 Xun Ma1
Xun Ma1 Xiaoqian Zhang2*
Xiaoqian Zhang2*- 1School of Clinical Medicine, Shandong Second Medical University, Weifang, China
- 2Department of Endocrinology and Metabology, The First Affiliated Hospital of Shandong First Medical University & Shandong Provincial Qianfoshan Hospital, Shandong Institute of Nephrology, Jinan, China
Diabetic osteoporosis (DOP) is a complex metabolic bone disorder characterized by impaired bone quality and increased fracture risk in patients with diabetes mellitus. The interplay between hyperglycemia, insulin resistance, and bone metabolism underscores the need for integrated therapeutic strategies that address both glycemic control and bone health. This review systematically examines the molecular mechanisms of glucose-lowering and bone-protective agents, highlighting their dual roles in managing DOP. We discuss the pathophysiological pathways underlying DOP, including insulin/IGF-1 deficiency, advanced glycation end products (AGEs) accumulation, oxidative stress, and vascular damage. Furthermore, we explore the mechanisms of action of antidiabetic drugs (e.g., metformin, GLP - 1 receptor agonists, SGLT2 inhibitors) and anti-osteoporotic agents (e.g., bisphosphonates, teriparatide, strontium ranelate), emphasizing their potential synergies and risks. Finally, we outline future directions for developing novel therapeutics and optimizing combination therapies to achieve dual metabolic and skeletal benefits in DOP patients.
1 Introduction
Diabetes and osteoporosis are globally prevalent chronic metabolic diseases whose pathological interplay in diabetic osteoporosis (DOP) poses a critical clinical concern. According to the International Diabetes Federation (IDF), approximately 537 million individuals aged 20 – 79 years were diagnosed with diabetes in 2021, a figure projected to rise to 783 million by 2045, with type 2 diabetes (T2DM) accounting for over 95% of cases. Concurrently, OP affects 19.7% of the global population, causing more than 8.9 million fractures annually, with hip fracture incidence expected to double by 2050 (1, 2). Epidemiological studies reveal that 50 – 66% of diabetic patients exhibit reduced bone mineral density (BMD), of whom 33% are diagnosed with DOP, underscoring its clinical significance as a major diabetic complication (3, 4). Despite normal or high BMD, T2DM patients face 40 - 70% higher fracture risk than non-diabetics, highlighting bone quality impairment and microstructural damage—not just bone loss—as key DOP characteristics (5, 6).
The pathophysiology of DOP involves multifaceted interactions. Chronic high blood sugar disrupts bone balance by causing oxidative stress, AGEs buildup, and inflammation, which weaken bone formation while increasing breakdown (7, 8). New research shows blood sugar fluctuations harm bone blood vessels and bone cells more than constant high sugar levels, mainly through oxidative stress, mitochondrial damage, and iron-related cell death (9). Ferroptosis, an iron-dependent lipid peroxidation-driven cell death mechanism, is amplified by glucolipotoxicity in T2DM, contributing to osteoblast dysfunction and osteocyte loss (10, 11). Furthermore, dysregulated autophagy (particularly mitophagy) and vitamin D receptor (VDR) gene polymorphisms (e.g., TaqI, EcoRV) impair skeletal homeostasis, while leptin and other adipokines secreted by perirenal adipose tissue exacerbate bone remodeling imbalance via pro-inflammatory pathways (12–14). Impaired osteogenic differentiation of bone marrow mesenchymal stem cells (BMSCs) and aberrant expression of key genes (e.g., FOXQ1) further underpin DOP pathogenesis (15– 16).
Emerging research emphasizes the dual mechanistic roles of glucose-lowering and anti-osteoporotic therapies. While conventional antidiabetic agents improve glycemic control, certain drugs (e.g., thiazolidinediones) may exacerbate bone loss by inhibiting osteoblast differentiation. In contrast, newer agents (e.g., GLP - 1 receptor agonists, SGLT2 inhibitors) demonstrate potential bone-protective effects via metabolic and anti-inflammatory modulation. Anti-resorptive therapies (e.g., bisphosphonates, RANKL inhibitors), though effective in suppressing bone resorption, exhibit variable efficacy in diabetic populations due to glucose metabolism dysregulation and vascular complications. Crucially, no comprehensive review has systematically integrated the pathophysiological interplay between these therapeutic classes in DOP management. This gap hinders the development of synergistic strategies to address concurrent glycemic and skeletal dysregulation.
In summary, the prevention and treatment strategies for DOP must be grounded in its multifactorial pathological mechanisms, with an emphasis on exploring individualized therapeutic targets. We systematically searched PubMed, Web of Science, and Embase (2018 – 2024) using MeSH terms: (diabetic osteoporosis OR diabetes bone disease) AND (pathogenesis OR drug therapy OR TCM). Inclusion criteria (1): Clinical/experimental studies on DOP mechanisms or treatments (2); English-language articles; (3) Prioritization of meta-analyses (e.g., Vestergaard, Diabetologia 2005) and guidelines (IOF, 2018). Exclusion criteria: (1) Case reports; (2) Non-diabetic osteoporosis studies; (3) Articles without mechanistic/clinical outcomes. From 238 initial results, 170 references were retained based on relevance and impact. This review systematically examines the pathophysiological network of DOP, the mechanisms of action of glucose-lowering and anti-osteoporotic drugs, and recent advances in prevention and treatment. It aims to provide a theoretical foundation for optimizing clinical practice and guiding future research directions. This integrative review holds significant translational value for improving the prognosis of DOP patients and reducing the burden on healthcare systems.
2 Diabetes and osteoporosis
2.1 Diabetes mellitus
Diabetes mellitus (DM) represents a group of metabolic disorders characterized by chronic hyperglycemia, arising from a combination of diverse etiologies. The core pathological mechanisms involve insufficient insulin secretion and/or impaired insulin action. Based on distinct pathogenic pathways, DM is primarily classified into type 1 diabetes (T1D) and type 2 diabetes (T2D). T1D is characterized by autoimmune destruction of pancreatic β-cells, leading to an absolute deficiency of insulin, whereas T2D is predominantly marked by insulin resistance accompanied by relative insulin deficiency (17). As a critical global public health issue, diabetes not only exhibits severe metabolic dysregulation but also induces multi-system damage due to prolonged hyperglycemia, resulting in various chronic complications. With the acceleration of population aging, lifestyle changes, and dietary shifts, diabetes has emerged as the third most prevalent non-communicable disease, following cardiovascular diseases and malignancies. According to the latest statistics from the International Diabetes Federation (IDF), the global diabetic population reached 530 million in 2021 and is projected to exceed 780 million by 2045 (18).
2.2 Osteoporosis
Osteoporosis (OP) is a systemic skeletal disorder characterized by reduced bone mass and deterioration of bone microarchitecture. The core problem is disrupted balance between bone breakdown and formation, causing thinner trabecular and cortical bones, reduced strength, and higher fracture risk. This progressive degeneration of bone tissue structure predisposes individuals to fragility fractures (FF) even under low-energy trauma (e.g., falls from standing height) or during routine daily activities. As the most prevalent non-communicable skeletal disease worldwide, the prevalence of osteoporosis increases significantly with age. Epidemiological data indicate that the overall prevalence of OP in individuals aged 50 years and older is 19.2%, with a markedly higher prevalence in women (32.1%) compared to men (6.9%). Among those aged 65 and older, the prevalence rises to 32%, with women disproportionately affected at 51.6%, compared to 10.7% in men (19). Globally, osteoporosis is responsible for over 8.9 million fractures annually, equating to one osteoporotic fracture occurring every 3 seconds (20). This imposes a substantial burden on both individual health and socioeconomic systems.
2.3 Diabetic osteoporosis
Diabetic osteoporosis (DOP) is a significant long-term complication of diabetes mellitus, classified under secondary osteoporosis. It is pathologically characterized by diabetes-induced reductions in bone mass and microstructural damage, leading to increased bone fragility and elevated fracture risk. Studies have demonstrated that both type 1 and type 2 diabetes are significantly associated with an increased risk of fractures. While microvascular and macrovascular complications are the most commonly recognized sequelae of diabetes, osteoporosis and its associated fracture risk also warrant considerable attention in clinical practice (21). Globally, over 90,000 osteoporotic fractures annually are linked to DOP, posing a substantial threat to human health and socioeconomic development (20).
Compared to the general population, diabetic patients exhibit a significantly higher fracture risk. Research by Vestergaard et al. revealed that the risk of fractures at any site in diabetic patients is 0.5 – 2 times higher than in controls, with odds ratios (ORs) of 1.3 (95% CI 1.2 – 1.5) for type 1 diabetes and 1.2 (95% CI 1.1 – 1.3) for type 2 diabetes. For site-specific fractures, type 1 diabetic patients have a notably elevated risk of hip fractures (OR = 1.7, 95% CI 1.3 – 2.2), while type 2 diabetic patients show a relatively lower but still significant risk (OR = 1.4, 95% CI 1.2 – 1.6) (22). A meta-analysis by Fan et al. found that the pooled relative risk of hip fractures in diabetic patients compared to non-diabetic individuals was 2.07 (95% CI 1.83 – 2.33) (23). Further research by Wang et al. elucidated the relationship between diabetes and site-specific fracture risks: overall, diabetes was significantly associated with increased risks of hip (RR 1.77, 95% CI 1.56 – 2.02), upper arm (RR 1.47, 95% CI 1.02 – 2.10), and ankle fractures (RR 1.24, 95% CI 1.10 – 1.40), but not with distal forearm fractures (RR 1.02, 95% CI 0.88 – 1.19). Notably, type 1 diabetic patients exhibited significantly higher risks of total hip, hip joint, and ankle fractures compared to type 2 diabetic patients (P-values: 0.002, <0.001, and 0.029, respectively) (24). These findings underscore the strong association between diabetes and osteoporosis, as well as the critical importance of recognizing and addressing DOP in clinical management. Figure 1 illustrated the intricate biological connections and clinical implications between Type 2 Diabetes Mellitus (T2DM) and osteoporosis.
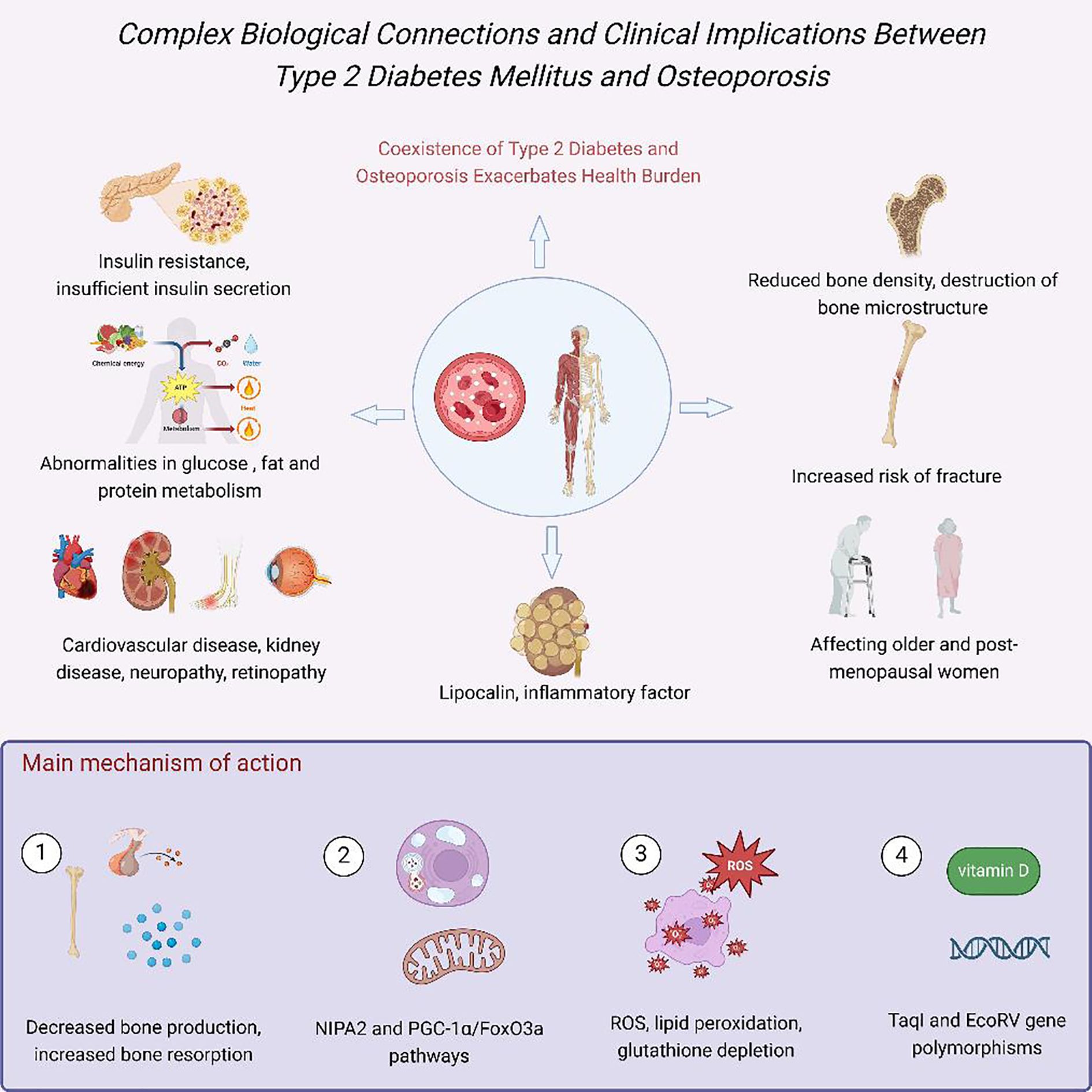
Figure 1. Complex biological connections and clinical implications between Type 2 Diabetes Mellitus (T2DM) and osteoporosis. Hyperglycemia, insulin resistance, and inflammation impair bone formation via AGEs, oxidative stress, and vascular dysfunction (25).
3 Pathophysiological mechanisms of diabetic osteoporosis
3.1 Insulin deficiency
Insulin, a pivotal metabolic regulatory hormone, exerts extensive anabolic effects in the body, with its role in osteoblast anabolism being particularly significant. Studies have shown that insulin primarily regulates blood glucose levels through the PI3K/Akt signaling pathway while promoting cell proliferation and osteogenic differentiation (26). Insulin acts on osteoblasts through membrane receptors that detect insulin and trigger intracellular signaling pathways, starting a series of biological responses (27, 28). Specifically, insulin stimulates DNA synthesis, induces cell proliferation, and promotes the synthesis of osteocalcin and collagen, which are essential precursors for bone formation. Additionally, insulin upregulates the expression of the RUNX2 gene, a key regulator of bone metabolism that facilitates osteoblast differentiation and bone matrix maturation (29).
The critical role of insulin in bone metabolism provides a theoretical basis for understanding the differences in bone mineral density (BMD) between patients with type 1 diabetes (T1D) and type 2 diabetes (T2D). In T1D, the autoimmune destruction of pancreatic β-cells during early childhood leads to an absolute insulin deficiency, which persists throughout adolescence and results in insufficient bone mineralization. Clinical observations indicate that insulin therapy can partially improve bone mineralization in T1D patients (29). In contrast, bone metabolism in T2D patients is more complex: in the early stages of the disease, systemic insulin resistance accompanied by compensatory hyperinsulinemia may lead to increased bone mineralization. However, as the disease progresses to advanced stages, pancreatic β-cell function declines, insulin secretion decreases, and BMD significantly diminishes. This dynamic interplay highlights the central role of insulin in diabetic bone metabolism disorders and provides a crucial theoretical foundation for the prevention and treatment of diabetic osteoporosis (30, 31).
3.2 Insulin-like growth factor 1 (deficiency
Insulin-like growth factor 1 (IGF - 1), structurally similar to insulin, exerts significant anabolic effects on osteoblasts. Specifically, IGF - 1 acts on osteoprogenitor cells, stimulating DNA synthesis and promoting osteoblast differentiation, thereby significantly enhancing osteoblast activity. Additionally, IGF - 1 facilitates the formation of bone collagen while inhibiting its degradation, ultimately promoting bone matrix formation and mineralization. IGF - 1 is primarily synthesized and secreted by hepatocytes; however, in diabetic patients, chronic hyperglycemia suppresses the synthesis and release of IGF - 1, thereby impairing its osteogenic effects. Studies have shown that serum IGF - 1 levels are significantly reduced in diabetic patients, leading to decreased bone formation, which may represent a key pathological mechanism underlying diabetic osteoporosis (32). Further clinical research has confirmed that in patients with type 2 diabetes, serum IGF - 1 levels are positively correlated with bone mineral density (BMD), and reduced IGF - 1 levels are a significant risk factor for increased fracture risk in these individuals (32). Insulin and IGF - 1 deficiency directly impair osteoblast function, which explains the generally reduced BMD observed in T1DM patients and underscores the importance of early (e.g., as recommended by International Society for Pediatric and Adolescent Diabetes, ISPAD in late adolescence) and regular BMD screening (following IOF guideline) in these patients.
3.3 Hyperglycemic environment
A hyperglycemic environment exerts significant adverse effects on various tissues and cells. Studies have shown that elevated glucose levels activate the non-canonical Wnt/protein kinase C pathway and upregulate the expression of peroxisome proliferator-activated receptor gamma (PPARγ), thereby promoting adipogenesis and exacerbating bone loss (33). Hyperglycemia directly inhibits osteoblast/osteocyte activity via cytotoxic effects (34, 35). Specifically, osteoblasts exposed to high glucose environments demonstrate reduced proliferation, slowed extracellular matrix synthesis, and delayed maturation and mineralization processes (36). Furthermore, hyperglycemia increases osteoblast apoptosis and accelerates cellular senescence, further impairing their function. Under diabetic conditions, the osteocyte lacunar-canalicular system, constructed by osteoblasts, undergoes significant microstructural damage, characterized by abnormally enlarged lacunae, reduced canalicular network density, and disrupted cellular projections. These structural alterations lead to abnormal bone remodeling and altered mechanical properties, accelerating the progression of diabetic bone disease (36).
On the other hand, glucose, as a fundamental energy substrate, stimulates osteoclast activity, and the bone-resorbing function of osteoclasts is glucose concentration-dependent. Consequently, under hyperglycemic conditions, enhanced bone resorption coupled with impaired bone formation collectively contribute to rapid bone loss (37). Figure 2 summarized the pathogenesis of diabetic osteoporosis induced by blood glucose fluctuations.
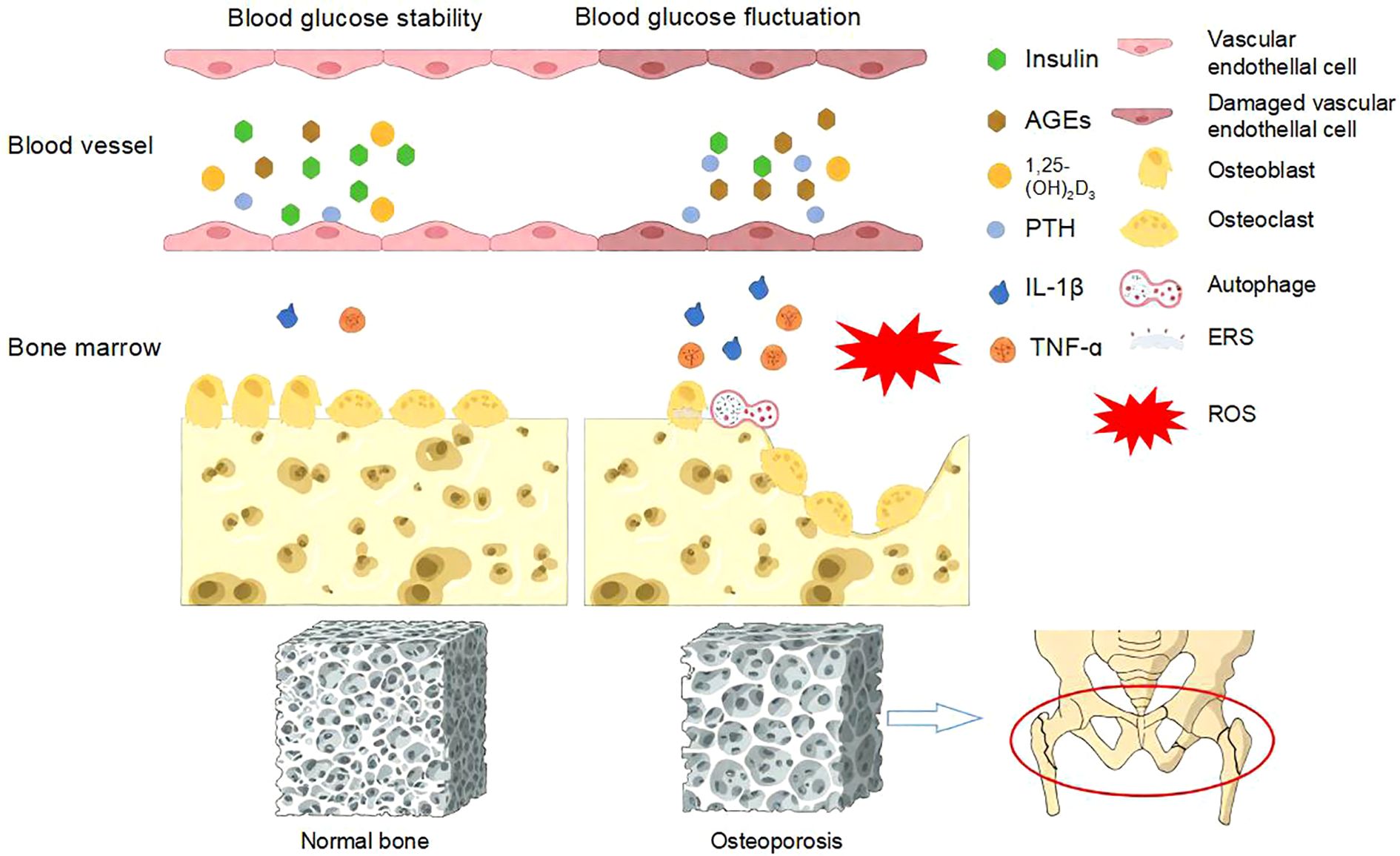
Figure 2. Pathogenesis of diabetic osteoporosis caused by blood glucose fluctuations. Blood glucose fluctuations can lead to oxidative stress, increased levels of advanced glycation end products and inflammatory mediators, reduced insulin secretion, decreased 1,25-(OH)2D3, elevated PTH levels, and the occurrence of ESR and autophagy. These factors, in turn, inhibit the function of osteoblasts and osteoclasts, leading to osteoporosis and osteoporotic fractures (38).
3.4 Accumulation of advanced glycation end products
Hyperglycemia may induce the formation of advanced glycation end products (AGEs) through non-enzymatic pathways. AGEs are generated via glycation reactions, which involve non-enzymatic interactions between ketones or aldehydes and protein amino groups, leading to protein dysfunction. This process occurs under both normoglycemic and hyperglycemic conditions, but its rate is influenced by substrate availability (39). Under normal physiological conditions, stable blood glucose levels result in a moderate rate of AGEs formation. However, in diabetic patients, significantly elevated glucose levels increase substrate availability, accelerating AGEs generation (39). Additionally, excess glucose can be converted to sorbitol via aldose reductase and subsequently to fructose by sorbitol dehydrogenase. Fructose and its metabolites, as potent glycating agents, further promote AGEs formation. AGEs exert multifaceted detrimental effects on the skeletal system, impairing extracellular matrix function and vascular structure.
3.4.1 The key mechanisms by which AGEs impair bone quality
One critical mechanism by which AGEs compromise bone quality is through abnormal cross-linking with collagen in the bone matrix. This abnormal cross-linking reduces collagen elasticity, whereas enzymatic cross-linking is essential for maintaining bone strength by increasing collagen stiffness. Non-enzymatic cross-linking, however, is detrimental to bone quality (40, 41). The abnormal cross-linking of AGEs with collagen alters the structural and functional properties of proteins, leading to reduced overall bone strength and increased fragility (42). Due to the lower bone turnover rate in diabetic patients, proteins have more time to accumulate AGEs, resulting in significantly higher AGEs levels compared to non-diabetic individuals (43). Chronic hyperglycemia not only promotes glycation of the bone matrix but also impairs collagen turnover and matrix renewal, ultimately leading to impaired bone formation and increased skeletal fragility. Studies have shown that elevated AGEs content per gram of collagen disrupts the bone matrix and alters its material properties, making it more susceptible to mechanical failure under physiological stress levels. Studies show higher pentosidine levels in diabetic patients’ blood and urine predict greater fracture risk, regardless of bone density (44–46). This finding provides critical insights into the phenomenon of increased bone fragility in diabetic patients despite normal BMD.
3.4.2 The inhibitory effects of AGEs on osteoblasts.
Advanced glycation end products (AGEs) exert significant inhibitory effects on osteoblast function. Studies have demonstrated that AGEs suppress osteoblast differentiation and reduce the expression of alkaline phosphatase, thereby impairing osteogenic activity (47, 48). Sun et al. found that AGE-RAGE signaling blocks bone formation, while blocking RAGE boosts bone-related gene and protein expression (49). Boosting glyoxalase-1 (Glo-1) levels can counteract the harmful effects of AGE-RAGE signaling on bone marrow stem cells by detoxifying AGE precursors. The receptor for advanced glycation end products (RAGE), a multi-ligand receptor, binds to various ligands, including AGEs, Mac-1, high-mobility group box 1 (HMGB1), and S100 calcium-binding proteins (50– 51). In vitro experiments have shown that the binding of AGEs to RAGE activates multiple signaling pathways, including p38 mitogen-activated protein kinase (p38 MAPK), extracellular signal-regulated kinase 1/2 (ERK1/2), Janus kinase (JAK), and Rho-GTPases (52– 53).
Furthermore, AGEs significantly inhibit osteoblast proliferation, promote osteoblast apoptosis, and increase the expression of sclerostin, a negative regulator of bone formation, while reducing the levels of receptor activator of nuclear factor kappa-B ligand (RANKL) (54, 55). In vitro data indicate that high glucose levels and AGEs markedly upregulate sclerostin expression in osteocytes, further impairing bone formation (54). This preclinical observation has been validated in clinical studies, which show that individuals with prediabetes exhibit significantly higher sclerostin levels compared to controls, and these levels are closely associated with insulin resistance (56).Research by Ge et al. further highlights that AGEs significantly inhibit both the proliferation and differentiation of osteoblasts (57). Additionally, AGEs suppress the secretion of parathyroid hormone (PTH) (58), leading to inhibited bone turnover and a low bone turnover state, which exacerbates the development of diabetic osteoporosis. These findings not only elucidate the central role of AGEs in diabetic bone disease but also provide a critical theoretical foundation for the prevention and treatment of this condition. The accumulation of AGEs and their abnormal cross-linking is a key mechanism leading to decreased bone quality and the “BMD-fracture risk dissociation” phenomenon in T2DM patients. This suggests that relying solely on BMD and traditional FRAX scores may underestimate fracture risk in these patients. Therefore, clinical guidelines (such as the IOF position statement) strongly recommend including tools for assessing bone microstructure, like TBS, in the fracture risk evaluation system for T2DM patients. They also advise considering stricter BMD diagnostic thresholds (e.g., T-score ≤ -2.0).
3.5 Chronic inflammation
Diabetic patients often experience a state of chronic low-grade inflammation, characterized by the excessive activation of inflammatory cytokines, including tumor necrosis factor-alpha (TNF-α) and interleukin-6 (IL - 6). This chronic inflammatory state is not only closely associated with microvascular and macrovascular complications in type 2 diabetes (T2DM) but also exerts significant negative effects on bone remodeling processes (59, 60). Furthermore, a hyperglycemic environment promotes the differentiation of bone marrow mesenchymal stem cells (BMSCs) toward adipogenesis, accompanied by increased bone marrow fat deposition and the release of large amounts of free fatty acids and inflammatory cytokines (61). Among these inflammatory factors, TNF-α plays a pivotal role in promoting bone resorption. It stimulates the proliferation and differentiation of osteoclast precursor cells in the bone marrow into mature osteoclasts, thereby accelerating bone resorption (62). IL - 6, a multifunctional cytokine secreted by various human cells, not only promotes the division and proliferation of osteoclast precursor cells but also directly participates in the differentiation and maturation of osteoclasts. Additionally, IL - 6 enhances bone resorption by increasing the release of collagenase and the degradation of the bone matrix (63). These inflammatory cytokines also trigger intracellular oxidative stress, further exacerbating bone metabolism disorders. Chronic inflammation and oxidative stress not only promote bone resorption but may also affect bone formation markers. While bone turnover markers (BTMs) have limited value in predicting fracture risk in diabetic osteoporosis (DOP), their pattern changes (such as low osteocalcin levels) might provide supplementary information for understanding individual bone metabolic status. However, diagnosis and risk stratification still primarily rely on tools like BMD, FRAX, and TBS.
3.6 Oxidative stress
The homeostasis of the human internal environment relies on a dynamic balance between the oxidative system (primarily reactive oxygen species, ROS) and the antioxidant defense system (including superoxide dismutase, SOD), catalase, and glutathione peroxidase, GPx). Excessive ROS production plays a critical role in various physiological and pathological processes (64), such as causing oxidative damage to DNA, lipid membranes, and proteins, disrupting the antioxidant defense system, activating downstream pathways related to oxidative stress, and inducing indirect cellular damage (65, 66). Studies have shown that glycemic variability significantly elevates ROS levels. In vitro experiments demonstrate that compared to human umbilical vein endothelial cells (HUVECs) cultured under sustained hyperglycemic conditions, those exposed to intermittent hyperglycemia exhibit markedly increased ROS generation, leading to exacerbated cell apoptosis (67, 68). In vivo experiments by Horvath et al. investigated the effects of glycemic variability on oxidative stress in diabetic rats. The rats were divided into a stable glucose group (receiving long-acting insulin daily) and a glucose fluctuation group (receiving long-acting insulin every other day). After 14 days, the glucose fluctuation group showed significantly elevated nitrotyrosine levels and endothelial dysfunction compared to rats with stable or normal glucose levels (69). Regarding the impact of glycemic variability on bone cells, Zhang et al. found that glucose fluctuations lead to a rapid increase in ROS levels and induce oxidative stress, thereby impairing osteoblast function, inhibiting their activity and proliferation, and ultimately promoting osteoblast apoptosis (70).
3.7 Vascular damage
The hyperglycemic state in diabetes leads to both microvascular and macrovascular complications. Microvascular damage harms small blood vessels, causing poor circulation, weakened vessel function, blood vessel loss, reduced new vessel growth, and leaky vessels. Chronic hyperglycemia causes endothelial damage in microvessels, leading to vasomotor dysfunction, vascular rarefaction, and hemodynamic abnormalities, which significantly reduce blood perfusion to bones and bone marrow. The impaired endothelial barrier function exacerbates vascular leakage, resulting in bone marrow microenvironment disturbances (e.g., hemorrhage, edema) and further disrupting bone homeostasis. These changes directly affect the blood supply to bones, as skeletal health relies on adequate blood flow to deliver oxygen, nutrients, and hormones while removing metabolic waste (71). The microvascular system in bone marrow is critical for bone development, repair, and regeneration.
In diabetic patients, the bone marrow microenvironment undergoes significant alterations, characterized by reduced hematopoietic tissue and increased adipocyte accumulation (marrow adiposity). Vascular rarefaction and endothelial dysfunction contribute to insufficient bone perfusion, leading to hypoxia and oxidative stress. Oxidative stress damages endothelial cells and reduces the production of nitric oxide (NO), a key mediator of vasodilation. The diminished NO-mediated vasodilatory response, coupled with increased activity of vasoconstrictors such as endothelin-1, predisposes bone arteries to a constricted state (72). Diabetes also reduces key angiogenesis factors like VEGF and HIF - 1α, worsening new blood vessel formation and repair. Macrovascular complications, including atherosclerosis and peripheral artery disease, exacerbate these issues by reducing overall skeletal blood supply and worsening local microcirculatory dysfunction. Diabetes-related vascular damage leads to inadequate bone perfusion and hypoxia, impairing bone formation and repair capacity. This highlights the importance of controlling cardiovascular risk factors (such as hypertension and dyslipidemia) in DOP management, which not only aligns with diabetes care guidelines (e.g., ADA standards) but may also indirectly improve bone health outcomes.
4 Pathophysiological mechanisms of glucose-lowering agents in osteoporosis treatment
Glucose-lowering agents play a significant role in the treatment of diabetic osteoporosis (DOP), not only by effectively controlling blood glucose levels but also by influencing bone metabolism through various mechanisms. These diabetes medications are mainly classified by their mechanisms into: thiazolidinediones (TZDs), sulfonylureas, metformin, incretin-based drugs (GLP - 1 agonists and DPP - 4 inhibitors), SGLT2 inhibitors, and insulin. These drugs regulate bone formation and resorption through different signaling pathways, thereby exerting either positive or negative effects on bone health. Table 1 provided a detailed summary of the functions and mechanisms of action (signaling pathways) of various glucose-lowering agents, offering important theoretical insights for the prevention and treatment of diabetic osteoporosis.
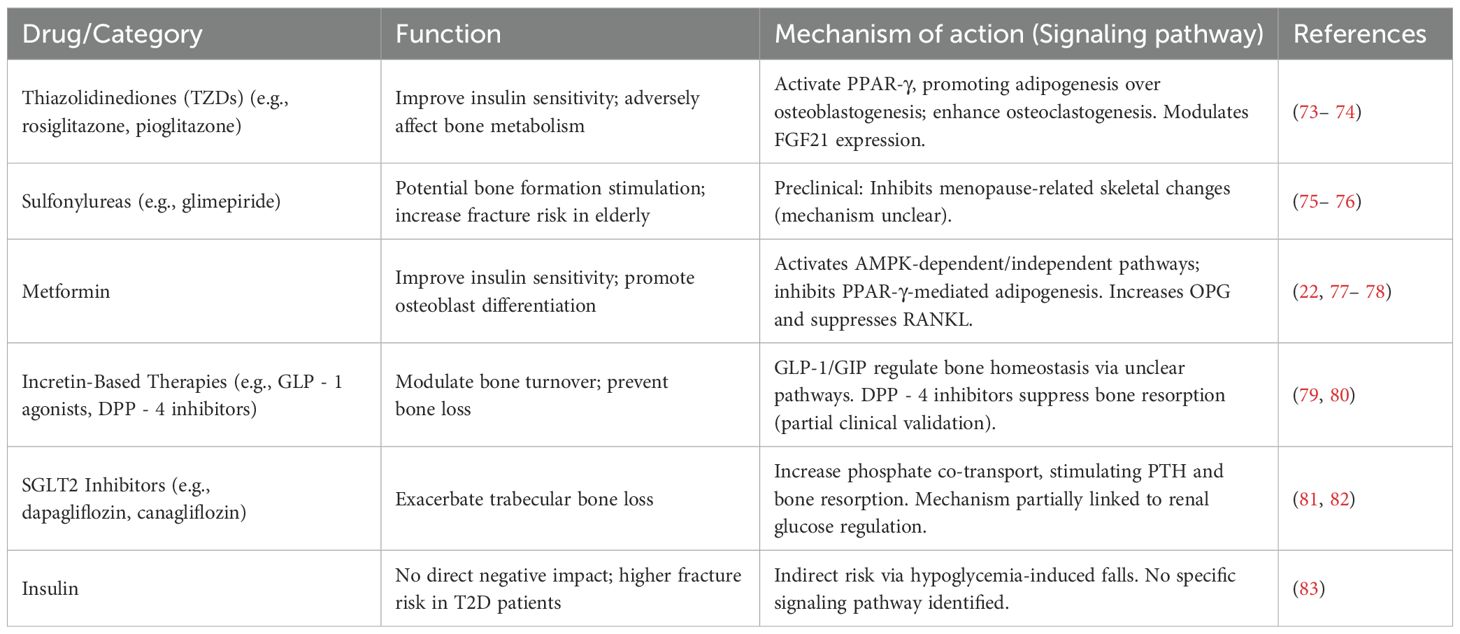
Table 1. Functions of glucose-lowering agents and mechanisms of action (signaling pathways) in the treatment of osteoporosis.
4.1 Thiazolidinediones
Thiazolidinediones (TZDs), represented by rosiglitazone and pioglitazone, are widely used antidiabetic drugs that primarily improve insulin sensitivity and reduce insulin resistance to achieve glycemic control. However, these drugs have significant adverse effects on bone metabolism. Studies have shown that TZDs impair osteoblast differentiation and reduce bone formation by modulating gene expression, leading to decreased bone mineral density (BMD) and increased fracture risk, particularly in female patients (73). Specifically, the use of rosiglitazone is associated with a 6 – 20% increase in bone resorption and a 4 – 13% reduction in bone formation markers in postmenopausal women, resulting in clinically significant bone loss (84). A pioglitazone meta-analysis reveals TZD treatment increases fracture risk in postmenopausal women, with longer treatment duration potentially heightening this risk.
The glucose-lowering effects of TZDs are primarily mediated through the activation of peroxisome proliferator-activated receptor gamma (PPAR-γ), which plays a critical role in glucose homeostasis and adipogenesis. PPAR-γ activation induces mesenchymal stem cells (MSCs) to differentiate into adipocytes rather than osteoblasts, thereby promoting adipogenesis at the expense of bone formation. This process disrupts skeletal metabolic balance. Additionally, PPAR-γ activation enhances osteoclastogenesis while suppressing osteoblastogenesis, leading to increased bone resorption and decreased bone formation (85– 86). TZDs also improve insulin resistance and sensitivity in diabetic patients by upregulating the expression of fibroblast growth factor 21 (FGF21), which enhances PPAR-γ transcriptional activity (87). However, excessive FGF21 expression contributes to bone loss. Research by Wei et al. demonstrates that rosiglitazone-induced bone loss can be prevented by inhibiting FGF21 function (74). Since TZDs are known to significantly increase bone loss and fracture risk (particularly in women), major osteoporosis and diabetes guidelines (including ADA, ESCEO, and AACE) all recommend avoiding or using these drugs with extreme caution in T2DM patients who already have osteoporosis or high fracture risk (as indicated by elevated FRAX assessment results).
4.2 Sulfonylureas
Sulfonylureas, a class of classic glucose-lowering agents, have shown inconsistent effects on bone metabolism in clinical studies. Current clinical data suggest that sulfonylureas do not significantly impact human bone health (75– 88). However, preclinical research indicates that certain sulfonylureas, particularly glimepiride, may have the potential to stimulate bone formation (89, 90). For instance, experimental data from ovariectomized rats demonstrate that glimepiride can inhibit menopause-related skeletal changes and promote bone formation. Despite these findings, sulfonylureas must be used cautiously due to their risk of inducing hypoglycemia, which is a major risk factor for fractures, especially in elderly and frail individuals (76, 91). A study on fracture risk in older men revealed that sulfonylurea users had a 66% higher risk of non-vertebral fractures compared to controls (91). Therefore, the use of sulfonylureas in elderly patients with osteoporosis requires careful consideration.
4.3 Metformin
Metformin, a classic oral hypoglycemic agent, not only improves insulin sensitivity to achieve glycemic control but also exhibits potential beneficial effects on bone health (22, 77). Preclinical studies have demonstrated that metformin promotes the differentiation of mesenchymal stem cells (MSCs) into osteoblasts while inhibiting adipogenesis and osteoclast differentiation (92– 93). Additionally, by reducing hepatic gluconeogenesis, metformin partially mitigates the negative impact of hyperglycemia on bone. Its mechanisms of action include both AMP-activated protein kinase (AMPK)-dependent and AMPK-independent pathways (94). Numerous in vitro studies have confirmed the osteogenic effects of metformin (95). Research by Zheng et al. further indicates that metformin regulates peroxisome proliferator-activated receptor gamma (PPAR-γ) through the AMPK pathway, thereby exerting protective effects on osteoblast differentiation and potentially counteracting osteoporosis (78).
Moreover, metformin inhibits osteoclast differentiation. Osteoblasts secrete receptor activator of nuclear factor kappa-B ligand (RANKL) and osteoprotegerin (OPG), which play critical roles in osteoclast differentiation. RANKL promotes osteoclast differentiation, while OPG, a decoy receptor for RANKL, inhibits this process. In vivo experiments have shown that metformin treatment increases OPG levels and suppresses RANKL expression, thereby reducing bone loss (96). However, not all studies support the positive effects of metformin on bone. For instance, research by Jeyabalan et al. found no significant impact of metformin on bone mass or fracture healing (97). Although clinical evidence for metformin’s bone-protective effects (Section 4.3) still requires confirmation through larger RCTs, its favorable preclinical data and relatively neutral clinical observations have made it one of the preferred insulin sensitizers for DOP patients (particularly those with fracture risk factors), and it is frequently recommended as first- or second-line therapy in clinical guidelines.
4.4 Incretin-based therapies
Incretin-based therapies, including glucagon-like peptide-1 (GLP - 1) receptor agonists and dipeptidyl peptidase-4 (DPP - 4) inhibitors, represent an important class of glucose-lowering agents. While GLP - 1 receptors are primarily expressed in pancreatic β-cells, their effects extend beyond glucose regulation. Studies suggest that GLP - 1 may modulate bone resorption through GLP - 2 and glucose-dependent insulinotropic polypeptide (GIP). In addition to promoting insulin secretion, GLP - 1 and GIP are involved in regulating bone turnover and maintaining bone homeostasis. Clinical studies have shown that treatments with exenatide and liraglutide effectively prevent bone loss associated with weight reduction, with liraglutide therapy increasing serum levels of total type I procollagen N-terminal propeptide (P1NP) by 16% (79, 80). However, findings on the impact of GLP - 1 agonists on fracture risk vary across clinical studies (98, 99), and no definitive conclusions have been reached. In mouse models, the DPP - 4 inhibitor sitagliptin has demonstrated inhibitory effects on bone resorption, a finding partially validated in clinical studies involving postmenopausal women (100, 101). Although some clinical studies suggest that DPP - 4 inhibitors may reduce fracture risk, a meta-analysis by Hidayat et al. did not support a significant association between DPP - 4 inhibitor use and fracture risk (102). GLP - 1 receptor agonists have demonstrated potential bone-protective effects, particularly during weight loss periods. Given their established cardiometabolic benefits, these agents may represent a preferred glucose-lowering option for DOP patients, especially those with high cardiovascular risk or obesity. However, current clinical guidelines have not yet specifically recommended them for bone protection purposes.
4.5 Sodium-glucose co-transporter-2 inhibitors
SGLT2 inhibitors represent a novel class of glucose-lowering agents. However, emerging evidence suggests that SGLT2 inhibitors may exacerbate trabecular bone loss (103). Although the precise mechanisms remain incompletely defined, this effect may be linked to the glucose-lowering mechanism of SGLT2 inhibitors. By reducing renal glucose reabsorption and sodium transport, SGLT2 inhibitors increase phosphate co-transport, which stimulates the parathyroid glands and enhances bone resorption (104). Treatment with dapagliflozin has been shown to elevate serum phosphate and parathyroid hormone (PTH) levels (105). In the Canagliflozin Cardiovascular Assessment Study (CANVAS), canagliflozin was associated with a higher incidence of fractures, primarily affecting the distal limbs, within just 12 weeks of treatment initiation compared to placebo (4.0% vs. 2.6%). For this reason, guidelines from organizations including ADA, EASD and ESCEO recommend cautious use of these medications in high fracture risk patients (such as elderly individuals, those with osteoporosis, fall history, or high FRAX scores), while prioritizing alternative glucose-lowering treatment options.
4.6 Insulin
As previously discussed, insulin plays a crucial role in bone metabolism, and insulin therapy itself is not considered to have a direct negative impact on bone health. However, some studies have reported that patients with type 2 diabetes receiving insulin therapy exhibit a higher risk of fractures compared to those not on insulin treatment (106). This observation may be influenced by the fact that patients requiring insulin typically have a longer disease duration and a higher prevalence of complications, which could independently contribute to increased fracture risk. Additionally, insulin-treated patients are at a higher risk of hypoglycemia, which may lead to an increased likelihood of falls and subsequent fractures (83). Current guidelines emphasize the need to optimize insulin regimens to reduce hypoglycemic episodes when treating elderly and osteoporotic patients, while also strengthening fall prevention measures (such as following the IOF six-step approach).
5 Pathophysiological mechanisms of anti-osteoporotic agents in osteoporosis treatment
The management of diabetic osteoporosis (DOP) should prioritize glycemic control, supplemented by anti-osteoporotic therapy (82). Insulin and glucose-lowering agents form the foundation of treatment for diabetic bone disease, but care must be taken to avoid medications with adverse skeletal effects, such as thiazolidinediones (TZDs). The pharmacological treatment of osteoporosis in diabetic patients is similar to that for other forms of osteoporosis and primarily includes agents that inhibit bone resorption, promote bone formation, and regulate bone metabolism. Table 2 summarized the functions and mechanisms of action (signaling pathways) of anti-osteoporosis drugs.
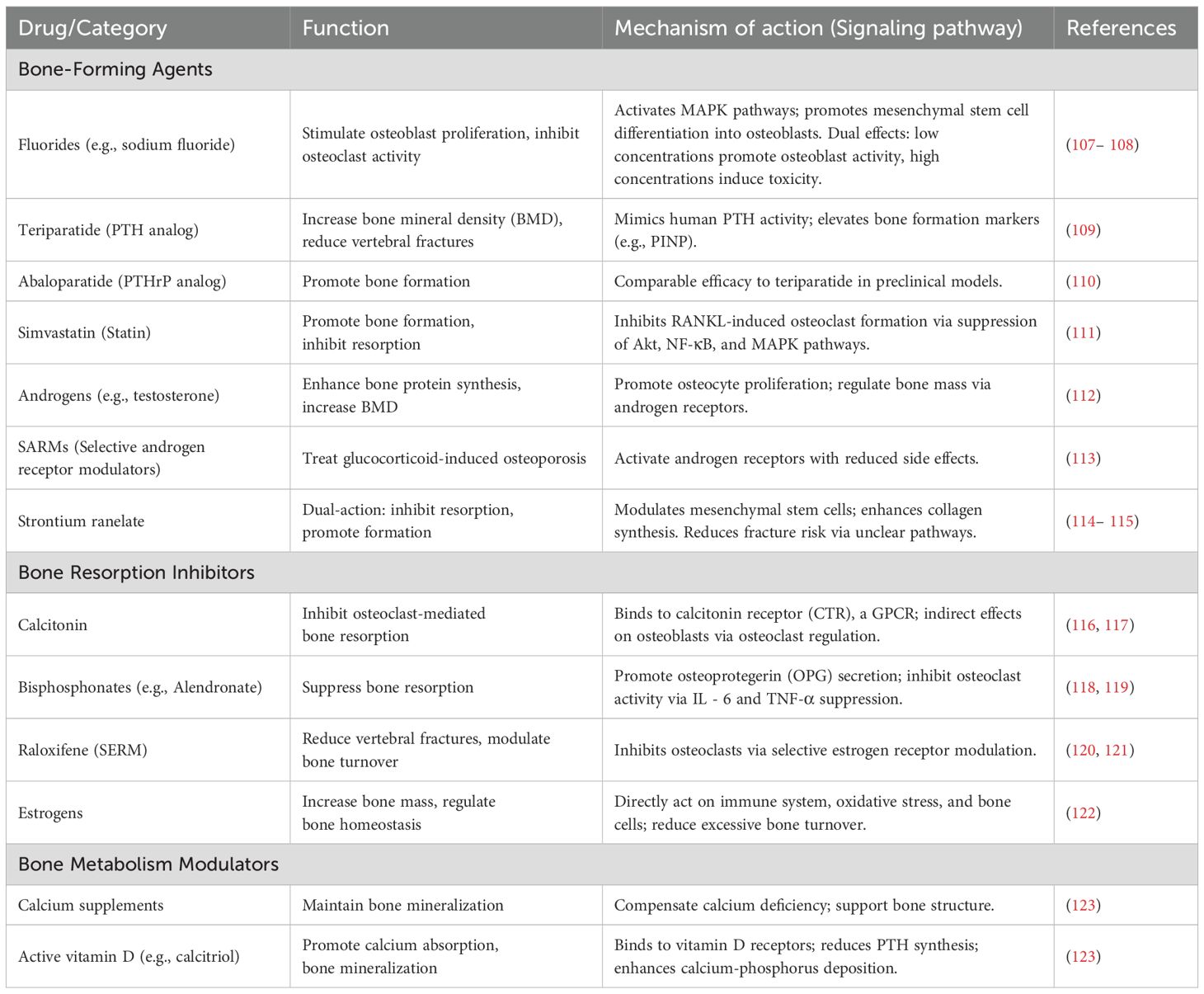
Table 2. Functions and mechanisms of action (signaling pathways) of anti-osteoporosis drugs.
5.1 Bone-forming agents
5.1.1 Fluorides
Fluorides, such as sodium fluoride and disodium monofluorophosphate, are essential trace nutrients for bone growth. Fluoride directly stimulates osteoblast proliferation and inhibits osteoclast activity, thereby increasing bone mass (107). It also induces bone formation by promoting the differentiation of mesenchymal stem cells into osteoblasts (124). Fluoride shows dual bone effects: low doses inhibit osteoblast phosphatases, activating growth pathways that boost bone-forming cell activity. At high concentrations, fluoride exerts toxic effects on osteoblasts while inhibiting osteoclast activity (108).
5.1.2 Parathyroid hormone analogs
Teriparatide, a recombinant human parathyroid hormone (33– 33) fragment, exhibits biological activity identical to human PTH. It significantly increases bone mineral density (BMD) in the lumbar spine and femoral neck over the long term and is an effective agent for reducing the risk of vertebral fractures in postmenopausal women with osteoporosis. Clinical trials have shown that serum levels of procollagen type I N-terminal propeptide (PINP), a marker of bone formation, increase consistently within three months of initiating teriparatide therapy (109). However, the high cost of teriparatide limits its widespread use. Abaloparatide, a novel parathyroid hormone-related protein (PTHrP) analog, has demonstrated comparable efficacy to teriparatide in mouse models at equivalent doses (110). Teriparatide represent potent therapeutic options for very high-risk patients (such as those with multiple vertebral fractures or extremely low BMD). While clinical data supporting their efficacy in DOP remains relatively limited but promising, current guidelines recommend their use in specific high-risk populations.
5.1.3 Statins
Statins exhibit dose-dependent effects on bone metabolism: low doses increase bone resorption, while high doses promote bone formation. Simvastatin, for instance, demonstrates anabolic effects on bone by inhibiting osteoblast apoptosis and suppressing osteoclast differentiation and activity (125). Research by Chowdhury et al. revealed that simvastatin inhibits RANKL-induced osteoclast formation and function by suppressing the Akt, NF-κB, and MAPK signaling pathways, leading to downregulation of key transcription factors such as c-Fos and NFATc1 during osteoclastogenesis (111).
5.1.4 Androgens and Prostaglandin E2
Androgens promote osteocyte proliferation, accelerate bone protein synthesis and mineralization, and increase trabecular bone volume and mass. Bone mineral density (BMD) in men is positively correlated with serum testosterone levels, which decline with age (112). Testosterone deficiency is a significant risk factor for male osteoporosis. Selective androgen receptor modulators (SARMs) have shown promising efficacy in treating glucocorticoid-induced osteoporosis in men (113). However, testosterone replacement therapy is associated with side effects such as acne and increased blood viscosity. Prostaglandin E2 (PGE2) is a potent bone anabolic agent that stimulates osteoblast differentiation and proliferation, promoting bone formation and increasing bone mass.
5.1.5 Strontium salts
Strontium salts promote osteoblast proliferation and differentiation while inhibiting osteoclast formation and differentiation, inducing osteoclast apoptosis, and reducing bone resorption. They enhance bone mechanical strength without altering bone structure (114), making them dual-action bone metabolism regulators. Strontium also modulates mesenchymal stem cells, increases bone formation rates, and promotes the synthesis of collagen and non-collagenous proteins in the bone matrix, exerting anti-osteoporotic effects (126).
Strontium ranelate, a representative strontium salt, reduces the risk of osteoporosis-related fractures by simultaneously inhibiting bone resorption and promoting bone formation. A study by Reginster et al. demonstrated that strontium ranelate treatment reduced the relative risk of all non-vertebral fractures by 16% and major fragility fractures by 19% compared to placebo (115). However, the use of strontium salts is associated with increased risks of myocardial infarction, thromboembolic events, and rare severe skin reactions (DRESS syndrome), leading to their non-approval in the United States (103).
5.2 Bone resorption inhibitors
5.2.1 Calcitonin
Calcitonin is a peptide hormone secreted by thyroid C cells and serves as a potent inhibitor of bone resorption. Its family peptide, amylin, can suppress osteoclast activity (127). The calcitonin receptor (CTR), a class B G protein-coupled receptor (GPCR), regulates calcium homeostasis and bone turnover upon activation by calcitonin, making it a therapeutic target for osteoporosis (116). Although calcitonin’s physiological role is to inhibit osteoclast-mediated bone resorption, its effects on osteoblasts are likely indirect and mediated through osteoclasts. Salmon calcitonin (MI), a peptide hormone with higher affinity for CTR than human calcitonin, is currently used as a clinical treatment for osteoporosis due to its ability to promote bone remodeling and inhibit bone resorption (117). However, calcitonin’s efficacy in fracture prevention is limited, and long-term use may increase the risk of cancer.
5.2.2 Bisphosphonates
Bisphosphonates (BPs), stable analogs of pyrophosphate characterized by a P-C-P group, are first-line drugs for the prevention and treatment of osteoporosis in postmenopausal women and men, as well as for alleviating bone pain and fractures. BPs promote osteoblast secretion of osteoprotegerin (OPG), and their mechanism of action in inhibiting bone resorption is twofold. On one hand, they regulate osteoclast proliferation, differentiation, and apoptosis; on the other hand, they suppress bone resorption by inhibiting osteoclast-mediated expression of cytokines such as interleukin-6 (IL - 6) and tumor necrosis factor-alpha (TNF-α) (118). Representative BPs include alendronate, zoledronate, and risedronate. While BPs are highly effective in treating osteoporosis, they are associated with side effects such as atypical femoral fractures and osteonecrosis of the jaw. For patients with severe osteoporosis and high fracture risk, long-term fracture prevention and bone mineral density (BMD) restoration may be challenging with BPs alone. A sequential approach—starting with bone-forming agents like teriparatide followed by long-term anti-resorptive therapy—may be beneficial (119).
5.2.3 Estrogens
Osteoporosis is closely associated with estrogen levels. Estrogens regulate bone homeostasis by directly acting on the immune system, oxidative stress, and bone cells, increasing bone mass, effectively modulating excessive bone turnover, and improving bone mineral density (BMD) (122). Raloxifene, the first selective estrogen receptor modulator (SERM) approved by the FDA for postmenopausal osteoporosis, is primarily used in postmenopausal women with significantly reduced estrogen levels. Raloxifene inhibits osteoclast activity through multiple signaling pathways without increasing the risk of endometrial cancer (120). It effectively reduces the risk of vertebral fractures in osteoporotic patients (121). Currently, most clinical treatments for postmenopausal osteoporosis are based on estrogen replacement therapy. However, long-term estrogen use increases the risk of breast, cervical, and endometrial cancers in women.
5.3 Bone metabolism modulators (active vitamin d and calcium supplements)
Calcium is a primary component of human bones and is essential for maintaining bone mineralization and metabolism. Osteoporosis is largely caused by calcium deficiency, which can be addressed by supplementing with calcium carbonate, calcium citrate, or calcium gluconate to increase calcium intake. Vitamin D promotes the absorption and storage of calcium ions in the body and directly influences calcium-phosphorus balance. Vitamin D metabolizes calcitriol, a circulating calcium-regulating hormone, accelerates gene synthesis in osteoblasts, and promotes bone mineralization (123). Active vitamin D directly acts on the parathyroid glands, reducing the synthesis and secretion of parathyroid hormone (PTH). It also binds to vitamin D receptors in osteoblasts and osteoclasts, ultimately promoting calcium and phosphorus deposition and mineralization in bones. However, severe adverse effects, such as hypercalcemia-induced renal dysfunction, may occur, necessitating regular monitoring of blood calcium levels.
5.4 Traditional Chinese medicine for osteoporosis treatment
Although currently approved anti-osteoporotic drugs can reduce the risk of fragility fractures, their long-term use is often limited by safety concerns, significant side effects, and low target specificity. In contrast, TCM, with its minimal side effects, offers unique advantages in the treatment of chronic diseases, including osteoporosis. TCM has a long history of use in preventing and treating osteoporosis (128). Based on modern medical understanding of osteoporosis pathology and clinical manifestations, TCM classifies osteoporosis as “bone impediment”, “bone wilting”, or “bone withering”. The primary pathogenesis involves kidney deficiency, blood stasis, and dual deficiency of qi and yin, with the disease located in the bones. The essence of osteoporosis in TCM is “kidney essence deficiency and spleen deficiency leading to malnourishment,” characterized by blood stasis obstruction (129). Table 3 summarized the active ingredients and mechanisms of action (signaling pathways) of commonly used traditional Chinese medicines.
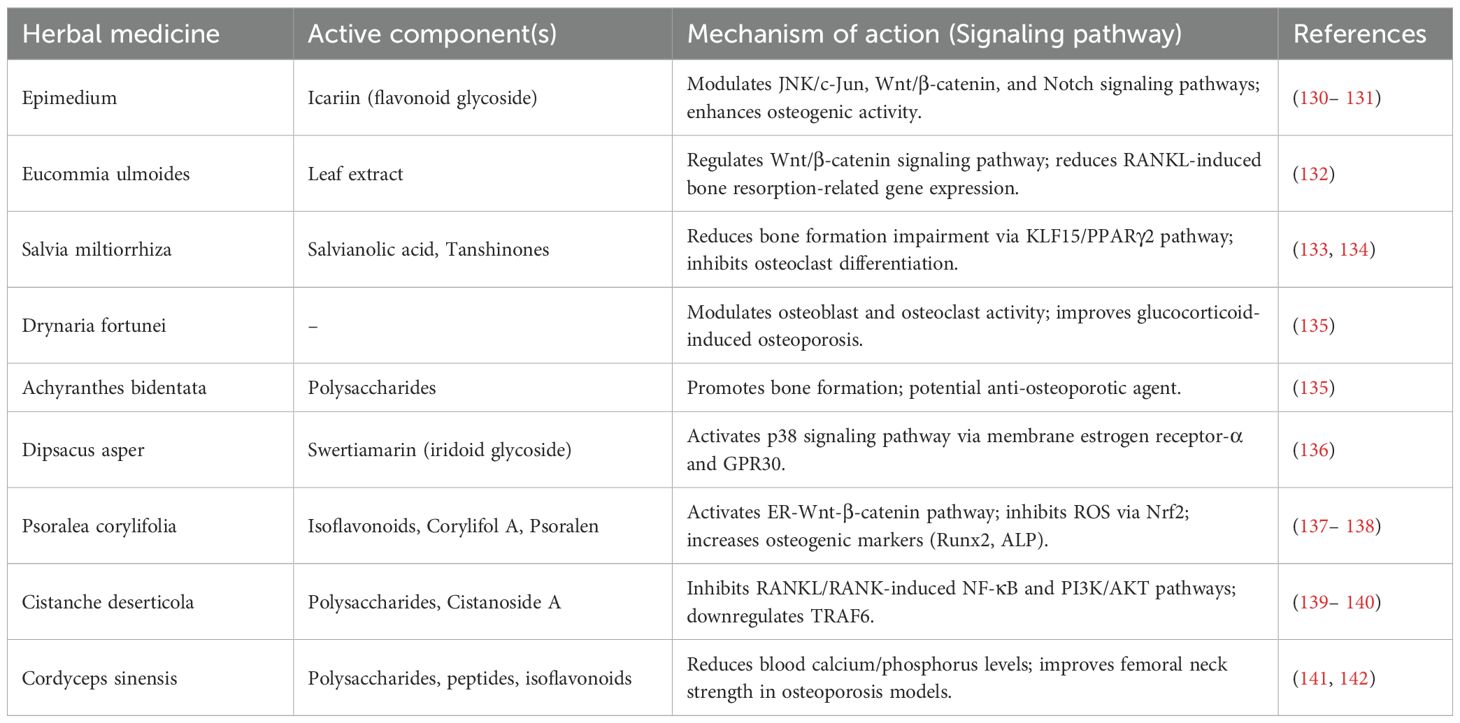
Table 3. Active components of commonly used traditional Chinese medicines and their mechanisms of action (signaling pathways).
According to TCM theory, bone health is closely related to kidney function. Kidney deficiency affects calcium and phosphorus metabolism, leading to reduced bone mineral density and impaired bone metabolism. Many kidney-nourishing herbs have been found to restore bone health and are used to treat bone-related diseases. TCM treatment primarily employs methods to tonify the kidneys and strengthen bones, focusing on replenishing kidney essence, while also supporting liver and spleen health, promoting blood circulation, and alleviating pain (143).
Herbal formulations, with their multi-component and multi-target advantages, have gained significant attention for the treatment of chronic diseases. Herbal medicines are derived from various parts of medicinal plants (e.g., leaves, stems, buds, flowers, or roots) and sometimes include non-plant components (e.g., insects, deer antlers, snakes, shells, and fossil powders). These ingredients can be used in their raw form or as water or alcohol extracts (144). They contain multiple active compounds, such as flavonoids, polysaccharides, saponins, and alkaloids, which exert therapeutic effects by regulating hormone levels, calcium-phosphorus metabolism, inflammatory responses, osteoclast activity, and osteoblast proliferation, ultimately improving bone microstructure.
Flavonoids, as the primary active components of many herbal medicines, play a significant role in treating osteoporosis. Epimedium, known for its kidney-tonifying and bone-strengthening properties, has a long history of use in treating bone diseases in China. Studies have shown that Epimedium, with its high flavonoid content, can effectively treat osteoporotic distal radius fractures (130). Icariin, the main active flavonoid glycoside in Epimedium, enhances osteogenic activity by modulating the JNK/c-Jun, Wnt/β-catenin, and Notch signaling pathways (131, 145).
Eucommia ulmoides leaf extract promotes osteogenic differentiation of bone marrow mesenchymal stem cells (BMSCs) by regulating the Wnt/β-catenin signaling pathway and reducing RANKL-induced bone resorption-related gene expression (132). Salvia miltiorrhiza has also been used historically to treat bone diseases (146). With advancements in modern analytical techniques, numerous compounds have been isolated from Salvia miltiorrhiza. Salvianolic acid, one of these compounds, plays a role in reducing bone formation impairment through the KLF15/PPARγ2 signaling pathway (133). Additionally, tanshinones have been shown to inhibit osteoclast differentiation and are considered potential candidates for osteoporosis treatment (134).
Drynaria fortunei improves glucocorticoid-induced osteoporosis by modulating osteoblast and osteoclast activity. Achyranthes bidentata, another traditional Chinese herb for osteoporosis, contains polysaccharides that have been extensively studied for their ability to promote bone formation, making them potential agents in anti-osteoporotic therapy (135).
Swertiamarin, a major active iridoid glycoside isolated from Dipsacus asper, exerts anti-osteoporotic effects by interacting with membrane estrogen receptor-α and GPR30, thereby activating the p38 signaling pathway (136). Asperosaponin VI, a triterpenoid saponin, exhibits anti-osteoclastic activity by inhibiting RANKL-induced osteoclast differentiation and function (147).
Certain compounds in Psoralea corylifolia exhibit anti-osteoporotic (OP) activity by activating the ER-Wnt-β-catenin signaling pathway, with isoflavonoids being the most potent (137). Corylifol A, a representative flavonoid, reduces reactive oxygen species (ROS) production by activating Nrf2, thereby inhibiting osteoclast formation and activation (148). Psoralen, another flavonoid from Psoralea corylifolia, increases the expression of osteogenic markers such as Runt-related transcription factor 2 (Runx2), osterix, type I collagen (Col1), and alkaline phosphatase (ALP), making it a targeted therapy for osteoblast-mediated OP (138).
Cistanche deserticola, an edible medicinal herb, inhibits RANKL/RANK-induced activation of downstream NF-κB and PI3K/AKT pathways and blocks the activity of key osteoclastogenic proteins, NFAT2 and c-Fos (139). Polysaccharides from Cistanche deserticola reduce RANKL-mediated ROS production in osteoclasts, impairing osteoclastogenesis and bone resorption (149). Cistanoside A, a phenylethanoid glycoside isolated from Cistanche deserticola, has potential in treating OP by downregulating TRAF6 (140).
Cordyceps sinensis contains active components such as polysaccharides, cordyceps peptides, isoflavonoids, amino acids, and alcohols. Cordyceps sinensis and its extracts have shown therapeutic effects on various types of osteoporosis. In a classic bilateral ovariectomy rat model simulating postmenopausal osteoporosis, Wei Qi et al. demonstrated that daily oral administration of 10 ml of Cordyceps sinensis mycelium extract significantly reduced blood calcium, blood phosphorus, and urinary calcium levels while increasing the mechanical strength of the femoral neck, effectively improving osteoporosis (141, 142).
In summary, many TCM formulations and their derived compounds demonstrate potential therapeutic effects in preventing and treating OP. Additionally, TCM therapies such as acupoint application, acupuncture, and massage can be used to improve bone mineral density.
6 Prevention and treatment of diabetic osteoporosis
The diagnosis of diabetic osteoporosis requires a comprehensive evaluation using multiple methods. Currently, clinical diagnostic techniques include dual-energy X-ray absorptiometry (DEXA), the Fracture Risk Assessment Tool (FRAX), trabecular bone score (TBS), and high-resolution peripheral quantitative computed tomography (HR-pQCT). DEXA, the gold standard for osteoporosis diagnosis, reveals that patients with type 1 diabetes (T1DM) exhibit significantly reduced bone mineral density (BMD), while those with type 2 diabetes (T2DM) show BMD values 5 – 10% higher than non-diabetic individuals (150). This paradoxical phenomenon may be related to the increased bone fragility in T2DM patients due to impaired bone quality. Despite higher BMD, T2DM patients exhibit elevated fracture risk due to trabecular microarchitecture deterioration, marrow adiposity, and AGEs-induced collagen brittleness. While FRAX includes T1DM as an osteoporosis risk factor, it substantially underestimates fracture risk in T2DM patients by 30 - 50%, according to studies (151). Therefore, a stricter diagnostic threshold (-2.0 vs. -2.5) is recommended for T2DM patients (152). Notably, FRAX assessments for T2DM patients require multi-parameter adjustments, including incorporating TBS to evaluate bone microstructure, adding rheumatoid arthritis as an input, reducing the femoral neck T-score by 0.5, and increasing the age parameter by 10 years (153). To more accurately assess fracture risk in diabetic patients, the combined use of FRAX and TBS is considered a more effective approach. By incorporating TBS as an adjustment factor for FRAX, clinicians can achieve more precise fracture risk stratification in diabetic patients, thereby better guiding decisions regarding osteoporosis treatment initiation (154). For example, a study conducted in Manitoba, Canada, compared four different methods for improving FRAX performance in type 2 diabetic patients (153). The results demonstrated that FRAX adjusted by TBS provided a more accurate fracture risk assessment in this population. Furthermore, TBS can identify elevated fracture risk in diabetic patients who have normal BMD but impaired bone microarchitecture. These patients might otherwise be overlooked if assessed solely using FRAX without TBS adjustment (155).
Advanced imaging techniques, such as TBS, analyze the spatial variation of grayscale in DEXA images to assess trabecular bone microstructure. A TBS value ≤1.2 indicates significant microstructural degradation and serves as a critical supplementary parameter for FRAX evaluation in T2DM patients. HR-pQCT provides three-dimensional volumetric BMD data, revealing that T1DM patients exhibit significant reductions in trabecular and cortical bone volume, closely associated with glycemic control and microvascular complications. In contrast, T2DM patients primarily show increased cortical bone porosity. Histomorphometric analysis demonstrates that even T1DM patients with good glycemic control exhibit normal bone formation markers but have significantly elevated abnormal deposition of advanced glycation end products (AGEs) in bone collagen. This non-enzymatic cross-linking, also observed in T2DM patients, may reduce bone matrix elasticity and increase fragility.
Bone is a dynamic tissue that undergoes continuous remodeling through bone formation (mediated by osteoblasts) and bone resorption (mediated by osteoclasts) (156). Bone turnover markers (BTMs) are biochemical byproducts released into blood or urine during these processes, reflecting the rate and balance of bone remodeling (157). In DOP patients, bone metabolism is often dysregulated. Notably, individuals with T2DM may exhibit normal or even elevated BMD while still having increased fracture risk-a phenomenon that cannot be explained by BMD measurements alone. This highlights the need for more nuanced indicators of bone health, and BTMs provide precisely such dynamic information. P1NP (procollagen type I N-terminal propeptide), a byproduct of type I collagen synthesis which is the primary structural protein of bone matrix, reflects osteoblastic activity in both healthy postmenopausal women and those with T2DM (158). Studies show reduced P1NP levels in diabetic patients. Osteocalcin, a non-collagenous protein secreted by osteoblasts that is critical for bone matrix mineralization, exists in an undercarboxylated form (ucOC) that may regulate glucose metabolism (158). This protein serves as a potential link between bone and glucose homeostasis, and its levels are consistently observed to be lower in diabetic populations. The combined evaluation of BTMs such as P1NP and osteocalcin with BMD and clinical tools like FRAX+TBS could enable earlier detection of skeletal fragility in diabetes, particularly in cases where BMD alone is misleading (159). The integrated application of these diagnostic tools enables a more accurate assessment of bone quality and fracture risk in diabetic patients, providing a critical foundation for individualized prevention and treatment strategies.
The International Osteoporosis Foundation (IOF) provides professional guidelines for the diagnosis and management of diabetic osteoporosis (160). For patients with type 2 diabetes (T2DM), baseline bone mineral density (BMD) testing using dual-energy X-ray absorptiometry (DEXA) is recommended five years after diagnosis, provided there are no additional risk factors (e.g., glucocorticoid use or advanced age). If the initial assessment indicates a low fracture risk, follow-up evaluations are advised every 3 – 5 years (160). For individuals with type 1 diabetes (T1DM), the International Society for Pediatric and Adolescent Diabetes (ISPAD) recommends BMD screening during late adolescence, particularly for those with celiac disease, poor long-term glycemic control, or existing microvascular complications.
In terms of prevention and management, lifestyle modifications to maintain a normal body mass index (BMI) are considered crucial. Overweight or obese patients should aim to reduce weight through calorie control and increased physical activity. Additionally, the IOF has proposed a six-step guide for fall prevention, which includes ensuring a safe home environment (e.g., adequate lighting and handrails), wearing slip-resistant footwear, correcting visual impairments, and maintaining healthy exercise and dietary habits. Diabetic patients should incorporate foods rich in vitamins, minerals, and protein into their daily diet. Diabetic patients should incorporate foods rich in vitamins, minerals, and protein into their daily diet to support bone health. Vitamin D levels should be maintained at ≥20 ng/mL. To achieve this, patients are advised to take 1000 – 2000 IU of vitamin D daily. If daily dietary calcium intake falls below 1.2 g, calcium supplements should be prescribed to ensure adequate bone mineralization and reduce fracture risk.A meta-analysis suggests that a Mediterranean diet based on fresh fruits, vegetables, and fish can reduce the risk of fractures and microvascular complications in T2DM patients (161).
7 Future perspectives
With the deepening understanding of the pathological mechanisms underlying diabetic osteoporosis (DOP), future research should focus on two major directions: the development of novel drugs and the systematic exploration of multi-dimensional interaction networks.
7.1 Development of novel antidiabetic drugs
While current glucose-lowering agents effectively improve glycemic control, their dual effects on bone metabolism require optimization. Emerging antidiabetic drugs, such as GLP - 1 receptor agonists and SGLT2 inhibitors, have demonstrated potential to enhance bone quality by modulating energy metabolism, inflammatory pathways, or oxidative stress. For instance, GLP - 1 receptor agonists may promote bone formation and inhibit bone resorption through AMPK signaling activation, while SGLT2 inhibitors could indirectly influence bone metabolism balance via the kidney-bone axis. Future studies should further elucidate the bone-protective mechanisms of these drugs and develop novel agents that combine glucose-lowering and bone-targeting functions, such as gut hormone-based peptide analogs or small-molecule compounds targeting bone microenvironment metabolic reprogramming. Additionally, the development of novel bone therapeutics targeting specific defects in bone quality—such as AGEs-RAGE pathway inhibitors, ferroptosis modulators, or mitochondrial function regulators—holds promise for overcoming the limitations of traditional anti-osteoporotic drugs and achieving precise intervention in diabetic bone disease.
7.2 Personalized medicine approaches
The heterogeneity of diabetic osteoporosis (DOP) demands a paradigm shift toward precision medicine, integrating molecular profiling, biomarker stratification, and tailored therapeutic interventions to optimize skeletal outcomes. Emerging evidence supports the use of genomic risk prediction, where polymorphisms in VDR (e.g., TaqI, EcoRV), RANKL/RANK/OPG signaling, and FOXQ1 expression patterns may identify patients at heightened risk for AGEs-mediated bone fragility or poor response to conventional therapies. Combining these genetic markers with clinical variables-such as diabetes duration, glycemic variability, and microvascular complications-could refine fracture risk assessment beyond traditional tools like FRAX, particularly in T2DM patients with preserved BMD but impaired bone quality. Biomarker-guided strategies hold promise for individualized treatment selection, leveraging serum pentosidine to quantify advanced glycation end-product burden or sclerostin levels to assess Wnt pathway suppression. Patients exhibiting low bone turnover (evidenced by depressed osteocalcin/P1NP ratios) may benefit preferentially from anabolic agents like teriparatide, while those with elevated resorption markers could derive greater protection from antiresorptives such as bisphosphonates or denosumab.
7.3 Systematic analysis of drug interaction networks and multi-target intervention strategies
Current research predominantly focuses on the mechanisms of individual drug classes, while the synergistic or antagonistic effects of combined “glucose-lowering and bone-protective” therapies in clinical practice remain poorly understood. Future efforts should systematically dissect the interaction networks between antidiabetic and anti-osteoporotic drugs at molecular, cellular, and systemic levels. For example, metformin and bisphosphonates may synergistically inhibit bone resorption through autophagy pathway regulation, whereas thiazolidinediones (TZDs) and RANKL inhibitors may exhibit reduced efficacy due to PPARγ signaling interference. Further exploration of key regulatory nodes, such as ferroptosis, VDR signaling, and Wnt/β-catenin pathways, will reveal the mechanisms underlying combined therapy efficacy and potential risks. Moreover, integrated multi-omics and artificial intelligence-based analyses can identify multi-target intervention strategies, such as dual-functional molecules that simultaneously inhibit AGEs accumulation and enhance bone formation, or multi-dimensional approaches targeting the bone-vascular-immune axis. These advancements will not only provide a theoretical foundation for optimizing clinical combination therapies but also pave the way for minimizing adverse drug reactions and developing personalized treatment regimens.
8 Conclusions
The prevention and treatment of diabetic osteoporosis (DOP) require a comprehensive approach based on its multifactorial pathological mechanisms, integrating metabolic regulation, bone microenvironment homeostasis, and systemic inflammation. This review systematically summarizes the pathophysiological network of DOP, encompassing core mechanisms such as insulin/IGF-1 deficiency, advanced glycation end products (AGEs) accumulation, oxidative stress, and vascular damage. It also elucidates the dual effects and potential risks of glucose-lowering and anti-osteoporotic drugs. Although current diagnostic and therapeutic approaches have significantly improved patient outcomes, precise interventions targeting bone quality deterioration remain challenging. Future research should focus on exploring novel drug targets, optimizing combination therapies, and advancing personalized treatment strategies through translational medicine. Ultimately, the goal is to achieve dual metabolic and skeletal benefits in diabetic bone disease.
Author contributions
XM: Writing – original draft. XZ: Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Xiao PL, Cui AY, Hsu CJ, Peng R, Jiang N, Xu XH, et al. Global, regional prevalence, and risk factors of osteoporosis according to the world health organization diagnostic criteria: A systematic review and meta-analysis. Osteoporosis Int. (2022) 33:2137–53. doi: 10.1007/s00198-022-06454-3
2. Sing CW, Lin TC, Bartholomew S, Bell JS, Bennett C, Beyene K, et al. Global epidemiology of hip fractures: secular trends in incidence rate, post-fracture treatment, and all-cause mortality. J Bone Mineral Res. (2023) 38:1064–75. doi: 10.1002/jbmr.4821
3. Cooper C, Campion G, and Melton LJ 3rd. Hip fractures in the elderly: A world-wide projection. Osteoporos Int. (1992) 2:285–9. doi: 10.1007/bf01623184
4. Shanbhogue VV, Hansen S, Frost M, Brixen K, and Hermann AP. Bone disease in diabetes: another manifestation of microvascular disease? Lancet Diabetes Endocrinol. (2017) 5:827–38. doi: 10.1016/s2213-8587(17)30134-1
5. Vestergaard P. Discrepancies in bone mineral density and fracture risk in patients with type 1 and type 2 diabetes–a meta-analysis. Osteoporos Int. (2007) 18:427–44. doi: 10.1007/s00198-006-0253-4
6. Janghorbani M, Van Dam RM, Willett WC, and Hu FB. Systematic review of type 1 and type 2 diabetes mellitus and risk of fracture. Am J Epidemiol. (2007) 166:495–505. doi: 10.1093/aje/kwm106
7. Belli M, Bellia A, Sergi D, Barone L, Lauro D, and Barillà F. Glucose variability: A new risk factor for cardiovascular disease. Acta Diabetol. (2023) 60:1291–9. doi: 10.1007/s00592-023-02097-w
8. Huang Y, Yue L, Qiu JH, Gao M, Liu SJ, and Wang JS. Endothelial dysfunction and platelet hyperactivation in diabetic complications induced by glycemic variability. Hormone Metab Res. (2022) 54:419–28. doi: 10.1055/a-1880-0978
9. Škrha J, Šoupal J, Škrha J Jr., and Prázný M. Glucose variability, hba1c and microvascular complications. Rev Endocr Metab Disord. (2016) 17:103–10. doi: 10.1007/s11154-016-9347-2
10. Liu BX, Sun W, and Kong XQ. Perirenal fat: A unique fat pad and potential target for cardiovascular disease. Angiology. (2019) 70:584–93. doi: 10.1177/0003319718799967
11. Kim JH, Han EH, Jin ZW, Lee HK, Fujimiya M, Murakami G, et al. Fetal topographical anatomy of the upper abdominal lymphatics: its specific features in comparison with other abdominopelvic regions. Anat Rec (Hoboken). (2012) 295:91–104. doi: 10.1002/ar.21527
12. Jespersen NZ, Feizi A, Andersen ES, Heywood S, Hattel HB, Daugaard S, et al. Heterogeneity in the perirenal region of humans suggests presence of dormant brown adipose tissue that contains brown fat precursor cells. Mol Metab. (2019) 24:30–43. doi: 10.1016/j.molmet.2019.03.005
13. Zhao W, Zhang W, Ma H, and Yang M. Nipa2 regulates osteoblast function by modulating mitophagy in type 2 diabetes osteoporosis. Sci Rep. (2020) 10:3078. doi: 10.1038/s41598-020-59743-4
14. Lemasters JJ. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. (2005) 8:3–5. doi: 10.1089/rej.2005.8.3
15. Sukumar D, Schlussel Y, Riedt CS, Gordon C, Stahl T, and Shapses SA. Obesity alters cortical and trabecular bone density and geometry in women. Osteoporosis Int. (2011) 22:635–45. doi: 10.1007/s00198-010-1305-3
16. Tarquini B, Navari N, Perfetto F, Piluso A, Romano S, and Tarquini R. Evidence for bone mass and body fat distribution relationship in postmenopausal obese women. Arch Gerontology Geriatrics. (1997) 24:15–21. doi: 10.1016/S0167-4943(96)00723-6
17. American Diabetes A. 2. Classification and diagnosis of diabetes: standards of medical care in diabetes—2021. Diabetes Care. (2020) 44:S15–33. doi: 10.2337/dc21-S002
18. Sun H, Saeedi P, Karuranga S, Pinkepank M, Ogurtsova K, Duncan BB, et al. Idf diabetes atlas: global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res Clin Pract. (2022) 183:109119. doi: 10.1016/j.diabres.2021.109119
19. Wang L, Yu W, Yin X, Cui L, Tang S, Jiang N, et al. Prevalence of osteoporosis and fracture in China: the China osteoporosis prevalence study. JAMA Netw Open. (2021) 4:e2121106. doi: 10.1001/jamanetworkopen.2021.21106
20. Johnell O and Kanis JA. An estimate of the worldwide prevalence and disability associated with osteoporotic fractures. Osteoporosis Int. (2006) 17:1726–33. doi: 10.1007/s00198-006-0172-4
21. Kurra S, Fink DA, and Siris ES. Osteoporosis-associated fracture and diabetes. Endocrinol Metab Clin North Am. (2014) 43:233–43. doi: 10.1016/j.ecl.2013.09.004
22. Vestergaard P, Rejnmark L, and Mosekilde L. Relative fracture risk in patients with diabetes mellitus, and the impact of insulin and oral antidiabetic medication on relative fracture risk. Diabetologia. (2005) 48:1292–9. doi: 10.1007/s00125-005-1786-3
23. Fan Y, Wei F, Lang Y, and Liu Y. Diabetes mellitus and risk of hip fractures: A meta-analysis. Osteoporos Int. (2016) 27:219–28. doi: 10.1007/s00198-015-3279-7
24. Wang H, Ba Y, Xing Q, and Du JL. Diabetes mellitus and the risk of fractures at specific sites: A meta-analysis. BMJ Open. (2019) 9:e024067. doi: 10.1136/bmjopen-2018-024067
25. Jiang L, Song X, Yan L, Liu Y, Qiao X, and Zhang W. Molecular insights into the interplay between type 2 diabetes mellitus and osteoporosis: implications for endocrine health. Front Endocrinol (Lausanne). (2024) 15:1483512. doi: 10.3389/fendo.2024.1483512
26. Wu K, Guo C, and Li R. Clinical characterization of icotinib-induced chemoresistance in erlotinib-treated lung adenocarcinoma patient with egfr mutations: A case report. Med (Baltimore). (2019) 98:e15489. doi: 10.1097/md.0000000000015489
27. Dixit M, Poudel SB, and Yakar S. Effects of gh/igf axis on bone and cartilage. Mol Cell Endocrinol. (2021) 519:111052. doi: 10.1016/j.mce.2020.111052
28. Yuan S, Wan ZH, Cheng SL, Michaëlsson K, and Larsson SC. Insulin-like growth factor-1, bone mineral density, and fracture: A mendelian randomization study. J Clin Endocrinol Metab. (2021) 106:e1552–e8. doi: 10.1210/clinem/dgaa963
29. Ouquerke A and Blulel J. Osteoblasts and insulin: an overview. . J Biol Regul Homeost Agents. (2020) 35.
30. Torres-Costoso A, Pozuelo-Carrascosa DP, Álvarez-Bueno C, Ferri-Morales A, Miota Ibarra J, Notario-Pacheco B, et al. Insulin and bone health in young adults: the mediator role of lean mass. PloS One. (2017) 12:e0173874. doi: 10.1371/journal.pone.0173874
31. Gilbert MP and Pratley RE. The impact of diabetes and diabetes medications on bone health. Endocr Rev. (2015) 36:194–213. doi: 10.1210/er.2012-1042
32. Govoni KE. Insulin-like growth factor-I molecular pathways in osteoblasts: potential targets for pharmacological manipulation. Curr Mol Pharmacol. (2012) 5:143–52. doi: 10.2174/1874467211205020143
33. Lorentzon M and Cummings SR. Osteoporosis: the evolution of a diagnosis. J Intern Med. (2015) 277:650–61. doi: 10.1111/joim.12369
34. Wang Y, Jiang C, Shang Z, Qiu G, Yuan G, Xu K, et al. Ages/rage promote osteogenic differentiation in rat bone marrow-derived endothelial progenitor cells via mapk signaling. J Diabetes Res. (2022) 2022:4067812. doi: 10.1155/2022/4067812
35. Li Z, Wang X, Hong TP, Wang HJ, Gao ZY, and Wan M. Advanced glycosylation end products inhibit the proliferation of bone-marrow stromal cells through activating mapk pathway. Eur J Med Res. (2021) 26:94. doi: 10.1186/s40001-021-00559-x
36. Li Y, Shrestha A, Zhang H, Li L, Li D, Fu T, et al. Impact of diabetes mellitus simulations on bone cell behavior through in vitro models. J Bone Miner Metab. (2020) 38:607–19. doi: 10.1007/s00774-020-01101-5
37. Kalaitzoglou E, Popescu I, Bunn RC, Fowlkes JL, and Thrailkill KM. Effects of type 1 diabetes on osteoblasts, osteocytes, and osteoclasts. Curr Osteoporos Rep. (2016) 14:310–9. doi: 10.1007/s11914-016-0329-9
38. Chen F, Wang P, Dai F, Zhang Q, Ying R, Ai L, et al. Correlation between blood glucose fluctuations and osteoporosis in type 2 diabetes mellitus. Int J Endocrinol. (2025) 2025:8889420. doi: 10.1155/ije/8889420
39. Fishman SL, Sonmez H, Basman C, Singh V, and Poretsky L. The role of advanced glycation end-products in the development of coronary artery disease in patients with and without diabetes mellitus: A review. Mol Med. (2018) 24:59. doi: 10.1186/s10020-018-0060-3
40. Karim L and Bouxsein ML. Effect of type 2 diabetes-related non-enzymatic glycation on bone biomechanical properties. Bone. (2016) 82:21–7. doi: 10.1016/j.bone.2015.07.028
41. Depalle B, Qin Z, Shefelbine SJ, and Buehler MJ. Influence of cross-link structure, density and mechanical properties in the mesoscale deformation mechanisms of collagen fibrils. J Mech Behav BioMed Mater. (2015) 52:1–13. doi: 10.1016/j.jmbbm.2014.07.008
42. Hough FS, Pierroz DD, Cooper C, and Ferrari SL. Mechanisms in endocrinology: mechanisms and evaluation of bone fragility in type 1 diabetes mellitus. Eur J Endocrinol. (2016) 174:R127–38. doi: 10.1530/eje-15-0820
43. Baynes JW. Role of oxidative stress in development of complications in diabetes. Diabetes. (1991) 40:405–12. doi: 10.2337/diab.40.4.405
44. Schwartz AV, Garnero P, Hillier TA, Sellmeyer DE, Strotmeyer ES, Feingold KR, et al. Pentosidine and increased fracture risk in older adults with type 2 diabetes. J Clin Endocrinol Metab. (2009) 94:2380–6. doi: 10.1210/jc.2008-2498
45. Neumann T, Lodes S, Kästner B, Franke S, Kiehntopf M, Lehmann T, et al. High serum pentosidine but not esrage is associated with prevalent fractures in type 1 diabetes independent of bone mineral density and glycaemic control. Osteoporos Int. (2014) 25:1527–33. doi: 10.1007/s00198-014-2631-7
46. Yamamoto M, Yamaguchi T, Yamauchi M, Yano S, and Sugimoto T. Serum pentosidine levels are positively associated with the presence of vertebral fractures in postmenopausal women with type 2 diabetes. J Clin Endocrinol Metab. (2008) 93:1013–9. doi: 10.1210/jc.2007-1270
47. Park SY, Choi KH, Jun JE, and Chung HY. Effects of advanced glycation end products on differentiation and function of osteoblasts and osteoclasts. J Korean Med Sci. (2021) 36:e239. doi: 10.3346/jkms.2021.36.e239
48. Ge W, Jie J, Yao J, Li W, Cheng Y, and Lu W. Advanced glycation end products promote osteoporosis by inducing ferroptosis in osteoblasts. Mol Med Rep. (2022) 25(4):140. doi: 10.3892/mmr.2022.12656
49. Sun Y, Zhu Y, Liu X, Chai Y, and Xu J. Morroniside attenuates high glucose-induced bmsc dysfunction by regulating the glo1/age/rage axis. Cell Prolif. (2020) 53:e12866. doi: 10.1111/cpr.12866
50. Hofmann MA, Drury S, Fu C, Qu W, Taguchi A, Lu Y, et al. Rage mediates a novel proinflammatory axis: A central cell surface receptor for S100/calgranulin polypeptides. Cell. (1999) 97:889–901. doi: 10.1016/s0092-8674(00)80801-6
51. Chavakis T, Bierhaus A, Al-Fakhri N, Schneider D, Witte S, Linn T, et al. The pattern recognition receptor (Rage) is a counterreceptor for leukocyte integrins: A novel pathway for inflammatory cell recruitment. J Exp Med. (2003) 198:1507–15. doi: 10.1084/jem.20030800
52. Grimm S, Ott C, Hörlacher M, Weber D, Höhn A, and Grune T. Advanced-glycation-end-product-induced formation of immunoproteasomes: involvement of rage and jak2/stat1. Biochem J. (2012) 448:127–39. doi: 10.1042/bj20120298
53. Tanikawa T, Okada Y, Tanikawa R, and Tanaka Y. Advanced glycation end products induce calcification of vascular smooth muscle cells through rage/P38 mapk. J Vasc Res. (2009) 46:572–80. doi: 10.1159/000226225
54. Tanaka K, Yamaguchi T, Kanazawa I, and Sugimoto T. Effects of high glucose and advanced glycation end products on the expressions of sclerostin and rankl as well as apoptosis in osteocyte-like mlo-Y4-A2 cells. Biochem Biophys Res Commun. (2015) 461:193–9. doi: 10.1016/j.bbrc.2015.02.091
55. Kume S, Kato S, Yamagishi S, Inagaki Y, Ueda S, Arima N, et al. Advanced glycation end-products attenuate human mesenchymal stem cells and prevent cognate differentiation into adipose tissue, cartilage, and bone. J Bone Miner Res. (2005) 20:1647–58. doi: 10.1359/jbmr.050514
56. Daniele G, Winnier D, Mari A, Bruder J, Fourcaudot M, Pengou Z, et al. Sclerostin and insulin resistance in prediabetes: evidence of a cross talk between bone and glucose metabolism. Diabetes Care. (2015) 38:1509–17. doi: 10.2337/dc14-2989
57. Xu Y, Ma X, Pan X, He X, Xiao Y, and Bao Y. Correlations between serum concentration of three bone-derived factors and obesity and visceral fat accumulation in a cohort of middle aged men and women. Cardiovasc Diabetol. (2018) 17:143. doi: 10.1186/s12933-018-0786-9
58. Kim HY, Choe JW, Kim HK, Bae SJ, Kim BJ, Lee SH, et al. Negative association between metabolic syndrome and bone mineral density in koreans, especially in men. Calcif Tissue Int. (2010) 86:350–8. doi: 10.1007/s00223-010-9347-2
59. Shahen VA, Gerbaix M, Koeppenkastrop S, Lim SF, McFarlane KE, Nguyen ANL, et al. Multifactorial effects of hyperglycaemia, hyperinsulinemia and inflammation on bone remodelling in type 2 diabetes mellitus. Cytokine Growth Factor Rev. (2020) 55:109–18. doi: 10.1016/j.cytogfr.2020.04.001
60. Adami G. Regulation of bone mass in inflammatory diseases. Best Pract Res Clin Endocrinol Metab. (2022) 36:101611. doi: 10.1016/j.beem.2021.101611
61. Palermo A, D’Onofrio L, Buzzetti R, Manfrini S, and Napoli N. Pathophysiology of bone fragility in patients with diabetes. Calcif Tissue Int. (2017) 100:122–32. doi: 10.1007/s00223-016-0226-3
62. Wang T and He C. Tnf-α and il-6: the link between immune and bone system. Curr Drug Targets. (2020) 21:213–27. doi: 10.2174/1389450120666190821161259
63. Sims NA. Cell-specific paracrine actions of il-6 family cytokines from bone, marrow and muscle that control bone formation and resorption. Int J Biochem Cell Biol. (2016) 79:14–23. doi: 10.1016/j.biocel.2016.08.003
64. Volpe CMO, Villar-Delfino PH, Dos Anjos PMF, and Nogueira-MaChado JA. Cellular death, reactive oxygen species (Ros) and diabetic complications. Cell Death Dis. (2018) 9:119. doi: 10.1038/s41419-017-0135-z
65. Ghosh N, Das A, Chaffee S, Roy S, and Sen CK. Chapter 4 - reactive oxygen species, oxidative damage and cell death. In: Chatterjee, Jungraithmayr W, and Bagchi D, editors. Immunity and inflammation in health and disease. Academic Press (2018). p. 45–55. doi: 10.1016/B978-0-12-805417-8.00004-4
66. Checa J and Aran JM. Reactive oxygen species: drivers of physiological and pathological processes. J Inflammation Res. (2020) 13:1057–73. doi: 10.2147/jir.S275595
67. Quagliaro L, Piconi L, Assaloni R, Martinelli L, Motz E, and Ceriello A. Intermittent high glucose enhances apoptosis related to oxidative stress in human umbilical vein endothelial cells: the role of protein kinase C and nad(P)H-oxidase activation. Diabetes. (2003) 52:2795–804. doi: 10.2337/diabetes.52.11.2795
68. Piconi L, Quagliaro L, Assaloni R, Da Ros R, Maier A, Zuodar G, et al. Constant and intermittent high glucose enhances endothelial cell apoptosis through mitochondrial superoxide overproduction. Diabetes Metab Res Rev. (2006) 22:198–203. doi: 10.1002/dmrr.613
69. Horváth EM, Benko R, Kiss L, Murányi M, Pék T, Fekete K, et al. Rapid ‘Glycaemic swings’ Induce nitrosative stress, activate poly(Adp-ribose) polymerase and impair endothelial function in a rat model of diabetes mellitus. Diabetologia. (2009) 52:952–61. doi: 10.1007/s00125-009-1304-0
70. Zhang P, Liao J, Wang X, and Feng Z. High glucose promotes apoptosis and autophagy of mc3t3-E1 osteoblasts. Arch Med Sci. (2023) 19:138–50. doi: 10.5114/aoms.2020.101307
71. Le T, Salas Sanchez A, Nashawi D, Kulkarni S, and Prisby RD. Diabetes and the microvasculature of the bone and marrow. Curr Osteoporos Rep. (2024) 22:11–27. doi: 10.1007/s11914-023-00841-3
72. Johnson DL, McAllister TN, and Frangos JA. Fluid flow stimulates rapid and continuous release of nitric oxide in osteoblasts. Am J Physiol. (1996) 271:E205–8. doi: 10.1152/ajpendo.1996.271.1.E205
73. Zhu ZN, Jiang YF, and Ding T. Risk of fracture with thiazolidinediones: an updated meta-analysis of randomized clinical trials. Bone. (2014) 68:115–23. doi: 10.1016/j.bone.2014.08.010
74. Wei W, Dutchak PA, Wang X, Ding X, Wang X, Bookout AL, et al. Fibroblast growth factor 21 promotes bone loss by potentiating the effects of peroxisome proliferator-activated receptor Γ. Proc Natl Acad Sci U.S.A. (2012) 109:3143–8. doi: 10.1073/pnas.1200797109
75. Gilbert MP, Marre M, Holst JJ, Garber A, Baeres FMM, Thomsen H, et al. Comparison of the long-term effects of liraglutide and glimepiride monotherapy on bone mineral density in patients with type 2 diabetes. Endocrine Pract. (2016) 22:406–11. doi: 10.4158/EP15758.OR
76. Lapane KL, Jesdale BM, Dubé CE, Pimentel CB, and Rajpathak SN. Sulfonylureas and risk of falls and fractures among nursing home residents with type 2 diabetes mellitus. Diabetes Res Clin Pract. (2015) 109:411–9. doi: 10.1016/j.diabres.2015.05.009
77. Melton LJ 3rd, Leibson CL, Achenbach SJ, Therneau TM, and Khosla S. Fracture risk in type 2 diabetes: update of a population-based study. J Bone Miner Res. (2008) 23:1334–42. doi: 10.1359/jbmr.080323
78. Zheng L, Shen X, Xie Y, Lian H, Yan S, and Wang S. Metformin promotes osteogenic differentiation and prevents hyperglycaemia-induced osteoporosis by suppressing pparγ Expression. Acta Biochim Biophys Sin (Shanghai). (2023) 55:394–403. doi: 10.3724/abbs.2023043
79. Bunck MC, Eliasson B, Cornér A, Heine RJ, Shaginian RM, Taskinen MR, et al. Exenatide treatment did not affect bone mineral density despite body weight reduction in patients with type 2 diabetes. Diabetes Obes Metab. (2011) 13:374–7. doi: 10.1111/j.1463-1326.2010.01355.x
80. Iepsen EW, Lundgren JR, Hartmann B, Pedersen O, Hansen T, Jørgensen NR, et al. Glp-1 receptor agonist treatment increases bone formation and prevents bone loss in weight-reduced obese women. J Clin Endocrinol Metab. (2015) 100:2909–17. doi: 10.1210/jc.2015-1176
81. Watts NB, Bilezikian JP, Usiskin K, Edwards R, Desai M, Law G, et al. Effects of canagliflozin on fracture risk in patients with type 2 diabetes mellitus. J Clin Endocrinol Metab. (2016) 101:157–66. doi: 10.1210/jc.2015-3167
82. Neal B, Perkovic V, Mahaffey KW, de Zeeuw D, Fulcher G, Erondu N, et al. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med. (2017) 377:644–57. doi: 10.1056/NEJMoa1611925
83. Schwartz AV, Vittinghoff E, Sellmeyer DE, Feingold KR, de Rekeneire N, Strotmeyer ES, et al. Diabetes-related complications, glycemic control, and falls in older adults. Diabetes Care. (2008) 31:391–6. doi: 10.2337/dc07-1152
84. Jawad AS. Osteoporosis in patients with type 2 diabetes mellitus. Saudi Med J. (2023) 44:1221. doi: 10.15537/smj.2023.12.20230714
85. Eller-Vainicher C, Cairoli E, Grassi G, Grassi F, Catalano A, Merlotti D, et al. Pathophysiology and management of type 2 diabetes mellitus bone fragility. J Diabetes Res. (2020) 2020:7608964. doi: 10.1155/2020/7608964
86. Shockley KR, Lazarenko OP, Czernik PJ, Rosen CJ, Churchill GA, and Lecka-Czernik B. Ppargamma2 nuclear receptor controls multiple regulatory pathways of osteoblast differentiation from marrow mesenchymal stem cells. J Cell Biochem. (2009) 106:232–46. doi: 10.1002/jcb.21994
87. Soccio RE, Chen ER, and Lazar MA. Thiazolidinediones and the promise of insulin sensitization in type 2 diabetes. Cell Metab. (2014) 20:573–91. doi: 10.1016/j.cmet.2014.08.005
88. Kohler S, Kaspers S, Salsali A, Zeller C, and Woerle HJ. Analysis of fractures in patients with type 2 diabetes treated with empagliflozin in pooled data from placebo-controlled trials and a head-to-head study versus glimepiride. Diabetes Care. (2018) 41:1809–16. doi: 10.2337/dc17-1525
89. Lecka-Czernik B. Diabetes, bone and glucose-lowering agents: basic biology. Diabetologia. (2017) 60:1163–9. doi: 10.1007/s00125-017-4269-4
90. Ma P, Gu B, Ma J, E L, Wu X, Cao J, et al. Glimepiride induces proliferation and differentiation of rat osteoblasts via the pi3-kinase/akt pathway. Metabolism. (2010) 59:359–66. doi: 10.1016/j.metabol.2009.08.003
91. Napoli N, Strotmeyer ES, Ensrud KE, Sellmeyer DE, Bauer DC, Hoffman AR, et al. Fracture risk in diabetic elderly men: the mros study. Diabetologia. (2014) 57:2057–65. doi: 10.1007/s00125-014-3289-6
92. Kheniser KG, Polanco Santos CM, and Kashyap SR. The effects of diabetes therapy on bone: A clinical perspective. J Diabetes Complications. (2018) 32:713–9. doi: 10.1016/j.jdiacomp.2018.04.005
93. Zhen D, Chen Y, and Tang X. Metformin reverses the deleterious effects of high glucose on osteoblast function. J Diabetes Complications. (2010) 24:334–44. doi: 10.1016/j.jdiacomp.2009.05.002
94. Yang Y, Zhao C, Liang J, Yu M, and Qu X. Effect of dipeptidyl peptidase-4 inhibitors on bone metabolism and the possible underlying mechanisms. Front Pharmacol. (2017) 8:487. doi: 10.3389/fphar.2017.00487
95. Sofer E and Shargorodsky M. Effect of metformin treatment on circulating osteoprotegerin in patients with nonalcoholic fatty liver disease. Hepatol Int. (2016) 10:169–74. doi: 10.1007/s12072-015-9649-6
96. Mai QG, Zhang ZM, Xu S, Lu M, Zhou RP, Zhao L, et al. Metformin stimulates osteoprotegerin and reduces rankl expression in osteoblasts and ovariectomized rats. J Cell Biochem. (2011) 112:2902–9. doi: 10.1002/jcb.23206
97. Jeyabalan J, Viollet B, Smitham P, Ellis SA, Zaman G, Bardin C, et al. The anti-diabetic drug metformin does not affect bone mass in vivo or fracture healing. Osteoporos Int. (2013) 24:2659–70. doi: 10.1007/s00198-013-2371-0
98. Mabilleau G, Mieczkowska A, and Chappard D. Use of glucagon-like peptide-1 receptor agonists and bone fractures: A meta-analysis of randomized clinical trials. J Diabetes. (2014) 6:260–6. doi: 10.1111/1753-0407.12102
99. Su B, Sheng H, Zhang M, Bu L, Yang P, Li L, et al. Risk of bone fractures associated with glucagon-like peptide-1 receptor agonists’ Treatment: A meta-analysis of randomized controlled trials. Endocrine. (2015) 48:107–15. doi: 10.1007/s12020-014-0361-4
100. Glorie L, Behets GJ, Baerts L, De Meester I, D’Haese PC, and Verhulst A. Dpp iv inhibitor treatment attenuates bone loss and improves mechanical bone strength in male diabetic rats. Am J Physiol Endocrinol Metab. (2014) 307:E447–55. doi: 10.1152/ajpendo.00217.2014
101. Hegazy SK. Evaluation of the anti-osteoporotic effects of metformin and sitagliptin in postmenopausal diabetic women. J Bone Miner Metab. (2015) 33:207–12. doi: 10.1007/s00774-014-0581-y
102. Hidayat K, Du X, and Shi BM. Risk of fracture with dipeptidyl peptidase-4 inhibitors, glucagon-like peptide-1 receptor agonists, or sodium-glucose cotransporter-2 inhibitors in real-world use: systematic review and meta-analysis of observational studies. Osteoporos Int. (2019) 30:1923–40. doi: 10.1007/s00198-019-04968-x
103. Thrailkill KM, Nyman JS, Bunn RC, Uppuganti S, Thompson KL, Lumpkin CK Jr., et al. The impact of sglt2 inhibitors, compared with insulin, on diabetic bone disease in a mouse model of type 1 diabetes. Bone. (2017) 94:141–51. doi: 10.1016/j.bone.2016.10.026
104. Taylor SI, Blau JE, and Rother KI. Possible adverse effects of sglt2 inhibitors on bone. Lancet Diabetes Endocrinol. (2015) 3:8–10. doi: 10.1016/s2213-8587(14)70227-x
105. Nauck MA, Del Prato S, Meier JJ, Durán-García S, Rohwedder K, Elze M, et al. Dapagliflozin versus glipizide as add-on therapy in patients with type 2 diabetes who have inadequate glycemic control with metformin: A randomized, 52-week, double-blind, active-controlled noninferiority trial. Diabetes Care. (2011) 34:2015–22. doi: 10.2337/dc11-0606
106. Monami M, Cresci B, Colombini A, Pala L, Balzi D, Gori F, et al. Bone fractures and hypoglycemic treatment in type 2 diabetic patients: A case-control study. Diabetes Care. (2008) 31:199–203. doi: 10.2337/dc07-1736
107. Liu S, Zhou H, Liu H, Ji H, Fei W, and Luo E. Fluorine-contained hydroxyapatite suppresses bone resorption through inhibiting osteoclasts differentiation and function in vitro and in vivo. Cell Prolif. (2019) 52:e12613. doi: 10.1111/cpr.12613
108. Ciosek Ż, Kot K, Kosik-Bogacka D, Łanocha-Arendarczyk N, and Rotter I. The effects of calcium, magnesium, phosphorus, fluoride, and lead on bone tissue. Biomolecules. (2021) 11(4):506. doi: 10.3390/biom11040506
109. Krege JH, Lane NE, Harris JM, and Miller PD. Pinp as a biological response marker during teriparatide treatment for osteoporosis. Osteoporos Int. (2014) 25:2159–71. doi: 10.1007/s00198-014-2646-0
110. Teitelbaum SL. Bone resorption by osteoclasts. Science. (2000) 289:1504–8. doi: 10.1126/science.289.5484.1504
111. Chowdhury K, Sharma A, Sharma T, Kumar S, and Mandal CC. Simvastatin and mbcd inhibit breast cancer-induced osteoclast activity by targeting osteoclastogenic factors. Cancer Invest. (2017) 35:403–13. doi: 10.1080/07357907.2017.1309548
112. Rinonapoli G, Ruggiero C, Meccariello L, Bisaccia M, Ceccarini P, and Caraffa A. Osteoporosis in men: A review of an underestimated bone condition. Int J Mol Sci. (2021) 22(4):2105. doi: 10.3390/ijms22042105
113. Vajda EG, Hogue A, Griffiths KN, Chang WY, Burnett K, Chen Y, et al. Combination treatment with a selective androgen receptor modulator Q(Sarm) and a bisphosphonate has additive effects in osteopenic female rats. J Bone Mineral Res. (2009) 24:231–40. doi: 10.1359/jbmr.081007
114. Chu JG, Dai MW, Wang Y, Tian FM, Song HP, Xiao YP, et al. Strontium ranelate causes osteophytes overgrowth in a model of early phase osteoarthritis. BMC Musculoskelet Disord. (2017) 18:78. doi: 10.1186/s12891-017-1399-2
115. Reginster JY, Seeman E, De Vernejoul MC, Adami S, Compston J, Phenekos C, et al. Strontium ranelate reduces the risk of nonvertebral fractures in postmenopausal women with osteoporosis: treatment of peripheral osteoporosis (Tropos) study. J Clin Endocrinol Metab. (2005) 90:2816–22. doi: 10.1210/jc.2004-1774
116. Lee SM, Jeong Y, Simms J, Warner ML, Poyner DR, Chung KY, et al. Calcitonin receptor N-glycosylation enhances peptide hormone affinity by controlling receptor dynamics. J Mol Biol. (2020) 432:1996–2014. doi: 10.1016/j.jmb.2020.01.028
117. Lee S. Development of high affinity calcitonin analog fragments targeting extracellular domains of calcitonin family receptors. Biomolecules. (2021) 11(9):1364. doi: 10.3390/biom11091364
118. Russell RG, Watts NB, Ebetino FH, and Rogers MJ. Mechanisms of action of bisphosphonates: similarities and differences and their potential influence on clinical efficacy. Osteoporos Int. (2008) 19:733–59. doi: 10.1007/s00198-007-0540-8
119. Lorentzon M. Treating osteoporosis to prevent fractures: current concepts and future developments. J Intern Med. (2019) 285:381–94. doi: 10.1111/joim.12873
120. Sverdlov O, Ryeznik Y, Anisimov V, Kuznetsova OM, Knight R, Carter K, et al. Selecting a randomization method for a multi-center clinical trial with stochastic recruitment considerations. BMC Med Res Method. (2024) 24:52. doi: 10.1186/s12874-023-02131-z
121. Ayub N, Faraj M, Ghatan S, Reijers JAA, Napoli N, and Oei L. The treatment gap in osteoporosis. J Clin Med. (2021) 10(13):3002. doi: 10.3390/jcm10133002
122. Weitzmann MN and Pacifici R. Estrogen deficiency and bone loss: an inflammatory tale. J Clin Invest. (2006) 116:1186–94. doi: 10.1172/jci28550
123. Al-Dujaili SA, Koh AJ, Dang M, Mi X, Chang W, Ma PX, et al. Calcium sensing receptor function supports osteoblast survival and acts as a co-factor in pth anabolic actions in bone. J Cell Biochem. (2016) 117:1556–67. doi: 10.1002/jcb.25447
124. Haguenauer D, Welch V, Shea B, Tugwell P, and Wells G. Fluoride for treating postmenopausal osteoporosis. Cochrane Database Syst Rev. (2000) 2000:Cd002825. doi: 10.1002/14651858.Cd002825
125. Guo Y, Huo J, Wu D, Hao H, Ji X, Zhao E, et al. Simvastatin Inhibits the Adipogenesis of Bone Marrow−Derived Mesenchymal Stem Cells through The downregulation of Chemerin/Cmklr1 Signaling. Int J Mol Med. (2020) 46:751–61. doi: 10.3892/ijmm.2020.4606
126. Kołodziejska B, Stępień N, and Kolmas J. The influence of strontium on bone tissue metabolism and its application in osteoporosis treatment. Int J Mol Sci. (2021) 22(12):6564. doi: 10.3390/ijms22126564
127. Naot D, Musson DS, and Cornish J. The activity of peptides of the calcitonin family in bone. Physiol Rev. (2019) 99:781–805. doi: 10.1152/physrev.00066.2017
128. Zhang ND, Han T, Huang BK, Rahman K, Jiang YP, Xu HT, et al. Traditional chinese medicine formulas for the treatment of osteoporosis: implication for antiosteoporotic drug discovery. J Ethnopharmacol. (2016) 189:61–80. doi: 10.1016/j.jep.2016.05.025
129. Mohammad NA, Razaly NI, Rani MDM, Aris MSM, and Effendy NM. An evidence-based review: the effects of Malaysian traditional herbs on osteoporotic rat models. Malays J Med Sci. (2018) 25:6–30. doi: 10.21315/mjms2018.25.4.2
130. Xue L, Jiang Y, Han T, Zhang N, Qin L, Xin H, et al. Comparative proteomic and metabolomic analysis reveal the antiosteoporotic molecular mechanism of icariin from epimedium brevicornu maxim. J Ethnopharmacol. (2016) 192:370–81. doi: 10.1016/j.jep.2016.07.037
131. Xu Y, Li L, Tang Y, Yang J, Jin Y, and Ma C. Icariin promotes osteogenic differentiation by suppressing notch signaling. Eur J Pharmacol. (2019) 865:172794. doi: 10.1016/j.ejphar.2019.172794
132. Cheng C-F, Chien-Fu Lin J, Tsai F-J, Chen C-J, Chiou J-S, Chou C-H, et al. Protective effects and network analysis of natural compounds obtained from radix dipsaci, eucommiae cortex, and rhizoma drynariae against rankl-induced osteoclastogenesis in vitro. J Ethnopharmacology. (2019) 244:112074. doi: 10.1016/j.jep.2019.112074
133. Yang YJ, Zhu Z, Wang DT, Zhang XL, Liu YY, Lai WX, et al. Tanshinol alleviates impaired bone formation by inhibiting adipogenesis via klf15/pparγ2 signaling in gio rats. Acta Pharmacol Sin. (2018) 39:633–41. doi: 10.1038/aps.2017.134
134. Lee SY, Choi DY, and Woo ER. Inhibition of osteoclast differentiation by tanshinones from the root of salvia miltiorrhiza bunge. Arch Pharm Res. (2005) 28:909–13. doi: 10.1007/bf02973876
135. Zhang S, Zhang Q, Zhang D, Wang C, and Yan C. Anti-osteoporosis activity of a novel achyranthes bidentata polysaccharide via stimulating bone formation. Carbohydr Polymers. (2018) 184:288–98. doi: 10.1016/j.carbpol.2017.12.070
136. Wu QC, Tang XY, Dai ZQ, Dai Y, Xiao HH, and Yao XS. Sweroside promotes osteoblastic differentiation and mineralization via interaction of membrane estrogen receptor-α and gpr30 mediated P38 signalling pathway on mc3t3-E1 cells. Phytomedicine. (2020) 68:153146. doi: 10.1016/j.phymed.2019.153146
137. Cai X-Y, Zhang Z-J, Xiong J-L, Yang M, and Wang Z-T. Experimental and molecular docking studies of estrogen-like and anti-osteoporosis activity of compounds in fructus psoraleae. J Ethnopharmacology. (2021) 276:114044. doi: 10.1016/j.jep.2021.114044
138. Yu T, Xiong Y, Luu S, You X, Li B, Xia J, et al. The shared kegg pathways between icariin-targeted genes and osteoporosis. Aging (Albany NY). (2020) 12:8191–201. doi: 10.18632/aging.103133
139. Zhang B, Yang LL, Ding SQ, Liu JJ, Dong YH, Li YT, et al. Anti-osteoporotic activity of an edible traditional chinese medicine cistanche deserticola on bone metabolism of ovariectomized rats through rankl/rank/traf6-mediated signaling pathways. Front Pharmacol. (2019) 10:1412. doi: 10.3389/fphar.2019.01412
140. Xu X, Zhang Z, Wang W, Yao H, and Ma X. Therapeutic effect of cistanoside a on bone metabolism of ovariectomized mice. Molecules. (2017) 22(2):197. doi: 10.3390/molecules22020197
141. Qi W, Yan YB, Wang PJ, and Lei W. The co-effect of cordyceps sinensis and strontium on osteoporosis in ovariectomized osteopenic rats. Biol Trace Elem Res. (2011) 141:216–23. doi: 10.1007/s12011-010-8711-4
142. Qi W, Wang PJ, Guo WJ, Yan YB, Zhang Y, and Lei W. The mechanism of cordyceps sinensis and strontium in prevention of osteoporosis in rats. Biol Trace Elem Res. (2011) 143:302–9. doi: 10.1007/s12011-010-8829-4
143. Yuan S, Zhang L, Ji L, Zhong S, Jiang L, Wan Y, et al. Foxo3a cooperates with runx1 to promote chondrogenesis and terminal hypertrophic of the chondrogenic progenitor cells. Biochem Biophys Res Commun. (2022) 589:41–7. doi: 10.1016/j.bbrc.2021.12.008
144. Liu Y, Liu JP, and Xia Y. Chinese herbal medicines for treating osteoporosis. Cochrane Database Syst Rev. (2014) 2014:Cd005467. doi: 10.1002/14651858.CD005467.pub2
145. Huang M, Wang Y, and Peng R. Icariin alleviates glucocorticoid-induced osteoporosis through ephb4/ephrin-B2 axis. Evid Based Complement Alternat Med. (2020) 2020:2982480. doi: 10.1155/2020/2982480
146. Guo Y, Li Y, Xue L, Severino RP, Gao S, Niu J, et al. Salvia miltiorrhiza: an ancient chinese herbal medicine as a source for anti-osteoporotic drugs. J Ethnopharmacol. (2014) 155:1401–16. doi: 10.1016/j.jep.2014.07.058
147. Liu K, Liu Y, Xu Y, Nandakumar KS, Tan H, He C, et al. Asperosaponin vi protects against bone destructions in collagen induced arthritis by inhibiting osteoclastogenesis. Phytomedicine. (2019) 63:153006. doi: 10.1016/j.phymed.2019.153006
148. Xu Y, Song D, Lin X, Peng H, Su Y, Liang J, et al. Corylifol a protects against ovariectomized-induced bone loss and attenuates rankl-induced osteoclastogenesis via ros reduction, erk inhibition, and nfatc1 activation. Free Radic Biol Med. (2023) 196:121–32. doi: 10.1016/j.freeradbiomed.2023.01.017
149. Song D, Cao Z, Liu Z, Tickner J, Qiu H, Wang C, et al. Cistanche deserticola polysaccharide attenuates osteoclastogenesis and bone resorption via inhibiting rankl signaling and reactive oxygen species production. J Cell Physiol. (2018) 233:9674–84. doi: 10.1002/jcp.26882
150. de L II, van der Klift M, de Laet CE, van Daele PL, Hofman A, and Pols HA. Bone mineral density and fracture risk in type-2 diabetes mellitus: the rotterdam study. Osteoporos Int. (2005) 16:1713–20. doi: 10.1007/s00198-005-1909-1
151. Giangregorio LM, Leslie WD, Lix LM, Johansson H, Oden A, McCloskey E, et al. Frax underestimates fracture risk in patients with diabetes. J Bone Miner Res. (2012) 27:301–8. doi: 10.1002/jbmr.556
152. Schwartz AV, Vittinghoff E, Bauer DC, Hillier TA, Strotmeyer ES, Ensrud KE, et al. Association of bmd and frax score with risk of fracture in older adults with type 2 diabetes. Jama. (2011) 305:2184–92. doi: 10.1001/jama.2011.715
153. Leslie WD, Johansson H, McCloskey EV, Harvey NC, Kanis JA, and Hans D. Comparison of methods for improving fracture risk assessment in diabetes: the manitoba bmd registry. J Bone Miner Res. (2018) 33:1923–30. doi: 10.1002/jbmr.3538
154. Chuang TL, Chuang MH, Wang YF, and Koo M. Comparison of Trabecular Bone Score-Adjusted Fracture Risk Assessment (Tbs-Frax) and Frax Tools for Identification of High Fracture Risk among Taiwanese Adults Aged 50 to 90 Years with or without Prediabetes and Diabetes. Medicina (Kaunas). (2022) 58(12):1766. doi: 10.3390/medicina58121766
155. Biamonte F, Pepe J, Colangelo L, Desideri G, Ettorre E, Nieddu L, et al. Assessment of trabecular bone score (Tbs) in the prediction of vertebral fracture in postmenopausal osteoporosis. Bone. (2025) 190:117307. doi: 10.1016/j.bone.2024.117307
156. Brown JP, Don-Wauchope A, Douville P, Albert C, and Vasikaran SD. Current use of bone turnover markers in the management of osteoporosis. Clin Biochem. (2022) 109 - 110:1–10. doi: 10.1016/j.clinbiochem.2022.09.002
157. Brunetti A, Cellini M, Vitale V, Gentile LMS, Birtolo MF, Vescini F, et al. A practical use of bone turnover markers in management of patients with skeletal fragility. Endocrine. (2025) 89:356–64. doi: 10.1007/s12020-025-04275-y
158. Wan Y, Hu C, Hou Y, Si C, Zhao Q, Wang Z, et al. Opg gene-modified adipose-derived stem cells improve bone formation around implants in osteoporotic rat maxillae. Heliyon. (2023) 9:e19474. doi: 10.1016/j.heliyon.2023.e19474
159. Holloway-Kew KL, De Abreu LLF, Kotowicz MA, Sajjad MA, and Pasco JA. Bone turnover markers in men and women with impaired fasting glucose and diabetes. Calcified Tissue Int. (2019) 104:599–604. doi: 10.1007/s00223-019-00527-y
160. Ferrari SL, Abrahamsen B, Napoli N, Akesson K, Chandran M, Eastell R, et al. Diagnosis and management of bone fragility in diabetes: an emerging challenge. Osteoporos Int. (2018) 29:2585–96. doi: 10.1007/s00198-018-4650-2
Keywords: diabetic osteoporosis, pathophysiological mechanisms, glucose-lowering agents, anti-osteoporotic agents, traditional Chinese medicine
Citation: Ma X and Zhang X (2025) Research progress of diabetic osteoporosis: a comprehensive review. Front. Endocrinol. 16:1595228. doi: 10.3389/fendo.2025.1595228
Received: 17 March 2025; Accepted: 17 August 2025;
Published: 02 September 2025.
Edited by:
Jiang Du, University of California, San Diego, United StatesReviewed by:
Yan Kai Dong, Lishui University, ChinaZitong Zhang, Icahn School of Medicine at Mount Sinai, United States
Copyright © 2025 Ma and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoqian Zhang, emhhbmd4aWFvcWlhbkBzZGZtdS5lZHUuY24=