Abstract
Diabetic patients have a higher tendency for vascular calcification (VC). This indicates a possible link between abnormal glucose metabolism and the development of VC. High glucose levels are a major cause of vascular calcification in diabetic patients. Vascular smooth muscle cells (VSMCs) are important functional units of the arterial media and show heterogeneity. Sustained hyperglycemia drives VSMCs to undergo a phenotypic transition from contractile state to osteo-/chondrogenic lineages through multiple pathophysiological mechanisms. Specifically, hyperglycemia stimulates metabolic reprogramming. This includes enhancing advanced glycation end products (AGEs), activating the diacylglycerol-dependent protein kinase C (PKC) pathway, disrupting the pentose phosphate flux (PPP), and dysregulating the hexosamine biosynthesis pathway (HBP). These changes trigger vesicles-mediated mineralization (including matrix/extracellular vesicles), oxidative stress, inflammatory cascades, and an imbalance between autophagy and apoptosis. This review systematically describes the metabolic remodelling induced by high glucose and its regulatory mechanisms in vascular calcification.
Highlights
What is currently known about this topic?
-
Diabetic patients face a higher risk of coronary artery calcification, which is related to hyperglycemia.
-
VSMCs play a key role in vascular calcification and can differentiate into osteoblast-like cells.
-
VSMCs osteogenic differentiation is likely driven by specific diabetes mellitus–associated mechanisms. These include oxidative stress, PKC activation, PPP, and HBP. These processes regulated by key enzymes such as hexokinase 2 (HK2), pyruvate kinase M (PKM), β-N-acetylglucosaminidase (OGA) and β-N-acetylglucosaminyltransferase (OGT).
What is the key research question?
-
How does hyperglycemia induce the osteogenic differentiation of VSMCs?
-
what are the underlying molecular mechanisms?
What is new?
-
Elucidation of the complex regulatory network of metabolic reprogramming in VSMCs under diabetic conditions.
-
Discovery of the role of various microRNAs and extracellular vesicles in regulating VSMCs osteogenic differentiation.
1 Introduction
Forecasts indicate that by 2030, 552 million people will be diabetes (1–3) Cardiovascular calcification was found in 81.2% of diabetic patients and 33.7% of nondiabetic patients. Therefore, diabetic patients are highly prone to VC (4, 5). A better understanding of the molecular processes between high glucose and VC may accelerate the development of new biomarkers and targeted drugs for calcification.
VC is mainly divided into initial and medial calcification (6). VSMCs are important components of the vascular media. In diabetes, hyperglycemia disrupts redox homeostasis and triggers inflammatory signaling, oxidative stress, formation of Ca-Pi crystals, O-GlcnAcylation, promoting the osteogenic differentiation of VSMCs (7–10).
During the osteogenic differentiation of VSMC, hyperglycemia upregulates expression of osteopontin (OPN) and osteoprotegerin (OPG) and then activates Msh homeobox-2 (MSX2) and Runt-related transcription factor 2 (RUNX2) through the wingless - type MMTV integration site family (WNT)/β-catenin and bone morphogenetic protein-2 (BMP-2) pathways (11–14). The mechanisms of VSMCs calcification caused by diabetes have been a hot topic of research in recent years, which mainly include oxidative stress, inflammation, death regulation, matrix vesicle formation, mineral deposition (15–17).Under the high glucose condition, glucose metabolic reprogramming is crucial for the phenotypic transformation of VSMCs and contributes to vascular remodeling (18–20). The complex interaction between metabolic reprogramming, signaling pathway activation, cell fate decisions, and post-transcriptional regulation underlies the osteogenic differentiation of VSMCs in diabetes. Besides, high glucose impairs the pyrophosphate-to-phosphate ratio and induce calcium deposition, disrupts extracellular pyrophosphate and calcium metabolism (21). Despite some progress in recent years, the impact of hyperglycemia on VSMCs osteogenic differentiation remains unclear. In-depth studies are needed to clarify the detailed mechanisms of VSMCs osteogenic differentiation in diabetes. Therefore, this reviewer pays attention to the pathophysiology of VSMCs osteogenic differentiation induced by hyperglycemia.
2 VSMCs’ osteogenic differentiation: a key event in VC
The etiology of VC involves the osteogenic differentiation of VSMCs (22). VSMCs exhibit phenotypic plasticity (23). With the development of high-throughput detection technologies such as single-cell and transcriptomics sequencing analysis, it is realized that VSMCs can be transformed into pro-inflammatory chemotactic like, macrophage-like/foam-like, and fibroblast/chondroblast-like smooth muscle cells (24–26). Among them, the osteogenic transformation of VSMCs is an important link in vascular calcification. During VSMCs’ osteogenic differentiation, the levels of MSX2, BMP2, RUNX2, osterix, sex-determining region Y-box 9 (SOX9) increase. These upregulate osteocytic and chondrocyte markers [such as OC (osteocalcin), Col1α1 (collagen type I α1), OPN, and alkaline phosphatase (ALP)] and downregulates contractile markers like smooth muscle 22 α (SM22α), calponin, and smooth muscle myosin heavy chain (SM-MHC) (27–31) (Figure 1). With the deepening of scientific research, a large number of regulatory factors will be discovered, which will provide new therapeutic targets for osteogenic transformation of VSMCs. For example, high-mobility group box-1 (HMGB-1) promotes vascular calcification in diabetic mice via endoplasmic reticulum stress (15). Non-POU domain-containing octamer-binding protein (NONO) or octamer-binding transcription factor 4 (OCT4) directly bound to the BMP2 promoter and inhibited BMP2 transcription, which protected against osteogenic differentiation of VSMCs (32, 33).
Figure 1

The role of VSMCs in Vascular calcification under the high glucose condition.
3 Glucose metabolic reprogramming and VSMCs osteogenic differentiation
Diabetic patients display elevated glucose in the blood, which lead to VSMCs dysfunction and significantly alter their metabolism. Previous studies have shown that in calcified VSMCs, glucose consumption and lactate generation increase due to a shift towards glycolysis (34, 35). During this process, high glucose promotes glucose uptake through glucose transporter 1 (GLUT1) and reprograms glucose metabolism by regulating activity of HK2, PDK4, 6-phosphofructokinase isozyme 1 (PFK1), PKM2, and lactate dehydrogenase A (LDHA) in VSMCs (12) (Figure 2). Therefore, clarifying these metabolic changes could help identify new therapeutic targets for vascular calcification.
Figure 2

The contribution of unscheduled glycolysis to osteogenic differentiation of vascular smooth muscle cells.
3.1 GLUT1: the gateway for glucose uptake in VSMCs
GLUT1 is the main isoform of the facilitative GLUT family and mediates glucose uptake in VSMCs (36). It is highly expressed in calcified VSMCs (37). In rodent human arterial smooth muscle cells (HASMCs) and aortic smooth muscle cell lines (A7r5), GLUT1 overexpression increased the intracellular glucose concentration by 44% and enhanced glycolytic flux and tricarboxylic acid cycle activity (TCA) (38–41). Hyperglycemia increase the GLUT1 expression (42). Downregulating GLUT can prevent excessive glucose influx, reducing intracellular protein glycation and free radical generation; both of these are harmful in the development of vascular disease in diabetes (43). High Mobility Group Box 2 (HMGB2) decreased GLUT1 expression and promoted GLUT4 translocation through PPAperoxisome proliferator-activated receptor-γ (PPAR-γ)/silent mating type information regulation 2 homolog 1(SIRT1) (44). Glycogen synthase kinase-3 (GSK-3), an enzyme that hinders the conversion of glucose to glycogen, can inhibit GLUT1 expression and glucose uptake through the tuberous sclerosis complex subunit 2 (TSC2)/mammalian target of rapamycin (mTOR) pathway (38). Ya-Rong Zhang et al. highlighted that intermedin alleviated diabetic vascular calcification by inhibiting GLUT1 through the activation of cyclic AMP (cAMP)/protein kinase A (PKA) signaling pathway (42)(Figure 3; Table 1). Therefore, GLUT1 serves as the first line of defense against VSMCs calcification induced by high glucose. Therefore, developing targets for inhibiting the regulatory expression of GLUT1 is of great clinical significance.
Figure 3

The role of AGEs/RAGE in mediating osteogenic differentiation of VSMCs under the high glucose condition.
Table 1
| Metabolic Pathways | Key Molecules/Enzymes | Biological Effect | Related Research Examples/Mechanisms |
|---|---|---|---|
| Glucose uptake | GLUT1 | Increased glucose uptake intracellular protein glycation and free radical generation |
1 HMGB2 decreased GLUT1 expression through PPAR-γ/SIRT1 (44). 2 GSK-3 can inhibit GLUT1 expression and glucose uptake throughTSC2/mTOR pathway (38). 3 Intermedin alleviated diabetic vascular calcification by inhibiting GLUT1 through the activation cAMP/PKA signaling pathway (42). |
| Pentose Phosphate Pathway (PPP) | HK2 | ROS MMP hyperpolarization apoptosis suppression |
1 PVT1 affects the glycolysis and phenotypic switch of VSMCs through HK2 (50). 2 Indirubin-3′-monoxime reduced HK2 expression and glycolysis in VSMCs induced by PDGF-BB (51). |
| G6PD | Activation of NADPH oxidase Inflammation Apoptosis inhibition |
1 Role of G6PD in tumor necrosis factor receptor-associated factor 6-induced SM22α ubiquitination and mitochondrial apoptosis inhibition through the voltage-dependent anion channel 1-Bcl-2 associated X protein pathway (54, 55). 2 Inhibiting Ca2+ uniporter or mitochondrial calcium uptake 1 overexpression on VSMCs from diabetic mice decreased activity of G6PD, and normalized cell proliferation (56). |
|
| Hexosamine Biosynthetic Pathway (HBP) | OGT, OGA | Upregulation of VCAM-1and RUNX2, Migration Autophagy inhibition |
1 Polymerase delta interacting protein 2 deficiency enhanced OGT-mediated protein O-GlcNAcylation (63). 2 STIM1 deficiency-induced impairment of calcium homeostasis and ER stress enhances protein O-GlcNAcylation in VSMCs (71). |
| Advanced Glycation End Products (AGEs) | AGEs, RAGE | ROS Autophagy and apoptosis |
1 Metformin inhibits upregulation of ALP and RUNX2 and downregulation of SM22α induced AGEs surplus (74, 80). 2 Intermedin exerts anti-calcification effects by inhibiting RAGEs via cAMP/PKA signaling pathway activation in diabetic vascular calcification (42). 3 USP10 alleviates CEL-induced vascular calcification and atherogenesis in diabetes mellitus by promoting AMPK activation (89). 4 The natural compound thonningianin A decreases expression of RUNX2, BMP2 and OPN via ATG7-dependent autophagy in HG-stimulated MASMCs (167). |
| Diacylglycerol-dependent PKC pathway activation | GADPH | Inducing intracellular Ca2+ Inflammation |
1 Activation of PKC-𝜁 inducing by AGE-RAGEs activates TGF-𝛽, NF-𝜅B, and p38 MAPK, inducing VSMCs to switch their phenotype into osteoblast-like (9, 29, 79, 95–98). |
| Aerobic glycolysis | LDHA | Inducing fission of mitochondria mitophagy inhibition |
1 Activation of κ-Opioid receptor impedes the calcification of VSMCs through decreasing lactate and PFKFB3 (116). 2 Prohibitin 2 deficiency facilitated PKM1/2 mRNA splicing and reversion from PKM1 to PKM2, and enhanced glycolysis in VSMCs (123). 3 GMRSP inhibits hnRNPA2B1-mediated alternative splicing PKM pre-mRNA, leading to reduced PKM2 production and glycolysis (124). 4 Deficiency of March2 lessened PKM2 dimer-to-tetramer conversion in VSMCs and promoted p53-driven apoptotic transcriptional response (125). |
Glucose metabolic pathways involved in VSMCs osteogenic differentiation.
VSMCs, Vascular smooth muscle cells; GSK-3, Glycogen synthase kinase-3; TSC2/mTOR, tuberous sclerosis complex subunit 2/mammalian target of rapamycin; cAMP/PKA, activation of cyclic AMP/protein kinase A; PVT1, Plasmacytoma variant translocation 1; PDGF-BB, platelet-derived growth factor-BB; PKM, pyruvate kinase M; HK2, Hexokinase 2; GLUT 1, Glucose transporter 1; G6PD, Glucose-6-phosphate dehydrogenase; AGEs, Advanced glycation end products; RAGE, Receptor for AGEs; MMP, mitochondrial me mbrane potential; AMPK, AMP-activated protein kinase; mTOR, mammalian target of rapamycin; ROS, Reactive Oxygen Species; TCA, Tricarboxylic Acid Cycle; ATP, Adenosine Triphosphate; NADPH, Nicotinamide adenine dinucleotide phosphate; HIF-1α, Hypoxia-Inducible Factor-1α; PPAR, Peroxisome Proliferator-Activated Receptor; SIRT, Sirtuin; PI3K, Phosphatidylinositol 3-Kinase; AKT, Protein Kinase B; MAPK, Mitogen-activated protein kinase; hnRNP, heterogeneous nuclear ribonucleoprotein; March2, membrane-associated RING finger protein 2; PFKFB3, Phosphofructo-2-kinase/fructose-2,6-biphosphatase 3; GMRSP, Glucose metabolism regulatory protein; LDHA, Lactate dehydrogenase A; GADPH, Glyceraldehyde-3-Phosphate Dehydrogenase; PVTC1, Plasmacytoma variant translocation 1; STIM1, Stromal interaction molecule 1; SM22α, Smooth muscle 22 α; Atg7, Autophagy Related 7; CEL, Carboxyethyl lysine; OGT, β-N-acetylglucosaminyltransferase; OGA, β-N-acetylglucosaminidase; UPS10, Ubiquitin-Specific Protease 10; VCAM-1, vascular cell adhesion molecule-1; TGF-β, Transforming growth factor-β.
3.2 Upstream glycolysis overload and VSMCs osteogenic differentiation
In the upper glycolysis process, hyperglycemia disrupts glycolytic flux, causing the accumulation of intermediate metabolites such as dihydroxyacetone phosphate (DHAP), glyceraldehyde-3-phosphate (GA3P), glucose-6-phosphate (G6P), and pyruvate. 6-Phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3), AMP-activated protein kinase (AMPK) strictly regulates glycolytic flux. PFKFB3 - mediated glycolysis enhances the osteogenic trans-differentiation of VSMCs by modulating Forkhead box O3(FoxO3) expression and lactate generation (45). AMPK regulates glucose metabolism reprogramming and osteogenic differentiation of VSMCs by affecting pathways like PPP and HBP and increasing lactate production. High risk human carotid atherosclerotic plaques, characterized as symptomatic, vulnerable, and inflamed, were reported to exhibit enhanced glycolysis and PPP pathways and elevated amino acid utilization (45). However, little is known about the mechanisms by how VSMCs regulate this complex network. The following part mainly introduces the main metabolic links of glucose metabolism and the influence of their interactions on the calcification process of vascular smooth muscle cells.
3.2.1 PPP and oxidative stress
In rat pulmonary artery smooth muscle cells (PASMCs) cultured in high glucose, the activation of the pentose phosphate pathway (PPP) affects upper glycolytic metabolites (46). Over-activation of the PPP is a key mechanism for the vascular damage related to hyperglycemia (47).
HK2, a key enzyme that phosphorylates glucose into glucose-6-phosphate, has become the predominant regulator of glycolysis in VSMCs. When high glucose levels exceed the catalytic ability of hexokinase, the surplus glucose enters the polyol pathway. These processes lead to the accumulation of reactive oxygen species (ROS), increasing oxidative stress (19, 48) (Figure 2). Over-expression of HK2 in human umbilical vein smooth muscle cells results in the mitochondrial membrane potential hyperpolarization and apoptosis suppression (49). Mengying Wu et al. showed that plasmacytoma variant translocation 1 (PVT1) affects the glycolysis and phenotypic switch of VSMCs through HK2 (50). Elke H. Heiss et al. found that indirubin-3′-monoxime reduced HK2 expression and glycolysis in VSMCs induced by platelet-derived growth factor (PDGF) (Table 1). Activation of signal transducer and activator of transcription (STAT) 3 could be identified as crucial event in upregulation of HK2 and glycolytic activity in PDGF-stimulated VSMCs (51). Xiao-Fei Gao et al. found m6A modification of profilin-1 interacted with the phosphorylation of ANXA2 (annexin A2) by recruiting SRC proto-oncogene, nonreceptor tyrosine kinase (Src), promoting the phosphorylation of signal transducer and activator of transcription 3 (STAT3) in VSMCs (52). Yanlin Huang et al. demonstrates that ANXA2 promotes osteogenic differentiation and inhibits cellular senescence of periodontal ligament cells (PDLCs) in high glucose conditions (53). The function of ANXA2/Src/STAT pathway on VSMCs calcification is need further study. Suppressing glycolytic enzymes such as phosphofructokinase (PFK)1 and PKM2 can redirect glycolysis towards the PPP (46). Glucose-6-phosphate dehydrogenase (G6PD) is PPP’s rate-limiting enzyme. Inhibition of G6PD in vascular cells not only prevents the over-activation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase but also reduces subsequent inflammation (47). Recent studies have highlighted the role of G6PD in tumor necrosis factor receptor-associated factor 6-induced SM22α ubiquitination and mitochondrial apoptosis inhibition through the voltage-dependent anion channel 1-Bcl-2 associated X protein (Bax) pathway (54, 55). Inhibiting Ca2+ uniporter or mitochondrial calcium uptake 1 overexpression on VSMCs from diabetic mice decreased activity of G6PD, and normalized cell proliferation (56; Table 1). Tumor protein 53 (p53) plays a role in inhibiting cancer cell proliferation and promoting apoptosis by inhibiting G6PD (57, 58). Additionally, p53 can regulate cell apoptosis by binding to VDAC1 (59, 60). However, it is still unclear whether p53 is involved in G6PD-VDAC1 mediated VSMC apoptosis.
Under high glucose conditions, the excessive activation of PPP is associated with the dysregulated regulation of HK2 and G6PD. Inhibition of 6-phosphofructokinase-1(PFK1) and PKM2 can redirect glycolysis to the PPP. Future studies could delve into the specific molecular mechanisms of ANXA2 and G6PD in glucose metabolism remodeling and calcification of VSMCs, as well as the synergistic regulatory network among key enzymes of different glucose metabolic pathways (Figure 2).
3.2.2 HBP and protein O-GlcNAcylation
The HBP can metabolize glucose into the active O-GlcNAcylation sugar donor UDP-β-D-N-acetylglucosamine. This pathway is dynamically regulated by OGT and OGA (61) (Figure 2). In hyperglycemic ApoE-/- mice fed a Western diet, deletion of smooth muscle-specific OGT prevents atherosclerosis development (62). F., et al. found polymerase delta interacting protein 2 (Poldip2) deficiency enhanced OGT-mediated protein O-GlcNAcylation, which inversely promoted myocardin, MRTFA (myocardin-related transcription factor A), and SRF (serum response factor) expressions (63; Table 1). These decreased the myocardin-dependent VSMCs marker gene expression and increased RUNX2-dependent osteogenic gene expression (64–67).
Under high glucose conditions, O-GlcNAcylation of nuclear factor-κB (NF-κB) in rat VSMCs induces upregulation of vascular cell adhesion molecule-1(VCAM-1) (68). Meanwhile, Barnes et al. demonstrated that higher O-GlcNAcylation of specificity protein 1 (sp1) in PASMCs promotes cell migration (69). Jack M Heath et al. demonstrated O-GlcNAcylation of protein kinase B (AKT) at two new sites, T430 and T479, promotes AKT phosphorylation, and then promotes RUNX2 trans-activity and VSMCs calcification (70). Besides, stromal interaction molecule 1 (STIM1) deficiency-induced impairment of calcium homeostasis and ER stress enhances protein O-GlcNAcylation in VSMC, which promotes VSMCs osteogenic differentiation and calcification in diabetes (71; Table 1). In other hand, O-GlcNAc signaling enhanced the osteogenic conversion of VSMCs through regulation of canonical Wnt/β-catenin pathway. Indeed, O-GlcNAcylation of β-catenin further increased it transcriptional activity in VSMCs (72). Besides, OGT enhances O- GlcNAcylation of kelch like ECH associated protein 1(KEAP1), leading to nuclear factor erythroid 2-related factor 2 (NRF2) degradation and subsequently inhibiting autophagy in VSMCs (73).
O-GlcNAcylation of cellular proteins such as Sp1, NF-κB, and Runx2 regulates VSMCs calcification, inflammation, migration, and the development of atherosclerosis. Future studies can further explore the specific targets and functions of O-GlcNAcylation modification, such as RUNX2.
3.2.3 Increased formation of AGEs
Notably, type II diabetes patients have much higher AGEs concentrations than non-diabetics (29–31). Accumulated studies have shown AGEs surplus promote osteogenic phenotype transformation of VSMCs by increasing the levels of ALP and RUNX2 and decreasing the expression of SM22α (74–79). This effect can be inhibited by metformin (80; Table 1). Poetsch, F., et al. reported that AGEs increased the expression of serum and glucocorticoid-inducible kinase 1 (SGK1) and induced the osteogenic trans-differentiation of VSMCs under high glucose conditions in a NF-κB-dependent manner (81). Besides, AGEs upregulate the expression of PDK4, which inhibits the conversion of pyruvate to acetyl-CoA, ultimately reducing Kreb’s cycle flux and exacerbating calcium deposition during VSMCs calcification (7) (Figures 2, 3). This effect further promotes the production of AGEs, thereby further expanding the role of AGEs in promoting VSMCs calcification.
AGEs bind to corresponding receptors, such as the receptor for AGEs (RAGE), AGE receptor 1(AGE - R1), AGE - R2, and AGE - R3, to trigger intracellular signaling cascades or exert their effects (82). RAGEs have been reported to exist in unstable plaques with microcalcifications by co-localizing with inflammatory cells and VSMCs undergoing osteochondrogenic differentiation (83). AGEs/RAGEs initiate the activation of PKC-ζ, which then activates the downstream signaling pathways mediated by p38 mitogen-activated protein kinase (p38 MAPK) and NF-κB (83, 84). (Figure 3). Intermedin exerts anti-calcification effects by inhibiting RAGEs via cAMP/PKA signaling pathway activation in diabetic vascular calcification (42; Table 1). AGEs/RAGE induce autophagy in HASMCs via the mechanistic target of rapamycin (mTOR) signaling pathway and trigger apoptosis, contributing to calcification (85). MTMR7 suppresses this effect (86). Recent study revealed that AGEs increased the activity of nuclear factor 90 (NF90), thereby promoting AGER1 degradation and ubiquitination via WD repeat domain-containing 7 and E3 ubiquitin ligase F-box’s mRNA stabilization in VSMCs induced by high glucose (87). (Figure 3) Therefore, AGEs receptors play important role in VSMCs calcification, death and inflammation. AGER and RAGEs may be targets to protect against VC.
Currently, carboxyethyl lysine (CEL), pentosidine, carboxymethyl-lysine (CML), and more than twenty other kinds of AGEs have been identified (88). CML was significantly increased in calcified ateries from diabetic atherosclerosis ApoE-/- mice fed with high-fat diets (89). The study found that CML promoted vascular calcification through different pathways in diabetes, including p38 MAPK pathway and the nuclear factor of activated T-cells 1 (NFATc1) (90, 91). In NFATc1pathway, Protein tyrosine kinase 2 (FAK) and SIRT3 affected the nuclear translocation of NFATc1 by regulating the acetylation-phosphorylation crosstalk. Besides, CML mediates vascular calcification in diabetic plaques by impaired osteoclastic bone resorption through NFATc1-N-acetylglucosamine-1-phosphate transferase (GNPTAB) (92). Ubiquitin-Specific Protease 10 (USP10) alleviates CEL-induced vascular calcification and atherogenesis in diabetes mellitus by promoting AMPK activation (89; Table 1). Furthermore, CML increased the expression of PDK4 by increasing ROS (35).
3.2.4 Diacylglycerol-dependent PKC pathway activation
Hyperglycemia induces accumulation of upper glycolytic intermediate glyceraldehyde-3-phosphate and increases diacylglycerol (DAG) production, which activates the PKC pathway, including PKCβ, PKCδ, and PKCα (93). G6PD can be activated by PKC to induce intracellular free Ca2+ to enhance the contraction of VSMCs (94). Additionally, activation of PKC-ζ inducing by AGE-RAGEs further activates transforming grow factor-β (TGF-β), NF-κB, and p38 MAPK, inducing VSMCs to switch their phenotype into osteoblast-like (9, 29, 79, 95–98) (Figure 3; Table 1).
3.3 Involvement of lower glycolysis overload in DR
Once pyruvate is generated, it can be converted to CO2 and acetyl-CoA, which enter the tricarboxylic apvt1cid cycle (TCA). Another fate of pyruvate is to be converted into lactate catalyzed by LDHA (99). The pyruvate dehydrogenase enzymatic complex (PDH) is an important link between glycolysis and the TCA (100). Reprogramming of glucose metabolism from mitochondrial oxidative phosphorylation (OXPHOS) to aerobic glycolysis has been observed during VSMCs phenotype switching (45). As regulators of mitochondrial glucose metabolism, PDKs inhibit the activity of PDH, slowing down the TCA and mediating the glucose metabolic switch of glucose metabolism from OXPHOS (100, 101). It has been made sure that PDK4 regulates VSMCs’ metabolic reprogramming towards a higher glycolysis rate, promoting lactic acid generation in the cytosol. This triggers mitochondrial dysfunction, calcium deposition and apoptosis and autophagy, inducing the osteogenic differentiation of VSMCs via direct SMAD1/5/8 phosphorylation and enhancing CBFα1 and BMP2 signaling (7, 35, 48, 102, 103).
3.3.1 LDHA
LDHA is key regulatory enzyme that catalyzes the production of lactate (100). Previous studies have shown that knockout of LDHA suppresses the survival, proliferation, migration, and invasive abilities of VSMCs (104, 105). Further investigations revealed that LDHA expression is regulated by forkhead box M1(FOXM1) via binding to the LDHA promoter (106). Recent study found that YTH N6-Methyladenosine RNA Binding Protein F1 (YTHDF1) recognized vir like M6A methyltransferase associated (KIAA1429)-methylated FOXM1 mRNA and raised FOXM1 stability. This promotes the levels of glycolysis-enhancing genes (GLUT1 and LDHA) and lactate production in multiple myeloma. There is no report on whether GLUT1/LDHA regulated by stabilizing FOXM1 mRNA is involved in the calcification of smooth muscle cells.
Upregulation of LDHA inducing surplus of lactate indue fission of mitochondria associated with dynamin-related protein 1 (Drp1) and impedes phosphatase and tensin homolog (PTEN)-induced mitophagy. PTEN-induced mitophagy mediate through putative kinase 1/parkin via the poly (ADP-ribose) polymerase 1 (PARP1)/DNA polymerase gamma (POLG)/uncoupling protein 2(UCP2) axis. Therefore, knockdown of UCP2 impedes fission of mitochondria as mediated by Drp1, while also partially restoring mitophagy via PTEN-induced putative kinase 1(PINK1)/Parkin in VSMC calcification (107) (Figure 4). Recent study found that PARP-1 is subjected to NEDD8 conjugation, leading to an increase in PARP-1 activity during VC. During this process, PARP-1 NEDDylation is mediated by the E3 ligase CBL proto-oncogene B (CBL-b) and is reversed by NEDD8-specific protease 1 (NEDP-1) during VC (108). Besides, lactate also accelerates VSMCs calcification through suppression of Bcl-2–19 kDa interacting protein (BNIP3)-mediated mitophagy (109). High glucose triggers lactate/G protein-coupled receptor 81(GPR81) and inhibits peroxisome proliferator-activated receptor-γ (PPAR-γ)/SIRT1 pathway, promoting vascular remodeling. HMGB2 promotes HASMC proliferation and vascular remodeling by regulating glucose metabolism through the PPAR-γ/SIRT1/PGC-1α pathway (44, 110–112). As downstream targets of the above two pathways, Peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) overexpression can reduce ROS-mediated VSMC migration. PGC-1α/NRF2/STAT3 is involved in decreasing VC by increasing superoxide dismutase 2 (SOD2) (110–112). PPAR-γ agonists alleviated periostin-promoted VSMCs calcification and corrected abnormal glycolysis and unbalanced mitochondrial homeostasis (113). 12,13-diHOME suppressed the up-regulation of carnitine O-palmitoyltransferase 1 (CPT1A) and CPT1A-induced succinylation of HMGB1. The succinylation of HMGB1 at the K90 promoted the protein stability and induced the enrichment of HMGB1 in cytoplasm, which induced the calcification in VSMCs (114). PFKFB3 is associated with diabetic atherosclerosis and vascular remodeling through increasing lactic acid and LDHA levels (115). Activation of κ-Opioid receptor impedes the calcification of VSMCs through decreasing lactate and PFKFB3, thus becoming a possible medicinal approach and target for vascular calcification treatment (116; Table 1). Besides, lactate induces lactylation at the K18 site of the H3 histone protein to up-regulate chitinase 3 like 1 (CHI3L1). CHI3L1 forms a polymer complex with interleukin 13 receptor subunit alpha 2 and interleukin 13 (IL-13), activates the janus kinase 1(JAK1)/STAT3/RUNX2 signaling pathway. These findings indicate that regulating lactylation and targeting inhibition of CHI3L1 and IL-13 represent a new therapeutic strategy to reduce arterial calcification in diabetes (117).
Figure 4

The fate and potential mechanism of VSMCs under the high glucose condition.
Therefore, as an important regulatory element in lactate metabolism regulation, PGC-1α serves as a hub for cell death regulation and is involved in apoptosis, senescence, and autophagy caused by mitochondrial dysfunction (Figure 4).
3.3.2 PKM2
The expression of PKM2 is greater in the VSMCs of atherosclerotic plaques than in those of in normal arteries (118). Tetrameric PKM2 is known to facilitate lactate production by regulating aerobic glycolysis, facilitating VSMC phenotypic switching (119). During this process, elevating crotonylation of PKM2 at K305 promotes the PKM2 dimeric form (120). Interestingly, nuclear dimeric PKM2 is implicated in the activation of the hypoxia-inducible factor-1 alpha (HIF-1α), STAT3, and β-catenin signaling pathways (121, 122). Prohibitin 2, through its C-terminus, directly interacts with heterogeneous nuclear ribonucleoprotein A1, a key modulator of PKM1/2 mRNA splicing that promotes PKM2 expression and glycolysis. Prohibitin 2 deficiency facilitated PKM1/2 mRNA splicing and reversion from PKM1 to PKM2, and enhanced glycolysis in VSMCs (123; Table 1). In PDGF-BB-induced synthetic VSMCs, PKM2 crotonylation (at K305) was upregulated and promotes its nuclear translocation, thereby facilitating the expression of GLUT1 and LDHA (120). Therefore, PKM2 crotonylation (at K305) may participate VSMCs calcification induced by high glucose. However, conclusive evidence on the role of PKM2 in VSMCs calcification induced by high glucose is lacking. Glucose metabolism regulatory protein (GMRSP) inhibits heterogeneous nuclear ribonucleoprotein (hnRNP) A2B1-mediated alternative splicing of pyruvate kinase M (PKM) pre-mRNA, leading to reduced PKM2 production and glycolysis. This reprogramming preserves the contractile phenotype of VSMCs and prevents their transition to a proliferative state (124; Table 1). Besides, membrane-associated RING finger protein 2 (March2) interacted with PKM2 to promote K33-linked polyubiquitination. Deficiency of March2 lessened PKM2 dimer-to-tetramer conversion in aortic aneurysm/dissection (AAD) and overtly exacerbated AAD-induced histone H3K18 lactylation in VSMCs by fostering glucose metabolism reprogramming, thereby promoting p53-driven apoptotic transcriptional response (Table 1). TEPP-46 (tetraethyl pyrophosphate), a PKM2-specific activator, pronouncedly alleviated March2 deficiency-deteriorated AAD pathology (125). Therefore, PKM2 dimer-to-tetramer conversion is one of important treatment targets.
4 Signaling pathways orchestrating VSMC osteogenic differentiation
The etiology of VC involves VSMCs’ osteogenic differentiation (22). The exposure of VSMCs to high glucose activates the WNT signaling pathway and BMPs via upregulation of transcription factors RUNX2 and MSX2 (126). Previous studies reveal that upregulation of MSX2 induced by high glucose increases expression of Wnt3a and Wnt7a and suppresses Dickkopf-1 (Dkk1) gene expression, which subsequently upregulates ALP and pituitary-specific positive transcription factor 1(PIT1) gene expression. These promote osteogenic trans-differentiation and calcification of VSMCs (127–130). Based on existing evidence, elevated levels of glucose participate in regulating phosphatidylinositol 3-kinase (PI3K), AMPK signaling pathway, cell death, oxidative stress and inflammation, microRNAs and extracellular vesicles, which enable chondrogenic/osteogenic phenotypic transition of VSMCs.
4.1 Vascular inflammation and oxidative stress
Streptozotocin-induced diabetes increases ROS and the adventitial inflammatory response (Tumor Necrosis Factor-α(TNF-α), interleukin IL-1β, IL-6, and IL-18), playing crucial roles in osteochondral differentiation of VSMCs (131–135). This increase MSX2, BMP2, Wnt7a, and Wnt3a expressions in the aorta and accelerates calcification of the aorta (136). PGC-1α/NRF2/STAT3 is involved in decreasing VC by increasing superoxide dismutase 2 (SOD2) (110–112). In contrast, in a diabetic mouse model, vascular parathyroid hormone receptor (PTH1R) activation can also partly restrict calcification via oxidant stress reduction (32). High glucose (HG)-induced VSMC inflammation increases the level of AKT/FoxO1/3, which leads the upregulation of RUNX2 via inhibiting RUNX2 ubiquitination and subsequent degradation (65) (Figure 5). Oxidative stress and inflammation participate VSMCs calcification in dependent NF-kB transcriptional activation in VSMCs cultured in high glucose (68, 81, 137, 138). Accumulated studies suggest that oxidative stress and inflammatory cytokines activate monocyte chemotactic protein-1(MCP-1)/chemokine (C-C motif) receptor 2 (CCR2) and receptor of nuclear factor-κB ligand (RANKL) through RUNX2 (65, 139–143). Recent study found the leucine-rich repeat-containing G-protein-coupled receptor 4 (LGR4), a novel receptor for RANKL, also regulates VSMCs calcification (144). Mice deficient in a decoy receptor for RANKL, osteoprotegerin (OPG), develop extensive vascular calcification which is reduced by OPG treatment (145). Palmdelphin (PALMD) promoted the adjustment of glycolysis and NF-κB-mediated inflammation (137, 138). Zinc elevated TNFα-induced protein 3 (TNFAIP3) expression that was dependent on NF-κB transcriptional activation in VSMCs cultured in high glucose. This change was inhibited by zinc-sensing receptor G protein-coupled receptor 39 (GPR39) silencing (146). Empagliflozin attenuated HG-induced osteogenic differentiation and calcium deposition by increasing Bhlhe40/NLRP3 in MOVASs (147). During this process, the stability and the nuclear translocation of Bhlhe40 protein is regulated by Long noncoding RNA SNHG1 (148).
Figure 5
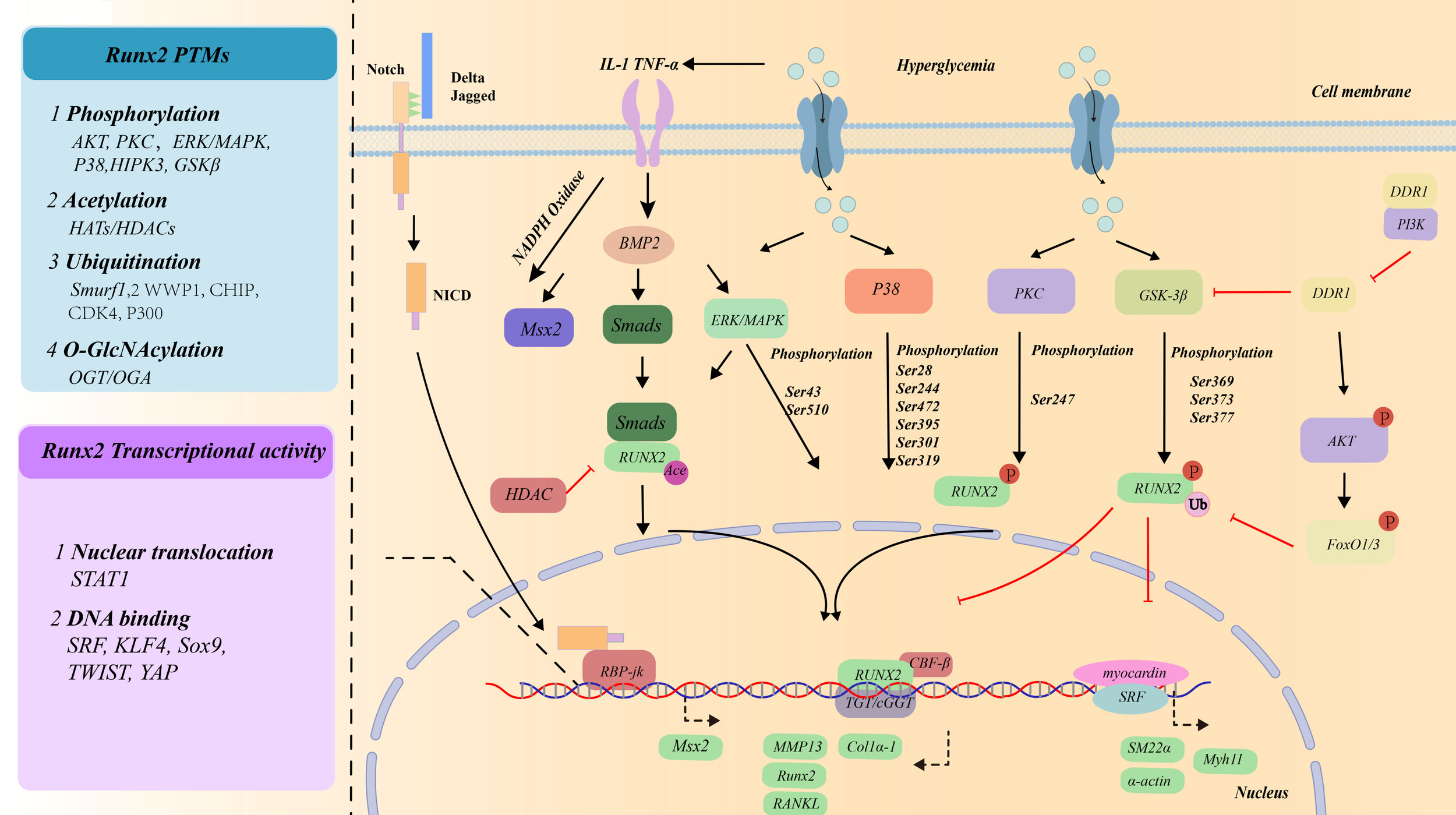
Transcriptional factors/co-factors mediating osteogenic differentiation of VSMCs under the high glucose condition.
4.2 AMPK
AMPK is a sophisticated regulator of diabetic atherosclerosis, especially in VSMCs (149). Under high glucose condition, AMPK strictly regulates glycolytic flux via glucose uptake and mitochondrial dysfunction in VSMCs (150). For example, intermedin inhibited the expression of RAGE and GLUT1 via the cAMP/PKA signaling pathway in diabetic VC (42). Mitochondrial fission and oxidative stress enhancing VC in diabetes are blocked by the AMPK activator metformin (151–153). The glucagon-like peptide-1 receptor (GLP-1R) agonist exendin-4 promotes mitophagy by activating AMPK, inhibiting the osteogenic phenotype switching of VSMCs (5). Based on the aforementioned discussions, AMPK signaling induction is an ideal strategy in improving glucose metabolism and ameliorating diabetic complications.
4.3 AKT signaling pathway
HG activates of PI3K/AKT/Glycogen Synthase Kinase 3 Beta (GSK3β) (154). This process is associated with discoidin domain receptor 1(DDR1). Restoring DDR1 expression in DDR1-null VSMCs rescues Akt activation (155). In contrast, DDR1-deficient VSMCs increase GSK-3β activation and impair microtubule organization, which may account for reduced RUNX2 activity and nuclear localization (155) (Figure 5). In diabetic mice, AKT activation via O-GlcNAcylation enables vascular calcification induced by hyperglycemia (70, 156). AKT O-GlcNAcylation at T479 and T430 has been identified as a potential regulator of diabetes-induced calcification (70). As the AKT target, FoxO1/3 promotes RUNX2 ubiquitination and subsequent degradation, inhibits VSMCs calcification (65) (Figure 5). Therefore, SMC-specific tensin homologue (PTEN) (a primary negative AKT regulator)/FOXO promotes RUNX2 upregulation and VSMCs calcification (157). Sal B exhibits substantial anti-inflammatory effects by modulating the miR-486a-5p/FOXO1 axis under HG conditions in VSMCs (158). Besides, GLP-1R mediates VSMCs calcification in diabetes through inhibiting ERK1/2 and PI3K/Akt signaling pathways (159).
4.4 Cell fate
High glucose triggers autophagy, apoptosis, defect mitophagy and senescence. This finding underscores the pivotal role of metabolic disturbances in vascular pathology (160). High glucose triggers autophagy in VSMCs through two distinct signaling pathways: HIF-1α/PDK4 axis and the cyclic guanosine monophosphate (cGMP)-protein kinase G (PKG) pathway (161, 162) (Figure 4). These partly contribute to AGEs accumulation (163, 164). Notably, Hu-Qiang He et al. confirmed that impaired autophagy effectively inhibited AGE-induced calcification in HASMCs (85). Xu, Z.J., et al. showed autophagic dysregulation is mechanistically also governed by the Notch receptor 3 (NOTCH3)/RAN binding protein 1(RANBP1) axis in high glucose-stimulated VSMCs (165). Dickkopf1 (Dkk1) slowed vascular calcification by promoting the degradation of PLD1 via the regulating autophagosome formation and maturation (166). The natural compound thonningianin A (TA) decreases expression of RUNX2, BMP2 and OPN via autophagy related 7 (ATG7)-dependent autophagy in HG-stimulated MASMCs (167; Table 1). AGEs also facilitate apoptosis to release more matrix vesicles and establish a calcium–phosphorus deposition microenvironment (168). The enhanced expression of HK2 in HASMCs results in the hyperpolarization of mitochondrial membrane potential and the suppression of apoptosis (49). PKM2 dimer-to-tetramer conversion foster glucose metabolism reprogramming and promote p53-driven apoptotic transcriptional response (125).
High glucose promotes surplus of lactate. Accumulated studies confirmed that lactate paly a core role in regulated cell fate in diabetes. In one hand, lactate accelerates Drp1-regulated fission of the mitochondria. On the other hand, lactate inhibits mitophagy via BNIP3 and PTEN-PINK1/Parkin. All promote the osteoblastic phenotype transition of VSMCs and calcium deposition (151). PARP1 knockdown inhibited Drp1-mediated mitochondrial fission and partially restored PINK1/Parkin-mediated mitophagy. Further study found lactate promote PARP1 expression and nuclear transfer and then activate POLG/UCP2 pathway, inhibiting mitochondrial DNA synthesis (107). Metformin reduces the advancement of diabetes-induced atherosclerosis by blocking Drp1-mediated mitochondrial fission (152) (Figure 4). The glucagon-like peptide-1 receptor (GLP-1R) agonist exendin 4 (EX4) inhibited osteogenic differentiation of HG/β-GP-induced VSMCs through promoting mitophagy by activating the AMPK signaling pathway (5).
VSMCs isolated from diabetic patients show elevated DNA damage and senescence. PGC-1α plays a causative role in the pathogenesis of senescence (169). Hyperglycaemic conditions induced DNA damage and enhanced senescence in VSMCs in vitro. CML caused these changes via stimulator of interferon response cyclic guanosine monophosphate - adenosine monophosphate (cGAMP) interactor 1 activation (170). DNA damage-induced calcification is accelerated within a diabetic environment and can be attenuated in vitro by SIRT1/ATM activation (171). DNA damage and senescence promote vascular calcification through SIRT1/PGC-1α pathway in VSMCs (44). PGC-1α and O- GlcNAcylation of KEAP1 lead to NRF2 degradation, thereby inhibiting the negative regulatory effect of NRF2 on the stability of STING mRNA and ultimately promoting STING expression (110–112, 172). Besides, Hyperglycemia stimulates vascular endothelial cells to upregulate cyclin-dependent kinase inhibitor 1A (p21) and p53, thereby exacerbating VSMC senescence and calcification (173).
4.5 miRNAs that regulates VSMC osteogenic differentiation
MicroRNAs (miRNAs) are small non-coding RNAs approximately 22 nucleotides in length. These molecules bind to complementary seed sequences located within the 3’ untranslated region (3’UTR) of target messenger RNAs (mRNAs). MicroRNAs have emerged as crucial post-transcriptional regulators in the process of VSMCs re-differentiation from contractile to osteoblast-like phenotype in diabetics (174).
miRNAs effectively silence gene expression, such as RUNX2, MSX2. Overexpression of miR-34a activates NF-κB, subsequently increasing the expression of RUNX2 and osteocalcin (175–177). Meanwhile, microRNA-126-3p impedes the osteogenic trans-differentiation of VSMCs by post-transcriptionally interfering with BMP2 gene expression (178). High glucose levels were shown to induce the upregulation of miR-32-5p by activating CCAAT/enhancer binding protein beta (CEBPB). Overexpression of GATA6 antagonized the effects of miR-32-5p on vascular calcification (179). By modulating the translation efficiency or stability of their target mRNAs, miRNAs are involved in key functions including cell growth, proliferation, differentiation, metabolic regulation, immune responses, and the regulation of cell death pathways (180). In high glucose conditions, the expression of miR142-3p (181), miR21-5p (182), and miR19a (183) are promoted. In contrast, miR24 (184) and miR9 (185) expression is inhibited. These miRNAs participate in regulating proliferation and migration of VSMCs. For example, miR-24 overexpression can mitigate proliferation of VSMCs in diabetic rats through the Wnt4/Dishevelled-1(Dvl-1)/β-catenin signaling pathway (186). Decreasing miR125a expression promotes VSMCs migration and proliferation by upregulating 3-hydroxy-3-methylglutaryl coenzyme A reductase (187). Additionally, miR210 inhibits the carbohydrate-responsive element-binding protein (ChREBP)/HIF-1α signaling pathway, regulating glycolysis and apoptosis (188). The binding of RNA-ES3 to Bhlhe40 suppresses the expression of miR-6776-5p, miR-95-5p, miR-4747-5p, and miR-3620-5p, leading to VSMCs senescence and calcification (189). In contrast, MiR-15a/15b/16 specifically target the 3’UTR of nuclear factor of activated T cells (3NFATc3) mRNA, and downregulate OCN expression. Therefore, MiR-15a/15b/16 inhibits human VSMCs osteogenic trans-differentiation (162).
Matrix vesicles and circulating miRNAs also play an important role in VSMCs calcification. A deficiency in Hsa_circRNA_0008028 exacerbates high glucose-induced calcification, autophagy, and proliferation in VSMCs by upregulating miR-182-5p. During this process, miR-182-5p targets tribbles pseudokinase 3 (TRIB3). TRIB3 plays a critical role in the induction and maintenance of contractile phenotype in VSMCs (180). Similarly, extracellular vesicles (EVs) carrying circ_0008362 elevated in diabetic patients. It increases the expression of RUNX2 through miR-1251-5p in VSMCs (190). Additionally, macrophage-derived miR-32 inhibits autophagy in type 2 diabetic mice’s arterial calcification (162). And MAEC-derived exosomal circHIPK3 increases high glucose-induced VSMCs proliferation via the miR-106a-5p/Foxo1/vascular cell adhesion molecule 1(VCAM1) pathway (190).
4.6 EVs
Mineral homeostasis disruption under high glucose conditions is closely associated with the dysregulation of EVs, including exosomes and matrix vesicles (MVs) (191). Emerging evidence highlights the critical role of EVs in mediating vascular calcification exacerbated by hyperglycemia (192). Therefore, EVs may be potential therapeutic targets in diabetic vascular complications.
4.6.1 EVs of VSMCs: high glucose-induced pro-calcific transformation
Extracellular vesicles coming from VSMCs contain calcification-promoting protein tissue-nonspecific alkaline phosphatase (TNAP) (193) and sortilin (194).The sortilin 1 (SORT1) and neutral sphingomyelinase 2 (nSMase2, also called sphingomyelin phosphodiesterase 3; SMPD3) are crucial enzymes in the generation of EVs and cargo sorting (195). Treatment with anti-sortilin antibodies may considerably lessen EV-induced VC (196). Inhibiting nSMase2 pharmacologically decreases VSMCs EVs secretion and VSMC-driven calcification (197, 198). High quantities of Nϵ-carboxymethyl-lysine increased release of EVs coming from VSMCs and recruitment of sortilin to EVs (196). Additionally, galectin-3 overexpression in macrophages, triggered by high glucose, facilitates the migration of VSMC-derived EVs to the intima, further exacerbating diabetic vascular intimal calcification (199).
4.6.2 EVs of ECs: bidirectional regulation under high glucose stimulation
Through EVs, endothelial cells (ECs) can communicate with VSMCs (200). High glucose-exposed human umbilical vein endothelial cells (HUVECs) secrete EVs that act as key mediators in VSMC calcification. These EVs deliver Notch3 to VSMCs via the mTOR signaling pathway, promoting VSMCs phenotypic transition towards an osteoblast-like state (201). HG-HUVECs-EVs also contain versican (VCAN), which induce mitochondrial dysfunction, oxidative stress and senescence, which accelerates VSMCs calcification (173). Moreover, high glucose induces the production of EC-derived EVs containing malondialdehyde (MDA), LDH, CircRNA-0077930, and circ_0008362, which promote VSMCs calcification (173, 190, 201–203). Notably, contrary to earlier research suggesting that AGEs were detrimental to vascular cells, Guo et al. demonstrated that AGEs might actually prevent diabetic media calcification by inducing HUVECs to release small EVs carrying abundant miR-126-5p. This miRNA targets bone morphogenetic protein receptor type 1 (BMPR1) and blocks the SMAD1/5/9 signaling pathway (203).
4.6.3 EVs of macrophages: molecular link between pro-inflammation and pro-calcification in high glucose environment
In the high glucose-induced inflammatory microenvironment, macrophage-derived EVs play a pivotal role in VSMCs osteogenic differentiation. MiR-17-5p, macrophage S100A9 and miR-32 are derived from EVs of macrophage. MiR-17-5p attenuates VSMCs osteogenesis through suppressing TGF-β signaling (204). Macrophage S100A9 controls diabetic vascular calcification via NRF-2 and NF-κB (205) (Figure 6). MiR-32 accelerates vascular calcification in type 2 diabetes by inhibiting VSMCs autophagy (162).
Figure 6

Potential mechanism of EVs regulating osteogenic differentiation of VSMCs under the high glucose condition.
5 Conclusions and future perspectives
Vascular calcification has emerged as a vital mortality or morbidity indicator in the cardiovascular system, particularly in diabetes. Osteogenic differentiation of VSMCs is considered a significant mechanism of diabetic macrovascular disease and the initiation mechanism of vascular calcification (65, 206–208). HG activates multiple signaling pathways, like Ca2+ signaling, Wnt/β-catenin, BMP/Smad, and Notch through AMPK, PI3K-AKT, oxidative stress and inflammation, microRNA, and EVs pathways (209–211). These activations contribute to glucose metabolic reprogramming such as (a) intracellular PKC overactivity; (b) increased polyol pathway flux; (c) increased the hexosamine pathway flux; and (d) generation of AGEs and other glycated compounds derived from both glucose oxidation in arterial VSMCs (212). These changes promote osteogenic transformation of VSMCs and the development of VC.
RUNX2 is the core of the regulatory network for VSMCs calcification. During glucose metabolism reprogramming, PPP, HBP, and AGEs/AGER activate ERK1/2, MAPK, AKT, NF-κB and BMP2 in VSMCs. These pathways mainly participate in RUNX2 expression and transcriptional activity through regulate RUNX2 protein structure, and the multilateral genetic, epigenetic and posttranslational modifications (PTMs) regulatory mechanisms control RUNX2 expression (213) (in Figure 2). Further investigations are warranted to address these unanswered questions, which should provide new breakthroughs in the understanding of RUNX2-dependent transcriptional reprogramming of vascular cells and their contributions to the development of arteriosclerosis. For example, it is unknown whether RUNX2 is upregulated in all VSMCs or exclusively in selective VSMCs subpopulations. Of particular interest, RUNX2-regulated expression of RANKL by VSMCs functions as a chemoattractant that induces macrophage/monocyte migration towards VSMCs; and further induces the differentiation of the macrophage/monocyte into bone-resorbing osteoclast-like cells.
Under the high glucose condition, the expression of GLUT, HK2, G6PD, PKM2 and LDHA increase, which enhances glycolytic activity and decreases OXPHOS in VSMCs. During VSMCs calcification, upregulation of PKM2 promote the expression of GLUT1 and LDHA (120). PDK4 inhibit the activity of PDH, slowing down the TCA and towards a higher glycolysis rate. Besides, GLUT1/LDHA may regulate aerobic glycolysis. These explain the preference of VSMCs for the less ATP-efficient metabolic mode of glycolysis. Future efforts will likely focus on identifying key regulatory points and interactions between mechanisms linked to VSMCs transformation that can be targeted to reduce calcification, and, thereby, improve vascular compliance and reduce cardiovascular risk.
There are still many difficult problems to be solved in future research. Specifically, the representation of cultured cells in in vivo studies is limited because of the lack of a suitable microenvironment, such as cell–cell interactions, extracellular elastin fibers, hemodynamic factors, and cytokines (214–216). In addition, animal models cannot successfully recapitulate the physiology and pathology in humans. Therefore, the trend of VSMCs transformation in diabetic patients and metabolic reprogramming remains to be further explored and has recently attracted attention.
Statements
Author contributions
JG: Writing – original draft, Writing – review & editing. LD: Funding acquisition, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. The study was funded by the High-Level Scientific and Technological Innovation Talent Cultivation Project of Henan University of Science and Technology First Affiliated Hospital.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1
Group, I.D.F.D.A . Update of mortality attributable to diabetes for the IDF Diabetes Atlas: Estimates for the year 2013. Diabetes Res Clin Pract. (2015) 109:461–5. doi: 10.1016/j.diabres.2015.05.037
2
Saeedi P Petersohn I Salpea P Malanda B Karuranga S Unwin N et al . Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9(th) edition. Diabetes Res Clin Pract. (2019) 157:107843. doi: 10.1016/j.diabres.2019.107843
3
Yao H Sun Z Zang G Zhang L Hou L Shao C et al . Epidemiological research advances in vascular calcification in diabetes. J Diabetes Res. (2021) 2021:4461311. doi: 10.1155/2021/4461311
4
Li Q Li P Xu Z Lu Z Yang C Ning J et al . Association of diabetes with cardiovascular calcification and all-cause mortality in end-stage renal disease in the early stages of hemodialysis: a retrospective cohort study. Cardiovasc Diabetol. (2024) 23:259. doi: 10.1186/s12933-024-02318-8
5
Chen K Jin HJ Wu ZH Zhang BF Wu J Huang ZY et al . Glucagon-like peptide-1 receptor agonist exendin 4 ameliorates diabetes-associated vascular calcification by regulating mitophagy through the AMPK signaling pathway. Mol Med. (2024) 30:58. doi: 10.1186/s10020-024-00817-8
6
Lee SJ Lee IK Jeon JH . Vascular calcification-new insights into its mechanism. Int J Mol Sci. (2020) 21:2685. doi: 10.3390/ijms21082685
7
Zhu Y Ma WQ Han XQ Wang Y Wang X Liu NF et al . Advanced glycation end products accelerate calcification in VSMCs through HIF-1α/PDK4 activation and suppress glucose metabolism. Sci Rep. (2018) 8:13730. doi: 10.1038/s41598-018-31877-6
8
Rasmussen LM Heickendorff L . Quantification of fibronectin in extracts of human aortae by an ELISA. Scand J Clin Lab Invest. (1989) 49:205–10. doi: 10.3109/00365518909089084
9
Tanikawa T Okada Y Tanikawa R Tanaka Y . Advanced glycation end products induce calcification of vascular smooth muscle cells through RAGE/p38 MAPK. J Vasc Res. (2009) 46:572–80. doi: 10.1159/000226225
10
Ghosh S Luo D He W Chen J Su X Huang H . Diabetes and calcification: The potential role of anti-diabetic drugs on vascular calcification regression. Pharmacol Res. (2020) 158:104861. doi: 10.1016/j.phrs.2020.104861
11
Takemoto M Yokote K Yamazaki M Ridall AL Butler WT Matsumoto T et al . Enhanced expression of osteopontin by high glucose. Involvement of osteopontin in diabetic macroangiopathy. Ann N Y Acad Sci. (2000) 902:357–63. doi: 10.1111/j.1749-6632.2000.tb06338.x
12
Shao JS Cheng SL Charlton-Kachigian N Loewy AP Towler DA . Teriparatide (human parathyroid hormone (1-34)) inhibits osteogenic vascular calcification in diabetic low density lipoprotein receptor-deficient mice. J Biol Chem. (2003) 278:50195–202. doi: 10.1074/jbc.M308825200
13
Chen NX Duan D O'Neill KD Moe SM . High glucose increases the expression of Cbfa1 and BMP-2 and enhances the calcification of vascular smooth muscle cells. Nephrol Dial Transplant. (2006) 21:3435–42. doi: 10.1093/ndt/gfl429
14
Liang X Li Y Wang P Liu H . Key regulators of vascular calcification in chronic kidney disease: Hyperphosphatemia, BMP2, and RUNX2. PeerJ. (2024) 12:e18063. doi: 10.7717/peerj.18063
15
Chen Z Li R Pei LG Wei ZH Xie J Wu H et al . High-mobility group box-1 promotes vascular calcification in diabetic mice via endoplasmic reticulum stress. J Cell Mol Med. (2021) 25:3724–34. doi: 10.1111/jcmm.16075
16
Wu CY Martel J Young JD . Ectopic calcification and formation of mineralo-organic particles in arteries of diabetic subjects. Sci Rep. (2020) 10:8545. doi: 10.1038/s41598-020-65276-7
17
Wang Z Wang Z Zhang L Sun Z Shao C Li Y Bao Z et al . Mechanisms of Matrix Vesicles Mediating calcification transition in diabetic plaque. Heart Lung Circ. (2020) 29:112–7. doi: 10.1016/j.hlc.2019.04.022
18
Bernal-Mizrachi C Bernal-Mizrachi C Gates AC Weng S Imamura T Knutsen RH DeSantis P et al . Vascular respiratory uncoupling increases blood pressure and atherosclerosis. Nature. (2005) 435:502–6. doi: 10.1038/nature03527
19
Docherty CK Carswell A Friel E Mercer JR . Impaired mitochondrial respiration in human carotid plaque atherosclerosis: A potential role for Pink1 in vascular smooth muscle cell energetics. Atherosclerosis. (2018) 268:1–11. doi: 10.1016/j.atherosclerosis.2017.11.009
20
Scheede-Bergdahl C Bergdahl A . Adaptation of mitochondrial expression and ATP production in dedifferentiating vascular smooth muscle cells. Can J Physiol Pharmacol. (2017) 95:1473–9. doi: 10.1139/cjpp-2017-0227
21
Flores-Roco A Lago BM Villa-Bellosta R . Elevated glucose levels increase vascular calcification risk by disrupting extracellular pyrophosphate metabolism. Cardiovasc Diabetol. (2024) 23:405. doi: 10.1186/s12933-024-02502-w
22
Sage AP Tintut Y Demer LL . Regulatory mechanisms in vascular calcification. Nat Rev Cardiol. (2010) 7:528–36. doi: 10.1038/nrcardio.2010.115
23
Miano JM Fisher EA Majesky MW . Fate and state of vascular smooth muscle cells in atherosclerosis. Circulation. (2021) 143:2110–6. doi: 10.1161/CIRCULATIONAHA.120.049922
24
Woo SH Kyung D Lee SH Park KS Kim M Kim K et al . TXNIP suppresses the osteochondrogenic switch of vascular smooth muscle cells in Atherosclerosis. Circ Res. (2023) 132:52–71. doi: 10.1161/CIRCRESAHA.122.321538
25
Qian Y Xiong S Li L Sun Z Zhang L Yuan W et al . Spatial multiomics atlas reveals smooth muscle phenotypic transformation and metabolic reprogramming in diabetic macroangiopathy. Cardiovasc Diabetol. (2024) 23:358. doi: 10.1186/s12933-024-02458-x
26
Yap C Mieremet A de Vries CJM Micha D de Waard V . Six shades of vascular smooth muscle cells illuminated by KLF4 (Krüppel-Like Factor 4). Arterioscler Thromb Vasc Biol. (2021) 41:2693–707. doi: 10.1161/ATVBAHA.121.316600
27
Bidder M Shao JS . Osteopontin transcription in aortic vascular smooth muscle cells is controlled by glucose-regulated upstream stimulatory factor and activator protein-1 activities. J Biol Chem. (2002) 277:44485–96. doi: 10.1074/jbc.M206235200
28
Durham AL Speer MY Scatena M Giachelli CM Shanahan CM . Role of smooth muscle cells in vascular calcification: implications in atherosclerosis and arterial stiffness. Cardiovasc Res. (2018) 114:590–600. doi: 10.1093/cvr/cvy010
29
Steitz SA Speer MY Curinga G Yang HY Haynes P Aebersold R et al . Smooth muscle cell phenotypic transition associated with calcification: upregulation of Cbfa1 and downregulation of smooth muscle lineage markers. Circ Res. (2001) 89:1147–54. doi: 10.1161/hh2401.101070
30
Rogers MA Aikawa E . Cardiovascular calcification: artificial intelligence and big data accelerate mechanistic discovery. Nat Rev Cardiol. (2019) 16:261–74. doi: 10.1038/s41569-018-0123-8
31
Faleeva M Ahmad S Theofilatos K Lynham S Watson G Whitehead M et al . Sox9 accelerates vascular aging by regulating extracellular matrix composition and stiffness. Circ Res. (2024) 134:307–24. doi: 10.1161/CIRCRESAHA.123.323365
32
Towler DA . Parathyroid hormone-PTH1R signaling in cardiovascular disease and homeostasis. Trends Endocrinol Metab. (2024) 35:648–60. doi: 10.1016/j.tem.2024.02.005
33
Ouyang L Su X Li W Tang L Zhang M Zhu Y et al . ALKBH1-demethylated DNA N6-methyladenine modification triggers vascular calcification via osteogenic reprogramming in chronic kidney disease. J Clin Invest. (2021) 131:e146985. doi: 10.1172/JCI146985
34
Idelevich A Rais Y Monsonego-Ornan E . Bone Gla protein increases HIF-1alpha-dependent glucose metabolism and induces cartilage and vascular calcification. Arterioscler Thromb Vasc Biol. (2011) 31:e55–71. doi: 10.1161/atvbaha.111.230904
35
Ma WQ Han XQ Wang Y Wang X Zhu Y Liu NF . Nϵ-carboxymethyl-lysine promotes calcium deposition in VSMCs via intracellular oxidative stress-induced PDK4 activation and alters glucose metabolism. Oncotarget. (2017) 8:112841–54. doi: 10.18632/oncotarget.22835
36
Hruz PW Mueckler MM . Structural analysis of the GLUT1 facilitative glucose transporter (review). Mol Membr Biol. (2001) 18:183–93. doi: 10.1080/09687680110072140
37
Rashdan NA Sim AM Cui L Phadwal K Roberts FL Carter R et al . Osteocalcin Regulates arterial calcification via altered Wnt signaling and glucose metabolism. J Bone Miner Res. (2020) 35:357–67. doi: 10.1002/jbmr.3888
38
Buller CL Loberg RD Fan MH Zhu Q Park JL Vesely E et al . A GSK-3/TSC2/mTOR pathway regulates glucose uptake and GLUT1 glucose transporter expression. Am J Physiol Cell Physiol. (2008) 295:C836–43. doi: 10.1152/ajpcell.00554.2007
39
Hall JL Chatham JC Eldar-Finkelman H Gibbons GH . Upregulation of glucose metabolism during intimal lesion formation is coupled to the inhibition of vascular smooth muscle cell apoptosis. Role GSK3beta Diabetes. (2001) 50:1171–9. doi: 10.2337/diabetes.50.5.1171
40
Lin CY Hsu SC Lee HS Lin SH Tsai CS Huang SM et al . Enhanced expression of glucose transporter-1 in vascular smooth muscle cells via the Akt/tuberous sclerosis complex subunit 2 (TSC2)/mammalian target of rapamycin (mTOR)/ribosomal S6 protein kinase (S6K) pathway in experimental renal failure. J Vasc Surg. (2013) 57:475–85. doi: 10.1016/j.jvs.2012.07.037
41
Zhou Q Xu J Liu M He L Zhang K Yang Y et al . Warburg effect is involved in apelin-13-induced human aortic vascular smooth muscle cells proliferation. J Cell Physiol. (2019) 234:14413–21. doi: 10.1002/jcp.28218
42
Zhang YR Liu SM Chen Y Zhang LS Ji DR Zhao J et al . Intermedin alleviates diabetic vascular calcification by inhibiting GLUT1 through activation of the cAMP/PKA signaling pathway. Atherosclerosis. (2023) 385:117342. doi: 10.1016/j.atherosclerosis.2023.117342
43
Totary-Jain H Naveh-Many T Riahi Y Kaiser N Eckel J Sasson S . Calreticulin destabilizes glucose transporter-1 mRNA in vascular endothelial and smooth muscle cells under high-glucose conditions. Circ Res. (2005) 97:1001–8. doi: 10.1161/01.RES.0000189260.46084.e5
44
Zheng Y Yao M Chen S Li J Wei X Qiu Z et al . HMGB2 promotes smooth muscle cell proliferation through PPAR-γ/PGC-1α pathway-mediated glucose changes in aortic dissection. Atherosclerosis. (2024) 399:119044. doi: 10.1016/j.atherosclerosis.2024.119044
45
Shi J Yang Y Cheng A Xu G He F . Metabolism of vascular smooth muscle cells in vascular diseases. Am J Physiol Heart Circ Physiol. (2020) 319:H613–h631. doi: 10.1152/ajpheart.00220.2020
46
Alamri A Burzangi AS Coats P Watson DG . Untargeted metabolic profiling cell-based approach of pulmonary artery smooth muscle cells in response to high glucose and the effect of the antioxidant vitamins D and E. Metabolites. (2018) 8:87. doi: 10.3390/metabo8040087
47
Peiró C Romacho T Azcutia V Villalobos L Fernández E Bolaños JP et al . Inflammation, glucose, and vascular cell damage: the role of the pentose phosphate pathway. Cardiovasc Diabetol. (2016) 15:82. doi: 10.1186/s12933-016-0397-2
48
Lee SJ Jeong JY Oh CJ Park S Kim JY Kim HJ et al . Pyruvate dehydrogenase kinase 4 promotes vascular calcification via SMAD1/5/8 phosphorylation. Sci Rep. (2015) 5:16577. doi: 10.1038/srep16577
49
Lambert CM Roy M Robitaille GA Richard DE Bonnet S . HIF-1 inhibition decreases systemic vascular remodelling diseases by promoting apoptosis through a hexokinase 2-dependent mechanism. Cardiovasc Res. (2010) 88:196–204. doi: 10.1093/cvr/cvq152
50
Wu M Liu W Huang H Chen Z Chen Y Zhong Y et al . PVT1/miR-145-5p/HK2 modulates vascular smooth muscle cells phenotype switch via glycolysis: The new perspective on the spiral artery remodeling. Placenta. (2022) 130:25–33. doi: 10.1016/j.placenta.2022.10.010
51
Heiss EH Schachner D Donati M Grojer CS Dirsch VM . Increased aerobic glycolysis is important for the motility of activated VSMC and inhibited by indirubin-3’-monoxime. Vascul Pharmacol. (2016) 83:47–56. doi: 10.1016/j.vph.2016.05.002
52
Gao XF Chen AQ Tang HY Kong XQ Zhang H Wang ZM et al . m(6)A Modification of Profilin-1 in vascular smooth muscle cells drives phenotype switching and neointimal hyperplasia via activation of the p-ANXA2/STAT3 pathway. Arterioscler Thromb Vasc Biol. (2024) 44:2543–59. doi: 10.1161/ATVBAHA.124.321399
53
Huang Y Wang J Jiang C Zheng M Han M Fang Q et al . ANXA2 promotes osteogenic differentiation and inhibits cellular senescence of periodontal ligament cells (PDLCs) in high glucose conditions. PeerJ. (2024) 12:e18064. doi: 10.7717/peerj.18064
54
Dong LH Li L Song Y Duan ZL Sun SG Lin YL et al . TRAF6-mediated SM22α K21 ubiquitination promotes G6PD activation and NADPH production, contributing to GSH homeostasis and VSMC survival in vitro and in vivo. Circ Res. (2015) 117:684–94. doi: 10.1161/CIRCRESAHA.115.306233
55
Zhang T Cao RJ Niu JL Chen ZH Mu SQ Cao T et al . G6PD maintains the VSMC synthetic phenotype and accelerates vascular neointimal hyperplasia by inhibiting the VDAC1-Bax-mediated mitochondrial apoptosis pathway. Cell Mol Biol Lett. (2024) 29:47. doi: 10.1186/s11658-024-00566-w
56
Gupte R Dhagia V Rocic P Ochi R Gupte SA . Glucose-6-phosphate dehydrogenase increases Ca(2+) currents by interacting with Ca(v)1.2 and reducing intrinsic inactivation of the L-type calcium channel. Am J Physiol Heart Circ Physiol. (2020) 319:H144–h158. doi: 10.1152/ajpheart.00727.2019
57
Li M He X Guo W Yu H Zhang S Wang N et al . Aldolase B suppresses hepatocellular carcinogenesis by inhibiting G6PD and pentose phosphate pathways. Nat Cancer. (2020) 1:735–47. doi: 10.1038/s43018-020-0086-7
58
Jiang P Du W Wang X Mancuso A Gao X Wu M et al . p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat Cell Biol. (2011) 13:310–6. doi: 10.1038/ncb2172
59
Mazure NM . News about VDAC1 in hypoxia. Front Oncol. (2016) 6:193. doi: 10.3389/fonc.2016.00193
60
Michels J Kepp O Senovilla L Lissa D Castedo M Kroemer G et al . Functions of BCL-X L at the interface between cell death and metabolism. Int J Cell Biol. (2013) 2013:705294. doi: 10.1155/2013/705294
61
Federici M Menghini R Mauriello A Hribal ML Ferrelli F Lauro D et al . Insulin-dependent activation of endothelial nitric oxide synthase is impaired by O-linked glycosylation modification of signaling proteins in human coronary endothelial cells. Circulation. (2002) 106:466–72. doi: 10.1161/01.CIR.0000023043.02648.51
62
Khanal S Bhavnani N Mathias A Lallo J Gupta S Ohanyan V et al . Deletion of smooth muscle O-GlcNAc transferase prevents development of atherosclerosis in western diet-fed hyperglycemic ApoE(-/-) mice in vivo. Int J Mol Sci. (2023) 24:7899. doi: 10.3390/ijms24097899
63
Paredes F Williams HC Quintana RA San Martin . Mitochondrial protein poldip2 (Polymerase Delta Interacting Protein 2) controls vascular smooth muscle differentiated phenotype by O-Linked GlcNAc (N-Acetylglucosamine) transferase-dependent inhibition of a ubiquitin proteasome system. Circ Res. (2020) 126:41–56. doi: 10.1161/CIRCRESAHA.119.315932
64
Chen Y Zhao X Wu H . Metabolic stress and cardiovascular disease in diabetes mellitus: The role of protein O-GlcNAc modification. Arterioscler Thromb Vasc Biol. (2019) 39:1911–24. doi: 10.1161/ATVBAHA.119.312192
65
Byon CH Javed A Dai Q Kappes JC Clemens TL Darley-Usmar VM et al . Oxidative stress induces vascular calcification through modulation of the osteogenic transcription factor Runx2 by AKT signaling. J Biol Chem. (2008) 283:15319–27. doi: 10.1074/jbc.M800021200
66
London GM Guérin AP Marchais SJ Métivier F Pannier B Adda H . Arterial media calcification in end-stage renal disease: impact on all-cause and cardiovascular mortality. Nephrol Dial Transplant. (2003) 18:1731–40. doi: 10.1093/ndt/gfg414
67
Chen J Yuan K Mao X Miano JM Wu H Chen Y . Serum response factor regulates bone formation via IGF-1 and Runx2 signals. J Bone Miner Res. (2012) 27:1659–68. doi: 10.1002/jbmr.1607
68
Yang WH Park SY Nam HW Kim DH Kang JG Kang ES et al . NFkappaB activation is associated with its O-GlcNAcylation state under hyperglycemic conditions. Proc Natl Acad Sci U.S.A. (2008) 105:17345–50. doi: 10.1073/pnas.0806198105
69
Barnes JW Tian L Krick S Helton ES Denson RS Comhair SAA et al . O-GlcNAc transferase regulates angiogenesis in idiopathic pulmonary arterial hypertension. Int J Mol Sci. (2019) 20:6299. doi: 10.3390/ijms20246299
70
Heath JM Sun Y Yuan K Bradley WE Litovsky S Dell'Italia LJ et al . Activation of AKT by O-linked N-acetylglucosamine induces vascular calcification in diabetes mellitus. Circ Res. (2014) 114:1094–102. doi: 10.1161/CIRCRESAHA.114.302968
71
Zhang W Sun Y Yang Y Chen Y . Impaired intracellular calcium homeostasis enhances protein O-GlcNAcylation and promotes vascular calcification and stiffness in diabetes. Redox Biol. (2023) 63:102720. doi: 10.1016/j.redox.2023.102720
72
Xu L Liu B Ma H Qi E Ma J Chang T et al . O-GlcNAc transferase promotes vascular smooth muscle calcification through modulating Wnt/β-catenin signaling. FASEB J. (2024) 38:e70271. doi: 10.1096/fj.202401649RR
73
Xu TH Du Y Sheng Z Li Y Qiu X Tian B et al . OGT-mediated KEAP1 glycosylation accelerates NRF2 degradation leading to high phosphate-induced vascular calcification in chronic kidney disease. Front Physiol. (2020) 11:1092. doi: 10.3389/fphys.2020.01092
74
Kennon AM Stewart JA Jr . RAGE differentially altered in vitro responses in vascular smooth muscle cells and adventitial fibroblasts in diabetes-induced vascular calcification. Front Physiol. (2021) 12:676727. doi: 10.3389/fphys.2021.676727
75
Hwang AR Nam JO Kang YJ . Fluvastatin inhibits advanced glycation end products-induced proliferation, migration, and extracellular matrix accumulation in vascular smooth muscle cells by targeting connective tissue growth factor. Korean J Physiol Pharmacol. (2018) 22:193–201. doi: 10.4196/kjpp.2018.22.2.193
76
Son M Oh S Jang JT Park CH Son KH Byun K . Attenuating effects of pyrogallol-phloroglucinol-6,6-bieckol on vascular smooth muscle cell phenotype changes to osteoblastic cells and vascular calcification induced by high fat diet. Nutrients. (2020) 12:2777. doi: 10.3390/nu12092777
77
Xu Z Liu X Wang Z Tao J Han Z Gu M et al . Effect of sirolimus on arteriosclerosis induced by advanced glycation end products via inhibition of the ILK/mTOR pathway in kidney transplantation recipients. Eur J Pharmacol. (2017) 813:1–9. doi: 10.1016/j.ejphar.2017.06.038
78
Tada Y Yano S Yamaguchi T Okazaki K Ogawa N Morita M et al . Advanced glycation end products-induced vascular calcification is mediated by oxidative stress: functional roles of NAD(P)H-oxidase. Horm Metab Res. (2013) 45:267–72. doi: 10.1055/s-0032-1329965
79
Kay AM Simpson CL Stewart JA Jr . The role of AGE/RAGE signaling in diabetes-mediated vascular calcification. J Diabetes Res. (2016) 2016:6809703. doi: 10.1155/2016/6809703
80
Molinuevo MS Cortizo AM Sedlinsky C . Effects of advanced glycation end-products, diabetes and metformin on the osteoblastic transdifferentiation capacity of vascular smooth muscle cells: In vivo and in vitro studies. J Diabetes Complications. (2023) 37:108626. doi: 10.1016/j.jdiacomp.2023.108626
81
Poetsch F Henze LA Estepa M Moser B Pieske B Lang F et al . Role of SGK1 in the osteogenic transdifferentiation and calcification of vascular smooth muscle cells promoted by hyperglycemic conditions. Int J Mol Sci. (2020) 21:7207. doi: 10.3390/ijms21197207
82
Sourris KC Watson A Jandeleit-Dahm K . Inhibitors of advanced glycation end product (AGE) formation and accumulation. Handb Exp Pharmacol. (2021) 264:395–423. doi: 10.1007/164_2020_391
83
Menini S Iacobini C Ricci C Blasetti Fantauzzi C Salvi L Pesce CM et al . The galectin-3/RAGE dyad modulates vascular osteogenesis in atherosclerosis. Cardiovasc Res. (2013) 100:472–80. doi: 10.1093/cvr/cvt206
84
Ott C Jacobs K Haucke E Navarrete Santos A Grune T Simm A . Role of advanced glycation end products in cellular signaling. Redox Biol. (2014) 2:411–29. doi: 10.1016/j.redox.2013.12.016
85
He HQ Qu YQ Kwan Law BY Qiu CL Han Y . AGEs-induced calcification and apoptosis in human vascular smooth muscle cells is reversed by inhibition of autophagy. Front Pharmacol. (2021) 12:692431. doi: 10.3389/fphar.2021.692431
86
Sun X Yang Y Zhao W Wang M Chen Y Wang J et al . MTMR7 suppresses the phenotypic switching of vascular smooth muscle cell and vascular intimal hyperplasia after injury via regulating p62/mTORC1-mediated glucose metabolism. Atherosclerosis. (2024) 390:117470. doi: 10.1016/j.atherosclerosis.2024.117470
87
Xie F Liu B Qiao W He JZ Cheng J Wang ZY et al . Smooth muscle NF90 deficiency ameliorates diabetic atherosclerotic calcification in male mice via FBXW7-AGER1-AGEs axis. Nat Commun. (2024) 15:4985. doi: 10.1038/s41467-024-49315-9
88
Perer J Jandova J Fimbres J Jennings EQ Galligan JJ Hua A et al . The sunless tanning agent dihydroxyacetone induces stress response gene expression and signaling in cultured human keratinocytes and reconstructed epidermis. Redox Biol. (2020) 36:101594. doi: 10.1016/j.redox.2020.101594
89
Mushajiang M Li Y Sun Z Liu J Zhang L Wang Z . USP10 alleviates Nϵ-carboxymethyl-lysine-induced vascular calcification and atherogenesis in diabetes mellitus by promoting AMPK activation. Cell Signal. (2024) 120:111211. doi: 10.1016/j.cellsig.2024.111211
90
Wang Z Li L Du R Yan J Liu N Yuan W et al . CML/RAGE signal induces calcification cascade in diabetes. Diabetol Metab Syndr. (2016) 8:83. doi: 10.1186/s13098-016-0196-7
91
Sun Z Zhang L Yin K Zang G Qian Y Mao X et al . SIRT3-and FAK-mediated acetylation-phosphorylation crosstalk of NFATc1 regulates N(ϵ)-carboxymethyl-lysine-induced vascular calcification in diabetes mellitus. Atherosclerosis. (2023) 377:43–59. doi: 10.1016/j.atherosclerosis.2023.06.969
92
Zhang L Wang Z Sun Z Pang M Shao C Li L . Nϵ-Carboxymethyl-Lysine mediates vascular calcification in diabetes caused by impaired osteoclastic resorption activity through NFATc1-GNPTAB. J Cardiovasc Transl Res. (2023) 16:233–43. doi: 10.1007/s12265-022-10300-6
93
Koya D King GL . Protein kinase C activation and the development of diabetic complications. Diabetes. (1998) 47:859–66. doi: 10.2337/diabetes.47.6.859
94
Ata H Rawat DK Lincoln T Gupte SA . Mechanism of glucose-6-phosphate dehydrogenase-mediated regulation of coronary artery contractility. Am J Physiol Heart Circ Physiol. (2011) 300:H2054–63. doi: 10.1152/ajpheart.01155.2010
95
Sakaguchi M Murata H Yamamoto K Ono T Sakaguchi Y Motoyama A et al . TIRAP, an adaptor protein for TLR2/4, transduces a signal from RAGE phosphorylated upon ligand binding. PloS One. (2011) 6:e23132. doi: 10.1371/journal.pone.0023132
96
Geraldes P King GL . Activation of protein kinase C isoforms and its impact on diabetic complications. Circ Res. (2010) 106:1319–31. doi: 10.1161/CIRCRESAHA.110.217117
97
Daffu G del Pozo CH O'Shea KM Ananthakrishnan R Ramasamy R Schmidt AM . Radical roles for RAGE in the pathogenesis of oxidative stress in cardiovascular diseases and beyond. Int J Mol Sci. (2013) 14:19891–910. doi: 10.3390/ijms141019891
98
Hu Y Chan E Wang SX Li B . Activation of p38 mitogen-activated protein kinase is required for osteoblast differentiation. Endocrinology. (2003) 144:2068–74. doi: 10.1210/en.2002-220863
99
Fantin VR St-Pierre J Leder P . Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell. (2006) 9:425–34. doi: 10.1016/j.ccr.2006.04.023
100
Jeong JY Jeoung NH Park KG Lee IK . Transcriptional regulation of pyruvate dehydrogenase kinase. Diabetes Metab J. (2012) 36:328–35. doi: 10.4093/dmj.2012.36.5.328
101
Kim JW Tchernyshyov I Semenza GL Dang CV . HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. (2006) 3:177–85. doi: 10.1016/j.cmet.2006.02.002
102
Wang F Bao Y Shen X Zengin G Lyu Y Xiao J et al . Niazirin from Moringa oleifera Lam. attenuates high glucose-induced oxidative stress through PKCζ/Nox4 pathway. Phytomedicine. (2021) 86:153066. doi: 10.1016/j.phymed.2019.153066
103
Ma WQ Sun XJ Zhu Y Liu NF . PDK4 promotes vascular calcification by interfering with autophagic activity and metabolic reprogramming. Cell Death Dis. (2020) 11:991. doi: 10.1038/s41419-020-03162-w
104
Chen S Chen H Yu C Lu R Song T Wang X et al . MiR-638 repressed vascular smooth muscle cell glycolysis by targeting LDHA. Open Med (Wars). (2019) 14:663–72. doi: 10.1515/med-2019-0077
105
Kim JH Bae KH Byun JK Lee S Kim JG Lee IK et al . Lactate dehydrogenase-A is indispensable for vascular smooth muscle cell proliferation and migration. Biochem Biophys Res Commun. (2017) 492:41–7. doi: 10.1016/j.bbrc.2017.08.041
106
Wu N Zhu Y . HNRNPC Regulates GLUT1/LDHA pathway by stabilizing FOXM1 mRNA to promote the progression and aerobic glycolysis of multiple myeloma. Ann Clin Lab Sci. (2024) 54:56–65.
107
Zhu Y Zhang JL Yan XJ Ji Y Wang FF . Exploring a new mechanism between lactate and VSMC calcification: PARP1/POLG/UCP2 signaling pathway and imbalance of mitochondrial homeostasis. Cell Death Dis. (2023) 14:598. doi: 10.1038/s41419-023-06113-3
108
Kwon DH Shin S Nam YS Choe N Lim Y Jeong A et al . CBL-b E3 ligase-mediated neddylation and activation of PARP-1 induce vascular calcification. Exp Mol Med. (2024) 56:2246–59. doi: 10.1038/s12276-024-01322-y
109
Zhu Y Ji JJ Yang R Han XQ Sun XJ Ma WQ et al . Lactate accelerates calcification in VSMCs through suppression of BNIP3-mediated mitophagy. Cell Signal. (2019) 58:53–64. doi: 10.1016/j.cellsig.2019.03.006
110
Feng H Wang JY Yu B Cong X Zhang WG Li L et al . Peroxisome Proliferator-Activated Receptor-γ coactivator-1α inhibits vascular calcification through Sirtuin 3-mediated reduction of mitochondrial oxidative stress. Antioxid Redox Signal. (2019) 31:75–91. doi: 10.1089/ars.2018.7620
111
Qu A Jiang C Xu M Zhang Y Zhu Y Xu Q et al . PGC-1alpha attenuates neointimal formation via inhibition of vascular smooth muscle cell migration in the injured rat carotid artery. Am J Physiol Cell Physiol. (2009) 297:C645–53. doi: 10.1152/ajpcell.00469.2008
112
Finck BN Kelly DP . PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. J Clin Invest. (2006) 116:615–22. doi: 10.1172/JCI27794
113
Zhu Y Ji JJ Wang XD Sun XJ Li M Wei Q et al . Periostin promotes arterial calcification through PPARγ-related glucose metabolism reprogramming. Am J Physiol Heart Circ Physiol. (2021) 320:H2222–h2239. doi: 10.1152/ajpheart.01009.2020
114
Li H Zou L Zheng J Yang T . 12,13-diHOME attenuates high glucose-induced calcification of vascular smooth muscle cells through repressing CPT1A-mediated HMGB1 succinylation. Exp Cell Res. (2024) 438:114031. doi: 10.1016/j.yexcr.2024.114031
115
Nomoto H Pei L Montemurro C Rosenberger M Furterer A Coppola G et al . Activation of the HIF1α/PFKFB3 stress response pathway in beta cells in type 1 diabetes. Diabetologia. (2020) 63:149–61. doi: 10.1007/s00125-019-05030-5
116
Niu J Wu C Zhang M Yang Z Liu Z Fu F et al . κ-opioid receptor stimulation alleviates rat vascular smooth muscle cell calcification via PFKFB3-lactate signaling. Aging (Albany NY). (2021) 13:14355–71. doi: 10.18632/aging.203050
117
Zhu Y Chen JC Zhang JL Wang FF Liu RP . A new mechanism of arterial calcification in diabetes: interaction between H3K18 lactylation and CHI3L1. Clin Sci (Lond). (2025) 139:115–30. doi: 10.1042/CS20243122
118
Zhao X Tan F Cao X Cao Z Li B Shen Z et al . PKM2-dependent glycolysis promotes the proliferation and migration of vascular smooth muscle cells during atherosclerosis. Acta Biochim Biophys Sin (Shanghai). (2020) 52:9–17. doi: 10.1093/abbs/gmz135
119
Liu H Takagaki Y Kumagai A Kanasaki K Koya D . The PKM2 activator TEPP-46 suppresses kidney fibrosis via inhibition of the EMT program and aberrant glycolysis associated with suppression of HIF-1α accumulation. J Diabetes Investig. (2021) 12:697–709. doi: 10.1111/jdi.13478
120
Cao SH Ma RY Cao T Hu T Yang S Ren ZY et al . PKM2 crotonylation reprograms glycolysis in VSMCs, contributing to phenotypic switching. Oncogene. (2025) 44:1990–2003. doi: 10.1038/s41388-025-03353-9
121
Alves-Filho JC Pålsson-McDermott EM . Pyruvate kinase M2: a potential target for regulating inflammation. Front Immunol. (2016) 7:145. doi: 10.3389/fimmu.2016.00145
122
Palsson-McDermott EM Curtis AM Goel G Lauterbach MA Sheedy FJ Gleeson LE et al . Pyruvate kinase M2 regulates Hif-1α activity and IL-1β induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab. (2015) 21:65–80. doi: 10.1016/j.cmet.2014.12.005
123
Jia Y Mao C Ma Z Huang J Li W Ma X et al . PHB2 maintains the contractile phenotype of VSMCs by counteracting PKM2 splicing. Circ Res. (2022) 131:807–24. doi: 10.1161/CIRCRESAHA.122.321005
124
Wang J Liu J Yang F Sun Y Chen J Liu J et al . GMRSP encoded by lncRNA H19 regulates metabolic reprogramming and alleviates aortic dissection. Nat Commun. (2025) 16:1719. doi: 10.1038/s41467-025-57011-5
125
Li YE Liu S Wang L Du Y Wu L Chen H et al . March2 alleviates aortic aneurysm/dissection by regulating PKM2 polymerization. Circ Res. (2025) 136:e73–93. doi: 10.1161/CIRCRESAHA.124.325049
126
Navas-Madroñal M Castelblanco E Camacho M Consegal M Ramirez-Morros A Sarrias MR et al . Role of the scavenger receptor CD36 in accelerated diabetic atherosclerosis. Int J Mol Sci. (2020) 21:7360. doi: 10.3390/ijms21197360
127
Shao JS Cheng SL Pingsterhaus JM Charlton-Kachigian N Loewy AP Towler DA . Msx2 promotes cardiovascular calcification by activating paracrine Wnt signals. J Clin Invest. (2005) 115:1210–20. doi: 10.1172/JCI24140
128
Cai T Sun D Duan Y Wen P Dai C Yang J et al . WNT/β-catenin signaling promotes VSMCs to osteogenic transdifferentiation and calcification through directly modulating Runx2 gene expression. Exp Cell Res. (2016) 345:206–17. doi: 10.1016/j.yexcr.2016.06.007
129
Yoshida T Yamashita M Horimai C Hayashi M . Smooth muscle-selective nuclear factor-κB inhibition reduces phosphate-induced arterial medial calcification in mice with chronic kidney disease. J Am Heart Assoc. (2017) 6:e007248. doi: 10.1161/JAHA.117.007248
130
Lee HL Woo KM Ryoo HM Baek JH . Tumor necrosis factor-alpha increases alkaline phosphatase expression in vascular smooth muscle cells via MSX2 induction. Biochem Biophys Res Commun. (2010) 391:1087–92. doi: 10.1016/j.bbrc.2009.12.027
131
Zhang L Zalewski A Liu Y Mazurek T Cowan S Martin JL et al . Diabetes-induced oxidative stress and low-grade inflammation in porcine coronary arteries. Circulation. (2003) 108:472–8. doi: 10.1161/01.CIR.0000080378.96063.23
132
Son DJ Jung YY Seo YS Park H Lee DH Kim S et al . Interleukin-32α inhibits endothelial inflammation, vascular smooth muscle cell activation, and atherosclerosis by upregulating Timp3 and Reck through suppressing microRNA-205 Biogenesis. Theranostics. (2017) 7:2186–203. doi: 10.7150/thno.18407
133
Yao L Chandra S Toque HA Bhatta A Rojas M Caldwell RB et al . Prevention of diabetes-induced arginase activation and vascular dysfunction by Rho kinase (ROCK) knockout. Cardiovasc Res. (2013) 97:509–19. doi: 10.1093/cvr/cvs371
134
Sánchez-Duffhues G García de Vinuesa A van de Pol V Geerts ME de Vries MR Janson SG et al . Inflammation induces endothelial-to-mesenchymal transition and promotes vascular calcification through downregulation of BMPR2. J Pathol. (2019) 247:333–46. doi: 10.1002/path.5193
135
Yang B Gao X Sun Y Zhao J Chen J Gao L et al . Dihydroartemisinin alleviates high glucose-induced vascular smooth muscle cells proliferation and inflammation by depressing the miR-376b-3p/KLF15 pathway. Biochem Biophys Res Commun. (2020) 530:574–80. doi: 10.1016/j.bbrc.2020.07.095
136
Al-Aly Z Shao JS Lai CF Huang E Cai J Behrmann A et al . Aortic Msx2-Wnt calcification cascade is regulated by TNF-alpha-dependent signals in diabetic Ldlr-/- mice. Arterioscler Thromb Vasc Biol. (2007) 27:2589–96. doi: 10.1161/ATVBAHA.107.153668
137
Wang S Yu H Gao J Chen J He P Zhong H et al . PALMD regulates aortic valve calcification via altered glycolysis and NF-κB-mediated inflammation. J Biol Chem. (2022) 298:101887. doi: 10.1016/j.jbc.2022.101887
138
Bossé Y Mathieu P Thériault S . PALMD as a novel target for calcific aortic valve stenosis. Curr Opin Cardiol. (2019) 34:105–11. doi: 10.1097/HCO.0000000000000605
139
Byon CH Sun Y Chen J Yuan K Mao X Heath JM et al . Runx2-upregulated receptor activator of nuclear factor κB ligand in calcifying smooth muscle cells promotes migration and osteoclastic differentiation of macrophages. Arterioscler Thromb Vasc Biol. (2011) 31:1387–96. doi: 10.1161/ATVBAHA.110.222547
140
Yoshida T Yamashita M Hayashi M . Kruppel-like factor 4 contributes to high phosphate-induced phenotypic switching of vascular smooth muscle cells into osteogenic cells. J Biol Chem. (2012) 287:25706–14. doi: 10.1074/jbc.M112.361360
141
Sun Y Byon CH Yuan K Chen J Mao X Heath JM et al . Smooth muscle cell-specific runx2 deficiency inhibits vascular calcification. Circ Res. (2012) 111:543–52. doi: 10.1161/CIRCRESAHA.112.267237
142
Lund SA Giachelli CM Scatena M . The role of osteopontin in inflammatory processes. J Cell Commun Signal. (2009) 3:311–22. doi: 10.1007/s12079-009-0068-0
143
Schoppet M Preissner KT Hofbauer LC . RANK ligand and osteoprotegerin: paracrine regulators of bone metabolism and vascular function. Arterioscler Thromb Vasc Biol. (2002) 22:549–53. doi: 10.1161/01.ATV.0000012303.37971.DA
144
Fernández-Villabrille S Martín-Vírgala J Martín-Carro B Baena-Huerta F González-García N Gil-Peña H et al . RANKL, but Not R-Spondins, Is involved in vascular smooth muscle cell calcification through LGR4 interaction. Int J Mol Sci. (2024) 25:5735. doi: 10.3390/ijms25115735
145
Harper E Forde H Davenport C Rochfort KD Smith D Cummins PM . Vascular calcification in type-2 diabetes and cardiovascular disease: Integrative roles for OPG, RANKL and TRAIL. Vascul Pharmacol. (2016) 82:30–40. doi: 10.1016/j.vph.2016.02.003
146
Henze LA Estepa M Pieske B Lang F Eckardt KU Alesutan I et al . Zinc ameliorates the osteogenic effects of high glucose in vascular smooth muscle cells. Cells. (2021) 10. doi: 10.3390/cells10113083
147
Li XX Chen ZD Sun XJ Yang YQ Jin H Liu NF . Empagliflozin ameliorates vascular calcification in diabetic mice through inhibiting Bhlhe40-dependent NLRP3 inflammasome activation. Acta Pharmacol Sin. (2024) 45:751–64. doi: 10.1038/s41401-023-01217-0
148
Li S Ni Y Li C Xiang Q Zhao Y Xu H et al . Long noncoding RNA SNHG1 alleviates high glucose-induced vascular smooth muscle cells calcification/senescence by post-transcriptionally regulating Bhlhe40 and autophagy via Atg10. J Physiol Biochem. (2023) 79:83–105. doi: 10.1007/s13105-022-00924-2
149
Herzig S Shaw RJ . AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol. (2018) 19:121–35. doi: 10.1038/nrm.2017.95
150
Entezari M Hashemi D Taheriazam A Zabolian A Mohammadi S Fakhri F et al . AMPK signaling in diabetes mellitus, insulin resistance and diabetic complications: A pre-clinical and clinical investigation. BioMed Pharmacother. (2022) 146:112563. doi: 10.1016/j.biopha.2021.112563
151
Zhu Y Han XQ Sun XJ Yang R Ma WQ Liu NF . Lactate accelerates vascular calcification through NR4A1-regulated mitochondrial fission and BNIP3-related mitophagy. Apoptosis. (2020) 25:321–40. doi: 10.1007/s10495-020-01592-7
152
Wang Q Zhang M Torres G Wu S Ouyang C Xie Z et al . Metformin suppresses diabetes-accelerated atherosclerosis via the inhibition of Drp1-mediated mitochondrial fission. Diabetes. (2017) 66:193–205. doi: 10.2337/db16-0915
153
Deng M Su D Xu S Little PJ Feng X Tang L et al . Metformin and vascular diseases: A focused review on smooth muscle cell function. Front Pharmacol. (2020) 11:635. doi: 10.3389/fphar.2020.00635
154
Wang L Zhu LH Jiang H Tang QZ Yan L Wang D et al . Grape seed proanthocyanidins attenuate vascular smooth muscle cell proliferation via blocking phosphatidylinositol 3-kinase-dependent signaling pathways. J Cell Physiol. (2010) 223:713–26. doi: 10.1002/jcp.22080
155
Lino M Wan MH Rocca AS Ngai D Shobeiri N Hou G et al . Diabetic vascular calcification mediated by the collagen receptor discoidin domain receptor 1 via the Phosphoinositide 3-kinase/Akt/Runt-related transcription factor 2 signaling axis. Arterioscler Thromb Vasc Biol. (2018) 38:1878–89. doi: 10.1161/ATVBAHA.118.311238
156
Yang Y Sun Y Chen J Bradley WE Dell'Italia LJ Wu H et al . AKT-independent activation of p38 MAP kinase promotes vascular calcification. Redox Biol. (2018) 16:97–103. doi: 10.1016/j.redox.2018.02.009
157
Deng L Huang L Sun Y Heath JM Wu H Chen Y . Inhibition of FOXO1/3 promotes vascular calcification. Arterioscler Thromb Vasc Biol. (2015) 35:175–83. doi: 10.1161/ATVBAHA.114.304786
158
Zhang ML Zhang MN Chen H Wang X Zhao K Li X et al . Salvianolic Acid B alleviates high glucose-induced vascular smooth muscle cell inflammation by upregulating the miR-486a-5p expression. Mediators Inflammation. (2024) 2024:4121166. doi: 10.1155/2024/4121166
159
Shi LL Hao M Jin ZY Peng GF Tang YY Kuang HY . Liraglutide alleviates diabetic atherosclerosis through regulating calcification of vascular smooth muscle cells. Dis Markers. (2022) 2022:5013622. doi: 10.1155/2022/5013622
160
Zhang M Li T Tu Z Zhang Y Wang X Zang D et al . Both high glucose and phosphate overload promote senescence-associated calcification of vascular muscle cells. Int Urol Nephrol. (2022) 54:2719–31. doi: 10.1007/s11255-022-03195-4
161
Yang R Zhu Y Wang Y Ma W Han X Wang X et al . HIF-1α/PDK4/autophagy pathway protects against advanced glycation end-products induced vascular smooth muscle cell calcification. Biochem Biophys Res Commun. (2019) 517:470–6. doi: 10.1016/j.bbrc.2019.07.102
162
Cao J Chen C Chen Q Gao Y Zhao Z Yuan Q et al . Extracellular vesicle miR-32 derived from macrophage promotes arterial calcification in mice with type 2 diabetes via inhibiting VSMC autophagy. J Transl Med. (2022) 20:307. doi: 10.1186/s12967-022-03502-8
163
Liu Y Wang WM Zhang XL He HQ Sun XL Zeng H et al . AGE/RAGE promotes thecalcification of human aortic smooth muscle cells via the Wnt/β-catenin axis. Am J Transl Res. (2016) 8:4644–56.
164
Shi L Hao M Qu G Xu Y Cui Z Geng L et al . The key role of Liraglutide in preventing autophagy of vascular smooth muscle cells in high glucose conditions. Balkan Med J. (2024) 41:54–63. doi: 10.4274/balkanmedj.galenos.2023.2023-8-44
165
Xu ZJ Xu J Lei WJ Wang X Zou QL Lv LC et al . RANBP1 regulates NOTCH3-mediated autophagy in high glucose-induced vascular smooth muscle cells. Front Biosci (Landmark Ed). (2025) 30:26850. doi: 10.31083/FBL26850
166
Li X Liu XL Li X Zhao YC Wang QQ Zhong HY et al . Dickkopf1 (Dkk1) alleviates vascular calcification by regulating the degradation of phospholipase D1 (PLD1). J Cardiovasc Transl Res. (2022) 15:1327–39. doi: 10.1007/s12265-022-10251-y
167
Shen J Zhang C Liu Y Zhao M Wang Q Li P et al . L-type calcium ion channel-mediated activation of autophagy in vascular smooth muscle cells via thonningianin A (TA) alleviates vascular calcification in type 2 diabetes mellitus. Eur J Pharmacol. (2023) 959:176084. doi: 10.1016/j.ejphar.2023.176084
168
Zhao Y Sun Z Li L Yuan W Wang Z . Role of collagen in vascular calcification. J Cardiovasc Pharmacol. (2022) 80:769–78. doi: 10.1097/FJC.0000000000001359
169
Xiong S Salazar G Patrushev N Ma M Forouzandeh F Hilenski L et al . Peroxisome proliferator-activated receptor γ coactivator-1α is a central negative regulator of vascular senescence. Arterioscler Thromb Vasc Biol. (2013) 33:988–98. doi: 10.1161/ATVBAHA.112.301019
170
Chen Z Li X Sun X Xiao S Chen T Ren L et al . STING1-accelerated vascular smooth muscle cell senescence-associated vascular calcification in diabetes is ameliorated by oleoylethanolamide via improved mitochondrial DNA oxidative damage. Free Radic Biol Med. (2024) 222:437–55. doi: 10.1016/j.freeradbiomed.2024.06.010
171
Bartoli-Leonard F Wilkinson FL Schiro A Serracino Inglott F Alexander MY Weston R . Loss of SIRT1 in diabetes accelerates DNA damage-induced vascular calcification. Cardiovasc Res. (2021) 117:836–49. doi: 10.1093/cvr/cvaa134
172
Liu H Yang P Chen S Wang S Jiang L Xiao X et al . Ncf1 knockout in smooth muscle cells exacerbates angiotensin II-induced aortic aneurysm and dissection by activating the STING pathway. Cardiovasc Res. (2024) 120:1081–96. doi: 10.1093/cvr/cvae081
173
Li S Zhan JK Wang YJ Lin X Zhong JY Wang Y et al . Exosomes from hyperglycemia-stimulated vascular endothelial cells contain versican that regulate calcification/senescence in vascular smooth muscle cells. Cell Biosci. (2019) 9:1. doi: 10.1186/s13578-018-0263-x
174
Leopold JA . MicroRNAs regulate vascular medial calcification. Cells. (2014) 3:963–80. doi: 10.3390/cells3040963
175
Toniolo A Warden EA Nassi A Cignarella A Bolego C . Regulation of SIRT1 in vascular smooth muscle cells from streptozotocin-diabetic rats. PloS One. (2013) 8:e65666. doi: 10.1371/journal.pone.0065666
176
Bartoli-Leonard F Wilkinson FL Schiro A Inglott FS Alexander MY Weston R . Suppression of SIRT1 in diabetic conditions induces osteogenic differentiation of human vascular smooth muscle cells via RUNX2 signalling. Sci Rep. (2019) 9:878. doi: 10.1038/s41598-018-37027-2
177
Badi I Mancinelli L Polizzotto A Ferri D Zeni F Burba I et al . miR-34a promotes vascular smooth muscle cell calcification by downregulating SIRT1 (Sirtuin 1) and Axl (AXL Receptor Tyrosine Kinase). Arterioscler Thromb Vasc Biol. (2018) 38:2079–90. doi: 10.1161/ATVBAHA.118.311298
178
Zeinali F Aghaei Zarch SM Jahan-Mihan A Kalantar SM Vahidi Mehrjardi MY Fallahzadeh H et al . Circulating microRNA-122, microRNA-126-3p and microRNA-146a are associated with inflammation in patients with pre-diabetes and type 2 diabetes mellitus: A case control study. PloS One. (2021) 16:e0251697. doi: 10.1371/journal.pone.0251697
179
Zhao Z Li A Zeng R Zeng Z Ou L Cao J et al . A CEBPB/miR-32-5p/GATA6 axis promotes vascular calcification in type 2 diabetes. Int J Biochem Cell Biol. (2024) 173:106613. doi: 10.1016/j.biocel.2024.106613
180
Shi L Li Y Shi M Li X Li G Cen J et al . Hsa_circRNA_0008028 deficiency ameliorates high glucose-induced proliferation, calcification, and autophagy of vascular smooth muscle cells via miR-182-5p/TRIB3 Axis. Oxid Med Cell Longev. (2022) 2022:5142381. doi: 10.1155/2022/5142381
181
Zhou DM Ran F Ni HZ Sun LL Xiao L Li XQ et al . Metformin inhibits high glucose-induced smooth muscle cell proliferation and migration. Aging (Albany NY). (2020) 12:5352–61. doi: 10.18632/aging.102955
182
Jia S Ma WD Zhang CY Zhang Y Yao ZH Quan XH et al . Tanshinone IIA attenuates high glucose induced human VSMC proliferation and migration through miR-21-5p-mediated tropomyosin 1 downregulation. Arch Biochem Biophys. (2019) 677:108154. doi: 10.1016/j.abb.2019.108154
183
Sun G Song H Wu S . miR−19a promotes vascular smooth muscle cell proliferation, migration and invasion through regulation of Ras homolog family member B. Int J Mol Med. (2019) 44:1991–2002. doi: 10.3892/ijmm.2019.4357
184
Zhang J Cai W Fan Z Yang C Wang W Xiong M et al . MicroRNA-24 inhibits the oxidative stress induced by vascular injury by activating the Nrf2/Ho-1 signaling pathway. Atherosclerosis. (2019) 290:9–18. doi: 10.1016/j.atherosclerosis.2019.08.023
185
Lu X Ma ST Zhou B Lí T . MiR-9 promotes the phenotypic switch of vascular smooth muscle cells by targeting KLF5. Turk J Med Sci. (2019) 49:928–38. doi: 10.3906/medical
186
Yang J Fan Z Yang J Ding J Yang C Chen L . MicroRNA-24 attenuates neointimal hyperplasia in the diabetic rat carotid artery injury model by inhibiting Wnt4 signaling pathway. Int J Mol Sci. (2016) 17. doi: 10.3390/ijms17060765
187
Ye D Lou GH Li AC Dong FQ Chen GP Xu WW et al . MicroRNA−125a−mediated regulation of the mevalonate signaling pathway contributes to high glucose−induced proliferation and migration of vascular smooth muscle cells. Mol Med Rep. (2020) 22:165–74. doi: 10.3892/mmr.2020.11077
188
Chen X Tian F Sun Z Zeng G Tang P . Elevation of circulating miR-210 participates in the occurrence and development of type 2 diabetes mellitus and its complications. J Diabetes Res. (2022) 2022:9611509. doi: 10.1155/2022/9611509
189
Zhong JY Cui XJ Zhan JK Wang YJ Li S Lin X et al . LncRNA-ES3 inhibition by Bhlhe40 is involved in high glucose-induced calcification/senescence of vascular smooth muscle cells. Ann N Y Acad Sci. (2020) 1474:61–72. doi: 10.1111/nyas.14381
190
Lin X He SQ Shan SK Xu F Wu F Li FX et al . Endothelial cells derived extracellular vesicles promote diabetic arterial calcification via circ_0008362/miR-1251-5p/Runx2 axial. Cardiovasc Diabetol. (2024) 23:369. doi: 10.1186/s12933-024-02440-7
191
Patidar A Singh DK Winocour P Farrington K Baydoun AR . Human uraemic serum displays calcific potential in vitro that increases with advancing chronic kidney disease. Clin Sci (Lond). (2013) 125:237–45. doi: 10.1042/CS20120638
192
Xie H Wang Z Zhang L Lei Q Zhao A Wang H et al . Extracellular Vesicle-functionalized decalcified bone matrix scaffolds with enhanced pro-angiogenic and pro-bone regeneration activities. Sci Rep. (2017) 7:45622. doi: 10.1038/srep45622
193
Goettsch C Hutcheson JD Aikawa M Iwata H Pham T Nykjaer A et al . Sortilin mediates vascular calcification via its recruitment into extracellular vesicles. J Clin Invest. (2016) 126:1323–36. doi: 10.1172/JCI80851
194
Itoh S Mizuno K Aikawa M Aikawa E . Dimerization of sortilin regulates its trafficking to extracellular vesicles. J Biol Chem. (2018) 293:4532–44. doi: 10.1074/jbc.RA117.000732
195
Shamseddine AA Airola MV Hannun YA . Roles and regulation of neutral sphingomyelinase-2 in cellular and pathological processes. Adv Biol Regul. (2015) 57:24–41. doi: 10.1016/j.jbior.2014.10.002
196
Jing L Li L Ren X Sun Z Bao Z Yuan G et al . Role of sortilin and matrix vesicles in Nϵ-Carboxymethyl-Lysine-induced diabetic atherosclerotic calcification. Diabetes Metab Syndr Obes. (2020) 13:4141–51. doi: 10.2147/DMSO.S273029
197
Kapustin AN Chatrou ML Drozdov I Zheng Y Davidson SM Soong D et al . Vascular smooth muscle cell calcification is mediated by regulated exosome secretion. Circ Res. (2015) 116:1312–23. doi: 10.1161/CIRCRESAHA.116.305012
198
Lallemand T Rouahi M Swiader A Grazide MH Geoffre N Alayrac P et al . nSMase2 (Type 2-Neutral Sphingomyelinase) deficiency or inhibition by GW4869 reduces inflammation and atherosclerosis in Apoe(-/-) Mice. Arterioscler Thromb Vasc Biol. (2018) 38:1479–92. doi: 10.1161/ATVBAHA.118.311208
199
Sun Z Li L Zhang L Yan J Shao C Bao Z et al . Macrophage galectin-3 enhances intimal translocation of vascular calcification in diabetes mellitus. Am J Physiol Heart Circ Physiol. (2020) 318:H1068–h1079. doi: 10.1152/ajpheart.00690.2019
200
Zhang YX Tang RN Wang LT Liu BC . Role of crosstalk between endothelial cells and smooth muscle cells in vascular calcification in chronic kidney disease. Cell Prolif. (2021) 54:e12980. doi: 10.1111/cpr.12980
201
Lin X Li S Wang YJ Wang Y Zhong JY He JY et al . Exosomal Notch3 from high glucose-stimulated endothelial cells regulates vascular smooth muscle cells calcification/aging. Life Sci. (2019) 232:116582. doi: 10.1016/j.lfs.2019.116582
202
Wang S Zhan J Lin X Wang Y Wang Y Liu Y . CircRNA-0077930 from hyperglycaemia-stimulated vascular endothelial cell exosomes regulates senescence in vascular smooth muscle cells. Cell Biochem Funct. (2020) 38:1056–68. doi: 10.1002/cbf.3543
203
Guo B Shan SK Xu F Lin X Li FX Wang Y et al . Protective role of small extracellular vesicles derived from HUVECs treated with AGEs in diabetic vascular calcification. J Nanobiotechnology. (2022) 20:334. doi: 10.1186/s12951-022-01529-z
204
Baba I Matoba T Katsuki S Koga JI Kawahara T Kimura M et al . EVs-miR-17-5p attenuates the osteogenic differentiation of vascular smooth muscle cells potentially via inhibition of TGF-β signaling under high glucose conditions. Sci Rep. (2024) 14:16323. doi: 10.1038/s41598-024-67006-9
205
Kawakami R Katsuki S Travers R Romero DC Becker-Greene D Passos LSA et al . S100A9-RAGE axis accelerates formation of macrophage-mediated extracellular vesicle microcalcification in diabetes mellitus. Arterioscler Thromb Vasc Biol. (2020) 40:1838–53. doi: 10.1161/ATVBAHA.118.314087
206
Carr JJ Register TC Hsu FC Lohman K Lenchik L Bowden DW et al . Calcified atherosclerotic plaque and bone mineral density in type 2 diabetes: the diabetes heart study. Bone. (2008) 42:43–52. doi: 10.1016/j.bone.2007.08.023
207
Raggi P Shaw LJ Berman DS Callister TQ . Prognostic value of coronary artery calcium screening in subjects with and without diabetes. J Am Coll Cardiol. (2004) 43:1663–9. doi: 10.1016/j.jacc.2003.09.068
208
Hoff JA Quinn L Sevrukov A Lipton RB Daviglus M Garside DB et al . The prevalence of coronary artery calcium among diabetic individuals without known coronary artery disease. J Am Coll Cardiol. (2003) 41:1008–12. doi: 10.1016/S0735-1097(02)02975-3
209
Ahn BY Jeong Y Kim S Zhang Y Kim SW Leem YE et al . Cdon suppresses vascular smooth muscle calcification via repression of the Wnt/Runx2 Axis. Exp Mol Med. (2023) 55:120–31. doi: 10.1038/s12276-022-00909-7
210
Kostina A Shishkova A Ignatieva E Irtyuga O Bogdanova M Levchuk K et al . Different Notch signaling in cells from calcified bicuspid and tricuspid aortic valves. J Mol Cell Cardiol. (2018) 114:211–9. doi: 10.1016/j.yjmcc.2017.11.009
211
Pustlauk W Westhoff TH Claeys L Roch T Geißler S Babel N . Induced osteogenic differentiation of human smooth muscle cells as a model of vascular calcification. Sci Rep. (2020) 10:5951. doi: 10.1038/s41598-020-62568-w
212
Ceriello A . Hyperglycaemia and the vessel wall: the pathophysiological aspects on the atherosclerotic burden in patients with diabetes. Eur J Cardiovasc Prev Rehabil. (2010) 17 Suppl 1:S15–9. doi: 10.1097/01.hjr.0000368193.24732.66
213
Chen Y Zhao X Wu H . Transcriptional programming in arteriosclerotic disease: A multifaceted function of the Runx2 (Runt-Related Transcription Factor 2). Arterioscler Thromb Vasc Biol. (2021) 41:20–34. doi: 10.1161/ATVBAHA.120.313791
214
Al-Lamki RS Bradley JR Pober JS . Human organ culture: Updating the approach to bridge the gap from in vitro to in vivo in inflammation, Cancer, and Stem Cell Biology. Front Med (Lausanne). (2017) 4:148. doi: 10.3389/fmed.2017.00148
215
Hortells L Sosa C Millán Á Sorribas V . Critical parameters of the in vitro method of vascular smooth muscle cell calcification. PLoS One. (2015) 10:e0141751. doi: 10.1371/journal.pone.0141751
216
Qiu J Zheng Y Hu J Liao D Gregersen H Deng X et al . Biomechanical regulation of vascular smooth muscle cell functions: from in vitro to in vivo understanding. J R Soc Interface. (2014) 11:20130852. doi: 10.1098/rsif.2013.0852
Summary
Keywords
atherosclerosis, calcification, osteogenic transformation, vascular smooth muscle cells, hyperglycemia research insights
Citation
Guo J and Du L (2025) Regulation of osteogenic differentiation in vascular smooth muscle cells under high-glucose condition. Front. Endocrinol. 16:1589160. doi: 10.3389/fendo.2025.1589160
Received
07 March 2025
Accepted
08 August 2025
Published
05 September 2025
Volume
16 - 2025
Edited by
Jaideep Menon, Amrita Vishwa Vidyapeetham University, India
Reviewed by
Michael Edwin Edmonds, King’s College Hospital NHS Foundation Trust, United Kingdom
Xuchang Zhou, The First Affiliated Hospital of Xiamen University, China
Louisa Bechohra, University of Science and Technology Houari Boumediene, Algeria
Updates
Copyright
© 2025 Guo and Du.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laijing Du, 17395953051@163.com
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.