 Esma Kaya Özdemir
Esma Kaya Özdemir Esra Döğer
Esra Döğer- Gazi University Faculty of Medicine, Department of Child Health and Diseases, Department of Pediatric Endocrinology, Ankara, Türkiye
Introduction: Short stature has many causes, including rare disorders of GH function. Bioinactive growth hormone (GH) refers to a phenotype characterized by immunoreactive but biologically ineffective GH. Importantly, it should not be regarded as a separate treatment but rather as a definable subgroup within the broader population of children receiving recombinant human growth hormone (rhGH) therapy. The aim of this study was to compare the growth response to rhGH among children with bioinactive GH, those born small for gestational age (SGA), and those with idiopathic short stature (ISS).
Methods: In this retrospective, single-center study, we reviewed the medical records of short-statured patients with a height ≤ –2 z-score, a normal peak GH response (≥10 ng/mL) to clonidine or L-dopa stimulation tests, and a history of rhGH treatment. Patients with chronic illness, malnutrition, syndromic or endocrine disorders, diabetes, metabolic disease, anemia, or prior pubertal suppression were excluded. Eligible patients meeting the definitions of bioinactive GH, SGA, or ISS were included.
Statistical Analysis: Data were analyzed with IBM SPSS Statistics 22.0 using parametric and non-parametric tests with Bonferroni correction; significance was set at p < 0.05.
Results: Among 170 patients screened, 109 fulfilled the criteria for analysis (bioinactive GH, n=8; SGA, n=27; ISS, n=74). Baseline Insulin-like Growth Factor 1 (IGF-1) and Insulin-like Growth Factor Binding Protein 3 (IGFBP-3) levels were markedly lower in the bioinactive GH group compared with SGA and ISS (p < 0.001). During rhGH therapy, patients with bioinactive GH exhibited the greatest gains in growth velocity and Δ height z-score, despite similar GH doses and a lower proportion of pubertal subjects. By final height, all patients with bioinactive GH achieved normal stature, with most surpassing target height, whereas fewer SGA and ISS patients reached their genetic potential.
Conclusion: Children with bioinactive GH form a biologically distinct and highly treatment-responsive subgroup of non-GHD short stature. Our findings highlight the diagnostic value of IGF-1 generation testing in this context. Future multicenter studies with genetic and bioactivity confirmation are essential to refine diagnostic criteria and establish international guidelines.
Introduction
Short stature is a common clinical issue in pediatric endocrinology with a diverse range of underlying causes, including both pathological conditions and constitutional factors. Although growth hormone deficiency (GHD) is a well-established and treatable cause of short stature, a subset of children exhibit significant growth failure despite having normal peak growth hormone (GH) levels on stimulation tests. This discrepancy reveals the limitations of current diagnostic approaches and draws attention to underrecognized GH dysfunctions, such as bioinactive GH. Bioinactive GH denotes a phenotype in which circulating GH is immunoreactive but biologically ineffective; it is not a distinct therapeutic intervention but rather an identifiable subgroup among children receiving recombinant human growth hormone (rhGH) (1–10).
Bioinactive GH is a rare but clinically important condition characterized by circulating GH that is immunoreactive but biologically inactive due to structural abnormalities of the GH molecule, often associated with pathogenic variants in the GH1 gene. These abnormal GH isoforms fail to effectively bind or activate the GH receptor, resulting in impaired activation of downstream signaling pathways, including JAK2–STAT5 signaling, which is essential for insulin-like growth factor-1 (IGF-1) production and linear growth. Despite normal GH secretion, affected children typically have significantly reduced serum IGF-1 and Insulin-like Growth Factor Binding Protein 3 (IGFBP-3) levels, along with a poor correlation between GH and IGF-1 concentrations, leading to short stature due to functional impairment of GH action (11–14). Initially described by Kowarski in 1978 (2) and further characterized at the molecular level by Takahashi in the 1990s (3), bioinactive GH remains underdiagnosed, with its true prevalence unknown due to limited awareness and the absence of standardized diagnostic tests. Recent reviews highlight the continued lack of standardized diagnostic tools and emphasize the need for harmonized international diagnostic criteria (15).
Previous studies have consistently shown that children with bioinactive GH respond well to exogenous rhGH therapy, despite normal endogenous GH secretion (15–17). Their growth patterns are largely comparable to those of patients with classical GHD, and in some cases, an even greater response has been observed (17). This paradox arises because exogenously administered rhGH retains full biological activity, effectively compensating for the functional defect of the endogenous hormone. Collectively, these findings indicate that bioinactive GH represents a GH-responsive phenotype with a pathophysiological profile more closely resembling GHD than other non-GHD etiologies of short stature.
Accordingly, the present study compares patients with bioinactive GH to two well-established non-GHD short stature cohorts: small for gestational age (SGA) and idiopathic short stature (ISS) (6, 8). Both SGA and ISS represent heterogeneous populations characterized by normal GH secretion but variable responsiveness to rhGH, thereby providing suitable comparator groups for evaluating the growth potential of bioinactive GH. Children born SGA typically experience intrauterine growth restriction (18–22) and demonstrate highly variable patterns of postnatal catch-up growth, whereas ISS is a diagnosis of exclusion applied to children with unexplained short stature in the absence of identifiable systemic, endocrine, or genetic abnormalities (8).
We hypothesize that children with bioinactive GH—representing a distinct subset of non-GHD short stature patients identified through IGF-1 generation testing—will demonstrate a stronger growth response to rhGH therapy, as measured by annual growth velocity, changes in height z-score, and final height z-score, compared with children diagnosed with SGA or ISS.
Patients and methods
Patients
Between January 2000 and January 2020, we conducted a retrospective review of medical records from patients treated with rhGH at our center who achieved a peak GH response ≥10 ng/mL on at least one conventional stimulation test with oral clonidine or L-dopa. Patients were excluded if they had chromosomal abnormalities, syndromic disorders, malnutrition, additional hormonal deficiencies (e.g., thyroxine or cortisol), insulin-dependent diabetes mellitus, bone disorders, chronic or metabolic diseases, anemia, or a history of pubertal suppression therapy. All patients underwent detailed physical examination and anthropometric assessment to rule out skeletal dysplasia; none showed disproportion or other physical findings suggestive of such conditions. At rhGH initiation, all patients had a height ≤ −2 z-score and a body mass index (BMI) > −2 z-score.
Definitions of study groups
IGF-1 and IGFBP-3 z-scores were calculated using sex- and age-specific reference data by Güven et al. (23). Patients with baseline IGF-1 ≤ −2 z-score underwent an IGF-1 generation test: subcutaneous rhGH 0.1 mg/kg/day for four consecutive days with serum IGF-1 and IGFBP-3 re-measured on day five. Patients demonstrating an increase of ≥40 ng/mL in IGF-1 and ≥400 ng/mL in IGFBP-3 were classified as having bioinactive GH (4). Infants with birth weight and/or length ≤ −2 z-score for gestational age were classified as SGA according to sex-specific standards (24). Patients with a height ≤ −2 z-score based on age- and sex-specific norms and without an identifiable cause were categorized as ISS (8).
Measurements and procedures
Height was measured with a wall-mounted stadiometer (Seca®; Seca GmbH & Co. KG, Hamburg, Germany) with patients barefoot and in a neutral stance. Body weight was measured using a digital scale (Fakir Hausgeräte) with a sensitivity range of 100 g to 150 kg while patients wore light clothing. Serum hormone levels were measured using a chemiluminescent immunoassay (Access 2 Immunoassay System; Beckman Coulter Inc., USA).
From medical records we extracted birth weight (BW), pre-treatment height, weight, BMI, and their z-scores; bone age (BA); IGF-1 and IGFBP-3 levels with corresponding z-scores; height and height z-scores at the first, second, and third years after treatment initiation; growth velocities (GV); and final height with z-score.
GV was defined as the annual height increment in cm/year. Pre-treatment GV was calculated from the 12-month interval immediately preceding rhGH initiation. Height age was defined as the age corresponding to the child’s current height on national reference curves. Final height was defined as height at growth cessation (BA ≥15 years in females and ≥16 years in males or GV <1 cm/year).
BMI was calculated as weight (kg) divided by height (m²). Height and BMI z-scores were assessed using Turkish reference data by Neyzi et al. (25). Target height was calculated as follows: females, [(mother’s height + father’s height − 13)/2]; males, [(mother’s height + father’s height + 13)/2]. Target height z-scores were derived relative to national reference standards Neyzi et al. (25). BA was evaluated using the Greulich–Pyle method.
Statistical analysis
Data were analyzed using IBM SPSS Statistics Version 22.0 (26). Normality was assessed with the Shapiro–Wilk and Kolmogorov–Smirnov tests, together with evaluation of skewness and kurtosis. Parametric data (mean ± SD) were compared using one-way ANOVA with Bonferroni post-hoc tests, while non-parametric data (median, Q1–Q3) were analyzed with the Kruskal–Wallis test, followed by Mann–Whitney U tests with Bonferroni correction when significant. Within-group longitudinal changes were assessed using repeated-measures ANOVA with Greenhouse–Geisser correction for normally distributed variables, followed by Bonferroni-adjusted pairwise comparisons. Time-point multiplicity was controlled using Bonferroni correction across time points. A p-value < 0.05 was considered statistically significant.
Height, growth velocity, and biochemical measurements were included in year-specific analyses only when complete data for that time point were available. Complete-case analyses were performed without imputation, and missingness was assumed to be missing completely at random (MCAR) or missing at random (MAR). The reduction in sample size in later treatment years reflects patients who either discontinued rhGH therapy, transferred follow-up to other institutions, or had incomplete documentation for that interval.
Results
In this study, out of 170 short-statured patients (height ≤ –2 z-score) who demonstrated a normal GH response (≥10 ng/mL) to clonidine or L-dopa stimulation tests and received rhGH treatment at our center, a total of 109 patients classified as bioinactive GH (n = 8), SGA (n = 27), and ISS (n = 74) according to the definitions provided in the Methods section were selected and included in the analysis. The selection of patients is summarized in Figure 1.
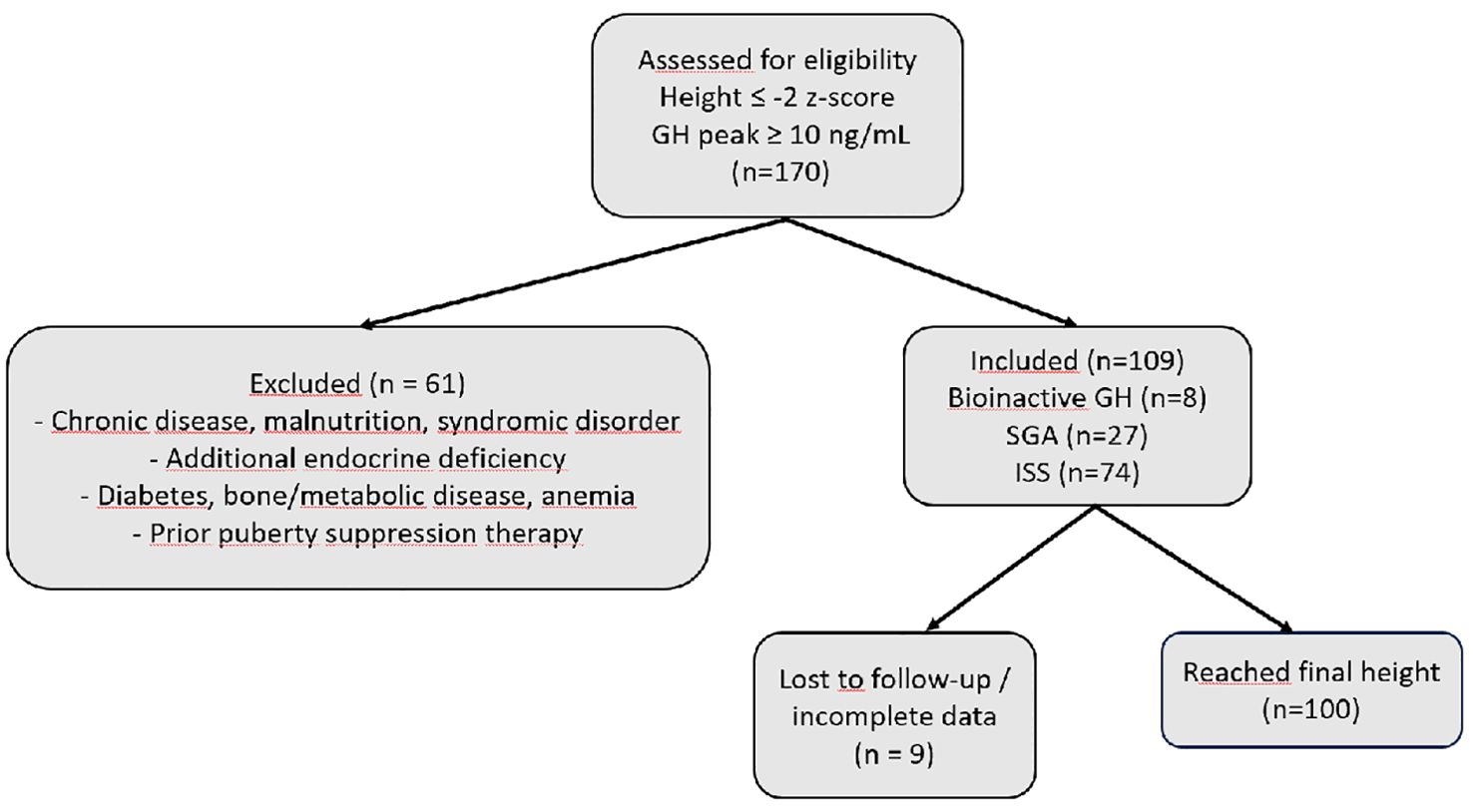
Figure 1. Flow diagram of patient selection and inclusion in the study. Abbreviations: GH, growth hormone; ISS, idiopathic short stature; SGA, small for gestational age; n, sample size; z-score = standard deviation score.
Baseline characteristics are shown in Table 1. Sex distribution was similar descriptively across groups (bioinactive GH: 38% male; SGA: 37% male; ISS: 40% male).
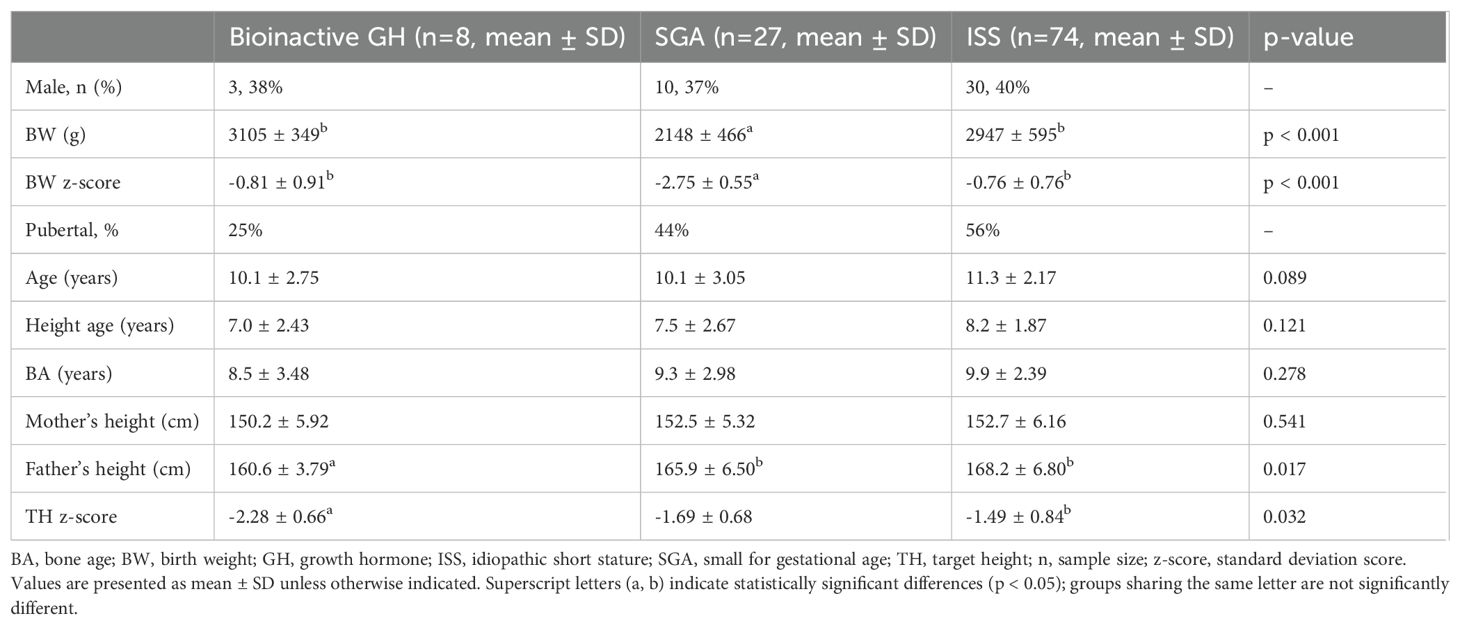
Table 1. Baseline anthropometric and parental characteristics of the study groups.
BW and BW z-score were significantly lower in the SGA group compared with both the bioinactive GH and ISS groups (p < 0.001). The mean BW was 2148 ± 466 g in the SGA group, 3105 ± 349 g in the bioinactive GH group, and 2947 ± 595 g in the ISS group. The mean BW z-score was –2.75 ± 0.55 in the SGA group, –0.81 ± 0.91 in the bioinactive GH group, and –0.76 ± 0.76 in the ISS group.
At treatment initiation, 25% of the bioinactive GH group, 44% of the SGA group, and 56% of the ISS group were pubertal.
No statistically significant differences were observed among the groups in terms of age, height age, BA, or maternal height. Paternal height differed significantly among groups (p = 0.017), being lowest in the bioinactive GH group (160.6 ± 3.79 cm) compared with SGA (165.9 ± 6.50 cm) and ISS (168.2 ± 6.80 cm). Similarly, target height z-score was significantly lower in the bioinactive GH group (–2.28 ± 0.66) than in the ISS group (–1.49 ± 0.84; p < 0.001).
Biochemical parameters related to the GH-IGF axis before treatment are shown in Table 2. All patients in the bioinactive GH group had IGF-1 and IGFBP-3 z-scores below –2 z-scores. Both the absolute levels and z-scores of IGF-1 and IGFBP-3 in this group were markedly lower compared with the SGA and ISS groups (p < 0.001 for all comparisons).
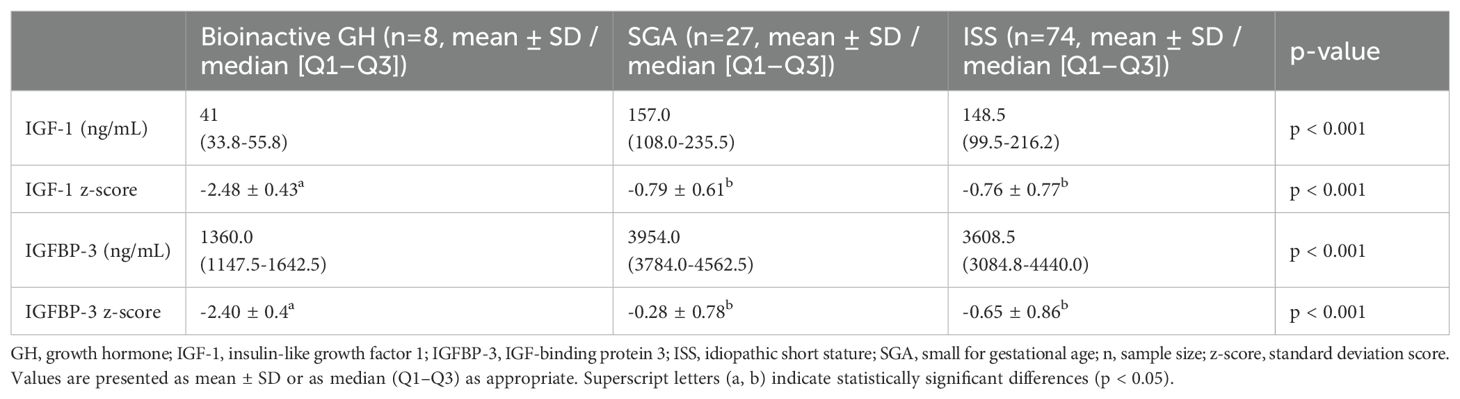
Table 2. Baseline IGF-1 and IGFBP-3 levels and z-scores.
There was no significant difference in pre-treatment GV among the groups (bioinactive GH: 3.7 ± 0.8 cm/year; SGA: 4.8 ± 1.7 cm/year; ISS: 4.7 ± 1.5 cm/year; p = 0.172).
During the first treatment year, GV increased substantially in all groups, with the highest mean GV observed in the bioinactive GH group (9.1 ± 1.4 cm/year) compared with SGA (8.2 ± 1.9 cm/year) and ISS (8.4 ± 1.9 cm/year; p = 0.504). GH doses were similar across groups (mean 0.032–0.033 mg/kg/day). Approximately half of the bioinactive GH group were pubertal at this stage, compared with 63% in the SGA group and 80% in the ISS group.
In the second year, the bioinactive GH group again showed the highest GV (8.6 ± 2.2 cm/year) relative to SGA (6.7 ± 2.1 cm/year) and ISS (7.5 ± 1.9 cm/year; p = 0.10). GH dose was comparable across groups (p = 0.31), and the proportion of pubertal patients increased in all cohorts, with ISS showing the highest rates.
In the third year, mean GV remained higher in the bioinactive GH group (8.2 ± 2.2 cm/year) than in the SGA (6.4 ± 1.7 cm/year) and ISS (6.9 ± 2.1 cm/year) groups (p = 0.372). GH doses did not differ significantly, and most patients in all groups were pubertal by this stage.
Final height analysis showed a greater total height gain in the bioinactive GH group (40.7 ± 12.5 cm) compared with SGA (28.2 ± 15.4 cm) and ISS (29.2 ± 11.0 cm), although this difference was not reach statistical significance (p = 0.071).
Longitudinal changes in height z-score from the initiation of rhGH therapy to final height are summarized in Table 3. At baseline, mean height z-scores were similar across groups (bioinactive GH: –3.14 ± 0.33; SGA: –3.02 ± 0.60; ISS: –2.92 ± 0.62; p = 0.572).
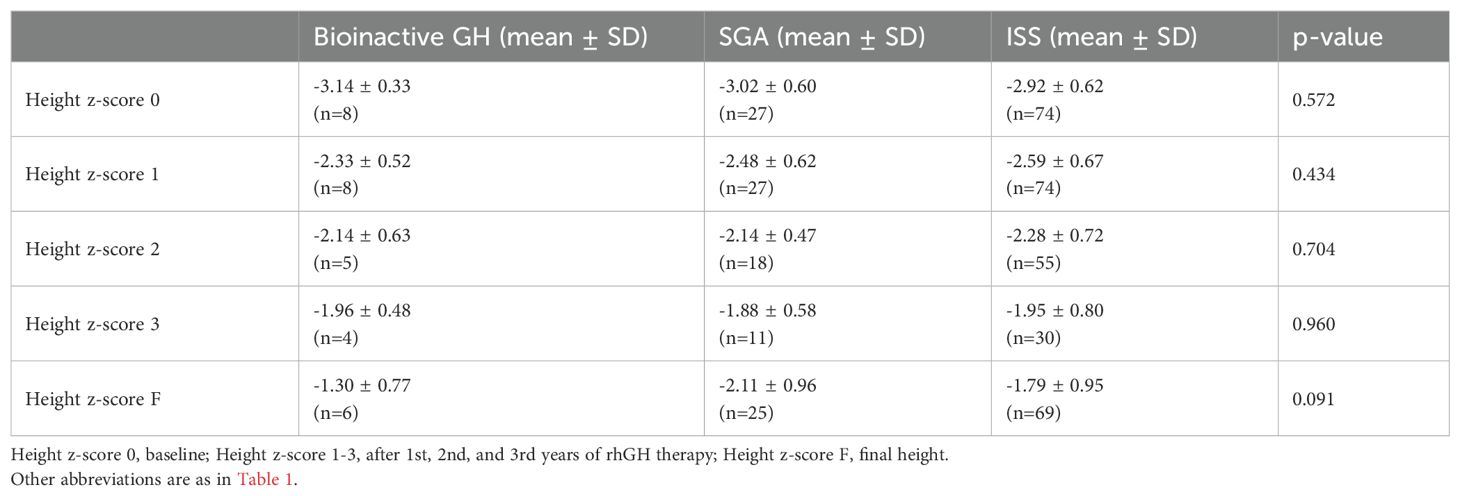
Table 3. Longitudinal changes in height z-score across study groups.
After the first year of treatment, height z-scores improved in all groups (bioinactive GH: –2.33 ± 0.52; SGA: –2.48 ± 0.62; ISS: –2.59 ± 0.67; p = 0.434). Incremental gains were maintained in the second and third treatment years, with no significant differences between groups (p = 0.704 and p = 0.960, respectively).
At final height, mean z-scores reached –1.30 ± 0.77 in the bioinactive GH group, –2.11 ± 0.96 in the SGA group, and –1.79 ± 0.95 in the ISS group (p = 0.091). Within-group analyses confirmed significant improvements in height z-scores over time in all cohorts (p < 0.001). Corresponding GH doses and pubertal distribution are presented in Table 4.
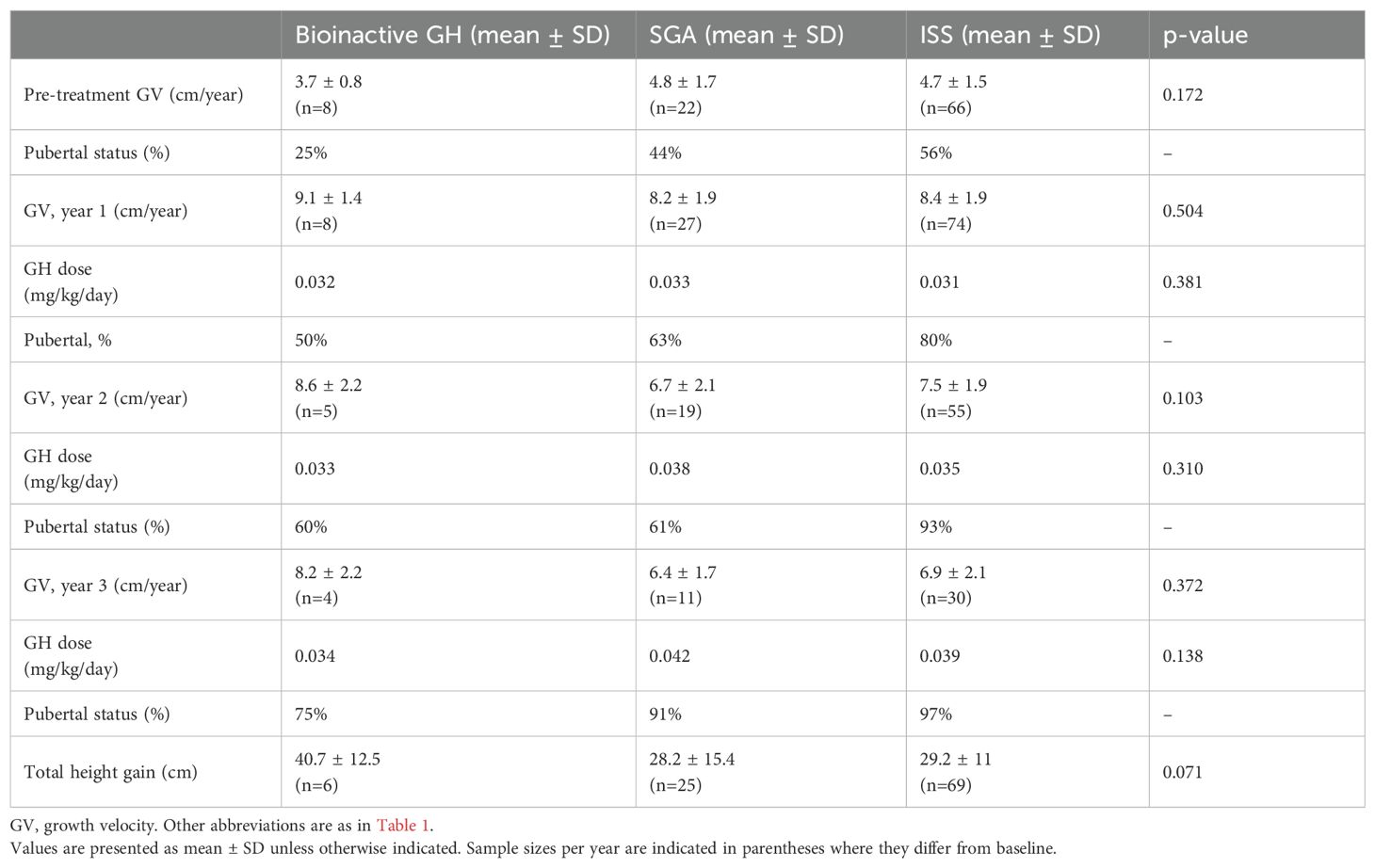
Table 4. Growth velocity, GH dose, and pubertal status before and during rhGH treatment.
Figure 2 illustrates longitudinal changes in height z-scores across the three study groups.
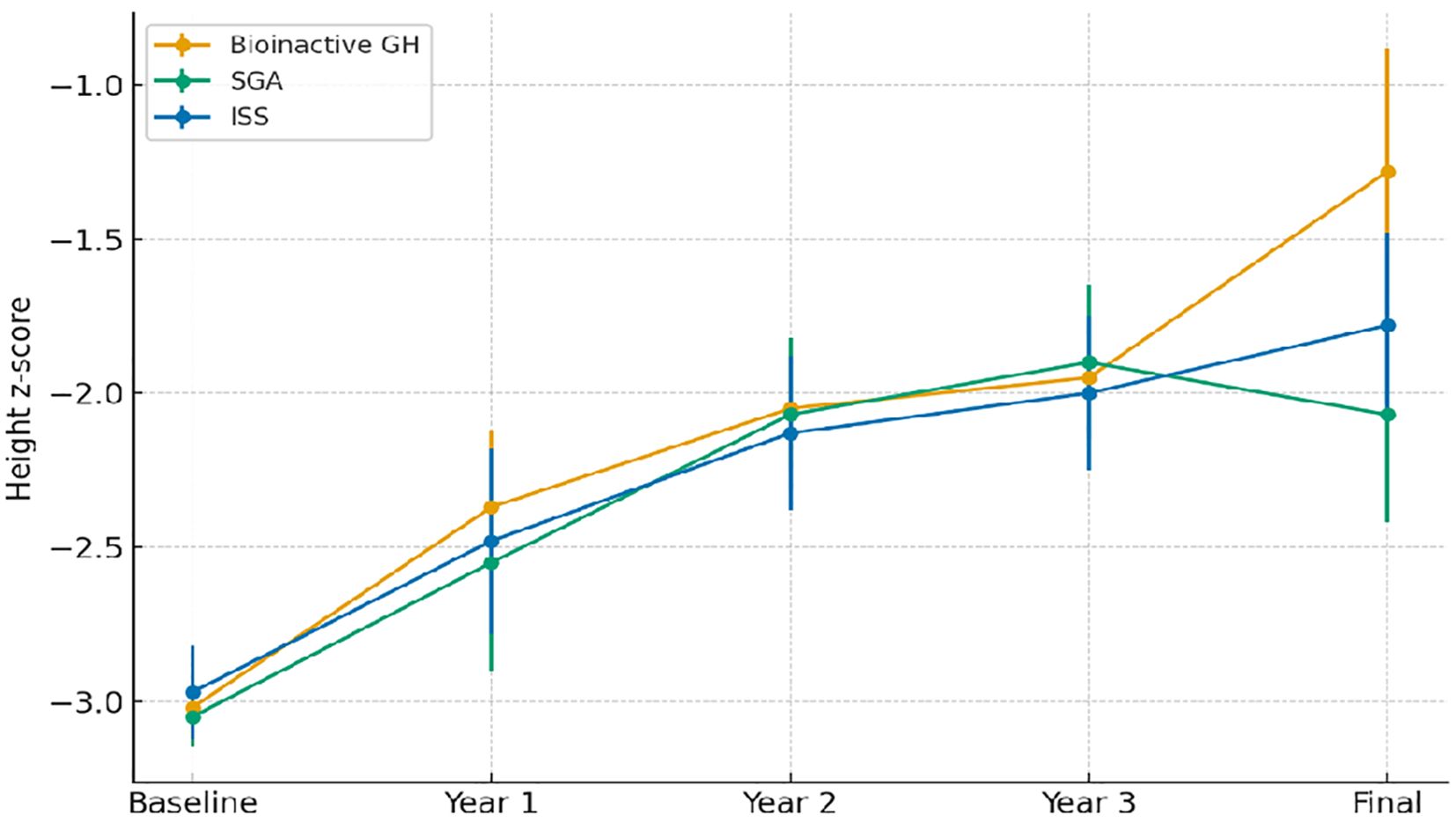
Figure 2. Longitudinal changes in height z-scores by study group. Abbreviations are as in Table 1.
Yearly and cumulative changes in height z-score (Δ height z-score), together with corresponding GH doses and pubertal distribution, are presented in Table 5.
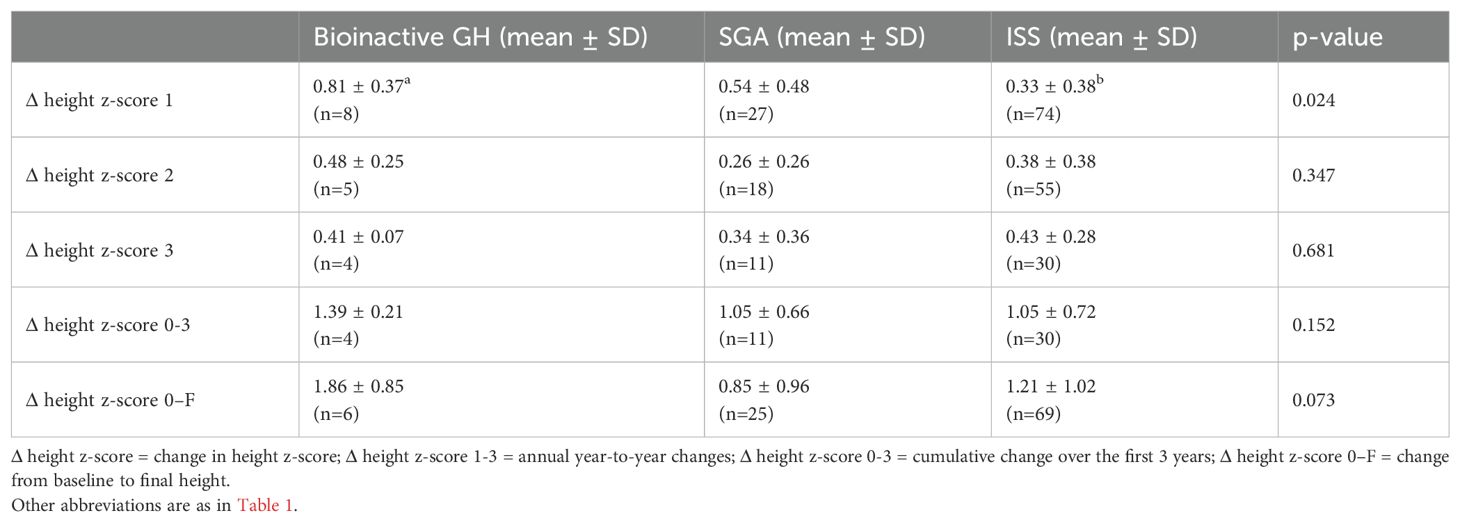
Table 5. Annual and cumulative Δ height z-score.
At the end of the first year, the mean Δ height z-score in the bioinactive GH group was 0.81 ± 0.37, higher than both the ISS group (0.33 ± 0.38, p = 0.024) and the SGA group (0.54 ± 0.48). GH dosing was comparable across groups, and the stronger response in the bioinactive GH group occurred despite a lower proportion of pubertal patients.
In the second year, the Δ height z-score remained higher in the bioinactive GH group (0.48 ± 0.25) compared with SGA (0.26 ± 0.26) and ISS (0.38 ± 0.38), though differences were not significant (p = 0.347). In the third year, Δ height z-scores were similar among groups.
Cumulatively over the first three years, the bioinactive GH group showed the highest total Δ height z-score (1.39 ± 0.21), compared with 1.05 in both SGA and ISS (p = 0.152). From baseline to final height, Δ height z-score was greatest in the bioinactive GH group (1.86 ± 0.85) versus SGA (0.85 ± 0.96) and ISS (1.21 ± 1.02), approaching statistical significance (p = 0.073).
Figure 3 shows the annual and cumulative Δ height z-score in the three groups—bioinactive GH, SGA, and ISS—following rhGH therapy.

Figure 3. Annual and cumulative changes in height z-scores by group. Abbreviations are as in Table 1 and Table 5.
Among the 100 patients who reached final height, 57 (57%) achieved a normal height (height > −2 z-score). In the bioinactive GH group, all 6 patients (100%) reached normal height, and 5 (83%) achieved their target height. In the SGA group, 11 out of 25 patients (44%) reached normal height and 9 (36%) reached target height, whereas in the ISS group, 40 out of 69 patients (58%) attained normal height and 29 (42%) reached target height.
Comparisons of target height z-scores and observed height z-scores at baseline, the third year of treatment, and final height are presented in Table 6. At baseline, the difference between target and observed height z-scores (Δ height z-score_target–observed) was significantly higher in all groups, indicating a marked initial height deficit relative to genetic potential. The largest discrepancy was observed in the ISS group (1.41 ± 0.90), followed by the SGA (1.30 ± 0.70) and bioinactive GH (0.94 ± 0.77) groups.
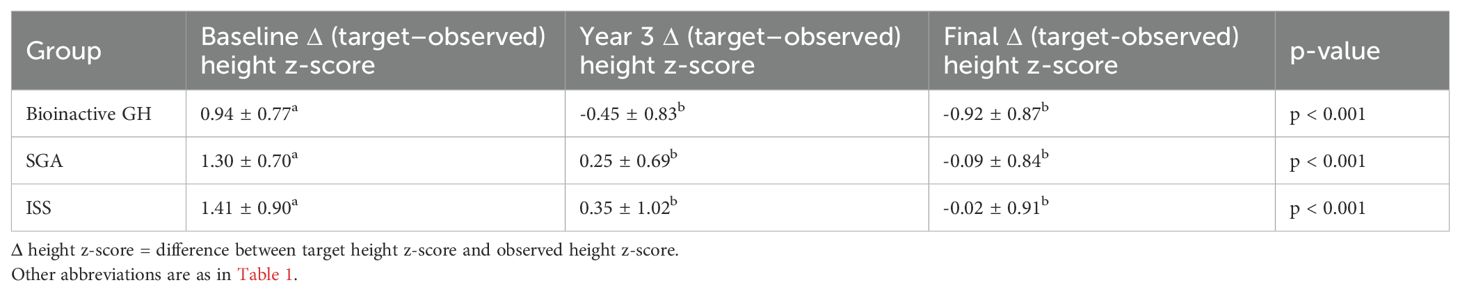
Table 6. Comparison of target and observed height z-scores at baseline, year 3, and final height.
After three years of rhGH therapy, all groups demonstrated significant improvement, with the gap between target and observed height z-scores narrowing considerably. In the bioinactive GH group, this difference became negative by year 3 (−0.45 ± 0.83) and further declined to −0.92 ± 0.87 at final height, indicating that most patients exceeded or approached their genetic potential. The SGA and ISS groups also showed progressive narrowing of the target–observed gap, reaching near parity by final height (−0.09 ± 0.84 and −0.02 ± 0.91, respectively), though less pronounced than in the bioinactive GH group.
Within-group changes over time were statistically significant in all three groups (p < 0.001).
Discussion
This study provides one of the most detailed comparative analyses to date of growth outcomes in children with bioinactive GH versus two major non-GH-deficient short stature populations: SGA and ISS. All participants exhibited normal peak GH responses on stimulation testing yet demonstrated distinct patterns of responsiveness to rhGH therapy. Children classified as having bioinactive GH based on IGF-1 generation test results demonstrated a more pronounced and consistent growth response than both the SGA and ISS groups, reflected in annual growth velocity, increases in height z-score, and attainment of higher final height z-scores. These findings highlight the clinical relevance of bioinactive GH as a distinct phenotype and reinforce the diagnostic utility of IGF-1 generation testing in selected patients with unexplained short stature.
A notable strength of this investigation is the direct, protocol-consistent comparison of three clinically meaningful subgroups treated in a single tertiary center, ensuring standardized dosing, monitoring, and anthropometric assessments. This methodological uniformity minimizes variability related to treatment approach or measurement techniques, thereby increasing the reliability of between-group comparisons. The consistent superiority of growth outcomes in the bioinactive GH group further supports the validity of this classification, despite the absence of confirmatory molecular or bioactivity assays.
Limitations and diagnostic challenges
The retrospective design and single-center setting of this study inherently constrain causal inference and limit external validity. The small number of patients with bioinactive GH (n = 8) reflects both the rarity of this phenotype and its probable underdiagnosis, resulting in reduced statistical power and wide confidence intervals of the estimated effects. While the low prevalence of bioinactive GH partly explains the limited cohort size, assembling a larger sample through coordinated multicenter collaboration would enhance statistical robustness, narrow confidence intervals, permit adequately powered subgroup analyses, and facilitate exploration of genotype–phenotype correlations.
Furthermore, the exclusive inclusion of Turkish patients treated at a single tertiary center introduces potential selection bias and may limit generalizability to populations with different genetic architectures, environmental exposures, or healthcare system structures. Prior studies have demonstrated that ethnic and environmental variability can significantly influence baseline growth patterns, pubertal timing, and responsiveness to rhGH therapy (27, 28), supporting the need for cautious extrapolation of the present findings to other contexts (4, 14, 29). Addressing these limitations through prospective, ethnically diverse, multicenter cohorts will be essential for establishing validated, widely applicable diagnostic and therapeutic recommendations for bioinactive GH.
The classification of bioinactive GH in this study was based solely on clinical and biochemical criteria, particularly IGF-1 generation testing, without molecular or in vitro bioactivity confirmation. GH immunoassays detect immunoreactive hormone but cannot differentiate structurally altered or functionally impaired isoforms, a discrepancy recognized for decades. Immunofunctional assays that quantify GH capable of bivalent receptor binding can enrich for biologically competent fractions, but their limited availability and imperfect correlation with in vivo signaling restrict clinical utility. Cell-based bioassays, such as the Nb2 rat lymphoma proliferation assay and Ba/F3–STAT5 luciferase reporter systems, differ in specificity and sensitivity; although the latter can detect markedly reduced bioactivity, both may fail to capture intermediate phenotypes and lack cross-platform comparability (12). Moreover, while several pathogenic GH1 variants (e.g., R77C, D112G) are known to produce biologically inactive GH, many patients with reduced GH bioactivity have no GH1 mutations, underscoring phenotypic heterogeneity and the need for multimodal confirmation (4, 30, 31). These diagnostic constraints may result in misclassification, potentially underestimating or overestimating the prevalence and treatment responsiveness of bioinactive GH in broader clinical populations.
Emerging evidence supports integrating next-generation sequencing (NGS) into the diagnostic work-up for growth disorders to identify GH1 and other relevant gene variants. However, interpretation of variants of uncertain significance (VUS) remains a challenge. Incorporating standardized immunofunctional and bioactivity assays alongside genetic testing would improve diagnostic certainty, refine patient stratification, and facilitate international comparability. Future prospective, multicenter studies should integrate these advanced modalities with phenotypic data to establish validated, widely applicable diagnostic algorithms for bioinactive GH, ultimately enabling more tailored therapeutic strategies (32).
Superior rhGH responsiveness in bioinactive GH
Children with bioinactive GH demonstrated markedly greater gains in height z-score and growth velocity throughout the treatment period compared with SGA and ISS peers, despite receiving similar GH doses and having a lower proportion of pubertal subjects during early therapy. Remarkably, all patients in this group achieved final heights within the normal range, and most exceeded their mid-parental height expectations, an outcome uncommon in other non-GHD populations. These findings align with reports by Binder et al. and Pagani et al., which documented striking catch-up growth in patients with reduced GH bioactivity but intact GH secretion (15, 16).
Mechanistically, bioinactive GH is characterized by immunoreactive hormone that is structurally or functionally impaired, often due to missense mutations or post-translational modifications in GH1 (3, 4, 7). This defective hormone fails to effectively activate GH receptors or downstream JAK2–STAT5 signaling, leading to low IGF-1 and IGFBP-3 levels despite normal circulating GH. Administration of exogenous rhGH—unaffected by these molecular defects—restores signaling pathways and drives robust linear growth. The substantial increases in IGF-1 and IGFBP-3 levels observed in our cohort after IGF-1 generation testing are consistent with this mechanism (33).
Distinction from SGA and ISS
While SGA and ISS patients also showed height improvements, these were generally less pronounced and more variable. SGA is frequently associated with intrauterine growth restriction and possible epigenetic modifications, placental insufficiency, or fetal programming that impair GH signaling at the receptor or post-receptor level (34, 35). Even with adequate GH secretion, residual effects of prenatal growth restriction can blunt catch-up growth, and responsiveness to rhGH often plateaus after 2–3 years in some cohorts (36, 37).
ISS represents a diagnosis of exclusion, encompassing etiologies from polygenic short stature to unrecognized syndromes and subtle endocrine dysfunctions (38–41). The heterogeneity of ISS explains the wide variation in rhGH responsiveness, as reflected by the large standard deviations in Δ height z-score in our ISS cohort. While ISS patients in our study achieved continued gains over time—possibly aided by IGF-1-guided dose adjustments and increased pubertal prevalence—fewer attained their target height compared with the bioinactive GH group. Collectively, these findings align with recent literature indicating that the rhGH response in SGA and ISS is largely determined by underlying pathophysiological mechanisms rather than GH secretion itself (17, 42).
Role of pubertal timing and height potential
Pubertal onset is a major determinant of growth rate and can confound the evaluation of rhGH efficacy. In our cohort, although the proportion of pubertal patients in the bioinactive GH group was lower during the first three years of treatment, greater increases in Δ height z-scores were achieved compared with the other groups. This suggests that their superior response is driven by treatment-specific factors rather than puberty-related acceleration. Furthermore, despite starting with the lowest target height z-score, these patients surpassed their predicted genetic height by final assessment—contrasting with SGA and ISS patients, who generally did not exceed mid-parental expectations. This reinforces the notion that bioinactive GH represents a biologically distinct and highly treatment-responsive subgroup.
Diagnostic and clinical implications
Bioinactive GH is rare and diagnostically challenging, as standard stimulation protocols assess immunoreactivity but not bioactivity, potentially leading to missed diagnoses. While GH1 sequencing, Nb2 proliferation, and Ba/F3–STAT5 assays can evaluate bioactivity, these remain largely confined to research settings and are not widely available (12–14). In this context, IGF-1 generation testing offers a practical, accessible diagnostic option when GH secretion is normal but IGF-1 is unexpectedly low.
Our results support incorporating IGF-1 generation testing into diagnostic algorithms for children with unexplained short stature, low baseline IGF-1, and normal stimulation test results. A robust rise in IGF-1 and IGFBP-3 following short-term rhGH administration should prompt consideration of bioinactive GH and can justify initiating therapy even without classical GH deficiency confirmation. Although genetic and bioactivity assays remain important confirmatory tools, IGF-1 generation testing is a valuable decision-making aid when these are unavailable.
Conclusion
Children with bioinactive GH form a biologically distinct and highly treatment-responsive subgroup of non-GHD short stature. Our findings highlight the diagnostic value of IGF-1 generation testing in this context. Future prospective, multicenter studies integrating genetic and bioactivity confirmation are essential to refine diagnostic criteria and establish internationally applicable guidelines for clinical management.
Data availability statement
The original contributions presented in the study are included in the article and its supplementary material. Further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by Gazi University Clinical Research Ethics Committee, Gazi University. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin.
Author contributions
EO: Writing – original draft, Writing – review & editing. ED: Writing – review & editing. MÇ: Writing – review & editing. AB: Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Parks JS. Gene sequence and production of recombinant MetGH/hGH. Pediatr Endocrinol Rev. (2018) 16:17–27. doi: 10.17458/per.vol16.2018.p.sequenceproductionmetgh
2. Kowarski AA, Schneider J, Ben-Galim E, Weldon VV, and Daughaday WH. Growth failure with normal serum RIA-GH and low somatomedin activity: somatomedin restoration and growth acceleration after exogenous GH. J Clin Endocrinol Metab. (1978) 47:461–4. doi: 10.1210/jcem-47-2-461
3. Takahashi Y, Kaji H, Okimura Y, Goji K, Abe H, and Chihara K. Short stature caused by a mutant growth hormone with an antagonistic effect. Endocr J. (1996) 43:S27–32. doi: 10.1507/endocrj.43.suppl_s27
4. Besson A, Salemi S, Deladoëy J, Vuissoz JM, Eblé A, Bidlingmaier M, et al. Short stature caused by a biologically inactive mutant growth hormone (GH-C53S). J Clin Endocrinol Metab. (2005) 90:2493–9. doi: 10.1210/jc.2004-1838
5. Karaoglan M. Short stature due to bioinactive growth hormone (Kowarski Syndrome). Endocr Pract. (2023) 29:902–11. doi: 10.1016/j.eprac.2023.08.013
6. Clayton PE, Cianfarani S, Czernichow P, Johannsson G, Rapaport R, and Rogol AD. Consensus statement: management of the child born small for gestational age through to adulthood. J Clin Endocrinol Metab. (2007) 92:804–10. doi: 10.1210/jc.2006-2017
7. Takahashi Y, Kojima I, Kanzaki S, Okimura Y, Kaji H, Abe H, et al. Biologically inactive growth hormone caused by a mutation in the GH1 gene. Endocr Rev. (2006) 27:168–77. doi: 10.1210/er.2005-00138
8. Cohen P, Rogol AD, Deal CL, Saenger P, Reiter EO, Ross JL, et al. 2007 ISS Consensus Workshop participants. Consensus statement on the diagnosis and treatment of children with idiopathic short stature. J Clin Endocrinol Metab. (2008) 93:4210–7. doi: 10.1210/jc.2008-0509
9. Cutfield WS and Albert B. Growth hormone treatment for idiopathic short stature. Pediatr Endocrinol Rev. (2018) 16:113–22. doi: 10.17458/per.vol16.2018.ca.ghidiopathicshortstature
10. Danowitz M and Grimberg A. Clinical indications for growth hormone therapy. Adv Pediatr. (2022) 69:203–17. doi: 10.1016/j.yapd.2022.03.005
11. Pagani S, Chaler EA, Meazza C, Maceiras M, Gonzalez ME, Rivarola MA, et al. Is BaF3 bioassay useful to identify patients with bioinactive growth hormone? J Pediatr Endocrinol Metab. (2010) 23:783–8. doi: 10.1515/jpem.2010.128
12. Bozzola M, Zecca M, Locatelli F, Radetti G, Pagani S, Autelli M, et al. Evaluation of growth hormone bioactivity using the Nb2 cell bioassay in children with growth disorders. J Endocrinol Invest. (1998) 21:765–70. doi: 10.1007/BF03348043
13. Dattani MT, Hindmarsh PC, Pringle PJ, Brook CGD, and Marshall NJ. The measurement of growth hormone bioactivity in patient serum using an eluted stain assay. J Clin Endocrinol Metab. (1995) 80:2675–83. doi: 10.1210/jcem.80.9.7545696
14. Strasburger CJ, Wu Z, Pflaum CD, and Dressendorfer RA. Immunofunctional assay of human growth hormone (hGH) in serum: a possible consensus for quantitative hGH measurement. J Clin Endocrinol Metab. (1996) 81:2613–20. doi: 10.1210/jcem.81.7.8675586
15. Grimberg A and Hawkes CP. Growth hormone treatment for non-GHD disorders: excitement tempered by biology. J Clin Endocrinol Metab. (2024) 109:e442–54. doi: 10.1210/clinem/dgad417
16. Pagani S, Meazza C, Laarej K, Cantoni F, and Bozzola M. Efficacy of long-term growth hormone therapy in short children with reduced growth hormone biological activity. J Endocrinol Invest. (2011) 34:366–9. doi: 10.1007/BF03347461
17. Binder G, Benz MR, Elmlinger M, Pflaum CD, Strasburger CJ, and Ranke MB. Reduced human growth hormone (hGH) bioactivity without a defect of the GH-1 gene in three patients with rhGH-responsive growth failure. Clin Endocrinol (Oxf). (1999) 51:89–95. doi: 10.1046/j.1365-2265.1999.00744.x
18. Saenger P, Czernichow P, Hughes I, and Reiter EO. Small for gestational age: short stature and beyond. Endocr Rev. (2007) 28:219–51. doi: 10.1210/er.2006-0039
19. Boguszewski M, Albertsson WK, Aronsson S, Gustafsson J, Hagenas L, Westgren U, et al. Growth hormone treatment of short children born small-for-gestational-age: the Nordic multicenter trial. Acta Pediatr. (1998) 87:257–63. doi: 10.1080/08035259850157282
20. Van PY, Mulder P, Houdijk M, Jansen M, Reeser M, and Hokken KA. Adult height after long-term, continuous growth hormone treatment in short children born small for gestational age: results of a randomized, double-blind, dose-response GH trial. J Clin Endocrinol Metab. (2003) 88:3584–90. doi: 10.1210/jc.2002-021172
21. Dahlgren J, Wikland KA, and Swedish Study Group for Growth Hormone Treatment. Final height in short children born small for gestational age treated with growth hormone. Pediatr Res. (2005) 57:216–22. doi: 10.1203/01.PDR.0000148716.71231.81
22. Kim S, Choi Y, Lee S, Ahn MB, Kim SH, Cho WK, et al. Growth patterns over 2 years after birth according to birth weight and length percentiles in children born preterm. Ann Pediatr Endocrinol Metab. (2020) 25:163–8. doi: 10.6065/apem.1938180.090
23. Guven B, Can M, Mungan G, and Acikgoz S. Reference values for serum levels of insulin-like growth factor 1 (IGF-1) and IGF-binding protein 3 (IGFBP-3) in the West Black Sea region of Turkey. Scand J Clin Lab Invest. (2013) 73:135–40. doi: 10.3109/00365513.2012.755739
24. Kurtoğlu S, Hatipoğlu N, Mazıcıoğlu MM, Akın MA, Çoban D, Gökoğlu S, et al. Body weight, length, and head circumference at birth in a cohort of Turkish newborns. J Clin Res Pediatr Endocrinol. (2012) 4:132–9. doi: 10.4274/jcrpe.693
25. Neyzi O, Bundak R, Gökçay G, Günöz H, Furman A, Darendeliler F, et al. Reference values for weight, height, head circumference, and body mass index in Turkish children. J Clin Res Pediatr Endocrinol. (2015) 7:280–93. doi: 10.4274/jcrpe.2183
27. Hawkes CP, Gunturi H, Dauber A, Hirschhorn JN, and Grimberg A. Racial/ethnic disparities in the investigation and treatment of growth hormone deficiency. J Pediatr. (2021) 236:238–45. doi: 10.1016/j.jpeds.2021.04.034
28. Soliman A, De Sanctis V, Elalaily R, and Bedair S. Advances in pubertal growth and factors influencing it: can we increase pubertal growth? Indian J Endocrinol Metab. (2014) 18:S53–8. doi: 10.4103/2230-8210.145075
29. Strasburger CJ. Methods in determining growth hormone concentrations: an immunofunctional assay available to purchase. Pediatrics. (1999) 104:1024–8. doi: 10.1542/peds.104.S5.1024
30. Takahashi Y, Shirono H, Arisaka O, Takahashi K, Yagi T, Koga J, et al. Biologically inactive growth hormone caused by an amino acid substitution. J Clin Invest. (1997) 100:1159–65. doi: 10.1172/JCI119627
31. Takahashi Y and Chihara K. Short stature by mutant growth hormones. Growth Horm IGF Res. (1999) 9:37–40; discussion 40-1. doi: 10.1016/s1096-6374(99)80079-3
32. Kim SJ, Joo E, Park J, Seol CA, and Lee J-E. Genetic evaluation using next-generation sequencing of children with short stature: a single tertiary-center experience. Ann Pediatr Endocrinol Metab. (2024) 29:38–45. doi: 10.6065/apem.2346036.018
33. Travaglino P, Buzi F, Meazza C, Pagani S, Tinelli C, Iughetti L, et al. Response to long-term growth hormone therapy in short children with reduced GH bioactivity. Horm Res. (2006) 66:189–94. doi: 10.1159/000094483
34. Lee HS, Kum CD, Rho JG, and Hwang JS. Long-term effectiveness of growth hormone therapy in children born small for gestational age: an analysis of LG growth study data. PloS One. (2022) 17:e0266329. doi: 10.1371/journal.pone.0266329
35. Thomas M, Beckers D, Brachet C, Dotremont H, Lebrethon MC, Lysy P, et al. Adult height after growth hormone treatment at pubertal onset in short adolescents born small for gestational age: results from a Belgian registry-based study. Int J Endocrinol. (2018) 2018:6421243. doi: 10.1155/2018/6421243
36. Ferrigno R, Savanelli MC, Cioffi D, Pellino V, and Klain A. Auxological and metabolic effects of long-term treatment with recombinant growth hormone in children born small for gestational age: a retrospective study. Endocrine. (2024) 84:213–22. doi: 10.1007/s12020-023-03665-4
37. Ferrigno R, Savage MO, Cioffi D, Pellino V, Savanelli MC, and Klain A. Effects of long-term treatment with recombinant growth hormone on growth outcome in children born small for gestational age: a systematic review. Rev Endocr Metab Disord. (2025) 26:147–59. doi: 10.1007/s11154-024-09911-y
38. Albertsson WK, Aronson AS, Gustafsson J, Hagenäs L, Ivarsson SA, Jonsson B, et al. Dose-dependent effect of growth hormone on final height in children with short stature without growth hormone deficiency. J Clin Endocrinol Metab. (2008) 93:4342–50. doi: 10.1210/jc.2008-0707
39. Ben AT, Chodick G, Shalev V, Goldstein D, Gomez R, and Landau Z. Real-world treatment patterns and outcomes of growth hormone treatment among children in Israel over the past decade (2004-2015). Front Pediatr. (2021) 9:711979. doi: 10.3389/fped.2021.711979
40. Im M, Kim YD, and Han HS. Effect of growth hormone treatment on children with idiopathic short stature and idiopathic growth hormone deficiency. Ann Pediatr Endocrinol Metab. (2017) 22:119–24. doi: 10.6065/apem.2017.22.2.119
41. Yang L and Yang F. Short-acting growth hormone supplementation for bone age and growth rate in children with idiopathic short stature: a meta-analysis. BMC Pediatr. (2025) 25:28. doi: 10.1186/s12887-024-05356-z
42. Jo HY, Jang HJ, Cheon CK, Yoon JY, Yoo S, Lee JH, et al. Comparison of growth hormone therapy response according to the presence of growth hormone deficiency in children born small for gestational age with short stature in Korea: a retrospective cohort study. BMC Pediatr. (2025) 25:89. doi: 10.1186/s12887-024-05339-0
Keywords: bioinactive growth hormone, small for gestational age, idiopathic short stature, recombinant human growth hormone, insulin-like growth factor 1, insulin-like growth factor binding protein-3, growth velocity, height z-score
Citation: Özdemir EK, Döğer E, Çamurdan MO and Bideci A (2025) Comparison of children with bioinactive growth hormone, small for gestational age, and idiopathic short stature. Front. Endocrinol. 16:1596976. doi: 10.3389/fendo.2025.1596976
Received: 20 March 2025; Accepted: 25 August 2025;
Published: 11 September 2025.
Edited by:
Anastasia Ibba, Pediatric Endocrinology Unit and Newborn Screening Center, ItalyReviewed by:
Ayca Altincik, Pamukkale University, TürkiyeKara Anderson, University of Virginia, United States
Kequan Lin, Zhejiang University, China
Copyright © 2025 Özdemir, Döğer, Çamurdan and Bideci. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Esma Kaya Özdemir, a2F5YWVzbWFnQGdtYWlsLmNvbQ==