 Changqing Liu
Changqing Liu Yuanyuan Long1
Yuanyuan Long1 Junjun Liu
Junjun Liu Yongfeng Song
Yongfeng Song- 1Endocrine and Metabolic Diseases Hospital of Shandong First Medical University, Shandong First Medical University and Shandong Academy of Medical Sciences, Jinan, Shandong, China
- 2Department of Endocrinology, Jinan Central Hospital Affiliated to Shandong First Medical University, Jinan, Shandong, China
- 3Shandong Clinical Research Center of Diabetes and Metabolic Diseases, Jinan, Shandong, China
- 4Shandong Institute of Endocrine and Metabolic Diseases, Shandong First Medical University, Jinan, Shandong, China
- 5Shandong Engineering Laboratory of Prevention and Control for Endocrine and Metabolic Diseases, Jinan, Shandong, China
The classical theory of the pituitary–target gland axis suggests that the hormones secreted by the pituitary gland only regulate the synthesis and secretion of target gland hormones, while the target gland hormones act on the tissues of the body to achieve biological functions. However, recent studies have shown that anterior pituitary hormone receptors are also expressed on the surface of hepatocytes. This suggests that anterior pituitary hormones may act directly on hepatocytes to exert regulatory effects independent of target hormones. The review systematically summarizes the mechanisms and effects of thyroid-stimulating hormone (TSH), follicle-stimulating hormone (FSH), luteinizing hormone (LH), prolactin (PRL), adrenocorticotropic hormone (ACTH), growth hormone (GH), and melanocyte-stimulating hormones (MSH) on liver metabolism and their roles in the pathogenesis of Metabolic-Associated Fatty Liver Disease (MAFLD). It is hoped that this will provide new insights into the prevention and treatment of MAFLD.
Introduction
As the central regulator of the body’s metabolism, the pituitary plays a crucial role in growth, development, immune function, energy metabolism and reproduction, among other functions. Each anterior pituitary hormone binds to its receptor in its classical target gland, for example the thyroid binds to TSH, to regulate physiological pathways. However, in recent decades, several reports have shown that anterior pituitary hormone receptors not only expressed in the target gland, but also expressed in the non-target gland, such as in the liver. Thus, each hormone exerts an unconventional biological effect in the liver. This review summarizes the functions of anterior pituitary hormones in the liver and their role in the pathogenesis of MAFLD. These findings extend our knowledge of the targets and functions of anterior pituitary hormones and provide new insights into the biological function of anterior pituitary hormones in the onset and development of MAFLD.
Overview of the MAFLD
MAFLD formerly known as Non-Alcoholic Fatty Liver Disease (NAFLD), has been repeatedly renamed by international medical organizations to reflect its involvement in various pathological conditions and has recently been redefined as Metabolic-Associated Steatotic Liver Disease (MASLD) (1–3). For the purposes of this article, we will refer to MAFLD. MAFLDs are among the most common liver diseases worldwide and are predicted to be the leading cause for end-stage liver disease in the coming decades (4). The prevalence of MAFLD in the global population is maybe as high as 25% (5), and the estimated combined prevalence of non-alcoholic steatohepatitis (NASH) in histologically confirmed MAFLD patients is 59% (6, 7). Importantly, there is strong clinical evidence that MAFLD is also associated with an increased risk of other diseases outside the liver, including cardiovascular disease and extra-hepatic malignancies (8), ultimately leading to death. MAFLD and its associated complications pose a significant burden on socioeconomic development and public health in China (9).
MAFLD is a chronic liver disease that is marked by intracytoplasmic lipid accumulation in hepatocytes, and hepatic inflammation and fibrosis (10, 11). And the occurrence of MAFLD is involved in multiple molecular pathways, including hepatic steatosis, insulin resistance, inflammatory cytokines and oxidative stress, apoptosis pathways and adipokines (12). Besides, MAFLD is closely linked to abnormalities in several metabolic processes (13), including glucose metabolism (14), lipid metabolism (15, 16)(Figure 1). Free fatty acid (FFA)-mediated-mediated lipotoxicity, cholesterol-induced toxicity and subsequent elevation of reactive oxygen species (ROS) in hepatocytes can promote the development of MALFD (17, 18).
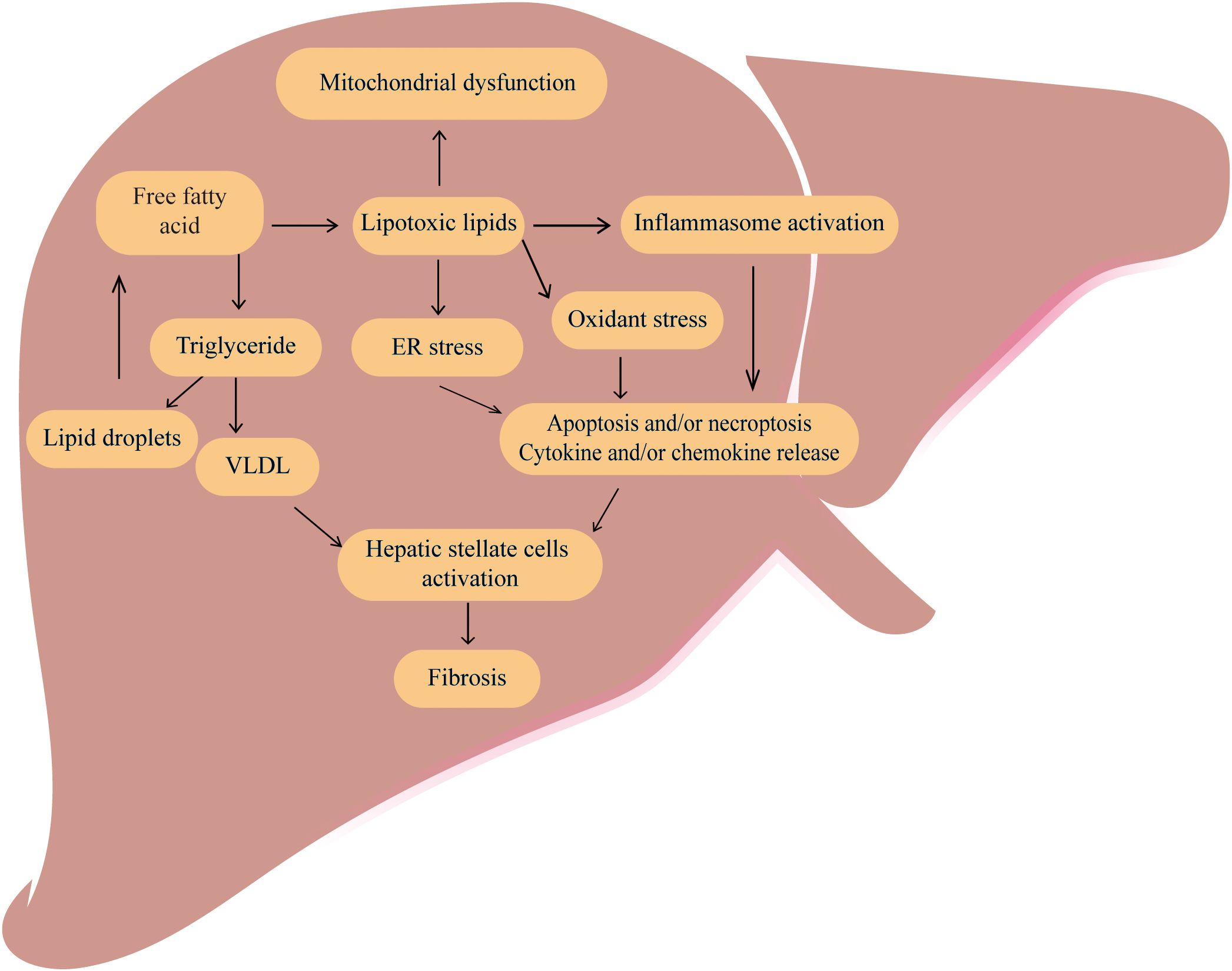
Figure 1. The classic pathogenesis of MAFLD MAFLD is caused by dysregulation of several metabolic processes especially an increased accumulation of free fatty acid and total triglycerides (TG) in the liver. Excessive free fatty acids can cause lipotoxic, ultimately leading to mitochondrial dysfunctions, inflammation, oxidative stress, and endoplasmic reticulum stress, promoting cell apoptosis, necrosis and cytokine release. The VLDL and cytokines can activate hepatic stellate cells and promote the development of liver fibrosis.
In addition to the above factors, endocrine disruption is an important contributor to hepatic metabolic abnormalities. With advancing age, the function of endocrine glands such as the thyroid and gonads gradually weakens. This leads to a progressive decline in thyroid and sex hormone levels, resulting in significant changes to lipid synthesis, metabolism and degradation processes and ultimately, lipid metabolism disorders. Furthermore, recent research has shown that anterior pituitary hormones can directly influence hepatic lipid metabolism, as well as the processes of inflammation and fibrosis in the liver, independently of the synthesis and secretion of target gland hormones. Therefore, this article will systematically summarize the onset and progression of MAFLD mediated by the imbalance of anterior pituitary hormones through multiple mechanisms, focusing on the following points.
General description of the anterior pituitary hormones
The pituitary plays a central role as a central regulator of metabolism in the body. The pituitary gland, included anterior pituitary and posterior pituitary gland (19). The anterior pituitary secretes TSH, FSH, LH, PRL, ACTH, GH, MSH. By contrast, the posterior pituitary gland secretes two nonapeptides, oxytocin (regulates parturition and lactation) and arginine vasopressin (AVP; controls water reabsorption in the kidney). Here we focus on anterior pituitary hormones. There are three main axis including hypothalamic-pituitary-gonadal (HPG) (20), hypothalamic-pituitary-thyroid (HPT) (21), and hypothalamic-pituitary-adrenal (HPA) axes (22). They influence different organs and tissues in the body by secreting variety of cytokines, growth factors and receptors. Specifically, the HPG axis primarily regulates processes such as sexual development, spermatogenesis and oocyte maturation. It contributes to the establishment and maintenance of the secondary sex characteristics, mainly through the release of LH and FSH. The HPT axis primarily regulates the levels of thyroid hormones and thus participates in regulating the overall metabolism, mainly through the secretion of TSH (23). The HPA axis is primarily involved in controlling stress and several physiological activities including digestion, mood and emotion, which include the main hormone is ACTH. As shown in Figure 2, in addition to the hormones secreted by the above three pituitary axes, PRL, GH and MSH are also secreted by the anterior pituitary (22).
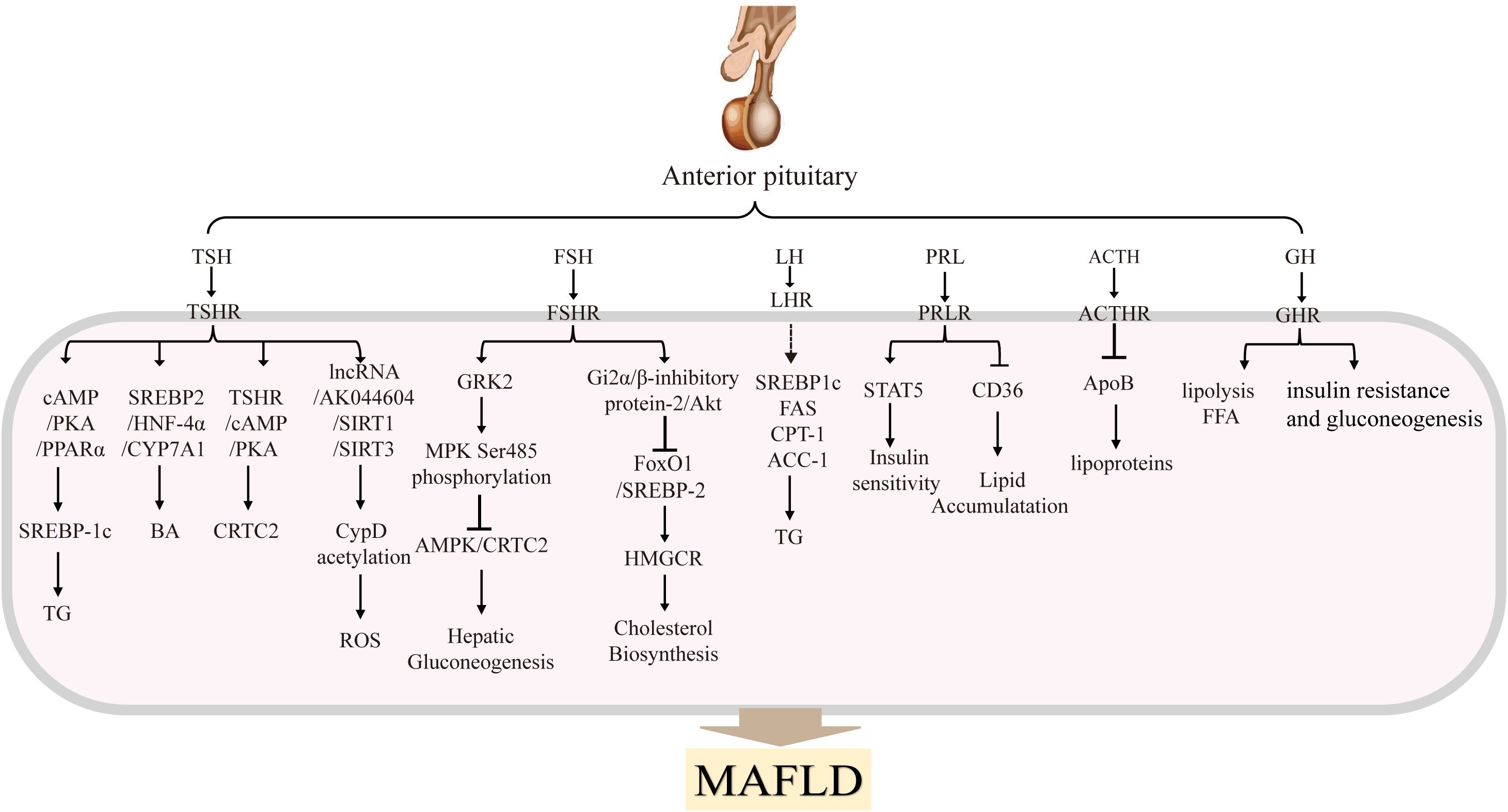
Figure 2. Anterior pituitary hormones and their main effects in MAFLDThe anterior pituitary gland secretes several hormones, including TSH, FSH, LH, PRL, ACTH, GH and MSH. These hormones are transported to the liver, where they bind to their respective receptors and activate signaling pathways that regulate hepatic glucolipid metabolism. These processes influence the progression of MAFLD.
The major effects of pituitary hormone in MAFLD
TSH
TSH is formed by α and β subunits, and it exerts its important functions by targeting the TSH receptor (TSHR), thereby regulating thyroid hormone levels. TSH plays an important role in metabolism, development, and growth (24). TSHRs are primarily found in the thyroid gland. However, researchers have also discovered their presence in various non-thyroid tissues, such as the liver, adipose tissue, myocardium, bone, thymus, and natural killer (NK) cells (25–31).
The different TSH levels reflect different thyroid status, and it is important to note that both subclinical hypothyroidism (SCH)and overt hypothyroidism are associated with NASH and advanced fibrosis (32–34). In biopsy-proven MAFLD patients with normal thyroid function, there is a clear relationship between high normal TSH levels and NASH, which may be related to the PNPLA3 G risk allele (35). Another study found a positive relationship between TSH levels and NASH in MAFLD patients with thyroid function normal (36). Meanwhile liver biopsies have revealed a significant correlation between TSH levels (32), including macrovesicular steatosis, inflammation, or hepatocyte balloon degeneration and steatosis. These clinical studies have shown that TSH levels are positively correlated with ALT and AST, with a significant reduction in risk when TSH is < 2.5 mIU/mL, but TSH concentrations >2.5mIU/L are associated with an increased risk of lipid and carbohydrate metabolism disorders (32). This has led to widespread discussion in clinical research as to whether a sufficiently low TSH level can prevent the development of MAFLD. As a result, TSH maybe as an independent risk factor for MAFLD, and the risk of MAFLD increases significantly with elevated TSH levels.
TSH in hepatocytes can regulate hepatic lipid and cholesterol metabolism, as shown in Figure 2. adenosine monophosphate-activated protein kinase (AMPK) is a key regulator of lipid and glucose metabolism, and reduced AMPK activity increases the expression of genes related to lipid synthesis and cholesterol biosynthesis. Our research has shown that TSH-TSHRs activate the cAMP/PKA/CREB signaling system in hepatocytes and directly promote cholesterol synthesis (37, 38). Furthermore, our research has demonstrated that abnormally elevated TSH, via TSHR, triggers SREBP-1c via the cAMP/PKA/PPARα pathway, thereby increasing hepatic lipid accumulation and ultimately leading to the onset of MAFLD (39). Cell experiments have also shown that TSH inhibits hepatic bile acid synthesis via the SREBP2/HNF-4α/CYP7A1 pathway (40). TSH can also attenuate hepatic fatty acid oxidation by decreasing the mitochondrial distribution of miR-449a/449b-5p/5194. This inhibits fatty acid (FA) cleavage and enhances triglyceride storage in hepatocytes (41). This is the most well-known pathogenic mechanism of TSH-mediated MAFLD.
In addition to affecting hepatic lipid and cholesterol metabolism, TSH may also contribute to the onset and progression of MAFLD by influencing glucose metabolism pathways. Previous reports have suggested that TSH acts directly on gluconeogenesis, with TSH directly regulating hepatic gluconeogenesis in HepG2 cells and Tshr-KO mice (42). More recent research has demonstrated that TSH can increase the expression of the CRTC2 gene and thereby upregulate hepatic gluconeogenic genes via the TSHR/cAMP/PKA pathway (43).
TSH may also mediate MAFLD via oxidative stress and inflammation. Animal studies using TSHR knockout and liver-specific knockout mice, revealed that TSH stimulates CypD acetylation in the liver via the lncRNA-AK044604/SIRT1/SIRT3 pathway (44). This study demonstrated the significant role of TSH in hepatic mitochondrial oxidative stress, leading to the generation of ROS and mediating the onset of MAFLD. In addition, TSH increases the secretion of exosomes by hepatocytes and alters their protein profile. Many of these proteins are required for metabolism, signal transduction, cell apoptosis and inflammation (45). In the obese mice, fed a high-fat diet (HFD), had significantly higher serum TSH levels. Elevated TSH levels lead to increased secretion of SPP1 in M1 macrophages and exacerbate lipid accumulation in hepatocytes (46).
Taken together, these findings suggest that TSH plays an intrinsic role in regulating liver lipid and cholesterol homeostasis. In addition to the liver, TSH may contribute to MAFLD through other organs and tissues. For instance, recent studies have shown that TSH can directly suppress the ATGL gene expression in mature mouse adipocytes via activating cAMP/PKA pathway, thereby inhibiting the basal breakdown of triglycerides (47). On the other hand, through the AMPK/PRDM16/PGC1α pathway, TSHR knockout induced a reduction in adiposity, increased energy expenditure, and promoted the development of beige adipocytes in mouse adipose tissue.
Therefore, TSH is a significant independent risk factor for MAFLD, with the risk increasing significantly with elevated TSH levels. TSH may mediate the onset and progression of MAFLD through various mechanisms, so controlling TSH levels should be taken into consideration as a possible future therapeutic strategy for MAFLD.
FSH
Both FSH and LH are gonadotropins, which play a role in promoting follicular development and maturation and stimulating estrogen secretion (48). FSH binds to the FSH receptor (FSHR) to exert its regulatory functions (48). FSH receptors (FSHR) are found not only in the ovaries and testes (49), but also in monocytes and osteoclasts (50), adipocytes (51), hepatocytes (52), blood vessels and maybe other tissues (53). In addition, FSH has been found to be associated with insulin sensitivity, bone metabolism, adipogenesis, inflammation, thermogenesis, osteogenesis and ovarian cancer (54). Most directly, liver and adipose can be directly regulated by FSH (55).
Numerous articles have examined the association between FSH and MAFLD in clinical investigations, producing conflicting results. For example, Wang et al. found that FSH levels were negatively associated with the prevalence of MAFLD in Chinese women over 55 years of age, independent of traditional metabolic risk factors such as BMI, glucose and lipids (56). Ge et al. also demonstrated that, in postmenopausal women with an average age of 60.22 ± 6.49 and type 2 diabetes mellitus, FSH was negatively and independently associated with MAFLD (57). A retrospective observational study of a Chinese elderly population, including both men and women aged 60–70 years and over 70 years, also showed a negative correlation between FSH and MAFLD (58). The above results showed that there is a negative correlation between FSH and MAFLD. However, another study of elderly Chinese men aged 80–98 showed a positive correlation between FSH and MAFLD (59), the study showed that a low FSH level may decrease the risk of MAFLD. Furthermore, another study showed that FSH acts on the pituitary corticotropes to inhibit corticosterone production and ultimately prevent hepatic steatosis, and that FSH administration is sufficient to improve metabolic disorders including hepatic steatosis in female mice (55). The reasons for the variation in research outcomes concerning the relationship between FSH and MAFLD are unclear. However, age may have some impact on these differences. Therefore, more well-designed clinical research studies are necessary to confirm the findings.
FSH exerts a significant influence on the body’s lipid metabolism (Figure 2). For example, Song et al. (2016) found that postmenopausal women with higher serum FSH (78.3 IU/L at baseline) had higher serum total cholesterol and LDL-C levels, and that there was a significant improvement in lipid levels after hormone replacement therapy or blocking FSH signaling by anti-FSHβ antibody or ablating the FSH receptor (FSHR) gene (52, 60). The main mechanism may be that FSH blocks FoxO1 binding to the sterol regulatory element-binding protein-2 (SREBP-2) promoter through activation of the Gi2α/β inhibitor protein-2/Akt pathway. This effect leads to an increase in SREBP-2, which drives de novo transcription of 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGCR) and increases cholesterol accumulation (60). These researches suggest that blocking FSH signaling may be a new strategy for treating menopausal hypercholesterolemia, particularly in peri-menopausal women characterized by elevated FSH levels.
FSH also plays a role in MAFLD by affecting hepatic gluconeogenesis in hepatocytes (Figure 2). Our researches provided evidence that FSH plays a direct role in causing gluconeogenesis, where FSH through FSHR targets GRK2 in the liver, increases AMPK Ser485 phosphorylation to inhibit AMPK activation, and then increases the transcription of hepatic PEPCK and G6pase through CRTC2, thereby enhancing hepatic gluconeogenesis independent of estrogen (61, 62). Beside we have identified a role for FSH in fasting serum glucose levels using FSH receptor knockout mice (61) and the association between FSH levels and insulin resistance has been confirmed in postmenopausal women (63). The relationship between IR and FSH levels may be mediated by regulation of glucocorticoid receptor (GCR) expression (64).
All this provides new insights into the role of FSH in lipid and glucose metabolism and demonstrates the direct involvement of FSH in MAFLD.
LH
LH stimulates the secretion of the testes and ovaries, whose the classical role is to regulate ovarian and testicular function (65–69)). The receptor of LH (LHR) was expressed in the pineal gland, pituitary, hypothalamus, gonad, kidney, brain, lymphatic tissue and lymphocytes (70, 71).
Although it has been shown that LH plays an important role in the reproductive, urinary and nervous system (72) (73), such as in the porcine oviduct, urinary tract, and hippocampus, there is no evidence of direct relationship between LH and liver or lipid metabolism. However, it is noteworthy that LH beta mRNA expression was elevated in fasted wild-type mice, but not in mice deficient in PPARalpha (74). This research suggests that LH may play a role in lipid metabolism, as PPAR-α is an important transcription factor involved in lipid metabolism.
Notably LH levels are significantly elevated in female populations with PCOS (polycystic ovary syndrome). And abnormal levels of phosphatidylcholine, FFAs and polyunsaturated fatty acid (PUFA) metabolites were found in patients with PCOS (75). Around 70-80% of patients with PCOS are obese. In addition, the expression of genes involved in lipid metabolism, such as FAS, SREBP1c, ACC-1 and CPT-1, is increased in patients with PCOS (76). From the above evidence we can infer that LH may be involved in lipid metabolism. However, there are no direct studies to prove this. Furthermore, elevated LH stimulates the luteal gland to produce progesterone and estrogen. Elevated estrogen is directly related to hepatic steatosis and insulin resistance (77). In addition, women with PCOS have androgen excess, IR, variable amounts of estrogen exposure, and many environmental factors, all of which can affect lipid metabolism (78) (79) (76). So further studies are needed to uncover the relationship between LH and lipid metabolism.
Nevertheless, we can infer that there may be a positive association between LH and MAFLD. LH may be related to metabolic diseases by playing an important role in energy metabolism. However, there is currently limited research on the relationship between LH and MAFLD. The specific relationship between LH and MAFLD should be further investigated in future studies.
PRL
Prolactin (PRL) is an important multifunctional pituitary hormone and involved in several biological functions (80), most notably the promotion of lactation (81). PRL can also stimulate β-cell proliferation and improve insulin secretion, as well as participating in the regulation of glucose metabolism (82, 83). Furthermore, PRL activates the peroxisome proliferator-activated receptor γ (PPARγ) to inhibit lipolysis and activate adipocyte differentiation. Wang et al. found that PRL levels are associated with the lipid metabolism and low-grade inflammatory markers in obesity (84). Studies in PRL receptor-deficient mice have shown increased oxidative stress, SIRT2 expression and apoptosis (85).
Accumulating evidence supports the idea that reduced PRL levels contribute to metabolic changes, and recent studies have found a strong association between PRL and the presence and development of MAFLD (86). Ping Xu et al. found that serum PRL may be a potential biomarker to prevent and treat MAFLD (87). Moderately high PRL levels, both within and above the physiological range, are metabolically beneficial, while extremely high (>100 mg/dL) and extremely low (<7 mg/dL) PRL levels are non-metabolically beneficial (88). In addition, PRL promotes hepatic insulin sensitivity and prevents hepatic steatosis (89–91). The study’s findings suggest that PRL is an important factor in the development of MAFLD.
Recent research has focused on the role of PRL in glucose and lipid metabolism, given its vital role in the maintenance of adipogenesis and adipocyte differentiation. Zhang’s team found that in MAFLD patients, peripheral blood PRL levels are reduced alongside downregulation of hepatic PRL receptor (PRLR) expression (89), while PRL intervention can increase PRLR expression in HepG2 cells. Further studies by Zhang et al. showed that PRL ameliorated hepatic steatosis by inhibiting CD36, suggesting that PRL may protect liver from lipid accumulation via inhibiting CD36 in liver cells, as shown in Figure 2 (89). Yan et al. reported that prolactin (PRL) acts as a protective factor in MAFLD and that increased PRL levels can improve liver insulin sensitivity. Conversely, decreased PRL secretion can lead to insulin resistance (92). Mechanistically, PRL targets the prolactin receptor (PRLR) and activates the downstream signal transducer and activator of transcription-5 (STAT5) to mediate insulin sensitization (93). Furthermore, PRLR expression is regulated by liver insulin resistance/sensitivity levels, and it is down-regulated in the insulin-resistant state and up-regulated in the insulin-sensitive state (90).
Overall, it can be concluded that PRL has a protective effect on the liver by improving its insulin sensitivity. It also prevents the fat accumulation in the liver. These findings have important implications for the potential use of PRL as a therapeutic target MAFLD.
ACTH
ACTH is also synthesized by the anterior pituitary gland. The effects of ACTH are mediated by ACTH receptors (ACTHRs), such as MC2R and MC5R.The expression of MC2R and MC5R proteins in mouse embryos was examined (94), and ACTHR mRNA was found to be present in various tissues, including mouse adipose tissue (95), skin (96, 97), mouse pituitary glands (98), rat sympathetic ganglia (99), mouse fetus and new-born testis (100) and human uterine endometrium (100). Besides ACTHR mRNA has been detected in human erythroblasts (101) and human bone marrow cells (102).
Relatively few articles directly describe the relationship between ACTH and MAFLD. It has been reported that three months of exposure to noise at 75 dB SPL was sufficient to exacerbate the progression of MAFLD in mice, with the activation of the hypothalamic–pituitary–adrenal (HPA) axis playing a critical role (103). In depression and CDAHFD-fed mice, hepatic steatosis was aggravated by activating the HPA axis (104) In clinical practice, in patients with insulin-dependent hyperglycemia and concomitant hyperprolactinemia, liver steatosis was reversed and triglyceride levels returned to normal or near-normal levels after treatment with the ACTH receptor antagonist mifepristone (105). Another clinical study also found that in male patients with idiopathic hypogonadotropic hypogonadism (IHH), there is an independent association between MAFLD and ACTH levels (106).
Basic experiments have shown that ACTH significantly inhibits high-density lipoprotein but has no effect on low-density lipoprotein. This suggests that ACTH primarily reduces intracellular lipoproteins (107, 108). ACTH plays an important role in controlling adrenal steroidogenesis. It also induces the expression of mitochondrial superoxide dismutase 2 (SOD2), which plays a role in the removal of ROS from the mitochondria (109). Thus, ACTH modulates the expression of enzymes involved in the biosynthesis of steroids and non-steroids, helping to prevent ROS-induced cell toxicity (110).
Under the stimulation of ACTH, free cholesterol is released from hydrolyzed lipid droplets, increasing steroid production. ACTH stimulates lipolysis, and ACTH treatment has a significant effect on lowering cholesterol levels (111). Current evidence suggests that the cholesterol-lowering effect of adrenal corticosteroid stimulation may be mediated by promoting the hepatic uptake of apolipoprotein B (ApoB)-rich lipoproteins. In HepG2 cells, Xu et al. found that ACTH could reduce the concentration of ApoB-containing lipoproteins in human plasma, suggesting that the main mechanism by which ACTH reduces cholesterol levels in vivo may be by reducing the rate of production of ApoB lipoproteins in the liver (112).
Thereby we can conclude that ACTH may play a role in MAFLD by regulating the lipoproteins.
GH
Growth hormone (GH) is essential for growth and development. It regulates growth, tissue remodeling, extracellular matrix formation and fibrosis. As a key regulator of body composition, GH plays a vital role in maintaining metabolic homeostasis in several organs, including the liver, skeletal muscle and adipose tissue. During fasting and stress, GH plays a key role in anabolic processes. GH acts in both direct and indirect ways: it binds to the growth hormone receptor (GHR) to active downstream signaling directly, it stimulates the expression of insulin-like growth factors (IGFs) and their binding proteins (IGFBP) indirectly to mediate metabolism (113, 114). Metabolically, GH can stimulate lipolysis in white adipose tissue and impair hepatic and peripheral insulin sensitivity (113).
The relationship between MAFLD and GH was investigated. Increased risk of MAFLD/NASH associated with growth hormone axis abnormalities (115). In healthy Asian individuals, individuals with low GH levels had a higher prevalence of MAFLD, there may be a negative correlation between GH levels and MAFLD (116). In line with this is MAFLD usually develops soon after the diagnosis of adult growth hormone deficiency (AGHD). It can progress to non-alcoholic steatohepatitis (NASH) with advanced fibrosis rapidly, and eventually require liver transplantation (117). Growth hormone deficient rats also have MAFLD (118). GH has been used as a drug in the clinic. Clinical studies have shown that administration of higher doses of GH can help overcome the GH resistance in cirrhotic patients and significantly increase serum IGF-I levels (119). Excess GH can reduce total fat mass and hepatic lipid content and induce insulin resistance.
GH can affect the liver by making it insulin resistant. Almost 90 years ago, the Argentinian clinical physician Bernardo Houssay discovered that GH can inhibit insulin action (120). In adipose tissue, GH can inhibit insulin action, thereby reducing glucose uptake, and promoting hepatic gluconeogenesis. Subsequent research has shown that GH has direct and indirect effects on glucose metabolism, with the indirect effects primarily being mediated by IGF-1 (121, 122). GH can also activate the JAK2-STAT2 signaling pathway by targeting the GH receptor, thereby stimulating the synthesis and secretion of insulin-like growth factors (123). Furthermore, GH is involved in pathways associated with late-stage fibrosis in MAFLD, including the TGF-β and MAPK pathways (124, 125).
The most prominent metabolic effects of GH are increased lipolysis and FFA levels. Further studies of longer duration are needed to confirm the potential effects of GH on MAFLD.
MSH
Melanocyte-stimulating hormone (MSH), which is derived from pro-opiomelanocortin (POMC), plays an important role in the regulation of metabolic functions (126). MSH can be divided into three subtypes: α-MSH, β-MSH, and γ-MSH (127). MSH binds to receptors on the surface cells, to stimulate melanocyte formation and activity of melanocytes, thereby promoting melanin synthesis and affecting the color of the skin, hair, and eye color. α-MSH is a peptide that can suppress people’s appetite and reduce food intake (128). However, in some obese individuals, lower levels of α-MSH expression may cause a disorder in energy balance, this may cause a disorder in energy balance (129), suggesting a potential link between α-MSH and obesity.
MSH also has various functions, most important, Melanocortin peptides have long been thought to be potent inhibitors of inflammation. They are a promising source of new anti-inflammatory and cytoprotective therapies (130). MSH’s effect on MAFLD may be due to its ability to alleviate oxidative stress and exert potent anti-inflammatory effects. α-MSH prevents liver inflammation and injury induced by LPS and paracetamol (131, 132). Furthermore, both in vitro and in vivo studies have confirmed that MSH reduces pro-inflammatory mediator levels by inducing cyclic adenosine monophosphate (cAMP) and inhibiting nuclear factors (133). Due to the antioxidant and anti-inflammatory characteristics of MSH, it can be used to promote melanin synthesis in adipose tissue. This reduces the generation of ROS and inflammation and prevents the sequelae of obesity. This is beneficial in the prevention of MAFLD.
There is increasing evidence that MSH may also have anti-fibrotic properties. For example, α-MSH has been shown to reduce endotoxin-induced liver inflammation (131), and in α-MSH gene-treated rats, hepatic stellate cells (HSC) and Kupffer cells were significantly inhibited (134). A recent report suggests that α-MSH can regulate collagen metabolism (135). Furthermore, α-MSH has been shown to increase collagen degradation by activating matrix metalloproteinase-1 (MMP-1) and MMP-2 (136, 137). Lee et al. recently found that MSH gene therapy can reverse liver fibrosis in mice treated with carbon tetrachloride for 10 weeks (138). In addition, MSH gene therapy increases the expression and/or activity of MMP-1, MMP -2 and MMP -8.
Furthermore, MSH treatment differentially regulates genes involved in lipid and carbohydrate metabolism in liver and adipose tissue based on their synthesis/degradation metabolic functions (139). Taken together, it can be concluded that MSH may have a beneficial impact on MAFLD through multiple effects, including alleviating oxidative stress and inflammation, scavenging reactive oxygen species, and regulating nutrient metabolism. These findings also provide a new therapeutic approach for treating human fibrotic diseases with MSH and related peptides.
Conclusion and future perspectives
MAFLD is a multifactorial disease characterized by excessive fat accumulation in the liver. The pathogenesis of MAFLD is complex and closely linked to glucose and lipid metabolism in the liver. In addition to disturbances in glucose and lipid metabolism, pituitary dysfunction is an important mechanism underlying the development of MAFLD. Recent studies have shown that anterior pituitary hormones may regulate hepatocytes independently of target gland hormones. These hormones may influence the development of MAFLD through various mechanisms. Besides, some current clinical therapeutic strategies for MAFLD can affect pituitary hormones. Glucagon-like peptide-1 receptor agonists (GLP-1 RAs) are renowned for their effectiveness in controlling blood sugar levels and managing weight. But the literature suggests that GLP-1Rs may modulate thyroid hormone production and secretion. For example, Ye et al. demonstrated that treatment with liraglutide reduced TSH levels and improved hepatic thyroid hormone resistance in a population of 49 diabetic patients with MAFLD (140). GLP-1 RAs may exert a direct inhibitory effect on the central nervous system, since GLP-1Rs are present in the paraventricular nucleus (PVN) of the hypothalamus, which contains thyrotropin-releasing hormone (TRH)-producing neurons. This suggests that GLP-1 RAs may influence TRH-producing neuron activity directly in the hypothalamus (141). Though, the causal relationship and underlying mechanisms between anterior pituitary dysfunction and MAFLD are still under investigation and require additional data from clinical and basic research. In the future, the impact of MAFLD clinical treatment on pituitary hormone secretion should also be considered, as this could have implications for the body’s endocrine system.
Author contributions
CL: Investigation, Writing – original draft. YL: Writing – original draft. JL: Investigation, Writing – original draft. YS: Funding acquisition, Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the National Natural Science Foundation (82270922) and Shandong Provincial Natural Science Foundation (ZR2023QH104) of China.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Eslam M, Sanyal AJ, George J, and Consensus International P. 1 MAFLD: A consensus-driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology. (2020) 158:1999–2014 e1. doi: 10.1053/j.gastro.2019.11.312
2. Eslam M, Anstee QM, Targher G, Romero-Gomez M, et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J Hepatol. (2020) 73:202–9. doi: 10.1016/j.jhep.2020.03.039
3. Hoofnagle JH and Doo E. Letter To The Editor: A multi-society delphi consensus statement on new fatty liver disease nomenclature. Hepatology. (2023). doi: 10.1097/HEP.0000000000000695
4. Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M, et al. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol. (2018) 15:11–20. doi: 10.1038/nrgastro.2017.109
5. Saiman Y, Duarte-Rojo A, and Rinella ME. Fatty liver disease: diagnosis and stratification. Annu Rev Med. (2022) 73:529–44. doi: 10.1146/annurev-med-042220-020407
6. Younossi Z, Tacke F, Arrese M, Sharma Chander B, Mostafa I, Bugianesi E, et al. Global perspectives on nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Hepatology. (2019) 69:2672–82. doi: 10.1002/hep.30251
7. Angulo P, Kleiner DE, Dam-Larsen S, Adams LA, Bjornsson ES, Charatcharoenwitthaya P, et al. Liver fibrosis, but no other histologic features, is associated with long-term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology. (2015) 149:389–97 e10. doi: 10.1053/j.gastro.2015.04.043
8. Aminian A, Al-Kurd A, Wilson R, Bena J, Fayazzadeh H, Singh T, et al. Association of bariatric surgery with major adverse liver and cardiovascular outcomes in patients with biopsy-proven nonalcoholic steatohepatitis. JAMA. (2021) 326:2031–42. doi: 10.1001/jama.2021.19569
9. Nasr P, Ignatova S, Kechagias S, and Ekstedt M. Natural history of nonalcoholic fatty liver disease: A prospective follow-up study with serial biopsies. Hepatol Commun. (2018) 2:199–210. doi: 10.1002/hep4.1134
10. Tiniakos DG, Vos MB, and Brunt EM. Nonalcoholic fatty liver disease: pathology and pathogenesis. Annu Rev Pathol. (2010) 5:145–71. doi: 10.1146/annurev-pathol-121808-102132
11. Bessone F, Razori MV, and Roma MG. Molecular pathways of nonalcoholic fatty liver disease development and progression. Cell Mol Life Sci. (2019) 76:99–128. doi: 10.1007/s00018-018-2947-0
12. Younossi ZM. Review article: current management of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis. Aliment Pharmacol Ther. (2008) 28:2–12. doi: 10.1111/j.1365-2036.2008.03710.x
13. Lim S, Kim JW, and Targher G. Links between metabolic syndrome and metabolic dysfunction-associated fatty liver disease. Trends Endocrinol Metab. (2021) 32:500–14. doi: 10.1016/j.tem.2021.04.008
14. Sakurai Y, Kubota N, Yamauchi T, and Kadowaki T. Role of insulin resistance in MAFLD. Int J Mol Sci. (2021) 22. doi: 10.3390/ijms22084156
15. Friedman SL, Neuschwander-Tetri BA, Rinella M, and Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med. (2018) 24:908–22. doi: 10.1038/s41591-018-0104-9
16. Heeren J and Scheja L. Metabolic-associated fatty liver disease and lipoprotein metabolism. Mol Metab. (2021) 50:101238. doi: 10.1016/j.molmet.2021.101238
17. Marra F and Svegliati-Baroni G. Lipotoxicity and the gut-liver axis in NASH pathogenesis. J Hepatol. (2018) 68:280–95. doi: 10.1016/j.jhep.2017.11.014
18. Dong J, Viswanathan S, Adami E, Singh BK, Chothani SP, Ng B, et al. Hepatocyte-specific IL11 cis-signaling drives lipotoxicity and underlies the transition from NAFLD to NASH. Nat Commun. (2021) 12:66. doi: 10.1038/s41467-020-20303-z
19. Zaidi M, Yuen T, and Kim SM. Pituitary crosstalk with bone, adipose tissue and brain. Nat Rev Endocrinol. (2023) 19:708–21. doi: 10.1038/s41574-023-00894-5
20. Abreu AP and Kaiser UB. Pubertal development and regulation. Lancet Diabetes Endocrinol. (2016) 4:254–64. doi: 10.1016/S2213-8587(15)00418-0
21. Zoeller RT, Tan SW, and Tyl RW. General background on the hypothalamic-pituitary-thyroid (HPT) axis. Crit Rev Toxicol. (2007) 37:11–53. doi: 10.1080/10408440601123446
22. Joseph DN and Whirledge S. Stress and the HPA axis: balancing homeostasis and fertility. Int J Mol Sci. (2017) 18. doi: 10.3390/ijms18102224
23. Ortiga-Carvalho TM, Chiamolera MI, Pazos-Moura CC, and Wondisford FE. Hypothalamus-pituitary-thyroid axis. Compr Physiol. (2016) 6:1387–428. doi: 10.1002/cphy.c150027
24. Szkudlinski MW, Fremont V, Ronin C, and Weintraub BD. Thyroid-stimulating hormone and thyroid-stimulating hormone receptor structure-function relationships. Physiol Rev. (2002) 82:473–502. doi: 10.1152/physrev.00031.2001
25. Klein JR. Physiological relevance of thyroid stimulating hormone and thyroid stimulating hormone receptor in tissues other than the thyroid. Autoimmunity. (2003) 36:417–21. doi: 10.1080/08916930310001603019
26. Zhang W, Tian LM, Han Y, Ma HY, Wang LC, Guo J, et al. Presence of thyrotropin receptor in hepatocytes: not a case of illegitimate transcription. J Cell Mol Med. (2009) 13:4636–42. doi: 10.1111/j.1582-4934.2008.00670.x
27. Lu S, Guan Q, Liu Y, Wang H, Xu W, Li X, et al. Role of extrathyroidal TSHR expression in adipocyte differentiation and its association with obesity. Lipids Health Dis. (2012) 11:17. doi: 10.1186/1476-511X-11-17
28. Alonso H, Fernandez-Ruocco J, Gallego M, Malagueta-Vieira LL, Rodriguez-de-Yurre A, Medei E, et al. Thyroid stimulating hormone directly modulates cardiac electrical activity. J Mol Cell Cardiol. (2015) 89:280–6. doi: 10.1016/j.yjmcc.2015.10.019
29. Ercolano MA, Drnovsek ML, Croome Silva MC, Moos M, Fuentes AM, Viale F, et al. Negative correlation between bone mineral density and TSH receptor antibodies in long-term euthyroid postmenopausal women with treated Graves’ disease. Thyroid Res. (2013) 6:11. doi: 10.1186/1756-6614-6-11
30. Stefan M, Wei C, Lombardi A, Li CW, Concepcion ES, Inabnet WB, et al. Genetic-epigenetic dysregulation of thymic TSH receptor gene expression triggers thyroid autoimmunity. Proc Natl Acad Sci U.S.A. (2014) 111:12562–7. doi: 10.1073/pnas.1408821111
31. Yang Q, Li J, Kou C, Zhang L, Wang X, Long Y, et al. Presence of TSHR in NK cells and action of TSH on NK cells. Neuroimmunomodulation. (2022) 29:77–84. doi: 10.1159/000516925
32. Kim D, Kim W, Joo SK, Bae JM, Kim JH, and Ahmed A. Subclinical hypothyroidism and low-normal thyroid function are associated with nonalcoholic steatohepatitis and fibrosis. Clin Gastroenterol Hepatol. (2018) 16:123–131 e1. doi: 10.1016/j.cgh.2017.08.014
33. Kim D, Yoo ER, Li AA, Fernandes CT, Tighe SP, Cholankeril G, et al. Low-normal thyroid function is associated with advanced fibrosis among adults in the United States. Clin Gastroenterol Hepatol. (2019) 17:2379–81. doi: 10.1016/j.cgh.2018.11.024
34. Chung GE, Kim D, Kim W, Yim JY, Park MJ, Kim YJ, et al. Non-alcoholic fatty liver disease across the spectrum of hypothyroidism. J Hepatol. (2012) 57:150–6. doi: 10.1016/j.jhep.2012.02.027
35. Hu DS, Zhu SH, Liu WY, Pan XY, Zhu PW, Li YY, et al. PNPLA3 polymorphism influences the association between high-normal TSH level and NASH in euthyroid adults with biopsy-proven NAFLD. Diabetes Metab. (2020) 46:496–503. doi: 10.1016/j.diabet.2020.02.001
36. Tao Y, Gu H, Wu J, and Sui J. Thyroid function is associated with non-alcoholic fatty liver disease in euthyroid subjects. Endocr Res. (2015) 40:74–8. doi: 10.3109/07435800.2014.952014
37. Tian L, Song Y, Xing M, Zhang W, Ning G, Li X, et al. A novel role for thyroid-stimulating hormone: up-regulation of hepatic 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase expression through the cyclic adenosine monophosphate/protein kinase A/cyclic adenosine monophosphate-responsive element binding protein pathway. Hepatology. (2010) 52:1401–9. doi: 10.1002/hep.23800
38. Zhang X, Song Y, Feng M, Zhou X, Lu Y, Gao L, et al. Thyroid-stimulating hormone decreases HMG-CoA reductase phosphorylation via AMP-activated protein kinase in the liver. J Lipid Res. (2015) 56:963–71. doi: 10.1194/jlr.M047654
39. Yan F, Wang Q, Lu M, Chen W, Song Y, Jing F, et al. Thyrotropin increases hepatic triglyceride content through upregulation of SREBP-1c activity. J Hepatol. (2014) 61:1358–64. doi: 10.1016/j.jhep.2014.06.037
40. Song Y, Xu C, Shao S, Liu J, Xing W, Xu J, et al. Thyroid-stimulating hormone regulates hepatic bile acid homeostasis via SREBP-2/HNF-4alpha/CYP7A1 axis. J Hepatol. (2015) 62:1171–9. doi: 10.1016/j.jhep.2014.12.006
41. Li J, Kong D, Gao X, Tian Z, Wang X, Guo Q, et al. TSH attenuates fatty acid oxidation in hepatocytes by reducing the mitochondrial distribution of miR-449a/449b-5p/5194. Mol Cell Endocrinol. (2021) 530:111280. doi: 10.1016/j.mce.2021.111280
42. Wang T, Xu J, Bo T, Zhou X, Jiang X, Gao L, et al. Decreased fasting blood glucose is associated with impaired hepatic glucose production in thyroid-stimulating hormone receptor knockout mice. Endocr J. (2013) 60:941–50. doi: 10.1507/endocrj.EJ12-0462
43. Li Y, Wang L, Zhou L, Song Y, Ma S, Yu C, et al. Thyroid stimulating hormone increases hepatic gluconeogenesis via CRTC2. Mol Cell Endocrinol. (2017) 446:70–80. doi: 10.1016/j.mce.2017.02.015
44. Wang X, Mao J, Zhou X, Li Q, Gao L, and Zhao J. Thyroid stimulating hormone triggers hepatic mitochondrial stress through cyclophilin D acetylation. Oxid Med Cell Longev. (2020) 2020:1249630. doi: 10.1155/2020/1249630
45. Ma S, Shao S, Yang C, Yao Z, Gao L, and Chen W. A preliminary study: proteomic analysis of exosomes derived from thyroid-stimulating hormone-stimulated HepG2 cells. J Endocrinol Invest. (2020) 43:1229–38. doi: 10.1007/s40618-020-01210-y
46. Huang B, Wen W, and Ye S. TSH-SPP1/TRbeta-TSH positive feedback loop mediates fat deposition of hepatocyte: Crosstalk between thyroid and liver. Front Immunol. (2022) 13:1009912. doi: 10.3389/fimmu.2022.1009912
47. Jiang D, Ma S, Jing F, Xu C, Yan F, Wang A, et al. Thyroid-stimulating hormone inhibits adipose triglyceride lipase in 3T3-L1 adipocytes through the PKA pathway. PloS One. (2015) 10:e0116439. doi: 10.1371/journal.pone.0116439
48. Simoni M, Gromoll J, and Nieschlag E. The follicle-stimulating hormone receptor: biochemistry, molecular biology, physiology, and pathophysiology. Endocr Rev. (1997) 18:739–73. doi: 10.1210/edrv.18.6.0320
49. Rannikki AS, Zhang FP, and Huhtaniemi IT. Ontogeny of follicle-stimulating hormone receptor gene expression in the rat testis and ovary. Mol Cell Endocrinol. (1995) 107:199–208. doi: 10.1016/0303-7207(94)03444-X
50. Robinson LJ, Tourkova I, Wang Y, Sharrow AC, Landau MS, Yaroslavskiy BB, et al. FSH-receptor isoforms and FSH-dependent gene transcription in human monocytes and osteoclasts. Biochem Biophys Res Commun. (2010) 394:12–7. doi: 10.1016/j.bbrc.2010.02.112
51. Cui H, Zhao G, Liu R, Zheng M, Chen J, and Wen J. FSH stimulates lipid biosynthesis in chicken adipose tissue by upregulating the expression of its receptor FSHR. J Lipid Res. (2012) 53:909–17. doi: 10.1194/jlr.M025403
52. Song Y, Wang ES, Xing LL, Shi S, Qu F, Zhang D, et al. Follicle-stimulating hormone induces postmenopausal dyslipidemia through inhibiting hepatic cholesterol metabolism. J Clin Endocrinol Metab. (2016) 101:254–63. doi: 10.1210/jc.2015-2724
53. Tan D, Zhao Y, Ma D, and Tong F. Role of FSH and FSH receptor on HUVECs migration. Gene Ther. (2021) 28:155–61. doi: 10.1038/s41434-020-00195-w
54. Crawford ED, Schally AV, Pinthus JH, Block NL, Rick FG, Garnick MB, et al. The potential role of follicle-stimulating hormone in the cardiovascular, metabolic, skeletal, and cognitive effects associated with androgen deprivation therapy. Urol Oncol. (2017) 35:183–91. doi: 10.1016/j.urolonc.2017.01.025
55. Qiao S, Alasmi S, Wyatt A, Wartenberg P, Wang H, Candlish M, et al. Intra-pituitary follicle-stimulating hormone signaling regulates hepatic lipid metabolism in mice. Nat Commun. (2023) 14:1098. doi: 10.1038/s41467-023-36681-z
56. Wang N, Li Q, Han B, Chen Y, Zhu C, Chen Y, et al. Follicle-stimulating hormone is associated with non-alcoholic fatty liver disease in Chinese women over 55 years old. J Gastroenterol Hepatol. (2016) 31:1196–202. doi: 10.1111/jgh.13271
57. Ge S, Zheng Y, Du L, Hu X, Zhou J, He Z, et al. Association between follicle-stimulating hormone and nonalcoholic fatty liver disease in postmenopausal women with type 2 diabetes mellitus. J Diabetes. (2023) 15:640–8. doi: 10.1111/1753-0407.13394
58. Li X, Xin N, Guo T, Wu Z, Zheng Y, Lin L, et al. Follicle-stimulating hormone is negatively associated with nonalcoholic fatty liver disease in a Chinese elderly population: a retrospective observational study. BMC Endocr Disord. (2023) 23:165. doi: 10.1186/s12902-023-01427-x
59. Zhu Y, Xu J, Zhang X, Ke Y, Fu G, Guo Q, et al. A low follicle-stimulating hormone level is a protective factor for non-alcoholic fatty liver disease in older men aged over 80. BMC Geriatr. (2021) 21:544. doi: 10.1186/s12877-021-02490-6
60. Guo Y, Zhao M, Bo T, Ma S, Yuan Z, Chen W, et al. Blocking FSH inhibits hepatic cholesterol biosynthesis and reduces serum cholesterol. Cell Res. (2019) 29:151–66. doi: 10.1038/s41422-018-0123-6
61. Qi X, Guo Y, Song Y, Yu C, Zhao L, Fang L, et al. Follicle-stimulating hormone enhances hepatic gluconeogenesis by GRK2-mediated AMPK hyperphosphorylation at Ser485 in mice. Diabetologia. (2018) 61:1180–92. doi: 10.1007/s00125-018-4562-x
62. Aydin BK, Stenlid R, Ciba I, Cerenius SY, Dahlbom M, Bergsten P, et al. High levels of FSH before puberty are associated with increased risk of metabolic syndrome during pubertal transition. Pediatr Obes. (2022) 17:e12906. doi: 10.1111/ijpo.12906
63. Stefanska A, Cembrowska P, Kubacka J, Kuligowska-Prusinska M, and Sypniewska G. Gonadotropins and their association with the risk of prediabetes and type 2 diabetes in middle-aged postmenopausal women. Dis Markers. (2019) 2019:2384069. doi: 10.1155/2019/2384069
64. Quinn MA, Xu X, Ronfani M, and Cidlowski JA. Estrogen deficiency promotes hepatic steatosis via a glucocorticoid receptor-dependent mechanism in mice. Cell Rep. (2018) 22:2690–701. doi: 10.1016/j.celrep.2018.02.041
65. Richards JS, Fitzpatrick SL, Clemens JW, Morris JK, Alliston T, and Sirois J. Ovarian cell differentiation: a cascade of multiple hormones, cellular signals, and regulated genes. Recent Prog Horm Res. (1995) 50:223–54. doi: 10.1016/b978-0-12-571150-0.50014-7
66. Catt KJ, Harwood JP, Clayton RN, Davies TF, Chan V, Katikineni M, et al. Regulation of peptide hormone receptors and gonadal steroidogenesis. Recent Prog Horm Res. (1980) 36:557–662. doi: 10.1016/B978-0-12-571136-4.50021-8
67. Filicori M. The role of luteinizing hormone in folliculogenesis and ovulation induction. Fertil Steril. (1999) 71:405–14. doi: 10.1016/S0015-0282(98)00482-8
68. Angelopoulos N, Goula A, and Tolis G. The role of luteinizing hormone activity in controlled ovarian stimulation. J Endocrinol Invest. (2005) 28:79–88. doi: 10.1007/BF03345534
69. Ayoub MA, Yvinec R, Jegot G, Dias JA, Poli SM, Poupon A, et al. Profiling of FSHR negative allosteric modulators on LH/CGR reveals biased antagonism with implications in steroidogenesis. Mol Cell Endocrinol. (2016) 436:10–22. doi: 10.1016/j.mce.2016.07.013
70. Ettinger AM, Gust SK, and Kutzler MA. Luteinizing hormone receptor expression by nonneoplastic and neoplastic canine lymphocytes. Am J Vet Res. (2019) 80:572–7. doi: 10.2460/ajvr.80.6.572
71. Vuorenoja S, Rivero-Muller A, Kiiveri S, Bielinska M, Heikinheimo M, Wilson DB, et al. Adrenocortical tumorigenesis, luteinizing hormone receptor and transcription factors GATA-4 and GATA-6. Mol Cell Endocrinol. (2007) 269:38–45. doi: 10.1016/j.mce.2006.11.013
72. Mey M, Bhatta S, and Casadesus G. Luteinizing hormone and the aging brain. Vitam Horm. (2021) 115:89–104. doi: 10.1016/bs.vh.2020.12.005
73. Burnham V, Sundby C, Laman-Maharg A, and Thornton J. Luteinizing hormone acts at the hippocampus to dampen spatial memory. Horm Behav. (2017) 89:55–63. doi: 10.1016/j.yhbeh.2016.11.007
74. Huang W, Acosta-Martinez M, Horton TH, and Levine JE. Fasting-induced suppression of LH secretion does not require activation of ATP-sensitive potassium channels. Am J Physiol Endocrinol Metab. (2008) 295:E1439–46. doi: 10.1152/ajpendo.90615.2008
75. Li S, Chu Q, Ma J, Sun Y, Tao T, Huang R, et al. Discovery of novel lipid profiles in PCOS: do insulin and androgen oppositely regulate bioactive lipid production? J Clin Endocrinol Metab. (2017) 102:810–21. doi: 10.1210/jc.2016-2692
76. Belani M, Deo A, Shah P, Banker M, Singal P, and Gupta S. Differential insulin and steroidogenic signaling in insulin resistant and non-insulin resistant human luteinized granulosa cells-A study in PCOS patients. J Steroid Biochem Mol Biol. (2018) 178:283–92. doi: 10.1016/j.jsbmb.2018.01.008
77. Chen CY, Li Y, Zeng N, He L, Zhang X, Tu T, et al. Inhibition of estrogen-related receptor alpha blocks liver steatosis and steatohepatitis and attenuates triglyceride biosynthesis. Am J Pathol. (2021) 191:1240–54. doi: 10.1016/j.ajpath.2021.04.007
78. Guo F, Gong Z, Fernando T, Zhang L, Zhu X, and Shi Y. The lipid profiles in different characteristics of women with PCOS and the interaction between dyslipidemia and metabolic disorder states: A retrospective study in chinese population. Front Endocrinol (Lausanne). (2022) 13:892125. doi: 10.3389/fendo.2022.892125
79. Zhu L, Brown WC, Cai Q, Krust A, Chambon P, McGuinness OP, et al. Estrogen treatment after ovariectomy protects against fatty liver and may improve pathway-selective insulin resistance. Diabetes. (2013) 62:424–34. doi: 10.2337/db11-1718
80. Binart N, Ormandy CJ, and Kelly PA. Mammary gland development and the prolactin receptor. Adv Exp Med Biol. (2000) 480:85–92. doi: 10.1007/0-306-46832-8_10
81. Macotela Y, Triebel J, and Clapp C. Time for a new perspective on prolactin in metabolism. Trends Endocrinol Metab. (2020) 31:276–86. doi: 10.1016/j.tem.2020.01.004
82. Nielsen JH, Galsgaard ED, Moldrup A, Friedrichsen BN, Billestrup N, Hansen JA, et al. Regulation of beta-cell mass by hormones and growth factors. Diabetes. (2001) 50 Suppl 1:S25–9. doi: 10.2337/diabetes.50.2007.S25
83. Baxter RC, Bryson JM, and Turtle JR. The effect of fasting on liver receptors for prolactin and growth hormone. Metabolism. (1981) 30:1086–90. doi: 10.1016/0026-0495(81)90052-4
84. Wang X, Ma B, Li G, Sheng C, Yang P, Gao J, et al. Glucose-lipid metabolism in obesity with elevated prolactin levels and alteration of prolactin levels after laparoscopic sleeve gastrectomy. Obes Surg. (2020) 30:4004–13. doi: 10.1007/s11695-020-04771-2
85. Melendez Garcia R, Zamarripa Arredondo D, Arnold E, Ruiz-Herrera X, Imm Noguez R, Cruz Baeza G, et al. Prolactin protects retinal pigment epithelium by inhibiting sirtuin 2-dependent cell death. EBioMedicine. (2016) 7:35–49. doi: 10.1016/j.ebiom.2016.03.048
86. Linher-Melville K, Zantinge S, Sanli T, Gerstein H, Tsakiridis T, and Singh G. Establishing a relationship between prolactin and altered fatty acid beta-oxidation via carnitine palmitoyl transferase 1 in breast cancer cells. BMC Cancer. (2011) 11:56. doi: 10.1186/1471-2407-11-56
87. Zhu C, Ma H, Huang D, Li G, Gao J, Cai M, et al. J-shaped relationship between serum prolactin and metabolic-associated fatty liver disease in female patients with type 2 diabetes. Front Endocrinol (Lausanne). (2022) 13:815995. doi: 10.3389/fendo.2022.815995
88. Ponce AJ, Galvan-Salas T, Lerma-Alvarado RM, Ruiz-Herrera X, Hernandez-Cortes T, Valencia-Jimenez R, et al. Low prolactin levels are associated with visceral adipocyte hypertrophy and insulin resistance in humans. Endocrine. (2020) 67:331–43. doi: 10.1007/s12020-019-02170-x
89. Zhang P, Ge Z, Wang H, Feng W, Sun X, Chu X, et al. Prolactin improves hepatic steatosis via CD36 pathway. J Hepatol. (2018) 68:1247–55. doi: 10.1016/j.jhep.2018.01.035
90. Yu J, Xiao F, Zhang Q, Liu B, Guo Y, Lv Z, et al. PRLR regulates hepatic insulin sensitivity in mice via STAT5. Diabetes. (2013) 62:3103–13. doi: 10.2337/db13-0182
91. Shao S, Yao Z, Lu J, Song Y, He Z, Yu C, et al. Ablation of prolactin receptor increases hepatic triglyceride accumulation. Biochem Biophys Res Commun. (2018) 498:693–9. doi: 10.1016/j.bbrc.2018.03.048
92. Macotela Y, Ruiz-Herrera X, Vazquez-Carrillo DI, Ramirez-Hernandez G, Escalera Martinez G, Clapp C, et al. The beneficial metabolic actions of prolactin. Front Endocrinol (Lausanne). (2022) 13:1001703. doi: 10.3389/fendo.2022.1001703
93. Xiao F, Xia T, Lv Z, Zhang Q, Xiao Y, Yu J, et al. Central prolactin receptors (PRLRs) regulate hepatic insulin sensitivity in mice via signal transducer and activator of transcription 5 (STAT5) and the vagus nerve. Diabetologia. (2014) 57:2136–44. doi: 10.1007/s00125-014-3336-3
94. Nimura M, Udagawa J, Hatta T, Hashimoto R, and Otani H. Spatial and temporal patterns of expression of melanocortin type 2 and 5 receptors in the fetal mouse tissues and organs. Anat Embryol (Berl). (2006) 211:109–17. doi: 10.1007/s00429-005-0066-9
95. Jun DJ, Na KY, Kim W, Kwak D, Kwon EJ, Yoon JH, et al. Melanocortins induce interleukin 6 gene expression and secretion through melanocortin receptors 2 and 5 in 3T3-L1 adipocytes. J Mol Endocrinol. (2010) 44:225–36. doi: 10.1677/JME-09-0161
96. Novoselova TV, Chan LF, and Clark AJL. Pathophysiology of melanocortin receptors and their accessory proteins. Best Pract Res Clin Endocrinol Metab. (2018) 32:93–106. doi: 10.1016/j.beem.2018.02.002
97. Park HJ, Kim HJ, Lee JY, Cho BK, Gallo RL, and Cho DH. Adrenocorticotropin hormone stimulates interleukin-18 expression in human HaCaT keratinocytes. J Invest Dermatol. (2007) 127:1210–6. doi: 10.1038/sj.jid.5700703
98. Morris DG, Kola B, Borboli N, Kaltsas GA, Gueorguiev M, McNicol AM, et al. Identification of adrenocorticotropin receptor messenger ribonucleic acid in the human pituitary and its loss of expression in pituitary adenomas. J Clin Endocrinol Metab. (2003) 88:6080–7. doi: 10.1210/jc.2002-022048
99. Nankova BB, Kvetnansky R, and Sabban EL. Adrenocorticotropic hormone (MC-2) receptor mRNA is expressed in rat sympathetic ganglia and up-regulated by stress. Neurosci Lett. (2003) 344:149–52. doi: 10.1016/S0304-3940(03)00361-6
100. Johnston H, King PJ, and O’Shaughnessy PJ. Effects of ACTH and expression of the melanocortin-2 receptor in the neonatal mouse testis. Reproduction. (2007) 133:1181–7. doi: 10.1530/REP-06-0359
101. Simamura E, Arikawa T, Ikeda T, Shimada H, Shoji H, Masuta H, et al. Melanocortins contribute to sequential differentiation and enucleation of human erythroblasts via melanocortin receptors 1, 2 and 5. PloS One. (2015) 10:e0123232. doi: 10.1371/journal.pone.0123232
102. Isales CM, Zaidi M, and Blair HC. ACTH is a novel regulator of bone mass. Ann N Y Acad Sci. (2010) 1192:110–6. doi: 10.1111/j.1749-6632.2009.05231.x
103. Luo J, Yan Z, Shen Y, Liu D, Su M, Yang J, et al. Exposure to low-intensity noise exacerbates nonalcoholic fatty liver disease by activating hypothalamus pituitary adrenal axis. Sci Total Environ. (2024) 906:167395. doi: 10.1016/j.scitotenv.2023.167395
104. Su M, Yan Z, Wang Y, Cai J, Dong J, Luo J, et al. Depression exacerbates hepatic steatosis in C57BL/6J mice by activating the hypothalamic-pituitary-adrenal axis. In Vivo. (2024) 38:1677–89. doi: 10.21873/invivo.13618
105. Parker JC, Moraitis AG, and Belanoff JK. Biochemical and radiological changes in liver steatosis following mifepristone treatment in patients with hypercortisolism. AACE Clin Case Rep. (2022) 8:25–9. doi: 10.1016/j.aace.2021.07.001
106. Wang WB, She F, Xie LF, Yan WH, Ouyang JZ, Wang BA, et al. Evaluation of basal serum adrenocorticotropic hormone and cortisol levels and their relationship with nonalcoholic fatty liver disease in male patients with idiopathic hypogonadotropic hypogonadism. Chin Med J (Engl). (2016) 129:1147–53. doi: 10.4103/0366-6999.181967
107. Rafnsson AT, Johannsson M, Olafsson I, Dallongeville J, Erfurth EM, Berg AL, et al. Effects of different doses of adrenocorticotrophic hormone on the serum lipoprotein profile in healthy subjects. Basic Clin Pharmacol Toxicol. (2005) 97:86–90. doi: 10.1111/j.1742-7843.2005.pto_108.x
108. Berg AL, Nilsson-Ehle P, and Arnadottir M. Beneficial effects of ACTH on the serum lipoprotein profile and glomerular function in patients with membranous nephropathy. Kidney Int. (1999) 56:1534–43. doi: 10.1046/j.1523-1755.1999.00675.x
109. Chinn AM, Ciais D, Bailly S, Chambaz E, LaMarre J, and Feige JJ. Identification of two novel ACTH-responsive genes encoding manganese-dependent superoxide dismutase (SOD2) and the zinc finger protein TIS11b [tetradecanoyl phorbol acetate (TPA)-inducible sequence 11b. Mol Endocrinol. (2002) 16:1417–27. doi: 10.1210/mend.16.6.0844
110. Lightman SL, Birnie MT, and Conway-Campbell BL. Dynamics of ACTH and cortisol secretion and implications for disease. Endocr Rev. (2020) 41. doi: 10.1210/endrev/bnaa002
111. Arnadottir M, Dallongeville J, Nilsson-Ehle P, and Berg AL. Effects of short-term treatment with corticotropin on the serum apolipoprotein pattern. Scand J Clin Lab Invest. (2001) 61:301–6. doi: 10.1080/00365510152379030
112. Xu N, Hurtig M, Ekstrom U, and Nilsson-Ehle P. Adrenocorticotrophic hormone retarded metabolism of low-density lipoprotein in rats. Scand J Clin Lab Invest. (2004) 64:217–22. doi: 10.1080/00365510410005730
113. Moller N and Jorgensen JO. Effects of growth hormone on glucose, lipid, and protein metabolism in human subjects. Endocr Rev. (2009) 30:152–77. doi: 10.1210/er.2008-0027
114. Postel-Vinay MC and Finidori J. Growth hormone receptor: structure and signal transduction. Eur J Endocrinol. (1995) 133:654–9. doi: 10.1530/eje.0.1330654
115. Xanthakos SA, Crimmins NA, and Chernausek SD. Abnormalities in the growth hormone axis and risk of nonalcoholic steatohepatitis: active player or innocent bystander? J Pediatr. (2014) 165:12–4. doi: 10.1016/j.jpeds.2014.03.031
116. Xu L, Xu C, Yu C, Miao M, Zhang X, Zhu Z, et al. Association between serum growth hormone levels and nonalcoholic fatty liver disease: a cross-sectional study. PloS One. (2012) 7:e44136. doi: 10.1371/journal.pone.0044136
117. Doycheva I, Erickson D, and Watt KD. Growth hormone deficiency and NAFLD: An overlooked and underrecognized link. Hepatol Commun. (2022) 6:2227–37. doi: 10.1002/hep4.1953
118. Nishizawa H, Takahashi M, Fukuoka H, Iguchi G, Kitazawa R, and Takahashi Y. GH-independent IGF-I action is essential to prevent the development of nonalcoholic steatohepatitis in a GH-deficient rat model. Biochem Biophys Res Commun. (2012) 423:295–300. doi: 10.1016/j.bbrc.2012.05.115
119. Donaghy A, Ross R, Wicks C, Hughes SC, Holly J, Gimson A, et al. Growth hormone therapy in patients with cirrhosis: a pilot study of efficacy and safety. Gastroenterology. (1997) 113:1617–22. doi: 10.1053/gast.1997.v113.pm9352864
120. Vitale G, Barbieri M, Kamenetskaya M, and Paolisso G. GH/IGF-I/insulin system in centenarians. Mech Ageing Dev. (2017) 165:107–14. doi: 10.1016/j.mad.2016.12.001
121. Kang DY, Sp N, Jo ES, Kim HD, Kim IH, Bae SW, et al. Non−toxic sulfur enhances growth hormone signaling through the JAK2/STAT5b/IGF−1 pathway in C2C12 cells. Int J Mol Med. (2020) 45:931–8. doi: 10.3892/ijmm.2019.4451
122. Berryman DE, Glad CA, List EO, and Johannsson G. The GH/IGF-1 axis in obesity: pathophysiology and therapeutic considerations. Nat Rev Endocrinol. (2013) 9:346–56. doi: 10.1038/nrendo.2013.64
123. Hwa V. Human growth disorders associated with impaired GH action: Defects in STAT5B and JAK2. Mol Cell Endocrinol. (2021) 519:111063. doi: 10.1016/j.mce.2020.111063
124. Hosui A, Kimura A, Yamaji D, Zhu BM, Na R, Hennighausen L, et al. Loss of STAT5 causes liver fibrosis and cancer development through increased TGF-beta and STAT3 activation. J Exp Med. (2009) 206:819–31. doi: 10.1084/jem.20080003
125. Jiang Q, Bai J, He M, Yuen KWY, and Wong AOL. Mechanisms underlying the synergistic action of insulin and growth hormone on IGF-I and -II expression in grass carp hepatocytes. Front Endocrinol (Lausanne). (2018) 9:336. doi: 10.3389/fendo.2018.00336
126. Roselli-Rehfuss L, Mountjoy KG, Robbins LS, Mortrud MT, Low MJ, Tatro JB, et al. Identification of a receptor for gamma melanotropin and other proopiomelanocortin peptides in the hypothalamus and limbic system. Proc Natl Acad Sci U.S.A. (1993) 90:8856–60. doi: 10.1073/pnas.90.19.8856
127. Yuan XC and Tao YX. Ligands for melanocortin receptors: beyond melanocyte-stimulating hormones and adrenocorticotropin. Biomolecules. (2022) 12. doi: 10.3390/biom12101407
128. Wu Q, Chen J, Hua T, and Cai J. Alpha-melanocyte-stimulating hormone-mediated appetite regulation in the central nervous system. Neuroendocrinology. (2023) 113:885–904. doi: 10.1159/000530804
129. Kravchychyn ACP, Campos R, Corgosinho FC, Masquio DCL, Vicente S, Ferreira YAM, et al. The long-term impact of high levels of alpha-melanocyte-stimulating hormone in energy balance among obese adolescents. Ann Nutr Metab. (2018) 72:279–86. doi: 10.1159/000488005
130. Singh M and Mukhopadhyay K. Alpha-melanocyte stimulating hormone: an emerging anti-inflammatory antimicrobial peptide. BioMed Res Int. (2014) 2014:874610. doi: 10.1155/2014/874610
131. Chiao H, Foster S, Thomas R, Lipton J, and Star RA. Alpha-melanocyte-stimulating hormone reduces endotoxin-induced liver inflammation. J Clin Invest. (1996) 97:2038–44. doi: 10.1172/JCI118639
132. Brzoska T, Luger TA, Maaser C, Abels C, and Bohm M. Alpha-melanocyte-stimulating hormone and related tripeptides: biochemistry, antiinflammatory and protective effects in vitro and in vivo, and future perspectives for the treatment of immune-mediated inflammatory diseases. Endocr Rev. (2008) 29:581–602. doi: 10.1210/er.2007-0027
133. Haycock JW, Wagner M, Morandini R, Ghanem G, Rennie IG, and Neil Mac S. Alpha-melanocyte-stimulating hormone inhibits NF-kappaB activation in human melanocytes and melanoma cells. J Invest Dermatol. (1999) 113:560–6. doi: 10.1046/j.1523-1747.1999.00739.x
134. Wang CC, Lin JW, Lee LM, Lin CM, Chiu WT, Pai HT, et al. alpha-melanocyte-stimulating hormone gene transfer attenuates inflammation after bile duct ligation in the rat. Dig Dis Sci. (2008) 53:556–63. doi: 10.1007/s10620-007-9901-6
135. Wang CH, Jawan B, Lee TH, Hung KS, Chou WY, Lu CN, et al. Single injection of naked plasmid encoding alpha-melanocyte-stimulating hormone protects against thioacetamide-induced acute liver failure in mice. Biochem Biophys Res Commun. (2004) 322:153–61. doi: 10.1016/j.bbrc.2004.07.091
136. Bohm M, Raghunath M, Sunderkotter C, Schiller M, Stander S, Brzoska T, et al. Collagen metabolism is a novel target of the neuropeptide alpha-melanocyte-stimulating hormone. J Biol Chem. (2004) 279:6959–66. doi: 10.1074/jbc.M312549200
137. Colombo G, Gatti S, Turcatti F, Sordi A, Fassati LR, Bonino F, et al. Gene expression profiling reveals multiple protective influences of the peptide alpha-melanocyte-stimulating hormone in experimental heart transplantation. J Immunol. (2005) 175:3391–401. doi: 10.4049/jimmunol.175.5.3391
138. Lee TH, Jawan B, Chou WY, Lu CN, Wu CL, Kuo HM, et al. Alpha-melanocyte-stimulating hormone gene therapy reverses carbon tetrachloride induced liver fibrosis in mice. J Gene Med. (2006) 8:764–72. doi: 10.1002/jgm.899
139. Barb CR, Hausman GJ, Rekaya R, Lents CA, Lkhagvadorj S, Qu L, et al. Gene expression in hypothalamus, liver, and adipose tissues and food intake response to melanocortin-4 receptor agonist in pigs expressing melanocortin-4 receptor mutations. Physiol Genomics. (2010) 41:254–68. doi: 10.1152/physiolgenomics.00006.2010
140. Ye J, Xu J, Wen W, and Huang B. Effect of liraglutide on serum TSH levels in patients with NAFLD and its underlying mechanisms. Int J Clin Pract. (2022) 2022:1786559. doi: 10.1155/2022/1786559
141. Farr OM, Sofopoulos M, Tsoukas MA, Dincer F, Thakkar B, Sahin-Efe A, et al. GLP-1 receptors exist in the parietal cortex, hypothalamus and medulla of human brains and the GLP-1 analogue liraglutide alters brain activity related to highly desirable food cues in individuals with diabetes: a crossover, randomised, placebo-controlled trial. Diabetologia. (2016) 59:954–65. doi: 10.1007/s00125-016-3874-y
Keywords: MAFLD, anterior pituitary hormone, lipid metabolism, metabolism dysfunction, metabolic mechanisms signalling
Citation: Liu C, Long Y, Liu J and Song Y (2025) Recent advances in anterior pituitary hormones and metabolic-associated fatty liver disease. Front. Endocrinol. 16:1600559. doi: 10.3389/fendo.2025.1600559
Received: 26 March 2025; Accepted: 11 June 2025;
Published: 04 July 2025.
Edited by:
Xiang'En Shi, Capital Medical University, ChinaReviewed by:
Hao Du, Yale University, United StatesShakun Chaudhary, Dr. Rajendra Prasad Government Medical College, India
Copyright © 2025 Liu, Long, Liu and Song. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Changqing Liu, Y2hxXzE2NkAxNjMuY29t; Yongfeng Song, c3lmMTk4NTA2QDE2My5jb20=