 Christianne Magee1
Christianne Magee1 Jennifer E. Kouri1
Jennifer E. Kouri1 Brian D. Cherrington2
Brian D. Cherrington2 Jeremy D. Cantlon1Dilyara A. Murtazina1Todd A. Farmerie1Meredith H. Davidsen1Terry M. Nett1
Jeremy D. Cantlon1Dilyara A. Murtazina1Todd A. Farmerie1Meredith H. Davidsen1Terry M. Nett1 Colin M. Clay1*
Colin M. Clay1*- 1Animal Reproduction and Biotechnology Laboratory, Department of Biomedical Sciences, Colorado State University, Fort Collins, CO, United States
- 2College of Agriculture, Life Sciences, and Natural Resources, Department of Zoology and Physiology, University of Wyoming, Laramie, WY, United States
Activin, GnRH, and estrogen are key endocrine inputs known to regulate the GnRH receptor (GnRHR) promoter; however, it has become increasingly evident that the mechanisms regulating the GnRHR promoter vary by model and species. To explore these differences, transgenic mice harboring either a wild-type mouse GnRHR (mGnRHR) or sheep (oGnRHR) GnRHR promoter fused to luciferase (-LUC) were infected with an adenovirus overexpressing follistatin, neutralizing activin and decreasing serum concentrations of FSH in both animal models. However, follistatin overexpression in the oGnRHR-LUC mouse more than doubled luciferase expression, whereas in the mGnRHR-LUC animals it led to a 40% decrease in luciferase expression. Thus, the divergent transcriptional responses of the mouse and sheep GnRHR genes to activin appear to be reliably recapitulated in transgenic mice. To further elucidate mechanisms regulating oGnRHR expression, a mouse with a mutated cyclic AMP response element (µCRE) in the proximal oGnRHR-LUC promoter was developed. Using an electrophoretic mobility shift assay, a specific and high affinity interaction of the ovine CRE with nuclear components exists, but these are not modified in the presence of E2, indicating that CRE binding protein (CREB) is necessary but not sufficient to mediate E2 input to oGnRHR expression.
Introduction
Reproductive function is dependent on the binding of Gonadotropin Releasing Hormone (GnRH) to GnRH receptors (GnRHR) located on gonadotropes in the anterior pituitary gland, resulting in the subsequent synthesis and secretion of Luteinizing Hormone (LH) and Follicle Stimulating Hormone (FSH). As an activator of gonadotropin synthesis and secretion, the pulsatile release of GnRH also stimulates transcription of its cognate receptor in all species studied to date (1–5). While the dynamic pulsatile release of GnRH from the hypothalamus is significant (6–9), the relative abundance of GnRH receptors in the pituitary is equally important. More than 200 genes are regulated by agonist binding to the GnRHR, and the amplitude of expression for those genes is highly dependent on the number of GnRHR present at the cell membrane (10). Therefore, our efforts to illuminate the required cascade of events that culminates in normal reproductive function converge upon the coordination of expression of GnRHR, which we know to be regulated by three primary inputs: GnRH, activin, and estrogen.
Both in vivo (11, 12) and in vitro models (2, 13) support the homologous regulation of GnRHR by GnRH. The ability to recapitulate GnRH regulation of the GnRHR expression in immortalized gonadotrope cell lines allowed for relatively rapid progress in characterizing the underlying molecular mechanisms. Study of the proximal GnRHR promoter has identified several cis-regulatory elements; the presence of these elements, their ability to regulate basal GnRHR expression and facilitate agonist-induced gene expression, varies by species (summarized in Figure 1). In the murine GnRHR (mGnRHR) promoter these cis regulatory elements include the Sequence Underlying Responsiveness to GnRH-1 (SURG-1) (14) as well as an activating protein-1 (AP-1) element that is known to bind transcription factors in the jun and fos families (15) and is critical for mediating GnRH input via MAP kinase signaling. The selective elimination of ERK1 and ERK2 in murine gonadotropes underlies the importance of GnRH induced JNK activation of the AP-1 site for maintaining mGnRHR and Fshb transcript levels (16). The complexity of AP-1 activation of the mGnRHR promoter has also been characterized in immortalized gonadotropes (17), where it is partially mediated by ERK-dependent activation (13) and participates in a tripartite enhancer that includes binding sites for nuclear receptor subfamily 5, Group A, Member 1 (NR5A1, previously known as steroidogenic factor-1), as well as an element referred to as the GnRH receptor activating sequence (GRAS) (12, 18). Furthermore, activin responsiveness of the mGnRHR promoter requires both GRAS as well as an additional cis-element located 16-bp 3’ of GRAS referred to as the downstream activin regulatory element (DARE) (19).
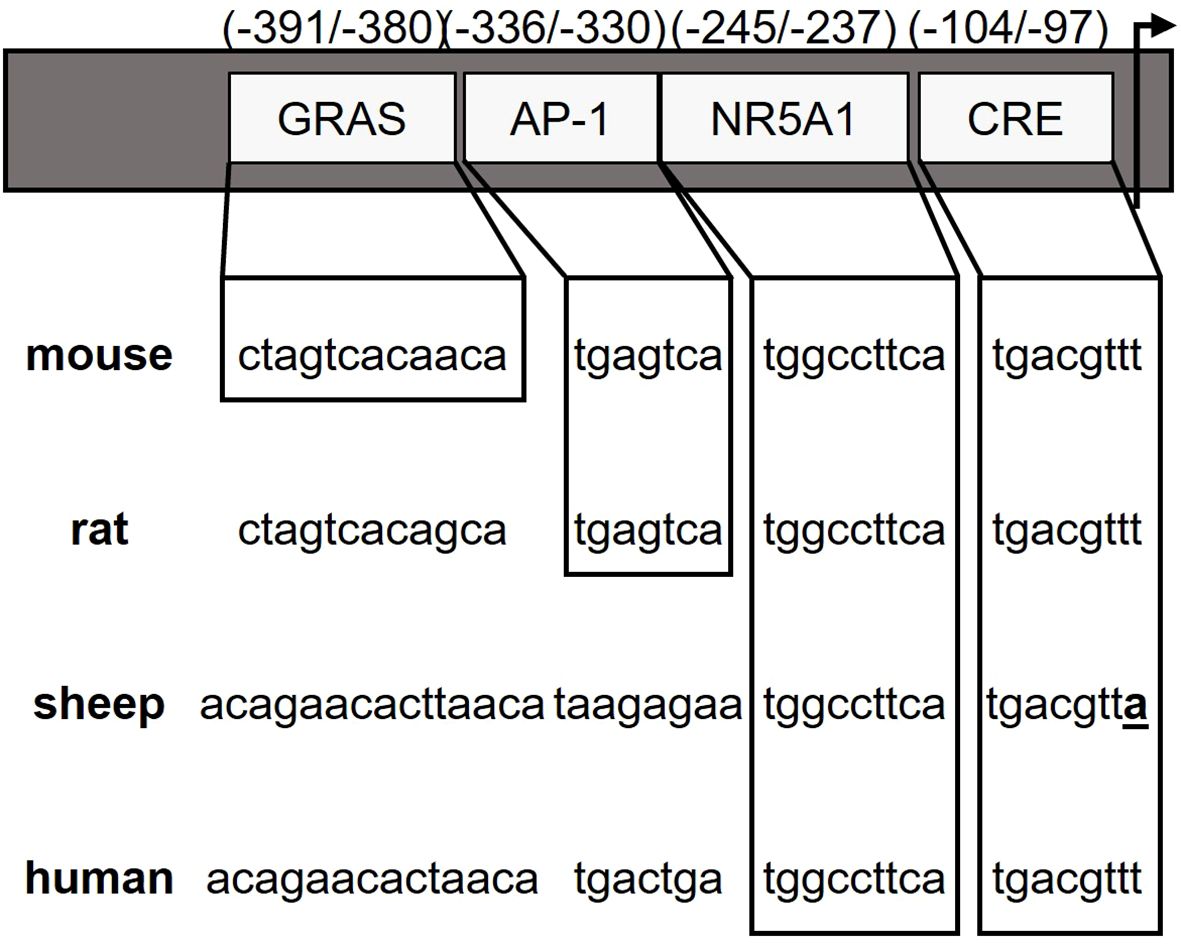
Figure 1. Species differences in GnRHR promoter elements. Promoter regions within -400 bp of the GnRHR start site (arrow) are indicated with sequence similarity between species presented by boxes for specific cis elements and divergence of the ovine CRE element is highlighted.
As a member of the transforming growth factor (TGFβ) family of growth and differentiation factors, activin contributes to GnRHR expression by gonadotropes (20–22). The activin binding protein, follistatin, acts as a primary modulator of the biological effects of activin by sequestering activin and preventing activin binding to its cognate receptor (23). However, divergent biological effects of activin are evident depending on species. In mice for example, activin stimulates both Fshb and mGnRHR expression (18, 19, 24); whereas in cultured ovine pituitary cells, activin selectively increases FSH secretion and decreases the number of GnRHR available for binding, though this effect on GnRHR is negated in the presence of estrogen (25). Much of this may have to do with the specific activin-responsive elements that are present in the GnRHR promoters for each species. Neither GRAS nor DARE has yet to be identified in any species other than the mouse, nor does GRAS confer activin responsiveness to the rat GnRHR promoter (18). The convergence of GnRH and activin initiating expression of the mGnRHR has been demonstrated by a non-consensus AP-1 binding site that overlaps with a putative SMAD-binding element (SBE) (26), but again, these findings are limited to the murine GnRHR promoter.
The consistent upregulation of GnRHR expression by GnRH is not at issue. It is the need to deconstruct the species-specific divergence of activin and estrogen regulation of GnRHR expression that has resulted in our use of transgenic mouse models to parse whether the in vivo findings are the result of transcriptional factors available in the gonadotrope cell, a matter of GnRHR promoter regulation, or some combination thereof. Herein we demonstrate that divergence in activin regulation is a result of species-specific differences in the promoter for GnRHR.
In many species, including humans and sheep, the increase in estradiol (E2) secreted by the pre-ovulatory follicle is obligatory to initiate an increase in GnRH secretion from the hypothalamus and the presence of GnRHR on the surface of the gonadotrope prior to the LH surge (27–31). However, a canonical estradiol response element (ERE) has not been identified in any GnRHR gene reported to date (32–36) and the identity of the transcriptional machinery by which E2 regulates GnRHR expression remains to be determined. Interestingly, a cyclic AMP response element (CRE) in the proximal promoter of GnRHR is highly conserved in all species except for the horse (37). Using an adenovirally delivered dominant-negative form of the CRE binding protein (ACREB), we have blocked E2 activation of GnRHR expression in primary cultures of ovine pituitary cell (38). Herein, we confine our studies of E2 regulation to the ovine GnRHR (oGnRHR) promoter as we have been unable to detect E2 regulation of the mGnRHR (39). With the use of a transgenic mouse expressing the oGnRHR promoter and ovine pituitary cells, we demonstrate that E2 activation of the oGnRHR promoter requires an intact CRE, and the interaction of CREB or phosphorylated CREB is independent of a canonical ERE. The overall goal of these studies was to provide additional mechanistic understanding of GnRHR regulation and continued evidence for the divergence in GnRHR regulation between the mouse and the ewe.
Methods and materials
Materials
Dr. Pamela Mellon (University of California San Diego, San Diego, CA) generously provided the αT3–1 cells. Dr. Wylie Vale (Salk Institute, La Jolla, CA) provided the adenoviral follistatin (AdCAFS288) and GFP (AdGFP) expression vectors (39). Amplification and purification of AdCAFS288 and AdGFP were performed as previously described (40). Continuous time-release estradiol-17β pellets (2.5 mg over 21 d, Catalogue #E-121) were obtained from Innovative Research of America (Sarasota, FL). Anti-GnRH serum (hereafter referred to as antiserum, AS) was prepared against keyhole limpet hemocyanin-conjugated GnRH in sheep and has been described (41). Restriction and modifying enzymes were purchased from New England Biolabs, Inc. (Beverly, MA) and were used according to supplier’s specifications.
Plasmids
Construction of plasmids containing 9,100 and 2,700 bp of proximal promoter regions for the oGnRHR fused to a luciferase (-LUC) expression vector (-9100LUC, -2700LUC) has been previously described (11). Plasmid -160LUC was constructed by PCR amplification of -2700LUC using the original antisense primer for -2700LUC (5’-GAGCTCGGCACTTCTGATGTT-3’) and a sense primer directed against the appropriate sequence in the 5’ flanking region (5’-TTAAATACAAAGTATCTCAGG -3’) and ligated into the SmaI site of the pGL3 vector (Promega Corp., Madison, WI). Additional -115LUC and -79LUC plasmids were similarly constructed by PCR amplification utilizing the -160pGL3, the GLprimer2 (5’-CTTTATGTTTTTGGCGTCTTCCA-3’, Promega Corp., Madison, WI) and a sense primer for either -115 bp (5’-CCTGTGACGTTACCAGCCA-3’) or -79 bp (5’-ACAGGACTCCAAGTGCAATTACA-3’). The CRE in the oGnRHR promoter is located within 160 bp of the transcriptional start site. A mutation of the oGnRHR promoter CRE (µCRE) was achieved by using a two-step PCR (42) to place an EcoRI recognition site in the context of the both the -160 and the -2700 oGnRHR-LUC constructs.
Cell culture, transient transfection, and luciferase assays
All cell cultures were maintained in a humidified atmosphere of 5% CO2 at 37°C. The αT3–1 cells were cultured in high-glucose DMEM containing 2 mM glutamine, 5% FBS, 5% horse serum, 100 U/ml penicillin, and 100 μg/ml streptomycin sulfate. These cells were transfected for 18 hours utilizing Lipofectamine™ (Thermo Fisher Scientific, Waltham, MA) with oGnRHR-LUC promoter constructs (-2700LUC, -160LUC, -115LUC, -79LUC) as well as the mutated CRE construct (-160µCRELUC) and promoterless control (LUC) (11, 43). To achieve a robust but not maximal response, cells were then treated for 5 h with 10 μM forskolin in DMSO (Sigma, St. Louis, Mo). At the time of sample collection, the media was aspirated, and the cells were washed twice with ice-cold PBS (pH 7.4). Cells were lysed in the wells by the addition of 200 μl lysis buffer (17). Lysates were collected and cellular debris was removed by centrifugation at 16,000 × g for 2 min. Cellular lysates were immediately assayed for luciferase activity by adding 20 µl lysate to 100 μl luciferin substrate (Promega Corp, Madison, WI) and measuring luminescence with a Turner model TD-20E luminometer set for a 5 sec delay and 10 sec integration. β-Galactosidase activity was measured in 50 μl of lysate using the luminescent assay system and substrate (Tropix, Bedford, MA) with the same luminometer set for a 10-sec delay and 5-sec integration following manufacturer’s instructions. Luciferase activity in transfection assays was normalized for transfection efficiency (% induction) by dividing the luciferase activity by β -galactosidase activity.
Generation and screening of transgenic mice
All animal work described herein was performed under veterinary supervision with approval from the Colorado State University Institutional Animal Care and Use Committee (IACUC) and in accordance with the NIH Animal Care and Use Guidelines. All mice used in these experiments were of the FVB inbred strain (44). Animals were maintained under a 14 h light, 10 h dark cycle, received food and water ad libitum, and all experiments were conducted using animals older than 6 weeks of age. Genomic DNA was extracted from tail or ear biopsies and analyzed for the presence of the transgene by slot blot hybridization (45, 46) or PCR amplification employing primers specific to luciferase (45) or the transgene. The generation of transgenic mice harboring either approximately 1,900 bp of proximal promoter from the murine GnRHR gene fused to the cDNA encoding for luciferase (mGnRHR-LUC) or 9,100 bp of proximal promoter from the ovine GnRHR gene fused to the cDNA encoding for luciferase (oGnRHR-LUC) has been described (11). However, cryopreserved embryos from the oGnRHR-LUC animals were thawed and transferred by The Jackson Laboratory (Bar Harbor, ME) and a single oGnRHR-LUC female was used to re-derive the wild-type (WT) promoter.
To generate animals containing a mutated CRE, a construct with an EcoRI site (µCRE) within the ovine -2700LUC plasmid (µCREoGnRHR-LUC) was digested with SacI and BamHI to release the transgene and purified by electroelution. Transgenic founder animals were generated using standard microinjection by the Transgenic and Gene Targeting Core at University of Colorado Denver-Anschutz Medical Campus (Denver, CO). Genomic DNA was extracted from tail biopsies with a REDExtract-N-Amp Kit (Sigma, St. Louis, MO) and analyzed for the presence of the transgene by PCR amplification using primers to luciferase. Four transgenic founder mice (3 males, A-C; 1 female, D) with the µCREoGnRHR-LUC gene were bred to non-transgenic FVB mice and offspring were genotyped specifically for the μCRE transgene. The female founder (Line D) was euthanized during parturition of her first litter as a result of dystocia.
Extensive tissue screening of gonad-intact males and females for all lines harboring the µCRE were performed to confirm tissue-specific expression of luciferase activity as compared to the animals harboring the WT promoter. Luciferase activity was measured from samples of the pituitary gland, brain, gonad, liver, kidney, lung, heart, and spleen.
Luciferase assays of tissue samples
Luciferase assays were performed as previously described (11). Briefly, tissue samples were prepared by homogenization in 200 μl cold lysis buffer (25 mm glycyl-glycine, pH 7.8; 1.0% Triton X-100, 10 mm MgSO4, and 1.0 mm dithiothreitol). Cellular debris was pelleted by microcentrifugation at 16,000 × g for 5 min at 4° C. Cellular lysates were immediately assayed for luciferase activity as above. Total protein was precipitated from lysates with 10% trichloroacetic acid and then dissolved in 0.1 n NaOH. Protein concentrations were determined using the bicinchoninic acid (BCA) Assay (Pierce Chemical Co., Rockford, IL), and luciferase activity was adjusted for protein content by dividing the arbitrary light units by the protein content in mg.
Radioimmunoassays
Concentrations for FSH were determined via radioimmunoassay (47). The reference standards NIH-rFSH-RP2, the antibody anti-rat FSH A621 (1:32,000), and iodinated rat FSH (125I-rFSH) were used. Intra-assay coefficient of variation ranged between 4.2 and 8.8%. The inter-assay coefficient of variation was 15.4%. The mean (± SEM) limit of detection for FSH was 43.3 ± 14.2 pg/200 μL.
Animal treatments
Experiment 1: AdCAFS288 and AdGFP infection of non-transgenic mice.
Male and female non-transgenic mice were gonadectomized. One week post-surgery, mice received a single intraperitoneal (IP) injection of adenovirus expressing either follistatin (AdCAFS288) or green fluorescent protein (AdGFP). Mice were randomly assigned to one of two doses of each virus; 3.9x1010 pfu/ml (AdGFP n=5; AdCAFS288 n=4) or 3.9x1011 pfu/ml (AdGFP n=5; AdCAFS288 n=3) in 200 μL of phosphate buffered saline (PBS). Three days following injection, trunk blood was collected and serum concentrations of FSH were determined by radioimmunoassay.
AdCAFS288 and AdGFP infection of mGNRHR-LUC and oGNRHR-LUC transgenic mice
Male and female mGnRHR-LUC (17) and oGnRHR-LUC (11) transgenic mice were gonadectomized. After 7 days, the mice received a single IP injection of 3.9x1011 pfu/ml of either AdGFP (mGnRHR-LUC n=8; oGnRHR-LUC n=7) or AdCAFS288 (mGnRHR-LUC n=8; oGnRHR-LUC n=6) in 200 μL of PBS. Three days post-injection, animals were sacrificed, and pituitary glands and livers were harvested for luciferase assay as described. Trunk blood samples were also collected for the determination of serum concentrations of FSH.
E2 Responsiveness of the oGnRHR Promoter in vivo
Extensive validation of the new µCREoGnRHR-LUC animals included luciferase assays from tissues pituitary (Pit), brain (B) gonad (G), liver (Lv), kidney (K), lung (Lg), heart (H), and spleen (S) obtained from transgenic male and female animals from each of the µCRE founder lines (µA n=12 male, n=8 female; µB n=8 male, n=14 female; µC n=13 male, n=5 female). For each founder line, animals that were PCR negative for the transgene were also evaluated (µA n=14 male, n=14 female; µB n=8 male, n=11 female; µC n=16 male, n=10 female). Average luciferase expression in non-transgenic animals (average + 2 standard deviations) was used to determine transgene expression for each of the tissues. Luciferase expression in transgenic (n=21) as compared to non-transgenic (n=19) WT oGnRHR-LUC animals was confirmed before initiating the E2 experiments.
To evaluate E2 responsiveness of the oGnRHR promoter, 7 days after OVX, transgenic females harboring the WT or µCREoGnRHR-LUC transgene were randomly assigned to one of three treatment groups: 1) no treatment (WT n=5; µA n=4; µB n=8; µC n=8); 2) AS but no E2 (WT n=8, µA n=7, µB n=10, µC n=8); and 3) AS and E2 implant (WT n=6; µA n=7; µB n=7; µC n=8). Those implanted with the 2.5 mg E2 pellet were housed separately from the sham (non-E2) OVX females. Five days later, females (± E2) were given GnRH antiserum (300 μl IP), and at 36–48 hr, tissues (pituitary gland, brain, liver) were harvested and assayed for luciferase activity.
Electrophoretic mobility shift assays
Ovine pituitary glands (n=2) were obtained from female sheep (ages 3–7 years) immediately following euthanasia and dissociation of ovine pituitary as previously described (48). Cells were treated with 10 nM E2 or an equivalent volume of vehicle (ethanol) in media for 30 min (38) before proceeding with nuclear extraction. Cell nuclear extracts were prepared using the Nuclear Extraction Kit (Signosis, Inc., Santa Clara, CA), and EMSAs were performed using the LightShift Chemiluminescent EMSA Kit (Thermo Fisher Scientific, Waltham, MA). Nuclear extracts were incubated at room temperature in 1x Binding buffer from Light Shift kit, 100 mM KCl, 5% (vol/vol) glycerol, 50 ng/μL poly(dI-dC), and 1 mM EDTA, pH 7.5 with unlabeled and biotinylated (probe) oligonucleotides. After incubation at room temperature, the samples were resolved on a 5% nondenaturing polyacrylamide gel prepared in 89 mM Tris-borate and 2 mM EDTA (TBE) buffer, pH 8.3. The specimens were electrotransferred onto a 0.45 μm Biodyne B nylon membrane (Thermo Fisher Scientific, Waltham, MA) at 380 mA for 30 min at 4°C, and crosslinked to the membrane using a CL-1000 UV Translinker and developed using a Chemiluminescent Nucleic Acid Detection Module Kit from Thermo Fisher Scientific (Waltham, MA). Biotinylated probe and unlabeled oligonucleotides (Table 1) were custom synthesized by Integrated DNA Technologies, Inc. (Coralville, Iowa). Goat polyclonal p-CREB-1 (Ser 133) (sc-7978) antibody and mouse monoclonal CREB-1 antibody (sc-240X) were purchased from Santa-Cruz Biotechnology (Dallas, TX).

Table 1. Oligonucleotides for EMSA.
Statistical analysis
Data are expressed as the mean ± the standard error of the mean (SEM). Only when evaluating the µCREoGnRHR-LUC transgene was sex considered a variable in the statistical analysis. For differences in luciferase expression or serum FSH concentration between mice infected with AdGFP versus AdCAFS288, a Student’s T-test was used for analysis. For the experiments involving the µCRE oGnRHR-LUC mice, all data were analyzed using the NCSS8 (Kaysville, UT) statistical software General Linear Model ANOVA, followed by a Tukey-Kramer Multiple Comparison Test. Significance, unless otherwise stated, was P < 0.05. Tissues expressing luciferase activity at levels above the mean + 2 Standard Deviations (2SD) of the values in tissues from non-transgenic animals in that population was considered positive for expression of the transgene. Where necessary, data were log transformed to correct for heterogeneity of variance. Means were separated using Student’s T-test and Dunnett’s (Figures 2–4) or Tukey’s (Figures 6, 7) methods of multiple comparisons.
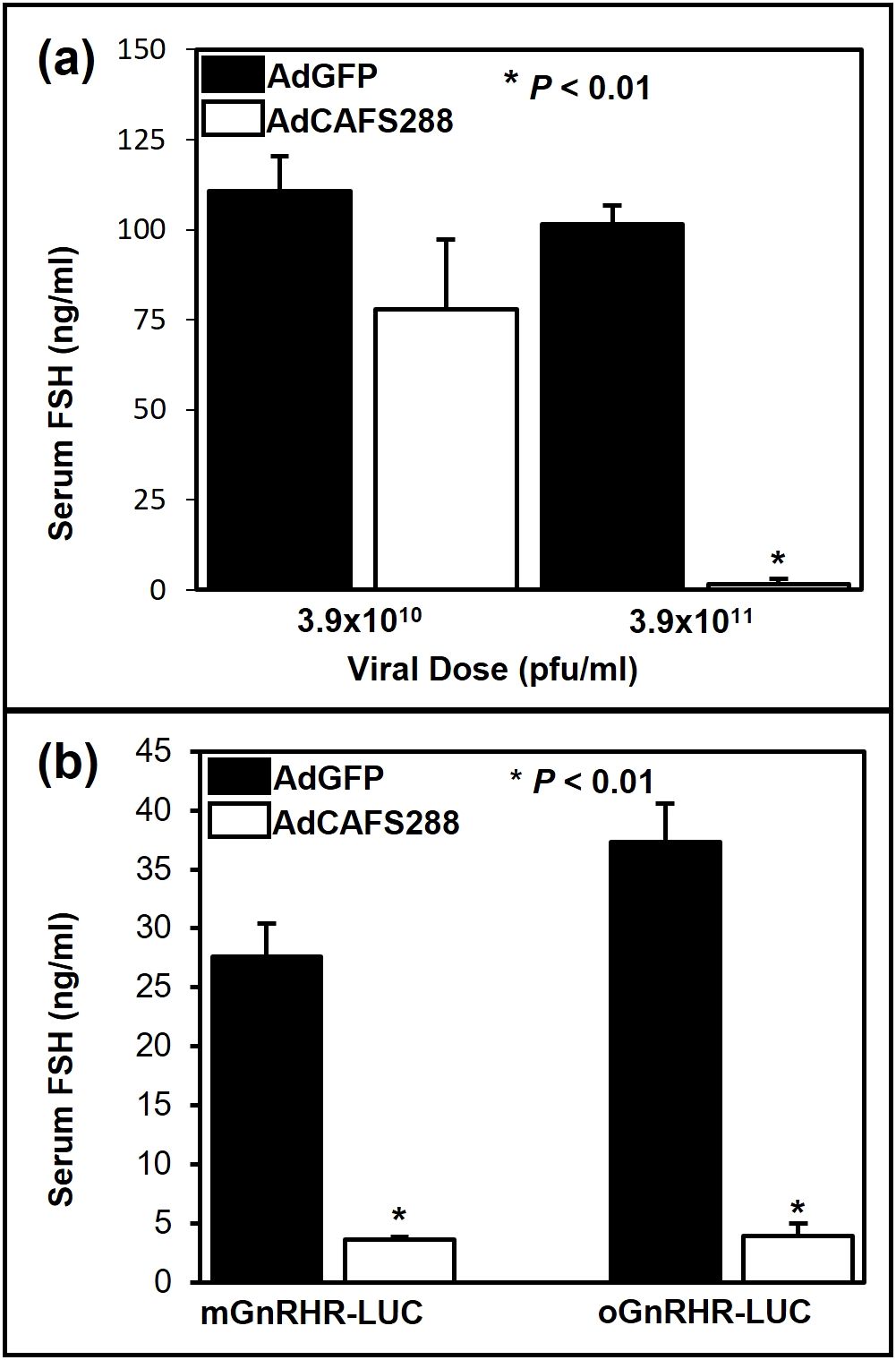
Figure 2. Adenoviral mediated over-expression of follistatin suppresses serum concentrations of FSH. (a) Non-transgenic FVB mice were gonadectomized and 7 d later infected with one of two doses of either AdGFP or AdCAFS288. At 3 d post-infection, trunk blood was collected and serum concentrations of FSH were determined by RIA. Values represent the mean + SEM, * (P<0.01) between AdGFP and AdCAFS288 infected mice for each dose. (b) Transgenic mice expressing either the wild-type mouse or sheep GNRHR promoters fused to luciferase were gonadectomized and infected as before with a single injection of 3.9 x 1011 pfu/ml of either AdGFP or AdCAFS288. At 3 d post-infection, trunk blood was collected and serum concentrations of FSH were determined by RIA. Values represent the mean + SEM. * (P<0.01) between AdGFP and AdCAFS288 infected animals for each line of transgenic mice.
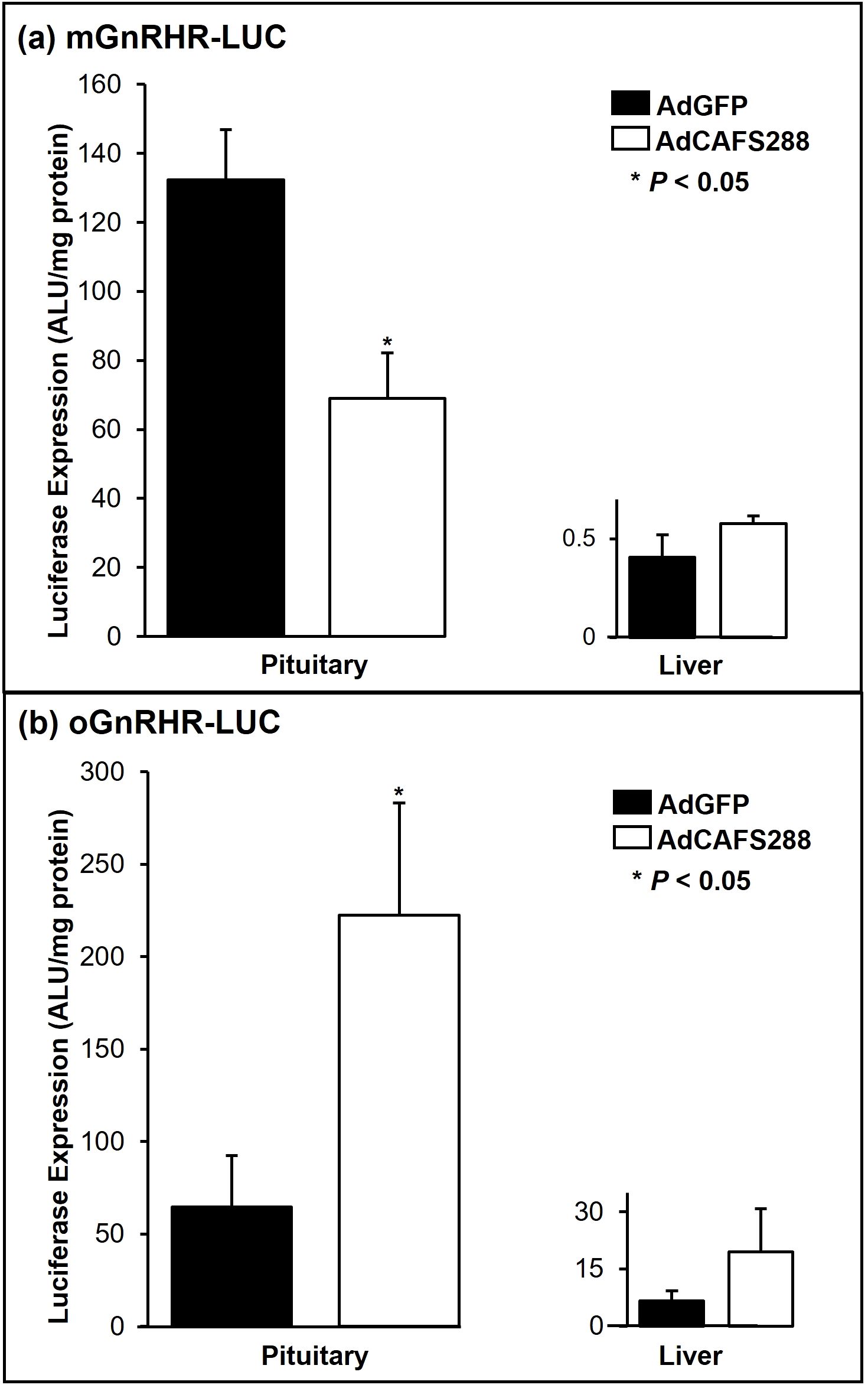
Figure 3. Divergence in follistatin mediated pituitary luciferase expression between the mouse and sheep GnRHR promoters. Transgenic mice expressing either the wild-type mouse (a) or sheep (b) promoters fused to luciferase were gonadectomized and 7 d later were infected with a single IP injection of 3.9 x 1011 pfu/ml of either AdGFP or AdCAFS288. At 3 d post-infection, the indicated tissues were harvested and assayed for luciferase activity. Luciferase values were adjusted for protein content and are expressed as arbitrary light units (ALU) per mg of protein. Values represent the mean + SEM. * (P<0.05) between AdGFP and AdCAFS288 for each transgenic line of mice.
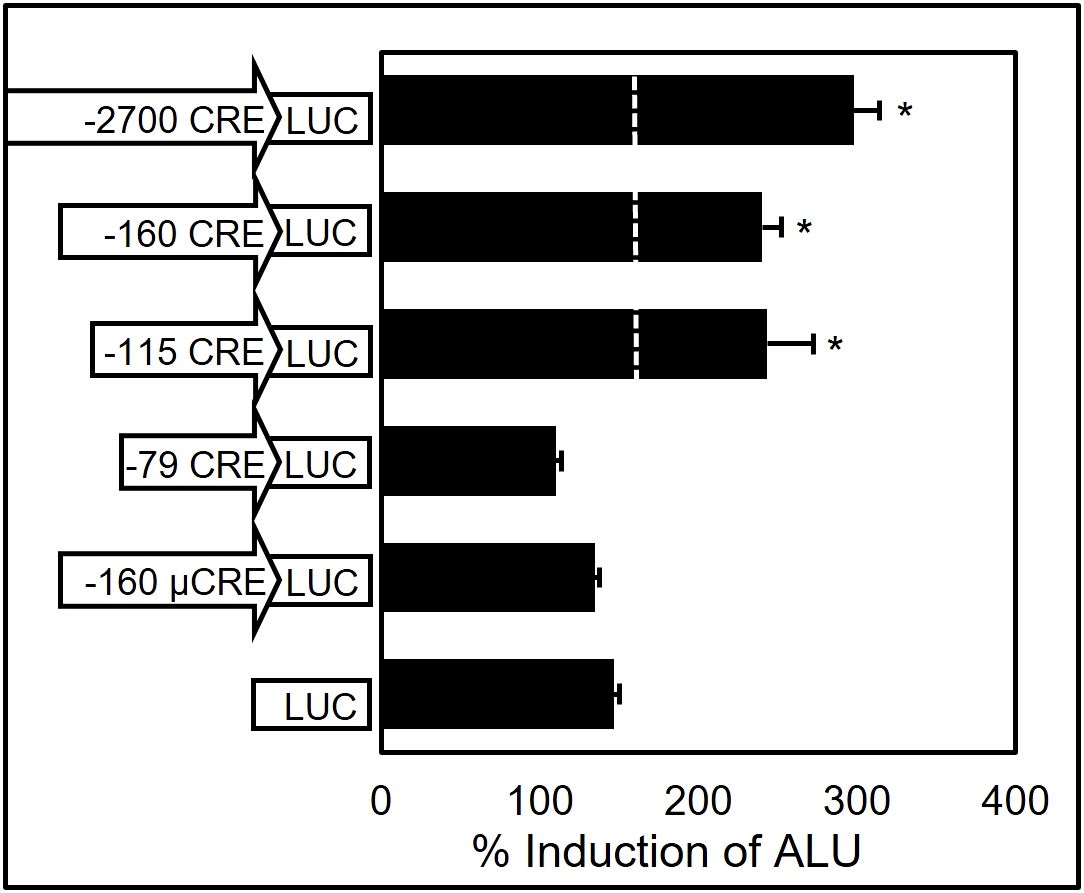
Figure 4. Transient transfection assay of αT3–1 cells with ovine GnRHR gene promoter deletion and mutation constructs reveals that the CRE confers responsiveness of the oGnRHR to forskolin. Cells were transfected for 18 h, then treated for 5 h with 10 μM forskolin in DMSO. Promoter regions with CRE or μCRE are indicated as fused to luciferase (LUC). Values represent mean + SEM percent (%) induction of adjusted luciferase units (ALU) over promoterless (LUC) control. * represents luciferase values significantly different from LUC only (P < 0.05).
Results
Divergent regulation of ovine and murine GnRHR gene expression by follistatin in transgenic mice
The ability of follistatin to attenuate transcriptional activity of the murine GnRHR gene promoter in αT3–1 cells has also been well established (18, 49) and more recently activin has been shown to induce mGnRHR transcription in vivo (50). Activin has been shown to decrease oGnRHR expression in primary ovine pituitary cells (25); however, this effect has never been studied in vivo. To address this question, we first tested the effect of increasing viral load on serum concentrations of FSH in non-transgenic mice of the same genetic background (FVB) in which our ovine and murine GnRHR-LUC transgenic lines were constructed. At the 3.9x1011 pfu/ml dose, FSH levels were reduced by approximately 90% (P < 0.01) in AdCAFS288 infected FVB mice compared to the AdGFP group (Figure 2a). Thus, as in rats (40), in vivo infection with AdCAFS288 effectively neutralizes activin input to gonadotropes in both male and female wild-type mice Subsequent studies in transgenic mice were conducted using the 3.9 x 1011 pfu/ml dose for AdCAFS288 and AdGFP. The adenoviral paradigm described for Figure 2a was then applied to mice harboring the transgene consisting of either approximately 1,900 bp of proximal promoter from the murine GnRHR gene fused to the cDNA encoding for luciferase (mGnRHR-LUC) or 9,100 bp of the proximal promoter from the ovine GnRHR gene fused to the luciferase cDNA (oGnRHR-LUC). The tissue-specific activity of these promoters has been previously established (11, 39). As above, in animals harboring both the ovine and murine GnRHR promoters, the AdCAFS288 infection significantly reduced serum FSH levels compared to AdGFP, thus confirming effective neutralization of activin in both transgenic models (Figure 2b). However, divergence in follistatin regulation of GnRHR expression was observed in pituitary expression of luciferase between the mouse and sheep GnRHR promoters (Figure 3). Pituitary luciferase expression in the mGnRHR-LUC animals was reduced by approximately 40% (P < 0.01) in animals receiving AdCAFS288 as compared to the AdGFP treatment group (Figure 3a). In alignment with what has been shown in primary ovine pituitary cells (25), pituitary luciferase expression in the oGnRHR-LUC animals was more than doubled (P < 0.05) in the animals receiving AdCAFS288 as compared to the AdGFP treatment group (Figure 3b). Follistatin-mediated reduction in luciferase expression in the mouse is equivalent to that which has been observed in vitro (49). The adenoviral constructs likely do not cross the blood-brain barrier, therefore evaluation of luciferase expression in the brain may not be biologically relevant and are therefore not shown. Hepatic expression of luciferase, included as a negative control tissue, was at or below detection limits in animals harboring the mouse promoter, but not different between AdCAFS288 as compared to the AdGFP for the promoter of either species (Figure 3). Sex differences were not observed in gonadectomized animals and therefore sex was not a variable included in the analysis.
The ovine GnRHR CRE confers responsiveness to cAMP and forskolin in αT3–1 cells
Despite the absence of significant basal activity of the ovine 5’ flanking region in the murine pituitary-derived αT3–1 cell line (11), there was still a distinct possibility that element(s) conferring responsiveness to endocrine mediators of GnRHR gene expression may be located within the promoter fragment and amenable to detection using transient transfection assays. Transfection of αT3–1 cells with oGnRHR promoter deletion vectors (-2700LUC, -160LUC, -115LUC, Figure 4) or a positive control vector of 1,500 bp of proximal promoter from the human glycoprotein hormone alpha-subunit gene (51–53), with subsequent treatment of the transfected cells with forskolin, led to robust induction (P < 0.05) of the oGnRHR promoter constructs. These data suggested that the presence of one or more elements located within 160 bp of proximal promoter was responding to cAMP. In fact, examination of this region had revealed the presence of a near-consensus cAMP response element (CRE; 5’TGACGTTA3’) (54) located between -111 and -104 relative to the start site of translation. To test the potential role of this element, two additional deletions were constructed that deleted (-79LUC) or mutated (-160μCRELUC) the CRE homolog. Forskolin treatment of αT3–1 cells transfected with ovine -2700, -160, and -115 constructs led to a significant increase in luciferase expression that was lost upon deletion or mutation (Figure 4).
Transgenic mice expressing the -9100 μCRE oGnRHR-LUC mice retain tissue specific luciferase expression
Amplification of DNA from the WT and μCRE lines (A-C) of mice, followed by EcoRI restriction enzyme digest of the PCR amplicon, confirmed the presence of the mutated CRE in the -9100 μCRE oGnRHR-LUC A-C lines of mice (Figure 5). To confirm tissue-specific luciferase expression in these animals, extensive tissue screens of luciferase-positive and negative animals were performed (Figures 6a–c). A confidence interval was constructed using the mean + 2SD from tissues collected from animals that were PCR-negative for the luciferase transgene from all 4 transgenic lines. Tissue luciferase activity in the μCRE animals was consistent with that of the WT animals, though the amplitude of expression was significantly lower in the μCRE lines (Figure 6d). In all lines, luciferase activity was significantly higher in the pituitary gland from transgenic females, the gonad from transgenic males, and in the hypothalamic region of the brain of both transgenic males and females; whereas the liver was consistently among the lowest luciferase-expressing samples. Interestingly, both transgenic Line B and WT females had increased luciferase activity in the heart.
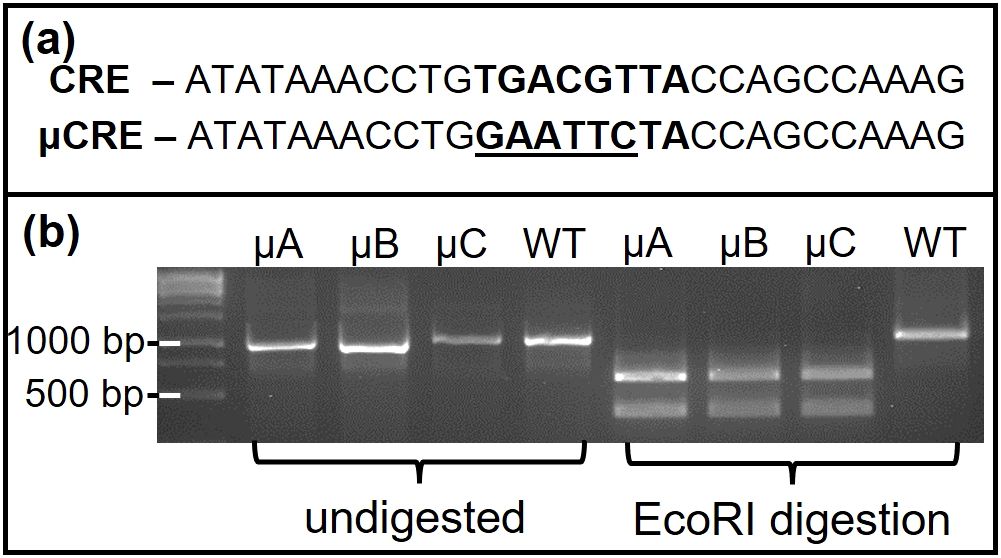
Figure 5. Mutation of CRE is present in µCRE oGnRHR-LUC founders but not WT oGnRHR-LUC animals. (a) The (5’-3’) conserved CRE site (TGACGTTA) and the EcoRI restriction site (GATTCC) introduced in -104/-97 bases of oGnRHR-LUC proximal to LUC. (b) PCR amplicon of genomic DNA spanning the oGnRHR promoter and LUC gene was digested with EcoRI to reveal the mutation in μCREoGnRHR-LUC founders A-C, but was not present in WT animals.
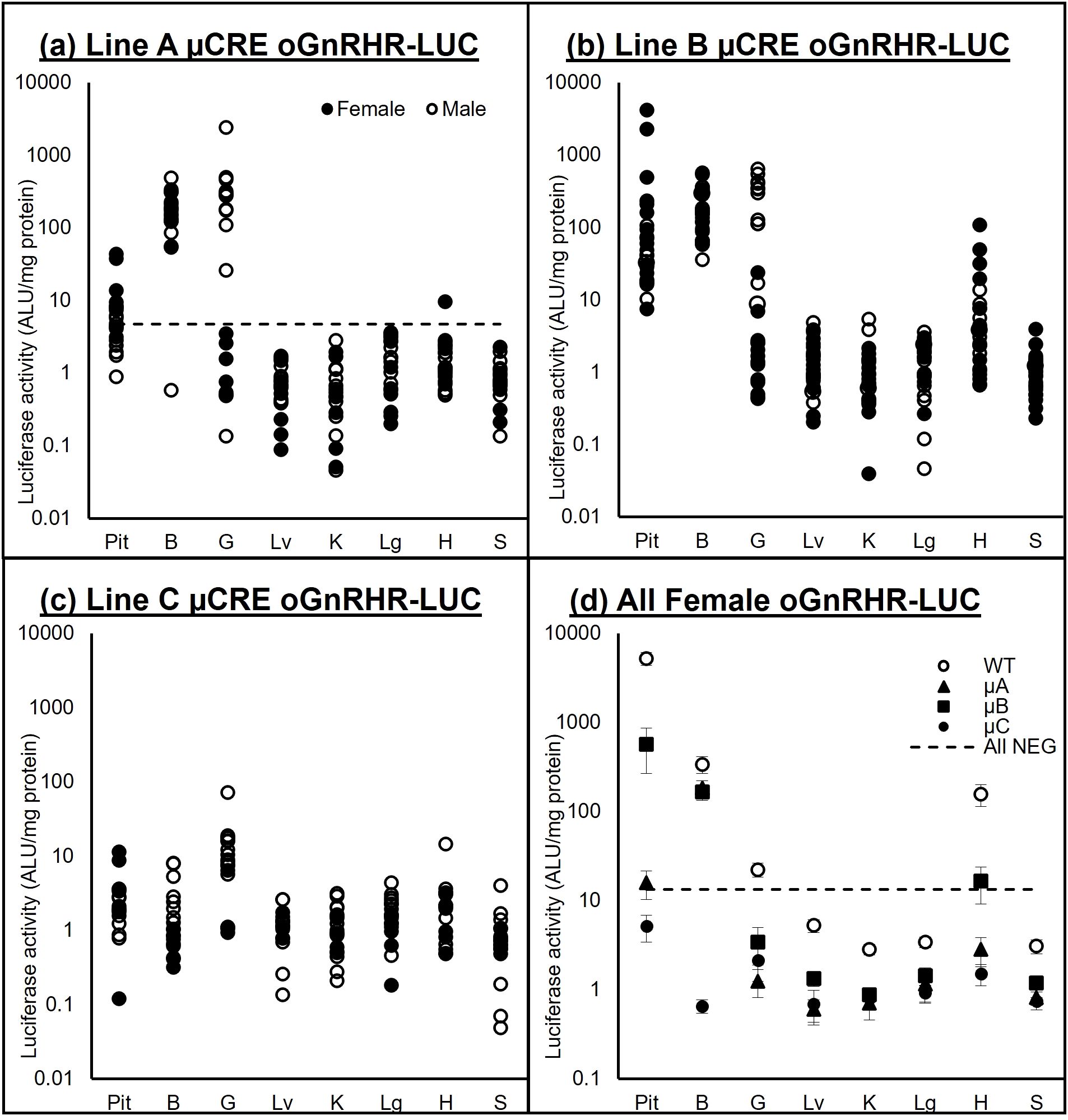
Figure 6. Luciferase activity (ALU/mg protein) in oGnRHR-LUC mice. (a–c) Samples from pituitary (Pit), brain (B) gonad (G), liver (Lv), kidney (K), lung (Lg), heart (H), and spleen (S) from males (○) and females (●) of Lines A-C of the µCREoGnRHR-LUC line of transgenic mice. (d) Mean (± SEM) tissue luciferase activity of transgenic females from each line (○WT, ▲µA, ■µB, ●µC) of oGnRHR-LUC. Tissue luciferase expression values from negative females from all 4 transgenic lines was averaged + 2 SD to determine the All NEG. Tissue luciferase activity in the μCRE animals was consistent with that of the WT animals, though the amplitude of expression was significantly lower in the μCRE lines. In all lines, luciferase activity was significantly higher in the pituitary gland from transgenic females, the gonad from transgenic males, and in the hypothalamic region of the brain of both transgenic males and females.
A functional CRE is necessary to confer estradiol responsiveness to the oGnRHR promoter
GnRH regulation of pituitary GnRHR expression was observed in transgenic mice with both the WT and µCRE versions of the oGnRHR promoter following treatment with GnRH AS. Specifically, in both the WT promoter and µCRE line B animals, blockade of GnRH with AS resulted in a significant decrease in pituitary luciferase expression (Figure 7b). In the same groups of animals, when compared with no treatment, treatment with AS and E2 resulted in a significant increase in pituitary luciferase expression in the animals with the WT oGnRHR promoter, but not in the µCRE line B oGnRHR promoter animals. While the changes in pituitary luciferase expression for the µCRE lines A and C following treatment with AS and/or E2 were not significantly different from the untreated animals, similar trends were observed (Figure 7b). There was no evidence of GnRH or E2 regulation of brain (Figure 7c) or liver (Figure 7d) luciferase expression in either the WT or µCRE oGnRHR promoter animals.
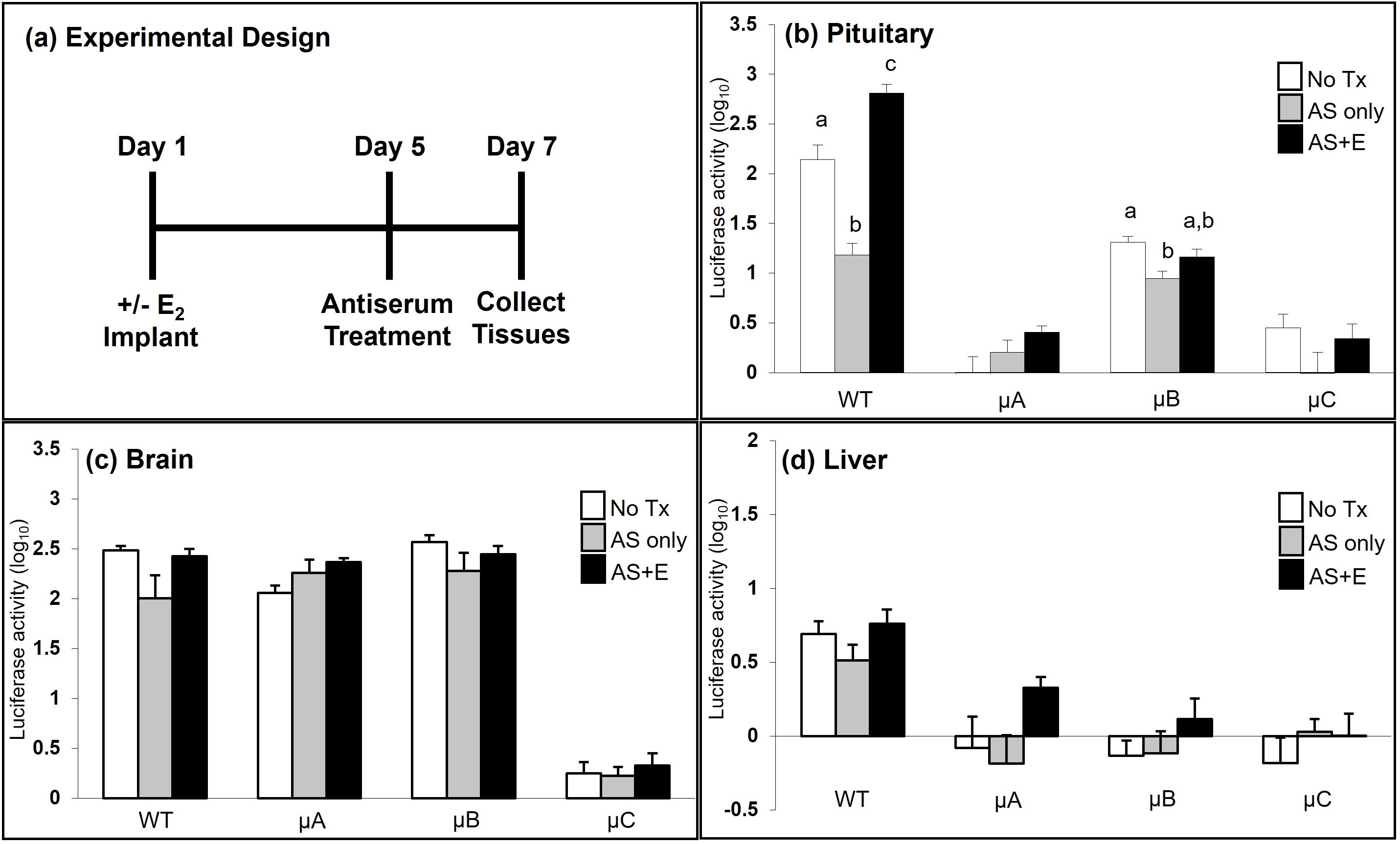
Figure 7. A functional CRE is necessary to confer estradiol responsiveness to the oGnRHR promoter. (a) Seven days after OVX, females (WT, and A-C of the µCRE -9100 oGnRHR-Luc) were randomly implanted with a 2.5 mg E2 pellet (Day 1) and housed separately from non-E2 (sham procedure) OVX females. Five days later (Day 5), females (± E2) were given GnRH antiserum (AS, 300 μl IP) or sham injection, and at 36–48 hr (Day 7) tissues were harvested for luciferase activity luciferase activity (ALU/mg protein). Data were log transformed and expression in the pituitary gland (b), brain (c), and liver (d) are presented. a,b,c indicate differences between treatment, within the WT or transgenic line for the tissue indicated, P < 0.05.
CREB binding is detected at the ovine CRE. While the transgenic mouse work indicates that a functional CRE binding domain of the ovine proximal GnRHR promoter is necessary to confer E2 input to the oGnRHR, the identity of the transcriptional machinery that mediates E2 input to the receptor in the ovine pituitary remains unknown. No canonical ERE is located within the 9,100 bases of the proximal oGnRHR promoter. EMSA using nuclear extract from ovine pituitary cells demonstrated competition of the biotinylated ovine CRE with increasing concentrations of ovine, human, and the consensus CRE oligonucleotide sequences, but not the non-specific CRE oligonucleotides (Figure 8a). Thus, CRE binding is specific with equivalent affinity across CRE oligonucelotide sequences. Further evaluation demonstrated that both CREB and phosphorylated-CREB (pCREB) are associated with elements in ovine nuclear extract, as demonstrated by the supershift that occurs with or without the presence of additional unlabeled oligonucelotides (Figure 8b). However, exposure of the ovine pituitary cells to E2 failed to result in any additional interactions or gel-shifts (data not shown). Thus, the identity of the element that confers E2 signaling to the ovine GnRHR remains unknown.
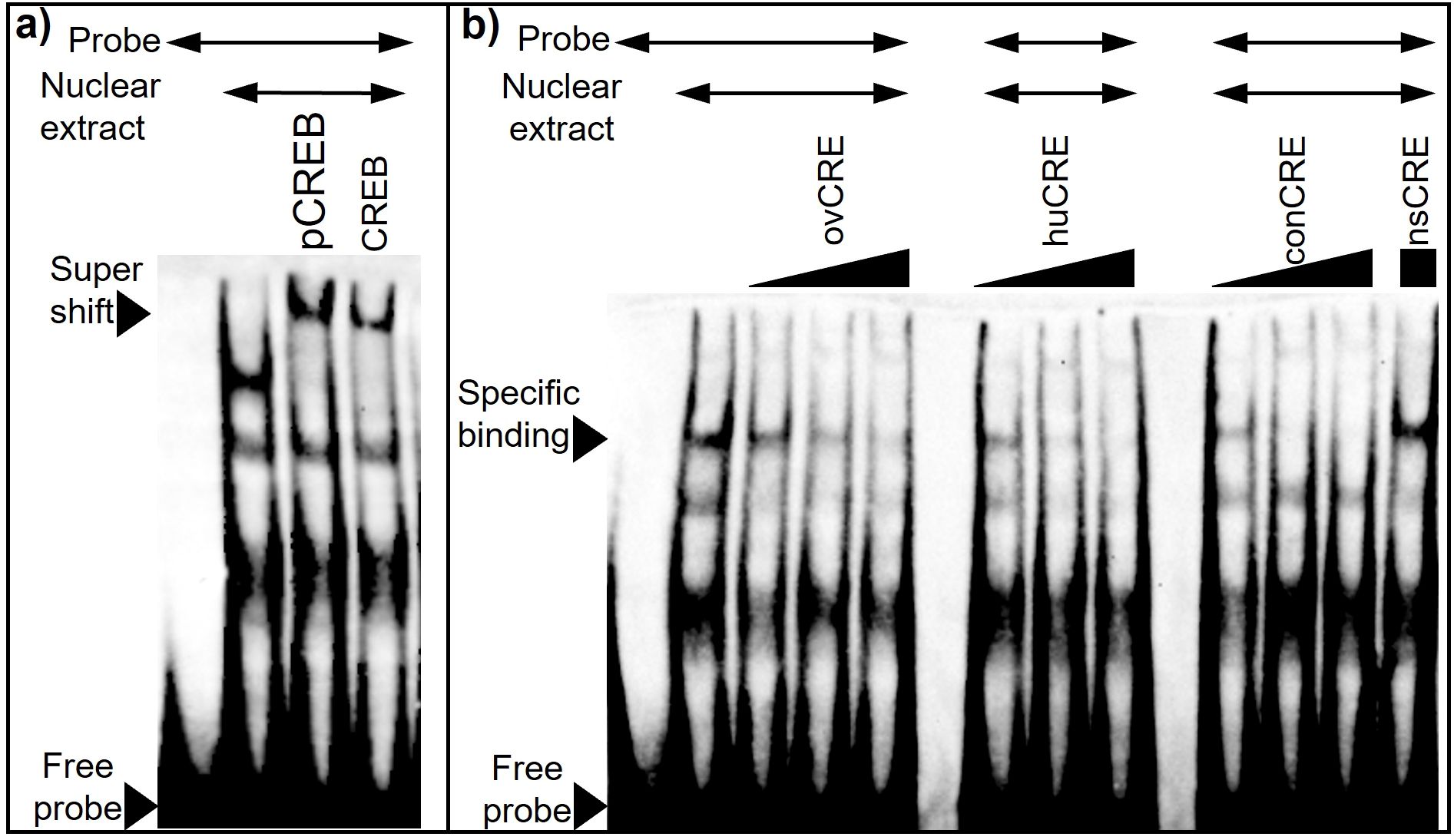
Figure 8. CREB binding is detected at the ovine CRE. (a) Both CREB and phosphorylated CREB (pCREB) are associated with DNA binding elements in ovine nuclear extract. Nuclear extract (5 ug) from ovine pituitary cells was incubated for 15 min with 1 µg of antibody for either CREB or pCREB, then incubated an additional 20 min with biotinylated oCRE oligonucelotide. (b) Human and consensus CRE elements compete for DNA binding elements in ovine pituitary nuclear extract. Nuclear extract (10 ug) was incubated for 20 min with biotinylated oCRE oligo. Specificity of DNA-protein interactions was assessed by competition with unlabeled oligonucleotides for ovine (ov), human (hu), and consensus (con) CRE at 10, 50, and 100 molar excess, and nonspecific (ns) CRE at 100 molar excess.
Discussion
The divergent mechanisms that regulate the female hypothalamic-pituitary-ovarian (HPO) axis demonstrate that there is no single best model or species. In the animal models predominantly used to study the HPO, GnRH input to the gonadotrope is unequivocally the most important and consistently positive endocrine regulator of GnRHR expression (55–61). However, the effect that the pulsatile nature of GnRH signaling has on the GnRHR and subsequent signaling pathways and transcriptional mechanisms that result in gonadotropin synthesis and secretion (62) confound the inherent effect of GnRH on GnRHR expression when gonadotropins or ovulation are used as the measurable factor. The initial murine-derived immortalized cell lines (i.e. αT3–1 cells, LβT-2) (63–65) are inherently valuable and have permitted innumerable experiments to study gonadotropes function and signaling. The more recently developed mPitA-12/3 cell line is presumed to be from a fully differentiated cell type and responds to GnRH, but not E2, treatment with an increase in GnRHR expression (66).
At issue is not the value of the model, but the diversity of the results gained from the different model systems. For example, myostatin as a regulator for FSH in rodents has been described, but the conservation of this mechanism in non-rodent species has yet to be determined (67). The study of activin and follistatin on mouse and rat GnRHR expression using immortalized cell lines and/or transgenic mice has consistently demonstrated distinct mechanisms (18, 50). While elements of these mechanisms, including FOXL2 (50) and AP-1 (43) sites are found in the human FSHB (68) and GnRHR (69) promoter, the GRAS (43) and DARE sequences found in rodents are not conserved in either the ovine or human GnRHR promoter (70). It is therefore not surprising that while follistatin overexpression is able to decrease serum FSH in transgenic mice expressing either the mouse or the sheep GnRHR fused to luciferase, we have demonstrated divergence in luciferase expression between the mouse and ovine GnRHR promoters (Figure 3). These findings are also consistent with treatment of ovine pituitary cells with either inhibin or activin-A (4, 25, 71).
Estradiol regulates oGnRHR expression through unresolved mechanisms. Work in mice (39) and sheep (11) consistently demonstrates that GnRH input to the GnRHR promoter is necessary to confer basal expression of GnRHR, but does not eliminate the input of E2 in the upregulation of luciferase expression in the oGNRHR promoter (11). The use of antiserum directed against GnRH effectively neutralizes GnRH input to the GnRHR promoter while not disrupting receptor expression. The indisputable ability of estradiol to enhance ovine pituitary sensitivity by increasing the number of GnRHR in vivo and in vitro (30, 58, 72–74) resulted in the initial use of transgenic mice expressing oGnRHR promoter constructs fused to luciferase expression vectors. These animals revealed E2 regulation of the oGnRHR that could not be elicited in gonadotrope-derived cell lines (11); although the LβT-2 cell demonstrates a moderate increase in GnRHR expression following E2 treatment (75), the αT3–1 cells actually demonstrate a decrease in GnRHR numbers and mRNA following E2 treatment (76, 77). The female rat also appears to be responsive to E2 in vivo (78, 79), but the changes in GnRHR number are most likely attributable to changes in GnRH secretion (3). Furthermore, GnRH (independent of E2) is able to activate an ERE driven luciferase reporter and transcriptional activation of the immediate early response gene fosB via an interaction of ERα with the histone acetyltransferase p300/CREB-binding protein-associated factor (PCAF) (80). While the fosB promoter contains an ERE, there is no canonical estradiol response element (ERE) in the rat, mouse, or sheep GnRHR promoter (36, 70, 81).
The data reported herein demonstrate that CRE in the oGnRHR promoter is necessary but not sufficient to mediate E2 input to the oGnRHR promoter. Unlike many other transcriptional factors, the CRE is conserved in the proximal region of the sheep, mouse, human, and rat GnRHR promoters (70). The oGnRHR promoter is responsive to forskolin in αT3–1 cells, but this responsiveness is lost with mutation of the CRE. The first indication of the importance of the CRE for conferring the E2 signal to the ovine oGnRHR came from adenoviral delivery of a dominant negative form of CREB (ACREB) eliminating the predicted E2 mediated increase in GnRHR number (38). In the reported EMSAs, there was no supershift observed in the presence of E2, but this is not conclusive evidence that CREB and E2 are not interacting. Converging mechanisms involving parallel enhanceosome or mediator complexes (82), including those that may be derived from a membrane site of action, may be involved. Treatment of ovariectomized ewes with an IM bolus of E2, or dispersed ovine pituitary cells with membrane-impermeable forms of E2, both result in a decrease in LH secretion followed by an increase in GnRHR number (38). However, in ovine pituitary cells, the dominant-negative form of ESR1 (L540Q), a mutation site known for co-activator binding (83, 84), is able to eliminate E2 activation of GnRHR (38). With mechanisms that clearly reside in different parts of the gonadotrope, it is altogether possible that the method by which these mechanisms converge has yet to be elucidated. This work also suggests that a membrane-associated ESR1 is the likely candidate to mediate E2 activation of the GnRHR promoter (38).
The µCRE oGnRHR-LUC mouse clearly demonstrates that CRE mutation does not eliminate GnRH input to luciferase expression but does eliminate E2 upregulation of luciferase. Therefore, future studies of the interaction of E2 with the CRE region of the proximal GnRHR promoter will require pituitary-specific elimination of ESR1 (85) in the context of a gonadotrope-specific ESR1 knockout (86) and the oGnRHR-LUC mouse to determine the functional contribution of ESR1 in vivo. Alternatively, the use of other technologies to eliminate the signal-to-noise ratio within the ovine pituitary gland (48) may be promising.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The animal study was approved by Colorado State University Institutional Animal Care and Use Committee. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
CM: Formal analysis, Investigation, Methodology, Writing – original draft, Data curation, Project administration, Supervision, Visualization, Writing – review & editing. JK: Data curation, Investigation, Methodology, Project administration, Writing – review & editing. BC: Formal analysis, Investigation, Writing – review & editing, Data curation, Writing – original draft. JC: Data curation, Formal analysis, Investigation, Methodology, Project administration, Supervision, Writing – review & editing, Validation, Visualization. DM: Data curation, Formal analysis, Investigation, Methodology, Validation, Writing – original draft, Writing – review & editing, Visualization. TF: Data curation, Formal analysis, Investigation, Methodology, Supervision, Visualization, Writing – review & editing. MD: Data curation, Formal analysis, Investigation, Visualization, Writing – review & editing. TN: Conceptualization, Formal analysis, Methodology, Writing – review & editing. CC: Conceptualization, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. Grant support: NIH R29 HD32416; NIH R01 HD065943.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Marian J, Cooper RL, and Conn PM. Regulation of the rat pituitary gonadotropin-releasing hormone receptor. Mol Pharmacol. (1981) 19:399–405. doi: 10.1016/S0026-895X(25)13699-7
2. Norwitz ER, Cardona GR, Jeong K-H, and Chin WW. Identification and characterization of the gonadotropin-releasing hormone response elements in the mouse gonadotropin-releasing hormone receptor gene. J Biol Chem. (1999) 274:867–80. doi: 10.1074/jbc.274.2.867
3. Kaiser UB, Jakubowiak A, Steinberger A, and Chin WW. Regulation of rat pituitary gonadotropin-releasing hormone receptor mRNA levels in vivo and in vitro. Endocrinology. (1993) 133:931–4. doi: 10.1210/endo.133.2.8393779
4. Wu JC, Sealfon SC, and Miller WL. Gonadal hormones and gonadotropin-releasing hormone (GnRH) alter messenger ribonucleic acid levels for GnRH receptors in sheep. Endocrinology. (1994) 134:1846–50. doi: 10.1210/endo.134.4.8137751
5. Mercer J and Chin WW. Regulation of pituitary gonadotrophin gene expression. Hum Reprod Update. (1995) 1:363–84. doi: 10.1093/humupd/1.4.363
6. Haisenleder D, Dalkin A, Ortolano GA, Marshall J, and Shupnik M. A pulsatile gonadotropin-releasing hormone stimulus is required to increase transcription of the gonadotropin subunit genes: evidence for differential regulation of transcription by pulse frequency in vivo*. Endocrinology. (1991) 128:509–17. doi: 10.1210/endo-128-1-509
7. Haisenleder DJ, Ortolano GA, Dalkin AC, Ellis TR, Paul SJ, and Marshall JC. Differential regulation of gonadotropin subunit gene expression by gonadotropin-releasing hormone pulse amplitude in female rats. Endocrinology. (1990) 127:2869–75. doi: 10.1210/endo-127-6-2869
8. Kaiser UB, Jakubowiak A, Steinberger A, and Chin WW. Differential effects of gonadotropin-releasing hormone (GnRH) pulse frequency on gonadotropin subunit and GnRH receptor messenger ribonucleic acid levels in vitro*. Endocrinology. (1997) 138:1224–31. doi: 10.1210/endo.138.3.4968
9. Papavasiliou SS, Zmeili S, Khoury S, Landefeld TD, Chin WW, and Marshall JC. Gonadotropin-releasing hormone differentially regulates expression of the genes for luteinizing hormone alpha and beta subunits in male rats. Proc Natl Acad Sci. (1986) 83:4026–9. doi: 10.1073/pnas.83.11.4026
10. Kakar SS, Winters SJ, Zacharias W, Miller DM, and Flynn S. Identification of distinct gene expression profiles associated with treatment of LβT2 cells with gonadotropin-releasing hormone agonist using microarray analysis. Gene. (2003) 308:67–77. doi: 10.1016/S0378-1119(03)00446-3
11. Duval DL, Farris AR, Quirk CC, Nett TM, Hamernik DL, and Clay CM. Responsiveness of the ovine gonadotropin-releasing hormone receptor gene to estradiol and gonadotropin-releasing hormone is not detectable in vitro but is revealed in transgenic mice. Endocrinology. (2000) 141:1001–10. doi: 10.1210/endo.141.3.7391
12. Ellsworth B, Burns A, Escudero K, Duval D, Nelson S, and Clay C. The gonadotropin releasing hormone (GnRH) receptor activating sequence (GRAS) is a composite regulatory element that interacts with multiple classes of transcription factors including Smads, AP-1 and a forkhead DNA binding protein. Mol Cell Endocrinol. (2003) 206:93–111. doi: 10.1016/S0303-7207(03)00235-1
13. White BR, Duval DL, Mulvaney JM, Roberson MS, and Clay CM. Homologous regulation of the gonadotropin-releasing hormone receptor gene is partially mediated by protein kinase C activation of an activator protein-1 element. Mol Endocrinol. (1999) 13:566–77. doi: 10.1210/mend.13.4.0262
14. Kam K-Y, Jeong K-H, Norwitz ER, Jorgensen EM, and Kaiser UB. Oct-1 and nuclear factor Y bind to the SURG-1 element to direct basal and gonadotropin-releasing hormone (GnRH)-stimulated mouse GnRH receptor gene transcription. Mol Endocrinol. (2005) 19:148–62. doi: 10.1210/me.2004-0025
15. Zhou H, Zarubin T, Ji Z, Min Z, Zhu W, Downey JS, et al. Frequency and distribution of AP-1 sites in the human genome(2005). Available online at: https://academic.oup.com/dnaresearch/article/12/2/139/360063 (Accessed July 2, 2025).
16. Bliss SP, Miller A, Navratil AM, Xie J, McDonough SP, Fisher PJ, et al. ERK signaling in the pituitary is required for female but not male fertility. Mol Endocrinol. (2009) 23:1092–101. doi: 10.1210/me.2009-0030
17. Ellsworth BS, White BR, Burns AT, Cherrington BD, Otis AM, and Clay CM. c-Jun N-terminal kinase activation of activator protein-1 underlies homologous regulation of the gonadotropin-releasing hormone receptor gene in αT3–1 cells. Endocrinology. (2003) 144:839–49. doi: 10.1210/en.2002-220784
18. Cherrington B, Farmerie T, Lents C, Cantlon J, Roberson M, and Clay C. Activin responsiveness of the murine gonadotropin-releasing hormone receptor gene is mediated by a composite enhancer containing spatially distinct regulatory elements. Mol Endocrinol. (2005) 19:898–912. doi: 10.1210/me.2004-0214
19. Cherrington BD, Farmerie TA, and Clay CM. A specific helical orientation underlies the functional contribution of the activin responsive unit to transcriptional activity of the murine gonadotropin-releasing hormone receptor gene promoter. Endocrine. (2006) 29:425–34. doi: 10.1385/ENDO:29:3:425
20. Fernández-Vázquez G, Kaiser UB, Albarracin CT, and Chin WW. Transcriptional activation of the gonadotropin-releasing hormone receptor gene by activin A. Mol Endocrinol. (1996) 10:356–66. doi: 10.1210/mend.10.4.8721981
21. Braden TD and Conn PM. Activin-A stimulates the synthesis of gonadotropin-releasing hormone receptors. Endocrinology. (1992) 130:2101–5. doi: 10.1210/endo.130.4.1312442
22. Braden TD, Farnworth PG, Burger HG, and Conn PM. Regulation of the synthetic rate of gonadotropin- releasing hormone receptors in rat pituitary cell cultures by inhibin*. Endocrinology. (1990) 127:2387–92. doi: 10.1210/endo-127-5-2387
23. Ying SY. Inhibins, activins, and follistatins: Gonadal proteins modulating the secretion of follicle-stimulating hormone. Endocr Rev. (1988) 9:267–93. doi: 10.1210/edrv-9-2-267
24. Bernard DJ and Tran S. Mechanisms of activin-stimulated FSH synthesis: the story of a pig and a FOX1. Biol Reprod. (2013) 88:1–10. doi: 10.1095/biolreprod.113.107797
25. Gregg DW, Schwall RH, and Nett TM. Regulation of gonadotropin secretion and number of gonadotropin-releasing hormone receptors by inhibin, activin-A, and estradiol. Biol Reprod. (1991) 44:725–32. doi: 10.1095/biolreprod44.4.725
26. Norwitz ER, Xu S, Xu J, Spiryda LB, Park JS, Jeong K-H, et al. Direct binding of AP-1 (Fos/Jun) proteins to a SMAD binding element facilitates both gonadotropin-releasing hormone (GnRH)- and activin-mediated transcriptional activation of the mouse GnRH receptor gene. J Biol Chem. (2002) 277:37469–78. doi: 10.1074/jbc.M206571200
27. Brinkley HJ. Endocrine signaling and female reproduction. Biol Reprod. (1981) 24:22–43. doi: 10.1095/biolreprod24.1.22
28. Pau KY, Berria M, Hess DL, and Spies HG. Preovulatory gonadotropin-releasing hormone surge in ovarian-intact rhesus macaques. Endocrinology. (1993) 133:1650–6. doi: 10.1210/endo.133.4.8404606
29. Moenter SM, Caraty A, and Karsch FJ. The estradiol-induced surge of gonadotropin-releasing hormone in the ewe. Endocrinology. (1990) 127:1375–84. doi: 10.1210/endo-127-3-1375
30. Crowder ME and Nett TM. Pituitary content of gonadotropins and receptors for gonadotropin-releasing hormone (GnRH) and hypothalamic content of GnRH during the periovulatory period of the ewe. Endocrinology. (1984) 114:234–9. doi: 10.1210/endo-114-1-234
31. Clarke IJ, Thomas GB, Yao B, and Cummins JT. GnRH secretion throughout the ovine estrous cycle. Neuroendocrinology. (1987) 46:82–8. doi: 10.1159/000124800
32. Clay CM, Nelson SE, DiGregorio GB, Campion CE, Wiedemann AL, and Nett RJ. Cell-specific expression of the mouse gonadotropin-releasing hormone (GnRH) receptor gene is conferred by elements residing within 500 bp of proximal 5′ flanking region. Endocrine. (1995) 3:615–22. doi: 10.1007/BF02953028
33. Kakar SS, Musgrove LC, Devor DC, Sellers JC, and Neill JD. Cloning, sequencing, and expression of human gonadotropin releasing hormone (GnRH) receptor. Biochem Biophys Res Commun. (1992) 189:289–95. doi: 10.1016/0006-291X(92)91556-6
34. Fan NC, Jeung E-B, Peng C, Olofsson JI, Krisinger J, and Leung PCK. The human gonadotropin-releasing hormone (GnRH) receptor gene: cloning, genomic organization and chromosomal assignment. Mol Cell Endocrinol. (1994) 103:R1–6. doi: 10.1016/0303-7207(94)90087-6
35. Albarracin CT, Kaiser UB, and Chin WW. Isolation and characterization of the 5’-flanking region of the mouse gonadotropin-releasing hormone receptor gene. Endocrinology. (1994) 135:2300–6. doi: 10.1210/endo.135.6.7988412
36. Campion CE, Turzillo AM, and Clay CM. The gene encoding the ovine gonadotropin-releasing hormone (GnRH) receptor: cloning and initial characterization. Gene. (1996) 170:277–80. doi: 10.1016/0378-1119(96)00042-X
37. Schang A. Inside and outside the pituitary: comparative analysis of Gnrhr expression provides insight into the mechanisms underlying the evolution of gene expression. J Neuroendocrinol. (2015) 27:177–86. doi: 10.1111/jne.12253
38. Davis TL, Whitesell JD, Cantlon JD, Clay CM, and Nett TM. Does a nonclassical signaling mechanism underlie an increase of estradiol-mediated gonadotropin-releasing hormone receptor binding in ovine pituitary cells? Biol Reprod. (2011) 85:770–8. doi: 10.1095/biolreprod.111.091926
39. McCue JM, Quirk CC, Nelson SE, Bowen RA, and Clay CM. Expression of a murine gonadotropin-releasing hormone receptor-luciferase fusion gene in transgenic mice is diminished by immunoneutralization of gonadotropin-releasing hormone. Endocrinology. (1997) 138:3154–60. doi: 10.1210/endo.138.8.5306
40. Leal AMO, Takabe K, Wang L, Donaldson CJ, MacConell LA, Bilezikjian LM, et al. Effect of adenovirus-mediated overexpression of follistatin and extracellular domain of activin receptor type II on gonadotropin secretion in vitro and in vivo. Endocrinology. (2002) 143:964–9. doi: 10.1210/endo.143.3.8667
41. Turzillo AM and Nett TM. Effects of bovine follicular fluid and passive immunization against gonadotropin-releasing hormone (GnRH) on messenger ribonucleic acid for GnRH receptor and gonadotropin subunits in ovariectomized Ewes1. Biol Reprod. (1997) 56:1537–43. doi: 10.1095/biolreprod56.6.1537
42. He T-C, Zhou S, da Costa LT, Yu J, Kinzler KW, and Vogelstein B. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci. (1998) 95:2509–14. doi: 10.1073/pnas.95.5.2509
43. Duval DL, Nelson SE, and Clay CM. The tripartite basal enhancer of the gonadotropin-releasing hormone (GnRH) receptor gene promoter regulates cell-specific expression through a novel GnRH receptor activating sequence. Mol Endocrinol. (1997) 11:1814–21. doi: 10.1210/mend.11.12.0020
44. Taketo M, Schroeder AC, Mobraaten LE, Gunning KB, Hanten G, Fox RR, et al. FVB/N: an inbred mouse strain preferable for transgenic analyses. Proc Natl Acad Sci. (1991) 88:2065–9. doi: 10.1073/pnas.88.6.2065
45. Duval DL, Farris AR, Quirk CC, Nett TM, Hamernik DL, and Clay CM. Responsiveness of the ovine gonadotropin-releasing hormone receptor gene to estradiol and gonadotropin-releasing hormone is not detectable in vitro but is revealed in transgenic mice 1. Endocrinology. (2000) 141:1001–10. doi: 10.1210/endo.141.3.7391
46. Ellsworth BS, White BR, Burns AT, Cherrington BD, Otis AM, and Clay CM. c-Jun N-terminal kinase activation of activator protein-1 underlies homologous regulation of the gonadotropin-releasing hormone receptor gene in alpha T3–1 cells. Endocrinology. (2003) 144:839–49. doi: 10.1210/en.2002-220784
47. L’hermite M, Niswender GD, Reichert LE, and Midgley AR. Serum follicle-stimulating hormone in sheep as measured by radioimmunoassay1. Biol Reprod. (1972) 6:325–32. doi: 10.1093/biolreprod/6.2.325
48. Murtazina DA, Arreguin-Arevalo JA, Cantlon JD, Ebrahimpour-Boroojeny A, Shrestha A, Hicks JA, et al. Enrichment of ovine gonadotropes via adenovirus gene targeting enhances assessment of transcriptional changes in response to estradiol-17 beta. Biol Reprod. (2020) 102:156–69. doi: 10.1093/biolre/ioz166
49. Duval DL, Ellsworth BS, and Clay CM. Is gonadotrope expression of the gonadotropin releasing hormone receptor gene mediated by autocrine/paracrine stimulation of an activin response element? Endocrinology. (1999) 140:1949–52. doi: 10.1210/endo.140.4.6780
50. Fortin J, Ongaro L, Li Y, Tran S, Lamba P, Wang Y, et al. Minireview: activin signaling in gonadotropes: what does the FOX say … to the SMAD? Mol Endocrinol. (2015) 29:963–77. doi: 10.1210/me.2015-1004
51. Silver BJ, Bokar JA, Virgin JB, Vallen EA, Milsted A, and Nilson JH. Cyclic AMP regulation of the human glycoprotein hormone alpha-subunit gene is mediated by an 18-base-pair element. Proc Natl Acad Sci. (1987) 84:2198–202. doi: 10.1073/pnas.84.8.2198
52. Jameson JL, Powerst AC, Gallagher GD, and Habener JF. Enhancer and promoter element interactions dictate cyclic adenosine monophosphate mediated and cell-specific expression of the glycoprotein hormone α-gene. Mol Endocrinol. (1989) 3:763–72. doi: 10.1210/mend-3-5-763
53. Kay TW and Jameson JL. Identification of a gonadotropin-releasing hormone-responsive region in the glycoprotein hormone alpha-subunit promoter. Mol Endocrinol. (1992) 6:1767–73. doi: 10.1210/mend.6.11.1282668
54. Montminy MR, Sevarino KA, Wagner JA, Mandel G, and Goodman RH. Identification of a cyclic-AMP-responsive element within the rat somatostatin gene. Proc Natl Acad Sci. (1986) 83:6682–6. doi: 10.1073/pnas.83.18.6682
55. Bjelobaba I, Janjic MM, Tavcar JS, Kucka M, Tomić M, and Stojilkovic SS. The relationship between basal and regulated Gnrhr expression in rodent pituitary gonadotrophs. Mol Cell Endocrinol. (2016) 437:302–11. doi: 10.1016/j.mce.2016.08.040
56. Cheon M, Park D, Park Y, Kam K, Park SD, and Ryu K. Homologous Upregulation of Gonadotropin-Releasing Hormone Receptor mRNA Occurs through Transcriptional Activation Rather than Modulation of mRNA Stability. Endocrine. (2000) 13:47–54. doi: 10.1385/ENDO:13:1:47
57. Cheon M, Park D, Kim K, Park SD, and Ryu K. Homologous upregulation of GnRH receptor mRNA by continuous GnRH in cultured rat pituitary cells. Endocrine. (1999) 11:49–56. doi: 10.1385/ENDO:11:1:49
58. Hamernik DL and Nett TM. Gonadotropin-releasing hormone increases the amount of messenger ribonucleic acid for gonadotropins in ovariectomized ewes after hypothalamic-pituitary disconnection. Endocrinology. (1988) 122:959–66. doi: 10.1210/endo-122-3-959
59. Adams BM, Sakurai H, and Adams TE. Concentrations of gonadotropin-releasing hormone (GnRH) receptor messenger ribonucleic acid in pituitary tissue of orchidectomized sheep: effect of estradiol and GnRH1. Biol Reprod. (1996) 54:407–12. doi: 10.1095/biolreprod54.2.407
60. Wise ME, Nieman D, Stewart J, and Nett TM. Effect of number of receptors for gonadotropin-releasing hormone on the release of luteinizing hormone. Biol Reprod. (1984) 31:1007–13. doi: 10.1095/biolreprod31.5.1007
61. Adams TE, Norman RL, and Spies HG. Gonadotropin-releasing hormone receptor binding and pituitary responsiveness in estradiol-primed monkeys. Science. (1979) 1981) 213:1388–90. doi: 10.1126/science.6267698
62. Thompson IR and Kaiser UB. GnRH pulse frequency-dependent differential regulation of LH and FSH gene expression. Mol Cell Endocrinol. (2014) 385:28–35. doi: 10.1016/j.mce.2013.09.012
63. Alarid ET, Windle JJ, Whyte DB, and Mellon PL. Immortalization of pituitary cells at discrete stages of development by directed oncogenesis in transgenic mice. Development. (1996) 122:3319–29. doi: 10.1242/dev.122.10.3319
64. Thomas P, Mellon PL, Turgeon J, and Waring DW. The L beta T2 clonal gonadotrope: a model for single cell studies of endocrine cell secretion. Endocrinology. (1996) 137:2979–89. doi: 10.1210/endo.137.7.8770922
65. Windle JJ, Weiner RI, and Mellon PL. Cell lines of the pituitary gonadotrope lineage derived by targeted oncogenesis in transgenic mice. Mol Endocrinol. (1990) 4:597–603. doi: 10.1210/mend-4-4-597
66. Kim GL, Wang X, Chalmers JA, Thompson DR, Dhillon SS, Koletar MM, et al. Generation of immortal cell lines from the adult pituitary: Role of cAMP on differentiation of SOX2-expressing progenitor cells to mature gonadotropes. PloS One. (2011) 6. doi: 10.1371/journal.pone.0027799
67. Ongaro L, Zhou X, Wang Y, Schultz H, Zhou Z, Buddle ERS, et al. Muscle-derived myostatin is a major endocrine driver of follicle-stimulating hormone synthesis. Science. (1979) 2025) 387:329–36. doi: 10.1126/science.adi4736
68. Corpuz PS, Lindaman LL, Mellon PL, and Coss D. FoxL2 Is required for activin induction of the mouse and human follicle-stimulating hormone beta-subunit genes. Mol Endocrinol. (2010) 24:1037–51. doi: 10.1210/me.2009-0425
69. Cheng KW, Ngan ESW, Kang SK, Chow BKC, and Leung PCK. Transcriptional down-regulation of human gonadotropin-releasing hormone (GnRH) receptor gene by GnRH: role of protein kinase C and activating protein 1*. Endocrinology. (2000) 141:3611–22. doi: 10.1210/endo.141.10.7730
70. Schang A-L, Quérat B, Simon V, Garrel G, Bleux C, Counis R, et al. Mechanisms underlying the tissue-specific and regulated activity of the Gnrhr promoter in mammals. Front Endocrinol (Lausanne). (2012) 3:162. doi: 10.3389/fendo.2012.00162
71. Sealfon SC, Laws SC, Wu JC, Gillo B, and Miller WL. Hormonal regulation of gonadotropin-releasing hormone receptors and messenger RNA activity in ovine pituitary culture. Mol Endocrinol. (1990) 4:1980–7. doi: 10.1210/mend-4-12-1980
72. Turzillo AM, DiGregorio GB, and Nett TM. Messenger ribonucleic acid for gonadotropin-releasing hormone receptor and numbers of gonadotropin-releasing hormone receptors in ovariectomized ewes after hypothalamic-pituitary disconnection and treatment with estradiol. J Anim Sci. (1995) 73:1784–88. doi: 10.2527/1995.7361784x
73. Kirkpatrick BL, Esquivel E, Moss GE, Hamernik DL, and Wise ME. Estradiol and gonadotropin-releasing hormone (GnRH) interact to increase GnRH receptor expression in ovariectomized ewes after hypothalamic-pituitary disconnection. Endocrine. (1998) 8:225–30. doi: 10.1385/ENDO:8:3:225
74. Hamernik DL, Clay CM, Turzillo A, Van Kirk EA, and Moss GE. Estradiol increases amounts of messenger ribonucleic acid for gonadotropin-releasing hormone receptors in sheep. Biol Reprod. (1995) 53:179–85. doi: 10.1095/biolreprod53.1.179
75. Turgeon JL, Kimura Y, Waring DW, and Mellon PL. Steroid and pulsatile gonadotropin-releasing hormone (GnRH) regulation of luteinizing hormone and GnRH receptor in a novel gonadotrope cell line. Mol Endocrinol. (1996) 10:439–50. doi: 10.1210/mend.10.4.8721988
76. Weiss JM, Polack S, Treeck O, Diedrich K, and Ortmann O. Regulation of GnRH I receptor gene expression by the GnRH agonist triptorelin, estradiol, and progesterone in the gonadotroph-derived cell line αT3-1. Endocrine. (2006) 30:139–44. doi: 10.1385/ENDO:30:1:139
77. McArdle CA, Schomerus E, Gröner I, and Poch A. Estradiol regulates gonadotropin-releasing hormone receptor number, growth and inositol phosphate production in αT3–1 cells. Mol Cell Endocrinol. (1992) 87:95–103. doi: 10.1016/0303-7207(92)90237-Z
78. Savoy-Moore RT, Schwartz NB, Duncan JA, and Marshall JC. Pituitary gonadotropin-releasing hormone receptors during the rat estrous cycle. Science. (1979) 1980) 209:942–4. doi: 10.1126/science.6250218
79. Bauer-Dantoin AC, Hollenberg AN, and Jameson JL. Dynamic regulation of gonadotropin-releasing hormone receptor mRNA levels in the anterior pituitary gland during the rat estrous cycle. Endocrinology. (1993) 133:1911–4. doi: 10.1210/endo.133.4.8404635
80. Chen J, An B-S, Cheng L, Hammond GL, and Leung PCK. Gonadotropin-releasing hormone-mediated phosphorylation of estrogen receptor-α Contributes to fosB expression in mouse gonadotrophs. Endocrinology. (2009) 150:4583–93. doi: 10.1210/en.2009-0455
81. Hapgood JP, Sadie H, Van Biljon W, and Ronacher K. Regulation of expression of mammalian gonadotrophin-releasing hormone receptor genes. J Neuroendocrinol. (2005) 17:619–38. doi: 10.1111/j.1365-2826.2005.01353.x
82. Vo N and Goodman RH. CREB-binding protein and p300 in transcriptional regulation. J Biol Chem. (2001) 276:13505–8. doi: 10.1074/jbc.R000025200
83. Ince BA, Zhuang Y, Wrenn CK, Shapiro DJ, and Katzenellenbogen BS. Powerful dominant negative mutants of the human estrogen receptor. J Biol Chem. (1993) 268:14026–32. doi: 10.1016/s0021-9258(19)85204-3
84. Acevedo ML, Lee KC, Stender JD, Katzenellenbogen BS, and Kraus WL. Selective recognition of distinct classes of coactivators by a ligand-inducible activation domain. Mol Cell. (2004) 13:725–38. doi: 10.1016/S1097-2765(04)00121-2
85. Gieske MC, Hyun JK, Legan SJ, Koo Y, Krust A, Chambon P, et al. Pituitary gonadotroph estrogen receptor-α is necessary for fertility in females. Endocrinology. (2008) 149:20–7. doi: 10.1210/en.2007-1084
Keywords: GnRH receptor, activin, estrogen gene expression, transgenic mice, follistatin
Citation: Magee C, Kouri JE, Cherrington BD, Cantlon JD, Murtazina DA, Farmerie TA, Davidsen MH, Nett TM and Clay CM (2025) Evidence for divergent endocrine regulation of the murine and ovine GnRH receptor gene promoters. Front. Endocrinol. 16:1597028. doi: 10.3389/fendo.2025.1597028
Received: 20 March 2025; Accepted: 23 June 2025;
Published: 29 July 2025.
Edited by:
Chun Peng, York University, CanadaReviewed by:
Gwen V. Childs, University of Arkansas for Medical Sciences, United StatesPatrick E Chappell, Oregon State University, United States
Copyright © 2025 Magee, Kouri, Cherrington, Cantlon, Murtazina, Farmerie, Davidsen, Nett and Clay. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Colin M. Clay, Y29saW4uY2xheUBjb2xvc3RhdGUuZWR1