 Maria Paula Bernal Zárate1,2
Maria Paula Bernal Zárate1,2 Daniel Felipe Mendivelso-Gonzalez2
Daniel Felipe Mendivelso-Gonzalez2 William Camilo Torres2Angelica Maria González Clavijo3
William Camilo Torres2Angelica Maria González Clavijo3 Diego Felipe Ballen3
Diego Felipe Ballen3 Rafael Parra Medina2
Rafael Parra Medina2 Julián C. Riaño-Moreno2,4,5*
Julián C. Riaño-Moreno2,4,5*- 1Department of Genetics, Fundación Universitaria de Ciencias de la Salud, Bogotá, Colombia
- 2Department of Pathology and Molecular Oncology, Instituto Nacional de Cancerología, Bogotá, D.C., Colombia
- 3Department of Endocrinology, Instituto Nacional de Cancerología, Bogotá, D.C., Colombia
- 4Faculty of Medicine, Universidad Cooperativa de Colombia, Villavicencio, Colombia
- 5Department of Clinical Oncology, Instituto Nacional de Cancerología, Bogotá, D.C., Colombia
Lynch syndrome (LS) is an autosomal dominant inherited disorder caused by pathogenic variants in DNA mismatch repair (MMR) genes, most commonly MLH1 and MSH2. LS significantly increases the risk of various cancers, including colorectal, endometrial, gastric, and ovarian malignancies. Neuroendocrine neoplasms (NENs) are rare tumors that arise from neuroendocrine cells, predominantly in the gastrointestinal tract, and are frequently associated with hereditary cancer syndromes such as multiple endocrine neoplasia types 1 and 2. While a definitive association between LS and NENs has not been established, isolated cases have been reported. We present the case of a 63-year-old woman with a history of colorectal cancer and a confirmed LS diagnosis, identified through genetic testing that revealed a pathogenic MLH1 variant. Years later, she developed a grade 2 non-functional neuroendocrine tumor (NET), likely of gastrointestinal origin. The patient underwent surgical resection, followed by treatment with somatostatin analogs. Due to this uncommon presentation, we conducted a literature review to explore the potential relationship between LS and NENs. Our analysis identified 13 additional cases of NENs in LS patients, encompassing NETs, neuroendocrine carcinomas (NECs), and mixed neuroendocrine non-neuroendocrine neoplasms (MiNENs). This growing body of evidence suggests that NENs may be part of the LS tumor spectrum. Further research is needed to elucidate the underlying mechanisms and determine whether LS predisposes individuals to NENs. Enhanced surveillance in LS patients could improve early detection of rare malignancies such as NENs, ultimately expanding our understanding of LS-associated cancer risks and guiding more effective clinical management.
Introduction
Lynch syndrome (LS), also known as hereditary non-polyposis colorectal cancer (HNPCC), is an autosomal dominant disorder caused by pathogenic variants in DNA mismatch repair (MMR) genes, including MLH1, MSH2, MSH6, PMS2, and, occasionally, EPCAM (1). These genes play a critical role in maintaining genomic integrity by correcting DNA replication errors. MLH1 and MSH2 variants account for most LS cases, with MLH1 pathogenic/likely pathogenic (P/LP) variants primarily associated with colorectal tumors, whereas MSH2 P/LP variants are linked to an increased risk of colorectal, endometrial, and ovarian cancers (2). LS is associated with a broad spectrum of malignancies, including colorectal (CRC), endometrial, gastric, ovarian, pancreatic, urothelial, biliary tract, and small intestinal cancers, as well as brain tumors (predominantly glioblastomas). Additionally, LS can manifest with sebaceous neoplasms (sebaceous adenomas, sebaceous carcinomas, and keratoacanthomas), particularly in Muir-Torre syndrome (3). Early identification of LS and genetic counseling are crucial for implementing targeted cancer prevention and surveillance strategies, improving patient prognosis and quality of life (4).
Neuroendocrine neoplasms (NENs) arise from neuroendocrine cells, which have both neural and endocrine properties, allowing them to respond to neuronal stimuli and secrete hormones or signaling molecules into the bloodstream. Due to the widespread distribution of neuroendocrine cells, NENs can develop in various organs, including the esophagus, stomach, pancreas, intestines, lungs, and endocrine glands (5). These tumors are characterized by the expression of neuroendocrine markers, such as synaptophysin and chromogranin A, and variable somatostatin receptor expression (6).
NENs are classified into three main categories: well-differentiated neuroendocrine tumors (NETs), poorly differentiated neuroendocrine carcinomas (NECs), and mixed neuroendocrine non-neuroendocrine neoplasms (MiNENs) (7). NETs are the most common and typically follow an indolent course, while NECs are highly aggressive. MiNENs combine a high-grade neuroendocrine component with a non-neuroendocrine malignancy, often exhibiting a poor prognosis. Most NETs (55%) originate in the gastrointestinal tract, with the small intestine being the most frequent site (45%), followed by the rectum (20%), appendix (16%), colon (11%), and stomach (7%) (8).
Approximately 20% of neuroendocrine tumors (NETs) are associated with hereditary cancer syndromes. The most well-established of these include multiple endocrine neoplasia types 1 and 2 (MEN1 and MEN2), as well as neurofibromatosis type 1 (NF1), Carney complex, Von Hippel-Lindau disease, and tuberous sclerosis complex, all of which are linked to an increased risk of NET development (9).
To date, no definitive association has been established between NENs and LS. These conditions are considered distinct clinical entities, characterized by different genetic alterations and pathogenic mechanisms. However, an increasing number of case reports have described NENs in patients with LS, suggesting a possible yet unconfirmed relationship.
In this study, we present a case of a NET in a patient with LS and conducted a literature analysis to explore the potential association between NENs and Lynch syndrome. Our review identified 13 additional cases with a clinical, molecular, or combined diagnosis of LS, including patients with NETs, NECs, and MiNENs. This is the first study to systematically compile the existing evidence on this association, highlighting the scarce but emerging data suggesting that NENs may be part of the LS tumor spectrum. However, due to the limited nature of the available evidence, further research is necessary to establish a definitive correlation.
Materials and methods
Case presentation
Our case describes a 50-year-old woman who presented to the emergency department in 2000 with hyporexia and postprandial liquid stools lasting three months, followed by an episode of acute abdominal pain requiring surgical exploration. Intraoperative findings revealed a colonic tumor mass, which was resected. She subsequently underwent eight cycles of adjuvant chemotherapy. Afterward, she was lost to follow-up until July 2015, when she was admitted to the Instituto Nacional de Cancerología in Bogotá, D.C. Colombia, at the age of 63 due to hematochezia. Colonoscopy identified a new lesion in the sigmoid colon, leading to a total colectomy with ileostomy and adjuvant chemotherapy. Pathological analysis confirmed a stage II rectosigmoid adenocarcinoma. Immunohistochemistry (IHC) demonstrated loss of MLH1 and PMS2 expression, with negative chromogranin staining (Figure 1). Molecular analysis of the rectosigmoid tumor by RT-PCR did not detect pathogenic variants in BRAF, KRAS, or NRAS.

Figure 1. Immunohistochemical studies (40X) showing (A) Chromogranin IHC in the affected lymph node, showing a positive staining. (B) Chromogranin IHC from the rectosigmoid adenocarcinoma, showing a negative staining. MMR IHC from this tumor shows no expression of MLH1 (C) and PMS2 (D), and an intact expression of MSH2 (E) and MSH6 (F).
Family history (Figure 2) revealed multiple first- and second-degree relatives with oncological conditions, including her mother (II-5), diagnosed with an unspecified malignancy at age 66; her sister (III-4), diagnosed with gastric cancer at age 63; and her niece (IV-3), diagnosed with CRC at age 38. Given her personal history of CRC and a family history of gastric and colorectal cancer, LS was suspected, and the patient was referred to the Medical Genetics Department at our institution for further evaluation.
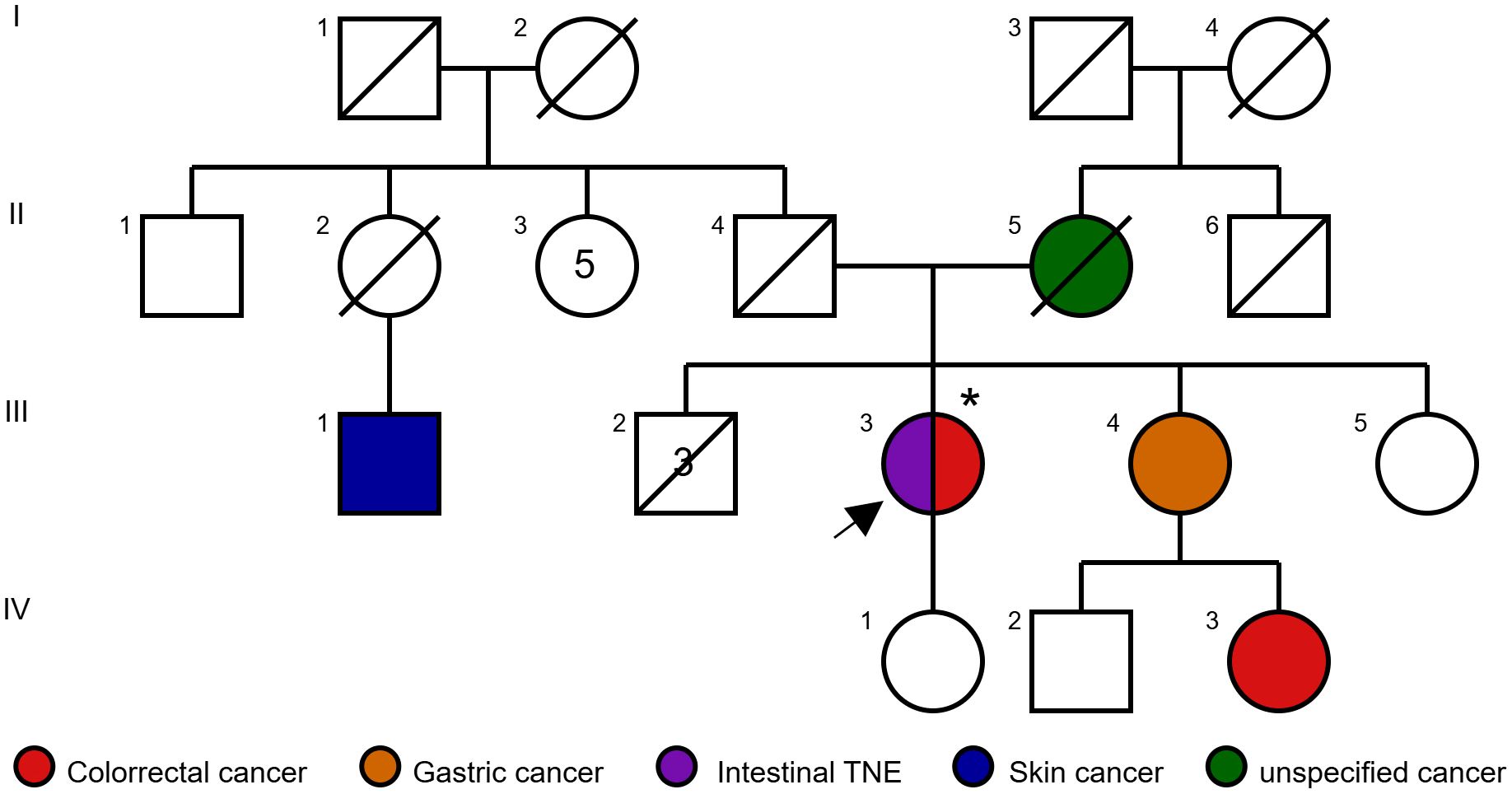
Figure 2. Pedigree of a family with Lynch syndrome, showing the proband with metachronous tumors (intestinal NET and colorectal cancer). *Documented carrier of pathogenic variant MLH1:c.1852_1854del; p.Lys618del.
Our patient met two out of five Bethesda criteria (1): CRC with high microsatellite instability (MSI-H) histology diagnosed before the age of 60 and (2) CRC or LS-associated tumors diagnosed at any age in at least two first- or second-degree relatives. She also met four out of five Amsterdam II criteria and fulfilled the National Comprehensive Cancer Network (NCCN) criteria for LS evaluation, which include clinical presentation in at least two generations, diagnosis of CRC or associated cancers before age 50, exclusion of familial adenomatous polyposis, and tumor verification through histopathology tests. Consequently, germline testing was performed using a Next-Generation Sequencing (NGS) multi-gene panel with the Illumina kit and the Canadian Consortia Inherited Cancer custom panel (Illumina, San Diego, CA), covering 105 genes associated with hereditary cancer syndromes, including mismatch repair (MMR) genes. Bioinformatic analysis was conducted using SOPHiA DDM® software with the GRCh37/hg19 human genome reference. A pathogenic heterozygous MLH1 (NM_000249.4) variant (c.1852_1854del; p.Lys618del) was identified, with a variant allele fraction of 44.21% and an allelic depth of 252X, confirming the diagnosis of LS.
In December 2016, as part of LS and CRC follow-up, an abdominal MRI revealed three hypervascular focal lesions with dominant arterial phase enhancement, located in segment III (21 mm), segment VII (15 mm), and segment V (8 mm). These lesions were initially suspected to be hepatic metastases from the rectosigmoid adenocarcinoma (Figure 3). The patient underwent a hepatic metastasectomy, and histopathological analysis, along with IHC, confirmed metastatic involvement from a newly identified grade 2 non-functional NET, likely of intestinal origin. IHC showed positive staining for CDX2, synaptophysin, and chromogranin A, while CK7, CK20, and TTF1 were negative. The Ki-67 index was >4%, supporting the diagnosis.
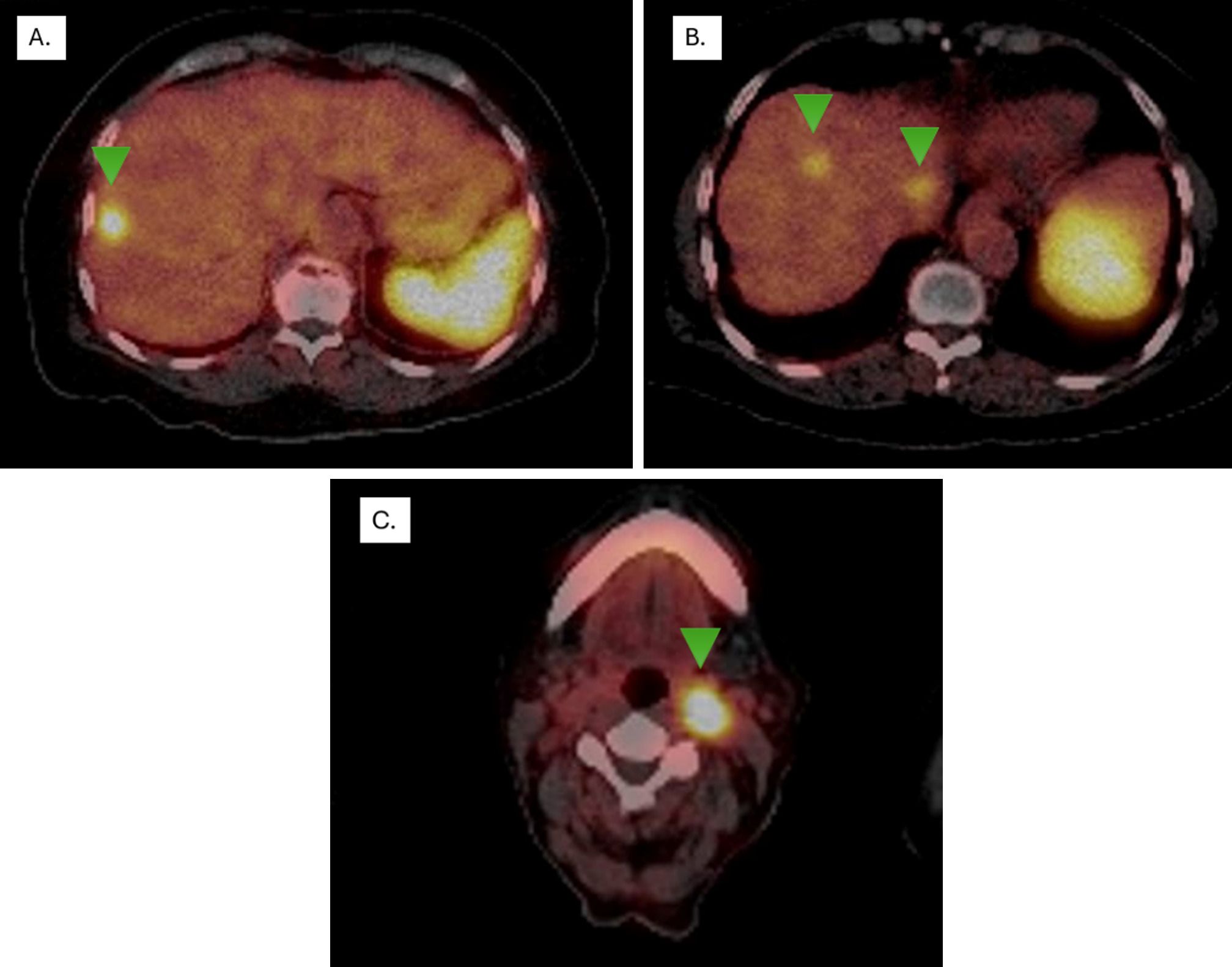
Figure 3. Ga-68 DOTA-péptido PETSCAN. Lesions with overexpression of somatostatin receptors (Green arrows) (A) segment VIII with a higher uptake, showing an SUVmax of 11.3 (previous study SUVmax 8.4), classified as Krenning 4, (B) segments VIII and I show SUVmax levels of 5.5 (previous study SUVmax 3.6) and 6.3 (previous study SUVmax 4.3), respectively, both classified as Krenning 3. (C) Lymph node in left station IIa, SUVmax of 40 (Krenning 4).
Given this finding, a PET-CT scan using 68Ga-labeled somatostatin analogues (SSAs) was performed, revealing high somatostatin receptor expression in liver lesions and a left cervical lymph node (Figure 1). A biopsy of the lymph node confirmed NET infiltration, with IHC showing positive staining for chromogranin and synaptophysin (Figure 3). Based on the metastasectomy findings and PET-CT results, which guided a tru-cut biopsy at the cervical level and confirmed a grade II NET, first-line treatment with somatostatin analogues was initiated. Unfortunately, MMR status or MSI testing in the NET could not be performed due to insufficient material in the tissue block.
The patient remains under follow-up at our institution, with stable NET findings. However, in 2023, imaging revealed a hypervascular nodule adjacent to the resection margin of the previous hepatic metastasectomy, suggesting possible tumor recurrence. Given the clinical and imaging findings, a multidisciplinary medical board determined that the tumor had not progressed, and no additional invasive biopsies were indicated. The patient continues treatment with subcutaneous lanreotide 120 mg every 28 days. Aside from CRC, the patient has not developed any other LS-related tumors. Following the LS diagnosis, she underwent a risk-reducing hysterectomy with bilateral salpingo-oophorectomy in late 2023. Segregation studies of the MLH1 germline variant could not be performed, as some family members were unreachable, while others declined testing.
Literature analysis
A literature search was performed in MEDLINE, EMBASE, and Web of Science. The search included articles published between January 2000 to December 2024, using key terms including “Lynch Syndrome” OR “hereditary nonpolyposis colorectal cancer (HNPCC)” AND “neuroendocrine neoplasm” OR “NEN” OR Neuroendocrine Tumor” OR “NET.” Additionally, we included searches for “MMR deficiency” OR “MSI” AND “Neuroendocrine neoplasm” OR “NEN” OR Neuroendocrine Tumor” OR “NET.”
We included articles reporting cases of NENs in patients with a clinical and molecular diagnosis of LS. Articles published in English and Spanish were considered. Only duplicate articles across databases were excluded; no exclusions were made based on article type. Ultimately, 11 articles were included for full-text analysis (Figure 4). Data extraction was conducted by MPBZ and DFMG, while AMGC and JCRM resolved any conflicts regarding case inclusion or data extraction.
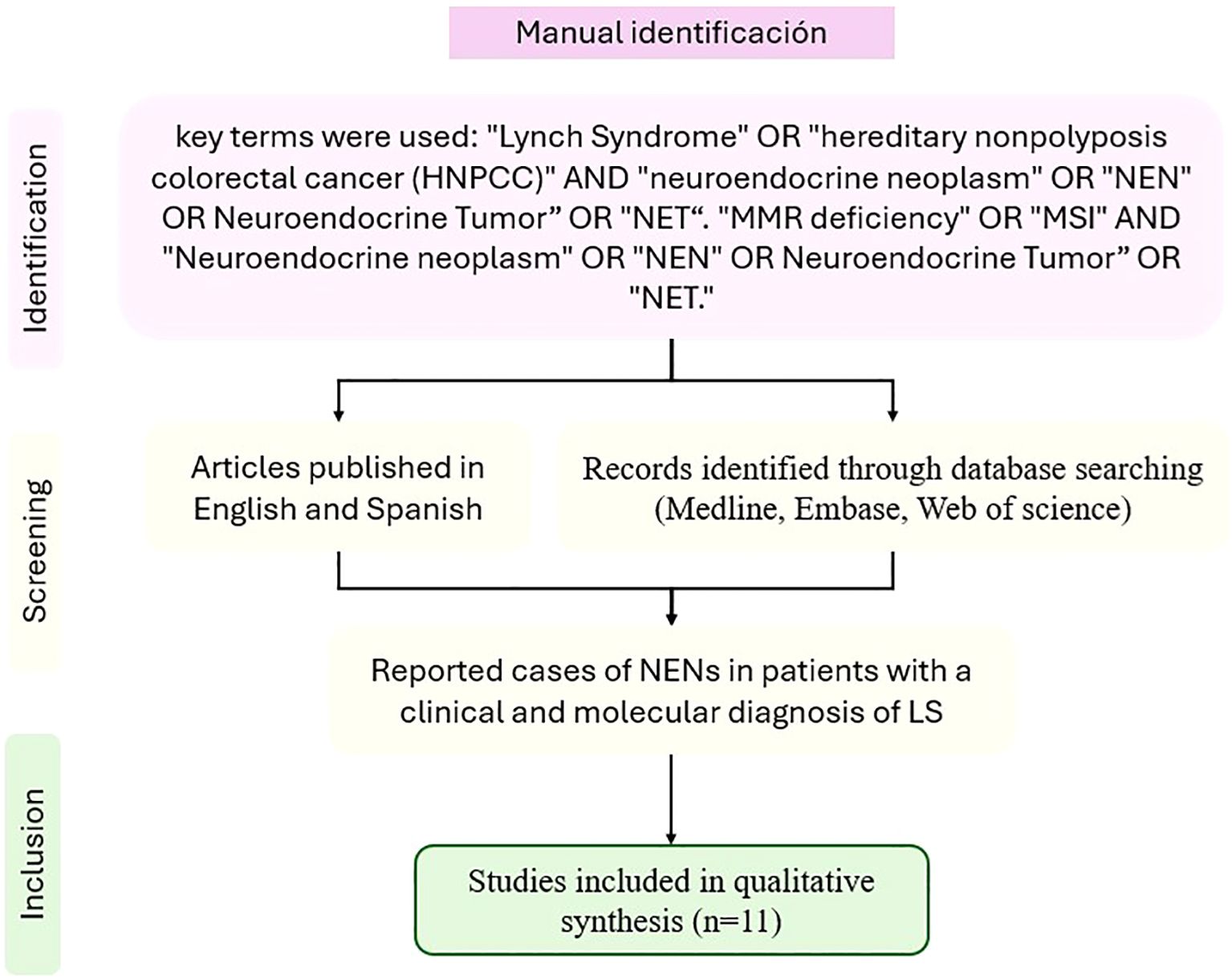
Figure 4. PRISMA flowchart for the manual literature review.
Results and discussion
LS confers an elevated risk for a spectrum of cancers (10), a spectrum that has broadened with enhanced disease surveillance (11). Prospective long-term follow-up studies have identified additional cancer types, including gastric, urinary tract, pancreaticobiliary, small intestinal, breast, and brain cancer. However, robust evidence supporting the efficacy of systematic screening for these less common LS-associated malignancies remains limited, with specialized screening primarily guided by strong familial aggregation of specific cancers (12). While NENs are not traditionally considered within the LS-associated cancer spectrum (3), we present a case of LS manifesting with metachronous rectosigmoid adenocarcinoma and a gastrointestinal NEN. This unusual presentation prompted a comprehensive literature review and analysis to further explore the potential biological and clinical relationship between LS and NENs.
Our literature review identified ten case reports and one case series, documenting thirteen unrelated cases (Table 1) describing the association between LS and NENs (13–17, 19–23) (summarized in Table 2). This cohort exhibited an equal sex distribution (50% female, 50% male). The mean age at first tumor presentation was 46.4 years, while the mean age at NEN diagnosis was 56.4 years. Of the fourteen individuals (including our case), seven received an LS diagnosis before the age of 50; however, only four developed a NEN at a similarly young age.

Table 1. Description of case reports on patients with neuroendocrine neoplasms associated with Lynch syndrome.
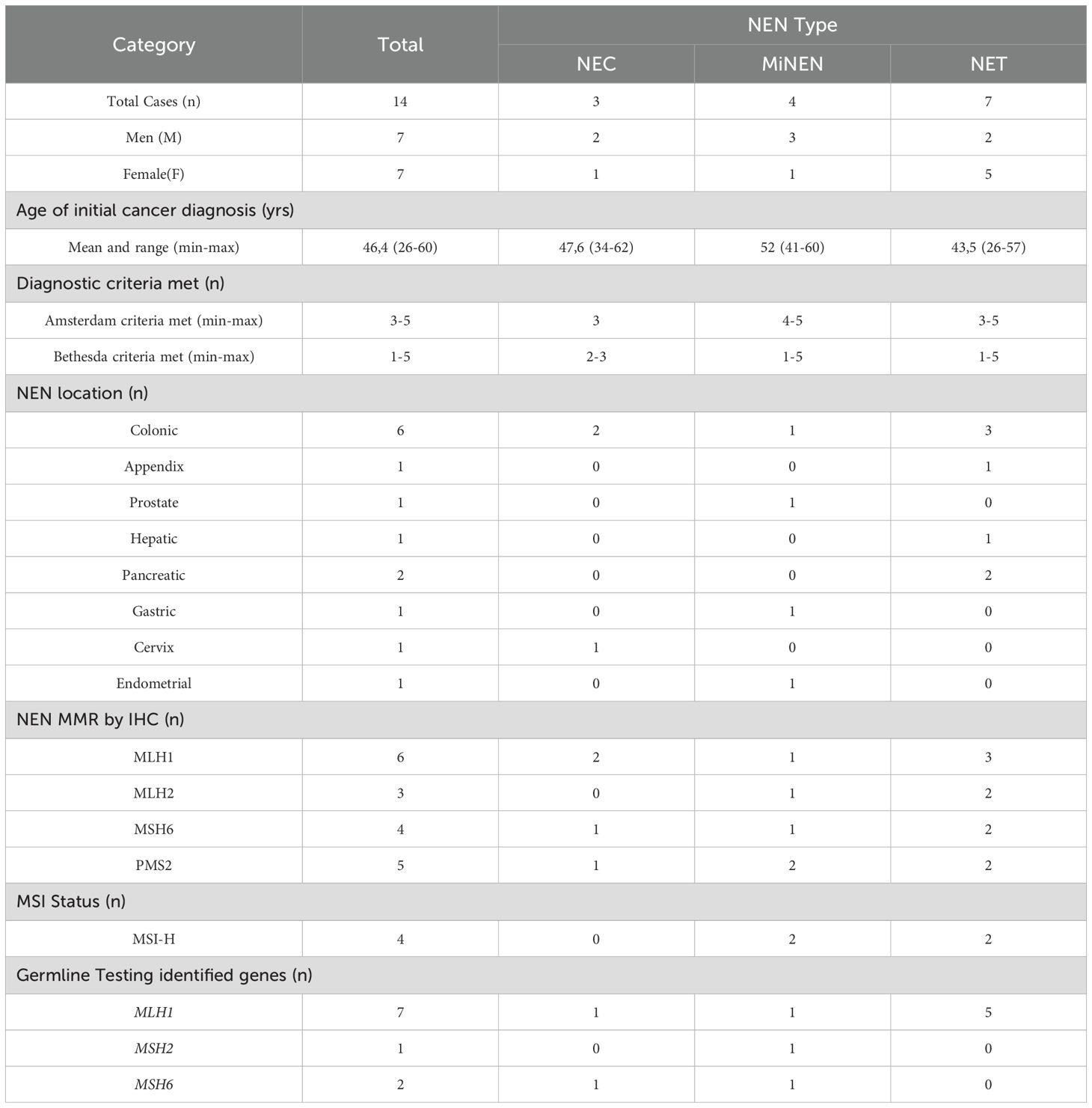
Table 2. Summary of reported cases of neuroendocrine neoplasms in Lynch syndrome patients.
All reported cases fulfilled at least one Bethesda criterion, although Amsterdam II criteria could not be fully assessed in two cases due to insufficient clinical information. Only three cases met all Amsterdam II criteria, while two cases satisfied all Bethesda criteria. Consequently, all cases identified can be considered LS-related based on established clinical guidelines. Notably, in three out of fourteen cases (21.4%), the NEN was the initial malignancy detected, preceding the diagnosis of LS.
The majority of reported NENs (11/14) originated within the gastrointestinal tract, with the colon being the most frequent primary site (6/11), followed by the pancreas (2/11), liver (1/11), stomach (1/11), and appendix (1/11). In our case, the NEN likely arose from an intestinal primary site. Other less common primary sites included the prostate and cervix, with presentations occurring either synchronously or metachronously with other LS-related tumors.
IHC analysis results for all four MMR proteins were available in only ten of the fourteen cases. Loss of MLH1 and PMS2 expression was the most frequent alteration (6/10 cases), followed by loss of MSH2 and MSH6 expression (2/10 cases). One case exhibited isolated MSH6 deficiency (a MiNEN), confirmed by germline molecular analysis, while another showed PMS2 loss (a MiNEN) without confirmatory somatic or germline studies. In the remaining cases, a single affected protein was reported without specifying the others, including MLH1 (2 cases) and MSH6 (1 case). Microsatellite instability (MSI) status was assessed in only four out of fourteen cases (two NETs and two MiNENs), all of which demonstrated high-frequency MSI (MSI-H).
The inherent heterogeneity of NENs complicates the establishment of direct and predictive correlations between MMR/MSI status, the specific molecular profile, and the NEN type. In Table 1 display seven cases exhibit an IHC dMMR pattern consistent with the molecular profile identified through germline genetic testing: (i) a high-grade hepatic NET with dMMR (MLH1) and a pathogenic germline variant in the MLH1 gene, (ii) a poorly differentiated colonic NEC with dMMR (MSH6) and a pathogenic germline variant in the MSH6 gene, (iii) a colonic NEC with dMMR (MLH1/PMS2) and a pathogenic germline variant in the MLH1 gene, (iv) a well-differentiated colonic NET with dMMR (MLH1/PMS2) and a pathogenic germline variant in the MLH1 gene, (v) a pancreatic NET with dMMR (MLH1/PMS2) and a pathogenic germline variant in the MLH1 gene, (vi) a colonic NEC with dMMR (MLH1/PMS2) and a pathogenic germline variant in the MLH1 gene, and (vii) a gastric NEN with dMMR (MSH6) and a pathogenic germline variant in the MSH6 gene. The observed correspondence between the IHC dMMR pattern and the germline molecular profile in these seven cases suggests the hypothesis that a portion of the pathogenic mechanisms underlying the development of a subgroup of NENs might be intrinsically linked to the pathophysiology of LS.
Regarding histology, seven out of fourteen cases were NETs, three were NECs, and four were classified as MiNENs, which included: high-grade prostatic intraepithelial neoplasia with neuroendocrine features (HGPIN-NE), poorly differentiated endometrial adenocarcinoma (80% large cell NET, 20% endometrioid adenocarcinoma), poorly differentiated gastric adenocarcinoma (30% large cell NET, 70% adenosquamous carcinoma), and colon adenocarcinoma with a large cell neuroendocrine carcinoma component. Notably, all NECs and MiNENs exhibited dMMR.
Several studies on non-LS-related NENs have indicated that dMMR/MSI-H is relatively common in colorectal NECs. MSI has been reported in three out of ten gastrointestinal NECs, with a prevalence ranging from 12.4% (24) to 30% (25). The most frequent finding is the loss of MLH1 and PMS2 expression, whereas dMMR/MSI-H remains highly uncommon in NENs originating from other anatomical sites. Interestingly, dMMR/MSI-H NECs exhibit distinct clinical behavior; none of the reported cases presented with distant metastases at diagnosis, and all were classified as stage II or III based on nodal involvement. The pathogenesis, clinicopathologic features, and molecular profiles of MSI-H NECs closely resemble those described for sporadic MSI-H adenocarcinomas of the stomach and colorectum.
Similar to MSI-H NECs, MSI-H colonic NETs exhibit pathogenetic and molecular characteristics comparable to MSI-H colonic adenocarcinomas (26). This similarity suggests that MSI-H colonic NETs may also benefit from early initiation of immune checkpoint inhibitor therapy. Although MMR status and MSI assessment could not be performed in our patient due to insufficient tissue material, the clinical presentation strongly suggests an MSI-H colonic NET phenotype, mimicking the behavior of MSI-H adenocarcinomas. Recognizing NENs as potential LS-related tumors could support the inclusion of MMR/MSI testing in clinical guidelines, facilitating early identification and tailored treatment approaches for LS patients. This would open new therapeutic avenues, with immunotherapy emerging as a promising strategy in this context.
Colorectal NECs and MiNENs are generally associated with poor prognosis (27). However, MSI or dMMR status has been linked to improved outcomes, likely because these tumors exhibit molecular and clinical behavior like MSI-positive adenocarcinomas (27). Due to this, some authors recommend routine IHC assessment of MMR proteins or MSI analysis in early-stage colorectal NECs and MiNENs to identify a subset of dMMR tumors with a significantly more favorable prognosis (28). Such testing should be conducted at the initial diagnosis to avoid delays in treatment decisions upon disease progression.
Given that colorectal NECs have a response rate of less than 50% to platinum-based chemotherapy and an overall survival of less than nine months, MSI testing at diagnosis could enable early initiation of immune checkpoint inhibitor therapy, mirroring treatment approaches used for MSI-H adenocarcinomas, which have shown clinical benefits in this setting (29).
In contrast, MSI is highly infrequent in non-LS-related NETs. However, studies analyzing larger tumor cohorts have reported MSI in 25.8% of colonic NETs and 13.3% of pancreatic NETs. It is important to note that these studies did not distinguish between patients with or without LS based on clinical or molecular criteria. In our review, dMMR was identified in six out of seven NETs (85.7%), with only one case being MMR-proficient (1/7), suggesting that LS could be associated with a distinct NET phenotype (18, 30).
Although our case and the other thirteen cases included in this analysis met the clinical criteria for LS, germline testing for MMR genes was reported in only ten out of fourteen cases. Specifically, five out of seven NETs (71.4%), three out of four MiNENs (75%), and two out of three NECs (66.7%) had available germline testing data. The most frequently affected gene was MLH1, identified in seven out of ten cases. These MLH1 variants included four deletions (three frameshift and one in-frame, the latter predicted to be pathogenic), two frameshift duplications, and one pathogenic missense variant. Additionally, two cases involved MSH6 frameshift variants (one duplication and one deletion), while one case reported a pathogenic variant in MSH2, though no further details were provided in the original report (Table 2). Notably, MiNEN and NEC cases exhibited heterogeneous germline results, with P/LP variants detected in MLH1, MSH2, and MSH6 (Table 2). In contrast, MLH1 variants were found in all tested NET cases, suggesting a potential genetic distinction between NETs and other NEN subtypes in LS.
Although the association between LS and NENs is uncommon, several mechanisms have been proposed to explain their potential relationship. First, P/LP variants in MMR genes, which define LS, lead to MSI. MSI, a hallmark of many LS-associated cancers, results in the accumulation of mutations throughout the genome, potentially increasing the risk of various malignancies, including rare tumors such as gastrointestinal NENs (31). Second, epigenetic alterations, such as promoter hypermethylation of tumor suppressor genes, are well-documented in LS-associated cancers. Similar epigenetic mechanisms may contribute to NEN development (32, 33). Lastly, individuals with LS may have an increased susceptibility to environmental carcinogens due to their deficient DNA repair system, potentially leading to the development of malignancies outside the classical LS spectrum, including NENs, particularly NETs (25, 34).
This evidence presents plausible mechanisms linking LS and NEN development, drawing upon established LS pathophysiology. The proposed mechanisms MSI-driven mutagenesis, epigenetic dysregulation, and increased susceptibility to environmental carcinogens, offer a biologically plausible rationale for this rare association. In our case, the presence of a P/LP variant in an MMR gene, confirming the LS diagnosis, directly supports MSI-driven mutagenesis. Determining the MSI status of both the adenocarcinoma and the NET in our case would further strengthen this link. While not investigated here, epigenetic alterations and environmental exposures remain potential contributing factors in LS-associated NENs. The metachronous presentation of two distinct tumor types within the LS spectrum, including a rare NEN, underscores the complex and potentially expanded tumor predisposition in individuals with germline MMR defects, highlighting the importance of comprehensive molecular characterization and long-term surveillance in LS patients, even for seemingly unrelated malignancies.
Conclusions
In the present study, we describe an unusual case of LS manifesting with metachronous tumors, specifically a rectosigmoid adenocarcinoma and a non-functional NET likely of gastrointestinal origin, which we compare with a compilation of reported cases of LS patients with NENs. To our knowledge, this represents the most extensive compilation of cases to date exploring the potential relationship between LS and NENs. Although the available data remain limited and primarily derived from isolated case reports, our clinical, pathological, and molecular analysis provides preliminary evidence supporting this possible association, albeit revealing significant heterogeneity in the presentation of NENs in patients with LS.
Our findings prompt us to suggest the incorporation of MMR/MSI testing in the analysis of patients with NENs, particularly in those with a family history of cancer suggestive of LS (meeting clinical criteria and/or with genetic confirmation). Furthermore, we propose considering the inclusion of clinical evaluation and, when appropriate, imaging studies directed at the detection of NENs in the surveillance protocol for patients with LS. Enhanced surveillance strategies for NENs, such as pNETs, may incorporate advanced imaging, including contrast-enhanced computed tomography (CECT) and apparent diffusion coefficient (ADC) with normalized values (35, 36).
The co-occurrence of NENs and LS, while rare, suggests a potential shared molecular pathway, at least in a subset of cases, influenced by dysfunction of the MMR system. The observed molecular similarities between dMMR/MSI-H gastrointestinal NENs and adenocarcinomas with the same profile, concerning prognosis and response to immunotherapy, warrants the prospective consideration of MMR/MSI status as a biomarker with prognostic, therapeutic, and predictive value in LS patients who develop NENs.
Despite the limited number of NEN cases documented in the literature within the context of LS, which hinders the identification of a definitive shared molecular pathway, this study constitutes a fundamental starting point for future investigations aimed at elucidating the nature of this interconnection and exploring potential causality. Large-scale prospective studies are necessary to investigate the prevalence of NENs in patients with LS and vice versa, as well as to characterize the specific molecular profile of these tumors and their potential response to targeted therapies, including immunotherapy.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/supplementary material.
Ethics statement
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
MB: Writing – original draft, Project administration, Formal Analysis, Conceptualization, Investigation. DM: Investigation, Conceptualization, Writing – review & editing. WT: Writing – review & editing, Investigation, Conceptualization, Supervision. AG: Writing – review & editing, Investigation, Conceptualization. DB: Writing – review & editing, Conceptualization. RP: Writing – review & editing, Conceptualization. JR: Writing – review & editing, Conceptualization, Formal Analysis, Project administration, Supervision, Writing – original draft.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. The Universidad Cooperativa de Colombia provided financial support for the publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Lippuner K, Grifone S, Schwenkglenks M, Schwab P, Popp AW, Senn C, et al. Comparative trends in hospitalizations for osteoporotic fractures and other frequent diseases between 2000 and 2008. Osteoporos Int. (2012) 23:829–39. doi: 10.1007/s00198-011-1660-8
2. Nolano A, Medugno A, Trombetti S, Liccardo R, De Rosa M, Izzo P, et al. Hereditary colorectal cancer: state of the art in Lynch syndrome. Cancers (Basel). (2022) 15:75. doi: 10.3390/cancers15010075
3. Weiss JM, Gupta S, Burke CA, Axell L, Chen LM, Chung DC, et al. NCCN guidelines® Insights: genetic/familial high-risk assessment: colorectal, version 1. 2021. J Natl Compr Canc Netw. (2021) 19:1122–32. doi: 10.1164/jnccn.2021.0048
4. Singh DN, Daripelli S, Elamin Bushara MO, Polevoy GG, and Prasanna M. Genetic testing for successful cancer treatment. Cureus. (2023) 15:e49889. doi: 10.7759/cureus.49889
5. Alexander ES and Ziv E. Neuroendocrine tumors: genomics and molecular biomarkers with a focus on metastatic disease. Cancers (Basel). (2023) 15:2249. doi: 10.3390/cancers15082249
6. Rindi G and Wiedenmann B. Neuroendocrine neoplasms of the gut and pancreas: new insights. Nat Rev Endocrinol. (2011) 8:54–64. doi: 10.1038/nrendo.2011.120
7. Rindi G and Wiedenmann B. Neuroendocrine neoplasia of the gastrointestinal tract revisited: towards precision medicine. Nat Rev Endocrinol. (2020) 16:590–607. doi: 10.1038/s41574-020-0391-3
8. Ahmed M. Gastrointestinal neuroendocrine tumors in 2020. World J Gastrointest Oncol. (2020) 12:791–807. doi: 10.4251/wjgo.v12.i8.791
9. Gut P, Komarowska H, Czarnywojtek A, Waligórska-Stachura J, Bączyk M, Ziemnicka K, et al. Familial syndromes associated with neuroendocrine tumours. Contemp Oncol (Pozn). (2015) 19:176–83. doi: 10.5114/wo.2015.52710
10. Dominguez-Valentin M, Sampson JR, Seppälä TT, Ten Broeke SW, Plazzer JP, Nakken S, et al. Cancer risks by gene, age, and gender in 6350 carriers of pathogenic mismatch repair variants: findings from the Prospective Lynch Syndrome Database. Genet Med. (2020) 22:15–25. doi: 10.1038/s41436-019-0596-9
11. Lynch HT, Snyder CL, Shaw TG, Heinen CD, and Hitchins MP. Milestones of lynch syndrome: 1895-2015. Nat Rev Cancer March. (2015) 15:181–94. doi: 10.1038/nrc3878
12. Yurgelun MB and Hampel H. Recent advances in Lynch syndrome: diagnosis, treatment, and cancer prevention. Am Soc Clin Oncol Educ Book. (2018) 38:101–9. doi: 10.1200/EDBK_208341
13. Wagner DG, Gatalica Z, Lynch HT, Kohl S, Johansson SL, and Lele SM. Neuroendocrine-type prostatic adenocarcinoma with microsatellite instability in a patient with Lynch syndrome. Int J Surg Pathol. (2010) 18:550–3. doi: 10.1177/1066896910379406
14. Sorscher S and Saroya B. A molecularly confirmed neuroendocrine tumor resulting from Lynch Syndrome. J Gastrointest Oncol. (2013) 4:95–6. doi: 10.3978/j.issn.2078-6891.2012.040
15. Karamurzin Y, Zeng Z, Stadler ZK, Zhang L, Ouansafi I, Al-Ahmadie HA, et al. Unusual DNA mismatch repair-deficient tumors in Lynch syndrome: a report of new cases and review of the literature. Hum Pathol. (2012) 43:1677–87. doi: 10.1016/j.humpath.2011.12.012
16. Yousef I, Siyam F, Layfield L, Freter C, and Sowers JR. Cervical neuroendocrine tumor in a young female with Lynch Syndrome. Neuro Endocrinol Lett. (2014) 35:89–94.
17. Kidambi TD, Pedley C, Blanco A, Bergsland EK, and Terdiman JP. Lower gastrointestinal neuroendocrine neoplasms associated with hereditary cancer syndromes: a case series. Fam Cancer. (2017) 16:537–43. doi: 10.1007/s10689-017-9979-8
18. Serracant Barrera A, Serra Pla S, Blázquez Maña CM, Salas RC, García Monforte N, Bejarano González N, et al. Pancreatic non-functioning neuroendocrine tumor: a new entity genetically related to Lynch syndrome. J Gastrointest Oncol. (2017) 8:E73–9. doi: 10.21037/jgo.2017.07.02
19. Whitman J, Kardosh A, Diaz L, Fong L, Hope T, Onodera C, et al. Complete response and immune-mediated adverse effects with checkpoint blockade: treatment of mismatch repair-deficient colorectal neuroendocrine carcinoma. JCO Precis Oncol. (2019) 3:1–7. doi: 10.1200/PO.19.00098
20. Dawley JC, Haroon Ahmad Y, Riall TS, and Sun BL. Gastric mixed large cell neuroendocrine-adenosquamous carcinoma with heterogeneous MSH6 loss in Lynch syndrome. Hum Pathol Rep. (2021) 26:300579. doi: 10.1016/j.hpr.2021.300579
21. Kobayashi N, Yoshida H, Kawaguchi S, Shiraso S, Nemoto N, Fujikawa N, et al. A case of strongly suspected Lynch syndrome with colorectal neuroendocrine carcinoma. Surg Case Rep. (2022) 8:114. doi: 10.1186/s40792-022-01471-0
22. Brignola C, Volorio S, De Vecchi G, Zaffaroni D, Dall’Olio V, Mariette F, et al. De novo germline pathogenic variant in Lynch Syndrome: A rare event or the tip of the iceberg? Tumori. (2024) 110:69–73. doi: 10.1177/03008916231197113
23. Siu WYS, Hong MK, and Ding DC. Neuroendocrine carcinoma of the endometrium concomitant with Lynch syndrome: A case report. World J Clin cases. (2023) 11:5160–6. doi: 10.12998/wjcc.v11.i21.5160
24. Sahnane N, Furlan D, Monti M, Romualdi C, Vanoli A, Vicari E, et al. Microsatellite unstable gastrointestinal neuroendocrine carcinomas: a new clinicopathologic entity. Endocr Relat Cancer. (2015) 22:35–45. doi: 10.1530/ERC-14-0410
25. Sorbye H, Welin S, Langer SW, Vestermark LW, Holt N, Osterlund P, et al. Predictive and prognostic factors for treatment and survival in 305 patients with advanced gastrointestinal neuroendocrine carcinoma (WHO G3): the NORDIC NEC study. Ann Oncol. (2013) 24:152–60. doi: 10.1093/annonc/mds276
26. Trifanov VS, Timoshkina NN, Gvaldin D, Mesheryakova M, Kolesnikov EN, Gaziev UM, et al. Study of microsatellite instability in neuroendocrine tumors of the pancreas and colon. JCO. (2021) 39:e16196–6. doi: 10.1200/JCO.2021.39.15_suppl.e16196
27. Lou L, Lv F, Wu X, Li Y, and Zhang X. Clinical implications of mismatch repair deficiency screening in patients with mixed neuroendocrine non-neuroendocrine neoplasms (MiNEN). Eur J Surg Oncol. (2021) 47:323–30. doi: 10.1016/j.ejso.2020.08.022
28. Luong TV, Nisa Z, Watkins J, and Hayes AR. Should immunohistochemical expression of mismatch repair (MMR) proteins and microsatellite instability (MSI) analysis be routinely performed for poorly differentiated colorectal neuroendocrine carcinomas? Endocrinol Diabetes Metab Case Rep. (2020) 2020:20–0058. doi: 10.1530/EDM-20-0058
29. Farha N, Hrabe J, Sleiman J, Beard J, Lyu R, Bhatt A, et al. Clinically actionable findings on surveillance EGD in asymptomatic patients with Lynch syndrome. Gastrointest Endosc. (2022) 95:105–14. doi: 10.1016/j.gie.2021.07.004
30. Helderman NC, Elsayed FA, van Wezel T, Terlouw D, Langers AMJ, van Egmond D, et al. Mismatch repair deficiency and MUTYH variants in small intestine-neuroendocrine tumors. Hum Pathol. (2022) 125:11–7. doi: 10.1016/j.humpath.2022.04.003
31. Boland CR and Goel A. Microsatellite instability in colorectal cancer. Gastroenterology. (2010) 138:2073–87.e3. doi: 10.1053/j.gastro.2009.12.064
32. Herman JG and Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. (2003) 349:2042–54. doi: 10.1056/NEJMra023075
33. Feinberg AP and Tycko B. The history of cancer epigenetics. Nat Rev Cancer. (2004) 4:143–53. doi: 10.1038/nrc1279
34. Lichtenstein P, Holm NV, Verkasalo PK, Iliadou A, Kaprio J, Koskenvuo M, et al. Environmental and heritable factors in the causation of cancer—analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med. (2000) 343:78–85. doi: 10.1056/NEJM200007133430201
35. Ren S, Guo K, Li Y, Cao Y, Wang Z, and Tian Y. Diagnostic accuracy of apparent diffusion coefficient to differentiate intrapancreatic accessory spleen from pancreatic neuroendocrine tumors. World J Gastrointestinal Oncol. (2023) 15:1051–61. doi: 10.4251/wjgo.v15.i6.1051
Keywords: lynch syndrome, neuroendocrine neoplasms (NENs), mismatch repair-deficient (dMMR), neuroendocrine tumor (NET), hereditary cancer
Citation: Bernal Zárate MP, Mendivelso-Gonzalez DF, Torres WC, González Clavijo AM, Ballen DF, Parra Medina R and Riaño-Moreno JC (2025) Neuroendocrine neoplasms as a lynch syndrome manifestation: a case report and comprehensive literature review. Front. Endocrinol. 16:1587889. doi: 10.3389/fendo.2025.1587889
Received: 05 March 2025; Accepted: 28 May 2025;
Published: 17 June 2025.
Edited by:
Alberto Bongiovanni, IRCCS Istituto Romagnolo per lo Studio dei Tumori (IRST) “Dino Amadori”, ItalyReviewed by:
Shuai Ren, Affiliated Hospital of Nanjing University of Chinese Medicine, ChinaMarie Louise Ndzie Noah, Shandong First Medical University, China
Copyright © 2025 Bernal Zárate, Mendivelso-Gonzalez, Torres, González Clavijo, Ballen, Parra Medina and Riaño-Moreno. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Julián C. Riaño-Moreno, anVsaWFuLmNhbWlsby5yaWFub0BnbWFpbC5jb20=