 Shilei Wang1
Shilei Wang1 Yuqing Shi
Yuqing Shi Jixiang Ren
Jixiang Ren- 1College of Traditional Chinese Medicine, Changchun University of Chinese Medicine, Changchun, Jilin, China
- 2Affiliated Hospital to Changchun University of Chinese Medicine, Changchun, Jilin, China
The association between metabolic syndrome (MetS) and Alzheimer’s disease (AD) has attracted widespread attention; nevertheless, the precise mechanism of action between the two is not yet fully elucidated. This review systematically explores the complex mechanisms of insulin resistance (IR) in MetS and AD. We first detail the intrinsic mechanisms of insulin resistance and emphasize its central role in the pathophysiology of MetS. Further, we reveal the underlying mechanisms by which insulin resistance in turn triggers AD through a multidimensional pathway that promotes the accumulation of pathological products, induces blood-brain barrier dysfunction, impairs neuroplasticity, induces neuroinflammatory responses, aberrantly activates the renin-angiotensin-aldosterone system, and exacerbates oxidative stress. In addition, we summarize potential strategies for targeting IR in AD treatment and demonstrate the promising prospects for improving insulin resistance in promoting cognitive recovery. This study offers a novel theoretical framework for elucidating the intricate relationship between MetS and AD. Furthermore, it provides a scientific foundation for the formulation of preventive and therapeutic strategies for metabolic and neurodegenerative diseases.
1 Introduction
Insulin resistance (IR), as a pathological state in which the body’s response to the physiological effects of insulin is reduced, has become a central issue in the field of modern metabolic diseases (1, 2). This metabolic disorder not only impairs the core physiological function of insulin in regulating glucose uptake and utilization but also extensively interferes with key metabolic pathways such as lipid metabolism and protein homeostasis, thus providing a common pathophysiological basis for numerous metabolic diseases (3).
With the acceleration of urbanization, the prevalence of sedentary behavior, and the global spread of high-calorie dietary patterns, the prevalence of MetS has continued to increase, and it has become a major challenge that threatens the security of global public health (4). Epidemiological research indicates a prevalence rate of MetS at 41.8% within the United States, while in China, the figure stands at 33.9% (5, 6), highlighting its widespread prevalence. MetS is a clinical syndrome with IR as the core pathogenesis, characterized by central obesity, elevated blood pressure, abnormal fasting glucose, and lipid metabolism disorders (7, 8). These metabolic abnormalities significantly increase the risk of cardiovascular disease (9–11).
With the acceleration of global population aging, the incidence of dementia, represented by Alzheimer’s disease (AD), has shown exponential growth. Epidemiological research reveals that presently, over 55 million individuals are afflicted with dementia globally, with projections anticipating that this figure will surpass 130 million by the year 2050 (https://www.who.int/zh). AD, as a major subtype of dementia, accounts for about 60-70% of all cases, and its progressive neurodegenerative lesions not only pose a serious threat to the quality of life of the elderly population but also become a significant burden to the public health system (12). However, the overall efficacy of current clinical therapeutic regimens is still unsatisfactory (13), making the identification and characterization of AD-related risk factors a priority for prevention and control strategies - targeting and modifying interventional risk factors, may provide a key target for intervention in the disease process. Notably, prior research have concentrated on the impact of an individual metabolic factor on AD, ignoring the fact that different metabolic factors might collectively or synergistically contribute to the heightened likelihood of developing AD (14). Therefore, studies targeting the MetS as a group of risk factors may be more helpful in the prevention and management of AD.
Accumulating epidemiological and clinical research evidence shows that MetS contributes significantly to the progression of AD (15–17). Particularly in older age groups, patients with MetS are more likely to develop AD, and this trend is more pronounced in women (18). The close relationship between these two diseases is centered on the fulcrum of IR (19), which is not only a core pathological mechanism of MetS, but also closely related to the pathological process of AD (20, 21). Therefore, this review aims to thoroughly analyze the multidimensional mechanism of IR as a key pathological hub in the association between MetS and AD. By integrating multidisciplinary evidence from molecular biology, neuropathology, and clinical medicine, the cascade response network between IR-MetS-AD will be systematically elucidated, and potential preventive and therapeutic strategies will be explored.
2 Methods
2.1 Literature search criteria
Databases: A systematic search was conducted in PubMed, Web of Science, and Scopus.
Search Terms:
PubMed: (“Insulin Resistance”[MeSH] OR insulin resistance) AND (“Metabolic Syndrome”[MeSH] OR metabolic syndrome) AND (“Alzheimer’s disease”[MeSH] OR Alzheimer’s disease) AND (“Cognitive impairment”[MeSH] OR cognitive impairment) AND (mechanism OR pathogenesis OR molecular pathway).
Web of Science: TS=(insulin resistance) AND TS=(metabolic syndrome) AND TS=(Alzheimer’s disease) AND TS=(mechanism OR pathogenesis).
Scopus: TITLE-ABS-KEY(insulin resistance) AND TITLE-ABS-KEY(metabolic syndrome) AND TITLE-ABS-KEY(Alzheimer disease) AND TITLE-ABS-KEY(cognitive impairment OR mild cognitive impairment OR cognitive dysfunction) AND TITLE-ABS-KEY(mechanism).
Language/Time: English-language studies published up to August 2025 were prioritized.
2.2 Inclusion/exclusion criteria
Inclusion criteria:
Original research articles, reviews, or meta-analyses that explore the role of insulin resistance (IR) as a bridge between metabolic syndrome (MetS) and Alzheimer’s disease (AD).
Studies involving IR and its relationship with cognitive function, neurodegeneration, or AD pathological mechanisms (e.g., Aβ deposition, Tau phosphorylation, blood-brain barrier dysfunction, RAAS system activation, oxidative stress, neuroinflammation, etc.).
Exclusion criteria:
Studies based on non-mammalian models.
Case reports, conference abstracts, and studies lacking full-text availability or complete data.
3 Pathophysiologic mechanisms of insulin resistance
3.1 Insulin signaling
The binding of insulin to the insulin receptor initiates a sequence of subsequent reactions, including the recruitment and phosphorylation of various proteins. The composition of these proteins is primarily constituted by IRS, PI3K, and AKT subtypes, which serve as the initiating agents for a series of insulin responses (22). The activation of AKT manifests diverse characteristics that result in varied distal signaling responses to insulin in target tissues (Figure 1). (a) In the complex metabolic regulatory network, AKT substrates include glycogen synthase kinase-3(GSK3), and phosphorylation-inactivation of GSK3 is a pivotal junction in the glycogen synthesis (23). Additionally, the transcription factor Forkhead Box O1 (FOXO1) undergoes nucleoplasmic shuttling facilitated by AKT kinase phosphorylation, leading to inhibition of its transcriptional activity, which represses the expression of glycoheterotrophic genes (24, 25). (b) Tuberous Sclerosis Complex 1/2 (TSC1/2) with Proline-Rich Akt Substrate 40 (PRAS40) acts as a negative regulator of the mTORC1 signaling pathway, modulating protein translation initiation and lipid biosynthesis pathways by inhibiting mTORC1 activity (26, 27). (c) Dephosphorylation of acetyl-CoA carboxylase (ACC) and activation of ATP citrate lyase (ACLY) phosphorylation lead to a constitutive increase in de novo lipogenesis (DNL) (28, 29). (d) Phosphodiesterase 3B (PDE3B) maintains lipid homeostasis and participates in the negative regulation of adipocyte lipolysis by inhibiting adipose triglyceride lipase (ATGL) and hormone-sensitive lipase (HSL) activities (30, 31).
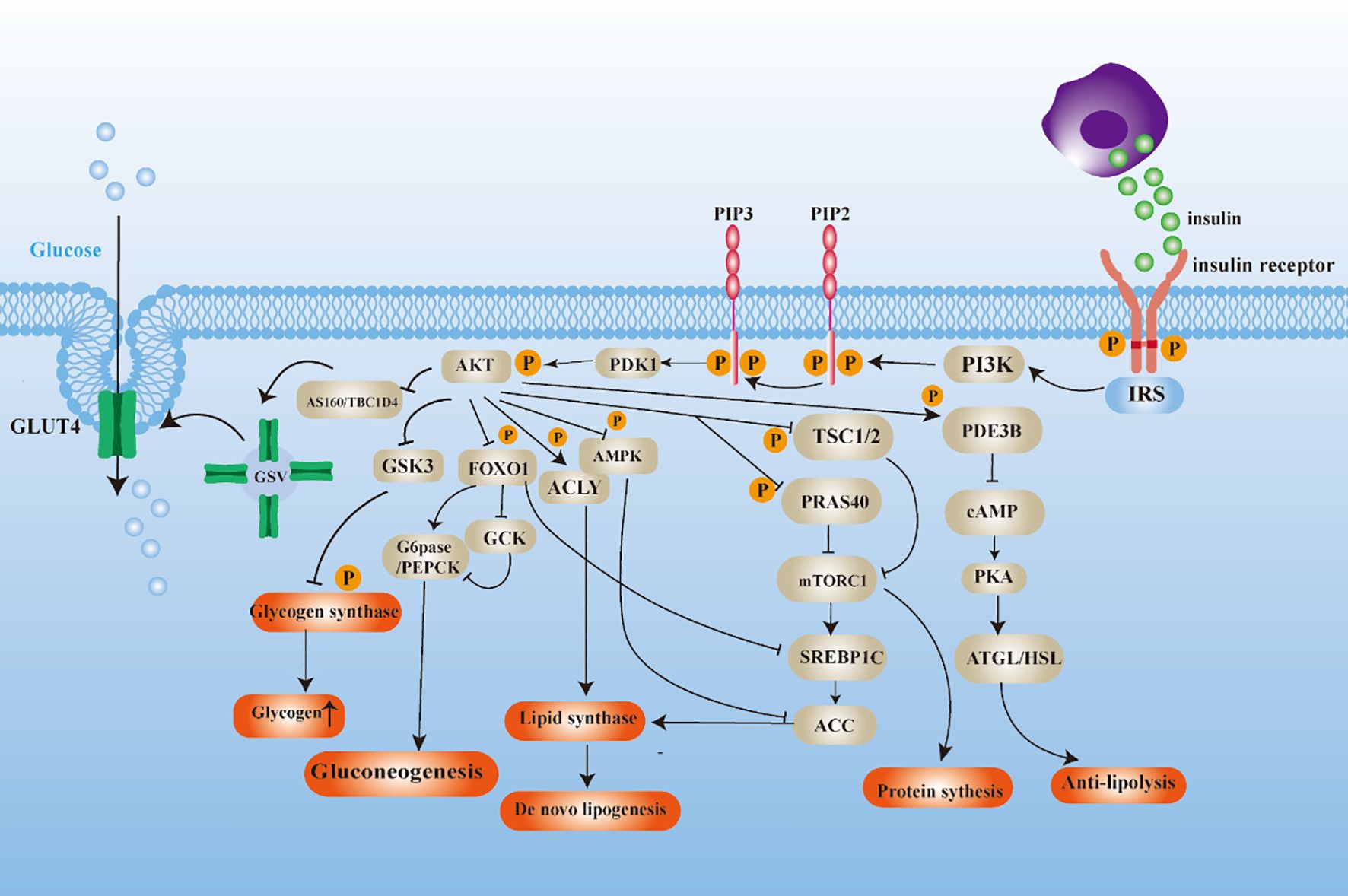
Figure 1. Diagram of insulin signaling mechanism.
In summary, insulin plays a pivotal role in the management of glucose and lipid balance. Following a meal, insulin secreted by pancreatic β-cells instigates anabolic programs while impeding catabolic pathways. In glucose metabolism, insulin stimulates glucose uptake in skeletal muscle, hepar, fatty tissue and other tissues, and accelerates glycogen and lipid synthesis. Furthermore, insulin effectively regulates glucose output from the liver by down-regulating gluconeogenic gene expression and inhibiting lipolysis, thereby maintaining energy balance in the body.
3.2 Mechanisms of insulin resistance
Insulin resistance is a complex pathophysiological process rooted in genetic susceptibility, initiated by core metabolic disturbances, and progressively amplified and sustained through cellular stress and systemic inflammation. Its underlying mechanisms can be systematically elaborated from the following three interrelated levels.
3.2.1 Core drivers: genetic susceptibility and ectopic lipid deposition
The development of insulin resistance (IR) is fundamentally grounded in genetic susceptibility. Clinical data from 220 Caucasian and 36 African American children and their parents revealed that children with at least one IR-affected parent exhibited significantly elevated insulin levels (32). Genetic background influences insulin sensitivity through multiple pathways, including: monogenic mutations (e.g., in AKT2 and INSR genes) that lead to severe insulin resistance phenotypes (33, 34); polygenic inheritance resulting from the cumulative small effects of multiple genes, such as PLA2G6 (which enhances sensitivity) and VGLL3 (which reduces sensitivity), identified through genome-wide association studies (35); and epigenetic modifications (e.g., DNA methylation) that regulate gene expression without altering the DNA sequence (36, 37).
Against this backdrop of genetic susceptibility, ectopic lipid deposition represents the most critical metabolic driver of insulin resistance. Although obesity is a well-established risk factor for type 2 diabetes mellitus (T2DM) (38), the crux of its pathophysiology lies in the abnormal accumulation of lipids in non-adipose tissues such as the liver and skeletal muscle (39–41). Studies have demonstrated a significant positive correlation between intrahepatic lipid content and the insulin resistance index (HOMA-IR), while weight loss interventions that reduce hepatic fat can effectively reverse hepatic insulin resistance and improve hyperglycemia (42). This lipotoxicity directly interferes with insulin signaling pathways, thereby constituting the initiating event in the development of insulin resistance.
3.2.2 Key nexus: cellular stress and dysfunction
Core metabolic disturbances triggered by ectopic lipid deposition and nutrient excess further activate cellular stress responses. These responses act as critical pathological hubs, closely linking upstream metabolic signals to downstream impairment of insulin signaling.
Mitochondrial Dysfunction and Oxidative Stress: Mitochondria are the primary source of superoxide and hydrogen peroxide (43). Excessive mitochondrial ROS production promotes a pro-oxidative shift in redox homeostasis, leading to disrupted redox signaling and oxidative damage, collectively termed oxidative stress (44), which contributes to the pathogenesis of insulin resistance and diabetes (45). A cross-sectional study involving overweight, obese, and normal-weight individuals demonstrated that oxidative stress levels were significantly positively correlated with the degree of insulin resistance, particularly in overweight and obese subjects. Specifically, oxidative stress markers such as total oxidant capacity (TOC) were positively correlated with the insulin resistance index (HOMA-IR), whereas total antioxidant capacity (TAC) showed a negative correlation with HOMA-IR (46).
Endoplasmic Reticulum Stress: The native structure of insulin is formed within the endoplasmic reticulum (ER); thus, proper maintenance of ER homeostasis is essential for the metabolic stability and function of pancreatic β-cells (47, 48). Studies using cell cultures and mouse models have revealed that obesity induces ER stress, which suppresses insulin receptor signaling through the overactivation of c-Jun N-terminal kinase (JNK) (49). Clinical research has further shown that serum ER stress markers are significantly elevated in patients with type 2 diabetes mellitus (T2DM) compared to healthy individuals, and their levels are positively correlated with the insulin resistance index (HOMA-IR) (50).
3.2.3 Systemic amplification: environmental factors and metaflammation
Cellular stress and dysfunction are not isolated events; rather, under the synergistic influence of adverse environmental factors, they are amplified into systemic metaflammation, ultimately forming a self-reinforcing vicious cycle.
3.2.3.1 Synergistic effects of environmental factors
Multiple environmental exposures can significantly exacerbate insulin resistance. For instance, Western dietary patterns—characterized by high consumption of red meat, sugar, and saturated fats—directly promote ectopic lipid deposition and inflammatory responses. In contrast, healthier dietary patterns such as the Mediterranean diet have demonstrated clear protective effects (42, 51, 52). Additionally, air pollution, including long-term exposure to particulate matter (e.g., PM2.5, PM10), is closely associated with the onset and progression of insulin resistance, with its adverse effects being partially mitigated by physical activity (53–55). Concurrently, stressors in the home, workplace, and community are linked to accelerated aging and alterations in metabolic and immune function (56). For example, a cohort study demonstrated an independent association between occupational stress and insulin resistance (57). Similarly, animal studies have revealed that stress increases both insulin secretion and insulin resistance (58).
3.2.3.2 Metaflammation
Metaflammation represents the core mechanism through which insulin resistance evolves from localized cellular damage into a systemic disease. Nutrient excess and adipocyte hypertrophy recruit and activate immune cells—such as macrophages and T cells—to infiltrate adipose tissue, where they secrete large quantities of pro-inflammatory cytokines (e.g., TNF-α, IL-1β, IL-6) (59, 60). This chronic, low-grade inflammatory state accelerates the spillover of lipids from adipose tissue into skeletal muscle and liver, leading to ectopic lipid deposition and insulin resistance in these tissues (61). Consequently, metaflammation acts not only as a downstream consequence of cellular stress but also as a key upstream driver that perpetuates and amplifies insulin resistance, establishing a difficult-to-break vicious cycle.
4 Insulin resistance and metabolic syndrome
The onset and progression of MetS is a multifactorial and complex process, in which IR plays a central role (3, 62), and there is a close correlation with other components of MetS.
4.1 Obesity
A vicious cycle has been established between IR and obesity, which are reciprocally causal. IR and its concomitant hyperinsulinemia trigger excessive fat storage, imbalanced energy metabolism, and disrupted neuroendocrine regulation. These factors act synergistically to promote the development and progression of obesity (63–65). A clinical study lends empirical support to this theory, demonstrating that specific, partial inhibition of insulin production is effective in alleviating high-fat diet-induced obesity (66). However, obesity itself has been shown to exacerbate IR (59), in which visceral adipose tissue plays a central role in the induction of IR (67), thereby consolidating a metabolic disorder that is difficult to reverse.
4.2 Hyperglycemia
Insulin acts a crucial regulatory part in the preservation of glucose homeostasis, including glucose uptake, gluconeogenesis and other physiological processes (68–70). However, in states of IR, the capacity of insulin to regulate blood glucose is markedly compromised (71). Specifically, impairment of the insulin signaling pathway hinders the translocation of glucose transporter proteins (e.g., GLUT4) to the cell membrane, which in turn reduces the efficiency of cellular glucose uptake (72). Furthermore, the liver’s increased production and secretion of glucose, prompted by IR, contributes to the exacerbation of blood glucose levels (73).
4.3 Abnormalities of lipid metabolism
In a state of IR, adipose tissue becomes less sensitive to insulin’s anti-lipolytic action, which consequently results in a heightened secretion of free fatty acids (FFA) (74). This increased influx of FFA into the liver and muscle tissue promotes triglyceride synthesis (75). A study conducted on non-obese men revealed that individuals with low insulin sensitivity and low lipocalin levels exhibited an elevated risk of developing fatty liver disease and dyslipidemia (76). A separate study further corroborates the notion that hepatic IR alone is sufficient to induce dyslipidemia, thereby accelerating the onset of atherosclerosis associated with the MetS (77). Conversely, abnormalities in lipid metabolism, especially the accumulation of intracellular triglycerides, activate novel protein kinase C, which impairs insulin signaling and has been implicated as one of the key mechanisms of IR (78).
4.4 Hypertension
IR impacts glucose and lipid metabolism, as well as the RAAS, which contributes to elevated vascular tension (79). A cross-sectional analysis indicated that Participants with prehypertension tend to have higher insulin levels and more significant IR compared to healthy individuals (80). In such cases, compensatory hyperinsulinemia in insulin-resistant individuals has been observed to intensify salt reabsorption in the renal tubules, leading to salt excess and increased vascular tension (81). Furthermore, excessive activation of RAAS has been demonstrated to exert inhibitory effects on insulin signaling, which may further exacerbate IR, thereby creating a vicious cycle (82).
In summary, IR occupies a central position in the pathophysiologic mechanisms of MetS. It exerts a substantial influence on the development of MetS components by broadly impacting numerous metabolic pathways and physiological processes. Moreover, it functions as a pivotal factor in perpetuating the metabolic disordered state. Consequently, the development of intervention strategies targeting IR is of particular importance, not only for the prevention of MetS but also for the treatment of MetS and its related diseases.
5 Association of metabolic syndrome with Alzheimer’s disease
5.1 Impact of metabolic syndrome on cognitive functioning
Several evidence indicates that metabolic syndrome (MetS) exerts a significant adverse impact on cognitive function. At the population level, a systematic review and dose-response meta-analysis of 30 studies provides robust support: 17 of these studies explicitly report that the presence of MetS accelerates cognitive decline, although a minority (2 studies) present opposing findings (83). A larger neuroimaging study involving over 37,000 participants further elucidates the potential neuropathological underpinnings, revealing significant associations between MetS and reduced brain volume, increased cerebrovascular pathology, and poorer performance across multiple cognitive domains (84). At the mechanistic level, animal model studies offer direct biological evidence. Research demonstrates that diet-induced MetS in animals not only leads to glucolipid metabolic disturbances but also results in significant impairments in learning and memory, accompanied by pathological alterations in the hippocampus (85, 86).
5.2 Epidemiologic studies of metabolic syndrome and Alzheimer’s disease risk
Multiple large-scale population studies have consistently demonstrated that metabolic syndrome and its individual components are significantly associated with an increased risk of dementia. Analyzing in depth a large dataset covering 466,788 individuals, the results of the study revealed a strong association between the MetS and its components with a markedly higher risk of dementia. Specifically, individuals with MetS faced a 25% increased risk of all-cause dementia, with a significant 50% increased risk of vascular dementia (VD) (87). In addition, a long-term follow-up study of nearly 200,000 elderly participants without dementia further confirmed that MetS was linked to a 12% elevated risk of dementia onset, and this risk association showed a more pronounced trend with longer follow-up (88). Another analysis based on large-scale health data from people aged 50 to 69 years in Korea also found that MetS and its early states were remarkably connected with an enhanced incidence of AD, with a 20% higher risk of developing AD in the early MetS group compared with the non-MetS group, and a further increase in risk to 39% in the MetS group (89). Collectively, these findings underscore the complexity of metabolic syndrome (MetS) as a risk factor for dementia. Not only does MetS independently influence Alzheimer’s disease (AD) and vascular dementia (VaD), but it may also play a central role in mixed dementia, where these two pathologies intersect. This critical area warrants further in-depth investigation in future research.
5.3 Direct and indirect associations between components of the metabolic syndrome and Alzheimer’s disease
5.3.1 Obesity and Alzheimer’s disease
Studies have shown that obesity in midlife increases the risk of developing AD independently of other factors. However, in the later stages of life, especially those obese individuals who are metabolically healthy may exhibit some protection against AD pathology (90). Obesity-induced chronic peripheral inflammation is not limited to other parts of the body, but may also spread to the brain, triggering a neuroinflammatory response, which in turn accelerates the course of cognitive impairment (91–93). In addition, obesity-induced leptin resistance further exacerbates Cognitive impairment and the advancement of AD (94). Nonetheless, existing studies present some complexity: some evidence suggests that obesity in midlife may play a role as a protective factor for AD, whereas midlife underweight status might contribute to an increased risk of dementia (95).
5.3.2 Hyperglycemia and Alzheimer’s disease
Cognitive dysfunction is increasingly being recognized by the academic community as an important comorbidity of diabetes (96). A long-term cohort analysis, upon correction for potential confounders like age, sex, and educational attainment, disclosed that diabetic patients had a 65% higher risk of developing AD compared to the non-diabetic group (97). Another cohort study further confirmed that the risk ratio (HR) for AD was significantly higher in diabetic patients, especially in the group of older diabetic women (98). In addition, the drugs traditionally used to treat type 2 diabetes showed activity in improving the cognitive health of patients with AD, which also confirmed the similarity of the pathogenesis of these two diseases (99). In recent years, with the advancing depth of research, there is more and more evidence that AD may be a brain-specific type of diabetes mellitus, the so-called “type 3 diabetes mellitus” (100, 101).
5.3.3 Dyslipidemia and Alzheimer’s disease
The connection between triglycerides and AD risk may be modulated by age, showing a complex nonlinear pattern. Specifically, higher triglyceride levels were markedly associated with an added risk of dementia in people under 60 years of age (102). However, in older populations, this relationship is reversed, and a reduced likelihood of AD has been associated with high triglyceride levels (102, 103). Furthermore, HDL can reduce the risk of AD due to its vasculoprotective function, which is mediated by mechanisms including increased Aβ clearance and induction of endothelial nitric oxide production (104). On the other hand, elevated cholesterol levels are regarded as a possible risk factor for the onset of AD (105). Cholesterol levels are generally higher in patients with AD compared to healthy individuals and are strongly associated with the accumulation of phosphorylated tau in the brain (106). Notably, an 8-year cohort study revealed an association between sustained statin use and a significantly lower risk of incident AD (107). However, there is also strong evidence that statin use is not effective in preventing memory disorders in the elderly at risk for vascular disease (108).
5.3.4 Hypertension and Alzheimer’s disease
Hypertension shows a positive correlation with neuropathological changes in Alzheimer’s disease, which is closely associated with an increase in plaques and neurofibrillary tangles in the brain, and has emerged as a significant contributing factor for the disease (109). Notably, hypertensive patients in the middle age period face a higher risk of cognitive impairment compared to the elderly population (110). Specifically, midlife stage 1 and stage 2 systolic hypertension are associated with an increased risk of AD of 18% and 25%, respectively (111). In response to this situation, the use of antihypertensive drugs is particularly important. Pertinent research has indicated that antihypertensive medications are efficacious in lowering the risk of AD among hypertensive individuals. To illustrate, a meta-analysis in a prospective cohort study revealed that compared with hypertensive people who did not use antihypertensive medication, those who regularly used antihypertensive medication had a 12% lower risk of dementia and a 16% lower risk of AD (112).
Overall, there is a clear link between components of the MetS and AD, with hyperglycemia having a particularly strong effect on AD. However, when assessing the impact of the MetS on AD risk, It is vital to take full account of the impact of age. In particular, when exploring the relationship between obesity dyslipidemia and AD, the influence of the age factor is particularly important and should not be ignored.
6 Insulin resistance and Alzheimer’s disease
6.1 Sources of insulin in the brain
Insulin levels in cerebrospinal fluid (CSF) are significantly lower than those in plasma, yet a strong correlation exists between the two, suggesting that brain insulin primarily originates from circulating pancreatic insulin (113). Insulin enters the central nervous system (CNS) via selective, saturable transport mechanisms across the blood-brain barrier (BBB) capillary endothelial cells (114, 115). Additionally, the choroid plexus—a key structure in CSF production—has been identified as another important source of insulin within the CNS (116). Recent studies have further detected insulin expression in the dorsal vagal complex (DVC) of the hindbrain (117), lending additional support to the possibility of local insulin synthesis in the brain.
6.2 Definition of brain insulin resistance
Brain insulin resistance is characterized by a diminished response to insulin signaling within the central nervous system (CNS), primarily involving neurons and/or glial cells. The underlying mechanisms include downregulation of insulin receptors, impaired insulin-receptor binding, or defective activation of insulin signaling cascades. At the cellular level, this dysfunction may manifest as impaired neuroplasticity, altered receptor modulation, or disrupted neurotransmitter release in neurons. Alternatively, it may directly impair insulin-dependent metabolic processes, such as glucose uptake or glycogen synthesis. Functionally, brain insulin resistance can present as dysregulation of central brain energy metabolism and peripheral glucolipid metabolism, or deficits in cognitive and emotional functions (118, 119).
6.3 Role of insulin signaling pathways in brain function
In the complex network of insulin signaling, phosphatidylinositol 3-kinase (PI3K) and protein kinase B (Akt) play indispensable roles as key kinases. They not only regulate neuronal plasticity and survival but also participate in neurotransmitter transport, which is essential for maintaining normal neural function (120, 121). In particular, brain areas closely connected with cognitive functions, such as the hippocampus, highly express insulin receptors, underscoring the importance of insulin in these regions (122, 123). The PI3K/Akt sequential response further influences various subsequent pathways, among them mTORC1, GSK3β, and the FoxO group of transcription factors, which are pivotal in in basic brain function (124). For instance, protein synthesis orchestrated by mTORC1 is pivotal for neural adaptability and the regulation of autophagic processes; however, misregulation of this process can precipitate neuronal apoptosis and the inception of neurodegenerative disorders (125–127). Furthermore, GSK3β is instrumental in governing various facets of neuronal activity, including neurogenesis and synaptic function (128, 129), and its ability to phosphorylate tau proteins and increase the amount of β-amyloid has been closely linked to the pathogenesis of AD (130, 131). FOXO3, a crucial component of the FOXO gene family, is extensively present in various organs and tissues of the human body. Changes in its protein expression level and post-translational modification status is crucial for upholding the stability of the body’s internal milieu and mitigating aging-related pathologies (132). In addition, insulin activates the MAPK cascade response, which regulates cell proliferation, differentiation, and apoptosis, and contributes to the maintenance of normal neuronal cell function and synaptic plasticity (133, 134). Collectively, these investigations indicate a profound and intricate association between the insulin signaling mechanism and the functionality of neurons and synapses. In summary, as described in the literature, insulin signaling is widely distributed throughout the brain and participates in multiple neurophysiological processes. It is closely associated with the functions of neurons, synapses, and neurotransmitters (135).
6.4 Insulin resistance in Alzheimer’s disease animal models
In the 3xTg-AD mouse model, Alzheimer’s disease neuropathology has been shown to impair insulin signaling pathways at the blood-brain barrier (BBB), specifically manifested as diminished activation of vascular insulin receptors (136). Further studies indicate that in this model, central nervous system insulin resistance emerges earlier and progresses more rapidly than peripheral insulin resistance (137). Notably, dietary restriction interventions can improve recognition deficits in 3xTg-AD mice. The underlying mechanism may involve reduced insulin secretion, which subsequently activates GSK-3β, thereby promoting hippocampal neuronal differentiation and maturation, ultimately contributing to the recovery of learning and memory functions (138). To more directly investigate the causal relationship between insulin resistance and AD, researchers developed the APP/IR-dKI double knockout mouse model through hybridization techniques. This model exhibits systemic insulin resistance without persistent hyperglycemia. Studies found that APP/IR-dKI mice display premature onset of cognitive dysfunction, providing compelling evidence for the hypothesis that “insulin resistance promotes cognitive impairment” (139). Additionally, in APP/PS1 transgenic mice, high-fat diets significantly accelerate cognitive deficits and AD-related pathology progression by inducing obesity and insulin resistance (140). Similarly, an insulin-resistant state has been observed in the brains of 5xFAD transgenic mice, with hippocampal levels of phosphorylated PI3K and phosphorylated AKT significantly lower than in wild-type controls (141). In chemically induced models, AD models established by intracerebroventricular injection of low-dose streptozotocin show significantly reduced expression of phosphorylated insulin receptors and phosphorylated Akt in the brain. Intranasal insulin therapy effectively ameliorates this insulin signaling impairment and associated cognitive deficits (142).
In summary, extensive animal model studies consistently demonstrate that Alzheimer’s disease neuropathology is frequently accompanied by brain insulin resistance, with substantial evidence supporting this association (143). However, despite this compelling evidence, establishing a definitive causal relationship between brain insulin resistance and AD neuropathology in animal models remains a formidable scientific challenge.
6.5 Regulation of insulin metabolism
The half-life of insulin in the human body is extremely short, only about 4–6 minutes, and its metabolic homeostasis is mainly regulated by the insulin-degrading enzyme (IDE) (144). Notably, amyloid-beta (Aβ) and insulin are both substrates of IDE, and when IDE dysfunction occurs, it not only leads to hyperinsulinemia and glucose intolerance but also triggers an abnormal accumulation of endogenous Aβ in the brain (145), which in turn affects normal neurological function.
6.6 Cell-type-specific effects of insulin resistance in the brain
6.6.1 Neurons
Insulin resistance leads to a significantly reduced responsiveness of neurons to insulin, manifested as mitochondrial dysfunction, decreased glucose metabolism, and elevated lactate levels. This metabolic abnormality prompts neurons to shift toward glycolysis for energy production, resulting in compensatory metabolic reprogramming. In the pathological progression of Alzheimer’s disease (AD), insulin resistance not only impairs neuronal insulin signaling but also exacerbates metabolic disturbances, thereby accelerating disease advancement (146). In vitro experiments further demonstrate that insulin resistance markedly compromises neuronal metabolic efficiency, leading to increased oxidative stress, reduced neuronal activity, and a decline in dopaminergic neuron count (147). Additionally, insulin resistance induces synaptic insulin resistance through a ubiquitination-dependent mechanism of synaptic protein degradation, which in turn impairs synaptic plasticity and cognitive function (148).
6.6.2 Microglia
Insulin resistance impairs the metabolic homeostasis and immunoregulatory functions of microglia. Studies have demonstrated that insulin signaling in microglia is essential for maintaining metabolic balance and immune regulation, whereas insulin resistance disrupts these critical functions, leading to reduced Aβ clearance and enhanced neuroinflammation. Additionally, insulin resistance induces metabolic reprogramming in microglia, characterized by increased glycolysis (149). In high-fat diet (HFD)-induced models of insulin resistance, microglial autophagy is significantly suppressed, indicating that insulin resistance exacerbates neurodegenerative pathology by interfering with cellular clearance mechanisms (150). Collectively, these findings reveal that insulin resistance affects microglial function through multiple pathways, thereby playing a pivotal role in the pathogenesis of Alzheimer’s disease.
6.6.3 Astrocytes
Astrocytes are essential for brain energy metabolism and exhibit a loss of homeostatic functions in Alzheimer’s disease (AD), which may contribute to neurodegeneration. Insulin resistance leads to impaired glucose uptake and reduced glycogen synthesis in astrocytes, thereby disrupting brain energy homeostasis (119). Furthermore, insulin resistance interferes with mitochondrial metabolism in astrocytes, manifested as abnormal mitochondrial responses to glucose. This dysfunction may result in neurovascular coupling impairment, further compromising the coordination between cerebral blood flow and glucose metabolism (151).
6.6.4 Endothelial cells
Endothelial cells in the brain are critical components of the blood-brain barrier (BBB), playing a central role in maintaining brain microenvironmental homeostasis, regulating substance transport, and controlling cerebral blood flow (152, 153). Damage to endothelial cells results in the loss of tight junctions and increased barrier permeability, leading to insufficient cerebral blood perfusion, microhemorrhages, and infiltration of neurotoxic substances (154). Such damage can manifest as early as the initial stages of Alzheimer’s disease (AD) (155). Insulin resistance affects the unique properties of brain endothelial cells, rendering them more susceptible to oxidative stress. While moderate oxidative stress is necessary for insulin signaling, excessive oxidative stress exerts detrimental effects (156).
In summary, insulin resistance comprehensively impairs brain cell function through the synergistic actions of multiple cell types and pathways—including metabolic reprogramming, immune dysregulation, barrier disruption, and proteostasis imbalance—thereby establishing a core pathological foundation for AD pathogenesis. Targeting cell-specific insulin signaling pathways may represent a key strategy for intervening in AD progression.
6.7 Possible mechanisms by which insulin resistance affects Alzheimer’s disease
6.7.1 Increase in pathologic products
Abnormal aggregation of Aβ and Tau proteins has become a typical pathological hallmark of AD (157–159). Insulin plays a key role in regulating amyloid precursor protein (APP) metabolism by promoting its degradation by the non-amyloid pathway, a process that involves both Gsk-3β-dependent and non-dependent mechanisms (160). However, IR is closely associated with amyloidosis and aberrant phosphorylation of Tau proteins in rodent and human brains (161). Research suggests a substantial association between heightened insulin resistance levels and augmented Pittsburgh compound B (PiB)uptake in the frontal and temporal regions, reflecting an increase in amyloid deposition (162). Further evidence suggests that IR may precede the onset of Aβ lesions, and a 15-year-long follow-up study revealed a significant association between midlife IR and increased cerebral amyloid load in later life (20). Furthermore, IR is not only associated with cognitive decline but also positively correlated with the accumulation of Tau biomarkers in cerebrospinal fluid (CSF) (163). Mechanistically, insulin affects Tau protein metabolism by regulating GSK3β activity, which in turn affects Tau protein levels (164). Notably, in IR states, peripheral hyperinsulinemia significantly inhibits insulin-degrading enzyme (IDE) activity, leading to an abnormal accumulation of Aβ oligomers in the brain, which further exacerbates the pathological process of AD (165).
6.7.2 Blood-brain barrier dysfunction
Blood-brain barrier (BBB) dysfunction is increasingly recognized as a critical factor in the pathophysiology of Alzheimer’s disease (AD), particularly during the early stages of the disease (166, 167). Insulin resistance, which is prevalent in AD patients, is closely associated with impaired BBB integrity. Studies have demonstrated a synergistic interaction between insulin resistance and APOE genotype, jointly influencing BBB permeability, which may represent a key component in AD pathogenesis (168). Conversely, functional deficits in insulin receptors (INSR) at the BBB are correlated with amyloid-β pathology, significantly contributing to cerebral insulin resistance in AD (136). These findings suggest that BBB dysfunction not only serves as an early biomarker of AD but may also play a pivotal role in disease progression by disrupting cerebral insulin signaling and amyloid-β metabolism.
6.7.3 Synaptic plasticity impairment
IR causes significant damage to synaptic plasticity and integrity. Obust evidence supporting this notion is furnished by the research conducted by Claudia A Grillo and colleagues, who adeptly constructed a model of hippocampus-specific insulin resistance. Their model efficiently attenuated insulin receptor expression within the rat hippocampus, leading to a notable decline in hippocampal neuroplasticity (169). In addition, it has been shown that insulin receptor signaling plays a key role in maintaining synaptic density, and once this signaling is impaired, it leads to the loss of synapses, a phenomenon that fully illustrates the crucial role of insulin in synapse development and maintenance (170).
6.7.4 Neuroinflammation
The persistence of neuroinflammation, one of the key pathological mechanisms of AD, has been shown to lead to neuronal damage, and synaptic dysfunction, and further exacerbate pathological changes in the brain (171, 172). In-depth studies revealed a substantial direct association between microglial activation and AD-related pathological products, a finding that clarifies the strong link between neuroinflammation and the severity of AD (173). By simulating the state of hyperinsulinemia through in vitro experiments, researchers treated primary cultured microglia and BV2 cell lines with insulin and found that hyperinsulinemia not only stimulated microglia proliferation and drove them to M1-type polarization by promoting the production of pro-inflammatory factors, but also caused significant impairment of the membrane translocation function of GLUT4 (174). In addition, aberrant activation of neuroinflammation in the context of IR has been shown to further accelerate the progression of AD (175).
6.7.5 Renin-angiotensin-aldosterone activation
IR activates RAAS, and excessive activity of this system in the brain is closely linked to the initiation and advancement of AD (176, 177). Studies have shown that angiotensin-converting enzyme (ACE) levels in cerebrospinal fluid are elevated among individuals affected by AD, a finding that further supports the important role of RAAS in AD pathology (178). In addition, patients taking angiotensin receptor blockers (ARBs) showed age-related reductions in cerebrospinal fluid Aβ levels, suggesting a potential role for ARBs in modulating markers of AD-related pathology (179). Strikingly, however, an independent study drew conclusions that differed significantly from the previous findings, demonstrating that cerebrospinal fluid and serum protein levels and activity of ACE tended to be reduced in patients with AD compared to controls (180). This paradoxical result not only reveals the complexity and diversity of the RAAS in the pathomechanism of AD but also implies the necessity of in-depth investigation of the mechanism of action of RAAS in AD and its potential therapeutic targets.
6.7.6 Oxidative stress
In the pathological context of IR, the levels of oxidative damage markers within the cerebral cortex are significantly elevated, a change that reveals an enhanced oxidative stress response (181). Of particular interest is the fact that the brain exhibits an extremely high sensitivity to free radical attacks compared to other body organs (182). Accordingly, the lesions presented in the brains of individuals with AD, such as DNA damage, protein oxidation, lipid peroxidation, and accumulation of advanced glycosylation end products, are closely related to free radical attack (183). In addition, antioxidants exhibit potential value in the treatment of AD by effectively reducing the generation of reactive oxygen species (184, 185).
Although significant progress has been made in elucidating individual pathological mechanisms in Alzheimer’s disease (AD), these studies often fail to fully account for the complexity and heterogeneity of its clinical manifestations, highlighting the limitations of a single-target approach.
For example, the correlation between amyloid-β (Aβ) pathology and cognitive decline is not always direct; some Aβ-positive individuals remain cognitively normal for extended periods, suggesting that Aβ deposition alone is insufficient to explain clinical heterogeneity (186). Moreover, therapeutic strategies targeting Aβ or tau protein in isolation have not yielded ideal outcomes in clinical trials (187, 188). Neuroinflammation is widely recognized as a key pathological feature of AD, yet its precise role remains incompletely understood. In particular, whether neuroinflammation acts as a primary driver or a secondary phenomenon in AD pathogenesis continues to be debated (189). Neuroinflammation exhibits a “double-edged sword” characteristic in AD: while moderate inflammatory responses may be protective, excessive suppression could be detrimental. For instance, microglial activation not only aids in Aβ clearance but may also exacerbate neuronal damage (190). The association between the renin-angiotensin-aldosterone system (RAAS) and AD pathology has been frequently proposed, but its underlying molecular mechanisms are not yet fully clarified. Some studies suggest that RAAS-targeting drugs (e.g., ARBs) may reduce AD risk (191, 192), while others report inconsistent findings. For example, although a clinical trial of losartan demonstrated reduced brain volume loss, it did not conclusively prove cognitive benefits (193). Furthermore, while oxidative damage is closely associated with Alzheimer’s disease (AD), antioxidant therapies have failed to achieve significant clinical efficacy, reflecting the complexity of oxidative stress in AD pathogenesis (194). Moreover, the causal relationships between oxidative stress and other pathological features—such as amyloid-β (Aβ) deposition and tau hyperphosphorylation—remain controversial. It remains challenging to determine whether oxidative stress acts as a primary event or a secondary phenomenon in the disease process.
Consequently, as noted by other researchers, AD remains a poorly understood disease with a complex, multifactorial pathogenesis (195). In this context, insulin resistance offers a theoretical bridge that closely links multiple mechanisms, including neuroinflammation, oxidative stress, and RAAS activation. Adopting this integrative perspective may deepen our understanding of AD pathophysiology, provide a more comprehensive view of the disease, and ultimately inform the development of more effective prevention and treatment strategies.
7 Potential therapeutic strategies
7.1 Pharmacologic interventions for insulin resistance
7.1.1 GLP-1 receptor agonists
Glucagon-like peptide-1 (GLP-1) receptor agonists are standard therapies for type 2 diabetes and obesity, lowering blood glucose and body weight by promoting insulin secretion, inhibiting glucagon release, and inducing satiety via central nervous system actions (196). In neuroprotection, liraglutide (a GLP-1 receptor agonist) shows therapeutic potential. Six months of treatment significantly enhances blood-brain barrier glucose transport, improves cerebral glucose metabolism, and reverses AD-related brain glucose transport abnormalities, supporting its role in brain energy metabolism and AD therapy (197). Additionally, 12 weeks of treatment improves brain connectivity in high-risk AD individuals (198). However, a systematic review notes that while GLP-1 receptor agonists offer metabolic and neuroprotective benefits, they do not significantly alter amyloid-β or tau biomarkers or improve cognitive function (199). Overall, liraglutide holds promise for cognitive function and neuroprotection, but its definitive efficacy and mechanisms require further validation through high-quality studies.
7.1.2 Intranasal insulin
Intranasal insulin administration is a non-invasive delivery method that directly transports insulin to the brain via the nasal mucosa, effectively bypassing the blood-brain barrier. This approach has shown potential value in treating neurological disorders such as Alzheimer’s disease (AD). One study reported that, compared to the placebo group, patients receiving conventional insulin therapy exhibited significant memory improvements at 2 and 4 months, with statistically significant differences (p < 0.03) (200). However, not all studies have observed similar cognitive benefits. For instance, a 12-month randomized controlled trial (RCT) demonstrated that intranasal insulin treatment did not lead to significant cognitive improvements in patients with mild cognitive impairment (MCI) or Alzheimer’s disease (201). Furthermore, a systematic review of seven RCTs involving patients with AD or MCI indicated that intranasal insulin therapy only modestly improved story recall performance in APOE4-negative (APOE4[-]) individuals, while its effects on other cognitive domains remained limited (202).
7.1.3 Metformin
Metformin, a drug widely used in the antidiabetic field, has received much attention in recent years for its potential role in the therapy of AD. A meta-analysis showed that long-term and high-dose use of metformin was significantly associated with a reduced risk of developing AD in elderly diabetic patients (203). It should be noted, however, that the results of several studies have shown that metformin treatment did not result in the expected significant improvement in patients’ cognitive performance, and some studies have suggested that it may increase the risk of developing AD (204–206). In particular, a meta-analysis of 10 studies found that metformin treatment may adversely affect the risk of developing AD in Asian populations (206). These conflicting findings suggest that the administration of metformin for AD should be evaluated more cautiously, taking into account the potential differences in different populations and the heterogeneity of their responses to the drug.
7.1.4 PPAR-γ agonists
Activation of PPAR-γ receptors plays multiple roles in regulating a wide range of biological processes, including lipid metabolism, inflammatory responses, and neuroprotection (207). Studies have shown that PPAR-γ agonists significantly improve spatial learning and memory in animal models, demonstrating their great potential in the therapeutic field of AD (208, 209). In particular, pioglitazone, a widely used PPAR-γ agonist, has shown potential benefits for AD patients in systematic evaluations, although these preliminary findings require further validation to establish their value for clinical application (210).
7.2 Lifestyle interventions
Studies have shown that limiting net carbohydrate intake to 130 grams per day may be effective in slowing the rate of cortical atrophy and contribute to lower insulin levels (211). Furthermore, in overweight and obese populations, regular exercise has shown significant effects in restoring insulin sensitivity in the brain (212). More importantly, comprehensive lifestyle Interventions markedly improve cognitive function in individuals with MCI or early AD, and may slow disease progression by improving IR and lowering insulin levels (213).
8 Conclusion
This review focuses on the central mechanism of IR in the association between MetS and AD. By systematically combing and analyzing the research findings of recent years, we demonstrate that IR plays a key bridging role between MetS and AD (Figure 2). On the one hand, there is a close association between IR and the components of the MetS; on the other hand, IR is a key factor in the pathogenesis of AD. However, current research exhibits significant limitations in elucidating this complex association. For instance, most studies overlook the impact of sex differences on the pathogenesis of metabolic syndrome (MetS) and Alzheimer’s disease (AD). As documented in the literature, women with MetS are more susceptible to AD than men (18), a disparity potentially amplified by insulin resistance-related MetS, as supported by previous findings (214, 215). Additionally, existing studies often treat mechanisms such as neuroinflammation, oxidative stress, overactivation of the renin-angiotensin-aldosterone system (RAAS), and mitochondrial dysfunction as independent factors, lacking in-depth critical analysis and cross-mechanistic integration. This “mechanism-listing” approach fails to adequately elucidate how these pathways dynamically interact, mutually amplify, and collectively drive neurodegeneration. For example, insulin resistance can directly induce central neuroinflammation, while inflammatory cytokines (e.g., TNF-α, IL-1β) released by activated glial cells further exacerbate peripheral and central insulin signaling impairment, creating a vicious cycle. The existence of such “crosstalk” and “positive feedback loops” among mechanisms implies that therapeutic strategies targeting a single pathway may yield limited efficacy or even be counteracted by compensatory activation of other pathways. Nevertheless, it is undeniable that the importance of IR in the pathogenesis of AD should not be underestimated, and considering it as a promising candidate for the intervention and management of AD holds great and far-reaching significance. Therefore, future studies should focus on the development of multidimensional intervention strategies that focus on the optimization of IR, to deeply analyze the complex pathological process of AD and further promote its application and development in the treatment of neurodegenerative diseases.
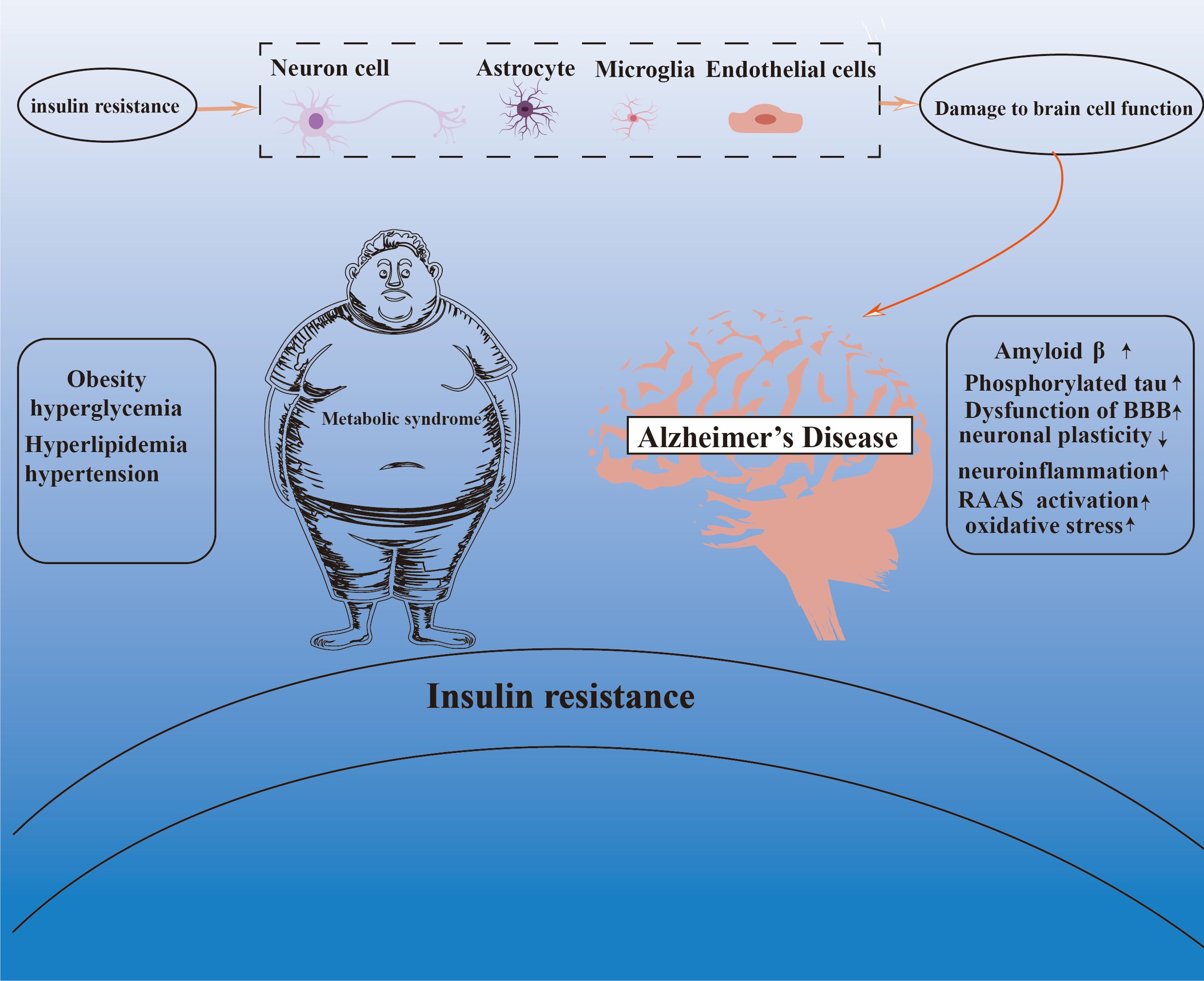
Figure 2. The central role of insulin resistance in the Mets-AD connection.
Author contributions
SW: Visualization, Conceptualization, Validation, Supervision, Writing – original draft, Writing – review & editing. YS: Data curation, Writing – original draft, Writing – review & editing. RX: Software, Resources, Writing – original draft, Supervision, Project administration. HK: Investigation, Writing – review & editing, Formal analysis, Data curation, Methodology. HX: Writing – original draft, Project administration, Resources. JR: Visualization, Conceptualization, Writing – review & editing, Writing – original draft.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was supported by the National Famous Elderly Traditional Chinese Medicine Inheritance Workshop Project; under Grant [(JCMD (2022) No. 48)]; 2022 State Administration of Traditional Chinese Medicine Qihuang Scholars Program under Grant [(National TCM Human Education Letter (2022) No. 256)]; and Jilin Science and Technology Development Program Project under Grant ((YDZJ202301ZYTS184)).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Petersen MC and Shulman GI. Mechanisms of insulin action and insulin resistance. Physiol Rev. (2018) 98:2133–223. doi: 10.1152/physrev.00063.2017
2. Kang S, Tsai LT-Y, and Rosen ED. Nuclear mechanisms of insulin resistance. Trends Cell Biol. (2016) 26:341–51. doi: 10.1016/j.tcb.2016.01.002
3. Zhao X, An X, Yang C, Sun W, Ji H, and Lian F. The crucial role and mechanism of insulin resistance in metabolic disease. Front Endocrinol (Lausanne). (2023) 14:1149239. doi: 10.3389/fendo.2023.1149239
4. Neeland IJ, Lim S, Tchernof A, Gastaldelli A, Rangaswami J, Ndumele CE, et al. Metabolic syndrome. Nat Rev Dis Primers. (2024) 10:77. doi: 10.1038/s41572-024-00563-5
5. Liang X, Or B, Tsoi MF, Cheung CL, and Cheung BMY. Prevalence of metabolic syndrome in the United States National Health and Nutrition Examination Survey 2011-18. Postgrad Med J. (2023) 99:985–92. doi: 10.1093/postmj/qgad008
6. Lu J, Wang L, Li M, Xu Y, Jiang Y, Wang W, et al. Metabolic syndrome among adults in China: the 2010 China noncommunicable disease surveillance. J Clin Endocrinol Metab. (2017) 102:507–15. doi: 10.1210/jc2016-2477
7. Saklayen MG. The global epidemic of the metabolic syndrome. Curr Hypertens Rep. (2018) 20:12. doi: 10.1007/s11906-018-0812-z
8. Grundy SM. Metabolic syndrome update. Trends Cardiovasc Med. (2016) 26:364–73. doi: 10.1016/j.tcm.2015.10.004
9. Zhang F, Liu L, Zhang C, Ji S, Mei Z, and Li T. Association of metabolic syndrome and its components with risk of stroke recurrence and mortality: A meta-analysis. Neurology. (2021) 97:e695–705. doi: 10.1212/WNL.0000000000012415
10. Gurka MJ, Guo Y, Filipp SL, and DeBoer MD. Metabolic syndrome severity is significantly associated with future coronary heart disease in Type 2 diabetes. Cardiovasc Diabetol. (2018) 17:17. doi: 10.1186/s12933-017-0647-y
11. DeBoer MD, Filipp SL, Sims M, Musani SK, and Gurka MJ. Risk of ischemic stroke increases over the spectrum of metabolic syndrome severity. Stroke. (2020) 51:2548–52. doi: 10.1161/STROKEAHA.120.028944
12. Kamatham PT, Shukla R, Khatri DK, and Vora LK. Pathogenesis, diagnostics, and therapeutics for Alzheimer's disease: Breaking the memory barrier. Ageing Res Rev. (2024) 20:102481. doi: 10.1016/j.arr.2024.102481
13. Congdon EE, Ji C, Tetlow AM, Jiang Y, and Sigurdsson EM. Tau-targeting therapies for Alzheimer disease: current status and future directions. Nat Rev Neurol. (2023) 19:715–36. doi: 10.1038/s41582-023-00883-2
14. Ezkurdia A, Ramírez MJ, and Solas M. Metabolic syndrome as a risk factor for Alzheimer’s disease: A focus on insulin resistance. Int J Mol Sci. (2023) 24:4354. doi: 10.3390/ijms24054354
15. Al-Kuraishy HM, Jabir MS, Albuhadily AK, Al-Gareeb AI, and Rafeeq MF. The link between metabolic syndrome and Alzheimer disease: A mutual relationship and long rigorous investigation. Ageing Res Rev. (2023) 91:102084. doi: 10.1016/j.arr.2023.102084
16. Navalón-Monllor V, Soriano-Romaní L, Silva M, de Las Hazas M-CL, Hernando-Quintana N, Suárez Diéguez T, et al. Microbiota dysbiosis caused by dietetic patterns as a promoter of Alzheimer’s disease through metabolic syndrome mechanisms. Food Funct. (2023) 14:7317–34. doi: 10.1039/d3fo01257c
17. Więckowska-Gacek A, Mietelska-Porowska A, Wydrych M, and Wojda U. Western diet as a trigger of Alzheimer’s disease: From metabolic syndrome and systemic inflammation to neuroinflammation and neurodegeneration. Ageing Res Rev. (2021) 70:101397. doi: 10.1016/j.arr.2021.101397
18. Vanhanen M, Koivisto K, Moilanen L, Helkala EL, Hänninen T, Soininen H, et al. Association of metabolic syndrome with Alzheimer disease: a population-based study. Neurology. (2006) 67:843–7. doi: 10.1212/01.wnl.0000234037.91185.99
19. Amin AM, Mostafa H, and Khojah HMJ. Insulin resistance in Alzheimer’s disease: The genetics and metabolomics links. Clin Chim Acta. (2023) 539:215–36. doi: 10.1016/j.cca.2022.12.016
20. Ekblad LL, Johansson J, Helin S, Viitanen M, Laine H, Puukka P, et al. Midlife insulin resistance, APOE genotype, and late-life brain amyloid accumulation. Neurology. (2018) 90:e1150–7. doi: 10.1212/WNL.0000000000005214
21. Pietilä E, Snellman A, Tuisku J, Helin S, Viitanen M, Jula A, et al. Midlife insulin resistance, APOE genotype, and change in late-life brain beta-amyloid accumulation - A 5-year follow-up [11C]PIB-PET study. Neurobiol Dis. (2024) 190:106385. doi: 10.1016/j.nbd.2023.106385
22. Haeusler RA, McGraw TE, and Accili D. Biochemical and cellular properties of insulin receptor signalling. Nat Rev Mol Cell Biol. (2018) 19:31–44. doi: 10.1038/nrm.2017.89
23. Beurel E, Grieco SF, and Jope RS. Glycogen synthase kinase-3 (GSK3): regulation, actions, and diseases. Pharmacol Ther. (2015) 148:114–31. doi: 10.1016/j.pharmthera.2014.11.016
24. Puigserver P, Rhee J, Donovan J, Walkey CJ, Yoon JC, Oriente F, et al. Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1alpha interaction. Nature. (2003) 423:550–5. doi: 10.1038/nature01667
25. Wu F, Lu F, Dong H, Hu M, Xu L, and Wang D. Oxyberberine inhibits hepatic gluconeogenesis via AMPK-mediated suppression of FoxO1 and CRTC2 signaling axes. Phytother Res. (2024) 83:153487. doi: 10.1002/ptr.8381
26. Buel GR, Dang HQ, Asara JM, Blenis J, and Mutvei AP. Prolonged deprivation of arginine or leucine induces PI3K/Akt-dependent reactivation of mTORC1. J Biol Chem. (2022) 298:102030. doi: 10.1016/j.jbc.2022.102030
27. Garami A, Zwartkruis FJT, Nobukuni T, Joaquin M, Roccio M, Stocker H, et al. Insulin activation of rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol Cell 11(6):1457–66. doi: 10.1016/s1097-2765(03)00220-x
28. Han Y, Hu Z, Cui A, Liu Z, Ma F, Xue Y, et al. Post-translational regulation of lipogenesis via AMPK-dependent phosphorylation of insulin-induced gene. Nat Commun. (2019) 10:623. doi: 10.1038/s41467-019-08585-4
29. Martinez Calejman C, Trefely S, Entwisle SW, Luciano A, Jung SM, Hsiao W, et al. mTORC2-AKT signaling to ATP-citrate lyase drives brown adipogenesis and de novo lipogenesis. Nat Commun. (2020) 11:575. doi: 10.1038/s41467-020-14430-w
30. Zhao W, Li A, Feng X, Hou T, Liu K, Liu B, et al. Metformin and resveratrol ameliorate muscle insulin resistance through preventing lipolysis and inflammation in hypoxic adipose tissue. Cell Signal. (2016) 28:1401–11. doi: 10.1016/j.cellsig.2016.06.018
31. Seo DH, Shin E, Lee Y-H, Park S-E, Nam KT, Kim J-W, et al. Effects of a phosphodiesterase inhibitor on the browning of adipose tissue in mice. Biomedicines. (2022) 10:1852. doi: 10.3390/biomedicines10081852
32. Pankow JS, Jacobs DR, Steinberger J, Moran A, and Sinaiko AR. Insulin resistance and cardiovascular disease risk factors in children of parents with the insulin resistance (Metabolic) syndrome. Diabetes Care. (2004) 27:775–80. doi: 10.2337/diacare.27.3.775
33. George S, Rochford JJ, Wolfrum C, Gray SL, Schinner S, Wilson JC, et al. A family with severe insulin resistance and diabetes due to a mutation in AKT2. Science. (2004) 304:1325–8. doi: 10.1126/science.1096706
34. Hacıhamdioğlu B, Baş EG, and Delil K. Homozygous mutation in the insulin receptor gene associated with mild type A insulin resistance syndrome: A case report. J Clin Res Pediatr Endocrinol. (2020) 13:100–3. doi: 10.4274/jcrpe.galenos.2020.2019.0213
35. DeForest N, Wang Y, Zhu Z, Dron JS, Koesterer R, Natarajan P, et al. Genome-wide discovery and integrative genomic characterization of insulin resistance loci using serum triglycerides to HDL-cholesterol ratio as a proxy. Nat Commun. (2024) 15:8068. doi: 10.1038/s41467-024-52105-y
36. Sharma NK, Comeau ME, Montoya D, Pellegrini M, Howard TD, Langefeld CD, et al. Integrative analysis of glucometabolic traits, adipose tissue DNA methylation, and gene expression identifies epigenetic regulatory mechanisms of insulin resistance and obesity in African Americans. Diabetes. (2020) 69:2779–93. doi: 10.2337/db20-0117
37. Krause C, Geißler C, Tackenberg H, El Gammal AT, Wolter S, Spranger J, et al. Multi-layered epigenetic regulation of IRS2 expression in the liver of obese individuals with type 2 diabetes. Diabetologia. (2020) 63:2182–93. doi: 10.1007/s00125-020-05212-6
38. Wang X, Wu Y, Wang Y, Zhou J, and Liu T. Relationship between metabolically healthy overweight/obesity and risk of type 2 diabetes in different ethnicity: a prospective cohort study in southwest China. BMC Public Health. (2024) 24:2798. doi: 10.1186/s12889-024-20254-w
39. Samuel VT and Shulman GI. Integrating mechanisms for insulin resistance: common threads and missing links. Cell. (2012) 148:852–71. doi: 10.1016/j.cell.2012.02.017
40. Park SS and Seo Y-K. Excess accumulation of lipid impairs insulin sensitivity in skeletal muscle. Int J Mol Sci. (2020) 21:1949. doi: 10.3390/ijms21061949
41. Larson-Meyer DE, Newcomer BR, Ravussin E, Volaufova J, Bennett B, Chalew S, et al. Intrahepatic and intramyocellular lipids are determinants of insulin resistance in prepubertal children. Diabetologia. (2011) 54:869–75. doi: 10.1007/s00125-010-2022-3
42. Barboza BP, Bricarello LP, Alves M de A, Tureck C, Retondario A, Longo GZ, et al. Dietary patterns and biochemical markers related to diabetes mellitus: an association analysis based on data from the Study of Cardiovascular Risk in Adolescents (ERICA). Nutrition. (2024) 118:112283. doi: 10.1016/j.nut.2023.112283
43. Shadel GS and Horvath TL. Mitochondrial ROS signaling in organismal homeostasis. Cell. (2015) 163:560–9. doi: 10.1016/j.cell.2015.10.001
44. Sies H, Berndt C, and Jones DP. Oxidative stress. Annu Rev Biochem. (2017) 86:715–48. doi: 10.1146/annurev-biochem-061516-045037
45. Kasai S, Kokubu D, Mizukami H, and Itoh K. Mitochondrial reactive oxygen species, insulin resistance, and Nrf2-mediated oxidative stress response-toward an actionable strategy for anti-aging. Biomolecules. (2023) 13:1544. doi: 10.3390/biom13101544
46. Kościuszko M, Buczyńska A, Łuka K, Duraj E, Żuk-Czerniawska K, Adamska A, et al. Assessing the impact of body composition, metabolic and oxidative stress parameters on insulin resistance as a prognostic marker for reactive hypoglycemia: a cross-sectional study in overweight, obese, and normal weight individuals. Front Pharmacol. (2024) 15:1329802. doi: 10.3389/fphar.2024.1329802
47. Lee J-H and Lee J. Endoplasmic reticulum (ER) stress and its role in pancreatic β-cell dysfunction and senescence in type 2 diabetes. Int J Mol Sci. (2022) 23:4843. doi: 10.3390/ijms23094843
48. Meyerovich K, Ortis F, Allagnat F, and Cardozo AK. Endoplasmic reticulum stress and the unfolded protein response in pancreatic islet inflammation. J Mol Endocrinol. (2016) 57:R1–R17. doi: 10.1530/JME-15-0306
49. Ozcan U, Cao Q, Yilmaz E, Lee A-H, Iwakoshi NN, Ozdelen E, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. (2004) 306:457–61. doi: 10.1126/science.1103160
50. Nourbakhsh M, Sharifi R, Heydari N, Nourbakhsh M, Ezzati-Mobasser S, and Zarrinnahad H. Circulating TRB3 and GRP78 levels in type 2 diabetes patients: crosstalk between glucose homeostasis and endoplasmic reticulum stress. J Endocrinol Invest. (2022) 45:649–55. doi: 10.1007/s40618-021-01683-5
51. Gower BA, Pearson K, Bush N, Shikany JM, Howard VJ, Cohen CW, et al. Diet pattern may affect fasting insulin in a large sample of black and white adults. Eur J Clin Nutr. (2021) 75:628–35. doi: 10.1038/s41430-020-00762-9
52. Yin X, Chen Y, Lu W, Jin T, and Li L. Association of dietary patterns with the newly diagnosed diabetes mellitus and central obesity: a community based cross-sectional study. Nutr Diabetes. (2020) 10:16. doi: 10.1038/s41387-020-0120-y
53. Gong X, Wang S, Wang X, Zhong S, Yuan J, Zhong Y, et al. Long-term exposure to air pollution and risk of insulin resistance: A systematic review and meta-analysis. Ecotoxicology Environ Saf. (2024) 271:115909. doi: 10.1016/j.ecoenv.2023.115909
54. Park H, Kim SY, Jang H, Ha YW, Yun YM, Kim KJ, et al. Impact of physical activity levels on the association between air pollution exposures and glycemic indicators in older individuals. Environ Health. (2024) 23:87. doi: 10.1186/s12940-024-01125-8
55. Jiménez-Chávez A, Morales-Rubio R, Sánchez-Gasca E, Rivera-Rosas M, Uribe-Ramírez M, Amador-Muñoz O, et al. Subchronic co-exposure to particulate matter and fructose-rich-diet induces insulin resistance in male Sprague Dawley rats. Environ Toxicol Pharmacol. (2023) 100:104115. doi: 10.1016/j.etap.2023.104115
56. Kivimäki M, Bartolomucci A, and Kawachi I. The multiple roles of life stress in metabolic disorders. Nat Rev Endocrinol. (2023) 19:10–27. doi: 10.1038/s41574-022-00746-8
57. Chen N, Wu L-J, Xiao H-B, Liu Y-H, Hu L-K, Ma L-L, et al. Occupational stress is associated with insulin resistance and incident type 2 diabetes: A prospective cohort study of functional community. Clin Chim Acta. (2023) 544:117356. doi: 10.1016/j.cca.2023.117356
58. Scanes CG, Pierzchała-Koziec K, and Gajewska A. Effects of restraint stress on circulating corticosterone and met enkephalin in chickens: induction of shifts in insulin secretion and carbohydrate metabolism. Anim (Basel). (2024) 14:752. doi: 10.3390/ani14050752
59. Wu H and Ballantyne CM. Metabolic inflammation and insulin resistance in obesity. Circ Res. (2020) 126:1549–64. doi: 10.1161/CIRCRESAHA.119.315896
60. Zatterale F, Longo M, Naderi J, Raciti GA, Desiderio A, Miele C, et al. Chronic adipose tissue inflammation linking obesity to insulin resistance and type 2 diabetes. Front Physiol. (2019) 10:1607. doi: 10.3389/fphys.2019.01607
61. Botchlett R, Woo S-L, Liu M, Pei Y, Guo X, Li H, et al. Nutritional approaches for managing obesity-associated metabolic diseases. J Endocrinol. (2017) 233:R145–71. doi: 10.1530/JOE-16-0580
62. Roberts CK, Hevener AL, and Barnard RJ. Metabolic syndrome and insulin resistance: underlying causes and modification by exercise training. Compr Physiol. (2013) 3:1–58. doi: 10.1002/cphy.c110062
63. Chen YY, Wang JP, Jiang YY, Li H, Hu YH, Lee KO, et al. Fasting Plasma Insulin at 5 Years of Age Predicted Subsequent Weight Increase in Early Childhood over a 5-Year Period—The Da Qing Children Cohort Study. PloS One. (2015) 10:e0127389. doi: 10.1371/journal.pone.0127389
64. Kolb H, Stumvoll M, Kramer W, Kempf K, and Martin S. Insulin translates unfavourable lifestyle into obesity. BMC Med. (2018) 16:232. doi: 10.1186/s12916-018-1225-1
65. Astley CM, Todd JN, Salem RM, Vedantam S, Ebbeling CB, Huang PL, et al. Genetic evidence that carbohydrate-stimulated insulin secretion leads to obesity. Clin Chem. (2018) 64:192–200. doi: 10.1373/clinchem.2017.280727
66. Page MM, Skovsø S, Cen H, Chiu AP, Dionne DA, Hutchinson DF, et al. Reducing insulin via conditional partial gene ablation in adults reverses diet-induced weight gain. FASEB J. (2018) 32:1196–206. doi: 10.1096/fj.201700518R
67. Neeland IJ, Ayers CR, Rohatgi AK, Turer AT, Berry JD, Das SR, et al. Associations of visceral and abdominal subcutaneous adipose tissue with markers of cardiac and metabolic risk in obese adults. Obes (Silver Spring). (2013) 21:E439–447. doi: 10.1002/oby.20135
68. Hatting M, Tavares CDJ, Sharabi K, Rines AK, and Puigserver P. Insulin regulation of gluconeogenesis. Ann N Y Acad Sci. (2018) 1411:21–35. doi: 10.1111/nyas.13435
69. Edgerton DS, Kraft G, Smith M, Farmer B, Williams PE, Coate KC, et al. Insulin’s direct hepatic effect explains the inhibition of glucose production caused by insulin secretion. JCI Insight. (2017) 2:e91863. doi: 10.1172/jci.insight.91863
70. Merz KE and Thurmond DC. Role of skeletal muscle in insulin resistance and glucose uptake. Compr Physiol. (2020) 10:785–809. doi: 10.1002/cphy.c190029
71. Czech MP. Insulin action and resistance in obesity and type 2 diabetes. Nat Med. (2017) 23:804–14. doi: 10.1038/nm.4350
72. Tokarz VL, Mylvaganam S, and Klip A. Palmitate-induced insulin resistance causes actin filament stiffness and GLUT4 mis-sorting without altered Akt signalling. J Cell Sci. (2023) 136:jcs261300. doi: 10.1242/jcs.261300
73. Titchenell PM, Chu Q, Monks BR, and Birnbaum MJ. Hepatic insulin signalling is dispensable for suppression of glucose output by insulin in vivo. Nat Commun. (2015) 6:7078. doi: 10.1038/ncomms8078
74. Nielsen S and Jensen MD. Insulin regulation of regional lipolysis in upper-body obese and lean humans. JCI Insight. (2024) 9:e175629. doi: 10.1172/jci.insight.175629
75. Bjornstad P and Eckel RH. Pathogenesis of lipid disorders in insulin resistance: A brief review. Curr Diabetes Rep. (2018) 18:127. doi: 10.1007/s11892-018-1101-6
76. Kiya M, Tamura Y, Takeno K, Someya Y, Kakehi S, Sato M, et al. Adipose insulin resistance and decreased adiponectin are correlated with metabolic abnormalities in nonobese men. J Clin Endocrinol Metab. (2021) 106:e2228–38. doi: 10.1210/clinem/dgab037
77. Biddinger SB, Hernandez-Ono A, Rask-Madsen C, Haas JT, Alemán JO, Suzuki R, et al. Hepatic insulin resistance is sufficient to produce dyslipidemia and susceptibility to atherosclerosis. Cell Metab. (2008) 7:125–34. doi: 10.1016/j.cmet.2007.11.013
78. Samuel VT, Petersen KF, and Shulman GI. Lipid-induced insulin resistance: unravelling the mechanism. Lancet. (2010) 375:2267–77. doi: 10.1016/S0140-6736(10)60408-4
79. Tagi VM, Mainieri F, and Chiarelli F. Hypertension in patients with insulin resistance: etiopathogenesis and management in children. Int J Mol Sci. (2022) 23:5814. doi: 10.3390/ijms23105814
80. Büyükkinaci M and Buyukkinaci M. A1272 Insulin resistance in patients with prehypertension. J Hypertension. (2018) 36:e118. doi: 10.1097/01.hjh.0000548472.26053.17
81. Soleimani M. Insulin resistance and hypertension: new insights. Kidney Int. (2015) 87:497–9. doi: 10.1038/ki.2014.392
82. Gutierrez-Rodelo C, Arellano-Plancarte A, Hernandez-Aranda J, Landa-Galvan HV, Parra-Mercado GK, Moreno-Licona NJ, et al. Angiotensin II inhibits insulin receptor signaling in adipose cells. Int J Mol Sci. (2022) 23:6048. doi: 10.3390/ijms23116048
83. Koutsonida M, Markozannes G, Bouras E, Aretouli E, and Tsilidis KK. Metabolic syndrome and cognition: A systematic review across cognitive domains and a bibliometric analysis. Front Psychol. (2022) 13:981379. doi: 10.3389/fpsyg.2022.981379
84. Qureshi D, Topiwala A, Al Abid SU, Allen NE, Kuźma E, and Littlejohns TJ. Association of metabolic syndrome with neuroimaging and cognitive outcomes in the UK biobank. Diabetes Care. (2024) 47:1415–23. doi: 10.2337/dc24-0537
85. Wang W, Li J, Cui S, Li J, Ye X, Wang Z, et al. Microglial Ffar4 deficiency promotes cognitive impairment in the context of metabolic syndrome. Sci Adv. (2024) 10:eadj7813. doi: 10.1126/sciadv.adj7813
86. Mulati A, Zhang X, Zhao T, Ren B, Wang L, Liu X, et al. Isorhamnetin attenuates high-fat and high-fructose diet induced cognitive impairments and neuroinflammation by mediating MAPK and NFκB signaling pathways. Food Funct. (2021) 12:9261–72. doi: 10.1039/d0fo03165h
87. Chen TS, Mi N-N, Lao HY, Wang C-Y, Lo WLA, Mao Y-R, et al. Investigating the nexus of metabolic syndrome, serum uric acid, and dementia risk: a prospective cohort study. BMC Med. (2024) 22:115. doi: 10.1186/s12916-024-03302-5
88. Qureshi D, Collister J, Allen NE, Kuźma E, and Littlejohns T. Association between metabolic syndrome and risk of incident dementia in UK Biobank. Alzheimers Dement. (2024) 20:447–58. doi: 10.1002/alz.13439
89. Yoo H, Kim H, Koh I, Lee K, and Ok J. Effect of metabolic syndrome on the incidence of dementia based on national insurance data in Korea. Metab Syndr Relat Disord. (2022) 20:29–35. doi: 10.1089/met.2021.0046
90. Sun Z, Wang Z-T, Sun F-R, Shen X-N, Xu W, Ma Y-H, et al. Late-life obesity is a protective factor for prodromal Alzheimer’s disease: a longitudinal study. Aging (Albany NY). (2020) 12:2005–17. doi: 10.18632/aging.102738
91. Rohm TV, Meier DT, Olefsky JM, and Donath MY. Inflammation in obesity, diabetes, and related disorders. Immunity. (2022) 55:31–55. doi: 10.1016/j.immuni.2021.12.013
92. Crispino M, Trinchese G, Penna E, Cimmino F, Catapano A, Villano I, et al. Interplay between peripheral and central inflammation in obesity-promoted disorders: the impact on synaptic mitochondrial functions. Int J Mol Sci. (2020) 21:5964. doi: 10.3390/ijms21175964
93. Guillemot-Legris O and Muccioli GG. Obesity-induced neuroinflammation: beyond the hypothalamus. Trends Neurosci. (2017) 40:237–53. doi: 10.1016/j.tins.2017.02.005
94. McGuire MJ and Ishii M. Leptin dysfunction and Alzheimer’s disease: evidence from cellular, animal, and human studies. Cell Mol Neurobiol. (2016) 36:203–17. doi: 10.1007/s10571-015-0282-7
95. Qizilbash N, Gregson J, Johnson ME, Pearce N, Douglas I, Wing K, et al. BMI and risk of dementia in two million people over two decades: a retrospective cohort study. Lancet Diabetes Endocrinol. (2015) 3:431–6. doi: 10.1016/S2213-8587(15)00033-9
96. Biessels GJ and Despa F. Cognitive decline and dementia in diabetes mellitus: mechanisms and clinical implications. Nat Rev Endocrinol. (2018) 14:591–604. doi: 10.1038/s41574-018-0048-7
97. Arvanitakis Z, Wilson RS, Bienias JL, Evans DA, and Bennett DA. Diabetes mellitus and risk of Alzheimer disease and decline in cognitive function. Arch Neurol. (2004) 61:661–6. doi: 10.1001/archneur.61.5.661
98. Wang K-C, Woung L-C, Tsai M-T, Liu C-C, Su Y-H, and Li C-Y. Risk of Alzheimer’s disease in relation to diabetes: a population-based cohort study. Neuroepidemiology. (2012) 38:237–44. doi: 10.1159/000337428
99. Du H, Meng X, Yao Y, and Xu J. The mechanism and efficacy of GLP-1 receptor agonists in the treatment of Alzheimer’s disease. Front Endocrinol (Lausanne). (2022) 13:1033479. doi: 10.3389/fendo.2022.1033479
100. de la Monte SM, Tong M, and Wands JR. The 20-year voyage aboard the journal of Alzheimer’s disease: docking at “Type 3 diabetes”, environmental/exposure factors, pathogenic mechanisms, and potential treatments. J Alzheimers Dis. (2018) 62:1381–90. doi: 10.3233/JAD-170829
101. Michailidis M, Moraitou D, Tata DA, Kalinderi K, Papamitsou T, and Papaliagkas V. Alzheimer’s disease as type 3 diabetes: common pathophysiological mechanisms between Alzheimer’s disease and type 2 diabetes. Int J Mol Sci. (2022) 23:2687. doi: 10.3390/ijms23052687
102. Gong J, Harris K, Peters SAE, and Woodward M. Serum lipid traits and the risk of dementia: A cohort study of 254,575 women and 214,891 men in the UK Biobank. EClinicalMedicine. (2022) 54:101695. doi: 10.1016/j.eclinm.2022.101695
103. Zhou Z, Ryan J, Tonkin AM, Zoungas S, Lacaze P, Wolfe R, et al. Association between triglycerides and risk of dementia in community-dwelling older adults: A prospective cohort study. Neurology. (2023) 101:e2288–99. doi: 10.1212/WNL.0000000000207923
104. Button EB, Gilmour M, Cheema HK, Martin EM, Agbay A, Robert J, et al. Vasoprotective functions of high-density lipoproteins relevant to Alzheimer’s disease are partially conserved in apolipoprotein B-depleted plasma. Int J Mol Sci. (2019) 20:462. doi: 10.3390/ijms20030462
105. Xue-shan Z, juan P, Qi W, Zhong R, Li-hong P, Zhi-han T, et al. Imbalanced cholesterol metabolism in Alzheimer’s disease. Clinica Chimica Acta. (2016) 456:107–14. doi: 10.1016/j.cca.2016.02.024
106. Popp J, Meichsner S, Kölsch H, Lewczuk P, Maier W, Kornhuber J, et al. Cerebral and extracerebral cholesterol metabolism and CSF markers of Alzheimer’s disease. Biochem Pharmacol. (2013) 86:37–42. doi: 10.1016/j.bcp.2012.12.007
107. Hendrie HC, Hake A, Lane K, Purnell C, Unverzagt F, Smith-Gamble V, et al. Statin use, incident dementia and Alzheimer disease in elderly African Americans. Ethn Dis. (2015) 25:345–54. doi: 10.18865/ed.25.3.345
108. McGuinness B, Craig D, Bullock R, and Passmore P. Statins for the prevention of dementia. Cochrane Database Syst Rev. (2016) 2016:CD003160. doi: 10.1002/14651858.CD003160.pub3
109. Abdulrahman H, van Dalen JW, den Brok M, Latimer CS, Larson EB, and Richard E. Hypertension and Alzheimer’s disease pathology at autopsy: A systematic review. Alzheimers Dement. (2022) 18:2308–26. doi: 10.1002/alz.12707
110. Ou Y-N, Tan C-C, Shen X-N, Xu W, Hou X-H, Dong Q, et al. Blood pressure and risks of cognitive impairment and dementia: A systematic review and meta-analysis of 209 prospective studies. Hypertension. (2020) 76:217–25. doi: 10.1161/HYPERTENSIONAHA.120.14993
111. Lennon MJ, Makkar SR, Crawford JD, and Sachdev PS. Midlife hypertension and Alzheimer’s disease: A systematic review and meta-analysis. J Alzheimers Dis. (2019) 71:307–16. doi: 10.3233/JAD-190474
112. Ding J, Davis-Plourde KL, Sedaghat S, Tully PJ, Wang W, Phillips C, et al. Anti-hypertensive medications and risk for incident dementia and Alzheimer’s disease: collaborative meta-analysis of individual participant data from prospective cohort studies. Lancet Neurol. (2020) 19:61–70. doi: 10.1016/S1474-4422(19)30393-X
113. Bromander S, Anckarsäter R, Ahrén B, Kristiansson M, Blennow K, Holmäng A, et al. Cerebrospinal fluid insulin during non-neurological surgery. J Neural Transm (Vienna). (2010) 117:1167–70. doi: 10.1007/s00702-010-0456-x
114. Rhea EM, Rask-Madsen C, and Banks WA. Insulin transport across the blood-brain barrier can occur independently of the insulin receptor. J Physiol. (2018) 596:4753–65. doi: 10.1113/JP276149
115. Nguyen V, Thomas P, Pemberton S, Babin A, Noonan C, Weaver R, et al. Central nervous system insulin signaling can influence the rate of insulin influx into brain. Fluids Barriers CNS. (2023) 20:28. doi: 10.1186/s12987-023-00431-6
116. Mazucanti CH, Liu Q-R, Lang D, Huang N, O’Connell JF, Camandola S, et al. Release of insulin produced by the choroid plexis is regulated by serotonergic signaling. JCI Insight. (2019) 4:e131682. doi: 10.1172/jci.insight.131682
117. Eerola K, Longo F, Reinbothe TM, Richard JE, Shevchouk OT, López-Ferreras L, et al. Hindbrain insulin controls feeding behavior. Mol Metab. (2022) 66:101614. doi: 10.1016/j.molmet.2022.101614
118. Arnold SE, Arvanitakis Z, Macauley-Rambach SL, Koenig AM, Wang H-Y, Ahima RS, et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: concepts and conundrums. Nat Rev Neurol. (2018) 14:168–81. doi: 10.1038/nrneurol.2017.185
119. Del Moro L, Pirovano E, and Rota E. Mind the Metabolic Gap: Bridging Migraine and Alzheimer’s disease through Brain Insulin Resistance. Aging Dis. (2024) 15:2526–53. doi: 10.14336/AD.2024.0351
120. Kleinridders A, Ferris HA, Cai W, and Kahn CR. Insulin action in brain regulates systemic metabolism and brain function. Diabetes. (2014) 63:2232–43. doi: 10.2337/db14-0568
121. van der Heide LP, Kamal A, Artola A, Gispen WH, and Ramakers GMJ. Insulin modulates hippocampal activity-dependent synaptic plasticity in a N-methyl-d-aspartate receptor and phosphatidyl-inositol-3-kinase-dependent manner. J Neurochem. (2005) 94:1158–66. doi: 10.1111/j.1471-4159.2005.03269.x
122. Shaughness M, Acs D, Brabazon F, Hockenbury N, and Byrnes KR. Role of insulin in neurotrauma and neurodegeneration: A review. Front Neurosci. (2020) 14:547175. doi: 10.3389/fnins.2020.547175
123. Xue C-Y, Gao T, Mao E, Kou Z-Z, Dong L, and Gao F. Hippocampus insulin receptors regulate episodic and spatial memory through excitatory/inhibitory balance. ASN Neuro. (2023) 15:17590914231206657. doi: 10.1177/17590914231206657
124. Fernandez AM and Torres-Alemán I. The many faces of insulin-like peptide signalling in the brain. Nat Rev Neurosci. (2012) 13:225–39. doi: 10.1038/nrn3209
125. Stoica L, Zhu PJ, Huang W, Zhou H, Kozma SC, and Costa-Mattioli M. Selective pharmacogenetic inhibition of mammalian target of Rapamycin complex I (mTORC1) blocks long-term synaptic plasticity and memory storage. Proc Natl Acad Sci U S A. (2011) 108:3791–6. doi: 10.1073/pnas.1014715108
126. Son JH, Shim JH, Kim KH, Ha JY, and Han JY. Neuronal autophagy and neurodegenerative diseases. Exp Mol Med. (2012) 44:89–98. doi: 10.3858/emm.2012.44.2.031
127. Caccamo A, Majumder S, Richardson A, Strong R, and Oddo S. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and Tau: effects on cognitive impairments. J Biol Chem. (2010) 285:13107–20. doi: 10.1074/jbc.M110.100420
128. Salcedo-Tello P, Ortiz-Matamoros A, and Arias C. GSK3 function in the brain during development, neuronal plasticity, and neurodegeneration. Int J Alzheimers Dis. (2011) 2011:189728. doi: 10.4061/2011/189728
129. Lauretti E, Dincer O, and Praticò D. Glycogen synthase kinase-3 signaling in Alzheimer’s disease. Biochim Biophys Acta Mol Cell Res. (2020) 1867:118664. doi: 10.1016/j.bbamcr.2020.118664
130. Zhao Z, Yuan Y, Li S, Wang X, and Yang X. Natural compounds from herbs and nutraceuticals as glycogen synthase kinase-3β inhibitors in Alzheimer’s disease treatment. CNS Neurosci Ther. (2024) 30:e14885. doi: 10.1111/cns.14885
131. Maqbool M, Mobashir M, and Hoda N. Pivotal role of glycogen synthase kinase-3: A therapeutic target for Alzheimer’s disease. Eur J Med Chem. (2016) 107:63–81. doi: 10.1016/j.ejmech.2015.10.018
132. Cao G, Lin M, Gu W, Su Z, Duan Y, Song W, et al. The rules and regulatory mechanisms of FOXO3 on inflammation, metabolism, cell death and aging in hosts. Life Sci. (2023) 328:121877. doi: 10.1016/j.lfs.2023.121877
133. Thomas GM and Huganir RL. MAPK cascade signalling and synaptic plasticity. Nat Rev Neurosci. (2004) 5:173–83. doi: 10.1038/nrn1346
134. Qin P, Ran Y, Liu Y, Wei C, Luan X, Niu H, et al. Recent advances of small molecule JNK3 inhibitors for Alzheimer’s disease. Bioorg Chem. (2022) 128:106090. doi: 10.1016/j.bioorg.2022.106090
135. Sullivan M, Fernandez-Aranda F, Camacho-Barcia L, Harkin A, Macrì S, Mora-Maltas B, et al. Insulin and disorders of behavioural flexibility. Neurosci Biobehav Rev. (2023) 150:105169. doi: 10.1016/j.neubiorev.2023.105169
136. Leclerc M, Bourassa P, Tremblay C, Caron V, Sugère C, Emond V, et al. Cerebrovascular insulin receptors are defective in Alzheimer’s disease. Brain. (2023) 146:75–90. doi: 10.1093/brain/awac309
137. Velazquez R, Tran A, Ishimwe E, Denner L, Dave N, Oddo S, et al. Central insulin dysregulation and energy dyshomeostasis in two mouse models of Alzheimer’s disease. Neurobiol Aging. (2017) 58:1–13. doi: 10.1016/j.neurobiolaging.2017.06.003
138. Li W, Wu M, Zhang Y, Wei X, Zang J, Liu Y, et al. Intermittent fasting promotes adult hippocampal neuronal differentiation by activating GSK-3β in 3xTg-AD mice. J Neurochem. (2020) 155:697–713. doi: 10.1111/jnc.15105
139. Izuo N, Watanabe N, Noda Y, Saito T, Saido TC, Yokote K, et al. Insulin resistance induces earlier initiation of cognitive dysfunction mediated by cholinergic deregulation in a mouse model of Alzheimer’s disease. Aging Cell. (2023) 22:e13994. doi: 10.1111/acel.13994
140. Lee Y-H, Hsu H-C, Kao P-C, Shiao Y-J, Yeh SH-H, Shie F-S, et al. Augmented insulin and leptin resistance of high fat diet-fed APPswe/PS1dE9 transgenic mice exacerbate obesity and glycemic dysregulation. Int J Mol Sci. (2018) 19:2333. doi: 10.3390/ijms19082333
141. Tang X, Kan Z, Li N, Huang J, Zhang J, Thompson HJ, et al. Mechanisms underlying large-leaf yellow tea mediated inhibition of cognitive impairment in the 5xFAD model of Alzheimer’s disease. Phytomedicine. (2023) 120:155030. doi: 10.1016/j.phymed.2023.155030
142. Kazkayasi I, Telli G, Nemutlu E, and Uma S. Intranasal metformin treatment ameliorates cognitive functions via insulin signaling pathway in ICV-STZ-induced mice model of Alzheimer’s disease. Life Sci. (2022) 299:120538. doi: 10.1016/j.lfs.2022.120538
143. Alves SS, Servilha-Menezes G, Rossi L, da Silva Junior RMP, and Garcia-Cairasco N. Evidence of disturbed insulin signaling in animal models of Alzheimer’s disease. Neurosci Biobehav Rev. (2023) 152:105326. doi: 10.1016/j.neubiorev.2023.105326
144. Hulse RE, Ralat LA, and Tang W-J. Structure, function, and regulation of insulin-degrading enzyme. Vitam Horm. (2009) 80:635–48. doi: 10.1016/S0083-6729(08)00622-5
145. Farris W, Mansourian S, Chang Y, Lindsley L, Eckman EA, Frosch MP, et al. Insulin-degrading enzyme regulates the levels of insulin, amyloid β-protein, and the β-amyloid precursor protein intracellular domain in vivo. Proc Natl Acad Sci U S A. (2003) 100:4162–7. doi: 10.1073/pnas.0230450100
146. Meng X, Zhang H, Zhao Z, Li S, Zhang X, Guo R, et al. Type 3 diabetes and metabolic reprogramming of brain neurons: causes and therapeutic strategies. Mol Med. (2025) 31:61. doi: 10.1186/s10020-025-01101-z
147. Zagare A, Kurlovics J, Almeida C, Ferrante D, Frangenberg D, Vitali A, et al. Insulin resistance compromises midbrain organoid neuronal activity and metabolic efficiency predisposing to Parkinson’s disease pathology. J Tissue Eng. (2025) 16:20417314241295928. doi: 10.1177/20417314241295928
148. Confettura AD, Cuboni E, Ammar MR, Jia S, Gomes GM, Yuanxiang P, et al. Neddylation-dependent protein degradation is a nexus between synaptic insulin resistance, neuroinflammation and Alzheimer’s disease. Transl Neurodegener. (2022) 11:2. doi: 10.1186/s40035-021-00277-8
149. Chen W, Liu X, Muñoz VR, and Kahn CR. Loss of insulin signaling in microglia impairs cellular uptake of Aβ and neuroinflammatory response exacerbating AD-like neuropathology. Proc Natl Acad Sci U S A. (2025) 122:e2501527122. doi: 10.1073/pnas.2501527122
150. González Pérez N, Bellotto M, Porte Alcón S, Bentivegna M, Arcucci L, Vinuesa Á, et al. Metformin protects against insulin resistance-related dementia risk involving restored microglial homeostasis. Life Sci. (2025) 378:123803. doi: 10.1016/j.lfs.2025.123803
151. Fernandez AM, Martinez-RaChadell L, Navarrete M, Pose-Utrilla J, Davila JC, Pignatelli J, et al. Insulin regulates neurovascular coupling through astrocytes. Proc Natl Acad Sci U S A. (2022) 119:e2204527119. doi: 10.1073/pnas.2204527119
152. Xiang Y, Gu Q, and Liu D. Brain endothelial cells in blood-brain barrier regulation and neurological therapy. Int J Mol Sci. (2025) 26:5843. doi: 10.3390/ijms26125843
153. Estudillo E, López-Ornelas A, Rodríguez-Oviedo A, Gutiérrez de la Cruz N, Vargas-Hernández MA, and Jiménez A. Thinking outside the black box: are the brain endothelial cells the new main target in Alzheimer’s disease? Neural Regener Res. (2023) 18:2592–8. doi: 10.4103/1673-5374.373672
154. Zhou H, Gao F, Yang X, Lin T, Li Z, Wang Q, et al. Endothelial BACE1 Impairs Cerebral Small Vessels via Tight Junctions and eNOS. Circ Res. (2022) 130:1321–41. doi: 10.1161/CIRCRESAHA.121.320183
155. Callewaert B, Gsell W, Lox M, Himmelreich U, and Jones EAV. A timeline study on vascular co-morbidity induced cerebral endothelial dysfunction assessed by perfusion MRI. Neurobiol Dis. (2024) 202:106709. doi: 10.1016/j.nbd.2024.106709
156. Banks WA and Rhea EM. The blood-brain barrier, oxidative stress, and insulin resistance. Antioxidants (Basel). (2021) 10:1695. doi: 10.3390/antiox10111695
157. Jia B, Xu Y, and Zhu X. Cognitive resilience in Alzheimer’s disease: Mechanism and potential clinical intervention. Ageing Res Rev. (2025) 106:102711. doi: 10.1016/j.arr.2025.102711
158. Pichet Binette A, Gaiteri C, Wennström M, Kumar A, Hristovska I, Spotorno N, et al. Proteomic changes in Alzheimer’s disease associated with progressive Aβ plaque and tau tangle pathologies. Nat Neurosci. (2024) 27:1880–91. doi: 10.1038/s41593-024-01737-w
159. d’Errico P and Meyer-Luehmann M. Mechanisms of pathogenic tau and Aβ Protein spreading in Alzheimer’s disease. Front Aging Neurosci. (2020) 12:265. doi: 10.3389/fnagi.2020.00265
160. Pandini G, Pace V, Copani A, Squatrito S, Milardi D, and Vigneri R. Insulin has multiple antiamyloidogenic effects on human neuronal cells. Endocrinology. (2013) 154:375–87. doi: 10.1210/en.2012-1661
161. Schubert M, Brazil DP, Burks DJ, Kushner JA, Ye J, Flint CL, et al. Insulin receptor substrate-2 deficiency impairs brain growth and promotes tau phosphorylation. J Neurosci. (2003) 23:7084–92. doi: 10.1523/JNEUROSCI.23-18-07084.2003
162. Willette AA, Johnson SC, Birdsill AC, Sager MA, Christian B, Baker LD, et al. Insulin resistance predicts brain amyloid deposition in late middle-aged adults. Alzheimers Dement. (2015) 11:504–510.e1. doi: 10.1016/j.jalz.2014.03.011
163. Laws SM, Gaskin S, Woodfield A, Srikanth V, Bruce D, Fraser PE, et al. Insulin resistance is associated with reductions in specific cognitive domains and increases in CSF tau in cognitively normal adults. Sci Rep. (2017) 7:9766. doi: 10.1038/s41598-017-09577-4
164. Schubert M, Gautam D, Surjo D, Ueki K, Baudler S, Schubert D, et al. Role for neuronal insulin resistance in neurodegenerative diseases. Proc Natl Acad Sci U S A. (2004) 101:3100–5. doi: 10.1073/pnas.0308724101
165. Sędzikowska A and Szablewski L. Insulin and insulin resistance in Alzheimer’s disease. Int J Mol Sci. (2021) 22:9987. doi: 10.3390/ijms22189987
166. Yamazaki Y and Kanekiyo T. Blood-brain barrier dysfunction and the pathogenesis of Alzheimer’s disease. Int J Mol Sci. (2017) 18:1965. doi: 10.3390/ijms18091965
167. Nation DA, Sweeney MD, Montagne A, Sagare AP, D’Orazio LM, Pachicano M, et al. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat Med. (2019) 25:270–6. doi: 10.1038/s41591-018-0297-y
168. Padovani A, Galli A, Bazzoli E, Tolassi C, Caratozzolo S, Gumina B, et al. The role of insulin resistance and APOE genotype on blood-brain barrier integrity in Alzheimer’s disease. Alzheimers Dement. (2025) 21:e14556. doi: 10.1002/alz.14556
169. Grillo CA, Piroli GG, Lawrence RC, Wrighten SA, Green AJ, Wilson SP, et al. Hippocampal insulin resistance impairs spatial learning and synaptic plasticity. Diabetes. (2015) 64:3927–36. doi: 10.2337/db15-0596
170. Chiu S-L, Chen C-M, and Cline HT. Insulin receptor signaling regulates synapse number, dendritic plasticity, and circuit function in vivo. Neuron. (2008) 58:708–19. doi: 10.1016/j.neuron.2008.04.014
171. Si Z-Z, Zou C-J, Mei X, Li X-F, Luo H, Shen Y, et al. Targeting neuroinflammation in Alzheimer’s disease: from mechanisms to clinical applications. Neural Regener Res. (2022) 18:708–15. doi: 10.4103/1673-5374.353484
172. Bairamian D, Sha S, Rolhion N, Sokol H, Dorothée G, Lemere CA, et al. Microbiota in neuroinflammation and synaptic dysfunction: a focus on Alzheimer’s disease. Mol Neurodegener. (2022) 17:19. doi: 10.1186/s13024-022-00522-2
173. Dani M, Wood M, Mizoguchi R, Fan Z, Walker Z, Morgan R, et al. Microglial activation correlates in vivo with both tau and amyloid in Alzheimer’s disease. Brain. (2018) 141:2740–54. doi: 10.1093/brain/awy188
174. Yang X, Xu Y, Gao W, Wang L, Zhao X, Liu G, et al. Hyperinsulinemia-induced microglial mitochondrial dynamic and metabolic alterations lead to neuroinflammation in vivo and in vitro. Front Neurosci. (2022) 16:1036872. doi: 10.3389/fnins.2022.1036872
175. Najem D, Bamji-Mirza M, Chang N, Liu QY, and Zhang W. Insulin resistance, neuroinflammation, and Alzheimer’s disease. Rev Neurosci. (2014) 25:509–25. doi: 10.1515/revneuro-2013-0050
176. De Dios L, Collazo C, and Inostroza-Nieves Y. Renin-angiotensin-system increases phosphorylated tau and Reactive Oxygen Species in human cortical neuron cell line. Biochem Biophys Rep. (2022) 32:101355. doi: 10.1016/j.bbrep.2022.101355
177. Loera-Valencia R, Eroli F, Garcia-Ptacek S, and Maioli S. Brain renin-angiotensin system as novel and potential therapeutic target for Alzheimer’s disease. Int J Mol Sci. (2021) 22:10139. doi: 10.3390/ijms221810139
178. Mateos L, Ismail M-A-M, Gil-Bea F-J, Leoni V, Winblad B, Björkhem I, et al. Upregulation of brain renin angiotensin system by 27-hydroxycholesterol in Alzheimer’s disease. J Alzheimers Dis. (2011) 24:669–79. doi: 10.3233/JAD-2011-101512
179. Nation DA, Ho J, Yew B, and Alzheimer’s Disease Neuroimaging Initiative. Older adults taking AT1-receptor blockers exhibit reduced cerebral amyloid retention. J Alzheimers Dis. (2016) 50:779–89. doi: 10.3233/JAD-150487
180. Jochemsen HM, Teunissen CE, Ashby EL, van der Flier WM, Jones RE, Geerlings MI, et al. The association of angiotensin-converting enzyme with biomarkers for Alzheimer’s disease. Alzheimers Res Ther. (2014) 6:27. doi: 10.1186/alzrt257
181. Maciejczyk M, Żebrowska E, Zalewska A, and Chabowski A. Redox balance, antioxidant defense, and oxidative damage in the hypothalamus and cerebral cortex of rats with high fat diet-induced insulin resistance. Oxid Med Cell Longev. (2018) 2018:6940515. doi: 10.1155/2018/6940515
182. Cobley JN, Fiorello ML, and Bailey DM. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. (2018) 15:490–503. doi: 10.1016/j.redox.2018.01.008
183. Christen Y. Oxidative stress and Alzheimer disease. Am J Clin Nutr. (2000) 71:621S–9S. doi: 10.1093/ajcn/71.2.621s
184. Tan X, Xu R, Li A-P, Li D, Wang Y, Zhao Q, et al. Antioxidant and anti-Alzheimer’s disease activities of 1,8-cineole and its cyclodextrin inclusion complex. BioMed Pharmacother. (2024) 175:116784. doi: 10.1016/j.biopha.2024.116784
185. Gao L, Wang B, Cui X, Xia L, Li X, Figueredo YN, et al. Neochlorogenic acid ameliorates Alzheimer’s disease-like pathology via scavenging oxidative stress and restoring blood-brain barrier function in Zebrafish. Prog Neuropsychopharmacol Biol Psychiatry. (2025), 138:111334. doi: 10.1016/j.pnpbp.2025.111334
186. Ioannou K, Bucci M, Tzortzakakis A, Savitcheva I, Nordberg A, Chiotis K, et al. Tau PET positivity predicts clinically relevant cognitive decline driven by Alzheimer’s disease compared to comorbid cases; proof of concept in the ADNI study. Mol Psychiatry. (2025) 30:587–99. doi: 10.1038/s41380-024-02672-9
187. Panza F, Lozupone M, Logroscino G, and Imbimbo BP. A critical appraisal of amyloid-β-targeting therapies for Alzheimer disease. Nat Rev Neurol. (2019) 15:73–88. doi: 10.1038/s41582-018-0116-6
188. Samudra N, Lane-Donovan C, VandeVrede L, and Boxer AL. Tau pathology in neurodegenerative disease: disease mechanisms and therapeutic avenues. J Clin Invest. (2023) 133:e168553. doi: 10.1172/JCI168553
189. Di Benedetto G, Burgaletto C, Bellanca CM, Munafò A, Bernardini R, and Cantarella G. Role of microglia and astrocytes in Alzheimer’s disease: from neuroinflammation to Ca2+ Homeostasis dysregulation. Cells. (2022) 11:2728. doi: 10.3390/cells11172728
190. Si Z-Z, Zou C-J, Mei X, Li X-F, Luo H, Shen Y, et al. Targeting neuroinflammation in Alzheimer’s disease: from mechanisms to clinical applications. Neural Regener Res. (2023) 18:708–15. doi: 10.4103/1673-5374.353484
191. Ouk M, Wu C-Y, Rabin JS, Jackson A, Edwards JD, Ramirez J, et al. The use of angiotensin-converting enzyme inhibitors vs. angiotensin receptor blockers and cognitive decline in Alzheimer’s disease: the importance of blood-brain barrier penetration and APOE ϵ4 carrier status. Alzheimers Res Ther. (2021) 13:43. doi: 10.1186/s13195-021-00778-8
192. Scotti L, Bassi L, Soranna D, Verde F, Silani V, Torsello A, et al. Association between renin-angiotensin-aldosterone system inhibitors and risk of dementia: A meta-analysis. Pharmacol Res. (2021) 166:105515. doi: 10.1016/j.phrs.2021.105515
193. Kehoe PG, Turner N, Howden B, Jarutyte L, Clegg SL, Malone IB, et al. Safety and efficacy of losartan for the reduction of brain atrophy in clinically diagnosed Alzheimer’s disease (the RADAR trial): a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet Neurol. (2021) 20:895–906. doi: 10.1016/S1474-4422(21)00263-5
194. Persson T, Popescu BO, and Cedazo-Minguez A. Oxidative stress in Alzheimer’s disease: why did antioxidant therapy fail? Oxid Med Cell Longev. (2014) 2014:427318. doi: 10.1155/2014/427318
195. Alves SS, da Silva-Junior RMP, Servilha-Menezes G, Homolak J, Šalković-Petrišić M, and Garcia-Cairasco N. Insulin resistance as a common link between current Alzheimer’s disease hypotheses. J Alzheimers Dis. (2021) 82:71–105. doi: 10.3233/JAD-210234
196. Tschöp M, Nogueiras R, and Ahrén B. Gut hormone-based pharmacology: novel formulations and future possibilities for metabolic disease therapy. Diabetologia. (2023) 66:1796–808. doi: 10.1007/s00125-023-05929-0
197. Gejl M, Brock B, Egefjord L, Vang K, Rungby J, and Gjedde A. Blood-brain glucose transfer in Alzheimer’s disease: effect of GLP-1 analog treatment. Sci Rep. (2017) 7:17490. doi: 10.1038/s41598-017-17718-y
198. Watson KT, Wroolie TE, Tong G, Foland-Ross LC, Frangou S, Singh M, et al. Neural correlates of liraglutide effects in persons at risk for Alzheimer’s disease. Behav Brain Res. (2019) 356:271–8. doi: 10.1016/j.bbr.2018.08.006
199. Liang Y, Doré V, Rowe CC, and Krishnadas N. Clinical evidence for GLP-1 receptor agonists in Alzheimer’s disease: A systematic review. J Alzheimers Dis Rep. (2024) 8:777–89. doi: 10.3233/ADR-230181
200. Craft S, Claxton A, Baker LD, Hanson AJ, Cholerton B, Trittschuh EH, et al. Effects of regular and long-acting insulin on cognition and Alzheimer’s disease biomarkers: A pilot clinical trial. J Alzheimers Dis. (2017) 57:1325–34. doi: 10.3233/JAD-161256
201. Craft S, Raman R, Chow TW, Rafii MS, Sun C-K, Rissman RA, et al. Safety, efficacy, and feasibility of intranasal insulin for the treatment of mild cognitive impairment and Alzheimer disease dementia: A randomized clinical trial. JAMA Neurol. (2020) 77:1099–109. doi: 10.1001/jamaneurol.2020.1840
202. Avgerinos KI, Kalaitzidis G, Malli A, Kalaitzoglou D, Myserlis PG, and Lioutas V-A. Intranasal insulin in Alzheimer’s dementia or mild cognitive impairment. A systematic review. J Neurol. (2018) 265:1497–510. doi: 10.1007/s00415-018-8768-0
203. Sluggett JK, Koponen M, Bell JS, Taipale H, Tanskanen A, Tiihonen J, et al. Metformin and risk of Alzheimer’s disease among community-dwelling people with diabetes: A national case-control study. J Clin Endocrinol Metab. (2020) 105:dgz234. doi: 10.1210/clinem/dgz234
204. Ha J, Choi D-W, Kim KJ, Cho SY, Kim H, Kim KY, et al. Association of metformin use with Alzheimer’s disease in patients with newly diagnosed type 2 diabetes: a population-based nested case-control study. Sci Rep. (2021) 11:24069. doi: 10.1038/s41598-021-03406-5
205. Tabatabaei Malazy O, Bandarian F, Qorbani M, Mohseni S, Mirsadeghi S, Peimani M, et al. The effect of metformin on cognitive function: A systematic review and meta-analysis. J Psychopharmacol. (2022) 36:666–79. doi: 10.1177/02698811211057304
206. Luo A, Ning P, Lu H, Huang H, Shen Q, Zhang D, et al. Association between metformin and Alzheimer’s disease: A systematic review and meta-analysis of clinical observational studies. J Alzheimers Dis. (2022) 88:1311–23. doi: 10.3233/JAD-220180
207. Kounatidis D, Vallianou NG, Rebelos E, Kouveletsou M, Kontrafouri P, Eleftheriadou I, et al. The many facets of PPAR-γ Agonism in obesity and associated comorbidities: benefits, risks, challenges, and future directions. Curr Obes Rep. (2025) 14:19. doi: 10.1007/s13679-025-00612-4
208. Li Z, Meng X, Ma G, Liu W, Li W, Cai Q, et al. Increasing brain glucose metabolism by ligustrazine piperazine ameliorates cognitive deficits through PPARγ-dependent enhancement of mitophagy in APP/PS1 mice. Alzheimers Res Ther. (2022) 14:150. doi: 10.1186/s13195-022-01092-7
209. He S, Shi J, Ma L, Pei H, Zhang P, Shi D, et al. Total ginsenosides decrease Aβ production through activating PPARγ. BioMed Pharmacother. (2024) 174:116577. doi: 10.1016/j.biopha.2024.116577
210. Basutkar RS, Sudarsan P, Robin SM, Bhaskar V, Viswanathan B, and Sivasankaran P. Drug repositioning of pioglitazone in management and improving the cognitive function among the patients with mild to moderate Alzheimer’s disease: A systematic review and meta-analysis. Neurol India. (2023) 71:1132–41. doi: 10.4103/0028-3886.391397
211. Bramen JE, Siddarth P, Popa ES, Kress GT, Rapozo MK, Hodes JF, et al. Impact of eating a carbohydrate-restricted diet on cortical atrophy in a cross-section of amyloid positive patients with Alzheimer’s disease: A small sample study. J Alzheimers Dis. (2023) 96:329–42. doi: 10.3233/JAD-230458
212. Kullmann S, Goj T, Veit R, Fritsche L, Wagner L, Schneeweiss P, et al. Exercise restores brain insulin sensitivity in sedentary adults who are overweight and obese. JCI Insight. (2022) 7:e161498. doi: 10.1172/jci.insight.161498
213. Ornish D, Madison C, Kivipelto M, Kemp C, McCulloch CE, Galasko D, et al. Effects of intensive lifestyle changes on the progression of mild cognitive impairment or early dementia due to Alzheimer’s disease: a randomized, controlled clinical trial. Alzheimers Res Ther. (2024) 16:122. doi: 10.1186/s13195-024-01482-z
214. Williamson J, James SA, Mukli P, Yabluchanskiy A, Wu DH, Sonntag W, et al. Sex difference in brain functional connectivity of hippocampus in Alzheimer’s disease. Geroscience. (2024) 46:563–72. doi: 10.1007/s11357-023-00943-x
Keywords: metabolic syndrome, insulin resistance, cognitive impairment, Alzheimer’s disease, mechanism research
Citation: Wang S, Shi Y, Xin R, Kang H, Xiong H and Ren J (2025) Exploring the role of insulin resistance in bridging the metabolic syndrome and Alzheimer’s disease-a review of mechanistic studies. Front. Endocrinol. 16:1614006. doi: 10.3389/fendo.2025.1614006
Received: 18 April 2025; Accepted: 23 September 2025;
Published: 08 October 2025.
Edited by:
Ramesh Kandimalla, Indian Institute of Chemical Technology (CSIR), IndiaReviewed by:
Jim R. Fadel, University of South Carolina, United StatesRui Milton Patricio da Silva-Junior, Fundació Clínic per a la Recerca Biomèdica, Spain
Copyright © 2025 Wang, Shi, Xin, Kang, Xiong and Ren. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jixiang Ren, cmVuangyMDAzQDE2My5jb20=