 Dana Bou Matar1
Dana Bou Matar1 Mahmoud Zhra2
Mahmoud Zhra2 Walid Khaled Nassar3
Walid Khaled Nassar3 Haifa Altemyatt3
Haifa Altemyatt3 Asfiya Naureen3Nada Abotouk3
Asfiya Naureen3Nada Abotouk3 Muhammad Affan Elahi3
Muhammad Affan Elahi3 Ahmad Aljada2*
Ahmad Aljada2*- 1Department of Physiology, College of Medicine, Alfaisal University, Riyadh, Saudi Arabia
- 2Department of Biochemistry and Molecular Medicine, College of Medicine, Alfaisal University, Riyadh, Saudi Arabia
- 3College of Medicine, Alfaisal University, Riyadh, Saudi Arabia
Background: Metabolic disease incidence continues rising globally. Adipose tissue dysfunction serves as a crucial pathophysiological mediator. We evaluate molecular mechanisms linking adipose dysfunction to metabolic dysregulation.
Methods: We systematically reviewed literature on adipose biology, stress mechanisms, inflammation, and metabolic networks. Analysis prioritized methodologically robust studies from the past decade.
Results: Adipose dysfunction disrupts metabolic homeostasis through complex molecular networks. Stressed adipocytes exhibit mitochondrial impairment and endoplasmic reticulum (ER) stress. These changes alter inflammatory mediators and adipokine secretion. Brown and beige adipose regulate energy balance via uncoupling protein 1 (UCP1)-mediated thermogenesis. Key transcriptional regulators, PGC-1α and PR domain containing 16 (PRDM16), control thermogenic adipocyte development. Cellular senescence contributes significantly to age-related adipose dysfunction through inflammatory secretory phenotypes. Brown fat also secretes specialized factors influencing whole-body metabolism, emphasizing adipose tissue’s endocrine function.
Conclusion: Adipose dysfunction represents a critical nexus in metabolic disease pathogenesis. Cellular stress, inflammation, and metabolic dysregulation converge at this point. Novel therapies targeting thermogenic activation and cellular senescence show promise. Despite advancing mechanistic understanding, developing effective interventions remains challenging due to adipose tissue’s complex roles in systemic metabolic regulation.
1 Introduction
Adipose tissue (AT) dysfunction represents a central pathophysiological process in obesity-related metabolic disorders. In health, AT maintains metabolic homeostasis by dynamically responding to energy demands through regulated lipid storage and mobilization. However, factors such as chronic nutrient excess, physical inactivity, genetic predisposition, and aging can drive AT dysfunction, overwhelming its adaptive capacity (1–14).
Multiple mechanisms underlie AT dysfunction, including mitochondrial impairment, ER stress, inflammatory pathway activation, and extracellular matrix (ECM) remodeling. These changes create self-reinforcing pathological cycles that worsen tissue function and promote systemic insulin resistance (15–17). Notably, AT dysfunction manifests across diverse metabolic disorders, from obesity-associated insulin resistance to lipodystrophies, highlighting that proper adipose function, not just presence, is essential for metabolic homeostasis.
AT comprises distinct depots (white, brown, beige, and pink) with unique physiological functions and metabolic characteristics (1, 18–21). White adipose tissue (WAT) controls lipolysis through Adipose Triglyceride Lipase (ATGL) and hormone-sensitive lipase (HSL) enzymes (19), while brown adipose tissue (BAT) expresses UCP1 for thermogenesis (20). Beige adipocytes demonstrate remarkable plasticity in response to environmental signals (1).
At the molecular level, adipose dysfunction involves cellular stress responses, aberrant inflammatory signaling, and dysregulated microRNA networks. These processes collectively compromise adipocyte metabolic and endocrine functions, disrupting inter-organ communication and systemic metabolism.
This review integrates current evidence on molecular mechanisms underlying AT dysfunction in metabolic disorders, emphasizing adipose-specific signaling networks. We examine dysfunction across various conditions and analyze emerging therapeutic approaches targeting thermogenic activation and cellular senescence pathways, providing an integrated framework for understanding metabolic disease pathogenesis and identifying novel intervention strategies.
2 Molecular and functional organization of adipose tissue
AT plays an essential role in metabolic regulation beyond simple energy storage (22). These distinct cell types (including the adipocytes at different stages of differentiation, resident immune cell types, and vascular elements) cooperatively organize into metabolically active tissue units in the extracellular space (23). Dense vascular beds and neural inputs regulate metabolic responses at both tissue and systemic levels.
Biochemical characteristics of AT have revealed complex regulatory networks that control energy metabolism. Key enzymes such as ATGL and HSL play a role in the storage and mobilization of lipids, and their dysregulation directly contributes to metabolic disorders (22). Several elements, such as hormonal levels, diet conditions, and the interaction of certain proteins, influence these pathways. AT acts as an endocrine organ and releases many bioactive substances that affect the overall metabolism. These adipokines control appetite, energy consumption, insulin sensitivity, and inflammation responses through a variety of signal pathways. Additionally, the tissue processes several key hormones via specific enzyme systems, modifying androgens, glucocorticoids, and thyroid hormones.
Anatomical distribution significantly impacts AT function, with distinct depots exhibiting unique molecular and metabolic profiles. Recent molecular analyses have identified four categories of AT - white, brown, beige, and pink - each serving specialized physiological roles (22, 23). These depot-specific characteristics stem from the differential expression of developmental transcription factors and metabolic enzymes.
The global rise in metabolic disease prevalence has spurred research into the molecular mechanisms underlying AT dysfunction. Analysis of AT from obese subjects reveals characteristic alterations in gene expression networks controlling metabolism, inflammation, and endocrine function. Recent studies focus on depot-specific transcriptional programs and their relationship to systemic metabolic regulation.
This article explores molecular control mechanisms in AT function and metabolic disease, based on evidence from biochemical, cellular, and clinical studies. Experimental findings highlight specific transcriptional networks and signaling pathways maintaining adipose homeostasis. Analysis of these molecular mechanisms provides critical insights for developing targeted therapeutic strategies.
3 AT types and their distinct functions
AT functions as a complex endocrine organ with diverse roles beyond simple energy storage (24). It encompasses multiple cell types and is classified into four distinct categories: white, brown, beige, and pink AT (14). AT’s plasticity and heterogeneous nature (Table 1) are fundamental to its role in energy homeostasis, metabolic regulation, and disease progression (18).
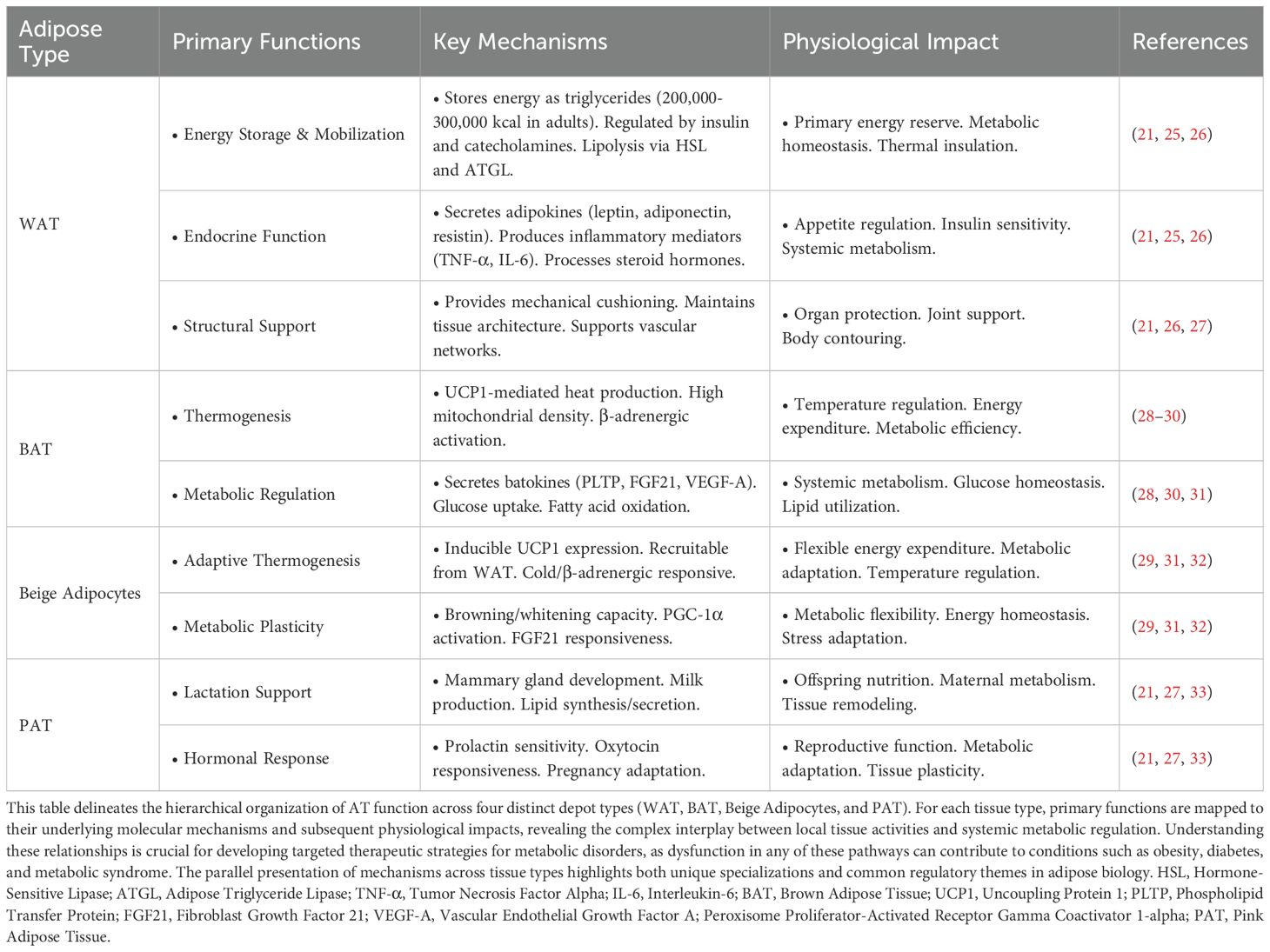
Table 1. Integrated analysis of AT functional networks.
3.1 White AT: energy storage, endocrine regulation, and metabolic homeostasis
White AT (WAT) regulates energy homeostasis through lipolysis, with ATGL and HSL mediating 95% of triglyceride catabolism (19). These enzymes exhibit complex regulation by hormonal and nutritional status (34), with disrupted lipolysis directly contributing to metabolic disorders (35, 36). WAT functions extend far beyond simple energy storage, secreting numerous bioactive adipokines including leptin, adiponectin, resistin, visfatin, Tumor necrosis factor-α (TNF-α), and various interleukins (37, 38). Each adipokine serves distinct metabolic roles. Leptin regulates appetite and energy consumption through the hypothalamic signal, while adiponectin increases insulin sensitivity and suppresses inflammation (39). Resistin and visfatin modulate glucose metabolism and immune responses (40).
WAT also processes several key hormones, transforms androgens into estrogens through aromatization activity, and mutates glucocorticoids through type 1 11β-hydroxysteroid dehydrogenase (11β-HSD1) (41). WAT metabolizes thyroid hormones, which regulate the lipogenic and lipolytic genes (42). Additionally, WAT facilitates thyroid hormone metabolism, thereby influencing lipogenic and lipolytic gene expression (42).
Structurally, WAT provides essential mechanical functions. The tissue acts as a protective cushion for internal organs, provides joint support, and generates thermal insulation. Particularly, subcutaneous WAT functions as an effective thermal buffer during cold environmental conditions.
3.2 Brown AT: thermogenic organ for heat production and energy expenditure
Brown AT (BAT) is characterized by the presence of UCP1 (thermogenin), a specialized mitochondrial protein that uncouples the electron transport chain from ATP synthesis by allowing protons to leak across the inner mitochondrial membrane, thereby dissipating the proton gradient as heat rather than using it for ATP production (20). Cold exposure triggers BAT activation through sympathetic nerve stimulation and β-adrenergic signaling (28). The tissue’s high metabolic activity drives systemic energy expenditure through rapid fatty acid oxidation and glucose uptake, significantly impacting basal metabolism and temperature regulation (43).
The discovery of active BAT in adult humans in 2009 fundamentally changed understanding of metabolic regulation (44). BAT’s beige/brite adipocytes display remarkable adaptation to environmental and physiological changes (45). Higher BAT activity correlates with reduced body fat mass, suggesting protective effects against obesity (46). BAT secretes specialized signaling factors - batokines - comprising peptides, metabolites, lipids, and regulatory RNAs (47). These molecules target metabolic processes in liver, heart, muscle, and WAT through multiple signaling pathways (48).
Batokines regulate whole-body metabolism by modifying glucose handling, insulin responses, and inflammatory signals (49). Key batokines include PLTP (Phospholipid Transfer Protein), FGF21 (Fibroblast Growth Factor 21), VEGF-A (Vascular Endothelial Growth Factor A), BMP8 (Bone Morphogenetic Protein 8), NRG-4 (Neuregulin 4), and IL-6, each controlling specific metabolic pathways (50–52). These factors enhance insulin sensitivity and substrate utilization across multiple tissues (53). Type 2 diabetes mellitus (T2DM) progression alters both batokine production and BAT function (54). Recent work identifies exosomal microRNAs as additional metabolic regulators (49). IL-6 shows tissue-specific effects - promoting glucose homeostasis and energy expenditure in BAT while potentially causing insulin resistance elsewhere (55, 56). Through these diverse endocrine functions, BAT emerges as a central coordinator of systemic metabolism.
3.3 Beige adipocytes: inducible thermogenic cells for metabolic flexibility
Beige adipocytes are a unique thermogenic cell population that emerge within WAT through a process called “browning” or “beiging,” triggered by various environmental stimuli such as cold exposure, β-adrenergic activation, and certain hormonal signals (1). These cells exhibit significant metabolic plasticity, expressing UCP1 and transitioning between energy storage and expenditure phenotypes in response to physiological demands (57). Beige adipocytes contribute to adaptive thermogenesis through both UCP1-dependent mechanisms and alternative pathways, such as calcium cycling and creatine-driven substrate cycling, thereby enhancing glucose homeostasis and lipid metabolism (58).
The molecular pathways governing beige adipose cells utilize specific transcriptional controllers for development and function. Each factor serves distinct roles: PPAR gamma coactivator 1 alpha (PGC-1α) initiates metabolic programs, PRDM16 (PR domain containing 16) modifies chromatin structure to enable differentiation, and FGF21 (fibroblast growth factor 21) enables heat production in mature cells (59). Recent studies demonstrate how muscle-derived myokines produced during exercise influence beige fat development. Thyroid hormone signaling pathways provide additional mechanisms for regulating beige adipocyte formation and activity (60). Systematic screening has identified numerous circulating molecules that control differentiation and thermogenic capacity in both brown and beige adipocytes, including irisin, FGF21, BMP8, NRG-4, and IL-6, establishing potential therapeutic targets for treating metabolic disorder (61).
3.4 Pink AT: dynamic plasticity in mammary gland development and lactation
Pink AT (PAT) showcases remarkable plasticity, forming from subcutaneous WAT during pregnancy and lactation. PAT is uniquely composed of mammary gland alveolar epithelial cells, known as pink adipocytes, which are specialized for milk production and secretion (21). PAT formation occurs through transdifferentiation, where white adipocytes undergo significant phenotypic and functional changes, including the development of milk-producing capabilities and alterations in lipid storage and secretion (21). Pink adipocytes possess distinct features that differentiate them from both white and brown adipocytes, including cellular machinery and molecular pathways specialized for lactation. The transdifferentiation process involves extensive genetic reprogramming, resulting in significant shifts in cellular identity and function (62, 63). Research into PAT and its transformation mechanisms provides valuable insights into adipose biology and cellular plasticity. Understanding these processes could lead to new therapies for metabolic disorders and breast cancer by leveraging cellular reprogramming and tissue adaptability.
4 Molecular mechanisms of AT dysfunction
4.1 Cellular stress pathways
Adipocyte dysfunction manifests through multiple disrupted cellular pathways (Table 2). Precise cellular changes compromise lipid metabolism, glucose transport, and inflammatory responses, generating cascading effects that extend beyond the local tissue environment (1–14). Recent research into obesity-associated adipose pathology has identified several key molecular signatures. Adipocyte hypertrophy, with cell diameters expanding up to 150-200 μm (94), leads to reduced oxygen availability and tissue hypoxia (95). This is accompanied by persistent inflammatory activation, marked by elevated levels of specific cytokines, including TNF-α, IL-6, and interleukin-1β (IL-1β) (96). Laboratory analyses reveal that these cellular perturbations alter essential signaling networks, as evidenced by abnormal adipokine profiles and increased immune cell presence. The subsequent release of free fatty acids (FFAs) into circulation impairs insulin signaling across multiple peripheral tissues (4–9).
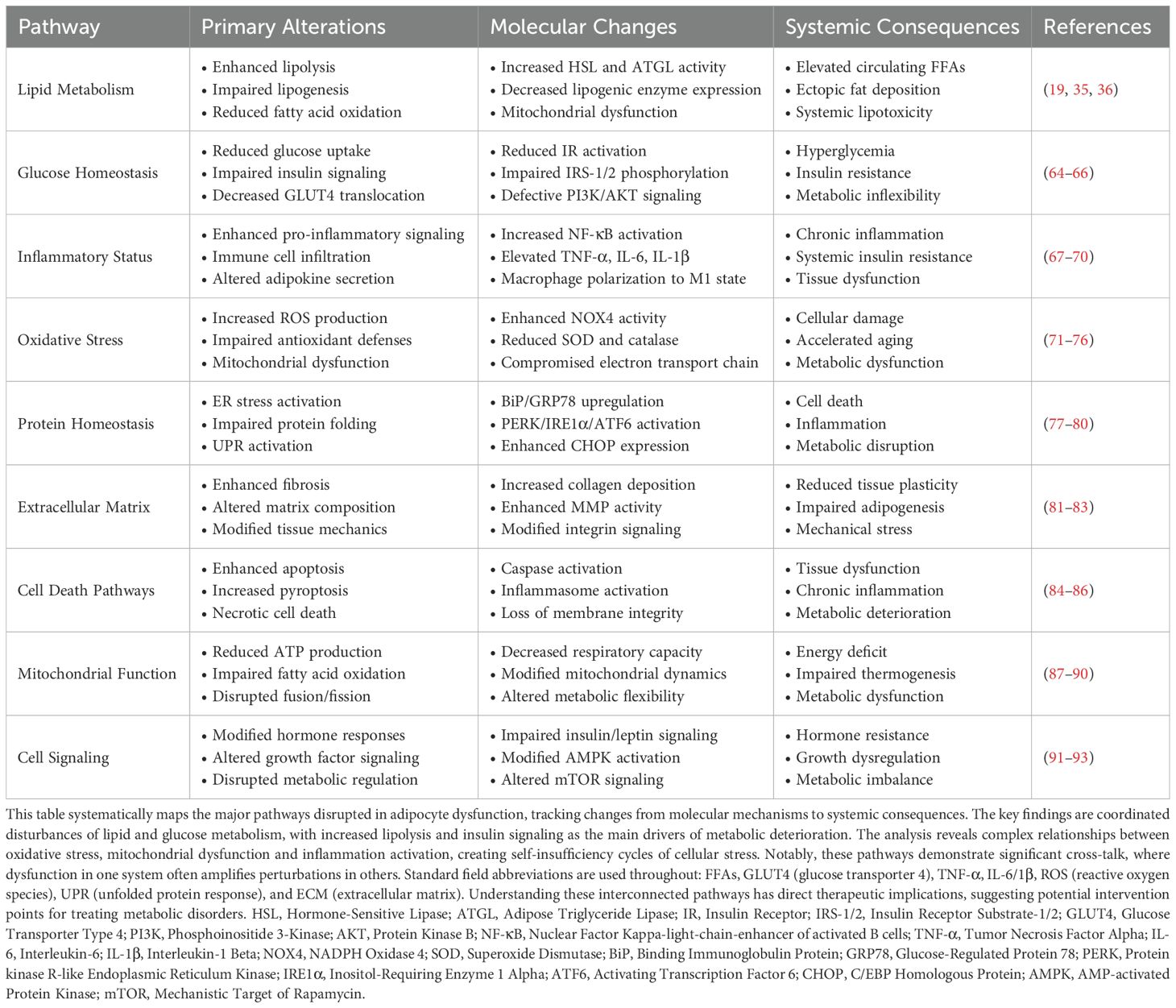
Table 2. Integrated analysis of adipocyte dysfunction: from molecular mechanisms to systemic impact.
Affected adipose depots develop sustained mitochondrial defects and heightened oxidative stress, which amplify existing metabolic disruptions (10). The location of adipose expansion critically influences disease progression, with visceral and ectopic fat deposits having particularly detrimental effects on systemic metabolism (11).
4.2 ER stress and the unfolded protein response
The ER contains numerous quality control pathways that regulate protein processing and maintain cellular homeostasis through distinct molecular mechanisms. Binding immunoglobulin protein (BiP) requires ATP for its chaperone function, recognizing exposed hydrophobic regions on unfolded proteins to prevent aggregation. X-box binding protein 1 (XBP1) transcriptionally regulates protein folding genes, while the unfolded protein response (UPR) activates when protein folding demands increase, reducing protein synthesis while expanding folding capacity (77).
The major UPR transducers operate through biochemically distinct mechanisms. Protein kinase R-like endoplasmic reticulum kinase (PERK) activation triggers eukaryotic initiation factor 2 alpha (eIF2α) phosphorylation specifically at serine 51, leading to selective mRNA translation despite global protein synthesis attenuation. The membrane-bound transcription factor ATF6 requires sequential proteolysis within Golgi compartments, generating an active nuclear form that transcriptionally upregulates ER-resident chaperones. The bifunctional enzyme inositol-requiring enzyme 1 α (IRE1α) exhibits both protein kinase and site-specific endoribonuclease activities, enabling XBP1 mRNA processing and targeted decay of ER-associated transcripts via Regulated IRE1-Dependent Decay (RIDD) (78). Analysis of metabolically compromised tissues reveals pronounced activation of these UPR components, particularly within AT, where UPR activation is significantly increased, with insulin upregulating the UPR dose-dependently over the entire physiological insulin range (from approximately 35 to 1,450 pmol/L) (97).
In AT, obesity induces pronounced ER stress that fundamentally disrupts metabolic homeostasis through multiple tissue-specific mechanisms (80, 98). Proteomic analyses of obese AT reveal significant upregulation of UPR-related proteins including calreticulin and protein disulfide-isomerase A3, with Glucose-Regulated Protein 78 (GRP78)/BiP expression increased 3–4 fold compared to lean controls (99, 100). This AT-specific ER stress directly impairs insulin signaling by promoting c-Jun N-terminal kinase (JNK)-mediated serine phosphorylation of insulin receptor substrate (IRS-1), reducing glucose uptake capacity by up to 60% (101). Moreover, UPR activation in adipocytes dramatically alters adipokine production patterns, with adiponectin secretion decreased while inflammatory cytokines including IL-6 and resistin are increased (101, 102).
The NOD-like receptor family, pyrin domain containing 3 (NLRP3) inflammasome represents a critical mediator of AT dysfunction during ER stress. Activated by lipotoxicity and ER stress signals, NLRP3 triggers IL-1β and IL-18 release specifically in adipocytes, promoting tissue inflammation and macrophage recruitment (103, 104). TNFα-induced NLRP3 activation in adipocytes causes mitochondrial dysfunction and exacerbates insulin resistance, while also impairing white adipocyte browning and thermogenic capacity (105, 106). The Toll-like receptor 4 (TLR4)/PI3K/Akt pathway converges with ER stress responses to amplify AT inflammation (107). Importantly, caloric restriction and exercise reduce AT NLRP3 expression and inflammation, suggesting therapeutic potential (104).
AT-specific UPR activation disrupts lipid metabolism through PERK-ATF4 pathway signaling. ATF4 regulates thermogenesis and lipolysis by influencing fatty acid utilization gene expression (108). The three UPR sensors - IRE1, PERK, and ATF6 - alter lipid enzyme function in adipocytes, affecting fatty acid synthesis and oxidation (109, 110). High-fat diets intensify ER stress in AT, compromising endocrine function and accelerating metabolic disease progression (78). The ER stress-induced transcription factor C/EBP homologous protein (CHOP) further drives AT dysfunction by promoting M1 macrophage polarization while suppressing anti-inflammatory Th2 cytokines (111).
Prolonged UPR activation fundamentally alters cellular function despite its initial adaptive purpose. Extended perturbation of ER function progressively compromises insulin biosynthetic capacity and ultimately triggers CHOP-dependent apoptotic programs through calcium-dependent mechanisms and mitochondrial dysfunction (112). These molecular alterations characterize both obesity and diabetes, where chronic ER dysfunction perpetuates inflammatory signaling networks and impairs IR signal transduction through multiple intersecting pathways (79, 80).
IRE1α activates JNK through TNF receptor-associated factor (TRAF2) binding, triggering inflammatory signaling cascades. Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) activation occurs through both PERK and IRE1α pathways, increasing inflammatory cytokine production. These pathways create a feed-forward cycle where altered lipid metabolism amplifies cellular stress and inflammation (79, 80). The ER integrates nutrient sensing across cell types through regulation of calcium fluxes and lipid metabolic programs, coordinating cellular responses to metabolic fluctuations (113). Post-transcriptional control through microRNA networks, including miR-211, miR-30c, and miR-34a, modulates the cellular response to protein folding stress, fine-tuning both adaptive and maladaptive responses (114). Experimental manipulation of ER protein folding capacity influences beige adipocyte differentiation programs and metabolic adaptation to high-fat feeding through coordinated effects on mitochondrial homeostasis and inflammatory tone in multiple tissues (115).
Therapeutic targeting of AT ER stress shows promise through chemical chaperone interventions. Studies utilizing 4-phenylbutyrate demonstrate reductions in adipose GRP78 expression concurrent with decreased plasma metabolites. Biochemical evaluation reveals 4-PBA mediated suppression of PERK and IRE1α phosphorylation cascades while maintaining IRS-1 function (116). Investigation of small molecule UPR modulators has yielded synergistic effects, with the selective IRE1α inhibitor HT-6184 enhancing semaglutide-mediated improvements in body composition and glucose homeostasis. Combined administration produces superior outcomes with enhanced preservation of lean mass and decreased ceramide accumulation in adipose depots (117). These findings establish AT ER stress as a central mechanism linking obesity to systemic metabolic disorders including T2DM and cardiovascular disease (118, 119).
4.3 The role of mitochondria in AT dysfunction and metabolic disorders
AT biopsies reveal substantial mitochondrial alterations at both structural and functional levels. Quantitative analysis of oxygen consumption demonstrates a 45% reduction in oxidative phosphorylation capacity, accompanied by dysregulated acyl-CoA oxidation and accumulation of oxidative stress markers, specifically 4-hydroxynonenal adducts within subcutaneous adipose deposits (87). Mass spectrometry-based metabolomic analyses have identified distinct fatty acid profiles characteristic of pathological tissue states.
ChIP-seq analysis has mapped PGC-1α binding sites across nuclear respiratory gene promoters, establishing direct transcriptional control mechanisms. Clinical specimens exhibit marked metabolic protein dysregulation - PGC-1α expression decreases 65% compared to controls, concurrent with diminished UCP1 and Carnitine palmitoyltransferase 1 (CPT1) protein levels as determined by immunoblot analysis (88). Tissue-specific deletion of PGC-1α in adipose reveals its metabolic regulatory functions. Adipose-selective PGC-1α ablation induces rapid insulin resistance alongside decreased thermogenic gene expression and mitochondrial dysfunction (120). This nuclear coactivator responds to metabolic signals to regulate oxidative phosphorylation genes through interactions with key transcription factors (121). Studies in WAT demonstrate PGC-1α’s requirement for both baseline and rosiglitazone-stimulated mitochondrial activity, though insulin sensitization by thiazolidinediones (TZDs) persists in its absence (122). Global PGC-1α knockout mice display unexpected metabolic phenotypes - reduced adiposity and increased physical activity despite compromised mitochondrial capacity (123). Moderate elevation of PGC-1α within physiological ranges augments fatty acid oxidative capacity and glucose transport in response to insulin (124). In cardiac tissue, PGC-1α collaborates with PGC-1β to preserve mitochondrial function as insulin resistance develops (125). Metabolic phenotyping studies demonstrate depot-specific requirements for PGC-1α, particularly in BAT where its absence severely impairs thermogenic capacity. PGC-1 coactivators regulate metabolism through coordinated binding to nuclear receptors, enabling adaptive responses to nutritional status and environmental cues. These molecular interactions establish distinct transcriptional programs across tissues to maintain metabolic homeostasis.
Dynamic imaging reveals fragmented mitochondrial networks and accelerated ROS generation in metabolically compromised adipocytes. Longitudinal clinical studies demonstrate progressive decline in PGC-1α activity correlating with heightened inflammatory markers and oxidative damage in AT specimens (120, 126). These mechanistic findings from both human pathology and experimental models highlight the therapeutic relevance of mitochondrial quality control mechanisms.
Biochemical analyses establish AMPK as a central regulator of mitochondrial biogenesis through direct modulation of lipid oxidation. Phosphoproteomic mapping in cardiac tissue reveals extensive AMPK-dependent signaling networks, where pathway perturbation produces severe metabolic consequences (16). Current therapeutic development focuses on mitochondrial function, with particular success seen in thiazolidinedione compounds that upregulate PGC-1α and mitochondria-targeted antioxidants (89, 90). Excessive mitochondrial ROS, especially superoxides (O2•-), hydrogen peroxides (H2O2) and hydroxides (•OH), characterize obesity adipocyte dysfunction. These cells show a significant reduction in antioxidant defense capacity, and reactive oxygen species (ROS) concentrations in cells usually go from physiological (100nM H2O2) to pathological (>500nM H2O2) (97, 98). NADPH oxidase 4 (NOX4) transfer electrons to molecular oxygen and acts as the main ROS generator. Redox homeostasis is controlled by a variety of cell defense mechanisms, including three different superoxide dismutase (SOD1 in cells, SOD2 in mitochondria, SOD3), glutathione peroxidase (GPX), and signal pathways Nuclear factor erythroid 2-related factor 2 (NRF2)- Kelch-like ECH-associated protein 1(KEAP1) (71).
Oxidative damage disrupts the potential of the mitochondrial membrane and inhibits the function of the I-IV electron transport chain. Due to the lack of control of fusion proteins (Mitofusin-1/2(MFN1/2), Optic atrophy 1(OPA1)) and fission proteins (Dynamin-related protein 1 (DRP1), mitochondrial fission 1 protein (FIS1), ROS accumulation changes the dynamics of mitochondrial networks (72–75). These changes reduce respiratory capacity by 50% and inhibit adipogenic differentiation (127, 128). The resulting metabolic disturbances trigger the production of adipocin, especially TNF-α, IL-6, and Monocyte Chemoattractant Protein-1 (MCP-1, also known as CCL2), to induce inflammatory mediators.
Cellular ROS concentrations require precise regulation through a complex interplay with calcium signaling and ER function. Both excessive and insufficient oxidative states compromise adipocyte function (76). Mitochondrial ROS serve as critical signaling molecules, regulating mitochondrial DNA transcription and modulating UCP1-dependent thermogenesis. These pathways directly influence cellular bioenergetics, with multiple metabolic processes depending on proper ROS signaling. Energy expenditure patterns show ROS-dependent regulation through AMPK pathway activation (129).
AT in obesity displays persistent inflammatory activation, with oxidative stress mechanisms operating continuously. These processes establish feed-forward cycles of metabolic dysfunction (130, 131). Multiple cellular ROS sources contribute to oxidative burden, with mitochondrial electron transport generating approximately 70% of total cellular ROS. NADPH oxidase complexes produce the remaining oxidative species, which activate NF-κB-dependent transcriptional programs (132). This activation results in a 5–10 fold increase in pro-inflammatory cytokine production.
Metabolic perturbations impair IR signaling cascades through JNK and IκB kinase-β (IKKβ) activation, leading to serine phosphorylation of IRS-1. They dysregulate lipid metabolism and storage, contributing to cardiovascular pathologies. Adipocyte hypertrophy triggers immune cell recruitment, with M1 macrophage infiltration maintaining the inflammatory state (133). Multiple pathological triggers initiate these cascades, including dietary lipid excess and altered microbiota composition, particularly reduced Bacteroidetes-to-Firmicutes ratio.
Oxidative stress synergizes with inflammatory processes in AT to severely disrupt glucose homeostasis. ROS directly modify insulin signaling proteins through oxidation of critical cysteine residues. Oxidative damage reduces glucose transporter type 4 (GLUT4) vesicle translocation by approximately 60%, as measured by membrane fraction analysis (133). Cellular glucose uptake capacity diminishes markedly, typically showing a 70-80% reduction compared to healthy adipocytes.
Current interventions target multiple pathways simultaneously for maximal effect. Currently, certain NOX4 inhibitors (GKT137831), as well as mitochondrial antioxidants (MitoQ, SS-31), of which some are in phase II clinical trials (134), fall into this category. Lifestyle changes, especially adherence to regular physical exercise and Mediterranean diet-as reflected by reduced oxidative stress of adipocytes (as measured by plasma levels of F2-isoprostanes and carbonyls of proteins)-showed positive effects (135, 136). Some of the important biomarkers to monitor are systemic oxidative stress (8-isoprostane, malondialdehyde), antioxidant capacity (Reduced glutathione/Oxidized glutathione (GSH/GSSG) ratios, SOD activity), inflammation (high-sensitivity C-reactive protein (CRP), IL-6), and metabolic function (adiponectin/leptin ratios) (137, 138).
Novel therapeutic approaches involve mitochondrial-targeted antioxidants, combination anti-inflammatory and antioxidant strategies, ER stress modulators, and microbiote-targeted therapies. These multi-targeted interventions try to restore metabolic homeostasis through multiple mechanisms targeting oxidative stress, inflammation, and cell metabolism simultaneously.
4.4 Inflammatory signaling
4.4.1 Immune cell infiltration in AT: mechanisms, consequences, and clinical implications
AT in obesity shows chronic low-grade inflammation. Multiple factors drive this inflammatory state. Enlarged adipocytes interact with infiltrating immune cells. Cellular stressors like mechanical strain from hypertrophy and local hypoxia initiate AT inflammation. Under stress, adipocytes release pro-inflammatory signals. MCP-1 acts as a key mediator that recruits immune cells to the tissue (139, 140). Macrophages are the first to respond, triggering self-perpetuating inflammatory cycles that increase cytokine production and alter tissue structure (133). These inflammatory changes progressively disrupt normal metabolism, leading to insulin resistance and numerous obesity complications (141). Adipocytes and immune cells establish complex interactions through adipokines and cytokines, affecting lipolysis regulation and maintaining a state of chronic inflammation in the tissue (142).
AT shows defined patterns as disease progresses (143). M2 macrophages shift to M1 type, gathering near dying fat cells. These M1 cells make crown structures and release TNF-α, IL-1β, and IL-6 (144, 145). Over time, these changes impair metabolism and cause insulin resistance (107). Multiple interconnected factors drive this dysfunction: continuing adipocyte hypertrophy, worsening tissue hypoxia, and elevated circulating FFAs create self-reinforcing inflammatory cycles. These changes result in increasingly severe metabolic dysregulation throughout the tissue (146).
The immune cell population in dysfunctional AT extends far beyond macrophages. The tissue accumulates CD8+ T cells, Th1 cells, natural killer cells, B cells, neutrophils, and mast cells in significant numbers (147). This diverse immune network amplifies inflammatory signaling through multiple pathways that disrupt normal AT function, impair insulin signaling cascades, and dramatically alter adipokine production patterns (133, 148). Ongoing inflammation changes tissue structure through fibrosis and altered function (149). Levels of MCP-1, C-C motif chemokine ligand 5/Regulated upon Activation, Normal T Cell Expressed and Secreted (CCL5/RANTES), IL-6, IFN-γ (interferon-γ), and TNF-α increase in the tissue (145). The effects reach beyond AT and disrupt multiple organ systems (133). Both B and T lymphocytes play essential regulatory roles in tissue inflammation and insulin sensitivity (150). The coordinated activities of macrophages, T cells, and NK cells drive substantial tissue remodeling and promote widespread metabolic dysfunction (151, 152).
4.4.2 Pro-inflammatory mediators
Pro-inflammatory Mediators Obesity-induced inflammation in AT involves macrophage infiltration and stress pathway activation, with NF-κB driving production of cytokines (TNF-α, IL-6, IL-1β) that disrupt insulin signaling (67, 68). Notably, C-C chemokine receptor type 2 (CCR2)-expressing M1 macrophages preferentially accumulate around necrotic adipocytes rather than arising through phenotypic conversion of resident M2 populations (68). Advanced single-cell RNA sequencing has uncovered previously unidentified AT macrophage subpopulations that transcend the traditional M1/M2 dichotomy (153). Mass spectrometry-based proteomic profiling has identified specific adipokine signatures that regulate macrophage chemotaxis and inflammatory activation states (140).
The progression of obesity involves multiple factors driving macrophage phenotypic alterations. Elevated FFAs activate inflammatory signaling cascades while complex cytokine networks orchestrate cellular responses (154, 155). The accumulation of M1 macrophages triggers activation of JNK and IKK pathways through pattern-recognition receptor engagement, promoting insulin resistance through stress kinase signaling mechanisms (69, 70). This process is exacerbated by concurrent reductions in anti-inflammatory mediators, particularly adiponectin, further disrupting AT metabolic homeostasis (156). Early immunological alterations include depletion of regulatory immune cell populations alongside enhanced activation of natural killer cells and CD8+ T lymphocytes (157). These inflammatory mechanisms progressively compromise multiple organ systems, accelerating the development of metabolic disease (5).
4.4.3 Chronic inflammation in AT: pathways to metabolic dysfunction
Self-perpetuating inflammatory circuits establish chronic AT dysfunction through interconnected molecular pathways. Initial inflammatory responses triggered by adipose expansion become self-sustaining through multiple feedback loops. The effective therapeutic targeting of these inflammation networks requires a detailed understanding of the pathological contributions to metabolic deterioration. Current research continues to uncover new regulatory mechanisms with potential therapeutic applications for obesity-related metabolic disorders. The development of targeted anti-inflammatory interventions may provide promising strategies to restore AT homeostasis.
4.5 Metabolic dysregulation
4.5.1 Insulin signaling pathways in obesity and metabolic disease
Metabolic tissues develop lipid intermediates alongside altered adipokine expression profiles and compromised mitochondrial function, progressively attenuating insulin responsiveness (64, 65). These changes compromise glucose homeostasis and create more widespread metabolic derangements associated with insulin-resistant conditions (158). Adipocytes use mechanistic target of rapamycin complex 1 (mTORC1) and mTORC2 for metabolic control (91). These complexes regulate the activities of IRS-1 and the growth factor receptor-bound protein 10 (Grb10) and fine-tune their insulin responses, from complex feedback circuits that ensure metabolic homeostasis in different nutrition environments (92). In AT, mTOR mediates multiple cellular processes via various downstream effectors controlling cellular growth, differentiation, and metabolic homeostasis. Conditional deletion of mTOR specifically from AT leads to decreased fat mass with increased systemic insulin resistance and hepatic lipid deposition (159).
In adipocytes, mTORC2 regulates both insulin-stimulated glucose transport and lipolysis rates (160). This regulation is achieved through Akt substrate of 160kDa (AS160) that produces cellular responses to both Akt and mTORC2 signals to regulate glucose transporter 4 (GLUT4) trafficking during insulin stimulation, and to maintain glucose homeostasis (161). The mTOR dependent mechanisms described give insights into selective insulin resistance in adipocytes and propose novel therapeutic strategies for interventions in the metabolic disease.
Inflammatory signaling via TNF-α disrupts insulin response pathways at multiple levels. TNF-α modifies IRS-1 through enhanced serine phosphorylation, inhibiting the tyrosine phosphorylation events essential for normal insulin signal transduction (162, 163). IκB kinase (IKK) phosphorylates specific serine residues on IRS-1, maintaining the insulin-resistant state throughout the inflammatory response (164). Suppressor of cytokine signaling (SOCS) proteins, most notably the SOCS-3, inhibit insulin signaling by preventing IRS protein activation through direct molecular interactions and altered protein stability in metabolically active tissues (165). TNF-α and SOCS3 establish a positive feedback loop that amplifies their expression and intensifies insulin resistance through sustained inflammatory pathway activation in AT (166) (Table 3). These molecular connections between inflammation and insulin resistance identify specific therapeutic targets for addressing metabolic disorders and their complications in obesity (93).
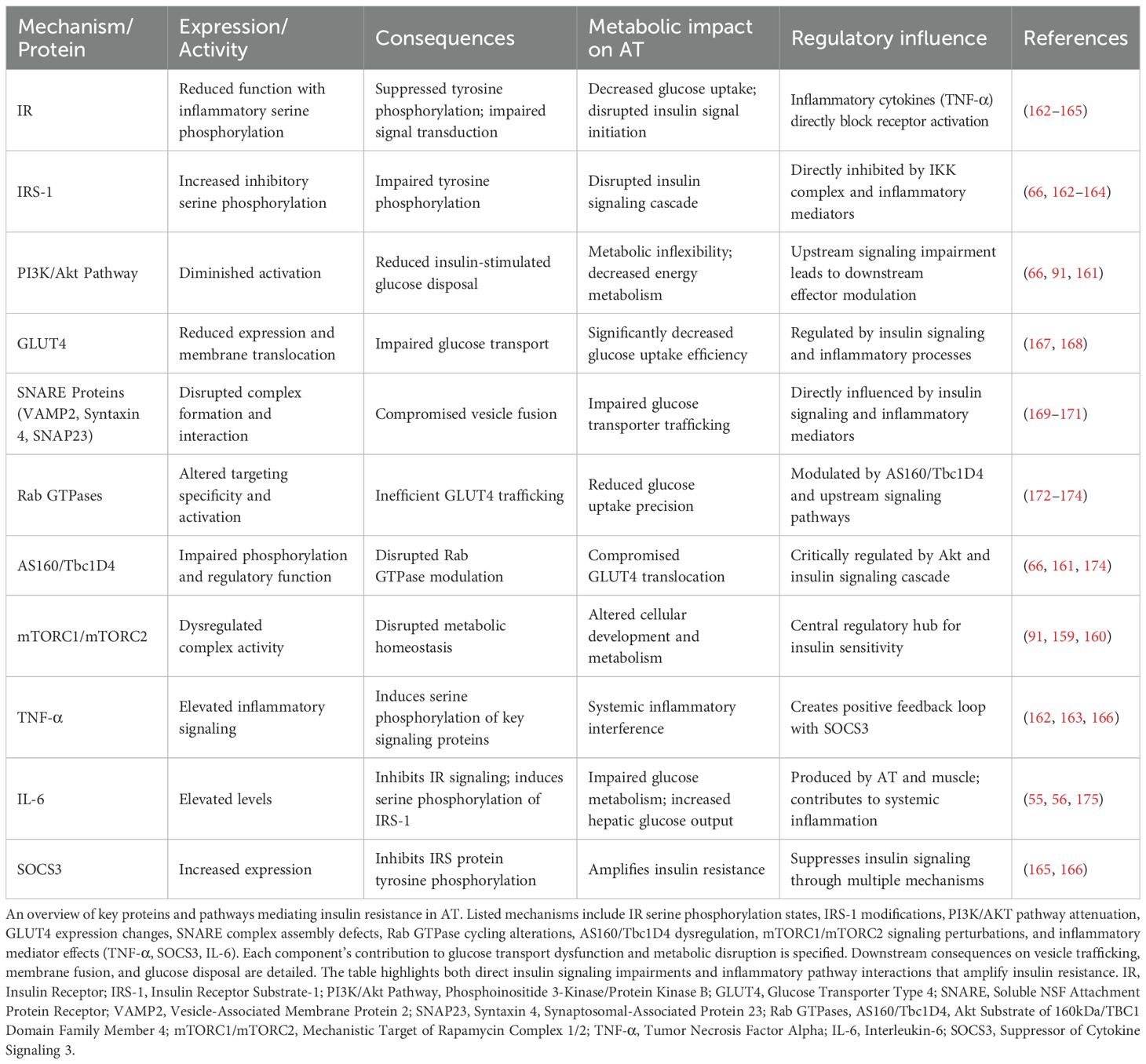
Table 3. Molecular mechanisms of insulin signaling disruption in AT.
4.5.2 Impaired glucose metabolism in AT
The adipose inflammatory milieu generates self-perpetuating cycles of metabolic deterioration that progressively advance insulin resistance and T2DM pathogenesis. Obesity-induced inflammation in AT leads to insulin resistance through various mechanisms. Proinflammatory macrophages secrete cytokines and microvesicles that impair insulin signaling and glucose uptake in adipocytes (176–178). This results in decreased GLUT4 translocation and reduced insulin-stimulated glucose uptake (167). Neuregulin 4 downregulation in adipocytes induces insulin resistance through inflammation and autophagic degradation of GLUT4 vesicles (179). The inflammatory milieu in obese AT disrupts normal function and leads to systemic insulin resistance (10).
Insulin-stimulated glucose uptake in AT involves the translocation of GLUT4 transporters from intracellular vesicles to the plasma membrane, mediated by soluble NSF attachment protein receptor (SNARE) proteins (168). The t-SNAREs syntaxin4 and synaptosomal-associated protein 23 (SNAP23) are essential for tethering GLUT4 vesicles to the plasma membrane, while the v-SNARE vesicle-associated membrane protein 2 (VAMP2) is crucial for fusion (169). These SNARE proteins are localized in lipid rafts, which may serve as platforms for GLUT4 vesicle fusion (170). Insulin stimulation increases syntaxin4-containing SNARE complex formation, possibly through phosphorylation of the regulatory protein Munc18c (171). Other regulatory factors, such as Rab GTPases, contribute to targeting specificity in the GLUT4 secretory pathway (172).
The insulin signaling network in metabolically compromised adipocytes manifests multiple molecular defects: impaired IR activation kinetics, substantial reduction in IRS-1 tyrosine phosphorylation, marked suppression of PI3K/Akt pathway signal propagation, and pathological elevation of inhibitory serine phosphorylation events on IRS-1 (66). These coordinated molecular perturbations manifest physiologically as systemic glucose disposal deficits, compensatory hyperinsulinemia, and progressive advancement toward metabolic syndrome.
Comprehensive understanding of these molecular regulatory networks offers critical insights for therapeutic development in insulin resistance and T2DM (Table 3).
4.6 Metabolic disruptions in lipid homeostasis
Peroxisome proliferator-activated receptor gamma (PPARγ) determines adipocyte identity while coordinating with sterol regulatory element-binding protein (SREBP1c) to regulate lipid homeostasis (180). ATGL and HSL mediate triglyceride breakdown through sequential enzymatic actions (181). Entry of fatty acids into cells requires cluster of differentiation 36 protein, followed by fatty acid binding protein 4-mediated transfer between cellular compartments. Metabolic disease states trigger enhanced activity of both lipases in AT, thereby increasing systemic fatty acid levels (182). Complex mechanisms control these changes through transcriptional regulation and post-translational modifications that alter protein function, while protein-protein interactions coordinate responses (183).
Insulin suppresses lipolysis through multiple mechanisms, with acute signaling cascades responding rapidly and transcriptional regulation occurring more slowly. ATGL expression shows particular insulin sensitivity (184). Perilipin proteins coat lipid droplets and regulate lipid storage tightly, with fatty acid release depending on perilipin function (185). Disrupted lipolytic balance promotes disease, as obesity develops progressively, leading to insulin resistance and frequently resulting in T2DM (186). Concurrent with enhanced lipolysis, adipocyte dysfunction involves impaired lipogenic capacity. PPARγ functions as a master regulator of adipocyte function and lipid homeostasis (187), with PPARγ2 specifically controlling AT lipid storage and metabolic flexibility (188). sterol regulatory element-binding protein 1c (SREBP1c) works in concert with PPARγ to regulate lipogenic gene expression, while fatty acid binding protein 4 (FABP4) and Cluster of differentiation 36 (CD36) facilitate fatty acid transport and metabolism. Compromised PPARγ function results in decreased expression of lipolytic genes and abnormal catecholamine-induced lipolysis (189). As AT serves as a critical buffer for daily lipid flux, its dysfunction can lead to ectopic fat accumulation and insulin resistance (190). PPARγ activation enhances AT function by modifying fat distribution, adipocyte phenotype, and lipid metabolism-related gene expression (191). Furthermore, liver X receptors (LXRs) collaborate with PPARγ in regulating hepatic and adipose lipogenesis during obesity and insulin resistance (Table 4).
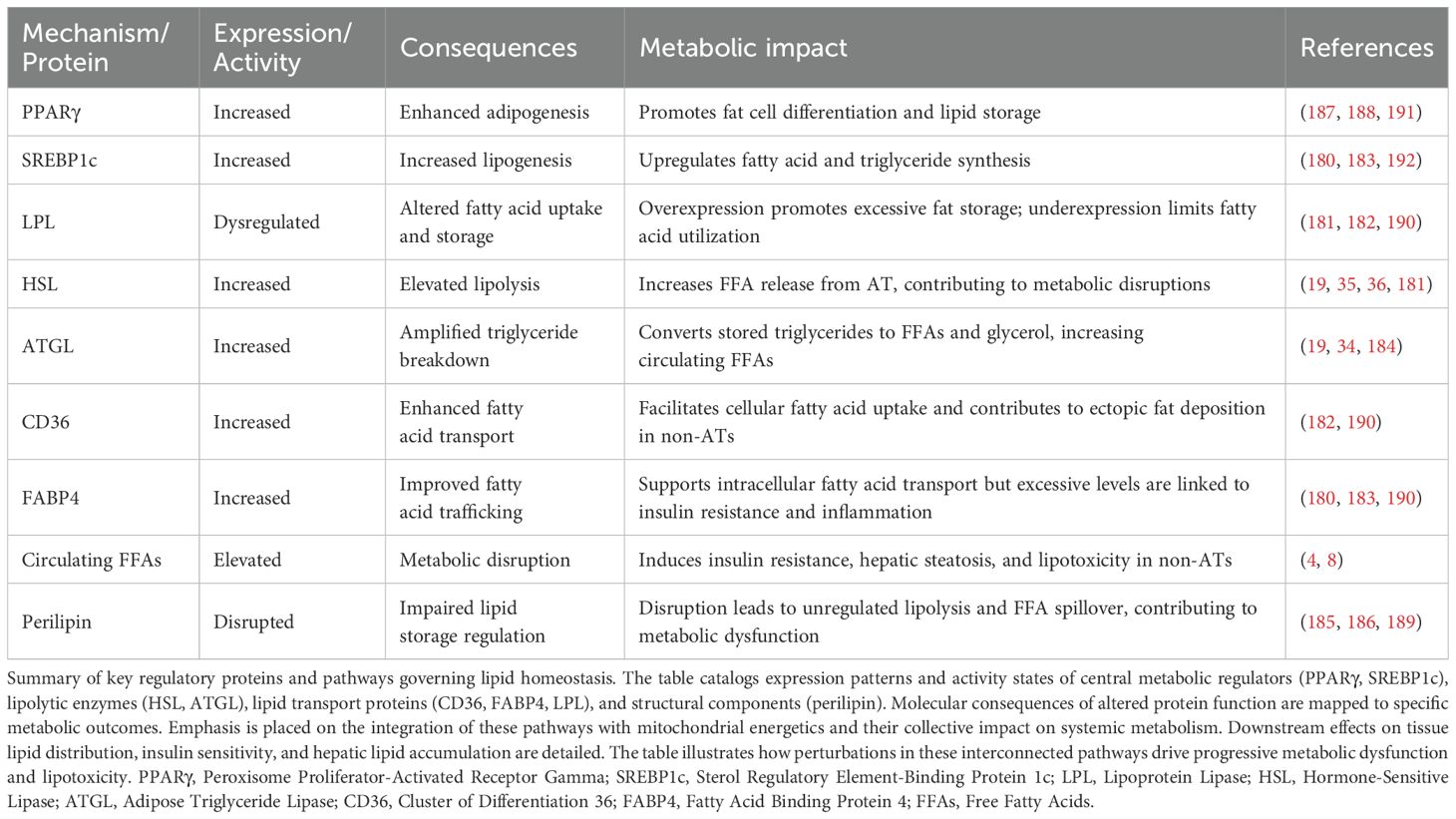
Table 4. Dysregulation of lipid metabolic networks in adipose dysfunction.
4.7 Metabolic regulation through protein farnesylation in AT
The post-translational addition of farnesyl groups to proteins plays a key role in AT metabolic regulation. Insulin triggers farnesyltransferase activity in adipocytes, leading to p21Ras modification and subsequent Mitogen-activated protein kinase (MAPK) pathway activation - a crucial sequence for metabolic signaling (193). Members of the Ras GTPase family exhibit distinct requirements for this lipid modification: H-Ras localizes exclusively to lipid rafts, while K-Ras4B shows plasma membrane preference through its polybasic domain. Data from adipocyte culture systems demonstrate insulin-stimulated Ras farnesylation drives both adipogenic differentiation programs and glucose transport mechanisms through extracellular signal-regulated kinase 1/2 (ERK1/2) activation (194, 195). Recent work has uncovered parallel MAPK activation pathways operating through farnesylation-independent mechanisms in mature adipocytes, highlighting the complexity of these signaling networks (196).
The Rho GTPase family undergoes similar prenyl modifications, though through more complex regulatory networks affecting glucose metabolism (197, 198). RhoA, Rac1, and Cdc42 require carefully balanced prenylation - both farnesyl and geranylgeranyl additions prove necessary for proper membrane targeting and effector interactions. Insulin signaling promotes Rab protein geranylgeranylation, particularly Rab4 and Rab11, facilitating GLUT4 vesicular trafficking in AT (173). Disrupting these modifications through statins or specific prenylation inhibitors blocks preadipocyte differentiation through impaired cytoskeletal remodeling (199) and compromises glucose-stimulated insulin release from pancreatic β-cells (200).
SREBP transcription factors, particularly SREBP-2, coordinate mevalonate pathway flux to generate prenylation substrates farnesyl pyrophosphate (FPP) and geranylgeranyl pyrophosphate (GGPP) through regulated intramembrane proteolysis (198, 201, 202). These regulatory proteins undergo cholesterol-dependent processing in the Golgi, releasing active nuclear forms that control both isoprenoid and fatty acid synthesis. SREBP-2 preferentially activates genes involved in cholesterol biosynthesis while modulating fatty acid synthesis through FPP-dependent mechanisms. Complex feedback loops connect SREBP activity to cellular sterol levels, prenylation substrate availability, and lipid metabolism through LXR-dependent pathways (201, 202).
Experimental manipulation of protein farnesylation reveals its broad metabolic impact. Loss of normal farnesylation disrupts IR trafficking dynamics and surface expression patterns through altered endosomal sorting mechanisms, contributing to cellular insulin resistance (203). The farnesylation machinery also influences GLUT4 movement through effects on cytoskeletal organization and membrane microdomain composition (174). Within pancreatic β-cells, farnesylation-dependent Raf/ERK signaling couples glucose sensing to insulin secretion through K-ATP channel regulation (204). These findings establish protein farnesylation as a central coordinator of AT function, with dysregulation leading to metabolic disease manifestations including impaired glucose uptake, altered lipid storage, and disrupted adipokine secretion (Table 5).
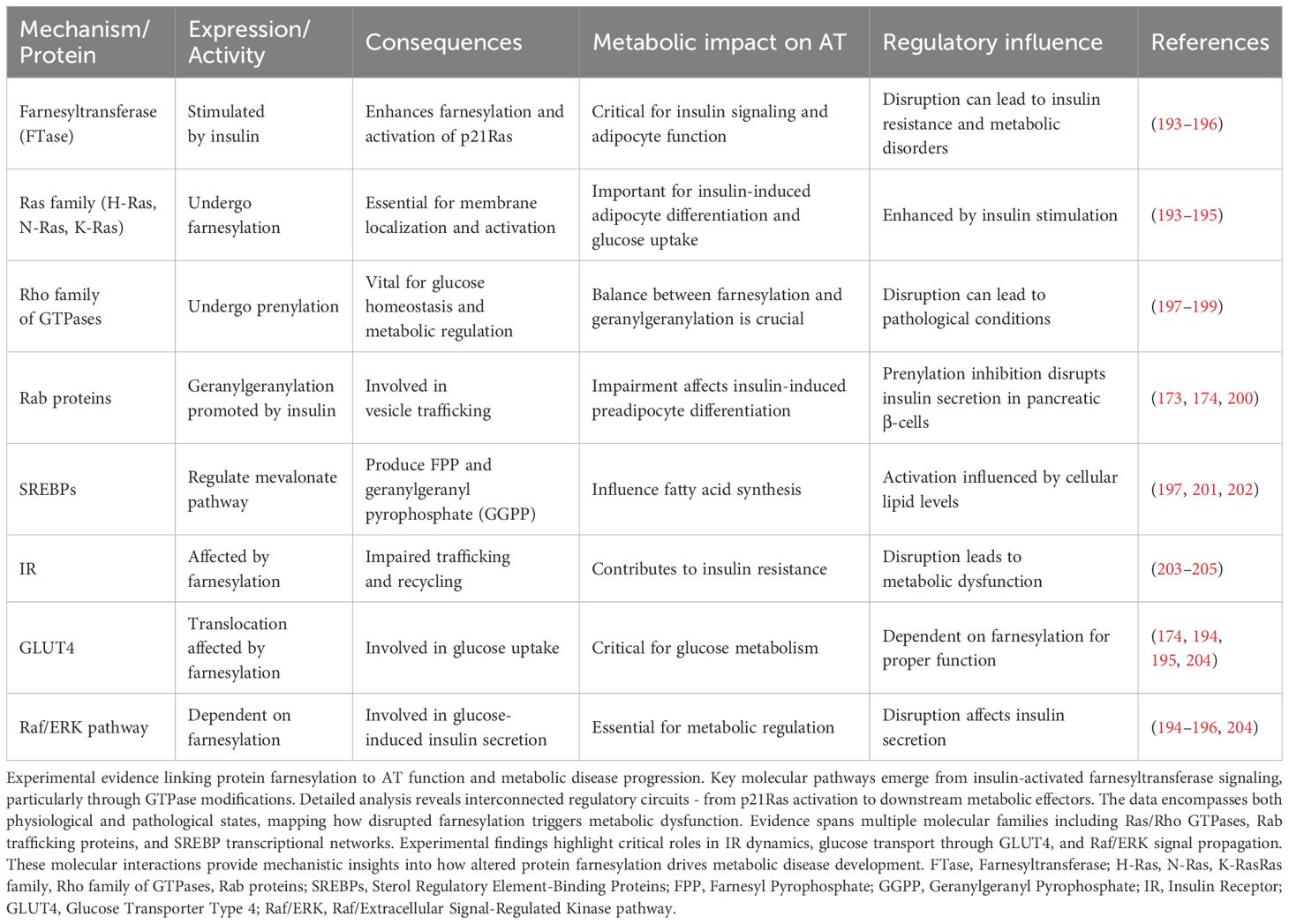
Table 5. Protein farnesylation networks in adipose metabolism and disease.
4.8 Lipid metabolism and trafficking
Disruption of normal lipid storage patterns frequently leads to accumulation within non-adipose tissues (non-ATs). This abnormal fat deposition triggers pathological cascades in cardiovascular and metabolic systems. The resulting tissue damage involves multiple molecular mechanisms (205, 206). Intracellular lipid homeostasis depends on precise transcriptional regulation. Recent studies have established the carbohydrate response element-binding protein (ChREBP) as essential in this process (192). The enzyme networks controlled by ChREBP regulate cellular lipid synthesis. Key components include fatty acid synthase (FAS), which generates palmitate molecules. Acetyl-CoA carboxylase (ACC) supplies critical malonyl-CoA building blocks. Stearoyl-CoA desaturase-1 (SCD1) creates specific double bonds in fatty acid chains (207). Each enzyme performs distinct catalytic roles in lipid metabolism. Additionally, emerging evidence suggests that circadian regulation of these enzymes significantly impacts their activity patterns (208).
In vulnerable tissues - particularly liver parenchyma, striated muscle, and myocardium - excess lipid burden initiates both cell death pathways and inflammatory responses (209). The buildup of specific lipid species, especially Diacylglycerol (DAGs) and ceramides, causes oxidative damage through ROS generation. These changes trigger progressive mitochondrial dysfunction (210). Impaired respiratory chain activity further increases ROS production. Local tissue inflammation worsens. A self-reinforcing cycle of metabolic deterioration emerges. Recent work has identified the endocannabinoid system (ECS) as a key mediator in lipotoxicity, with Cannabinoid receptor type 1 (CB1) activation promoting lipogenesis and inflammation (211). The newly characterized role of mitochondrial-associated membranes (MAMs) in lipid trafficking adds another layer of complexity to this pathological cascade. MAMs regulate lipid metabolism, calcium homeostasis, and ROS generation (212). Disruption of MAM integrity can lead to increased ROS production, mitochondrial damage, and activation of inflammatory pathways (213).
The effects manifest differently across organ systems. Hepatocytes develop steatosis and advance toward non-alcoholic fatty liver disease (NAFLD). Skeletal muscle fibers show severe insulin resistance. Cardiac function deteriorates. Pancreatic β-cells display significant secretory deficits (214, 215). Despite tissue-specific manifestations, common pathogenic mechanisms exist: disrupted AT function floods circulation with FFAs, overwhelming normal lipid processing pathways (215, 216). The resulting cellular stress responses - from ER protein folding defects to mitochondrial dysfunction - amplify metabolic disruption. Recent research highlights the crucial role of EVs in inter-organ metabolic communication and disease progression. EVs, including exosomes, mediate intercellular and inter-organ crosstalk by carrying bioactive molecules such as proteins, lipids, and microRNA (miRNAs) (217). These mechanistic insights into ectopic fat accumulation highlight several therapeutic targets. Pharmacological regulation of lipogenic enzymes, enhancement of FA oxidation, and suppression of inflammatory mediators could interrupt disease progression. The increasing prevalence of these disorders necessitates rapid therapeutic development. Success will likely require concurrent targeting of both cellular stress responses and systemic metabolic dysfunction.
4.9 Environmental obesogens and their impact on adipogenic regulation
Environmental obesogens represent endocrine-disrupting chemicals that promote obesity through alterations in adipogenesis and metabolic homeostasis (218, 219). Tributyltin (TBT), a well-characterized obesogen, activates retinoid X receptor (RXR) and PPARγ-key transcriptional regulators of adipocyte differentiation (218, 220). Critical developmental exposure to TBT enhances adipocyte differentiation, modifies gene expression profiles, and induces persistent obesogenic phenotypes (218, 221). The spectrum of identified obesogens encompasses bisphenol A, phthalates, and perfluorinated compounds (219).
The obesogen hypothesis proposes that prenatal exposure reprograms stem cells toward preferential adipocyte differentiation, potentially establishing transgenerational inheritance patterns (222). Recent investigations demonstrate that developmental obesogen exposure increases AT formation and fat storage capacity in offspring, with effects persisting across generations (223, 224). These compounds, prevalent in pesticides, food packaging, and cosmetics, reprogram adipose stem cells through epigenetic mechanisms including DNA methylation alterations and chromatin remodeling (225–228). Transgenerational consequences include increased white adipose depot weights, adipocyte hyperplasia and hypertrophy, and hepatic lipid accumulation (223, 229).
Population biomonitoring reveals widespread exposure to bisphenol A (BPA) and its structural analogues-bisphenol S (BPS) and bisphenol F (BPF)-detected in consumer products and biological fluids (230–233). These substitutes exhibit comparable or enhanced endocrine-disrupting potential relative to BPA (232). In vitrostudies demonstrate bisphenol-mediated promotion of adipogenesis and lipid accumulation in human adipocytes through interference with adipogenic gene expression and metabolic pathways (234–236).
Epidemiological investigations establish significant associations between BPA exposure and metabolic perturbations. Meta-analyses report increased obesity odds ratios of 1.40-1.76 correlating with elevated BPA concentrations (237–240). BPA exposure correlates with abdominal obesity risk (odds ratios: 1.31-1.62) and increased BMI and waist circumference (239–242). Mechanistic evidence suggests BPA functions as an obesogen through hormonal receptor modulation and metabolic syndrome promotion (243). However, cross-sectional study designs and single-point measurements limit causal inference capabilities (244).
Emerging evidence indicates BPA alternatives, including Bisphenol AF (BPAF), demonstrate similar endocrine-disrupting profiles associated with obesity, glucose dysregulation, and cardiovascular abnormalities (245, 246). Mechanistic investigations reveal these compounds activate PPARγ pathways, stimulate adipocyte hypertrophy, and dysregulate adipogenic networks (234, 236). Exposure induces lipid accumulation, pro-inflammatory cytokine expression, and impaired insulin signaling in human adipocytes (235, 247, 248). BPAF specifically compromises mitochondrial function and promotes adipose inflammation (249).
Current research priorities encompass: identifying adipose-specific molecular targets through single-cell genomics; characterizing critical developmental windows via longitudinal cohorts; and elucidating transgenerational effects using multi-generational models. Advanced analytical platforms reveal novel obesogenic compounds, necessitating regulatory reassessment. Intervention strategies under investigation include targeted nutritional approaches and exposure reduction protocols. Understanding gene-environment interactions, particularly metabolic gene polymorphisms, may facilitate personalized prevention strategies for preserving AT function.
4.10 Lifestyle interventions and environmental factors in AT function
AT plays a crucial role in regulating whole-body metabolism and energy homeostasis (250). Exercise and physical activity significantly impact AT function through multiple mechanisms, including enhanced mitochondrial biogenesis, increased oxidative capacity, and reduced inflammation (251, 252). Regular exercise promotes AT remodeling, improves metabolic flexibility, and stimulates the browning of WAT (251, 253). These effects contribute to improved insulin sensitivity and reduced risk of cardiometabolic disorders (254). AT dysfunction, often associated with obesity and aging, can lead to various metabolic disorders (3). However, exercise-induced changes in adipokine secretion and lipid composition can positively influence other organs and tissues, promoting overall metabolic health (255, 256).
Exercise-induced browning of WAT has emerged as a promising mechanism for improving metabolic health. Physical activity stimulates the release of myokines like irisin and FGF21 from skeletal muscle, which promote WAT browning (257–259). This process involves increased expression of UCP1 and enhanced mitochondrial function, leading to improved thermogenesis and energy expenditure. High-intensity exercise appears more effective in inducing WAT browning compared to low-intensity exercise (260). The browning effect is mediated through various pathways, including β-adrenergic signaling, ROS, and exerkines (261). Irisin, in particular, plays a crucial role by binding to integrin αV/β5 receptors and promoting WAT browning (262). Furthermore, irisin supplementation or exercise-induced irisin activation may offer therapeutic potential for metabolic disorders (263).
Dietary interventions beyond caloric restriction can significantly impact AT function and inflammation. The Mediterranean diet, rich in monounsaturated fats and polyphenols, reduces adipose inflammation by suppressing NF-κB and MAPK pathways while activating AMPK (264, 265). Intermittent fasting and ketogenic diets improve mitochondrial function, reduce inflammation, and enhance autophagy in AT (266, 267). These diets modulate gut microbiota composition, decreasing lipopolysaccharide-producing bacteria and inflammatory signaling in monocytes (266). Caloric restriction and low-fat diets both promote weight loss and reduce macrophage infiltration in AT, with caloric restriction showing superior effects on mitochondrial metabolism (268). The Mediterranean diet supplemented with almonds improves AT biology by promoting angiogenesis, adipogenesis, and M2-like macrophage polarization (269). These dietary interventions offer promising strategies for managing obesity-related inflammation and metabolic dysfunction.
Environmental factors, particularly cigarette smoking, significantly impact AT homeostasis and function. Smoking induces AT dysfunction through multiple pathways, including increased lipolysis, inflammation, and insulin resistance (270, 271). Nicotine activates AMPKα2 in adipocytes, leading to increased lipolysis and free fatty acid release (271). Smoke-derived oxidants promote adipose inflammation by recruiting macrophages and increasing pro-inflammatory cytokine production (272, 273). This chronic low-grade inflammation disrupts insulin signaling, contributing to insulin resistance. Smoking-induced adipose dysfunction is characterized by altered adipocyte differentiation, impaired insulin action, and dysregulated adipokine secretion (274). These effects extend beyond AT, impacting whole-body metabolism and increasing the risk of various metabolic disorders.
Chronic alcohol consumption and obesity significantly disrupt AT homeostasis, leading to fibrosis and metabolic dysfunction. Alcohol impairs adipocyte differentiation, reduces adiponectin expression, and promotes inflammation. It also increases lipolysis and ectopic fat deposition, contributing to fatty liver disease (275). Obesity-induced AT fibrosis involves complex cellular interplays, including macrophage infiltration and preadipocyte activation (276, 277). The Hippo pathway, in conjunction with Transforming growth factor- β (TGF-β) signaling, plays a crucial role in adipocyte plasticity and fibrosis development (278). TGF-β superfamily members regulate adipocyte differentiation, fibrosis, and metabolic functions (279). Autophagy dysregulation in AT may contribute to alcohol-induced liver injury (280).
Sleep deprivation significantly impacts AT function and adipokine secretion patterns. Reduced sleep duration is associated with increased leptin and visfatin levels, potentially contributing to inflammation and insulin resistance (281). Sleep loss also decreases adiponectin levels, which may lead to metabolic dysregulation (282). The circadian clock plays a crucial role in regulating adipokine secretion, as demonstrated by the blunted metabolic response to sleep restriction in Per1/2 mutant mice (283). Interestingly, chronic sleep deprivation is associated with higher adiponectin levels in patients with endocrine metabolic disorders, possibly as a compensatory mechanism (284). The day/night pattern of leptin is influenced by both the endogenous circadian pacemaker and behavioral factors such as sleep and food intake (285).
The mechanisms underlying metabolic dysfunction in AT extend beyond cellular senescence to include multiple interconnected pathways. Chronic overnutrition leads to adipocyte hypertrophy and tissue hypoxia, while persistent psychological stress activates glucocorticoid signaling and promotes visceral fat accumulation. Circadian rhythm disruption alters adipose metabolic gene expression patterns, affecting normal metabolic oscillations. Environmental pollutant exposure (as discussed in section 4.9) contributes to adipose dysfunction through multiple mechanisms. Additionally, gut microbiome dysbiosis influences adipose inflammation through altered production of short-chain fatty acids, establishing a complex gut-adipose axis in metabolic regulation.
Understanding these modifiable factors provides critical opportunities for preventing and treating adipose dysfunction through comprehensive lifestyle interventions rather than relying solely on pharmacological approaches. Future research should focus on personalized lifestyle prescriptions based on individual AT characteristics and metabolic phenotypes.
4.11 Regulatory networks of small RNAs in adipose metabolism
MicroRNA-mediated regulation of gene expression occurs through binding to partially complementary sequences primarily in the 3’ untranslated regions (3’ UTRs) of target messenger RNAs, though binding can also occur in 5’ UTRs and coding regions. This interaction typically leads to translational repression and/or mRNA decay, with the relative contribution of each mechanism varying by cellular context and the degree of complementarity. The regulatory influence of miRNAs spans multiple cellular pathways, with current bioinformatic predictions and experimental evidence suggesting that miRNAs may regulate the majority (estimated 60-90%) of mammalian protein-coding genes through complex regulatory networks (286–288).
AT function depends on precise microRNA-mediated regulation. Distinct microRNA populations control white adipocyte differentiation and brown/beige adipocyte thermogenic programming (289, 290). The impact of these regulatory RNAs extends to metabolic tissues including pancreatic β-cells, hepatocytes, and skeletal muscle (291). High-throughput sequencing has revealed tissue-specific expression patterns that coordinate systemic metabolic responses (292).
The let-7 family highlights the complexity of microRNA-mediated metabolic control. These regulators target key components of glucose homeostasis and insulin signaling networks, with their expression significantly diminished in diabetic tissues (293, 294). Obesity alters microRNA profiles across metabolic organs, though these changes can normalize following weight reduction (295, 296). Notably, adiponectin regulation involves miR-193b activity, linking obesity-associated decreases in this microRNA to broader metabolic dysfunction (297, 298).
Analysis of AT from obese subjects reveals characteristic alterations in microRNA expression. Fat depot expansion correlates with increased miR-221 and altered patterns of miR-17-5p and miR-132 across anatomical locations (299–301). Detection of these molecules in blood points to their role in systemic metabolic regulation (302, 303). MiR-223 exhibits key functions in metabolic homeostasis. Its abundance increases in subcutaneous fat during insulin resistance development (304) and shapes inflammatory responses in tissue macrophages (305, 306). Notably, circulating miR-223 decreases as obesity progresses toward T2DM (307).
The miR-130 family members suppress adipogenesis through PPARγ inhibition (308) and mediate inflammatory signaling (309). Their reduced expression in obese subcutaneous AT (310) affects inflammatory responses and insulin sensitivity through altered immune cell function, with implications for metabolic syndrome progression (292, 302).
Recent advances highlight emerging regulatory mechanisms in adipose dysfunction. Epigenetic modifications play a crucial role in regulating stem cell fate and function, particularly in adipose-derived stem cells (ADSCs). DNA methylation, histone modifications, and chromatin remodeling fundamentally reprogram cell fate decisions and metabolic capacity (311, 312). These epigenetic mechanisms influence ADSC differentiation into various lineages, including osteogenic and adipogenic pathways (313). In the context of obesity and type 2 diabetes, DNA methylation events are associated with altered AT function and gene regulation (314, 315). Novel signaling crosstalk between AMP-activated protein kinase (AMPK) and TBC1 domain family member ¼ (TBC1D1) reveals additional layers of insulin-independent glucose uptake regulation (316), while tissue-specific microRNA networks, particularly miR-223 and miR-130, regulate complex inflammatory and metabolic responses through post-transcriptional control (310, 317, 318). These emerging pathways provide new therapeutic targets beyond traditional approaches.
4.12 Disrupted adipogenesis in metabolic disease
Transcription of adipogenic genes begins with chromatin binding of CCAAT/enhancer-binding protein (C/EBPβ) at target promoters. DNA binding sites for PPARγ and C/EBPα become accessible during subsequent phases, allowing transcriptional activation of differentiation factors. DNA accessibility changes through histone modifications at adipogenic gene promoters. The differentiation program incorporates additional layers of control through methylation patterns, chromatin structure alterations, and specific microRNA expression. Transcriptional networks coordinate with chromatin remodeling complexes to establish cell-type specific gene expression patterns (319–321).
Bone morphogenetic protein signaling activates early commitment factors in mesenchymal precursor cells. Wnt pathway activation modifies chromatin structure at adipogenic loci, enabling progression toward the adipocyte phenotype. Further maturation yields functional fat cells containing characteristic lipid stores and metabolic enzymes. Disruption of these molecular pathways impairs AT development and function, leading to systemic metabolic deterioration. The transition between developmental stages requires precise temporal control of multiple signaling cascades. Defects in these regulatory networks prevent proper adipocyte maturation (322, 323).
4.13 ECM remodeling
4.13.1 Matrix composition changes
AT matrix undergoes substantial reorganization during obesity, marked by elevated deposition of fibrillar collagens and advancing fibrosis. Analysis of matrix composition reveals that increased collagen VI levels regulate both metabolic function and inflammatory states. Molecular studies show collagen VI deposition initiates cellular responses including enhanced inflammatory mediator production and disrupted insulin signaling networks (97). Proteomic profiling reveals elevated expression of multiple matrix metalloproteinase (MMPs), including MMP-2, -3, -12, -14, -19, alongside increased TIMP-1 levels within obese adipose samples, reflecting dynamic matrix restructuring (81). While obesity shifts MMP/TIMP (tissue inhibitor of metalloproteinases) ratios toward enhanced degradation, this compensatory response fails to prevent progressive fibrosis (82). Multiple matrix components including distinct collagen types, fibronectin molecules, and hyaluronan networks interact with cellular receptors such as integrins and Cluster of differentiation 44 (CD44), activating signaling cascades that regulate cellular metabolism and inflammatory pathways (324). Research demonstrates that adipocyte differentiation requires coordinated matrix remodeling, as experimental MMP inhibition disrupts normal adipogenesis (82). Detailed characterization of matrix regulation pathways (Table 6) suggests therapeutic opportunities through targeted modification of specific matrix components and their regulatory enzymes.
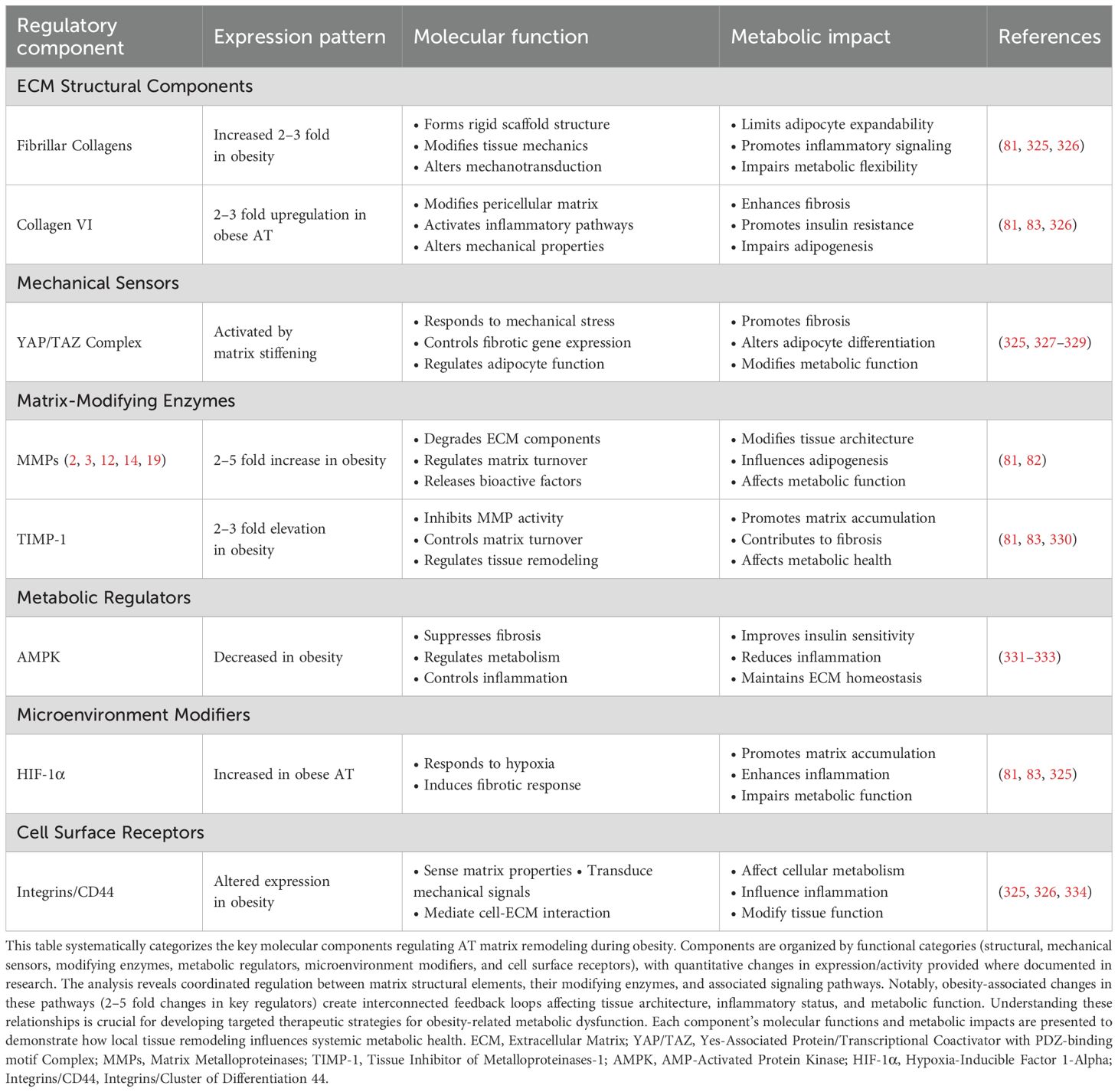
Table 6. Molecular architecture of matrix remodeling in obese AT.
4.13.2 Mechanical stress
Mechanical testing demonstrates increased tissue stiffness that limits adipocyte expansion and triggers mechanotransduction through focal adhesion complexes (327, 328). Research demonstrates that adipocytes sense elevated matrix rigidity through mechanosensitive pathways, resulting in increased pro-fibrotic gene transcription and accelerated matrix protein synthesis. These mechanical signals operate via organized actin cytoskeletal networks and mechanosensitive transcription factors, particularly the (Yes-associated protein/Transcriptional coactivator with PDZ-binding motif (YAP/TAZ) complex (329). Expanding adipose depots develop localized hypoxia that activates hypoxia-inducible factor 1- α (HIF-1α)-dependent signaling cascades, establishing self-perpetuating cycles of matrix accumulation. These matrix modifications propagate beyond AT to influence systemic metabolism, demonstrating the central role of matrix remodeling in obesity pathophysiology (325).
4.13.3 Fibrosis development
Matrix protein dynamics emerge as key determinants of AT plasticity during metabolic disease progression. Studies reveal that obesity-driven matrix deposition creates fibrotic microenvironments that sustain inflammatory responses and compromise insulin signaling pathways (83). Analysis shows that metformin modulates metabolism through AMPK activation across multiple tissues, though the precise mechanisms linking AMPK activation to improved insulin sensitivity remain an area of active investigation (331). Metformin treatment actively suppresses matrix buildup and fat tissue scarring that typically accompany obesity-driven insulin resistance, operating through multiple cellular mechanisms. At the molecular level, metformin triggers AMPK activation, which interferes with TGF-β1/Smad3 signaling - a key driver of tissue fibrosis. This interference reduces collagen formation and dials down genes involved in the scarring process (332). Metformin also modulates other critical pathways, dampening integrin/ERK signaling, limiting matrix-degrading enzymes, and protecting enlarged fat cells from premature death (334). These molecular mechanisms help maintain appropriate matrix elasticity during tissue expansion (328). Additionally, AMPK pathway activation in AT suppresses inflammatory signaling networks while improving insulin sensitivity (333). The demonstrated effects of metformin on both AMPK signaling and matrix remodeling provide multiple therapeutic targets for treating obesity-related metabolic disorders.
4.14 The role of autophagy in AT function and metabolic disorders
Autophagy maintains AT homeostasis by regulating adipocyte development, metabolism, and inflammatory status. This cellular recycling pathway removes damaged proteins and organelles, supporting proper cell function. Impaired autophagy in AT drives metabolic disease. Studies link autophagy defects to obesity and insulin resistance. The progression to T2DM accelerates when autophagy fails (335–337).
Autophagy regulates adipose function through distinct mechanisms. It controls lipid droplets via lipophagy pathways. The process degrades specific proteins to modulate adipokine release. Mitochondrial quality depends on mitophagy-mediated turnover. Mouse models reveal the metabolic impact of autophagy. Adipose-specific deletion of Atg7 reduces white fat mass. These mice show poor adipogenesis and whole-body insulin resistance (338, 339).
Obese AT exhibits heightened autophagy markers, yet suppressing this pathway yields metabolic benefits (340). While obesity initially triggers increased autophagy as an adaptive response, chronic metabolic stress leads to autophagy dysfunction. Single-cell transcriptomics and proteomics studies reveal that suppressing excessive autophagy in established obesity paradoxically improves metabolic outcomes by reducing inflammatory activation. The autophagic machinery influences adipocyte phenotype transitions between white and brown states, affecting whole-body energy balance (341). Understanding autophagy regulation in mature fat cells remains incomplete, highlighting the need for mechanistic studies to guide therapy development.
Defective autophagy pathways alter adipocyte differentiation, lipid handling, and insulin responses (335). Body fat distribution patterns, especially visceral depot expansion, compound these metabolic perturbations. This inflammatory state within AT raises the likelihood of developing metabolic diseases, including insulin resistance and cardiovascular problems (342). The autophagic machinery helps regulate immune responses at multiple levels - from bacterial clearance to immune cell activation and inflammatory mediator production (343). When autophagy fails, inappropriate inflammatory activation accelerates disease progression (344). Sustained inflammation suppresses autophagy function, establishing self-perpetuating pathological cycles (345). In acute kidney injury models, autophagy attenuates inflammatory damage via mTOR and AMPK signaling networks (346). Clinical intervention strategies targeting autophagy - through dietary modification, exercise programs, and drug development - aim to restore AT health and metabolic function.
4.15 Metabolic regulation by adipose-derived extracellular vesicles
Secretion of extracellular vesicles (EVs) from WAT represents a key mechanism in metabolic regulation between organs (347, 348). Analysis of vesicular content has identified specific metabolic enzymes and adipose hormones, alongside regulatory RNAs that influence target cell signaling pathways (349, 350).
Electron microscopy coupled with nanoparticle tracking analysis distinguishes vesicle subpopulations through unique biophysical properties and specific protein markers. Endosomal sorting complex required for transport (ESCRT)-dependent exosome formation generates 30–150 nm particles characterized by CD63, CD81, and tumor susceptibility gene 101 (TSG101) expression. Calcium-dependent membrane budding produces larger 100–1000 nm microvesicles expressing annexin V and selectins. Membrane phospholipid redistribution during apoptosis yields >1000 nm apoptotic bodies marked by phosphatidylserine externalization (351). Each population exhibits distinct membrane protein topology and internal cargo composition, allowing targeted isolation and characterization. These distinct vesicle populations serve unique roles in intercellular communication and metabolic regulation.
The vesicular miRNA cargo plays a crucial role in metabolic regulation. Studies investigating vesicular RNA content have demonstrated functional consequences in metabolic tissues. Through direct modulation of glucose transporter expression, miR-222 regulates skeletal muscle metabolism, concurrent with miR-23b-mediated effects on insulin signaling proteins (350, 352). The resulting perturbations in glucose homeostasis and hepatic lipid handling establish a mechanistic framework for vesicle-mediated metabolic regulation (350, 353).
Obesity alters both the protein composition and signaling effects of adipose-derived vesicles. Mass spectrometry has identified increased inflammatory mediators and decreased adiponectin levels, along with changes in lipid transport proteins (351, 354). These alterations promote inflammatory responses in AT and disrupt glucose homeostasis in liver and muscle (348). The dysregulation of vesicle secretion and composition represents a fundamental mechanism linking obesity to systemic metabolic dysfunction.
Pathological changes in adipocytes influence vesicle composition through specific molecular mechanisms (351). Hyperglycemia-stressed adipocytes release vesicles enriched in pro-inflammatory miRNAs that activate macrophage responses and tissue inflammation. These vesicles interact with cellular targets through EphB2-ephrinB1 binding, affecting insulin signaling and lipid trafficking pathways (350). BAT vesicles show unique properties through miR-92a and BMP7 enrichment, supporting thermogenic programming and metabolic balance (355). The differential regulation of vesicle production and content between white and BAT highlights their specialized roles in metabolic control.
The molecular characteristics of adipose-derived vesicles enable their use as diagnostic tools and therapeutic vectors in metabolic disease management (356). Their composition serves as biomarkers for metabolic syndrome, cardiovascular disease, and cancer progression (357). Clinical applications include longitudinal disease monitoring and therapeutic response assessment (358). Current research focuses on engineering vesicles for targeted drug delivery and metabolic modulation through cargo manipulation. These tissue-specific modifications provide mechanistic insights while suggesting novel therapeutic approaches for metabolic disorders. The development of standardized isolation protocols and stability enhancement methods addresses key challenges in clinical translation.
5 Systemic impact of adipose dysfunction
AT dysfunction serves as the primary driver of systemic metabolic dysregulation, orchestrating metabolic perturbations across liver, skeletal muscle, pancreas, and other organs through integrated molecular mechanisms. The following sections detail how compromised AT propagates dysfunction through altered adipokine secretion, excessive free fatty acid release, inflammatory mediator production, and EV signaling, establishing self-perpetuating cycles that progressively worsen whole-body metabolic homeostasis.
5.1 Endocrine disruption
5.1.1 Adipokine dysregulation
Beyond storing energy, AT actively produces proteins called adipokines. These factors act throughout the body, profoundly affecting metabolism, immune function, and insulin signaling pathways (359, 360). Within healthy AT, specific adipokines work together to regulate appetite, energy use, and tissue responses to insulin (361). AT produces several metabolically critical proteins, including the energy-regulating hormone leptin and the insulin-sensitizing factor adiponectin. Additional secreted factors like resistin, visfatin, and retinol binding protein 4 (RBP4) also shape metabolic outcomes. Targeted gene deletions in mouse models highlight their unique functions - leptin knockouts develop severe obesity with decreased energy expenditure, while mice lacking adiponectin exhibit profound insulin resistance. Similarly, genetic ablation of resistin, visfatin or RBP4 leads to complex changes in inflammatory signaling networks and systemic glucose regulation (361, 362).
Obesity and T2DM trigger pronounced alterations in AT structure and function, disrupting normal adipokine production patterns (363). Recent studies demonstrate depot-specific adipokine secretion patterns, with visceral fat showing distinct inflammatory profiles compared to subcutaneous deposits. These depot-specific differences contribute significantly to metabolic outcomes. The resulting secretory profile shows suppressed levels of metabolically protective factors like adiponectin alongside increased inflammatory mediators TNF-α and IL-6 (2, 250). This adipokine imbalance initiates and maintains chronic low-grade inflammation, progressively impairing insulin signaling across multiple tissue beds (175).
The inflammatory adipokines resistin and visfatin compromise metabolic health through multiple mechanisms: disrupting IR activation, amplifying inflammatory protein production, and perturbing glucose homeostasis (360). These molecular derangements create recurring cycles of metabolic dysfunction and inflammation. Given their central regulatory roles, adipokine pathways represent attractive therapeutic targets for obesity-related disorders. Current therapeutic development focuses on strategies to normalize adipokine profiles through direct pathway modulation or broader improvements in AT function.
5.1.2 Hormone resistance
Adipose dysfunction manifests through cellular defects and altered tissue architecture. Limited formation of new adipocytes leads to pathological expansion of existing fat cells, reducing lipid storage capacity (364). Tissue hypoxia develops alongside reduced blood vessel formation, triggering inflammatory cascades (365). Analysis of subcutaneous fat reveals altered BMP-4 signaling networks and increased gremlin-1 protein levels in hypertrophic obesity (366, 367). These molecular alterations modify adipokine production profiles, enhance lipolytic activity, and maintain chronic inflammatory states (342).
Loss of IR signaling in fat cells triggers lipase activation, with increased hydrolytic activity. The resulting release of stored lipids promotes fat accumulation in nonadipose organs, disrupting metabolic homeostasis. Ectopic lipid deposition in liver, skeletal muscle, and pancreatic tissue progressively impairs systemic glucose regulation and insulin sensitivity (368).
5.1.3 Metabolic consequences
Pro-inflammatory signals block insulin pathways (4), while hypertrophic fat cells show reduced metabolic responses and trigger fat accumulation in other tissues (369). Inflammatory mediators, oxidative damage, and excess lipids suppress adipocyte formation (364). The limitation in adipose expansion drives metabolic disease progression through multiple mechanisms. Macrophages and T lymphocytes within AT suppress formation of new adipocytes while stimulating connective tissue production. The resulting accumulation of collagen and other matrix proteins creates physical constraints on AT growth and cellular differentiation. Local tissue inflammation and matrix remodeling establish a microenvironment that perpetuates dysfunction through impaired adipogenesis (370).
5.2 AT receptor networks and metabolic regulation
AT receptor systems form interconnected signaling networks that regulate metabolic responses. Severe metabolic disruption and lipodystrophy emerge when insulin receptor (IR) or insulin-like growth factor 1 receptor (IGF1R) signaling fails, highlighting their central role in adipose development (371). Beyond simple fat storage, AT demonstrates remarkable plasticity through two distinct growth mechanisms. While both hyperplastic and hypertrophic expansion increase adipose mass, new adipocyte formation through hyperplasia appears to preserve metabolic health during obesity (372). AT dysfunction manifests when expansion limits are reached, forcing lipid accumulation in non-ATs and triggering systemic metabolic deterioration (37, 261).
TLR2 and TLR4 expression patterns shift dramatically in obese AT, establishing these pattern recognition receptors as central inflammatory mediators. Beyond pathogen sensing, these receptors respond to elevated fatty acids and other obesity-associated molecular signals (373). Subsequent NF-κB pathway activation drives macrophage recruitment and amplifies inflammatory cytokine production (374). This inflammatory cascade particularly affects visceral fat deposits, which show heightened susceptibility compared to subcutaneous stores.
AT dysfunction orchestrates systemic metabolic perturbations through distinct molecular mechanisms targeting peripheral organs. Dysfunctional adipocytes release excessive FFAs through uncontrolled lipolysis, which directly impair hepatic insulin signaling by inducing diacylglycerol accumulation and protein kinase C (PKC) activation, leading to hepatic steatosis and gluconeogenesis dysregulation (8, 375). In skeletal muscle, these circulating FFAs promote intramyocellular lipid accumulation, disrupting insulin-stimulated glucose uptake through ceramide-mediated inhibition of Akt phosphorylation (376, 377). Moreover, aberrant adipokine secretion profiles, characterized by elevated TNF-α and IL-6 alongside diminished adiponectin, propagate inflammatory signaling that impairs both hepatic and muscle insulin sensitivity (175, 378). Adipose-derived EV carrying specific miRNA signatures (miR-222, miR-23b) further mediate inter-organ communication, directly altering glucose transporter expression in skeletal muscle and lipid metabolism in hepatocytes (350, 352). This molecular crosstalk establishes a self-perpetuating cycle where adipose dysfunction progressively compromises peripheral tissue metabolic homeostasis.
PPARs regulates complex metabolic networks while directing adipocyte differentiation programs (379). Chromatin landscape remodeling by PPARγ proves essential for adipogenesis (380). These nuclear receptors undergo sophisticated post-translational control, with disrupted modifications linked to obesity and metabolic dysfunction (381, 382). Each PPAR subtype serves specialized metabolic functions - PPARα coordinates fatty acid oxidation pathways while PPARβ/δ enhances lipid metabolism (383). Pharmacological modulation of PPAR receptors represents a therapeutic avenue for metabolic disease intervention, though challenges remain in targeting specificity (384). Within AT, complex signaling networks emerge from adipocyte and macrophage interactions, generating localized inflammation marked by specific immune cell populations and altered cytokine profiles (385). The resulting metabolic stress triggers phosphorylation cascades through several pathways. Experimental evidence points to IKK/NF-κB signaling as a central mediator, while PI3K/Akt and MAP kinase activation leads to serine/threonine modifications of insulin receptor substrates (IRSs), ultimately disrupting glucose homeostasis (386). Pattern recognition receptors and inflammasome complexes sustain this inflammatory state (69). Disrupted adipokine secretion patterns and altered lipid handling further compound insulin resistance development.
Anatomically distinct fat deposits display unique receptor expression patterns and metabolic properties. Upper body and visceral deposits correlate with increased metabolic risk, while gluteofemoral fat appears protective (387). These depot-specific differences stem from varied cellular composition, gene regulatory networks, and physiological functions - including distinct steroid receptor profiles, adipokine signatures, and metabolic activities (388, 389). Recent work has expanded this heterogeneity concept to brown, beige, and ectopic fat, with each depot’s molecular profile differently impacting systemic metabolism (390, 391).
5.3 Distinct mechanisms of adipocyte elimination in metabolic disease
AT undergoes several forms of cellular elimination under metabolic stress: mitochondrial-dependent apoptotic pathways, inflammatory cell death via receptor-interacting serine/threonine-protein kinase 3 (RIPK3)/Mixed lineage kinase domain-like protein (MLKL) signaling, and inflammasome-triggered membrane disruption through specific molecular cascades (84). High-resolution microscopy demonstrates characteristic morphological changes: distended ER, damaged mitochondrial networks, and oxidative modifications within hypertrophied adipocytes. These conditions promote NLRP3 inflammasome assembly, triggering caspase-1 activation and subsequent plasma membrane permeabilization (85). Infiltrating macrophages organize around dying adipocytes, forming characteristic inflammatory structures termed crown-like structures (CLS) within affected regions (86). During cell death, released cellular components - including DNA fragments, bioactive lipids, and pro-inflammatory mediators - amplify both local and systemic metabolic disruption through well-defined pathways (392). Adipocyte survival regulation involves intricate crosstalk between death receptor signaling and mitochondrial pathways.
The consequences of adipocyte death extend beyond local tissue dysfunction, driving organ-specific metabolic disruptions through precisely characterized molecular pathways. Released cellular components from dying adipocytes, including mitochondrial DNA fragments and oxidized lipids, trigger hepatic Kupffer cell activation via TLR9 signaling, promoting liver inflammation and fibrosis (393). In skeletal muscle, these damage-associated molecular patterns activate resident macrophages, inducing myocyte insulin resistance through paracrine IL-1β secretion (394). Furthermore, the compensatory hyperinsulinemia resulting from adipose dysfunction stimulates de novolipogenesis in hepatocytes while simultaneously impairing muscle glucose uptake, creating a pathological metabolic state that propagates systemic insulin resistance (157). Advanced imaging studies demonstrate that adipocyte-derived ceramides specifically accumulate in hepatic and muscle tissue, directly inhibiting insulin receptor substrate phosphorylation and disrupting mitochondrial function (395, 396). Key molecular executioners, particularly caspase-1 and MLKL, orchestrate distinct death programs through specific biochemical mechanisms (397). These detailed molecular insights suggest new therapeutic approaches.
Adipocyte elimination triggers extensive signaling network perturbations. Specific molecular signals from dying cells guide macrophage recruitment and inflammatory focus development. Notably, adipocyte size expansion beyond critical thresholds activates death pathways even in lean tissue by compromising phosphatidylserine-dependent clearance mechanisms. Recruited macrophages secrete elevated levels of TNF-α and IL-6 through persistent NF-κB activation (398, 399). The combination of altered adipokine profiles and disrupted adipogenic transcription compromises metabolic homeostasis (141). Continuous cycles of cell death and immune cell accumulation create an inflammatory environment that impairs insulin signaling (86). This adipose dysfunction increases lipid flux to peripheral organs, particularly through portal circulation to the liver, accelerating broader metabolic disease progression (400).
6 Molecular mechanisms of age-related AT dysfunction: from cellular senescence to systemic impact
Advancing age fundamentally alters AT biology, initiating molecular and cellular cascades that drive metabolic perturbations and age-associated pathologies. Primary mechanistic drivers encompass adipogenic dysregulation, cellular senescence programs, and aberrant adipokine signaling networks (401, 402). The senescence-associated secretory phenotype (SASP) emerges as a central regulator, propagating chronic inflammatory states and metabolic dysfunction (403). Age progression correlates with significant adipose depot redistribution patterns, coupled to accumulation of senescent cell populations and progressive mitochondrial deficits (404). These alterations manifest across diverse adipose-resident cell populations - mature adipocytes, immune cell subsets, and progenitor compartments (405) - culminating in systemic inflammatory activation, insulin resistance development, and accelerated aging phenotypes (406). Current therapeutic strategies targeting these age-related perturbations include senolytic compounds, nutritional interventions, physical activity protocols, and heterochronic parabiosis approaches (407).
At the molecular level, Sirtuins (silent mating type information regulation 2 homolog) emerge as crucial regulators in this complex landscape. These NAD+-dependent deacetylases coordinate AT function and metabolism during aging and obesity (408). Through precise regulation of lipid metabolism, inflammation, and fibrosis in AT, sirtuins fundamentally shape energy homeostasis and metabolic health (409). The aging process modulates AT function through sirtuin-dependent pathways, driving changes in fat distribution, adipogenesis, and inflammatory responses (401). SIRT1 activation enhances fatty acid oxidation and lipid mobilization, potentially protecting against obesity-linked metabolic disorders (410). Notably, obesity disrupts both adipose NAD+homeostasis and sirtuin enzymatic function, leading to mitochondrial deficits and metabolic complications (411).
The aging process induces dramatic changes in AT distribution and function, with significant systemic consequences. A hallmark of these changes is the reduction in subcutaneous fat accompanied by an increase in visceral fat and ectopic lipid deposition (412). The cellular composition of aging AT undergoes substantial alterations, characterized by diminished preadipocyte function and increased presence of senescent cells (401). These changes manifest in impaired adipogenesis, persistent inflammation, and dysregulated adipokine production, all contributing to insulin resistance and metabolic disorders (407). The decline in BAT activity with age further compromises metabolic homeostasis (406), although some fat redistribution patterns may serve protective functions in extreme old age (413).
Aging leads to notable alterations in the regulation of adipogenesis at the transcriptional level. Studies indicate a decline in the expression of essential adipogenic regulators such as C/EBPα and PPARγ, alongside an elevation in inhibitory factors, including C/EBPβ-LIP and CHOP (414, 415). These changes occur alongside alterations in miRNA regulation, particularly miR-143, which affects the ERK5-PPARγ axis crucial for adipocyte differentiation (416). Accumulated oxidative stress in aging AT impairs preadipocyte differentiation through cell cycle regulatory disruption (417), while enhanced SASP factor and proinflammatory cytokine production sustains chronic inflammation and insulin resistance (401).
Cellular senescence programming, characterized by permanent cell cycle arrest and SASP development, represents a fundamental mechanism driving age-related adipose dysfunction. SASP encompasses secretion of diverse factors - inflammatory mediators, growth factors, and matrix components (418) - regulated through NF-κB, C/EBPβ, and Janus kinase/Signal transducer and activator of transcription (JAK/STAT) signaling networks (419, 420). While SASP activation supports tumor suppression and tissue repair processes, it simultaneously promotes chronic inflammatory states and age-related functional decline (421). JAK pathway targeting presents therapeutic potential for addressing SASP-mediated inflammation and frailty in aging populations (422), driving research into SASP-modulating therapeutic approaches including senolytic and senomorphic compounds (423).
The systemic impact of age-related AT dysfunction extends beyond local effects, contributing to widespread metabolic dysfunction and chronic low-grade inflammation (401). These changes encompass fat deposit redistribution, reduced adipogenesis, senescent cell accumulation, and altered immune cell composition (424). The dysregulated secretion of adipokines leads to insulin resistance and increased inflammation (406), while the intricate interplay between AT and the immune system becomes crucial in age-related metabolic decline, mirroring patterns observed in obesity (425). The disruption of inter-organ communication due to AT dysfunction accelerates the aging process and increases metabolic disease risk (426). Notably, both obesity and aging share key features in AT, including elevated visceral-to-subcutaneous fat ratios and pro-inflammatory immune cell phenotypes (Figure 1) (427).
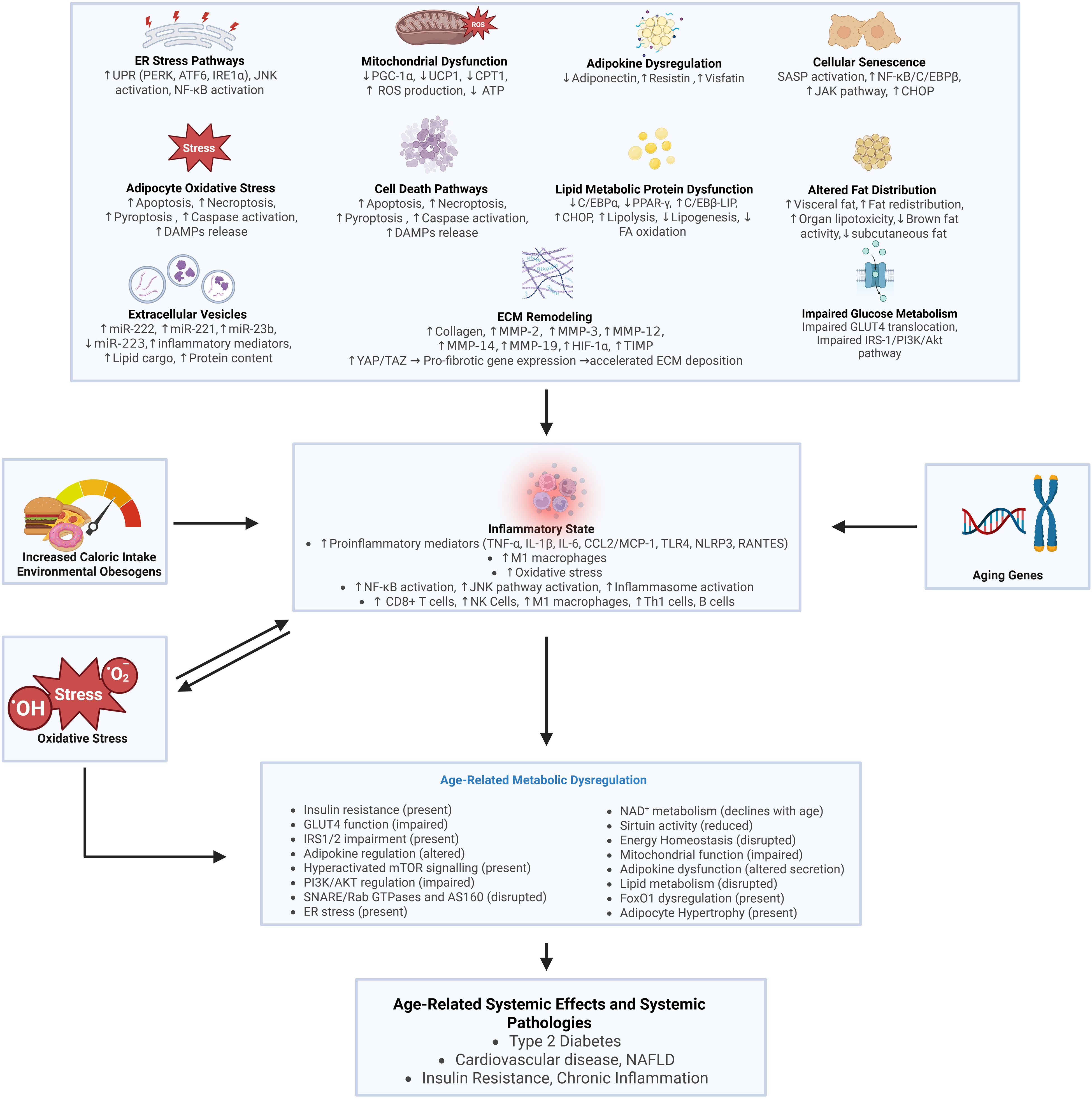
Figure 1. Age-related AT dysfunction arises through dysregulation of diverse molecular pathways. The schematic illustrates cellular stress responses including protein misfolding within the ER, compromised mitochondrial oxidative phosphorylation, and accelerated cellular senescence programs. Extensive ECM remodeling alters tissue architecture while impaired progenitor populations severely limit regenerative capacity. Complex metabolic perturbations emerge through increased PPAR-γ signaling cascades coupled with defective lipid oxidation pathways. Pathological fat redistribution occurs through visceral and ectopic lipid accumulation alongside progressive subcutaneous depot depletion. Adipocyte hypertrophy predominates over healthy hyperplastic expansion due to compromised progenitor function. Systemic metabolic dysregulation manifests through altered insulin sensitivity, disrupted NAD+ metabolism, and impaired mitochondrial function. The resultant disruption of glucose regulation and energy homeostasis promotes cardiometabolic disorders including T2DM and NAFLD through mechanisms spanning cellular to organismal scales. These molecular and physiological changes create integrated pathways of dysfunction affecting multiple metabolic systems.
7 Therapeutic advances in metabolic AT dysfunction
AT thermogenesis presents a compelling therapeutic avenue for obesity and metabolic disorders. Brown and beige adipocytes drive energy expenditure through UCP1-mediated non-shivering thermogenesis, with activation achievable via cold exposure, β3-adrenergic receptor agonists, or exercise-induced pathways (428, 429). While BAT activation demonstrates significant metabolic benefits in experimental models, sustained clinical weight loss remains challenging, and cardiovascular complications continue to limit therapeutic applications (428). Current therapeutic development encompasses multiple parallel approaches: novel pharmacological agents targeting thermogenic pathways, cell-based interventions to enhance brown adipose function, and genetic modifications designed to amplify thermogenic capacity in existing AT (430).
Adipokine signaling modulation represents another key therapeutic direction in metabolic disease treatment. Leptin sensitization strategies combat cellular resistance mechanisms, while newly developed adiponectin mimetics boost insulin sensitivity and reduce inflammatory cascades throughout metabolic tissues. The therapeutic targeting of adipokines has yielded particularly promising results for obesity-related metabolic conditions (431). These approaches span multiple molecular interventions: engineered adiponectin mimetics, synthetic leptin sensitizers, and targeted anti-inflammatory compounds, each aiming to recalibrate adipokine signaling networks and downstream metabolic parameters (432, 433). Modifying the adipose inflammasome architecture and tissue-specific inflammatory pathways shows substantial potential for attenuating chronic inflammation and restoring metabolic equilibrium across multiple tissue types.
Recent genetic and RNA-based therapeutic approaches have advanced substantially in treating metabolic dysfunction. CRISPR-Cas9 applications include generating precise lipid gene knockouts and enhancing human adipocyte browning capacity, demonstrating significant metabolic improvements in preclinical models (434). Targeted CRISPR interference against FABP4 expression in WAT reduces obesity progression and inflammatory markers in murine models (435). RNA therapeutics, including modified antisense oligonucleotides and siRNA constructs, show particular efficacy in modulating genes central to lipoprotein metabolism (436, 437). Advanced lipid nanoparticle delivery systems extend therapeutic possibilities beyond traditional hepatic targets, enabling tissue-specific intervention (438).
Diacylglycerol acyltransferase (DGAT) enzymes regulate triglyceride synthesis and lipid metabolism through multiple cellular pathways. DGAT inhibition shows therapeutic potential by limiting triglyceride synthesis and storage in metabolic tissues (439). PPARα and AMPK activation enhances fatty acid oxidation rates, reducing ectopic lipid accumulation across tissues. PPARα forms functional heterodimers with RXRβ to activate numerous genes involved in fatty acid oxidation cascades. Polyunsaturated fatty acids serve as natural PPAR activators through direct binding interactions (440). PPARβ/δ activation prevents high-fat diet-induced AMPK suppression and amplifies the PGC-1α-Lipin 1-PPARα signaling axis, substantially boosting fatty acid oxidation capacity (441). Current clinical applications include Fibrates for PPARα activation and TZDs as PPARγ agonists in dyslipidemia and diabetes treatment protocols.
Multipotent stem cells derived from AT have proven valuable in regenerative therapy. These ADSCs differentiate along distinct lineages - producing adipocytes, osteocytes, chondrocytes, and neurons under controlled conditions (442). The advantageous accessibility of ADSCs through minimally invasive lipoaspiration protocols distinguishes them from conventional mesenchymal stem cell sources (443). Their therapeutic mechanism encompasses both immunomodulatory functions and targeted trophic factor secretion, which synergistically enhance tissue repair processes (444). While clinical studies have validated ADSC efficacy across multiple pathological conditions (445), fundamental questions persist regarding lineage-specific differentiation mechanisms and delivery optimization. Elucidating the molecular determinants of cell fate decisions and characterizing the temporal dynamics of ADSC-mediated tissue regeneration remains crucial for therapeutic advancement. Contemporary investigations concentrate on decoding the complex interplay between signal transduction networks, chromatin modifications, and microenvironmental cues that regulate ADSC functionality across diverse therapeutic applications.
Nutritional interventions targeting Polyunsaturated fatty acids (PUFAs) and gut microbiota demonstrate substantial metabolic benefits through multiple mechanisms. Omega-3 PUFAs reduce AT inflammation, enhance cellular energy metabolism, and upregulate BAT thermogenic markers through direct and indirect pathways (446). These compounds modify gut microbiota composition through specific prebiotic effects on bacterial populations (447). Probiotic and prebiotic interventions favorably alter host lipid metabolism and tissue fatty acid profiles across multiple organ systems (448). Omega-3 PUFA supplementation improves circulating lipid profiles, glycemic control parameters, and hepatic fat content in various metabolic disorders. Individual genetic background and obesity status significantly influence intervention efficacy, necessitating personalized therapeutic approaches.
Molecular analyses of FGF21 and GLP-1 pathways underscore their central roles in metabolic control. Laboratory analyses of FGF21 demonstrate multifaceted effects on hepatic glucose output, AT lipid handling, and whole-body energy expenditure (449). Administration of engineered FGF21 variants yields metabolic improvements spanning glycemic control, lipid homeostasis, and body weight regulation in species ranging from mice to nonhuman primates (450). Recent clinical trials combining GLP-1 pathway activators with modified FGF21 molecules and thyroid receptor β-selective compounds show enhanced efficacy against fatty liver disease, suggesting pathway convergence in metabolic regulation (451).
The accumulation of senescent cells emerges as a critical factor in age-related metabolic decline. These cells, characterized by permanent cell cycle arrest, establish inflammatory microenvironments within adipose and other metabolic tissues. Elimination of senescent cell populations through targeted molecular approaches improves glucose homeostasis and insulin action in experimental obesity models (452). Evidence from aged mouse studies demonstrates that senescent cell removal enhances both metabolic parameters and physical function (453). Senolytic therapy shows particular promise for treating obesity-related metabolic dysfunction and numerous T2DM complications through senescent cell clearance (454, 455).
AT mitochondrial dysfunction correlates strongly with obesity progression and T2DM development, significantly affecting adipocyte differentiation capacity, cellular lipid metabolism, and insulin signaling responses. Mitochondrial function enhancement through targeted antioxidant compounds and exercise protocols improves multiple metabolic parameters (456). Complex autophagy and mitophagy pathways maintain essential adipocyte function and cellular identity. Novel antifibrotic agents target obesity-associated AT fibrosis, which contributes substantially to metabolic dysfunction through altered tissue architecture (457).
These therapeutic approaches target multiple fundamental aspects of adipose biology, including cellular energetics, lipid metabolism pathways, inflammatory cascades, and essential cellular maintenance processes. Combined, they represent significant advances in treatment options for metabolic diseases and systemic metabolic regulation across multiple tissue types and pathways (Figure 2). Current clinical investigations explore multimodal treatment combinations targeting severe metabolic disorders through integrated pathway modulation. Ongoing research continues to reveal additional molecular targets and regulatory pathways crucial for metabolic disease treatment strategies.
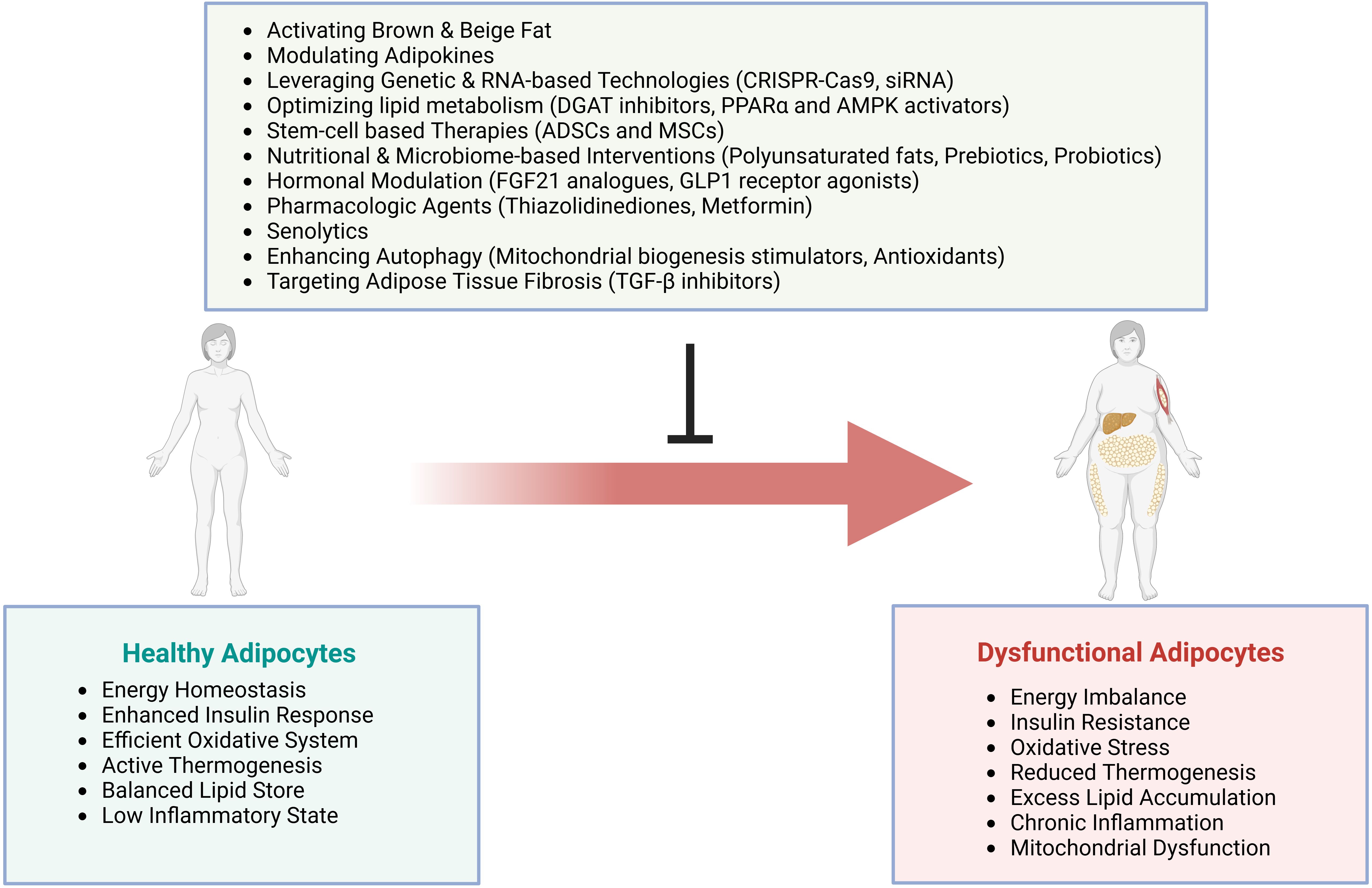
Figure 2. Therapeutic strategies for AT dysfunction. Key therapeutic approaches for addressing dysfunctional AT.
8 Conclusion
AT dysfunction disrupts metabolic homeostasis through complex interactions of cellular stress, inflammatory processes, and regulatory mechanisms. Our analysis reveals critical molecular pathways, including NF-κB and JNK signaling, that fundamentally compromise metabolic function.
Inflammatory pathways and oxidative stress critically impact metabolic health. Specific molecular markers like TNF-α, IL-6, and increased ROS demonstrate how AT depots generate unique cellular responses that influence systemic metabolism.
Molecular alterations in receptor functionality, particularly IR signaling and GLUT4 translocation, generate metabolic disruptions through complex inter-organ communication. Adipokine interactions, including dysregulated leptin and adiponectin profiles, reveal complex signaling networks between AT and metabolic systems.
Emerging therapeutic strategies target specific molecular mechanisms, including PPAR-γ pathway modulation, BAT activation through UCP1 targeting, AMPK pathway interventions, senolytic approaches to eliminate dysfunctional adipose cells, and precise genomic and RNA-based interventions.
Critical knowledge gaps persist in our understanding of AT dysfunction, particularly regarding depot-specific molecular heterogeneity, temporal dynamics of dysfunction progression, and sex-specific differences in adipose pathophysiology. The field requires comprehensive single-cell resolution mapping of AT microenvironments, longitudinal studies tracking dysfunction development from early metabolic stress to established disease, and systems-level integration of multi-omics data to identify causal relationships in metabolic regulation.
Key research priorities include: (1) elucidating the molecular determinants of adipose depot specialization and their therapeutic potential; (2) characterizing the temporal sequence of cellular events during dysfunction progression to identify critical intervention windows; (3) investigating sex hormone influences on adipose immune cell trafficking and inflammatory resolution; (4) developing novel imaging technologies for non-invasive assessment of AT health; and (5) designing targeted delivery systems for adipose-specific therapeutic agents that avoid systemic effects.
Future investigations should prioritize translational approaches bridging mechanistic discoveries to clinical applications. Personalized medicine strategies incorporating AT biomarkers, genetic risk profiles, and metabolic phenotyping may enable early intervention before irreversible dysfunction occurs. Integration of artificial intelligence with multi-omics analyses could reveal previously unrecognized regulatory networks and therapeutic targets. Ultimately, addressing these knowledge gaps through coordinated research efforts will advance our ability to combat the global epidemic of metabolic disease.
Author contributions
DM: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. MZ: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. WN: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. HA: Conceptualization, Data curation, Investigation, Methodology, Resources, Validation, Writing – original draft, Writing – review & editing. AN: Investigation, Methodology, Resources, Validation, Visualization, Writing – original draft, Writing – review & editing. NA: Conceptualization, Funding acquisition, Investigation, Resources, Software, Validation, Visualization, Writing – original draft, Writing – review & editing. ME: Data curation, Investigation, Resources, Software, Validation, Visualization, Writing – original draft, Writing – review & editing. AA: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Sponton CHG and Kajimura S. Multifaceted roles of beige fat in energy homeostasis beyond ucp1. Endocrinology. (2018) 159:2545–53. doi: 10.1210/en.2018-00371
2. Waki H and Tontonoz P. Endocrine functions of adipose tissue. Annu Rev Pathol. (2007) 2:31–56. doi: 10.1146/annurev.pathol.2.010506.091859
3. Choe SS, Huh JY, Hwang IJ, Kim JI, and Kim JB. Adipose tissue remodeling: its role in energy metabolism and metabolic disorders. Front Endocrinol (Lausanne). (2016) 7:30. doi: 10.3389/fendo.2016.00030
4. Blüher M. Adipose tissue dysfunction contributes to obesity related metabolic diseases. Best Pract Res Clin Endocrinol Metab. (2013) 27:163–77. doi: 10.1016/j.beem.2013.02.005
5. Matafome PN and Seiça R. Function and dysfunction of adipose tissue. Adv Neurobiol. (2017) 19:3–31. doi: 10.1007/978-3-319-63260-5_1
6. Blüher M. Adipose tissue dysfunction in obesity. Exp Clin Endocrinol Diabetes: Off J German Soc Endocrinol German Diabetes Assoc. (2009) 117:241–50. doi: 10.1055/s-0029-1192044
7. Shah A, Mehta NN, and Reilly MP. Adipose inflammation, insulin resistance, and cardiovascular disease. JPEN J Parenteral Enteral Nutr. (2008) 32:638–44. doi: 10.1177/0148607108325251
8. Ahmed B, Sultana R, and Greene MW. Adipose tissue and insulin resistance in obese. Biomed Pharmacother. (2021) 137:111315. doi: 10.1016/j.biopha.2021.111315
9. Mlinar B and Marc J. New insights into adipose tissue dysfunction in insulin resistance. Clin Chem Lab Med (CCLM). (2011) 49:1925–35. doi: 10.1515/CCLM.2011.697
10. McArdle MA, Finucane OM, Connaughton RM, McMorrow AM, and Roche HM. Mechanisms of obesity-induced inflammation and insulin resistance: insights into the emerging role of nutritional strategies. Front Endocrinol. (2013) 4. doi: 10.3389/fendo.2013.00052
11. Gaggini M, Saponaro C, and Gastaldelli A. Not all fats are created equal: adipose vs. Ectopic fat, implication in cardiometabolic diseases. Hormone Mol Biol Clin Invest. (2015) 22:18–7. doi: 10.1515/hmbci-2015-0006
12. Longo M, Zatterale F, Naderi J, Parrillo L, Formisano P, Raciti GA, et al. Adipose tissue dysfunction as determinant of obesity-associated metabolic complications. Int J Mol Sci. (2019) 20(9):2358. doi: 10.3390/ijms20092358
13. Samuel VT and Shulman GI. The pathogenesis of insulin resistance: integrating signaling pathways and substrate flux. J Clin Invest. (2016) 126:12–22. doi: 10.1172/JCI77812
14. Morigny P, Boucher J, Arner P, and Langin D. Lipid and glucose metabolism in white adipocytes: pathways, dysfunction and therapeutics. Nat Rev Endocrinol. (2021) 17:276–95. doi: 10.1038/s41574-021-00471-8
15. Ma K, Zhang Y, Zhao JY, Zhou L, and Li M. Endoplasmic reticulum stress: bridging inflammation and obesity-associated adipose tissue. Front Immunol. (2024) 15:1381227. doi: 10.3389/fimmu.2024.1381227
16. Wu S-N and Zou M-H. Ampk, mitochondrial function, and cardiovascular disease. Int J Mol Sci. (2020) 21(14):4987. doi: 10.3390/ijms21144987
17. Fernando R, Wardelmann K, Deubel S, Kehm R, Jung T, Mariotti M, et al. Low steady-state oxidative stress inhibits adipogenesis by altering mitochondrial dynamics and decreasing cellular respiration. Redox Biol. (2020) 32:101507. doi: 10.1016/j.redox.2020.101507
18. Harvey I, Boudreau A, and Stephens JM. Adipose tissue in health and disease. Open Biol. (2020) 10(12):200291. doi: 10.1098/rsob.200291
19. Schweiger M, Schreiber R, Haemmerle G, Lass A, Fledelius C, Jacobsen P, et al. Adipose triglyceride lipase and hormone-sensitive lipase are the major enzymes in adipose tissue triacylglycerol catabolism. J Biol Chem. (2006) 281:40236–41. doi: 10.1074/jbc.M608048200
20. Bargut TCL, Aguila MB, and Mandarim-de-Lacerda CA. Brown adipose tissue: updates in cellular and molecular biology. Tissue Cell. (2016) 48:452–60. doi: 10.1016/j.tice.2016.08.001
21. Giordano A, Smorlesi A, Frontini A, Barbatelli G, and Cinti S. White, brown and pink adipocytes: the extraordinary plasticity of the adipose organ. Eur J Endocrinol. (2014) 170:R159–71. doi: 10.1530/EJE-13-0945
22. Guo G, Wang W, Tu M, Zhao B, Han J, Li J, et al. Deciphering adipose development: function, differentiation and regulation. Dev Dyn. (2024) 253:956–97. doi: 10.1002/dvdy.708
23. Jia Z, Wang Z, Pan H, Zhang J, Wang Q, Zhou C, et al. Crosstalk between fat tissue and muscle, brain, liver, and heart in obesity: cellular and molecular perspectives. Eur J Med Res. (2024) 29:637. doi: 10.1186/s40001-024-02176-w
24. Frühbeck G. Overview of adipose tissue and its role in obesity and metabolic disorders. Methods Mol Biol. (2008) 456:1–22. doi: 10.1007/978-1-59745-245-8_1
25. Kershaw EE and Flier JS. Adipose tissue as an endocrine organ. J Clin Endocrinol Metab. (2004) 89:2548–56. doi: 10.1210/jc.2004-0395
26. Rosen ED and Spiegelman BM. What we talk about when we talk about fat. Cell. (2014) 156:20–44. doi: 10.1016/j.cell.2013.12.012
27. Zwick RK, Guerrero-Juarez CF, Horsley V, and Plikus MV. Anatomical, physiological, and functional diversity of adipose tissue. Cell Metab. (2018) 27:68–83. doi: 10.1016/j.cmet.2017.12.002
28. Cannon B and Nedergaard J. Brown adipose tissue: function and physiological significance. Physiol Rev. (2004) 84:277–359. doi: 10.1152/physrev.00015.2003
29. Kajimura S, Spiegelman BM, and Seale P. Brown and beige fat: physiological roles beyond heat generation. Cell Metab. (2015) 22:546–59. doi: 10.1016/j.cmet.2015.09.007
30. Villarroya F, Cereijo R, Villarroya J, and Giralt M. Brown adipose tissue as a secretory organ. Nat Rev Endocrinol. (2017) 13:26–35. doi: 10.1038/nrendo.2016.136
31. Wu J, Bostrom P, Sparks LM, Ye L, Choi JH, Giang AH, et al. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell. (2012) 150:366–76. doi: 10.1016/j.cell.2012.05.016
32. Pellegrinelli V, Carobbio S, and Vidal-Puig A. Adipose tissue plasticity: how fat depots respond differently to pathophysiological cues. Diabetologia. (2016) 59:1075–88. doi: 10.1007/s00125-016-3933-4
33. Cinti S. Pink adipocytes. Trends Endocrinol Metab. (2018) 29:651–66. doi: 10.1016/j.tem.2018.05.007
34. Cerk IK, Wechselberger L, and Oberer M. Adipose triglyceride lipase regulation: an overview. Curr Protein Pept Sci. (2017) 19:221–33. doi: 10.2174/1389203718666170918160110
35. Bolsoni-Lopes A and Alonso-Vale MIC. Lipolysis and lipases in white adipose tissue-an update. Arch Endocrinol Metab. (2015) 59:335–42. doi: 10.1590/2359-3997000000067
36. Jaworski K, Sarkadi-Nagy E, Duncan RE, Ahmadian M, and Sul HS. Regulation of triglyceride metabolism. IV. Hormonal regulation of lipolysis in adipose tissue. Am J Physiol Gastrointest Liver Physiol. (2007) 293(1):G1–4. doi: 10.1152/ajpgi.00554.2006
37. Sahu B, Tikoo O, Pati B, Senapati U, and Bal NC. Role of distinct fat depots in metabolic regulation and pathological implications. Rev Physiol Biochem Pharmacol. (2023) 186:135–76. doi: 10.1007/112_2022_73
38. López-Yus M, Hörndler C, Borlan S, Bernal-Monterde V, and Arbonés-Mainar JM. Unraveling adipose tissue dysfunction: molecular mechanisms, novel biomarkers, and therapeutic targets for liver fat deposition. Cells. (2024) 13(5):380. doi: 10.3390/cells13050380
39. Valjevac A. The endocrine function of adipose tissue. In Začiragić A, editor Meta‑Inflammation and Obesity. Bentham Science Publishers (2020) 23–43. doi: 10.2174/9789811479656120010004
40. Harwood HJ. The adipocyte as an endocrine organ in the regulation of metabolic homeostasis. Neuropharmacology. (2012) 63:57–75. doi: 10.1016/j.neuropharm.2011.12.010
41. Dieudonné MN, Sammari A, Santos ED, Leneveu MC, Giudicelli Y, and Pecquery R. Sex steroids and leptin regulate 11β-hydroxysteroid dehydrogenase I and P450 aromatase expressions in human preadipocytes: sex specificities. J Steroid Biochem Mol Biol. (2006) 99:189–96. doi: 10.1016/j.jsbmb.2006.01.007
42. Weiner J, Hankir MK, Heiker JT, Fenske WK, and Krause K. Thyroid hormones and browning of adipose tissue. Mol Cell Endocrinol. (2017) 458:156–9. doi: 10.1016/j.mce.2017.01.011
43. Chechi K, Carpentier AC, and Richard D. Understanding the brown adipocyte as a contributor to energy homeostasis. Trends Endocrinol Metab. (2013) 24:408–20. doi: 10.1016/j.tem.2013.04.002
44. Saito M, Okamatsu-Ogura Y, Matsushita M, Watanabe K, Yoneshiro T, Nio-Kobayashi J, et al. High incidence of metabolically active brown adipose tissue in healthy adult humans: effects of cold exposure and adiposity. Diabetes. (2009) 58:1526–31. doi: 10.2337/db09-0530
45. Yoneshiro T and Saito M. Activation and recruitment of brown adipose tissue as anti-obesity regimens in humans. Ann Med. (2015) 47:133–41. doi: 10.3109/07853890.2014.911595
46. Fenzl A and Kiefer FW. Brown adipose tissue and thermogenesis. Hormone Mol Biol Clin Invest. (2014) 19:25–37. doi: 10.1515/hmbci-2014-0022
47. Yang FT and Stanford KI. Batokines: mediators of inter-tissue communication (a mini-review). Curr Obes Rep. (2022) 11:1–9. doi: 10.1007/s13679-021-00465-7
48. Ziqubu K, Dludla PV, Mabhida SE, Jack BU, Keipert S, Jastroch M, et al. Brown adipose tissue-derived metabolites and their role in regulating metabolism. Metabol: Clin Exp. (2023) 150:155709. doi: 10.1016/j.metabol.2023.155709
49. Lee M-W, Lee M, and Oh K-J. Adipose tissue-derived signatures for obesity and type 2 diabetes: adipokines, batokines and micrornas. J Clin Med. (2019) 8(6):854. doi: 10.3390/jcm8060854
50. Martins FF, Souza-Mello V, Aguila MB, and Mandarim-de-Lacerda CA. Brown adipose tissue as an endocrine organ: updates on the emerging role of batokines. Hormone Mol Biol Clin Invest. (2022) 44:219–27. doi: 10.1515/hmbci-2022-0044
51. Gavalda-Navarro A, Villarroya J, Cereijo R, Giralt M, and Villarroya F. The endocrine role of brown adipose tissue: an update on actors and actions. Rev Endocr Metab Disord. (2022) 23:31–41. doi: 10.1007/s11154-021-09640-6
52. Sponton CH, Hosono T, Taura J, Jedrychowski MP, Yoneshiro T, Wang Q, et al. The regulation of glucose and lipid homeostasis via pltp as a mediator of bat-liver communication. EMBO Rep. (2020) 21:e49828. doi: 10.15252/embr.201949828
53. Nicholson TA, Church CD, Baker DJ, and Jones SW. The role of adipokines in skeletal muscle inflammation and insulin sensitivity. J Inflammation (London England). (2018) 15:9. doi: 10.1186/s12950-018-0185-8
54. Ziqubu K, Dludla PV, Moetlediwa MT, Nyawo TA, Pheiffer C, Jack BU, et al. Disease progression promotes changes in adipose tissue signatures in type 2 diabetic (Db/db) mice: the potential pathophysiological role of batokines. Life Sci. (2022) 313:121273. doi: 10.1016/j.lfs.2022.121273
55. Wueest S and Konrad D. The controversial role of il-6 in adipose tissue on obesity-induced dysregulation of glucose metabolism. Am J Physiol Endocrinol Metab. (2020) 319(3):E607–13. doi: 10.1152/ajpendo.00306.2020
56. Pal M, Febbraio MA, and Whitham M. From cytokine to myokine: the emerging role of interleukin-6 in metabolic regulation. Immunol Cell Biol. (2014) 92(4):331–9. doi: 10.1038/icb.2014.16
57. Ruan H-B. Developmental and functional heterogeneity of thermogenic adipose tissue. J Mol Cell Biol. (2020) 12:775–84. doi: 10.1093/jmcb/mjaa029
58. Kiefer FW. Browning and thermogenic programing of adipose tissue. Best Pract Res Clin Endocrinol Metab. (2016) 30:479–85. doi: 10.1016/j.beem.2016.09.003
59. Vergnes L and Reue K. Adaptive thermogenesis in white adipose tissue: is lactate the new brown(Ing)? Diabetes. (2014) 63:3175–6. doi: 10.2337/db14-0815
60. Phillips KJ. Beige fat, adaptive thermogenesis, and its regulation by exercise and thyroid hormone. Biology. (2019) 8(3):57. doi: 10.3390/biology8030057
61. Ludwig RG, Rocha AL, and Mori MA. Circulating molecules that control brown/beige adipocyte differentiation and thermogenic capacity. Cell Biol Int. (2018) 42(6):701–10. doi: 10.1002/cbin.10946
62. Valencak TG, Osterrieder A, and Schulz TJ. Sex matters: the effects of biological sex on adipose tissue biology and energy metabolism. Redox Biol. (2017) 12:806–13. doi: 10.1016/j.redox.2017.04.012
63. Cinti S. Ucp1 protein: the molecular hub of adipose organ plasticity. Biochimie. (2017) 134:71–6. doi: 10.1016/j.biochi.2016.09.008
64. Saini V. Molecular mechanisms of insulin resistance in type 2 diabetes mellitus. World J Diabetes. (2010) 1:68–75. doi: 10.4239/wjd.v1.i3.68
65. Cheng Z, Tseng YD, and White MF. Insulin signaling meets mitochondria in metabolism. Trends Endocrinol Metab. (2010) 21:589–98. doi: 10.1016/j.tem.2010.06.005
66. Sano H, Kane SE, Sano E, Mîinea CP, Asara JM, Lane WAS, et al. Insulin-stimulated phosphorylation of a rab gtpase-activating protein regulates glut4 translocation. J Biol Chem. (2003) 278:14599–602. doi: 10.1074/jbc.C300063200
67. Varra F-N, Varras M, Varra V-K, and Theodosis-Nobelos P. Molecular and pathophysiological relationship between obesity and chronic inflammation in the manifestation of metabolic dysfunctions and their inflammation−Mediating treatment options (Review). Mol Med Rep. (2024) 29(6):95. doi: 10.3892/mmr.2024.13219
68. Lumeng CN, Delproposto JB, Westcott DJ, and Saltiel AR. Phenotypic switching of adipose tissue macrophages with obesity is generated by spatiotemporal differences in macrophage subtypes. Diabetes. (2008) 57:3239–46. doi: 10.2337/db08-0872
69. Tanti J-F, Ceppo F, Jager J, and Berthou F. Implication of inflammatory signaling pathways in obesity-induced insulin resistance. Front Endocrinol. (2013) 3. doi: 10.3389/fendo.2012.00181
70. Zatterale F, Longo M, Naderi J, Raciti GA, Desiderio A, Miele C, et al. Chronic adipose tissue inflammation linking obesity to insulin resistance and type 2 diabetes. Front Physiol. (2020) 10. doi: 10.3389/fphys.2019.01607
71. Nicze M, Dec A, Borowka M, Krzyzak D, Boldys A, Buldak L, et al. Molecular mechanisms behind obesity and their potential exploitation in current and future therapy. Int J Mol Sci. (2024) 25(15):8202. doi: 10.3390/ijms25158202
72. Park J, Lee J, and Choi C. Mitochondrial network determines intracellular ros dynamics and sensitivity to oxidative stress through switching inter-mitochondrial messengers. PloS One. (2011) 6:e23211. doi: 10.1371/journal.pone.0023211
73. Li C, Liu Q, Chang Q, Xie M, Weng J, Wang X, et al. Role of mitochondrial fusion proteins mfn2 and opa1 on lung cellular senescence in chronic obstructive pulmonary disease. Respir Res. (2023) 24:319. doi: 10.1186/s12931-023-02634-9
74. Robert P, Nguyen PMC, Richard A, Grenier C, Chevrollier A, Munier M, et al. Protective role of the mitochondrial fusion protein opa1 in hypertension. FASEB J. (2021) 35:e21678. doi: 10.1096/fj.202000238RRR
75. Loson OC, Song Z, Chen H, and Chan DC. Fis1, mff, mid49, and mid51 mediate drp1 recruitment in mitochondrial fission. Mol Biol Cell. (2013) 24:659–67. doi: 10.1091/mbc.E12-10-0721
76. Castro JP, Grune T, and Speckmann B. The two faces of reactive oxygen species (Ros) in adipocyte function and dysfunction. Biol Chem. (2016) 397:709–24. doi: 10.1515/hsz-2015-0305
77. Lee J and Ozcan U. Unfolded protein response signaling and metabolic diseases. J Biol Chem. (2013) 289:1203–11. doi: 10.1074/jbc.R113.534743
78. Hu Y-R, Chen Y, and Liu Y. Role of endoplasmic reticulum stress response in regulation of adipose tissue metabolism. Sheng Li Xue Bao. (2021) 73:115–25.
79. Ajoolabady A, Lebeaupin C, Wu NN, Kaufman RJ, and Ren J. Er stress and inflammation crosstalk in obesity. Med Res Rev. (2022) 43:30–5. doi: 10.1002/med.21921
80. Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. (2010) 140:900–17. doi: 10.1016/j.cell.2010.02.034
81. Berg G, Barchuk M, and Miksztowicz V. Behavior of metalloproteinases in adipose tissue, liver and arterial wall: an update of extracellular matrix remodeling. Cells. (2019) 8(2):158. doi: 10.3390/cells8020158
82. Chavey C, Mari B, Monthouel MN, Bonnafous S, Anglard P, Van Obberghen E, et al. Matrix metalloproteinases are differentially expressed in adipose tissue during obesity and modulate adipocyte differentiation. J Biol Chem. (2003) 278:11888–96. doi: 10.1074/jbc.M209196200
83. Ruiz-Ojeda FJ, Plaza-Díaz J, Anguita-Ruiz A, Mendez-Gutierrez A, and Aguilera CM. Adipose extracellular matrix remodeling in obesity and insulin resistance. Cell Biochem Mech Obes. (2021) 20(19):4888. doi: 10.3390/ijms20194888
84. Ketelut-Carneiro N and Fitzgerald KA. Apoptosis, pyroptosis, and necroptosis-oh my! The many ways a cell can die. J Mol Biol. (2021) 434(4):167378. doi: 10.1016/j.jmb.2021.167378
85. Giordano A, Murano I, Mondini E, Perugini J, Smorlesi A, Severi I, et al. Obese adipocytes show ultrastructural features of stressed cells and die of pyroptosis. J Lipid Res. (2013) 54:2423–36. doi: 10.1194/jlr.M038638
86. Kuroda M and Sakaue H. Adipocyte death and chronic inflammation in obesity. J Med Investigation: JMI. (2017) 64:4. doi: 10.2152/jmi.64.193
87. Das S, Mukhuty A, Mullen GP, and Rudolph MC. Adipocyte mitochondria: deciphering energetic functions across fat depots in obesity and type 2 diabetes. Int J Mol Sci. (2024) 25(12):6681. doi: 10.3390/ijms25126681
88. Rius-Pérez S, Torres-Cuevas I, Millán I, Ortega ÁL, and Pérez S. Pgc-1α, inflammation, and oxidative stress: an integrative view in metabolism. Oxid Med Cell Longevity. (2020) 2020:1452696. doi: 10.1155/2020/1452696
89. Bhatti JS, Bhatti GK, and Reddy PH. Mitochondrial dysfunction and oxidative stress in metabolic disorders-a step towards mitochondria based therapeutic strategies. Biochim Biophys Acta Mol Basis Dis. (2017) 1863:1066–77. doi: 10.1016/j.bbadis.2016.11.010
90. Rocha M, Apostolova N, Herance JR, Rovira-Llopis S, Hernández-Mijares A, and Víctor VM. Perspectives and potential applications of mitochondria-targeted antioxidants in cardiometabolic diseases and type 2 diabetes. Med Res Rev. (2014) 34(1):160–89. doi: 10.1002/med.2014.34.issue-1
91. Lee PL, Jung SM, and Guertin DA. The complex roles of mechanistic target of rapamycin in adipocytes and beyond. Trends Endocrinol Metab. (2017) 28:319–39. doi: 10.1016/j.tem.2017.01.004
92. Yoon M-S. The role of mammalian target of rapamycin (Mtor) in insulin signaling. Nutrients. (2017) 9(11):1176. doi: 10.3390/nu9111176
93. Sykiotis GP and Papavassiliou AG. Serine phosphorylation of insulin receptor substrate-1: A novel target for the reversal of insulin resistance. Mol Endocrinol. (2001) 15:1864–9. doi: 10.1210/mend.15.11.0725
94. Horwitz A and Birk R. Adipose tissue hyperplasia and hypertrophy in common and syndromic obesity-the case of bbs obesity. Nutrients. (2023) 15(15):3445. doi: 10.3390/nu15153445
95. Goossens GH, Bizzarri A, Venteclef N, Essers Y, Cleutjens JP, Konings E, et al. Increased adipose tissue oxygen tension in obese compared with lean men is accompanied by insulin resistance, impaired adipose tissue capillarization, and inflammation. Circulation. (2011) 124:67–76. doi: 10.1161/CIRCULATIONAHA.111.027813
96. Jo J, Gavrilova O, Pack S, Jou W, Mullen S, Sumner AE, et al. Hypertrophy and/or hyperplasia: dynamics of adipose tissue growth. PloS Comput Biol. (2009) 5:e1000324. doi: 10.1371/journal.pcbi.1000324
97. Boden G, Cheung P, Salehi S, Homko C, Loveland-Jones C, Jayarajan S, et al. Insulin regulates the unfolded protein response in human adipose tissue. Diabetes. (2014) 63:912–22. doi: 10.2337/db13-0906
98. Gregor MF and Hotamışlıgil GS. Thematic review series: adipocyte biology. Adipocyte stress: the endoplasmic reticulum and metabolic disease published, jlr papers in press, may 9, 2007. J Lipid Res. (2007) 48:1905–14. doi: 10.1194/jlr.R700007-JLR200
99. Sharma NK, Das SK, Mondal AK, Hackney OG, Chu WS, Kern PA, et al. Endoplasmic reticulum stress markers are associated with obesity in nondiabetic subjects. J Clin Endocrinol Metab. (2008) 93:4532–41. doi: 10.1210/jc.2008-1001
100. Boden G, Duan X, Homko C, Molina EJ, Song W, Perez O, et al. Increase in endoplasmic reticulum stress-related proteins and genes in adipose tissue of obese, insulin-resistant individuals. Diabetes. (2008) 57:2438–44. doi: 10.2337/db08-0604
101. Xu L, Spinas GA, and Niessen M. Er stress in adipocytes inhibits insulin signaling, represses lipolysis, and alters the secretion of adipokines without inhibiting glucose transport. Horm Metab Res. (2010) 42:643–51. doi: 10.1055/s-0030-1255034
102. Mondal AK, Das SK, Varma V, Nolen GT, McGehee RE, Elbein SC, et al. Effect of endoplasmic reticulum stress on inflammation and adiponectin regulation in human adipocytes. Metab Syndr Relat Disord. (2012) 10:297–306. doi: 10.1089/met.2012.0002
103. Menu P, Mayor A, Zhou R, Tardivel A, Ichijo H, Mori K, et al. Er stress activates the nlrp3 inflammasome via an upr-independent pathway. Cell Death Dis. (2012) 3:e261. doi: 10.1038/cddis.2011.132
104. Vandanmagsar B, Youm Y, Ravussin A, Galgani JE, Stadler K, Mynatt RL, et al. The nalp3/nlrp3 inflammasome instigates obesity-induced autoinflammation and insulin resistance. Nat Med. (2010) 17:179–88. doi: 10.1038/nm.2279
105. Javaid HMW, Ko E, Joo EJ, Kwon SH, Park J-H, Shin S, et al. Tnfα-induced nlrp3 inflammasome mediates adipocyte dysfunction and activates macrophages through adipocyte-derived lipocalin 2. Metabol: Clin Exp. (2023) 142:155527. doi: 10.1016/j.metabol.2023.155527
106. Okla M, Zaher W, Alfayez M, and Chung S. Inhibitory effects of toll-like receptor 4, nlrp3 inflammasome, and interleukin-1beta on white adipocyte browning. Inflammation. (2018) 41:626–42. doi: 10.1007/s10753-017-0718-y
107. Engin A. The pathogenesis of obesity-associated adipose tissue inflammation. Adv Exp Med Biol. (2017) 960:221–45. doi: 10.1007/978-3-319-48382-5
108. Wang C, Huang Z, Du Y, Cheng Y, Chen S, and Guo F. Atf4 regulates lipid metabolism and thermogenesis. Cell Res. (2010) 20:174–84. doi: 10.1038/cr.2010.4
109. Moncan M, Mnich K, Blomme A, Almanza A, Samali A, and Gorman AM. Regulation of lipid metabolism by the unfolded protein response. J Cell Mol Med. (2021) 25:1359–70. doi: 10.1111/jcmm.16255
110. Lee AH and Glimcher LH. Intersection of the unfolded protein response and hepatic lipid metabolism. Cell Mol Life Sci. (2009) 66:2835–50. doi: 10.1007/s00018-009-0049-8
111. Suzuki T, Gao J, Ishigaki Y, Kondo K, Sawada S, Izumi T, et al. Er stress protein chop mediates insulin resistance by modulating adipose tissue macrophage polarity. Cell Rep. (2017) 18:2045–57. doi: 10.1016/j.celrep.2017.01.076
112. Meyerovich K, Ortis F, Allagnat F, and Cardozo AK. Endoplasmic reticulum stress and the unfolded protein response in pancreatic islet inflammation. J Mol Endocrinol. (2016) 57:R1–R17. doi: 10.1530/JME-15-0306
113. Lemmer IL, Willemsen N, Hilal N, and Bartelt A. A guide to understanding endoplasmic reticulum stress in metabolic disorders. Mol Metab. (2021) 47:101169. doi: 10.1016/j.molmet.2021.101169
114. Menikdiwela KR, Tôrres Guimarães JP, Ramalingam L, Kalupahana NS, Dufour JM, Washburn RL, et al. Mechanisms linking endoplasmic reticulum (Er) stress and micrornas to adipose tissue dysfunction in obesity. Crit Rev Biochem Mol Biol. (2021) 56:455–81. doi: 10.1080/10409238.2021.1925219
115. Lee J-M, Park S, Lee D, Ginting RP, Lee MR, Lee M-W, et al. Reduction in endoplasmic reticulum stress activates beige adipocytes differentiation and alleviates high fat diet-induced metabolic phenotypes. Biochim Biophys Acta Mol Basis Dis. (2021) 1867(5):166099. doi: 10.1016/j.bbadis.2021.166099
116. Basseri S, Lhotak S, Sharma AM, and Austin RC. The chemical chaperone 4-phenylbutyrate inhibits adipogenesis by modulating the unfolded protein response. J Lipid Res. (2009) 50:2486–501. doi: 10.1194/jlr.M900216-JLR200
117. Engin F, Yermalovich A, Nguyen T, Hummasti S, Fu W, Eizirik DL, et al. Restoration of the unfolded protein response in pancreatic beta cells protects mice against type 1 diabetes. Sci Transl Med. (2013) 5:211ra156. doi: 10.1126/scitranslmed.3006534
118. Cnop M, Foufelle F, and Velloso LA. Endoplasmic reticulum stress, obesity and diabetes. Trends Mol Med. (2012) 18:59–68. doi: 10.1016/j.molmed.2011.07.010
119. Khan S and Wang CH. Er stress in adipocytes and insulin resistance: mechanisms and significance (Review). Mol Med Rep. (2014) 10:2234–40. doi: 10.3892/mmr.2014.2532
120. Kleiner S, Mepani RJ, Laznik D, L-f Y, Jurczak MJ, Jornayvaz FR, et al. Development of insulin resistance in mice lacking pgc-1α in adipose tissues. Proc Natl Acad Sci. (2012) 109:9635–40. doi: 10.1073/pnas.1207287109
121. Puigserver P and Spiegelman BM. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (Pgc-1 alpha): transcriptional coactivator and metabolic regulator. Endocrine Rev. (2003) 24:78–90. doi: 10.1210/er.2002-0012
122. Enguix N, Pardo R, Gonzalez A, Lopez VM, Simó R, Kralli A, et al. Mice lacking pgc-1β in adipose tissues reveal a dissociation between mitochondrial dysfunction and insulin resistance. Mol Metab. (2013) 2:215–26. doi: 10.1016/j.molmet.2013.05.004
123. Lin JD, Wu P-H, Tarr PT, Lindenberg KS, St-Pierre J, Zhang C-Y, et al. Defects in adaptive energy metabolism with cns-linked hyperactivity in pgc-1α Null mice. Cell. (2004) 119:121–35. doi: 10.1016/j.cell.2004.09.013
124. Benton CR, Wright DC, and Bonen A. Pgc-1alpha-mediated regulation of gene expression and metabolism: implications for nutrition and exercise prescriptions. Appl Physiol Nutrition Metab. (2008) 33:843–62. doi: 10.1139/H08-074
125. Mitra R, Nogee DP, Zechner JF, Yea K, Gierasch CM, Kovács A, et al. The transcriptional coactivators, pgc-1α and β, cooperate to maintain cardiac mitochondrial function during the early stages of insulin resistance. J Mol Cell Cardiol. (2012) 52:701–10. doi: 10.1016/j.yjmcc.2011.10.010
126. Shen S-H, Singh SP, Raffaele M, Waldman M, Hochhauser E, Ospino J, et al. Adipocyte-specific expression of pgc1α Promotes adipocyte browning and alleviates obesity-induced metabolic dysfunction in an ho-1-dependent fashion. Antioxidants. (2022) 11(6):1147. doi: 10.3390/antiox11061147
127. Keuper M, Jastroch M, Yi CX, Fischer-Posovszky P, Wabitsch M, Tschop MH, et al. Spare mitochondrial respiratory capacity permits human adipocytes to maintain atp homeostasis under hypoglycemic conditions. FASEB J. (2014) 28:761–70. doi: 10.1096/fj.13-238725
128. Al-Mansoori L, Al-Jaber H, Prince MS, and Elrayess MA. Role of inflammatory cytokines, growth factors and adipokines in adipogenesis and insulin resistance. Inflammation. (2022) 45:31–44. doi: 10.1007/s10753-021-01559-z
129. Ortega SP, Chouchani ET, and Boudina S. Stress turns on the heat: regulation of mitochondrial biogenesis and ucp1 by ros in adipocytes. Adipocyte. (2017) 6:56–61. doi: 10.1080/21623945.2016.1273298
130. Maslov LN, Naryzhnaya NV, Boshchenko AA, Popov SV, Ivanov VV, and Oeltgen PR. Is oxidative stress of adipocytes a cause or a consequence of the metabolic syndrome? J Clin Trans Endocrinol. (2018) 15:1–5. doi: 10.1016/j.jcte.2018.11.001
131. Yadav UCS, Rani V, Deep G, Singh RK, and Palle K. Oxidative stress in metabolic disorders: pathogenesis, prevention, and therapeutics. Oxid Med Cell Longevity. (2016) 2016:183–93. doi: 10.1155/2016/9137629
132. Park J, Choe SS, Choi AH, Kim KH, Yoon MJ, Suganami T, et al. Increase in glucose-6-phosphate dehydrogenase in adipocytes stimulates oxidative stress and inflammatory signals. Diabetes. (2006) 55:2939–49. doi: 10.2337/db05-1570
133. Kawai T, Autieri MV, and Scalia R. Adipose tissue inflammation and metabolic dysfunction in obesity. Am J Physiol Cell Physiol. (2020) 320(3):C375–91. doi: 10.1152/ajpcell.00379.2020
134. Sassetti E, Clausen MH, and Laraia L. Small-molecule inhibitors of reactive oxygen species production. J Med Chem. (2021) 64:5252–75. doi: 10.1021/acs.jmedchem.0c01914
135. Meng Q and Su CH. The impact of physical exercise on oxidative and nitrosative stress: balancing the benefits and risks. Antioxid (Basel). (2024) 13(5):573. doi: 10.3390/antiox13050573
136. Bianchi E, Erbasan H, Riso P, and Perna S. Impact of the mediterranean diet on athletic performance, muscle strength, body composition, and antioxidant markers in both athletes and non-professional athletes: A systematic review of intervention trials. Nutrients. (2024) 16(20):3454. doi: 10.3390/nu16203454
137. Zhao RZ, Jiang S, Zhang L, and Yu ZB. Mitochondrial electron transport chain, ros generation and uncoupling (Review). Int J Mol Med. (2019) 44:3–15. doi: 10.3892/ijmm.2019.4188
138. Dhama K, Latheef SK, Dadar M, Samad HA, Munjal A, Khandia R, et al. Biomarkers in stress related diseases/disorders: diagnostic, prognostic, and therapeutic values. Front Mol Biosci. (2019) 6:91. doi: 10.3389/fmolb.2019.00091
139. Dommel S and Blüher M. Does C-C motif chemokine ligand 2 (Ccl2) link obesity to a pro-inflammatory state? Int J Mol Sci. (2021) 22(3):1500. doi: 10.3390/ijms22031500
140. Bai Y and Sun Q. Macrophage recruitment in obese adipose tissue. Obes Rev. (2015) 16(2):127–36. doi: 10.1111/obr.2015.16.issue-2
141. Maurizi G, Della Guardia L, Maurizi A, and Poloni A. Adipocytes properties and crosstalk with immune system in obesity-related inflammation. J Cell Physiol. (2018) 233:88–97. doi: 10.1002/jcp.v233.1
142. Grant RW and Stephens JM, eds. Fat in flames: influence of cytokines and pattern recognition receptors on adipocyte lipolysis. Am J Physiol Endocrinol Metab. (2015) 309(3):E205–E213. doi: 10.1152/ajpendo.00053.2015
143. Vieira-Potter VJ. Inflammation and macrophage modulation in adipose tissues. Cell Microbiol. (2014) 16(10):1484–92. doi: 10.1111/cmi.2014.16.issue-10
144. Thomas DD and Apovian CM. Macrophage functions in lean and obese adipose tissue. Metabol: Clin Exp. (2017) 72:120–43. doi: 10.1016/j.metabol.2017.04.005
145. Guzik TJ, Skiba D, Touyz RM, and Harrison DG. The role of infiltrating immune cells in dysfunctional adipose tissue. Cardiovasc Res. (2017) 113:1009–23. doi: 10.1093/cvr/cvx108
146. Začiragić A. The Role of Meta Inflammation in the Adipose Tissue Dysfunction and Obesity. In: Meta Inflammation and Obesity. Bentham Science Publishers (2020), 63–80. doi: 10.2174/9789811479656120010006
147. Huh JY, Park YJ, Ham M, and Kim JB. Crosstalk between adipocytes and immune cells in adipose tissue inflammation and metabolic dysregulation in obesity. Mol Cells. (2014) 37:365–71. doi: 10.14348/molcells.2014.0074
148. Unamuno X, Gómez-Ambrosi J, Rodríguez A, Becerril S, Frühbeck G, and Catalán V. Adipokine dysregulation and adipose tissue inflammation in human obesity. Eur J Clin Invest. (2018) 48(9):e12997. doi: 10.1111/eci.2018.48.issue-9
149. Crewe C, An YA, and Scherer PE. The ominous triad of adipose tissue dysfunction: inflammation, fibrosis, and impaired angiogenesis. J Clin Invest. (2017) 127:74–82. doi: 10.1172/JCI88883
150. Winer S and Winer DA. The adaptive immune system as a fundamental regulator of adipose tissue inflammation and insulin resistance. Immunol Cell Biol. (2012) 90(8):755–62. doi: 10.1038/icb.2011.110
151. Mráz M and Haluzík M. The role of adipose tissue immune cells in obesity and low-grade inflammation. J Endocrinol. (2014) 222:R113–27. doi: 10.1530/JOE-14-0283
152. Exley MA, Hand LE, O’Shea D, and Lynch L. Interplay between the immune system and adipose tissue in obesity. J Endocrinol. (2014) 223:R41–8. doi: 10.1530/JOE-13-0516
153. Nance SA, Muir LA, and Lumeng CN. Adipose tissue macrophages: regulators of adipose tissue immunometabolism during obesity. Mol Metab. (2022) 66:101642. doi: 10.1016/j.molmet.2022.101642
154. Olefsky JM and Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol. (2010) 72:219–46. doi: 10.1146/annurev-physiol-021909-135846
155. Tateya S, Kim F, and Tamori Y. Recent advances in obesity-induced inflammation and insulin resistance. Front Endocrinol. (2013) 4. doi: 10.3389/fendo.2013.00093
156. Burhans MS, Hagman DK, Kuzma JN, Schmidt KA, and Kratz M. Contribution of adipose tissue inflammation to the development of type 2 diabetes mellitus. Compr Physiol. (2018) 9:1–58. doi: 10.1002/j.2040-4603.2019.tb00055.x
157. Wensveen FM, Valentić S, Šestan M, Turk Wensveen T, and Polić B. The “Big bang” in obese fat: events initiating obesity-induced adipose tissue inflammation. Eur J Immunol. (2015) 45(9):2446–56. doi: 10.1002/eji.201545502
158. Rajan MR, Nyman E, Kjølhede P, Cedersund G, and Strålfors P. Systems-wide experimental and modeling analysis of insulin signaling through forkhead box protein O1 (Foxo1) in human adipocytes, normally and in type 2 diabetes. J Biol Chem. (2016) 291:15806–19. doi: 10.1074/jbc.M116.715763
159. Shan T, Zhang P, Jiang Q, Xiong Y, Wang Y, and Kuang S. Adipocyte-specific deletion of mtor inhibits adipose tissue development and causes insulin resistance in mice. Diabetologia. (2016) 59:1995–2004. doi: 10.1007/s00125-016-4006-4
160. Kumar A, Lawrence JC, Jung DY, Ko HJ, Keller SR, Kim JK, et al. Fat cell–specific ablation of rictor in mice impairs insulin-regulated fat cell and whole-body glucose and lipid metabolism. Diabetes. (2010) 59:1397–406. doi: 10.2337/db09-1061
161. Sakamoto K and Holman GD. Emerging role for as160/tbc1d4 and tbc1d1 in the regulation of glut4 traffic. Am J Physiol Endocrinol Metab. (2008) 295:E29–37. doi: 10.1152/ajpendo.90331.2008
162. Ishizuka K, Usui I, Kanatani Y, Bukhari A, He J, Fujisaka S, et al. Chronic tumor necrosis factor-alpha treatment causes insulin resistance via insulin receptor substrate-1 serine phosphorylation and suppressor of cytokine signaling-3 induction in 3t3-L1 adipocytes. Endocrinology. (2007) 148:2994–3003. doi: 10.1210/en.2006-1702
163. Kanety H, Feinstein R, Papa MZ, Hemi R, and Karasik A. Tumor necrosis factor alpha-induced phosphorylation of insulin receptor substrate-1 (Irs-1). Possible mechanism for suppression of insulin-stimulated tyrosine phosphorylation of irs-1. J Biol Chem. (1995) 270:23780–4. doi: 10.1074/jbc.270.40.23780
164. Gao Z, Hwang DH, Bataille F, Lefevre M, York DA, Quon M, et al. Serine phosphorylation of insulin receptor substrate 1 by inhibitor kappa B kinase complex. J Biol Chem. (2002) 277:48115–21. doi: 10.1074/jbc.M209459200
165. Ueki K, Kondo T, and Kahn CR. Suppressor of cytokine signaling 1 (Socs-1) and socs-3 cause insulin resistance through inhibition of tyrosine phosphorylation of insulin receptor substrate proteins by discrete mechanisms. Mol Cell Biol. (2004) 24:5434–46. doi: 10.1128/MCB.24.12.5434-5446.2004
166. Jiang Y, Biswas SK, and Steinle JJ. Serine 307 on insulin receptor substrate 1 is required for socs3 and tnf-α Signaling in the rmc-1 cell line. Mol Vision. (2014) 20:1463–70.
167. Ducluzeau PH, Fletcher LM, Vidal H, Laville M, and Tavaré JM. Molecular mechanisms of insulin-stimulated glucose uptake in adipocytes. Diabetes Metab. (2002) 28:85–92.
168. Cheatham B, Volchuk A, Kahn CR, Wang L, Rhodes CJ, and Klip A. Insulin-stimulated translocation of glut4 glucose transporters requires snare-complex proteins. Proc Natl Acad Sci United States America. (1996) 93:15169–73. doi: 10.1073/pnas.93.26.15169
169. Kawaguchi T, Tamori Y, Kanda H, Yoshikawa M, Tateya S, Nishino N, et al. The T-Snares Syntaxin4 and Snap23 but Not V-Snare Vamp2 Are Indispensable to Tether Glut4 Vesicles at the Plasma Membrane in Adipocyte. Biochem Biophys Res Commun. (2010) 391:1336–41. doi: 10.1016/j.bbrc.2009.12.045
170. Chamberlain LH and Gould GW. The vesicle- and target-snare proteins that mediate glut4 vesicle fusion are localized in detergent-insoluble lipid rafts present on distinct intracellular membranes. J Biol Chem. (2002) 277:49750–4. doi: 10.1074/jbc.M206936200
171. Kioumourtzoglou D, Gould GW, and Bryant NJ. Insulin stimulates syntaxin4 snare complex assembly via a novel regulatory mechanism. Mol Cell Biol. (2014) 34:1271–9. doi: 10.1128/MCB.01203-13
172. Grusovin J and Macaulay SL. Snares for glut4–mechanisms directing vesicular trafficking of glut4. Front Biosci. (2003) 8:d620–41. doi: 10.2741/1052
173. Goalstone ML, Leitner JW, Golovchenko I, Stjernholm MR, Cormont M, Le Marchand-Brustel Y, et al. Insulin promotes phosphorylation and activation of geranylgeranyltransferase ii. J Biol Chem. (1999) 274:2880–4. doi: 10.1074/jbc.274.5.2880
174. van Gerwen J, Amber S-SS, and Fazakerley DJ. Insulin signalling and glut4 trafficking in insulin resistance. Biochem Soc Trans. (2023) 51:1057–69. doi: 10.1042/BST20221066
175. Falcão-Pires I, Castro-Chaves P, Miranda-Silva D, Lourenço AP, and Leite-Moreira AF. Physiological, pathological and potential therapeutic roles of adipokines. Drug Discov Today. (2012) 17:880–9. doi: 10.1016/j.drudis.2012.04.007
176. Zhang Y, Shi L, Mei H, Zhang J, Zhu Y, Han X, et al. Inflamed macrophage microvesicles induce insulin resistance in human adipocytes. Nutr Metab (Lond). (2015) 12:21. doi: 10.1186/s12986-015-0016-3
177. Lumeng CN, Deyoung SM, and Saltiel AR. Macrophages block insulin action in adipocytes by altering expression of signaling and glucose transport proteins. Am J Physiol Endocrinol Metab. (2007) 292:E166–74. doi: 10.1152/ajpendo.00284.2006
178. Mleczko J, Ortega FJ, Falcon-Perez JM, Wabitsch M, Fernandez-Real JM, and Mora S. Extracellular vesicles from hypoxic adipocytes and obese subjects reduce insulin-stimulated glucose uptake. Mol Nutr Food Res. (2018) 62(5):1700917. doi: 10.1002/mnfr.201700917
179. Diaz-Saez F, Blanco-Sinfreu C, Archilla-Ortega A, Sebastian D, Romero M, Hernandez-Alvarez MI, et al. Neuregulin 4 downregulation induces insulin resistance in 3t3-L1 adipocytes through inflammation and autophagic degradation of glut4 vesicles. Int J Mol Sci. (2021) 22(23):12960. doi: 10.3390/ijms222312960
180. Kim JB, Wright HM, Wright M, and Spiegelman BM. Add1/srebp1 activates pparγ through the production of endogenous ligand. Proc Natl Acad Sci United States America. (1998) 95:4333–7. doi: 10.1073/pnas.95.8.4333
181. Watt MJ. Triglyceride lipases alter fuel metabolism and mitochondrial gene expression. Appl Physiol Nutrition Metab. (2009) 34:340–7. doi: 10.1139/H09-019
182. Gaidhu MP, Anthony NM, Patel P, Hawke TJ, and Ceddia RB. Dysregulation of lipolysis and lipid metabolism in visceral and subcutaneous adipocytes by high-fat diet: role of atgl, hsl, and ampk. Am J Physiol Cell Physiol. (2010) 298:C961–71. doi: 10.1152/ajpcell.00547.2009
183. Nielsen TS, Jessen N, Jørgensen JOL, Møller N, and Lund S. Dissecting adipose tissue lipolysis: molecular regulation and implications for metabolic disease. J Mol Endocrinol. (2014) 52:R199–222. doi: 10.1530/JME-13-0277
184. Chakrabarti P and Kandror KV. Adipose triglyceride lipase: A new target in the regulation of lipolysis by insulin. Curr Diabetes Rev. (2011) 7:270–7. doi: 10.2174/157339911796397866
185. Tansey JT, Sztalryd C, Hlavin EM, Kimmel AR, and Londos C. The central role of perilipin a in lipid metabolism and adipocyte lipolysis. IUBMB Life. (2004) 56(7):379–85. doi: 10.1080/15216540400009968
186. Watt MJ and Steinberg GR. Regulation and function of triacylglycerol lipases in cellular metabolism. Biochem J. (2008) 414:313–25. doi: 10.1042/BJ20080305
187. Christodoulides C and Vidal-Puig A. Ppars and adipocyte function. Mol Cell Endocrinol. (2010) 318:61–8. doi: 10.1016/j.mce.2009.09.014
188. Virtue S, Petkevicius K, Moreno-Navarrete JM, Jenkins BJ, Hart D, Dale M, et al. Peroxisome proliferator-activated receptor Γ2 controls the rate of adipose tissue lipid storage and determines metabolic flexibility. Cell Rep. (2018) 24:2005–12.e7. doi: 10.1016/j.celrep.2018.07.063
189. Rodriguez-Cuenca S, Carobbio S, Velagapudi VR, Barbarroja N, Moreno-Navarrete JM, Tinahones FJ, et al. Peroxisome proliferator-activated receptor Γ-dependent regulation of lipolytic nodes and metabolic flexibility. Mol Cell Biol. (2012) 32:1555–65. doi: 10.1128/MCB.06154-11
190. Frayn KN. Adipose tissue as a buffer for daily lipid flux. Diabetologia. (2002) 45:1201–10. doi: 10.1007/s00125-002-0873-y
191. Sharma AM and Staels B. Review: peroxisome proliferator-activated receptor gamma and adipose tissue–understanding obesity-related changes in regulation of lipid and glucose metabolism. J Clin Endocrinol Metab. (2007) 92:386–95. doi: 10.1210/jc.2006-1268
192. Denechaud P-D, Dentin R, Girard J, and Postic C. Role of chrebp in hepatic steatosis and insulin resistance. FEBS Lett. (2008) 582(1):582. doi: 10.1016/j.febslet.2007.07.084
193. Goalstone ML, Carel K, Leitner JW, and Draznin B. Insulin stimulates the phosphorylation and activity of farnesyltransferase via the ras-mitogen-activated protein kinase pathway. Endocrinology. (1997) 138:5119–24. doi: 10.1210/endo.138.12.5621
194. Nair AS and Saha B. Regulation of ras-gtpase signaling and localization by post-translational modifications. Kinases Phosphatases. (2023) 1(2):97–116. doi: 10.3390/kinasesphosphatases1020007
195. Satoh T. Molecular mechanisms for the regulation of insulin-stimulated glucose uptake by small guanosine triphosphatases in skeletal muscle and adipocytes. Int J Mol Sci. (2014) 15:18677–92. doi: 10.3390/ijms151018677
196. Carel K, Kummer JL, Schubert C, Leitner W, Heidenreich KA, and Draznin B. Insulin stimulates mitogen-activated protein kinase by a ras-independent pathway in 3t3-L1 adipocytes. J Biol Chem. (1996) 271:30625–30. doi: 10.1074/jbc.271.48.30625
197. Møller LLV, Klip A, and Sylow L. Rho gtpases—Emerging regulators of glucose homeostasis and metabolic health. Cells. (2019) 8(5):434. doi: 10.3390/cells8050434
198. Xu N, Shen N, Wang X, Jiang S, Xue B, and Li C. Protein prenylation and human diseases: a balance of protein farnesylation and geranylgeranylation. Sci China Life Sci. (2015) 58(4):328–35. doi: 10.1007/s11427-015-4836-1
199. Solomon C, Leitner JW, and Goalstone ML. Dominant negative α-subunit of farnesyl- and geranylgeranyl-transferase I inhibits insulin-induced differentiation of 3t3-L1 pre-adipocytes. Int J Obes. (2003) 27:40–7. doi: 10.1038/sj.ijo.0802189
200. Kowluru A and Kowluru RA. Protein prenylation in islet β-cell function in health and diabetes: putting the pieces of the puzzle together. Biochem Pharmacol. (2015) 98:363–70. doi: 10.1016/j.bcp.2015.07.004
201. Ye J and DeBose-Boyd RA. Regulation of cholesterol and fatty acid synthesis. Cold Spring Harbor Perspect Biol. (2011) 3(7):a004754. doi: 10.1101/cshperspect.a004754
202. Murthy S, Tong H, and Hohl RJ. Regulation of fatty acid synthesis by farnesyl pyrophosphate. J Biol Chem. (2005) 280:41793–804. doi: 10.1074/jbc.M504101200
203. Chen Y, Huang L-L, Qi X, and Chen C. Insulin receptor trafficking: consequences for insulin sensitivity and diabetes. Int J Mol Sci. (2019) 20(20):5007. doi: 10.3390/ijms20205007
204. Kowluru A, Veluthakal R, Rhodes CJ, Kamath VG, Syed I, and Koch BJ. Protein farnesylation–dependent raf/extracellular signal–related kinase signaling links to cytoskeletal remodeling to facilitate glucose-induced insulin secretion in pancreatic β-cells. Diabetes. (2010) 59:967–77. doi: 10.2337/db09-1334
205. Ferrara D, Montecucco F, Dallegri F, and Carbone F. Impact of different ectopic fat depots on cardiovascular and metabolic diseases. J Cell Physiol. (2019) 234:21630–41. doi: 10.1002/jcp.v234.12
206. Gustafson B, Hedjazifar S, Gogg S, Hammarstedt A, and Smith U. Insulin resistance and impaired adipogenesis. Trends Endocrinol Metab. (2015) 26:193–200. doi: 10.1016/j.tem.2015.01.006
207. Iizuka K, Takao K, and Yabe D. Chrebp-mediated regulation of lipid metabolism: involvement of the gut microbiota, liver, and adipose tissue. Front Endocrinol. (2020) 11. doi: 10.3389/fendo.2020.587189
208. Liu M, Zhang Z, Chen Y, Feng T, Zhou Q, and Tian X. Circadian clock and lipid metabolism disorders: A potential therapeutic strategy for cancer. Front Endocrinol. (2023) 14. doi: 10.3389/fendo.2023.1292011
209. Schaffer JE. Lipotoxicity: when tissues overeat. Curr Opin Lipidol. (2003) 14:281–7. doi: 10.1097/00041433-200306000-00008
210. Snel M, Jonker JT, Schoones JW, Lamb HJ, Roos A, Pijl H, et al. Ectopic fat and insulin resistance: pathophysiology and effect of diet and lifestyle interventions. Int J Endocrinol. (2012) 2012:983814. doi: 10.1155/2012/983814
211. Alswat KA. The role of endocannabinoids system in fatty liver disease and therapeutic potentials. Saudi J Gastroenterol: Off J Saudi Gastroenterol Assoc. (2013) 19:144–51. doi: 10.4103/1319-3767.114505
212. Giorgi C, Missiroli S, Patergnani S, Duszyński J, Wieckowski MR, and Pinton P. Mitochondria-associated membranes: composition, molecular mechanisms, and physiopathological implications. Antioxid Redox Signaling. (2015) 22:995–1019. doi: 10.1089/ars.2014.6223
213. Degechisa ST, Dabi YT, and Gizaw ST. The mitochondrial associated endoplasmic reticulum membranes: A platform for the pathogenesis of inflammation-mediated metabolic diseases. Immun Inflammation Dis. (2022) 10(7):e647. doi: 10.1002/iid3.v10.7
214. Gusdon AM, Song KX, and Qu S. Nonalcoholic Fatty liver disease: pathogenesis and therapeutics from a mitochondria-centric perspective. Oxid Med Cell Longev. (2014) 2014:637027. doi: 10.1155/2014/637027
215. Byrne CD and Targher G. Ectopic fat, insulin resistance, and nonalcoholic fatty liver disease: implications for cardiovascular disease. Arteriosclerosis Thrombosis Vasc Biol. (2014) 34:1155–61. doi: 10.1161/ATVBAHA.114.303034
216. Borén J, Taskinen M-R, Olofsson S-O, and Levin MC. Ectopic lipid storage and insulin resistance: A harmful relationship. J Internal Med. (2013) 274(1):25–40. doi: 10.1111/joim.2013.274.issue-1
217. Hu S, Hu Y, and Yan W. Extracellular vesicle-mediated interorgan communication in metabolic diseases. Trends Endocrinol Metabol: TEM. (2023) 34(9):571–82. doi: 10.1016/j.tem.2023.06.002
218. Grun F and Blumberg B. Environmental obesogens: organotins and endocrine disruption via nuclear receptor signaling. Endocrinology. (2006) 147:S50–5. doi: 10.1210/en.2005-1129
219. Grün F. Obesogens. Curr Opin Endocrinol Diabetes Obes. (2010) 17:453–9. doi: 10.1097/MED.0b013e32833ddea0
220. Janesick A and Blumberg B. Minireview: ppargamma as the target of obesogens. J Steroid Biochem Mol Biol. (2011) 127:4–8. doi: 10.1016/j.jsbmb.2011.01.005
221. Blumberg B. Obesogens, stem cells and the maternal programming of obesity. J Dev Origins Health Dis. (2010) 2:3–8. doi: 10.1017/S2040174410000589
222. Janesick AS and Blumberg B. Obesogens: an emerging threat to public health. Am J Obstet Gynecol. (2016) 214:559–65. doi: 10.1016/j.ajog.2016.01.182
223. Chamorro-Garcia R, Sahu M, Abbey RJ, Laude J, Pham N, and Blumberg B. Transgenerational inheritance of increased fat depot size, stem cell reprogramming, and hepatic steatosis elicited by prenatal exposure to the obesogen tributyltin in mice. Environ Health Perspect. (2013) 121:359–66. doi: 10.1289/ehp.1205701
224. Janesick A and Blumberg B. Endocrine disrupting chemicals and the developmental programming of adipogenesis and obesity. Birth Defects Res C Embryo Today. (2011) 93:34–50. doi: 10.1002/bdrc.20197
225. King SE, Nilsson E, Beck D, and Skinner MK. Adipocyte epigenetic alterations and potential therapeutic targets in transgenerationally inherited lean and obese phenotypes following ancestral exposures. Adipocyte. (2019) 8:362–78. doi: 10.1080/21623945.2019.1693747
226. Stel J and Legler J. The role of epigenetics in the latent effects of early life exposure to obesogenic endocrine disrupting chemicals. Endocrinology. (2015) 156:3466–72. doi: 10.1210/en.2015-1434
227. Lecoutre S, Kwok KHM, Petrus P, Lambert M, and Breton C. Epigenetic programming of adipose tissue in the progeny of obese dams. Curr Genomics. (2019) 20:428–37. doi: 10.2174/1389202920666191118092852
228. Nunez-Sanchez MA, Jimenez-Mendez A, Suarez-Cortes M, Martinez-Sanchez MA, Sanchez-Solis M, Blanco-Carnero JE, et al. Inherited epigenetic hallmarks of childhood obesity derived from prenatal exposure to obesogens. Int J Environ Res Public Health. (2023) 20(6):4711. doi: 10.3390/ijerph20064711
229. Janesick A and Blumberg B. Obesogens, stem cells and the developmental programming of obesity. Int J Androl. (2012) 35:437–48. doi: 10.1111/j.1365-2605.2012.01247.x
230. Ye X, Wong LY, Kramer J, Zhou X, Jia T, and Calafat AM. Urinary concentrations of bisphenol a and three other bisphenols in convenience samples of U.S. Adults during 2000-2014. Environ Sci Technol. (2015) 49:11834–9. doi: 10.1021/acs.est.5b02135
231. Lehmler HJ, Liu B, Gadogbe M, and Bao W. Exposure to bisphenol a, bisphenol F, and bisphenol S in U.S. Adults and children: the national health and nutrition examination survey 2013-2014. ACS Omega. (2018) 3:6523–32. doi: 10.1021/acsomega.8b00824
232. Chen D, Kannan K, Tan H, Zheng Z, Feng Y-L, Wu Y, et al. Bisphenol analogues other than bpa: environmental occurrence, human exposure, and toxicity-a review. Environ Sci Technol. (2016) 50:5438–53. doi: 10.1021/acs.est.5b05387
233. Zhou X, Kramer JP, Calafat AM, and Ye X. Automated on-Line Column-Switching High Performance Liquid Chromatography Isotope Dilution Tandem Mass Spectrometry Method for the Quantification of Bisphenol a, Bisphenol F, Bisphenol S, and 11 Other Phenols in Urine. J Chromatogr B Analytical Technol Biomed Life Sci. (2014) 944:152–6. doi: 10.1016/j.jchromb.2013.11.009
234. Schaffert A, Krieg L, Weiner J, Schlichting R, Ueberham E, Karkossa I, et al. Alternatives for the worse: molecular insights into adverse effects of bisphenol a and substitutes during human adipocyte differentiation. Environ Int. (2021) 156:106730. doi: 10.1016/j.envint.2021.106730
235. Reina-Pérez I, Olivas-Martínez A, Mustieles V, Ruiz-Ojeda FJ, Molina-Molina JM, Olea N, et al. Bisphenol F and bisphenol S promote lipid accumulation and adipogenesis in human adipose-derived stem cells. Food Chem Toxicol. (2021) 152:112216. doi: 10.1016/j.fct.2021.112216
236. Verbanck M, Canouil M, Leloire A, Dhennin V, Coumoul X, Yengo L, et al. Low-dose exposure to bisphenols a, F and S of human primary adipocyte impacts coding and non-coding rna profiles. PloS One. (2017) 12:e0179583. doi: 10.1371/journal.pone.0179583
237. Wu W, Li M, Liu A, Wu C, Li D, Deng Q, et al. Bisphenol a and the risk of obesity a systematic review with meta-analysis of the epidemiological evidence. Dose Response. (2020) 18:1559325820916949. doi: 10.1177/1559325820916949
238. Kim KY, Lee E, and Kim Y. The association between bisphenol a exposure and obesity in children-a systematic review with meta-analysis. Int J Environ Res Public Health. (2019) 16(14):2521. doi: 10.3390/ijerph16142521
239. Do MT, Chang VC, Mendez MA, and de Groh M. Urinary bisphenol a and obesity in adults: results from the canadian health measures survey. Health Promot Chronic Dis Prev Can. (2017) 37:403–12. doi: 10.24095/hpcdp.37.12.02
240. Carwile JL and Michels KB. Urinary bisphenol a and obesity: nhanes 2003-2006. Environ Res. (2011) 111:825–30. doi: 10.1016/j.envres.2011.05.014
241. Wang T, Li M, Chen B, Xu M, Xu Y, Huang Y, et al. Urinary bisphenol a (Bpa) concentration associates with obesity and insulin resistance. J Clin Endocrinol Metab. (2012) 97:E223–7. doi: 10.1210/jc.2011-1989
242. Xiao T, Huang Z, Zheng C-J, Quach B, Zhu Y, Li F, et al. Associations of bisphenol a exposure with metabolic syndrome and its components: A systematic review and meta-analysis. Obes Rev. (2024) 25(6):e13738. doi: 10.1111/obr.13738
243. Garcia Garcia M, Pico Y, and Morales-Suarez-Varela M. Effects of bisphenol a on the risk of developing obesity. Nutrients. (2024) 16(21):3740. doi: 10.3390/nu16213740
244. LaKind JS, Goodman M, and Mattison DR. Bisphenol a and indicators of obesity, glucose metabolism/type 2 diabetes and cardiovascular disease: A systematic review of epidemiologic research. Crit Rev Toxicol. (2014) 44:121–50. doi: 10.3109/10408444.2013.860075
245. Varghese SV and Hall JM. Bisphenol a substitutes and obesity: A review of the epidemiology and pathophysiology. Front Endocrinol (Lausanne). (2023) 14:1155694. doi: 10.3389/fendo.2023.1155694
246. Alharbi HF, Algonaiman R, Alduwayghiri R, Aljutaily T, Algheshairy RM, Almutairi AS, et al. Exposure to bisphenol a substitutes, bisphenol S and bisphenol F, and its association with developing obesity and diabetes mellitus: A narrative review. Int J Environ Res Public Health. (2022) 19(23):15918. doi: 10.3390/ijerph192315918
247. Menale C, Piccolo MT, Cirillo G, Calogero RA, Papparella A, Mita L, et al. Bisphenol a effects on gene expression in adipocytes from children: association with metabolic disorders. J Mol Endocrinol. (2015) 54:289–303. doi: 10.1530/JME-14-0282
248. Kulsange SE, Sharma M, Sonawane B, Jaiswal MR, Kulkarni MJ, and Santhakumari B. Swath-ms reveals that bisphenol a and its analogs regulate pathways leading to disruption in insulin signaling and fatty acid metabolism. Food Chem Toxicol. (2024) 188:114667. doi: 10.1016/j.fct.2024.114667
249. Chernis N, Masschelin P, Cox AR, and Hartig SM. Bisphenol af promotes inflammation in human white adipocytes. Am J Physiol Cell Physiol. (2020) 318:C63–72. doi: 10.1152/ajpcell.00175.2019
250. Luo L and Liu M. Adipose tissue in control of metabolism. J Endocrinol. (2016) 231:R77–99. doi: 10.1530/JOE-16-0211
251. Garritson JD and Boudina S. The effects of exercise on white and brown adipose tissue cellularity, metabolic activity and remodeling. Front Physiol. (2021) 12:772894. doi: 10.3389/fphys.2021.772894
252. Thompson D, Karpe F, Lafontan M, and Frayn KN. Physical activity and exercise in the regulation of human adipose tissue physiology. Physiol Rev. (2012) 92:157–91. doi: 10.1152/physrev.00012.2011
253. Jia D, Zhang H, Liu T, and Wang R. Exercise alleviates aging of adipose tissue through adipokine regulation. Metabolites. (2024) 14(3):135. doi: 10.3390/metabo14030135
254. Abedpoor N, Taghian F, and Hajibabaie F. Physical activity ameliorates the function of organs via adipose tissue in metabolic diseases. Acta Histochem. (2022) 124:151844. doi: 10.1016/j.acthis.2022.151844
255. Leiria LO and Tseng YH. Lipidomics of brown and white adipose tissue: implications for energy metabolism. Biochim Biophys Acta Mol Cell Biol Lipids. (2020) 1865:158788. doi: 10.1016/j.bbalip.2020.158788
256. Boudina S and Graham TE. Mitochondrial function/dysfunction in white adipose tissue. Exp Physiol. (2014) 99(9):1168–78. doi: 10.1113/expphysiol.2014.081414
257. Shi H, Hao X, Sun Y, Zhao Y, Wang Y, Cao X, et al. Exercise-inducible circulating extracellular vesicle irisin promotes browning and the thermogenic program in white adipose tissue. Acta Physiol. (2024) 240(3):240. doi: 10.1111/apha.v240.3
258. Severinsen MCK, Schéele C, and Pedersen BK. Exercise and browning of white adipose tissue-a translational perspective. Curr Opin Pharmacol. (2020) 52:18–24. doi: 10.1016/j.coph.2020.04.004
259. Flori L, Testai L, and Calderone V. The “Irisin system”: from biological roles to pharmacological and nutraceutical perspectives. Life Sci. (2020) 267:118954. doi: 10.1016/j.lfs.2020.118954
260. Tanimura R, Kobayashi L, Shirai T, and Takemasa T. Effects of exercise intensity on white adipose tissue browning and its regulatory signals in mice. Physiol Rep. (2022) 10:e15205. doi: 10.14814/phy2.15205
261. Ceccarini G, Magno S, Gilio D, Pelosini C, and Santini F. Autoimmunity in lipodystrophy syndromes. Presse Med. (2021) 50:104073. doi: 10.1016/j.lpm.2021.104073
262. Liu S, Cui F, Ning K, Wang Z, Fu P, Wang D, et al. Role of irisin in physiology and pathology. Front Endocrinol (Lausanne). (2022) 13:962968. doi: 10.3389/fendo.2022.962968
263. Di Maio G, Alessio N, Peluso G, Perrotta S, Monda M, and Di Bernardo G. Molecular and physiological effects of browning agents on white adipocytes from bone marrow mesenchymal stromal cells. Int J Mol Sci. (2022) 23(20):12151. doi: 10.3390/ijms232012151
264. Siriwardhana N, Kalupahana NS, Cekanova M, LeMieux MJ, Greer BP, and Moustaid-Moussa N. Modulation of adipose tissue inflammation by bioactive food compounds. J Nutr Biochem. (2013) 24:613–23. doi: 10.1016/j.jnutbio.2012.12.013
265. Nani A, Murtaza B, Sayed Khan A, Khan NA, and Hichami A. Antioxidant and anti-inflammatory potential of polyphenols contained in mediterranean diet in obesity: molecular mechanisms. Molecules. (2021) 26(4):985. doi: 10.3390/molecules26040985
266. Guevara-Cruz M, Hernández-Gómez KG, Condado-Huerta C, González-Salazar LE, Peña-Flores AK, Pichardo-Ontiveros E, et al. Intermittent fasting, calorie restriction, and a ketogenic diet improve mitochondrial function by reducing lipopolysaccharide signaling in monocytes during obesity: A randomized clinical trial. Clin Nutr. (2024) 43:1914–28. doi: 10.1016/j.clnu.2024.06.036
267. Barrea L, Caprio M, Watanabe M, Cammarata G, Feraco A, Muscogiuri G, et al. Could very low-calorie ketogenic diets turn off low grade inflammation in obesity? Emerging evidence. Crit Rev Food Sci Nutr. (2022) 63:8320–36. doi: 10.1080/10408398.2022.2054935
268. Hoevenaars FP, Keijer J, Herreman L, Palm I, Hegeman MA, Swarts HJ, et al. Adipose tissue metabolism and inflammation are differently affected by weight loss in obese mice due to either a high-fat diet restriction or change to a low-fat diet. Genes Nutr. (2014) 9:391. doi: 10.1007/s12263-014-0391-9
269. Osorio-Conles O, Olbeyra R, Moize V, Ibarzabal A, Giro O, Viaplana J, et al. Positive effects of a mediterranean diet supplemented with almonds on female adipose tissue biology in severe obesity. Nutrients. (2022) 14(13):2617. doi: 10.3390/nu14132617
270. Wang Z, Wang D, and Wang Y. Cigarette smoking and adipose tissue: the emerging role in progression of atherosclerosis. Mediators Inflammation. (2017) 2017:3102737. doi: 10.1155/2017/3102737
271. Wu Y, Song P, Zhang W, Liu J, Dai X, Liu Z, et al. Activation of ampkα2 in adipocytes is essential for nicotine-induced insulin resistance in vivo. Nat Med. (2015) 21:373–82. doi: 10.1038/nm.3826
272. Wang L, van Iersel LEJ, Pelgrim CE, Lu J, van Ark I, Leusink-Muis T, et al. Effects of cigarette smoke on adipose and skeletal muscle tissue: in vivoand in vitrostudies. Cells. (2022) 11(18):2893. doi: 10.3390/cells11182893
273. Mukharjee S, Bank S, and Maiti S. Chronic tobacco exposure by smoking develops insulin resistance. Endocrine Metab Immune Disord Drug Targets. (2020) 20(6):869–77. doi: 10.2174/1871530320666200217123901
274. Regnier SM and Sargis RM. Adipocytes under assault: environmental disruption of adipose physiology. Biochim Biophys Acta. (2014) 1842:520–33. doi: 10.1016/j.bbadis.2013.05.028
275. Steiner JL and Lang CH. Alcohol, adipose tissue and lipid dysregulation. Biomolecules. (2017) 7(1):16. doi: 10.3390/biom7010016
276. Keophiphath M, Achard V, Henegar C, Rouault C, Clement K, and Lacasa D. Macrophage-secreted factors promote a profibrotic phenotype in human preadipocytes. Mol Endocrinol. (2009) 23:11–24. doi: 10.1210/me.2008-0183
277. Marcelin G, Silveira ALM, Martins LB, Ferreira AVM, and Clément K. Deciphering the cellular interplays underlying obesity-induced adipose tissue fibrosis. J Clin Invest. (2019) 129(10):4032–40. doi: 10.1172/JCI129192
278. Shen H, Huang X, Zhao Y, Wu D, Xue K, Yao J, et al. The hippo pathway links adipocyte plasticity to adipose tissue fibrosis. Nat Commun. (2022) 13:6030. doi: 10.1038/s41467-022-33800-0
279. Lee M-J. Transforming growth factor beta superfamily regulation of adipose tissue biology in obesity. Biochim Biophys Acta Mol Basis Dis. (2018) 1864–4 Pt A:1160–71. doi: 10.1016/j.bbadis.2018.01.025
280. Li Y and Ding WX. Adipose tissue autophagy and homeostasis in alcohol-induced liver injury. Liver Res. (2017) 1:54–62. doi: 10.1016/j.livres.2017.03.004
281. Hayes AL, Xu F, Babineau DC, and Patel SR. Sleep duration and circulating adipokine levels. Sleep. (2011) 34:147–52. doi: 10.1093/sleep/34.2.147
282. Padilha HG, Crispim CA, Crispim CA, Zimberg IZ, De-Souza DA, Waterhouse J, et al. A link between sleep loss, glucose metabolism and adipokines. Braz J Med Biol Res. (2011) 44:992–9. doi: 10.1590/S0100-879X2011007500113
283. Husse J, Hintze SC, Eichele G, Lehnert H, and Oster H. Circadian clock genes per1 and per2 regulate the response of metabolism-associated transcripts to sleep disruption. PloS One. (2012) 7:e52983. doi: 10.1371/journal.pone.0052983
284. Oliveira RF, Daniele T, Facanha CFS, Forti ACE, Bruin PFC, and Bruin VMS. Adiponectin levels and sleep deprivation in patients with endocrine metabolic disorders. Rev Assoc Med Bras (1992). (2018) 64:1122–8. doi: 10.1590/1806-9282.64.12.1122
285. Shea S, Shea S, Hilton MF, Hilton MF, Orlova C, Ayers RT, et al. Independent circadian and sleep/wake regulation of adipokines and glucose in humans. J Clin Endocrinol Metab. (2005) 90:2537–44. doi: 10.1210/jc.2004-2232
286. Solaroglu I and Zhang JH. The micrornas: small size, big value,…. Surg Neurol Int. (2010) 1:45. doi: 10.4103/2152-7806.68706
287. Felekkis K, Touvana E, Ch S, and Deltas C. Micrornas: A newly described class of encoded molecules that play a role in health and disease. Hippokratia. (2010) 14:236–40.
288. Freedman JE and Tanriverdi K. Defining mirna targets: balancing simplicity with complexity. Circulation. (2013) 127:2075–7. doi: 10.1161/CIRCULATIONAHA.113.003058
289. Guo Y, Zhang X, Huang W, and Miao X. Recent advances of mirnas in adipose tissues. Chin J Biotechnol. (2016) 32:151–63.
290. Hilton C, Neville MJ, and Karpe F. Micrornas in adipose tissue: their role in adipogenesis and obesity. Int J Obes. (2013) 37:325–32. doi: 10.1038/ijo.2012.59
291. Vienberg SG, Geiger J, Madsen SN, and Dalgaard LT. Micrornas in metabolism. Acta Physiol (Oxford England). (2016) 219:346–61. doi: 10.1111/apha.2017.219.issue-2
292. Kuryłowicz A. Micrornas in human adipose tissue physiology and dysfunction. Cells. (2021) 10(12):3342. doi: 10.3390/cells10123342
293. Frost RJA and Olson EN. Control of glucose homeostasis and insulin sensitivity by the let-7 family of micrornas. Proc Natl Acad Sci. (2011) 108:21075–80. doi: 10.1073/pnas.1118922109
294. Brennan EP, Wang B, McClelland AD, Mohan M, Marai M, Beuscart O, et al. Protective effect of let-7 mirna family in regulating inflammation in diabetes-associated atherosclerosis. Diabetes. (2017) 66:2266–77. doi: 10.2337/db16-1405
295. Landrier J-F, Derghal A, and Mounien L. Micrornas in obesity and related metabolic disorders. Cells. (2019) 8(8):859. doi: 10.3390/cells8080859
296. Cruz KJC, de Oliveira ARS, Morais JBS, Severo JS, and Marreiro PD. Role of micrornas on adipogenesis, chronic low-grade inflammation, and insulin resistance in obesity. Nutrition. (2017) 35:28–35. doi: 10.1016/j.nut.2016.10.003
297. Belarbi Y, Mejhert N, Lorente-Cebrián S, Dahlman I, Arner P, Rydén M, et al. Microrna-193b controls adiponectin production in human white adipose tissue. J Clin Endocrinol Metab. (2015) 100:E1084–8. doi: 10.1210/jc.2015-1530
298. Price NL and Fernández-Hernando C. Mirna regulation of white and brown adipose tissue differentiation and function. Biochim Biophys Acta. (2016) 1861–12 Pt B:2104–10. doi: 10.1016/j.bbalip.2016.02.010
299. Meerson A, Traurig MT, Ossowski VM, Fleming J, Mullins M, and Baier LJ. Human adipose microrna-221 is upregulated in obesity and affects fat metabolism downstream of leptin and tnf-α. Diabetologia. (2013) 56:1971–9. doi: 10.1007/s00125-013-2950-9
300. Heneghan HM, Miller N, Mcanena OJ, O’Brien T, and Kerin MJ. Differential mirna expression in omental adipose tissue and in the circulation of obese patients identifies novel metabolic biomarkers. J Clin Endocrinol Metab. (2011) 96:E846–50. doi: 10.1210/jc.2010-2701
301. Iacomino G and Siani A. Role of micrornas in obesity and obesity-related diseases. Genes Nutr. (2017) 12:23. doi: 10.1186/s12263-017-0577-z
302. Heyn GS, Corrêa LH, and Magalhães KG. The impact of adipose tissue–derived mirnas in metabolic syndrome, obesity, and cancer. Front Endocrinol. (2020) 11. doi: 10.3389/fendo.2020.563816
303. Lorente-Cebrián S, González-Muniesa P, Milagro FI, and Martínez JA. Micrornas and other non-coding rnas in adipose tissue and obesity: emerging roles as biomarkers and therapeutic targets. Clin Sci. (2019) 133:23–40. doi: 10.1042/CS20180890
304. Chuang TY, Wu H-L, Chen C-C, Gamboa GM, Layman LC, Diamond MP, et al. Microrna-223 expression is upregulated in insulin resistant human adipose tissue. J Diabetes Res. (2015) 2015:943659. doi: 10.1155/2015/943659
305. Zhuang G, Meng C, Guo X, Cheruku PS, Shi L, Xu H, et al. A novel regulator of macrophage activation: mir-223 in obesity-associated adipose tissue inflammation. Circulation. (2012) 125:2892–903. doi: 10.1161/CIRCULATIONAHA.111.087817
306. Deiuliis JA, Syed R, Duggineni D, Rutsky J, Rengasamy P, Zhang J, et al. Visceral adipose microrna 223 is upregulated in human and murine obesity and modulates the inflammatory phenotype of macrophages. PloS One. (2016) 11(11):e0165962. doi: 10.1371/journal.pone.0165962
307. Williams MD and Mitchell GM. Micrornas in insulin resistance and obesity. Exp Diabetes Res. (2012) 2012:484696. doi: 10.1155/2012/484696
308. Li H, Xue M, Xu J, and Qin X. Mir-301a is involved in adipocyte dysfunction during obesity-related inflammation via suppression of pparγ. Die Pharmazie. (2016) 71:84–8.
309. Kim C, Lee H, Cho YM, Kwon OJ, Kim W, and Lee EK. Tnfα-induced mir-130 resulted in adipocyte dysfunction during obesity-related inflammation. FEBS Lett. (2013) 587(23):3853–8. doi: 10.1016/j.febslet.2013.10.018
310. Zhang M, Zhou Z-Q, Wang J, and Li S-F. Mir-130b promotes obesity associated adipose tissue inflammation and insulin resistance in diabetes mice through alleviating M2 macrophage polarization via repression of ppar-Γ. Immunol Lett. (2016) 180:1–8. doi: 10.1016/j.imlet.2016.10.004
311. Tollervey JR and Lunyak VV. Epigenetics: judge, jury and executioner of stem cell fate. Epigenetics. (2012) 7:823–40. doi: 10.4161/epi.21141
312. Ryall JG, Cliff T, Dalton S, and Sartorelli V. Metabolic reprogramming of stem cell epigenetics. Cell Stem Cell. (2015) 17:651–62. doi: 10.1016/j.stem.2015.11.012
313. Mortada I and Mortada R. Epigenetic changes in mesenchymal stem cells differentiation. Eur J Med Genet. (2017) 61:114–8. doi: 10.1016/j.ejmg.2017.10.015
314. Barilla S, Treuter E, and Venteclef N. Transcriptional and epigenetic control of adipocyte remodeling during obesity. Obesity. (2021) 29:2013–25. doi: 10.1002/oby.v29.12
315. Ma X and Kang S. Functional implications of DNA methylation in adipose biology. Diabetes. (2019) 68:871–8. doi: 10.2337/dbi18-0057
316. Thomas EC, Hook SC, Gray A, Chadt A, Carling D, Al-Hasani H, et al. Isoform-specific ampk association with tbc1d1 is reduced by a mutation associated with severe obesity. Biochem J. (2018) 475:2969–83. doi: 10.1042/BCJ20180475
317. Bauernfeind F, Rieger A, Schildberg FA, Knolle PA, Schmid-Burgk JL, and Hornung V. Nlrp3 inflammasome activity is negatively controlled by mir-223. J Immunol. (2012) 189:4175–81. doi: 10.4049/jimmunol.1201516
318. Lee EK, Lee MJ, Abdelmohsen K, Kim W, Kim MM, Srikantan S, et al. Mir-130 suppresses adipogenesis by inhibiting peroxisome proliferator-activated receptor Γ Expression. Mol Cell Biol. (2010) 31:626–38. doi: 10.1128/MCB.00894-10
319. Mota de Sá P, Richard AJ, Hang H, and Stephens JM. Transcriptional regulation of adipogenesis. Compr Physiol. (2017) 7:635–74. doi: 10.1002/j.2040-4603.2017.tb00753.x
320. Guo L, Li X, and Tang Q-Q. Transcriptional regulation of adipocyte differentiation: A central role for ccaat/enhancer-binding protein (C/ebp) β. J Biol Chem. (2014) 290:755–61. doi: 10.1074/jbc.R114.619957
321. Lee J-E, Schmidt H, Lai B, and Ge K. Transcriptional and epigenomic regulation of adipogenesis. Mol Cell Biol. (2019) 39(11):e00601-18. doi: 10.1128/MCB.00601-18
322. Ambele MA, Dhanraj P, Giles R, and Pepper MS. Adipogenesis: A complex interplay of multiple molecular determinants and pathways. Int J Mol Sci. (2020) 21(12):4283. doi: 10.3390/ijms21124283
323. Kim H-Y, Jang H-J, Muthamil S, Shin UC, Lyu J-H, Kim S-W, et al. Novel insights into regulators and functional modulators of adipogenesis. Biomed Pharmacother. (2024) 177:117073. doi: 10.1016/j.biopha.2024.117073
324. Musale V, Wasserman DH, and Kang L. Extracellular matrix remodelling in obesity and metabolic disorders. Life Metab. (2023) 2(4):load021. doi: 10.1093/lifemeta/load021
325. Sun K, Li X, and Scherer PE. Extracellular matrix (Ecm) and fibrosis in adipose tissue: overview and perspectives. Compr Physiol. (2023) 13:4387–407. doi: 10.1002/j.2040-4603.2023.tb00254.x
326. Pasarica M, Gowronska-Kozak B, Burk D, Remedios I, Hymel D, Gimble J, et al. Adipose tissue collagen vi in obesity. J Clin Endocrinol Metab. (2009) 94:5155–62. doi: 10.1210/jc.2009-0947
327. Lecoutre S, Lambert M, Drygalski K, Dugail I, Maqdasy S, Hautefeuille M, et al. Importance of the microenvironment and mechanosensing in adipose tissue biology. Cells. (2022) 11(15):2310. doi: 10.3390/cells11152310
328. Ruiz-Ojeda FJ, Mendez-Gutierrez A, Aguilera CM, and Plaza-Díaz J. Extracellular matrix remodeling of adipose tissue in obesity and metabolic diseases. Int J Mol Sci. (2019) 20(19):4888. doi: 10.3390/ijms20194888
329. Di Caprio N and Bellas E. Collagen stiffness and architecture regulate fibrotic gene expression in engineered adipose tissue. Adv Biosyst. (2020) 4(6):e1900286. doi: 10.1002/adbi.201900286
330. Kralisch S, Lossner U, Bluher M, Paschke R, Stumvoll M, and Fasshauer M. Tissue inhibitor of metalloproteinase 1 expression and secretion are induced by beta-adrenergic stimulation in 3t3-L1 adipocytes. J Endocrinol. (2006) 189:665–70. doi: 10.1677/joe.1.06645
331. Misra P. Amp activated protein kinase: A next generation target for total metabolic control. Expert Opin Ther Targets. (2008) 12:100–91. doi: 10.1517/14728222.12.1.91
332. Luo T, Nocon A, Fry JL, Sherban A, Rui X, Jiang B, et al. Ampk activation by metformin suppresses abnormal extracellular matrix remodeling in adipose tissue and ameliorates insulin resistance in obesity. Diabetes. (2016) 65:2295–310. doi: 10.2337/db15-1122
333. Bijland S, Mancini SJ, and Salt IP. Role of amp-activated protein kinase in adipose tissue metabolism and inflammation. Clin Sci. (2013) 124:491–507. doi: 10.1042/CS20120536
334. Malekpour-Dehkordi Z, Teimourian S, Nourbakhsh M, Naghiaee Y, Sharifi R, and Mohiti-Ardakani J. Metformin reduces fibrosis factors in insulin resistant and hypertrophied adipocyte via integrin/erk, collagen vi, apoptosis, and necrosis reduction. Life Sci. (2019) 233:116682. doi: 10.1016/j.lfs.2019.116682
335. Clemente-Postigo M, Tinahones A, El Bekay R, Malagón MM, and Tinahones FJ. The role of autophagy in white adipose tissue function: implications for metabolic health. Metabolites. (2020) 10(5):179. doi: 10.3390/metabo10050179
336. Ferhat M, Funai K, and Boudina S. Autophagy in adipose tissue physiology and pathophysiology. Antioxid Redox Signaling. (2019) 31(6):487–501. doi: 10.1089/ars.2018.7626
337. Altshuler-Keylin S and Kajimura S. Mitochondrial homeostasis in adipose tissue remodeling. Sci Signaling. (2017) 10(468):eaai9248. doi: 10.1126/scisignal.aai9248
338. Zhang Y, Goldman SJ, Baerga R, Zhao Y, Komatsu M, and Jin S. Adipose-specific deletion of autophagy-related gene 7 (Atg7) in mice reveals a role in adipogenesis. Proc Natl Acad Sci. (2009) 106:19860–5. doi: 10.1073/pnas.0906048106
339. Goldman SJ, Zhang Y, and Jin S. Autophagy and adipogenesis: implications in obesity and type ii diabetes. Autophagy. (2010) 6:179–81-5. doi: 10.4161/auto.6.1.10814
340. Ro S-H, Jang Y, Bae J, Kim IM, Schaecher C, and Shomo ZD. Autophagy in adipocyte browning: emerging drug target for intervention in obesity. Front Physiol. (2019) 10. doi: 10.3389/fphys.2019.00022
341. Zhang Y, Zeng X, and Jin S. Autophagy in adipose tissue biology. Pharmacol Res. (2012) 66:505–12. doi: 10.1016/j.phrs.2012.09.004
342. Morais JBS, Dias TMS, Cardoso BEP, de Paiva Sousa M, Sousa TGV, Araújo DSC, et al. Adipose tissue dysfunction: impact on metabolic changes? Hormone Metab Res. (2022) 54:785–94. doi: 10.1055/a-1922-7052
343. Gan T, Qu S, Zhang H, and Zhou X-J. Modulation of the immunity and inflammation by autophagy. MedComm. (2023) 4(4):e311. doi: 10.1002/mco2.v4.4
344. Friuli M, Sepe C, Panza E, Travelli C, Paterniti I, and Romano A. Autophagy and inflammation an intricate affair in the management of obesity and metabolic disorders: evidence for novel pharmacological strategies? Front Pharmacol. (2024) 15. doi: 10.3389/fphar.2024.1407336
345. Pang Y, Wu L, Tang C, Wang H, and Wei Y. Autophagy-inflammation interplay during infection: balancing pathogen clearance and host inflammation. Front Pharmacol. (2022) 13. doi: 10.3389/fphar.2022.832750
346. Gong L, Pan Q, and Yang N. Autophagy and inflammation regulation in acute kidney injury. Front Physiol. (2020) 11. doi: 10.3389/fphys.2020.576463
347. Huang-Doran I, Zhang C-Y, and Vidal-Puig A. Extracellular vesicles: novel mediators of cell communication in metabolic disease. Trends Endocrinol Metab. (2017) 28:3–18. doi: 10.1016/j.tem.2016.10.003
348. Akbar N, Azzimato V, Choudhury RP, and Aouadi M. Extracellular vesicles in metabolic disease. Diabetologia. (2019) 62:2179–87. doi: 10.1007/s00125-019-05014-5
349. Camino T, Lago-Baameiro N, Martís-Sueiro A, Couto I, Santos FF, Baltar J, et al. Deciphering adipose tissue extracellular vesicles protein cargo and its role in obesity. Int J Mol Sci. (2020) 21(24):9366. doi: 10.3390/ijms21249366
350. Sandoval-Bórquez A, Carrión P, Hernández MP, Pérez JA, Tapia-Castillo A, Vecchiola A, et al. Adipose tissue dysfunction and the role of adipocyte-derived extracellular vesicles in obesity and metabolic syndrome. J Endocrine Soc. (2024) 8(8):bvae126. doi: 10.1210/jendso/bvae126
351. Bond ST, Calkin AC, and Drew BG. Adipose-derived extracellular vesicles: systemic messengers and metabolic regulators in health and disease. Front Physiol. (2022) 13. doi: 10.3389/fphys.2022.837001
352. Kim Y and Kim O-K. Potential roles of adipocyte extracellular vesicle–derived mirnas in obesity-mediated insulin resistance. Adv Nutr. (2020) 12:566–74. doi: 10.1093/advances/nmaa105
353. Mori MA, Ludwig RG, Garcia-Martin R, Brandao BBC, and Kahn R. Extracellular mirnas: from biomarkers to mediators of physiology and disease. Cell Metab. (2019) 30(4):656–73. doi: 10.1016/j.cmet.2019.07.011
354. Le Lay S, Rome S, Loyer X, and Nieto L. Adipocyte-derived extracellular vesicles in health and diseases: nano-packages with vast biological properties. FASEB BioAdvances. (2021) 3:407–19. doi: 10.1096/fba.2020-00147
355. Li C, Fang Q, Liu M-L, and Lin J-N. Current understanding of the role of adipose-derived extracellular vesicles in metabolic homeostasis and diseases: communication from the distance between cells/tissues. Theranostics. (2020) 10:7422–35. doi: 10.7150/thno.42167
356. Yadav SK and Kaur AK. Unlocking the potential of extracellular vesicles for therapeutic and diagnostic application. Int J Pharma Professional’s Res (IJPPR). (2024) 21(4):513–27. doi: 10.69580/IJPPR.15.2.2024.32-51
357. Qian F, Huang Z, Zhong H, Lei Q, Ai Y, Xie Z, et al. Analysis and biomedical applications of functional cargo in extracellular vesicles. ACS Nano. (2022) 16:19980–20001. doi: 10.1021/acsnano.2c11298
358. Lawson C, Vicencio JM, Yellon DM, and Davidson SM. Microvesicles and exosomes: new players in metabolic and cardiovascular disease. J Endocrinol. (2016) 228:R57–71. doi: 10.1530/JOE-15-0201
359. Ahima RS and Osei SY. Adipokines in obesity. Front Hormone Res. (2008) 36:182–97. doi: 10.1159/000115365
360. Stofkova A. Resistin and visfatin: regulators of insulin sensitivity, inflammation and immunity. Endocrine Regulations. (2010) 44:25–36. doi: 10.4149/endo_2010_01_25
361. Booth AD, Magnuson AM, Fouts JK, and Foster MT. Adipose tissue: an endocrine organ playing a role in metabolic regulation. Hormone Mol Biol Clin Invest. (2016) 26:25–42. doi: 10.1515/hmbci-2015-0073
362. Arner P. Insulin resistance in type 2 diabetes – role of the adipokines. Curr Mol Med. (2005) 5:333–9. doi: 10.2174/1566524053766022
363. Maury E and Brichard SM. Adipokine dysregulation, adipose tissue inflammation and metabolic syndrome. Mol Cell Endocrinol. (2010) 314:1–16. doi: 10.1016/j.mce.2009.07.031
364. Al-Sulaiti H, Dömling A, and Elrayess MA. Mediators of impaired adipogenesis in obesity-associated insulin resistance and T2dm. Adipose Tissue-An Update. (2019). doi: 10.5772/intechopen.88746
365. Goossens GH. The role of adipose tissue dysfunction in the pathogenesis of obesity-related insulin resistance. Physiol Behav. (2008) 94:206–18. doi: 10.1016/j.physbeh.2007.10.010
366. Smith U and Kahn BB. Adipose tissue regulates insulin sensitivity: role of adipogenesis, de novolipogenesis and novel lipids. J Internal Med. (2016) 280:465–75. doi: 10.1111/joim.2016.280.issue-5
367. Hammarstedt A, Gogg S, Hedjazifar S, Nerstedt A, and Smith U. Impaired adipogenesis and dysfunctional adipose tissue in human hypertrophic obesity. Physiol Rev. (2018) 98:1911–41. doi: 10.1152/physrev.00034.2017
368. Gustafson B and Smith U. Regulation of white adipogenesis and its relation to ectopic fat accumulation and cardiovascular risk. Atherosclerosis. (2015) 241:27–35. doi: 10.1016/j.atherosclerosis.2015.04.812
369. Lafontan M. Adipose tissue and adipocyte dysregulation. Diabetes Metab. (2014) 40:16–28. doi: 10.1016/j.diabet.2013.08.002
370. Nunn ER, Shinde AB, and Zaganjor E. Weighing in on adipogenesis. Front Physiol. (2022) 13. doi: 10.3389/fphys.2022.821278
371. Sakaguchi M, Fujisaka S, Cai W, Winnay JN, Konishi M, O’Neill BT, et al. Adipocyte dynamics and reversible metabolic syndrome in mice with an inducible adipocyte-specific deletion of the insulin receptor. Cell Metab. (2017) 25:448–62. doi: 10.1016/j.cmet.2016.12.008
372. De Fano M, Bartolini D, Tortoioli C, Vermigli C, Malara M, Galli F, et al. Adipose tissue plasticity in response to pathophysiological cues: A connecting link between obesity and its associated comorbidities. Int J Mol Sci. (2022) 23(10):5511. doi: 10.3390/ijms23105511
373. Ahmad R, Al-Mass A, Atizado V, Al-Hubail A, Al-Ghimlas F, Al-Arouj M, et al. Elevated expression of the toll like receptors 2 and 4 in obese individuals: its significance for obesity-induced inflammation. J Inflammation (Lond). (2012) 9:48. doi: 10.1186/1476-9255-9-48
374. Suganami T, Tanimoto-Koyama K, Nishida J, Itoh M, Yuan X, Mizuarai S, et al. Role of the toll-like receptor 4/nf-&Kgr;B pathway in saturated fatty acid–induced inflammatory changes in the interaction between adipocytes and macrophages. Arteriosclerosis Thrombosis Vasc Biol. (2007) 27:84–91. doi: 10.1161/01.ATV.0000251608.09329.9a
375. Birkenfeld AL and Shulman GI. Nonalcoholic fatty liver disease, hepatic insulin resistance, and type 2 diabetes. Hepatology. (2014) 59:713–23. doi: 10.1002/hep.26672
376. Park SS and Seo YK. Excess accumulation of lipid impairs insulin sensitivity in skeletal muscle. Int J Mol Sci. (2020) 21(6):1949. doi: 10.3390/ijms21061949
377. Mahfouz R, Khoury R, Blachnio-Zabielska A, Turban S, Loiseau N, Lipina C, et al. Characterising the inhibitory actions of ceramide upon insulin signaling in different skeletal muscle cell models: A mechanistic insight. PloS One. (2014) 9:e101865. doi: 10.1371/journal.pone.0101865
378. Stefanyk LE and Dyck DJ. The interaction between adipokines, diet and exercise on muscle insulin sensitivity. Curr Opin Clin Nutr Metab Care. (2010) 13:255–9. doi: 10.1097/MCO.0b013e328338236e
379. Sun C, Mao S, Chen S, Zhang W, and Liu C. Ppars-orchestrated metabolic homeostasis in the adipose tissue. Int J Mol Sci. (2021) 22(16):8974. doi: 10.3390/ijms22168974
380. Eeckhoute J, Oger F, Staels B, and Lefebvre P. Coordinated regulation of pparγ Expression and activity through control of chromatin structure in adipogenesis and obesity. PPAR Res. (2012) 2012:164140. doi: 10.1155/2012/164140
381. Brunmeir R and Xu F. Functional regulation of ppars through post-translational modifications. Int J Mol Sci. (2018) 19(6):1738. doi: 10.3390/ijms19061738
382. Corrales P, Vidal-Puig A, and Medina-Gómez G. Ppars and metabolic disorders associated with challenged adipose tissue plasticity. Int J Mol Sci. (2018) 19(7):19. doi: 10.3390/ijms19072124
383. Desvergne B, Michalik L, and Wahli W. Be fit or be sick: peroxisome proliferator-activated receptors are down the road. Mol Endocrinol. (2004) 18:1321–32. doi: 10.1210/me.2004-0088
384. Weiss LM. Faculty opinions recommendation of ppars and metabolic disorders associated with challenged adipose tissue plasticity. Faculty Opin – Post-Publication Peer Rev Biomed Literature. (2019) 19(7):2124. doi: 10.3390/ijms19072124
385. Glass CK and Olefsky JM. Inflammation and lipid signaling in the etiology of insulin resistance. Cell Metab. (2012) 15:635–45. doi: 10.1016/j.cmet.2012.04.001
386. Ruan H and Pownall HJ. The adipocyte ikk/nfkappab pathway: A therapeutic target for insulin resistance. Curr Opin Investigational Drugs. (2009) 10:346–52.
387. Hill J, Solt CM, and Foster MT. Obesity associated disease risk: the role of inherent differences and location of adipose depots. Hormone Mol Biol Clin Invest. (2018) 33(2):/j/hmbci.2018.33.issue-2/hmbci-2018-0012/hmbci-2018-0012.xml. doi: 10.1515/hmbci-2018-0012
388. Schleinitz D, Krause K, Wohland T, Gebhardt C, Linder N, Stumvoll M, et al. Identification of distinct transcriptome signatures of human adipose tissue from fifteen depots. Eur J Hum Genet. (2020) 28:1714–25. doi: 10.1038/s41431-020-0681-1
389. Kwok KHM, Lam KSL, and Xu A. Heterogeneity of white adipose tissue: molecular basis and clinical implications. Exp Mol Med. (2016) 48(3):e215. doi: 10.1038/emm.2016.5
390. Schöttl T, Fischer IP, and Ussar S. Heterogeneity of adipose tissue in development and metabolic function. J Exp Biol. (2018) 221(Pt Suppl 1):jeb162958. doi: 10.1242/jeb.162958
391. Roca-Rivada A, Alonso J, Al-Massadi O, Castelao C, Peinado JR, Seoane LM, et al. Secretome analysis of rat adipose tissues shows location-specific roles for each depot type. J Proteomics. (2011) 74:1068–79. doi: 10.1016/j.jprot.2011.03.010
392. Hildebrandt X, Ibrahim M, and Peltzer N. Cell death and inflammation during obesity: “Know my methods, wat(Son). Cell Death Differentiation. (2022) 30:279–92. doi: 10.1038/s41418-022-01062-4
393. An P, Wei LL, Zhao S, Sverdlov DY, Vaid KA, Miyamoto M, et al. Hepatocyte mitochondria-derived danger signals directly activate hepatic stellate cells and drive progression of liver fibrosis. Nat Commun. (2020) 11:2362. doi: 10.1038/s41467-020-16092-0
394. Yuzefovych LV, Pastukh VM, and Rachek LI. Mitochondrial DNA damps induce inflammation and insulin resistance. Diabetes. (2018) 67(suppl_1):1783 P. doi: 10.2337/db18-1783-P
395. Roszczyc-Owsiejczuk K and Zabielski P. Sphingolipids as a culprit of mitochondrial dysfunction in insulin resistance and type 2 diabetes. Front Endocrinol (Lausanne). (2021) 12:635175. doi: 10.3389/fendo.2021.635175
396. Kim JK, Fillmore JJ, Chen Y, Yu C, Moore IK, Pypaert M, et al. Tissue-specific overexpression of lipoprotein lipase causes tissue-specific insulin resistance. Proc Natl Acad Sci U.S.A. (2001) 98:7522–7. doi: 10.1073/pnas.121164498
397. Wallach D, Kang T-B, Dillon CP, and Green DR. Programmed necrosis in inflammation: toward identification of the effector molecules. Science. (2016) 352(6281):aaf2154. doi: 10.1126/science.aaf2154
398. Strissel KJ, Stancheva ZS, Miyoshi H, Perfield JW, Defuria J, Jick Z, et al. Adipocyte death, adipose tissue remodeling, and obesity complications. Diabetes. (2007) 56:2910–8. doi: 10.2337/db07-0767
399. Lindhorst A, Raulien N, Wieghofer P, Eilers J, Rossi FMV, Bechmann I, et al. Adipocyte death triggers a pro-inflammatory response and induces metabolic activation of resident macrophages. Cell Death Dis. (2021) 12(6):579. doi: 10.1038/s41419-021-03872-9
400. Makki K, Froguel P, and Wolowczuk I. Adipose tissue in obesity-related inflammation and insulin resistance: cells, cytokines, and chemokines. ISRN Inflammation. (2013) 2013:139239. doi: 10.1155/2013/139239
401. Ou M-Y, Zhang H, P-c T, Zhou S, and Li Q-F. Adipose tissue aging: mechanisms and therapeutic implications. Cell Death Dis. (2022) 13(4):300. doi: 10.1038/s41419-022-04752-6
402. Liu Z, Wu KK-L, Jiang X, Xu A, and Cheng KK-Y. The role of adipose tissue senescence in obesity- and ageing-related metabolic disorders. Clin Sci. (2020) 134:315–30. doi: 10.1042/CS20190966
403. Stout MB, Justice JN, Nicklas BJ, and Kirkland JL. Physiological aging: links among adipose tissue dysfunction, diabetes, and frailty. Physiology. (2017) 32:9–19. doi: 10.1152/physiol.00012.2016
404. de Lange P, Lombardi A, Silvestri E, Cioffi F, Giacco A, Iervolino S, et al. Physiological approaches targeting cellular and mitochondrial pathways underlying adipose organ senescence. Int J Mol Sci. (2023) 24(14):11676. doi: 10.3390/ijms241411676
405. Tchkonia T, Morbeck DE, von Zglinicki T, van Deursen JM, Lustgarten J, Scrable HJ, et al. Fat tissue, aging, and cellular senescence. Aging Cell. (2010) 9:667–84. doi: 10.1111/j.1474-9726.2010.00608.x
406. Mancuso P and Bouchard B. The impact of aging on adipose function and adipokine synthesis. Front Endocrinol. (2019) 10. doi: 10.3389/fendo.2019.00137
407. Cai Z and He B. Adipose tissue aging: an update on mechanisms and therapeutic strategies. Metabol: Clin Exp. (2022) 138:155328. doi: 10.1016/j.metabol.2022.155328
408. Chen J-L, Lou R, Zhou F, Li D, Peng C, and Lin L. Sirtuins: key players in obesity-associated adipose tissue remodeling. Front Immunol. (2022) 13. doi: 10.3389/fimmu.2022.1068986
409. Lomb DJ, Laurent G, and Haigis MC. Sirtuins regulate key aspects of lipid metabolism. Biochim Biophys Acta. (2010) 1804:1652–7. doi: 10.1016/j.bbapap.2009.11.021
410. Yamaguchi S and Yoshino J. Adipose tissue nad+ Biology in obesity and insulin resistance: from mechanism to therapy. BioEssays: News Rev Mol Cell Dev Biol. (2017) 39(5):10.1002/bies.201600227. doi: 10.1002/bies.201600227
411. Jokinen R, Pirnes-Karhu S, Pietiläinen KH, and Pirinen E. Adipose tissue nad+-homeostasis, sirtuins and poly(Adp-ribose) polymerases -important players in mitochondrial metabolism and metabolic health. Redox Biol. (2017) 12:246–63. doi: 10.1016/j.redox.2017.02.011
412. Palmer AK and Kirkland JL. Aging and adipose tissue: potential interventions for diabetes and regenerative medicine. Exp Gerontol. (2016) 86:97–105. doi: 10.1016/j.exger.2016.02.013
413. Conte M, Martucci M, Sandri M, Franceschi C, and Salvioli S. The dual role of the pervasive “Fattish” Tissue remodeling with age. Front Endocrinol. (2019) 10. doi: 10.3389/fendo.2019.00114
414. Karagiannides I, Tchkonia T, Dobson D, Steppan CM, Cummins P, Chan GHH, et al. Altered expression of C/ebp family members results in decreased adipogenesis with aging. Am J Physiol Regulatory Integr Comp Physiol. (2001) 280:R1772–80. doi: 10.1152/ajpregu.2001.280.6.R1772
415. Karagiannides I, Thomou T, Tchkonia T, Pirtskhalava T, Kypreos KE, Cartwright A, et al. Increased cug triplet repeat-binding protein-1 predisposes to impaired adipogenesis with aging. J Biol Chem. (2006) 281:23025–33. doi: 10.1074/jbc.M513187200
416. Fei J, Tamski H, Cook CR, and Santanam N. Microrna regulation of adipose derived stem cells in aging rats. PloS One. (2013) 8(3):e59238. doi: 10.1371/journal.pone.0059238
417. Findeisen HM, Pearson KJ, Gizard F, Zhao Y-Y, Qing H, Jones KL, et al. Oxidative stress accumulates in adipose tissue during aging and inhibits adipogenesis. PloS One. (2011) 6(4):e18532. doi: 10.1371/journal.pone.0018532
418. Lopes-Paciencia S, Saint-Germain E, Rowell M-C, Ruiz AF, Kalegari P, and Ferbeyre G. The senescence-associated secretory phenotype and its regulation. Cytokine. (2019) 117:15–22. doi: 10.1016/j.cyto.2019.01.013
419. Salminen A, Kauppinen A, and Kaarniranta K. Emerging role of nf-κb signaling in the induction of senescence-associated secretory phenotype (Sasp). Cell Signalling. (2012) 24:835–45. doi: 10.1016/j.cellsig.2011.12.006
420. Xu M, Tchkonia T, and Kirkland JL. Perspective: targeting the jak/stat pathway to fight age-related dysfunction. Pharmacol Res. (2016) 111:152–4. doi: 10.1016/j.phrs.2016.05.015
421. Han X, Lei Q, Xie J, Liu H, Sun H, Jing L, et al. Potential regulators of the senescence-associated secretory phenotype during senescence and ageing. J Gerontol Ser A Biol Sci Med Sci. (2022) 77(11):2207–18. doi: 10.1093/gerona/glac097
422. Xu M, Tchkonia T, Ding H, Ogrodnik M, Lubbers ER, Pirtskhalava T, et al. Jak inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc Natl Acad Sci. (2015) 112:E6301–E10. doi: 10.1073/pnas.1515386112
423. Cuollo L, Antonangeli F, Santoni A, and Soriani A. The senescence-associated secretory phenotype (Sasp) in the challenging future of cancer therapy and age-related diseases. Biology. (2020) 9(12):485. doi: 10.3390/biology9120485
424. Zhang YX, Ou M-Y, Yang Z, Sun Y, Li Q-F, and Zhou S. Adipose tissue aging is regulated by an altered immune system. Front Immunol. (2023) 14. doi: 10.3389/fimmu.2023.1125395
425. Trim WV, Turner JE, and Thompson D. Parallels in immunometabolic adipose tissue dysfunction with ageing and obesity. Front Immunol. (2018) 9. doi: 10.3389/fimmu.2018.00169
426. Armutcu F and Ozen OA. Inter-organ crosstalk and the effect on the aging process in obesity. Curr Aging Sci. (2023) 16(2):97–111. doi: 10.2174/1874609816666230223110458
427. Reyes-Farias M, Fos-Domènech J, Serra D, Herrero L, and Sánchez-Infantes D. White adipose tissue dysfunction in obesity and aging. Biochem Pharmacol. (2021) 192:114723. doi: 10.1016/j.bcp.2021.114723
428. Chen KY, Brychta RJ, Abdul Sater Z, Cassimatis TM, Cero C, Fletcher LA, et al. Opportunities and challenges in the therapeutic activation of human energy expenditure and thermogenesis to manage obesity. J Biol Chem. (2019) 295:1926–42. doi: 10.1074/jbc.REV119.007363
429. McMillan AC and White MD. Induction of thermogenesis in brown and beige adipose tissues: molecular markers, mild cold exposure and novel therapies. Curr Opin Endocrinol Diabetes Obes. (2015) 22:347–52. doi: 10.1097/MED.0000000000000191
430. Cheng Y, Liang S, Zhang S, and Hui X. Thermogenic fat as a new obesity management tool: from pharmaceutical reagents to cell therapies. Biomedicines. (2024) 12(7):1474. doi: 10.3390/biomedicines12071474
431. Kralisch S, Bluher M, Paschke R, Stumvoll M, and Fasshauer M. Adipokines and adipocyte targets in the future management of obesity and the metabolic syndrome. Mini Rev Medicinal Chem. (2007) 7:39–45. doi: 10.2174/138955707779317821
432. Blüher M. Adipokines – removing road blocks to obesity and diabetes therapy. Mol Metab. (2014) 3:230–40. doi: 10.1016/j.molmet.2014.01.005
433. Padmalayam I and Suto MJ. Role of adiponectin in the metabolic syndrome: current perspectives on its modulation as a treatment strategy. Curr Pharm Design. (2013) 19:5755–63. doi: 10.2174/13816128113199990360
434. Tsagkaraki E, Nicoloro SM, DeSouza T, Solivan-Rivera J, Desai A, Lifshitz LM, et al. Crispr-enhanced human adipocyte browning as cell therapy for metabolic disease. Nat Commun. (2021) 12(1):6931. doi: 10.1038/s41467-021-27190-y
435. Chung JY, Ain Q, Song Y, Yong SB, and Kim Y-H. Targeted delivery of crispr interference system against fabp4 to white adipocytes ameliorates obesity, inflammation, hepatic steatosis, and insulin resistance. Genome Res. (2019) 29:1442–52. doi: 10.1101/gr.246900.118
436. Valanti E-K, Dalakoura-Karagkouni K, Siasos G, Kardassis D, Eliopoulos AG, and Sanoudou D. Advances in biological therapies for dyslipidemias and atherosclerosis. Metabol: Clin Exp. (2020) 116:154461. doi: 10.1016/j.metabol.2020.154461
437. De Giorgi M and Lagor WR. Gene delivery in lipid research and therapies. Methodist DeBakey Cardiovasc J. (2019) 15:62–9. doi: 10.14797/mdcj-15-1-62
438. Kulkarni JA, Cullis PR, and van der Meel R. Lipid nanoparticles enabling gene therapies: from concepts to clinical utility. Nucleic Acid Ther. (2018) 28:146–57. doi: 10.1089/nat.2018.0721
439. King AJ, Judd AS, and Souers AJ. Inhibitors of diacylglycerol acyltransferase: A review of 2008 patents. Expert Opin Ther Patents. (2010) 20:19–29. doi: 10.1517/13543770903499305
440. Forman BM, Chen J, and Evans RM. Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids are ligands for peroxisome proliferator-activated receptors α and Δ. Proc Natl Acad Sci United States America. (1997) 94:4312–7. doi: 10.1073/pnas.94.9.4312
441. Barroso E, Rodríguez-Calvo R, Serrano-Marco L, Astudillo AM, Balsinde J, Palomer X, et al. editors. The ppar/activator gw 501516 prevents the down-regulation of ampk caused by a high-fat diet in liver and amplifies the pgc-1-lipin 1-ppar pathway leading to increased fatty acid oxidation. Endocrinology. (2011) 152(5):1848–59. doi: 10.1210/en.2010-1468
442. Zhang J, Liu Y, Chen Y, Yuan L, Liu H, Wang J, et al. Adipose-derived stem cells: current applications and future directions in the regeneration of multiple tissues. Stem Cells Int. (2020) 2020:8810813. doi: 10.1155/2020/8810813
443. Gimble JM and Guilak F. Adipose-derived adult stem cells: isolation, characterization, and differentiation potential. Cytotherapy. (2003) 5:362–9. doi: 10.1080/14653240310003026
444. Ceccarelli S, Pontecorvi P, Anastasiadou E, Napoli C, and Marchese C. Immunomodulatory effect of adipose-derived stem cells: the cutting edge of clinical application. Front Cell Dev Biol. (2020) 8. doi: 10.3389/fcell.2020.00236
445. Foti R, Storti G, Palmesano M, Scioli MG, Fiorelli E, Terriaca S, et al. Senescence in adipose-derived stem cells: biological mechanisms and therapeutic challenges. Int J Mol Sci. (2024) 25(15):8390. doi: 10.3390/ijms25158390
446. Kalupahana NS, Goonapienuwala BL, and Moustaid-Moussa N. Omega-3 fatty acids and adipose tissue: inflammation and browning. Annu Rev Nutr. (2020) 40:25–49. doi: 10.1146/annurev-nutr-122319-034142
447. Djuricić I and Calder PC. Polyunsaturated fatty acids and metabolic health: novel insights. Curr Opin Clin Nutr Metab Care. (2022). 25(6):436–42 doi: 10.1097/MCO.0000000000000865
448. Wall R, Ross P, Shanahan F, Quigley EMM, Dinan TG, Cryan JF, et al. Influence of gut microbiota and manipulation by probiotics and prebiotics on host tissue fat: potential clinical implications. Lipid Technol. (2012) 24:227–9. doi: 10.1002/lite.201200232
449. Chen Z, Yang L, Liu Y, Huang P, Song H, and Zheng P. The potential function and clinical application of fgf21 in metabolic diseases. Front Pharmacol. (2022) 13. doi: 10.3389/fphar.2022.1089214
450. Gimeno RE and Moller DE. Fgf21-based pharmacotherapy – potential utility for metabolic disorders. Trends Endocrinol Metab. (2014) 25:303–11. doi: 10.1016/j.tem.2014.03.001
451. Chui ZSW, Xue Y, and Xu A. Hormone-based pharmacotherapy for metabolic dysfunction-associated fatty liver disease. Med Rev. (2024) 4:158–68. doi: 10.1515/mr-2024-0007
452. Palmer AK, Xu M, Zhu Y, Pirtskhalava T, Weivoda MM, Hachfeld CM, et al. Targeting senescent cells alleviates obesity-induced metabolic dysfunction. Aging Cell. (2019) 18(3):e12950. doi: 10.1111/acel.2019.18.issue-3
453. Xu M, Pirtskhalava T, Farr JN, Weigand BM, Palmer AK, Weivoda MM, et al. Senolytics improve physical function and increase lifespan in old age. Nat Med. (2018) 24:1246–56. doi: 10.1038/s41591-018-0092-9
454. Palmer AK, Tchkonia T, and Kirkland JL. Senolytics: potential for alleviating diabetes and its complications. Endocrinology. (2021) 162(8):bqab058. doi: 10.1210/endocr/bqab058
455. Chaib S, Tchkonia T, and Kirkland JL. Obesity, senescence, and senolytics. Handb Exp Pharmacol. (2021) 274:165–80. doi: 10.1007/164_2021_555
456. Lee JH, Park A, Oh K-J, Lee SC, Kim WK, and Bae K-H. The role of adipose tissue mitochondria: regulation of mitochondrial function for the treatment of metabolic diseases. Int J Mol Sci. (2019) 20(19):4924. doi: 10.3390/ijms20194924
Keywords: adipose tissue dysfunction, metabolic disorders, insulin resistance, inflammatory pathways, adipokine dysregulation, oxidative stress, mitochondrial dysfunction, cellular senescence
Citation: Bou Matar D, Zhra M, Nassar WK, Altemyatt H, Naureen A, Abotouk N, Elahi MA and Aljada A (2025) Adipose tissue dysfunction disrupts metabolic homeostasis: mechanisms linking fat dysregulation to disease. Front. Endocrinol. 16:1592683. doi: 10.3389/fendo.2025.1592683
Received: 12 March 2025; Accepted: 02 June 2025;
Published: 24 June 2025.
Edited by:
Lynda Bourebaba, Wroclaw University of Environmental and Life Sciences, PolandReviewed by:
Klaus-Dieter Schlüter, University of Giessen, GermanyNawel Adjeroud, University of Béjaïa, Algeria
Copyright © 2025 Bou Matar, Zhra, Nassar, Altemyatt, Naureen, Abotouk, Elahi and Aljada. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ahmad Aljada, YWFsamFkYUBhbGZhaXNhbC5lZHU=