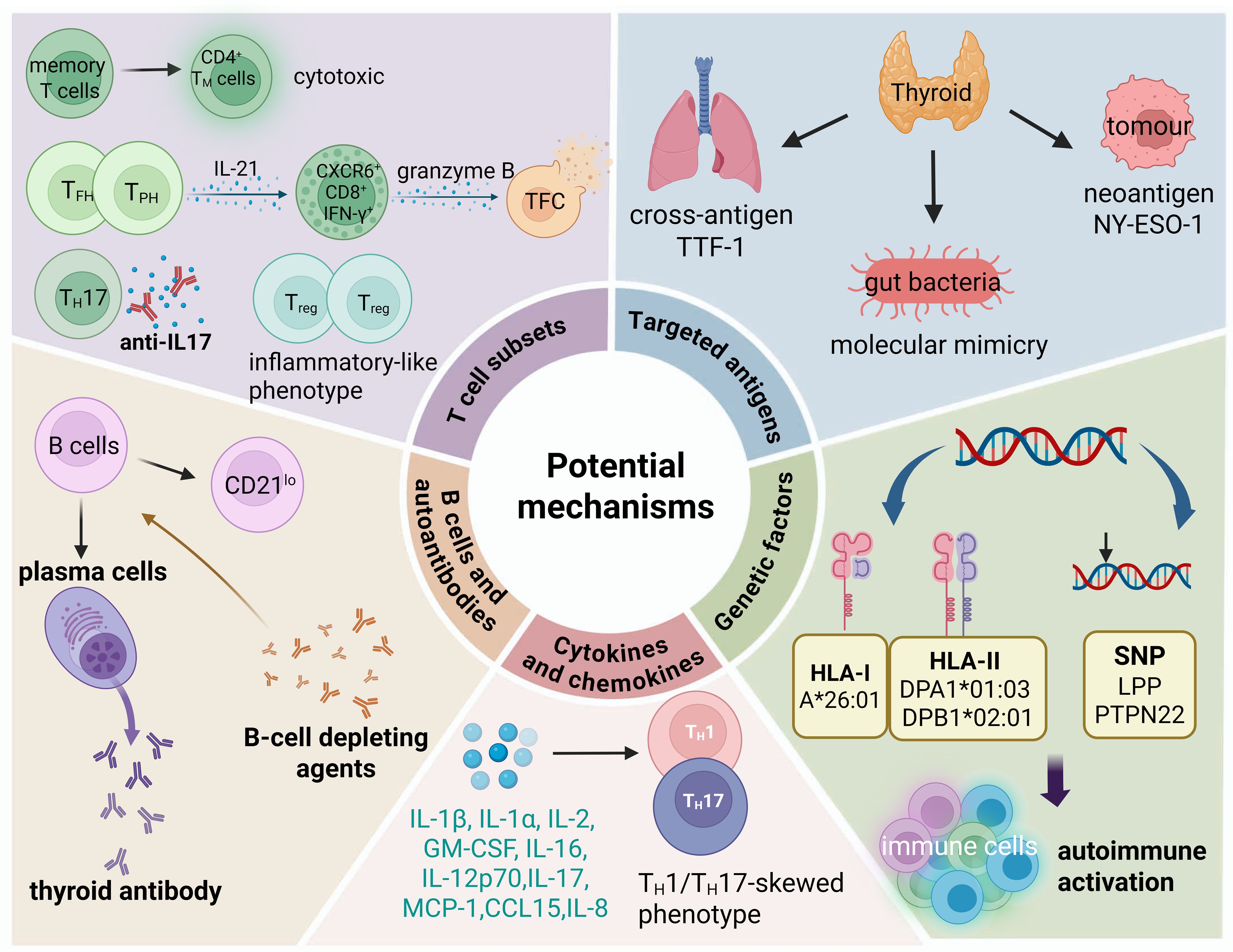
Figure 2. Possible pathogenic mechanisms of ICI-induced thyroiditis. Potential mechanisms include shared antigens, activation and expansion of memory T cells and effector T cells, B cells and autoantibodies, cytokine and chemokine activity, and genetic susceptibility.
Targeted antigens
The initiation of specific autoimmune responses in irAEs is mediated by T-cell clones that recognize cross-antigens present in both normal and tumor tissues (41, 42). For instance, the existence of cross-antigens encoded by the MYH6 gene between cardiomyocytes and tumor cells serves as an autoantigen targeted by CD8+ T cells in ICI-induced myocarditis (43). Berner et al. (44) compared peripheral blood mononuclear cells (PBMCs), tumor biopsy specimens, and biopsies from sites of immune-associated skin toxicity in non-small cell lung cancer patients treated with anti-PD-1 through T-cell receptor sequencing (TCRseq). They identified nine identical T-cell antigens shared by tumor tissues and skin, which were capable of stimulating CD8+ and CD4+ T cells in vitro (44). In tissue TCRseq studies of colitis and pneumonia irAEs, identical T-cell clones were detected at the lesion site of the irAEs and at the site of tumor origin (45, 46). However, there are few reports on ICI-induced thyroiditis-related antigens. One study found that in non-small cell lung cancer (NSCLC) patients treated with anti-PD-1 therapy, the expression of thyroid transcription factor-1 (TTF-1) in tumor tissue has been significantly correlated with both the incidence of ICI-induced thyroiditis and the clinical efficacy of ICI therapy (47). TTF-1 is a protein that is widely expressed in the lung and thyroid (48). The findings suggest that TTF-1 may function as a shared antigen between thyroid and lung cancer tissues, potentially contributing to the dual regulation of antitumor immune responses and thyroid autoimmunity. However, the observed association between TTF-1 expression and the incidence of ICI-induced thyroiditis has not yet been definitively attributed to antigenic cross-reactivity. Therefore, further investigation is needed to validate TTF-1 as a potential cross-reactive antigen.
Cancer neoantigens are aberrant peptide fragments arising from tumor-specific mutations that can be recognized by the immune system, thereby eliciting antigen-specific T cell responses directed against early-stage tumors. These neoantigens are detected by the immune system, initiating a tumor-specific immune response, and serve as pivotal targets in the development of cancer immunotherapeutic strategies (48–50). Notably, the potent anti-tumor immunity driven by neoantigens may be accompanied by the risk of autoimmune damage. Vita et al. (51) reported a case involving a 32-year-old female patient with synovial sarcoma who developed GD following administration of the NY-ESO-1 cancer vaccine. This case revealed that multiple epitope regions of the tumor neoantigen NY-ESO-1 exhibit sequence homology with thyroid autoantigens, including TSHR, Tg and TPO, suggesting a mechanism of cross-reactivity (51). Such cross-reactive immune responses may result in T cells targeting both tumor and thyroid tissues. Moreover, following the release of T cell inhibition by ICIs, neoantigen-activated T cells may initiate recognition of additional sequestered thyroid antigens through epitope spreading (49). Thus, the overlap between tumor neoantigens and thyroid autoantigens, coupled with ICI-driven immune activation, provides a plausible mechanism underlying immune-related thyroid dysfunction in cancer immunotherapy.
Emerging evidence indicates that dysregulation of the gut microbiota plays a pivotal role in the development of autoimmune diseases (52). ICI changes immune microenvironment of gut, consequently disrupt the gut microbial community, which can lead to the overactivation of both innate and adaptive immune responses in local tissues, ultimately resulting in systemic immune imbalance (53). Several mechanisms have been proposed to explain the gut microbiota–thyroid axis dysregulation, including molecular mimicry, microbial translocation due to compromised gut barrier integrity, and immune modulation triggered by microbial metabolites (53). Among these, molecular mimicry is increasingly recognized as a critical factor linking gut microbiota to autoimmune dysregulation (53). This mechanism involves structural similarities between microbial peptides and host antigens, which may lead to cross-reactive immune responses. Studies have suggested that vaccines and commensal microbes may influence the development of irAEs through molecular mimicry and related pathways (54). Associations between molecular mimicry and irAEs have been reported in multiple organ systems, including the heart and gastrointestinal tract (55). In a study exploring the mechanisms of gut microbiota’s effects on lung cancer, it was discovered that molecular mimicry might contribute to the development of irAEs and the efficacy of ICIs (54). Notably, certain strains of Lactobacillus and Bifidobacterium have been shown to induce the production of antibodies that cross-react with thyroid-specific antigens such as TPO and Tg, implicating microbial mimicry in thyroid autoimmunity (56, 57). Similarly, Yersinia enterocolitica in small intestinal colitis also exhibits molecular mimicry and may promote the generation and maturation of antibodies against the TSHR (58). Collectively, these findings suggest the overlap between gut microbiota and abundant autoimmune antigenic epitopes may play a part in the onset of irAEs.
The mechanisms of cross-antigenicity and molecular mimicry are involved in ICI-induced thyroiditis remains unclear. Antigens shared between tumors and healthy tissues are marked by high specificity and strong immunogenicity. Identifying cross-antigens not only aids in predicting autoimmune toxic effects but also uncovers potential targets for novel tumor antigens. Therefore, a key challenge lies in distinguishing neoantigens capable of eliciting potent tumor-specific immune responses from those that avoid inducing immune toxicity. Optimizing this balance is essential for improving the efficacy and safety of ICI therapies.
T-cell subsets
The over-stimulation of TCR signaling associated with ICIs can disrupt the maintenance of peripheral immune tolerance and trigger the activation of potentially autoreactive T-cell clones (59). The loss of T-cell tolerance and the subsequent expansion of autoreactive T-cell clones are among the mechanisms underlying irAEs (60). Given that ICIs exert their effects by enhancing T-cell activity through modulation of T-cell immune checkpoints, T-cell-mediated mechanisms are considered the principal drivers of irAEs. These mechanisms have been shown to involve the activation of targeted memory T cells, the clonal expansion and effector functions of CD8+ T cells, the proliferation of T helper (TH)17 cells, and the dysregulation of Treg cells.
Activation of CD4+ memory T cells
Memory T cells (TM) can be broadly categorized into three subsets: central memory T (TCM) cells, effector memory T (TEM) cells, and tissue-resident memory T (TRM) cells (61). Despite their significance in immune responses, the role of memory T cells in ICI-induced thyroiditis remains poorly understood, primarily due to the limitations inherent in diagnostic biopsies (62). TRM cells, particularly in mucosal barrier organs such as the colon and skin, can be reactivated upon ICIs, leading to inflammation (62). In a study by Lozano et al. (63), multi-omics analysis of early blood samples from melanoma patients revealed a correlation between the abundance of activated CD4+ TM cells and increased TCR diversity with the development of severe irAE. Moreover, PD-1, a checkpoint molecule highly expressed on resting TM cells, appears to play a crucial role in regulating TM cell activation (64). This suggests that ICIs may directly affect TM cells during the early stages of irAEs. Evidence from murine models supports this hypothesis. In these models, administration of PD-1 inhibitors following Tg immunization resulted in the infiltration of granzyme B-expressing CD4+ TEM cells within the thyroid, indicating their cytotoxic role in thyroid inflammation (65). Clinically, patients with ICI-induced thyroiditis were found to have significantly elevated levels of CD27+ CD4+ TEM cells in their PBMCs, compared to those without thyroid involvement (65). These findings collectively highlight the pathogenic role of activated TM cells in driving thyroid autoimmunity and suggest that they may serve as early biomarkers for ICI-induced thyroiditis.
Cytotoxic activity of CD8+ T cells
CD8+ T cells are well recognized for their direct cytotoxic activity against target cells, and their expansion and activation play pivotal roles in the development of irAEs, including ICI-induced thyroiditis (66, 67). Recent studies have revealed distinct immunological mechanisms underlying ICI-induced thyroiditis. Single-cell RNA (scRNAseq) sequencing analysis of thyroid specimens from patients with ICI-thyroiditis demonstrated a predominant infiltration of clonally expanded cytotoxic CD8+ T cells, particularly a subset characterized by CXCR6 expression (67). These CXCR6+CD8+ effector T cells exhibited elevated production of IFN-γ and granzyme B, suggesting their crucial role in thyroid tissue destruction (67). Importantly, IL-21, secreted by intrathyroidal T follicular helper (TFH) and T peripheral helper (TPH) cells, was identified as a key driver of this pathogenic CD8+ T cell differentiation (67). In vitro experiments confirmed that IL-21 stimulation promoted CD8+ T cells to acquire an activated effector phenotype, marked by increased CXCR6 expression, enhanced IFN-γ production, and elevated granzyme B levels, collectively contributing to thyroid toxicity (66). Based on this, it can be hypothesized that CXCR6+CD8+ T cells may constitute the main subset of effector CD8+ T cells that respond to irAEs. Additionally, Wu et al. (68) demonstrated in the in vitro experiment part, human normal thyroid cells (NTHY) treated with nivolumab (NIVO) and CD8+ T cells were cultured. Through a series of experiments, the key protein AKT1 was screened out, and it was found that NIVO could enhance the immune sensitivity of thyroid cells by downregulating AKT1-SKP2 pathway, thereby promoting the killing of thyroid cells by CD8+ T cells (68). However, within the context of this study, the in vivo experiment did not verify the killing effect of CD8+ T cells. In one of the previously mentioned mouse experiments, pre-depletion of CD4+ T cells could completely prevent thyroiditis, while depletion of CD8+ T cells could partially prevent thyroiditis. The result indicates that CD4+ T cells may assist CD8+ T cell activation, highlighting immune collaboration in ICI-induced thyroiditis.
Pro-inflammatory effect of TH17 cells
Recent evidence highlights the critical role of TH17 cells and their associated cellular and secreted components in the pathogenesis and progression of AITD and irAEs (69, 70). TH17 cells, characterized by their production of IL-17A, are recognized for their pro-inflammatory effects and have been implicated in various autoimmune conditions (71). A scRNAseq analysis of peripheral circulating T cells from patients with tumors treated with ICIs revealed a significant increase in the abundance of CD4+ TH17 cells in those who developed ICI-induced thyroiditis compared to control patients (70). This finding underscores the association between TH17 cell expansion and the development of thyroid autoimmune responses in the context of ICI therapies. In addition to clinical findings, studies in tumor-bearing non-obese diabetic (NOD) mouse models have further supported the involvement of TH17 cells in ICI-induced thyroiditis (72). Additionally, targeting the TH17 and γδT17 cell axis with interleukin-17A (IL-17A) may reduce irAEs without diminishing the antitumor efficacy of ICIs (72).
Targeting the TH17 cell axis, particularly through the inhibition of IL-17A, has emerged as a promising strategy to mitigate irAEs without compromising the efficacy of ICIs in tumor control. In a clinical trial involving stage IV melanoma patients who developed multiple concurrent irAEs, including myocarditis, colitis, and rash, the administration of anti-IL-17A therapy resulted in significant regression of these adverse events, alongside an improvement in patient symptoms (73). Importantly, the use of anti-IL-17A therapy did not impair the antitumor effects of ICIs, demonstrating its potential as an effective approach to managing immune-related toxicities while preserving therapeutic efficacy.
Dysfunction of Treg cells
The activation and proliferation of autoreactive T cells are considered crucial in the development of irAEs. Treg cells play a central role in suppressing the activation of peripheral autoreactive T cells. They maintain immune homeostasis by expressing immune checkpoints that can inhibit the activation and function of other leukocytes (74, 75). Grigoriou et al. (76) discovered that in the peripheral blood of advanced melanoma patients who developed irAEs (including those with ICI-induced thyroiditis) after anti-PD-1 treatment, CD4+CD25+CD127+ Treg cells are expanded and highly express PD-1 and CTLA-4. Moreover, the peripheral Treg cells exhibit characteristic inflammatory transcriptional products, such as IFNG, STAT1, RORC and STAT3. It is evident that Treg cells exhibit an inflammatory-like phenotype in irAEs. Their stability and immunosuppressive function can be maintained through a feedback increase mechanism. This suggests that Treg cells are intricately involved in the immune response related to irAEs and may be a key factor in modulating the occurrence and development of these adverse events.
B cells proliferation and autoantibodies production
Beyond T cell-mediated mechanisms, emerging evidence highlights the critical involvement of B cells and autoantibodies in irAEs, positioning them as predictive biomarkers (77–79). Studies have demonstrated that early reductions in circulating B cells, alongside elevations in CD21lo B cells and plasma cells, correlate with irAE development (80). Moreover, the severity of the early decline in B cell counts following treatment is directly correlated with both the duration of toxicity episodes and the maximum toxicity grade (80). CD21lo subset, a unique memory B cell population implicated in plasma cell differentiation, may serve as an early predictor of ICI-induced thyroiditis (81). Consequently, early changes in CD21lo B cells may be able to act as one of the predictors of ICI-induced thyroiditis. The aforementioned scRNA-seq analyses of thyroid tissues from ICI-induced thyroiditis patients revealed infiltrating TFH and TPH cells driving thyroid cell destruction (67). Given the established presence of ectopic GCs in GD and HT (82, 83), it is plausible that similar GC-driven antibody production occurs in ICI-associated thyroid inflammation. Furthermore, clinical trials combining B-cell-depleting agents (e.g., rituximab) with ICIs reported reduced hypothyroidism incidence compared to ICI monotherapy, likely attributable to suppressed plasma cell differentiation and antibody synthesis (84). Therefore, it is evident that B cells can be used as a monitoring indicator for ICI-induced thyroiditis and may represent a novel therapeutic strategy for preventing ICI-induced thyroiditis.
The progression of ICI-induced thyroiditis is often associated with the presence of thyroid autoantibodies, which can manifest in three patterns: pre-existing thyroid autoantibody positivity, the emergence of new antibodies post-treatment, or an increase in antibody levels during treatment. Previous studies have shown that patients receiving PD-1 inhibitors are at higher risk for thyroid disease, particularly when thyroid antibodies (TPOAb and TgAb) are present either at baseline or develop during treatment (2). In a study examining the relationship between antithyroid antibodies and ICI-induced thyroiditis, the authors suggested that the presence or increase in TPOAb and TgAb during treatment could serve as reliable biomarkers for identifying patients at higher risk of thyroiditis (85). Furthermore, Ghosh et al. (86) employed autoantigen microarrays to detect 120 autoantibodies (including thyroid antibodies) in patients with advanced melanoma. They found that patients with fewer baseline autoantibodies experienced earlier onset of irAEs (86). This study diverges from prior research in that the authors suggest patients with higher baseline autoantibodies possess a tolerance mechanism that “protects” the body from ICI toxicity (86). These conflicting results underscore the uncertainty surrounding the role of baseline antibodies in predicting thyroid status. Consequently, further in-depth research is required to clarify this issue.
Increased secretions of cytokines and chemokines
The interplay between cytokines and chemokines in ICI-induced thyroiditis involves complex signaling cascades that amplify immune cell infiltration and thyroid tissue damage (87, 88). Given that organ-specific irAEs may exhibit distinct cytokine profiles, investigating the cytokine signatures associated with ICI-induced thyroiditis is crucial.
A prospective study that evaluated peripheral blood cytokines and chemokines in patients with advanced malignancies undergoing ICI treatment demonstrated that elevated baseline serum levels of IL-1β, IL-2, and granulocyte-macrophage colony-stimulating factor (GM-CSF), along with early decreases in the levels of IL-8, granulocyte colony-stimulating factor (G-CSF), and monocyte chemoattractant protein-1 (MCP-1), were significantly correlated with the development of ICI-induced thyroiditis (89). Liu et al. (90) discovered that early IL-16, IL-12p70, IL-17, IL-1α and C-C motif chemokine ligand 15 (CCL15) could potentially serve as predictive biomarkers for anti-PD-1-induced thyroiditis. The cytokines involved in these studies (IL-1β, IL-1α, IL-2, GM-CSF, IL-16, IL-12p70, and IL-17) are known to have pro-inflammatory effects in autoimmune diseases (91–97). IL-2, primarily produced by CD4+ T cells, plays a key role in promoting TH1 polarization (93). IL-16 is a chemokine involved in the recruitment and activation of CD4+ T cells at inflammatory sites (98). IL-16 enhances TH1 polarization and increases the production of the TH1 effector cytokine IFN-γ (99). IL-12p70 is a key cytokine that responds to infection and induces TH1 responses (100). It can be inferred that ICI-induced thyroiditis is associated with a TH1-skewed immune phenotype, a hypothesis supported by recent studies (90). In contrast, MCP-1, which regulates the polarization of TH0 cells toward a TH2 phenotype, decreases in the context of ICI-induced thyroiditis, further supporting the TH1/TH2 imbalance (101). Moreover, IL-1β plays a vital role in the differentiation of TH17 cells, which are known to drive inflammation, autoantibody production, and immune tolerance disruption in autoimmune thyroid diseases by secreting cytokines like IL-17 and IL-21 (102). The presence of cytokines and chemokines promoting TH1/TH2 imbalance, a TH17-skewed phenotype and CD8+ T effect, may contribute to the development of ICI-induced thyroiditis, as reflected by the observed cytokine profile.
Cytokine inhibitors have transformed the treatment landscape of various autoimmune diseases, underscoring their potential role in suppressing irAEs. Among the most studied cytokine inhibitors are those targeting IL-6, IL-1, IL-17, IL-23, and IL-27 (89). Recent studies in animal models have demonstrated that inhibiting IL-17A can reduce the severity of ICI-induced thyroiditis without compromising the antitumor efficacy of ICIs (72). However, further clinical trials are needed to evaluate the safety and effectiveness of cytokine inhibitors in patients with ICI-induced thyroiditis. It is important to note that the cytokine profile associated with ICI-induced immunotoxicity may differ from that of the tumor microenvironment (TME) (103). Consequently, targeting cytokines involved in irAEs may not necessarily impair ICI-mediated antitumor immunity, as the immune mechanisms driving tumor progression differ from those involved in irAEs.
Genetic factors
The impact of genetic factors on ICI-induced thyroiditis is complex and multifaceted. Research has shown that the HLA system, a key genetic marker, is associated with the development of irAEs (104–106). Both classical HLA-I and HLA-II genes are frequently implicated as drivers of this association. These molecules play a crucial role in presenting antigenic peptides to T cells, enabling the immune system to differentiate between self and non-self-antigens (107). Akturk et al. (105) conducted an HLA-DR genetic analysis in 132 advanced melanoma patients receiving ICIs and found that specific HLA-DR alleles were linked to the development of certain irAEs. Notably, 50% of patients with hypothyroidism carried the DR8 allele (105). Sasaki et al. (108) performed HLA genotyping on 71 cancer patients treated with ICIs and identified HLA alleles A*26:01, DPA1*01:03, and DPB1*02:01 as being associated with ICI-induced thyroiditis. Specifically, A*26:01 and DPB1*02:01 were unique to the thyroid irAE group, while DPA1*01:03 was common across multiple irAE groups. These findings highlight specific HLA genotypes as key factors in the development of ICI-induced thyroiditis. Although the precise mechanisms by which HLA allele variations contribute to irAEs remain unclear, it is known that certain alleles, such as HLA-DR3, are associated with increased susceptibility to AITD like GD and HT (109–111). The amino acid residues linked to disease risk are typically located in the peptide-binding groove or functional pockets of HLA molecules, suggesting their role in antigen presentation and protein stability (109). Therefore, alleles such as A*26:01, DPA1*01:03, and DPB1*02:01 may facilitate the binding of self-proteins, particularly thyroid-derived peptides, leading to a misperception of thyroid tissue as foreign. This misrecognition can trigger immune responses, resulting in ICI-induced thyroiditis. It is important to note that AITD, including HT and GD, have established genetic susceptibilities, with several key loci involved, including HLA, CTLA-4, PTPN22, TSHR, and FCRL3 (112–114). The overlap in genetic susceptibility between AITD and ICI-induced thyroiditis suggests that the latter is not merely a “side effect” of ICI treatment, but rather an exacerbation of pre-existing autoimmune tendencies under conditions of immune activation.
Single nucleotide polymorphisms (SNPs) are DNA sequence variations that arise from single-nucleotide changes at the genomic level, contributing to individual differences (115). Groha et al. (116) identified an IL-7 SNP associated with an increased risk of irAEs, noting that patients with IL-7 germline variants exhibited greater lymphocyte stability following ICI treatment. This suggests that individuals harboring SNPs linked to abnormal autoimmune activation or disrupted signal transduction pathways may be more prone to localized hyperimmune responses, such as those in the thyroid gland, which could lead to ICI-induced thyroiditis. Khan et al. (117) summarized data from seven Phase III clinical trials involving atezolizumab and chemotherapy, using a genome-wide association study (GWAS) approach to identify shared genetic factors for hypothyroidism. They constructed a polygenic risk score (PRS) for validation, which revealed that risk loci in the intronic region of the LPP gene and the PTPN22 rs2476601 (R620W) missense variant were key contributors (117). According to studies, LPP gene and PTPN22 are known to disrupt immune tolerance by modulating T/B cell receptor signaling (117). Additionally, the results highlighted other critical genes involved in T cell initiation (CTLA4) and activation (CD69) (117). These findings underscore the potential for genetic markers to optimize immunotherapy risk assessment, paving the way for personalized approaches in cancer immunotherapy. However, the molecular mechanisms underlying susceptibility SNPs in ICI-induced thyroiditis remain incompletely understood, necessitating further research to clarify their role in the development of these adverse events.
Conclusion
Among endocrine glands, the thyroid is the most commonly affected by autoimmune disorders, and during ICI treatment, it is particularly susceptible to toxicity. ICI-induced thyroiditis is a complex immunological process primarily driven by the activation of memory T cells upon ICI exposure, which triggers a cascade of downstream events. This leads to thyroid cell destruction through the recruitment of cytotoxic inflammatory cells and the release of inflammatory mediators. Consequently, the follicular structure of the thyroid is disrupted, and the inflammatory response intensifies. Importantly, ICI-induced thyroiditis is typically irreversible. While levothyroxine replacement therapy can effectively restore thyroid function during the hypothyroid phase, it does not address the underlying immune-mediated thyroid tissue damage nor prevent the thyrotoxic phase. Additionally, lifelong hormone replacement therapy places a considerable burden on patients and does not alleviate the potential severity of acute thyroiditis, such as cardiovascular complications. Therefore, more effective treatment strategies are needed to target the key immune mechanisms underlying ICI-induced thyroiditis and prevent the onset of irAEs.
Based on current insights into the mechanisms underlying ICI-induced thyroiditis, potential intervention strategies can be developed at multiple levels. First, preventive monitoring, including regular testing of thyroid function and autoantibody levels at baseline and during treatment, can facilitate the early identification of high-risk patients. Second, targeting key pathogenic pathways offers a foundation for developing targeted therapies. For example, blocking the IL-17A signaling pathway has been shown to alleviate thyroiditis in mouse models while preserving the anti-tumor effects of ICIs, as demonstrated by the use of anti-IL-17 monoclonal antibodies. Additionally, B-cell depletion therapies, such as rituximab, have shown promise in reducing the incidence of thyroiditis in clinical trials. Further approaches include modulating the gut microbiota to inhibit cross-immune reactions driven by molecular mimicry and utilizing HLA genotype or SNP analysis to identify susceptible populations and optimize ICI treatment regimens. The development of predictive models such as cytokine and chemokine profiles offer a new avenue for personalized interventions. Future research also should focus on understanding microbiome-immune interactions, conducting single-cell multi-omics analyses of dynamic changes in the thyroid microenvironment, and developing novel immune modulators that specifically block thyroid autoantigen presentation. These efforts may lead to precise prevention and treatment strategies for ICI-related thyroid toxicity.
Author contributions
XM: Writing – original draft, Writing – review & editing, Investigation, Methodology, Software, Validation, Visualization. CM: Writing – original draft, Writing – review & editing, Conceptualization, Data curation, Project administration, Resources, Validation. JL: Writing – original draft, Writing – review & editing, Conceptualization, Data curation, Formal Analysis, Project administration, Resources. XW: Writing – review & editing, Data curation, Methodology, Project administration, Writing – original draft. YM: Writing – review & editing, Writing – original draft.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by grants from the National Natural Science Foundation of China (grant numbers 81370889) and funded by the key research and development plan of Zhenjiang city (grant number SH2022046, YLJ202101 and JDYY2023006).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Iwama S, Kobayashi T, Yasuda Y, and Arima H. Immune checkpoint inhibitor-related thyroid dysfunction. Best Pract Res Clin Endocrinol Metab. (2022) 36:101660. doi: 10.1016/j.beem.2022.101660
2. Postow MA, Sidlow R, and Hellmann MD. Immune-related adverse events associated with immune checkpoint blockade. N Engl J Med. (2018) 378:158–68. doi: 10.1056/NEJMra1703481
3. Sosa A, Lopez Cadena E, Simon Olive C, Karachaliou N, and Rosell R. Clinical assessment of immune-related adverse events. Ther Adv Med Oncol. (2018) 10:175883591876462. doi: 10.1177/1758835918764628
4. Darnell EP, Mooradian MJ, Baruch EN, Yilmaz M, and Reynolds KL. Immune-related adverse events (irAEs): diagnosis, management, and clinical pearls. Curr Oncol Rep. (2020) 22:39. doi: 10.1007/s11912-020-0897-9
5. Wright JJ, Powers AC, and Johnson DB. Endocrine toxicities of immune checkpoint inhibitors. Nat Rev Endocrinol. (2021) 17:389–99. doi: 10.1038/s41574-021-00484-3
6. Fletcher K and Johnson DB. Chronic immune-related adverse events arising from immune checkpoint inhibitors: an update. J Immunother Cancer. (2024) 12:e008591. doi: 10.1136/jitc-2023-008591
7. Zhan L, Feng H, Liu H, Guo L, Chen C, Yao X, et al. Immune checkpoint inhibitors-related thyroid dysfunction: epidemiology, clinical presentation, possible pathogenesis, and management. Front Endocrinol. (2021) 12:649863. doi: 10.3389/fendo.2021.649863
8. Haanen J, Obeid M, Spain L, Carbonnel F, Wang Y, Robert C, et al. Management of toxicities from immunotherapy: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up☆. Ann Oncol. (2022) 33:1217–38. doi: 10.1016/j.annonc.2022.10.001
9. Torphy RJ, Schulick RD, and Zhu Y. Newly emerging immune checkpoints: Promises for future cancer therapy. Int J Mol Sci. (2017) 18:2642. doi: 10.3390/ijms18122642
10. Paluch C, Santos AM, Anzilotti C, Cornall RJ, and Davis SJ. Immune checkpoints as therapeutic targets in autoimmunity. Front Immunol. (2018) 9:2306. doi: 10.3389/fimmu.2018.02306
11. Zhang Y and Zheng J. Functions of immune checkpoint molecules beyond immune evasion. In: Xu J, editor. Regulation of Cancer Immune Checkpoints: Molecular and Cellular Mechanisms and Therapy. Springer, Singapore (2020). p. 201–26. doi: 10.1007/978-981-15-3266-5_9
12. Makkouk A and Weiner GJ. Cancer immunotherapy and breaking immune tolerance: New approaches to an old challenge. Cancer Res. (2015) 75:5–10. doi: 10.1158/0008-5472.CAN-14-2538
13. Mi Y, Han J, Zhu J, and Jin T. Role of the PD-1/PD-L1 signaling in multiple sclerosis and experimental autoimmune encephalomyelitis: recent insights and future directions. Mol Neurobiol. (2021) 58:6249–71. doi: 10.1007/s12035-021-02495-7
14. Chen L and Han X. Anti–PD-1/PD-L1 therapy of human cancer: past, present, and future. J Clin Invest. (2015) 125:3384–91. doi: 10.1172/JCI80011
15. Gao M, Shi J, Xiao X, Yao Y, Chen X, Wang B, et al. PD-1 regulation in immune homeostasis and immunotherapy. Cancer Lett. (2024) 588:216726. doi: 10.1016/j.canlet.2024.216726
16. Tang Q, Chen Y, Li X, Long S, Shi Y, Yu Y, et al. The role of PD-1/PD-L1 and application of immune-checkpoint inhibitors in human cancers. Front Immunol. (2022) 13:964442. doi: 10.3389/fimmu.2022.964442
17. Hossen MM, Ma Y, Yin Z, Xia Y, Du J, Huang JY, et al. Current understanding of CTLA-4: from mechanism to autoimmune diseases. Front Immunol. (2023) 14:1198365. doi: 10.3389/fimmu.2023.1198365
18. Wei SC, Duffy CR, and Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. (2018) 8:1069–86. doi: 10.1158/2159-8290.CD-18-0367
19. Topalian SL, Taube JM, Anders RA, and Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer. (2016) 16:275–87. doi: 10.1038/nrc.2016.36
20. Moslehi J, Lichtman AH, Sharpe AH, Galluzzi L, and Kitsis RN. Immune checkpoint inhibitor–associated myocarditis: manifestations and mechanisms. J Clin Invest. (2021) 131:e145186. doi: 10.1172/JCI145186
21. Johnson DB, Nebhan CA, Moslehi JJ, and Balko JM. Immune-checkpoint inhibitors: long-term implications of toxicity. Nat Rev Clin Oncol. (2022) 19:254–67. doi: 10.1038/s41571-022-00600-w
22. Carlino MS, Larkin J, and Long GV. Immune checkpoint inhibitors in melanoma. Lancet. (2021) 398:1002–14. doi: 10.1016/S0140-6736(21)01206-X
23. Boutros C, Tarhini A, Routier E, Lambotte O, Ladurie FL, Carbonnel F, et al. Safety profiles of anti-CTLA-4 and anti-PD-1 antibodies alone and in combination. Nat Rev Clin Oncol. (2016) 13:473–86. doi: 10.1038/nrclinonc.2016.58
24. Barroso-Sousa R, Barry WT, Garrido-Castro AC, Hodi FS, Min L, Krop IE, et al. Incidence of endocrine dysfunction following the use of different immune checkpoint inhibitor regimens: A systematic review and meta-analysis. JAMA Oncol. (2018) 4:173–82. doi: 10.1001/jamaoncol.2017.3064
25. McLachlan SM and Rapoport B. Breaking tolerance to thyroid antigens: Changing concepts in thyroid autoimmunity. Endocr Rev. (2014) 35:59–105. doi: 10.1210/er.2013-1055
26. Wang SJ, Dougan SK, and Dougan M. Immune mechanisms of toxicity from checkpoint inhibitors. Trends Cancer. (2023) 9:543–53. doi: 10.1016/j.trecan.2023.04.002
27. Marinkovic T, Garin A, Yokota Y, Fu Y-X, Ruddle NH, Furtado GC, et al. Interaction of mature CD3+CD4+ T cells with dendritic cells triggers the development of tertiary lymphoid structures in the thyroid. J Clin Invest. (2006) 116:2622–32. doi: 10.1172/JCI28993
28. Zhao L, Jin S, Wang S, Zhang Z, Wang X, Chen Z, et al. Tertiary lymphoid structures in diseases: immune mechanisms and therapeutic advances. Sig Transduct Target Ther. (2024) 9:1–43. doi: 10.1038/s41392-024-01947-5
29. Dieu-Nosjean M-C, Goc J, Giraldo NA, Sautès-Fridman C, and Fridman WH. Tertiary lymphoid structures in cancer and beyond. Trends Immunol. (2014) 35:571–80. doi: 10.1016/j.it.2014.09.006
30. Sato Y, Silina K, van den Broek M, Hirahara K, and Yanagita M. The roles of tertiary lymphoid structures in chronic diseases. Nat Rev Nephrol. (2023) 19:525–37. doi: 10.1038/s41581-023-00706-z
31. Li S, Zhang N, Zhang H, Yang Z, Cheng Q, Wei K, et al. Deciphering the role of LGALS2: insights into tertiary lymphoid structure-associated dendritic cell activation and immunotherapeutic potential in breast cancer patients. Mol Cancer. (2024) 23:216. doi: 10.1186/s12943-024-02126-4
32. Matsubara S, Seki M, Suzuki S, Komori T, and Takamori M. Tertiary lymphoid organs in the inflammatory myopathy associated with PD-1 inhibitors. J Immunother Cancer. (2019) 7:256. doi: 10.1186/s40425-019-0736-4
33. Schumacher TN and Thommen DS. Tertiary lymphoid structures in cancer. Science. (2022) 375:eabf9419. doi: 10.1126/science.abf9419
34. Merrill SJ and Minucci SB. Thyroid autoimmunity: An interplay of factors. Vitam Horm. (2018) 106:129–45. doi: 10.1016/bs.vh.2017.07.001
35. Merrill SJ and Mu Y. Thyroid autoimmunity as a window to autoimmunity: An explanation for sex differences in the prevalence of thyroid autoimmunity. J Theor Biol. (2015) 375:95–100. doi: 10.1016/j.jtbi.2014.12.015
36. Álvarez-Sierra D, Rodríguez-Grande J, Gómez-Brey A, Bello I, Caubet E, González Ó, et al. Single cell transcriptomic analysis of Graves’ disease thyroid glands reveals the broad immunoregulatory potential of thyroid follicular and stromal cells and implies a major re-interpretation of the role of aberrant HLA class II expression in autoimmunity. J Autoimmun. (2023) 139:103072. doi: 10.1016/j.jaut.2023.103072
37. Kawashima A, Tanigawa K, Akama T, Yoshihara A, Ishii N, and Suzuki K. Innate immune activation and thyroid autoimmunity. J Clin Endocrinol Metab. (2011) 96:3661–71. doi: 10.1210/jc.2011-1568
38. Elgueta R, Benson MJ, De Vries VC, Wasiuk A, Guo Y, and Noelle RJ. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunological Rev. (2009) 229:152–72. doi: 10.1111/j.1600-065X.2009.00782.x
39. Khan S and Gerber DE. Autoimmunity, checkpoint inhibitor therapy and immune-related adverse events: A review. Semin Cancer Biol. (2020) 64:93–101. doi: 10.1016/j.semcancer.2019.06.012
40. Angell TE, Min L, Wieczorek TJ, and Hodi FS. Unique cytologic features of thyroiditis caused by immune checkpoint inhibitor therapy for Malignant melanoma. Genes Dis. (2018) 5:46–8. doi: 10.1016/j.gendis.2017.11.002
41. Cottrell T, Zhang J, Zhang B, Kaunitz GJ, Burman P, Chan H-Y, et al. Evaluating T-cell cross-reactivity between tumors and immune-related adverse events with TCR sequencing: pitfalls in interpretations of functional relevance. J Immunother Cancer. (2021) 9:e002642. doi: 10.1136/jitc-2021-002642
42. Tison A, Garaud S, Chiche L, Cornec D, and Kostine M. Immune-checkpoint inhibitor use in patients with cancer and pre-existing autoimmune diseases. Nat Rev Rheumatol. (2022) 18:641–56. doi: 10.1038/s41584-022-00841-0
43. Gergely TG, Drobni ZD, Sayour NV, Ferdinandy P, and Varga ZV. Molecular fingerprints of cardiovascular toxicities of immune checkpoint inhibitors. Basic Res Cardiol. (2024) 120:187–205. doi: 10.1007/s00395-024-01068-8
44. Berner F, Bomze D, Diem S, Ali OH, Fässler M, Ring S, et al. Association of checkpoint inhibitor–induced toxic effects with shared cancer and tissue antigens in non–small cell lung cancer. JAMA Oncol. (2019) 5:1043–7. doi: 10.1001/jamaoncol.2019.0402
45. Rapisuwon S, Izar B, Batenchuk C, Avila A, Mei S, Sorger P, et al. Exceptional response and multisystem autoimmune-like toxicities associated with the same T cell clone in a patient with uveal melanoma treated with immune checkpoint inhibitors. J immunotherapy Cancer. (2019) 7:61. doi: 10.1186/s40425-019-0533-0
46. Läubli H, Koelzer VH, Matter MS, Herzig P, Schlienger BD, Wiese MN, et al. The T cell repertoire in tumors overlaps with pulmonary inflammatory lesions in patients treated with checkpoint inhibitors. Oncoimmunology. (2017) 7:e1386362. doi: 10.1080/2162402X.2017.1386362
47. Koyama J, Horiike A, Yoshizawa T, Dotsu Y, Ariyasu R, Saiki M, et al. Correlation between thyroid transcription factor-1 expression, immune-related thyroid dysfunction, and efficacy of anti-programmed cell death protein-1 treatment in non-small cell lung cancer. J Thorac Dis. (2019) 11:1919–28. doi: 10.21037/jtd.2019.04.102
48. Schumacher TN and Schreiber RD. Neoantigens in cancer immunotherapy. Science. (2015) 348:69–74. doi: 10.1126/science.aaa4971
49. Desrichard A, Snyder A, and Chan TA. Cancer neoantigens and applications for immunotherapy. Clin Cancer Res. (2016) 22:807–12. doi: 10.1158/1078-0432.CCR-14-3175
50. Bassani-Sternberg M and Coukos G. Mass spectrometry-based antigen discovery for cancer immunotherapy. Curr Opin Immunol. (2016) 41:9–17. doi: 10.1016/j.coi.2016.04.005
51. Vita R, Guarneri F, Agah R, and Benvenga S. Autoimmune thyroid disease elicited by NY-ESO-1 vaccination. Thyroid. (2014) 24:390–4. doi: 10.1089/thy.2013.0170
52. Chervonsky AV. Microbiota and autoimmunity. Cold Spring Harb Perspect Biol. (2013) 5:a007294. doi: 10.1101/cshperspect.a007294
53. Zhang X, Chen B, Zhao L, and Li H. The gut microbiota: emerging evidence in autoimmune diseases. Trends Mol Med. (2020) 26:862–73. doi: 10.1016/j.molmed.2020.04.001
54. Liu X, Lu B, Tang H, Jia X, Zhou Q, Zeng Y, et al. Gut microbiome metabolites, molecular mimicry, and species-level variation drive long-term efficacy and adverse event outcomes in lung cancer survivors. eBioMedicine. (2024) 109:105427. doi: 10.1016/j.ebiom.2024.105427
55. Won T, Kalinoski HM, Wood MK, Hughes DM, Jaime CM, Delgado P, et al. Cardiac myosin-specific autoimmune T cells contribute to immune-checkpoint-inhibitor-associated myocarditis. Cell Rep. (2022) 41:111611. doi: 10.1016/j.celrep.2022.111611
56. Benvenga S and Guarneri F. Molecular mimicry and autoimmune thyroid disease. Rev Endocr Metab Disord. (2016) 17:485–98. doi: 10.1007/s11154-016-9363-2
57. Fröhlich E and Wahl R. Microbiota and thyroid interaction in health and disease. Trends Endocrinol Metab. (2019) 30:479–90. doi: 10.1016/j.tem.2019.05.008
58. Ludgate ME, Masetti G, and Soares P. The relationship between the gut microbiota and thyroid disorders. Nat Rev Endocrinol. (2024) 20:511–25. doi: 10.1038/s41574-024-01003-w
59. Mueller DL. Mechanisms maintaining peripheral tolerance. Nat Immunol. (2010) 11:21–7. doi: 10.1038/ni.1817
60. Casagrande S, Sopetto GB, Bertalot G, Bortolotti R, Racanelli V, Caffo O, et al. Immune-related adverse events due to cancer immunotherapy: immune mechanisms and clinical manifestations. Cancers. (2024) 16:1440. doi: 10.3390/cancers16071440
61. Jameson SC and Masopust D. Understanding subset diversity in T cell memory. Immunity. (2018) 48:214–26. doi: 10.1016/j.immuni.2018.02.010
62. Zhao Y and Wucherpfennig KW. Tissue-resident T cells in clinical response and immune-related adverse events of immune checkpoint blockade. Clin Cancer Res. (2024) 30:5527–34. doi: 10.1158/1078-0432.CCR-23-3296
63. Lozano AX, Chaudhuri AA, Nene A, Bacchiocchi A, Earland N, Vesely MD, et al. T cell characteristics associated with toxicity to immune checkpoint blockade in patients with melanoma. Nat Med. (2022) 28:353–62. doi: 10.1038/s41591-021-01623-z
64. Fanelli G, Romano M, Nova-Lamperti E, Sunderland MW, Nerviani A, Scottà C, et al. PD-L1 signaling on human memory CD4+ T cells induces a regulatory phenotype. PloS Biol. (2021) 19:e3001199. doi: 10.1371/journal.pbio.3001199
65. Yasuda Y, Iwama S, Sugiyama D, Okuji T, Kobayashi T, Ito M, et al. CD4 + T cells are essential for the development of destructive thyroiditis induced by anti–PD-1 antibody in thyroglobulin-immunized mice. Sci Transl Med. (2021) 13:eabb7495. doi: 10.1126/scitranslmed.abb7495
66. Koh C-H, Lee S, Kwak M, Kim B-S, and Chung Y. CD8 T-cell subsets: heterogeneity, functions, and therapeutic potential. Exp Mol Med. (2023) 55:2287–99. doi: 10.1038/s12276-023-01105-x
67. Lechner MG, Zhou Z, Hoang AT, Huang N, Ortega J, Scott LN, et al. Clonally expanded, thyrotoxic effector CD8+ T cells driven by IL-21 contribute to checkpoint inhibitor thyroiditis. Sci Trans Med. (2023) 15:eadg0675. doi: 10.1126/scitranslmed.adg0675
68. Wu Y, Li J, Yang X, Hou B, and Qiao H. Immunosensitivity mediated by downregulated AKT1-SKP2 induces anti-PD-1-associated thyroid immune injury. Int Immunopharmacol. (2023) 121:110452. doi: 10.1016/j.intimp.2023.110452
69. Wang Y, Fang S, and Zhou H. Pathogenic role of Th17 cells in autoimmune thyroid disease and their underlying mechanisms. Best Pract Res Clin Endocrinol Metab. (2023) 37:101743. doi: 10.1016/j.beem.2023.101743
70. Bukhari S, Henick BS, Winchester RJ, Lerrer S, Adam K, Gartshteyn Y, et al. Single-cell RNA sequencing reveals distinct T cell populations in immune-related adverse events of checkpoint inhibitors. CR Med. (2023) 4:100868. doi: 10.1016/j.xcrm.2022.100868
71. Schnell A, Littman DR, and Kuchroo VK. TH17 cell heterogeneity and its role in tissue inflammation. Nat Immunol. (2023) 24:19–29. doi: 10.1038/s41590-022-01387-9
72. Lechner MG, Cheng MI, Patel AY, Hoang AT, Yakobian N, Astourian M, et al. Inhibition of IL-17A Protects against Thyroid Immune-Related Adverse Events while Preserving Checkpoint Inhibitor Antitumor Efficacy. J Immunol. (2022) 209:696–709. doi: 10.4049/jimmunol.2200244
73. Dimitriou F, Cheng PF, Saltari A, Schaper-Gerhardt K, Staeger R, Haunerdinger V, et al. A targetable type III immune response with increase of IL-17A expressing CD4+ T cells is associated with immunotherapy-induced toxicity in melanoma. Nat Cancer. (2024) 5:1390–408. doi: 10.1038/s43018-024-00810-4
74. Lu L, Barbi J, and Pan F. The regulation of immune tolerance by FOXP3. Nat Rev Immunol. (2017) 17:703–17. doi: 10.1038/nri.2017.75
75. Hu W, Wang G, Wang Y, Riese MJ, and You M. Uncoupling therapeutic efficacy from immune-related adverse events in immune checkpoint blockade. iScience. (2020) 23:101580. doi: 10.1016/j.isci.2020.101580
76. Grigoriou M, Banos A, Hatzioannou A, Kloetgen A, Kouzis P, Aggouraki D, et al. Regulatory T-cell transcriptomic reprogramming characterizes adverse events by checkpoint inhibitors in solid tumors. Cancer Immunol Res. (2021) 9:726–34. doi: 10.1158/2326-6066.CIR-20-0969
77. Taylor J, Gandhi A, Gray E, and Zaenker P. Checkpoint inhibitor immune-related adverse events: A focused review on autoantibodies and B cells as biomarkers, advancements and future possibilities. Front Immunol. (2023) 13:991433. doi: 10.3389/fimmu.2022.991433
78. Liudahl SM and Coussens LM. B cells as biomarkers: predicting immune checkpoint therapy adverse events. J Clin Invest. (2018) 128:577–9. doi: 10.1172/JCI99036
79. Labadzhyan A, Wentzel K, Hamid O, Chow K, Kim S, Piro L, et al. Endocrine autoantibodies determine immune checkpoint inhibitor-induced endocrinopathy: A prospective study. J Clin Endocrinol Metab. (2022) 107:1976–82. doi: 10.1210/clinem/dgac161
80. Das R, Bar N, Ferreira M, Newman AM, Zhang L, Bailur JK, et al. Early B cell changes predict autoimmunity following combination immune checkpoint blockade. J Clin Invest. (2018) 128:715–20. doi: 10.1172/JCI96798
81. Lau D, Lan LY-L, Andrews SF, Henry C, Rojas KT, Neu KE, et al. Low CD21 expression defines a population of recent germinal center graduates primed for plasma cell differentiation. Sci Immunol. (2017) 2:eaai8153. doi: 10.1126/sciimmunol.aai8153
82. Gensous N, Charrier M, Duluc D, Contin-Bordes C, Truchetet M-E, Lazaro E, et al. T follicular helper cells in autoimmune disorders. Front Immunol. (2018) 9:1637. doi: 10.3389/fimmu.2018.01637
83. Zhang Q-Y, Ye X-P, Zhou Z, Zhu C-F, Li R, Fang Y, et al. Lymphocyte infiltration and thyrocyte destruction are driven by stromal and immune cell components in hashimoto’s thyroiditis. Nat Commun. (2022) 13:775. doi: 10.1038/s41467-022-28120-2
84. Risbjerg RS, Hansen MV, Sørensen AS, and Kragstrup TW. The effects of B cell depletion on immune related adverse events associated with immune checkpoint inhibition. Exp Hematol Oncol. (2020) 9:9. doi: 10.1186/s40164-020-00167-1
85. Muir CA, Wood CCG, Clifton-Bligh RJ, Long GV, Scolyer RA, Carlino MS, et al. Association of antithyroid antibodies in checkpoint inhibitor–associated thyroid immune–related adverse events. J Clin Endocrinol Metab. (2022) 107:e1843–9. doi: 10.1210/clinem/dgac059
86. Ghosh N, Postow M, Zhu C, Jannat-Khah D, Li Q-Z, Vitone G, et al. Lower baseline autoantibody levels are associated with immune-related adverse events from immune checkpoint inhibition. J Immunother Cancer. (2022) 10:e004008. doi: 10.1136/jitc-2021-004008
87. Mikoś H, Mikoś M, Obara-Moszyńska M, and Niedziela M. The role of the immune system and cytokines involved in the pathogenesis of autoimmune thyroid disease (AITD). Endokrynologia Polska. (2014) 65:150–5. doi: 10.5603/EP.2014.0021
88. Conti P, Youinou P, and Theoharides TC. Modulation of autoimmunity by the latest interleukins (with special emphasis on IL-32). Autoimmun Rev. (2007) 6:131–7. doi: 10.1016/j.autrev.2006.08.015
89. Kurimoto C, Inaba H, Ariyasu H, Iwakura H, Ueda Y, Uraki S, et al. Predictive and sensitive biomarkers for thyroid dysfunctions during treatment with immune-checkpoint inhibitors. Cancer Sci. (2020) 111:1468–77. doi: 10.1111/cas.14363
90. Liu J, Chen M, Li S, Cai L, Ma L, Yang Q, et al. Biomarkers in the early stage of PD-1 inhibitor treatment have shown superior predictive capabilities for immune-related thyroid dysfunction. Front Immunol. (2024) 15:1458488. doi: 10.3389/fimmu.2024.1458488
91. Gabay C, Lamacchia C, and Palmer G. IL-1 pathways in inflammation and human diseases. Nat Rev Rheumatol. (2010) 6:232–41. doi: 10.1038/nrrheum.2010.4
92. Wicks IP and Roberts AW. Targeting GM-CSF in inflammatory diseases. Nat Rev Rheumatol. (2016) 12:37–48. doi: 10.1038/nrrheum.2015.161
93. Spolski R, Li P, and Leonard WJ. Biology and regulation of IL-2: from molecular mechanisms to human therapy. Nat Rev Immunol. (2018) 18:648–59. doi: 10.1038/s41577-018-0046-y
94. Cavalli G, ColaFrancesco S, Emmi G, Imazio M, Lopalco G, Maggio MC, et al. Interleukin 1α: a comprehensive review on the role of IL-1α in the pathogenesis and treatment of autoimmune and inflammatory diseases. Autoimmun Rev. (2021) 20:102763. doi: 10.1016/j.autrev.2021.102763
95. Seegert D, Rosenstiel P, Pfahler H, Pfefferkorn P, Nikolaus S, and Schreiber S. Increased expression of IL-16 in inflammatory bowel disease. Gut. (2001) 48:326–32. doi: 10.1136/gut.48.3.326
96. Tugues S, Burkhard SH, Ohs I, Vrohlings M, Nussbaum K, vom Berg J, et al. New insights into IL-12-mediated tumor suppression. Cell Death Differ. (2015) 22:237–46. doi: 10.1038/cdd.2014.134
97. Mills KHG. IL-17 and IL-17-producing cells in protection versus pathology. Nat Rev Immunol. (2023) 23:38–54. doi: 10.1038/s41577-022-00746-9
98. Cho M-L, Jung YO, Kim K-W, Park M-K, Oh H-J, Ju J-H, et al. IL-17 induces the production of IL-16 in rheumatoid arthritis. Exp Mol Med. (2008) 40:237–45. doi: 10.3858/emm.2008.40.2.237
99. Wen Z, Liu T, Xu X, Acharya N, Shen Z, Lu Y, et al. Interleukin-16 enhances anti-tumor immune responses by establishing a Th1 cell-macrophage crosstalk through reprogramming glutamine metabolism in mice. Nat Commun. (2025) 16:2362. doi: 10.1038/s41467-025-57603-1
100. Posseme C, Llibre A, Charbit B, Bondet V, Rouilly V, Saint-André V, et al. Early IFNβ secretion determines variable downstream IL-12p70 responses upon TLR4 activation. Cell Rep. (2022) 39:110989. doi: 10.1016/j.celrep.2022.110989
101. Singh S, Anshita D, and Ravichandiran V. MCP-1: Function, regulation, and involvement in disease. Int Immunopharmacol. (2021) 101:107598. doi: 10.1016/j.intimp.2021.107598
102. Li Q, Wang B, Mu K, and Zhang J-A. The pathogenesis of thyroid autoimmune diseases: New T lymphocytes – Cytokines circuits beyond the Th1–Th2 paradigm. J Cell Physiol. (2019) 234:2204–16. doi: 10.1002/jcp.27180
103. Kang JH, Bluestone JA, and Young A. Predicting and preventing immune checkpoint inhibitor toxicity: targeting cytokines. Trends Immunol. (2021) 42:293–311. doi: 10.1016/j.it.2021.02.006
104. Hasan Ali O, Berner F, Bomze D, Fässler M, Diem S, Cozzio A, et al. Human leukocyte antigen variation is associated with adverse events of checkpoint inhibitors. Eur J Cancer. (2019) 107:8–14. doi: 10.1016/j.ejca.2018.11.009
105. Akturk HK, Couts KL, Baschal EE, Karakus KE, Van Gulick RJ, Turner JA, et al. Analysis of human leukocyte antigen DR alleles, immune-related adverse events, and survival associated with immune checkpoint inhibitor use among patients with advanced Malignant melanoma. JAMA Netw Open. (2022) 5:e2246400. doi: 10.1001/jamanetworkopen.2022.46400
106. Abed A, Law N, Calapre L, Lo J, Bhat V, Bowyer S, et al. Human leucocyte antigen genotype association with the development of immune-related adverse events in patients with non-small cell lung cancer treated with single agent immunotherapy. Eur J Cancer. (2022) 172:98–106. doi: 10.1016/j.ejca.2022.05.021
107. Dendrou CA, Petersen J, Rossjohn J, and Fugger L. HLA variation and disease. Nat Rev Immunol. (2018) 18:325–39. doi: 10.1038/nri.2017.143
108. Sasaki E, Natori Y, Tokuda E, Kimura-Tsuchiya R, Suga J, Kanazawa K, et al. Association between specific human leukocyte antigen alleles and development of thyroid immune-related adverse event. Immunotherapy. (2024) 16:723–32. doi: 10.1080/1750743X.2024.2353539
109. Inaba H, De Groot LJ, and Akamizu T. Thyrotropin receptor epitope and human leukocyte antigen in graves’ Disease. Front Endocrinol. (2016) 7:120. doi: 10.3389/fendo.2016.00120
110. Shin D-H, Baek I-C, Kim HJ, Choi E-J, Ahn M, Jung MH, et al. HLA alleles, especially amino-acid signatures of HLA-DPB1, might contribute to the molecular pathogenesis of early-onset autoimmune thyroid disease. PloS One. (2019) 14:e0216941. doi: 10.1371/journal.pone.0216941
111. Manger I, Schmitt C, Berking C, French LE, Vera-Gonzalez J, and Heinzerling L. Association of HLA-A*02:01 type with efficacy and toxicity of immune checkpoint inhibitor therapy in melanoma patients: a retrospective cohort study. BMC Cancer. (2025) 25:565. doi: 10.1186/s12885-025-13857-y
112. Eriksson N, Tung JY, Kiefer AK, Hinds DA, Francke U, Mountain JL, et al. Novel associations for hypothyroidism include known autoimmune risk loci. PloS One. (2012) 7:e34442. doi: 10.1371/journal.pone.0034442
113. Taylor PN, Medici MM, Hubalewska-Dydejczyk A, and Boelaert K. Hypothyroidism. Lancet. (2024) 404:1347–64. doi: 10.1016/S0140-6736(24)01614-3
114. Grixti L, Lane LC, and Pearce SH. The genetics of Graves’ disease. Rev Endocr Metab Disord. (2024) 25:203–14. doi: 10.1007/s11154-023-09848-8
115. Cai L, Lyu Z, Zhang Y, Xie K, and Chen M. Association between programmed death protein 1-related single-nucleotide polymorphisms and immune-related adverse events induced by programmed death protein 1 inhibitors—a pilot study. Int Immunopharmacol. (2024) 143:113269. doi: 10.1016/j.intimp.2024.113269
116. Groha S, Alaiwi SA, Xu W, Naranbhai V, Nassar AH, Bakouny Z, et al. Germline variants associated with toxicity to immune checkpoint blockade. Nat Med. (2022) 28:2584–91. doi: 10.1038/s41591-022-02094-6
Keywords: immune-related adverse events, immune checkpoint inhibitors, immune checkpoint inhibitor-induced thyroiditis, thyrotoxicosis, hypothyroidism
Citation: Mao X, Mao C, Liu J, Wang X and Mao Y (2025) Immune checkpoint inhibitor-induced thyroiditis and its potential mechanisms. Front. Endocrinol. 16:1584675. doi: 10.3389/fendo.2025.1584675
Received: 27 February 2025; Accepted: 19 May 2025;
Published: 04 June 2025.
Edited by:
Rosa Maria Paragliola, Saint Camillus International University of Health and Medical Sciences, ItalyReviewed by:
Mohamed Essameldin Abdelgawad, Wake Forest University, United StatesJian Zhao, Shanghai Changzheng Hospital, China
Gianluca Cera, Catholic University of the Sacred Heart, Italy
Copyright © 2025 Mao, Mao, Liu, Wang and Mao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chaoming Mao, anExMDAxQHVqcy5lZHUuY24=; Yufei Mao, MTAwMDAxMjk4NEB1anMuZWR1LmNu