 Salwa A. Musa1,2*
Salwa A. Musa1,2* Mohamed A. Abdullah2,3
Mohamed A. Abdullah2,3 Samar S. Hassan2
Samar S. Hassan2 Eliane Streiff4Franziska Lange4Omer O. Babiker5Areej A. Ibrahim2Hiba A. Elshafie6Angela Huebner4
Eliane Streiff4Franziska Lange4Omer O. Babiker5Areej A. Ibrahim2Hiba A. Elshafie6Angela Huebner4 Katrin Koehler4
Katrin Koehler4 Friederike Quitter4*
Friederike Quitter4*- 1Department of Pediatrics and Child Health, Faculty of Medicine, Al-Neelain University, Khartoum, Sudan
- 2Department of Pediatric Endocrinology and Diabetes, Gaafar Ibn Auf Pediatric Tertiary Hospital, Khartoum, Sudan
- 3Department of Pediatrics and Child Health, Faculty of Medicine, University of Khartoum, Khartoum, Sudan
- 4Division of Pediatric Endocrinology and Diabetes, Department of Pediatrics, Faculty of Medicine and University Hospital Carl Gustav Carus, Technische Universität Dresden, Dresden, Germany
- 5Department of Pediatrics, Faculty of Medicine and Health Sciences, Omdurman Islamic University, Khartoum, Sudan
- 6School of Medicine, Ahfad University for Women, Khartoum, Sudan
Introduction: Triple A syndrome (OMIM*231550) is a rare autosomal recessive disorder characterized by achalasia, alacrima, adrenal insufficiency, and neurological features. It is caused by functional impairment of the nucleoporin ALADIN due to mutations in the AAAS gene. Limited data exists on triple A syndrome from Sub-Saharan African and Arab countries. Our objective is to perform a comprehensive clinical and genetic study in Sudanese patients diagnosed with triple A syndrome.
Methods: The clinical diagnoses were based on characteristic clinical and laboratory findings. Genetic testing was conducted in 20 families, encompassing 31 patients, revealing six different AAAS mutations.
Results: A previously described mutation in exon 9 (c.934C>T) was present in 35%, and the known Arabic founder mutation c.1331+1G>A (intron 14) was found in 30% of the families. In addition, two novel mutations, including an 8 bp-deletion at the exon 4/intron 4 junction (c.394_399+2delCTGTCTGT) and a 1 bp-deletion in exon 9 (c.852delG) were identified.
Discussion: Genotype-phenotype analyses highlighted significant variability in symptom occurrence, age of onset, and disease severity. Consistent with the high consanguinity rates in Sudan, most mutations (95%) occurred in a homozygous state. In conclusion, triple A syndrome is likely underdiagnosed in Sudan and exhibits significant variability in phenotypic presentation even among affected individuals within the same family or mutation.
1 Introduction
Triple A syndrome, also known as Allgrove syndrome (OMIM#231550), is an autosomal recessive multisystemic disorder. It is a very rare disease with an extremely low prevalence of fewer than 1 in 1 000 000 (www.orpha.net). The condition is initially characterized by the clinical triad of alacrima, achalasia, and adrenal insufficiency (1). Over time, two-thirds of the patients present with progressive neurological impairment affecting the central, peripheral, and autonomic nervous systems, prompting some authors to suggest the term 4A syndrome (2, 3). Additional symptoms include a dysmorphic facial appearance, xerostomia, dental caries, palmar and plantar hyperkeratosis, gait disturbances, microcephaly, developmental delays, and delayed puberty (4, 5). Patients with triple A syndrome exhibit significant variability in clinical presentation, including the age of symptom onset, the frequency and severity, and the discordance between phenotypes and genotypes. This variability is evident even among affected members from the same family or mutation, further contributing to the heterogeneity of this syndrome. Alacrima is the earliest and most consistent symptom, occurring in over 90% of patients, whereas achalasia is present in 75-85% of the patients and primary adrenal insufficiency (PAI) in about 85% (3, 6).
Triple A syndrome is caused by mutations in the AAAS gene on chromosome 12q13, which consists of 16 exons encoding a 546 amino acid protein termed ALADIN (Alacrima–Achalasia–Adrenal Insufficiency Neurologic disorder). ALADIN is a protein of the nuclear pore complex that controls the transport of macromolecules between the nucleus and the cytoplasm. Most mutations in AAAS result in a delocalization of ALADIN to the cytoplasm (7), which affects the nucleocytoplasmic transport of specific proteins, including DNA repair proteins (8). This renders cells more vulnerable to oxidative stress, leading to selective tissue degeneration (9–11). The precise role of the ALADIN protein remains largely unknown, but studies investigating the pathogenic mechanisms of various mutations have shown an impaired nuclear import of DNA repair proteins and increased oxidative stress within the cell. ALADIN I482S fibroblasts were found to be more vulnerable to oxidative stress, which combined with impaired import of DNA repair proteins, led to increased cell death. This may contribute to the progressive nature of the disease (9).
The AAAS gene is expressed in a wide range of human tissues, leading to a variety of symptoms in the affected patients. Notably, there is a particularly high expression in the adrenal gland, gastrointestinal tract, and brain (12, 13). To date, 91 unique mutations have been identified across the AAAS gene according to “The Human Gene Mutation Database” (https://www.hgmd.cf.ac.uk/ac/gene.php?gene=AAAS). The majority of the mutations are nonsense and frameshift mutations, which are likely to result in a truncated, non-functional protein, followed by missense and splice-site mutations (14). Mutations were identified across the entire AAAS gene, indicating the absence of specific hotspots. However, several of the most frequently observed mutations cluster in specific ethnicities. For instance, mutations c.1432C>T (p.Arg478*) and c.787T>C (p.Ser263Pro) are common in Europe, c.771delG (p.Arg258fs) is prevalent in China, c.762delC (p.Ser255fs) is found in India, while the widely reported splice-site mutation c.1331+1G>A is noted in Africa and the USA (15). The c.1331+1G>A substitution was first identified in unrelated North African patients with triple A syndrome, indicating a founder effect (12).
Many studies have been conducted worldwide; however, apart from a few studies from North Africa, reports from Sub-Saharan Africa and Arab countries are limited, primarily consisting of small studies or case reports (16, 17). Here, we report clinical and genetic characterization of 31 children from 20 Sudanese families. To our knowledge, no studies have simultaneously examined both aspects in a sizable cohort from Sub-Saharan Africa and Arab countries to date.
2 Materials and methods
2.1 Subjects and DNA extraction
The clinical and biochemical presentations of all patients with clinically suspected triple A syndrome who presented to Gaafar Ibn Auf (GIA) Children’s Tertiary Hospital in Khartoum, Sudan, between 2018 and 2023 were reviewed. The initial diagnosis of triple A syndrome was based on characteristic clinical signs and symptoms, including alacrima, achalasia, adrenal insufficiency, as well as autonomic and neurological features. Adrenal insufficiency was confirmed using the following criteria: Early morning serum cortisol levels below the reference range (< 6 μg/dL), elevated ACTH levels (> 100 pg/mL), and cortisol levels below 18 μg/dL following an ACTH stimulation test (18). Additional diagnostic support was provided by pathological findings on barium swallow studies and positive Schirmer’s test results.
The total number of patients who fulfilled the clinical and biochemical criteria for triple A syndrome was 48 patients from 32 families. From those, 31 patients from 20 families were included in this study, as other patients were not available or had passed away before the genetic testing was performed. Clinical data for those patients included personal demographics, age at symptom onset, symptom duration, growth parameters, parental consanguinity, family history of similar conditions or sibling deaths, as well as the diagnostic workup and management provided (Figure 1). EDTA blood samples were collected from 31 patients from 20 families, along with their siblings and parents who consented to participate in the study. Genomic DNA was extracted from blood cells using the QIAamp DNA Mini Kit (QIAGEN GmbH, Hilden, Germany) according to standard protocols. The study adhered to the principles outlined in the Declaration of Helsinki.
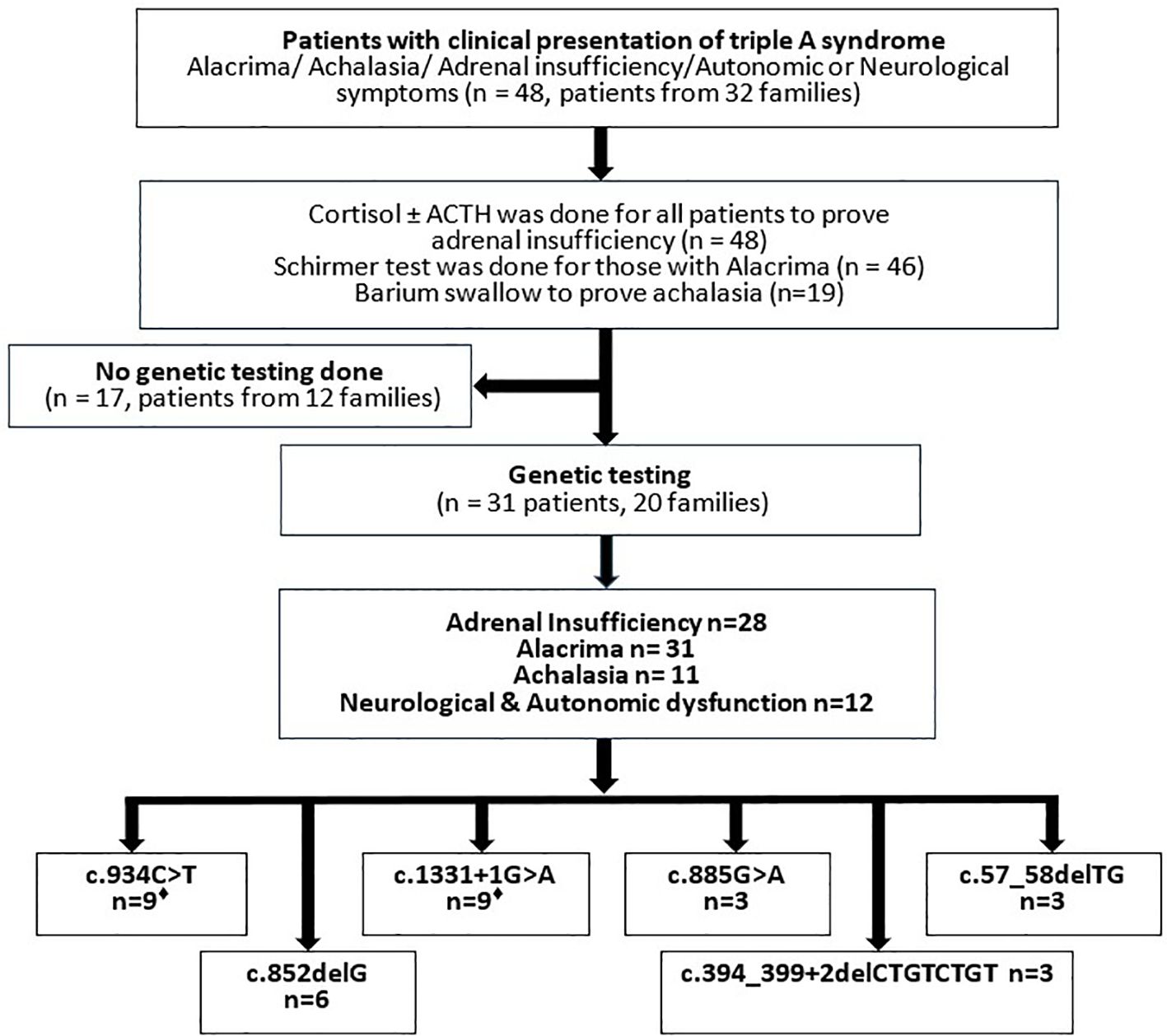
Figure 1. Flowchart showing the course of the investigation of Sudanese patients admitted with triple A syndrome. Laboratory and instrumentational examinations, result of genetic testing of the Sudanese cohort. ♦Two patients carry the mutations in a heterozygous form.
2.2 PCR and sanger sequencing
Coding sequences of the 16 exons of AAAS, including exon-intron boundaries, were amplified from genomic DNA by polymerase chain reaction (PCR) using gene-specific primers (Eurofins Genomics Germany GmbH, Ebersberg, Germany) (Table 1) and PCR standard protocols.
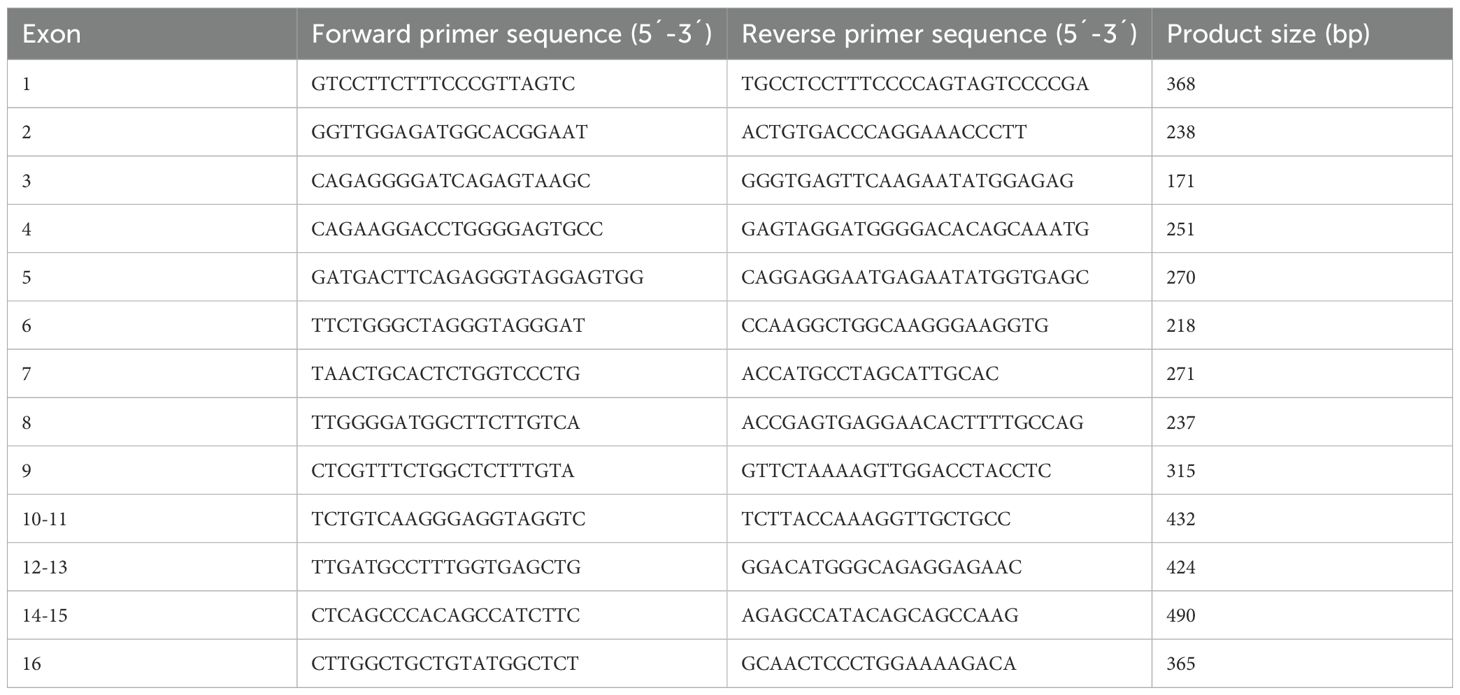
Table 1. Primer sequences for amplification of genomic DNA of AAAS.
PCR products were purified using AcroPrep 96-well filter plates filled with hydrated illustra™ Sephadex® G-50 DNA grade F chromatography resin (Sigma-Aldrich Chemie GmbH, Taufkirchen, Germany), and sequenced on an ABI 3130XL genetic analyzer using BigDye Terminator Cycle Sequencing Kit 1.1 (Applied Biosystems Inc., Foster City, CA). Data were analyzed using Mutation Surveyor V5.1.2 software (SoftGenetics, LLC. State College, PA, USA).
2.3 RNA analysis
For RNA preparation, the blood of relevant patients (Patients 23, 24) and healthy controls was collected in PAXgene Blood RNA tubes (BD, Heidelberg, Germany). Total RNA was prepared using PAXgene Blood RNA Kit (QIAGEN GmbH, Hilden Germany). After reverse transcription of messenger ribonucleic acid (mRNA) with Go Script™ Reverse Transcription System (Promega GmbH, Mannheim, Germany), the sequences of exons 2 to 6 were amplified using the forward primer (exon 2) 5’-GATCCCCTAAAGACCCCTGG-3’ and the reverse primer (exon 6) 5’-CTGCTGGCATTATACACACG-3’.
3 Results
3.1 Patients
We studied 20 Sudanese families, including 31 patients (15 male, 16 female) from Gaafar Ibn Auf Pediatric Hospital, the main tertiary hospital in Khartoum, Sudan. All of the probands exhibited at least two symptoms of the diagnostic triad: achalasia, alacrima, and adrenal insufficiency. The medical diagnosis was confirmed by the detection of a mutation in the AAAS gene in all 31 patients. Clinical and genetic details are summarized in Table 2.
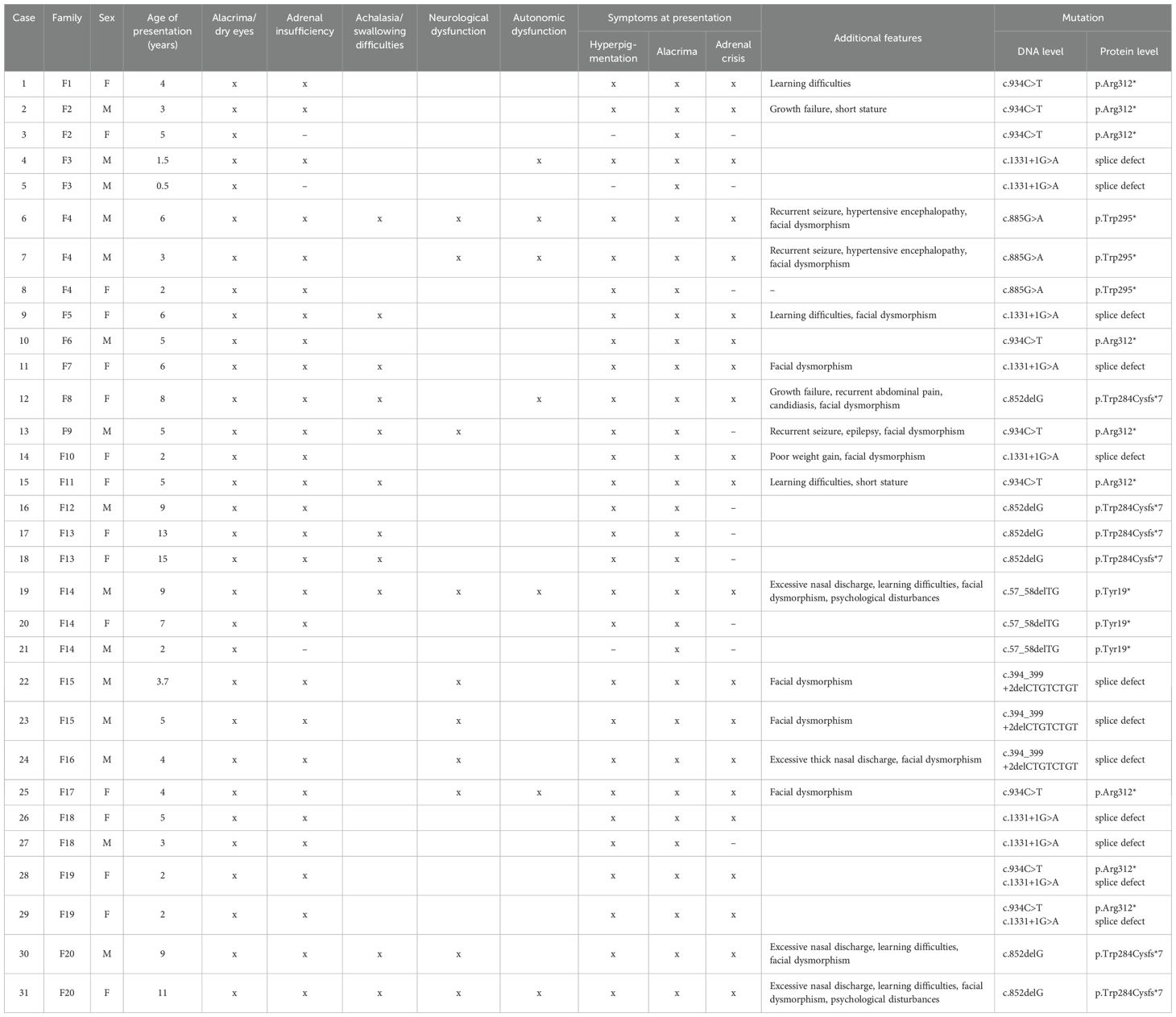
Table 2. Clinical findings and genetic analysis of 31 patients with triple A syndrome (M, male; F, female).
3.2 Clinical findings
The gender distribution in our cohort was balanced (48.4% male, 51.6% female). The median age at presentation was 5.5 years (range 6 months - 15 years). Alacrima was consistently observed in all 31 patients, while hyperpigmentation indicating the presence of ACTH-resistant PAI was present in 28/31 patients (90%) at the time of presentation. The primary presenting symptom in the index patients was adrenal insufficiency, manifesting as adrenal crises, hypoglycemic seizures, and hyperpigmentation. The other three patients presented with alacrima as the only symptom, with a family history of a confirmed affected sibling with triple A syndrome that warranted genetic testing to prove the diagnosis.
The duration of PAI symptoms before medical consultation ranged from one month to 36 months. Swallowing difficulties were elicited in 11 patients (35%), with the youngest being five years old. These patients underwent barium swallow studies to confirm the achalasia diagnosis. The triad of alacrima, achalasia, and adrenal insufficiency was detected in 11 patients (35%), while alacrima and adrenal insufficiency were detected in 28 patients (90%) at presentation. Twelve patients (39%) exhibited autonomic and neurological features, including orthostatic hypotension, increased sweating, gait abnormalities, progressive neurological dysfunction, and recurrent convulsions. Other clinical findings detected at presentation include facial dysmorphism in 14/31 patients (45%) and growth failure or short stature in 12 patients (39%). Other rare clinical aspects included two patients with hypertensive encephalopathy requiring intensive care unit admission, four patients (13%) reported unexplained excessive thick nasal mucus, psychological disturbances (seen in one patient) and learning difficulties in six patients (19%).
Consanguinity was observed in 16 families (80%). Additionally, eight families (40%) reported premature deaths of one or more siblings who had shown similar symptoms and died with symptoms suggestive of adrenal crisis. Cortisol was low (< 6 mcg/dl) in 25 out of 26 patients who had serum cortisol tested (mean = 1.7 mcg/dl), and borderline low in one patient (7.9 mcg/dl), while ACTH levels were high (> 100 pg/ml) (mean = 1535 pg/ml) in 23 patients who were tested. The patient with borderline cortisol levels had high ACTH in addition to a genetic diagnosis and an affected family member. The five patients who did not undergo cortisol testing had alacrima, a genetic diagnosis, and a family history of a confirmed triple A diagnosis. Three of those five did not have PAI symptoms, which makes future follow-up examinations necessary. The same applies to the eight patients who did not undergo ACTH testing, three of whom did not have PAI and the remaining five exhibited low cortisol levels (< 6 mcg/dl) in addition to genetic diagnosis of triple A syndrome. Additionally, Schirmer tests were positive for all patients, indicating alacrima or hypolacrima to varying degrees. Hydrocortisone treatment was initiated for 11 patients (mean dosage per day 12–15 mg), while 17 older patients were prescribed prednisolone due to the unavailability of hydrocortisone. Three patients undergo annual monitoring of their cortisol levels to evaluate for potential PAI. Three patients, unfortunately died at different ages despite receiving medical management. One male patient (F4) passed away at the age of 9 years following severe hypertensive encephalopathy that required prolonged pediatric intensive care unit admission. Patient F8, a 16-year-old female who suffered from severe achalasia and experienced multiple adrenal crises despite adequate steroid therapy, also succumbed. The third case was an 8-year-old male patient (F15), whose family had discontinued hydrocortisone treatment for some time due to unavailability, leading to a fatal adrenal crisis.
3.3 Sanger sequencing
Sequencing of the coding exons and adjacent intronic regions of the AAAS gene (OMIM *605378, Transcript-ID ENST00000209873; GenBank NM_015665) revealed six different AAAS mutations in this cohort, which are summarized in Table 3. Two of the mutations are novel AAAS mutations (c.394_399+2delCTGTCTGT and c.852delG) that have not been previously described in any other case of triple A syndrome nor in the variant aggregation database GnomAD 4.1 (https://gnomad.broadinstitute.org/). The four other mutations are previously recognized mutations (12, 19, 20).
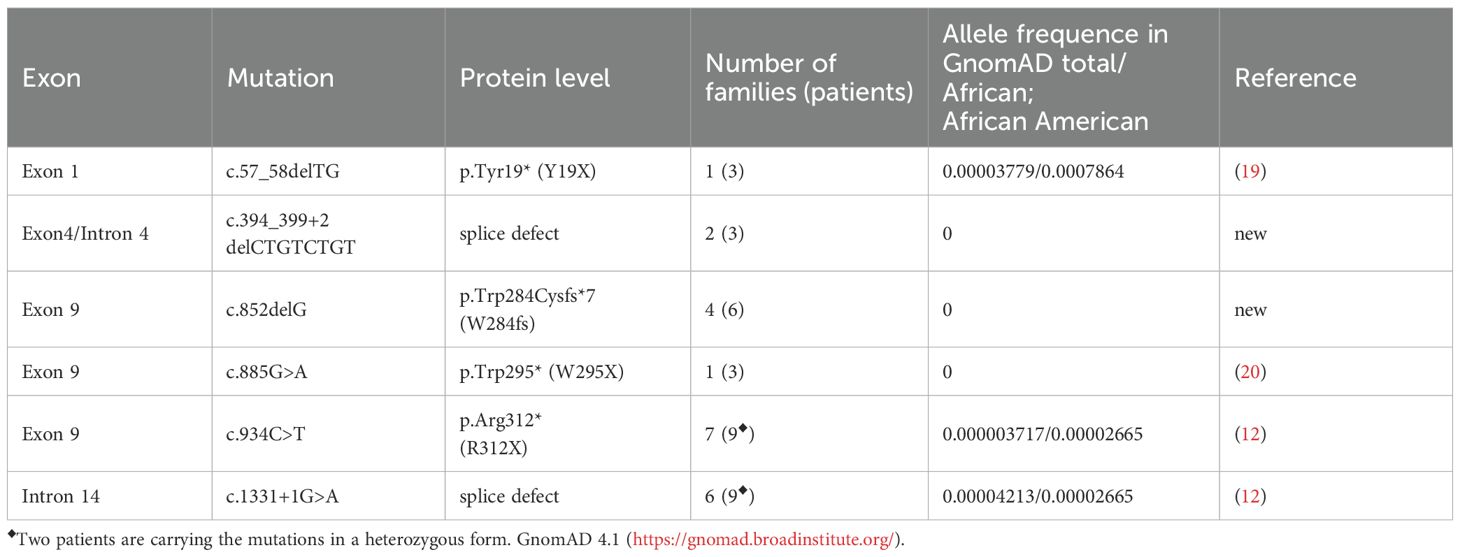
Table 3. Spectrum of AAAS variants in Sudanese patients with triple A syndrome.
3.4 RNA analysis
To investigate how the genetic alteration at donor splice site of exon 4 impact mRNA processing we used RNA from PAX-blood from two patients (patient 23 and 24) and a healthy control from family 15 and two independent controls. We revealed that cDNA with the mutation c.394_399+2delCTGTCTGT caused exon 4 to be skipped during RNA translation due to the splice mutation (Figure 2). As a result, this mutation leads to a frameshift after amino acid 103 and introduces a premature stop codon, which likely causes a nonsense-mediated RNA decay or impairs the function of the truncated ALADIN protein. We were able to confirm this splice result using the latest prediction tool SpliceAI (21).
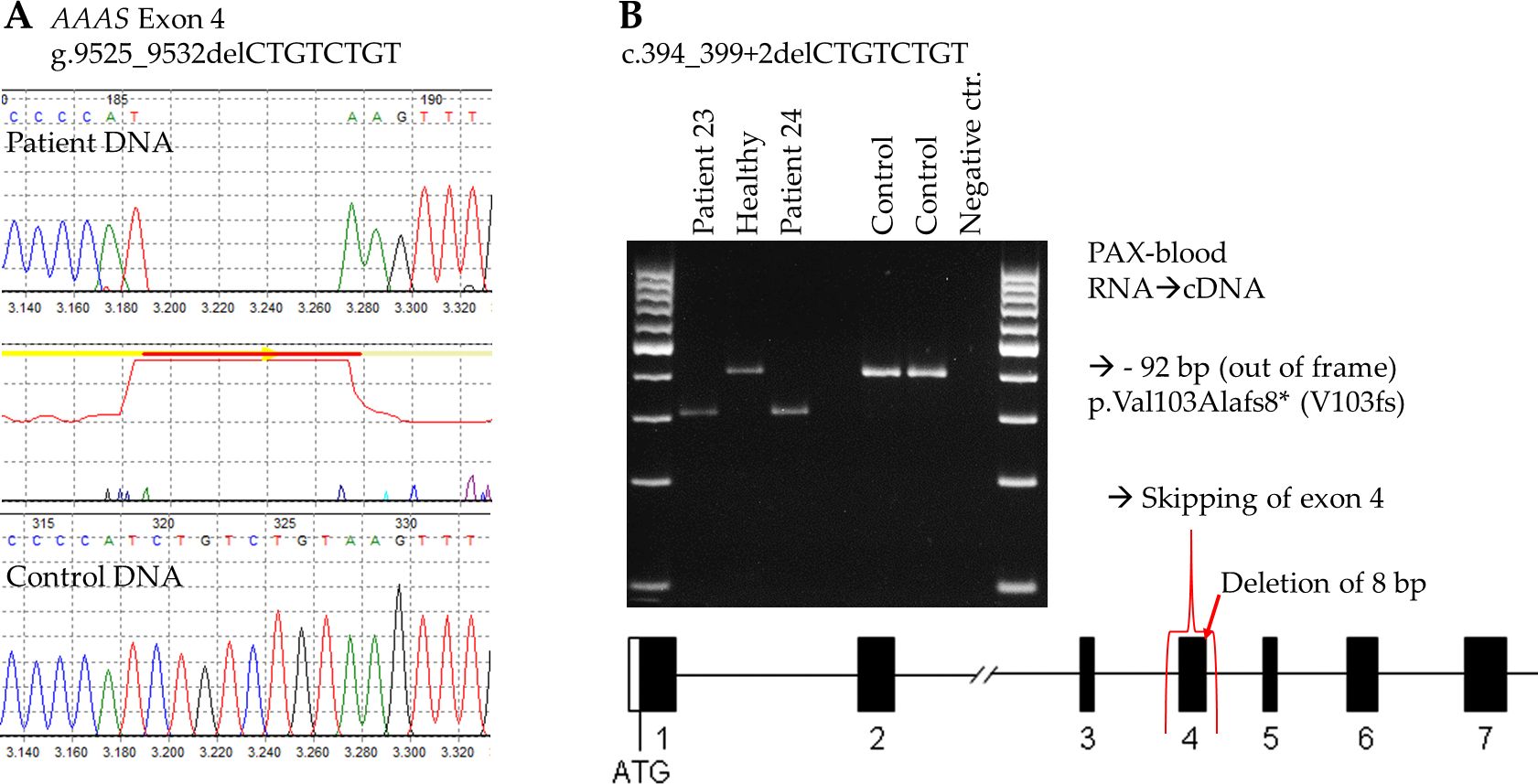
Figure 2. Investigation of the novel AAAS mutation c.394_399+2delCTGTCTGT at DNA and RNA level. (A) Electropherogram of WT and patient DNA sequencing showing the deletion of 8 base pairs at the border of exon 4 and intron 4. (B) RT-PCR of two patients showing that exon 4 is missing due to the 8 bp deletion, as compared to the RT-PCR of a healthy family member and two independent controls.
4 Discussion
Sudan is an African and Arab country with multiple tribes, diverse ethnic groups, and a high rate of consanguinity, an environment that leads to a high prevalence of autosomal recessive conditions. While awareness of genetic disorders is growing, many remain underdiagnosed due to the varying nonspecific symptoms that overlap with more common childhood diseases. This is the first comprehensive report on Sudanese patients with triple A syndrome. In this study, genetic analysis of 31 patients revealed six different mutations in the AAAS gene associated with significant clinical variability even among family members with the same mutation(s), consistent with previous studies (4, 14, 22). The consanguinity rate in this cohort was 80%, which aligns with findings from similar studies (23). Notably, there were no sex-based differences in the presentation of triple A syndrome, with a nearly balanced gender distribution (male-to-female ratio of 0.94). The main presenting symptom was adrenal insufficiency manifested as severe adrenal crisis and hypoglycemic seizure, while alacrima was present in all patients upon evaluation. Alacrima is typically the first and most common symptom to be reported in triple A syndrome patients, although it is often overlooked, despite being present from birth (24, 25). Since isolated alacrima is rare in children, its diagnosis should prompt careful evaluation for triple A syndrome, particularly when accompanied by additional symptoms or a family history of unexplained sibling deaths (26, 27). Adrenal insufficiency is the leading cause of mortality in undiagnosed triple A syndrome patients, which is usually a consequence of ACTH insensitivity and is typically associated with hypoglycemia, chronic vomiting, hyperpigmentation, and, in some cases, sudden death due to adrenal crisis (28). This condition affected 28 (90%) of our patients and was the primary presentation leading to triple A syndrome diagnosis. In our cohort, all patients with adrenal insufficiency exhibited generalized or localized hyperpigmentation, 75% experienced severe adrenal crises, and eight families reported sibling deaths likely attributable to adrenal crisis. The age of PAI onset varied widely, even within the same affected family (F2, F3, F14, F18), and 90% percent of the children in this cohort exhibited PAI symptoms within the first decade of life, with a mean age of onset at 5.5 years. 55% developed PAI symptoms before the age of five, highlighting an early onset of the disease and the need for early recognition and management. Triple A syndrome is the second most common cause of primary adrenal insufficiency (PAI) in Sudan, possibly due to distinctive features that facilitate clinical diagnosis once adrenal insufficiency is recognized (23). Mineralocorticoid deficiency, reported in up to 15% of patients in demographically similar cohorts, is typically associated with the degeneration of the zona glomerulosa. Variability in prevalence across different studies underscores the need for further investigation into this component of the syndrome’s clinical presentation (22, 29). Achalasia was observed in 11 patients (35%) in this cohort, a lower prevalence than reported in some larger studies, potentially due to the younger age of the study participants. Two patients were diagnosed with both achalasia and adrenal insufficiency concurrently, while others developed achalasia later, consistent with findings from other studies (15, 22).
Neurological dysfunction in triple A syndrome often emerges in the 2nd or the 3rd decade(s) of life, indicating the neurodegenerative nature of the disease. However, early presentation in childhood was also observed in our cohort, with 80% of patients presented before the age of 10 years. This may be due to recurrent hypoglycemic seizures or other mechanisms that have yet to be fully elucidated (7, 30). Early diagnosis and treatment appeared to slow down neurological progression, as observed in some families in this study. In these cases, late presenters showed severe progressive neurological symptoms (Patients no. 4, 6, 7, 19) whereas early diagnosed siblings (Patients no. 5, 8, 20, 21) showed milder progression and preserved neurological functions. Long-term follow-up and further studies are needed to elucidate the mechanisms leading to this observation.
Hypertensive encephalopathy, linked to autonomic dysfunction, is a rare complication of triple A syndrome (31). We reported two male siblings who presented with fever, hypertensive crisis, and convulsions. One fully recovered after three weeks in intensive care, while the other progressed to a complete neurovegetative state and died at the age of nine years, highlighting the clinical variability among affected siblings.
Excessive thick nasal mucus discharge, a newly documented feature in this cohort, was observed in 13% of our patients who were associated with three different mutations. One family sought a CT scan, revealing adenoid enlargement and a complicated middle concha bullosa. The other patients had similar symptoms for years, often misattributed to chronic respiratory tract bacterial infections. This discharge may be linked to autonomic dysfunction, but the exact mechanism remains unclear and is yet to be uncovered.
In our Sudanese cohort, six different AAAS gene mutations were identified, four of which were previously described. In general, triple A syndrome-associated mutations can occur in all 16 exons of the AAAS gene in a homozygous or compound heterozygous form (5, 32, 33). In our cohort, mutations were identified in exons 1, 4, 9, and 14. These mutations included predominantly homozygous point mutations and small deletions.
The c.934C>T mutation, one of the most common in our cohort (29%), was first described in an Algerian patient by Tullio-Pelet et al. in the year 2000 (12). All nine affected patients presented with alacrima and adrenal insufficiency, except for patient 3, who had alacrima and a confirmed genetic diagnosis of triple A syndrome but has not developed adrenal insufficiency to date.
The common Arabic founder mutation c.1331+1G>A was also present in 29% of our patients. It is one of the most common AAAS gene mutations worldwide and is most prevalent in North Africa (17). The mutation was initially identified in multiple consanguineous North African triple A patients, and it may represent a regional founder mutation (12). This variant is a substitution at the start of intron 14 that disrupts the splice donor site leading to abnormal transcripts (34). Patients with c.1331+1G>A mutation presented alacrima and adrenal insufficiency except for patient 5, who exhibited alacrima only. Achalasia was observed in just two patients (Patients 9 and 11). Ten percent of the cohort had the p.Tyr19* (Y19X) mutation (F14), resulting in a non-functional protein (19). Despite carrying the same mutation, siblings in this family exhibited different clinical symptoms (Table 2). The lack of adrenal insufficiency in the youngest sibling may be age-related, as the condition can develop later in life.
Another set of siblings (F4) carried the c.885G>A (p.Trp295* or W295X) mutation, first described in 2001 (20). This mutation creates a premature stop codon, causing mRNA degradation (35). Both male siblings from this family developed hypertensive crisis and early neurological and autonomic dysfunction, while the female sibling with the same mutation had preserved neurological function.
Six patients from four families (Patients 12, 16, 17, 18, 30, 31) were identified with a novel c.852delG mutation. This mutation, a deletion of one base pair, leads to a predicted frameshift and a premature stop codon at the protein level (p.Trp284Cysfs*7). All truncating mutations in ALADIN up to amino acid 478 impair the normal function of the protein and, in the homozygous form, lead to triple-A syndrome (7). Interestingly, despite sharing the same mutations, these families (F8, F12, F13) who are from the same tribe and geographical area showed all three cardinal symptoms of triple A syndrome and no neurological involvement as of date. While another family (F20) with two affected siblings presented with full picture of triple A syndrome, including early neurological involvement, learning difficulties and thick nasal discharge, a previously unreported symptom.
Another novel mutation, c.394_399+2delCTGTCTGT, was identified in three male patients from two families (Patients 22, 23, 24) located at the border of exon 4 and intron 4 of the AAAS gene. This mutation affects the splicing of exon 4 and results on RNA-level in skipping of complete exon 4. On protein level skipping of exon 4 leads to an amino acid change of valine at position 103 to alanine and a frameshift with a premature stop codon eight amino acids later (p.Val103Alafs8*). Those patients from unrelated families and the same tribal ethnicity and geographical area had a similar set of symptoms, including alacrima, adrenal insufficiency, and neurological dysfunction, and no achalasia, possibly due to a younger age at presentation. One was also presented with excessive nasal discharge. The similarity in clinical symptoms among patients with the same novel mutation from the same tribe and geographical region suggests a possible role of genetic background in disease manifestation. However, this is contradicted by the significant clinical heterogeneity in symptoms observed among siblings from the same family, despite sharing the same mutation and tribal ancestry.
Triple A syndrome is a rare condition, with limited data from Sub-Saharan Africa and Arab countries. This study presents the largest comprehensive cohort of triple A patients from these countries. It highlights a wide range of clinical symptoms, including newly reported features and identifies a greater number of genetic mutations than previous studies from the region. In a Tunisian study, 26 patients were investigated, with 25 carrying the common Arabic founder mutation c.1331+1G>A, and one patient having the R286X mutation (16). A 2024 study of 10 Moroccan patients from seven families by Belmokhtar et al. found that all were homozygous for the c.1331+1G>A mutation (36). Other non-recurrent mutations such as c.1024C>T, c.1292-1294delTTCinsA, and c.1304delA have been found in North African patients (13, 37, 38).
In conclusion, this study is the first to provide both clinical and genetic insights into a large cohort of Sudanese patients with triple A syndrome. It sheds light on genetic mutations specific to Sub-Saharan Africa and the Arab population, where only a few reports have previously been reported from the region. This could enhance recognition and representation in similar ethnic and geographical populations where the condition is likely underdiagnosed due to limited awareness and diagnostic resources. Our findings also highlight a significant interfamilial and intrafamilial clinical variability, even among patients with the same mutation, suggesting that unlinked genetic and environmental factors may modulate the expression of the mutant genotype. This research fills a critical gap in understanding the diagnosis and genetic spectrum of this rare condition in a country with a high rate of consanguinity. Early diagnosis and treatment could potentially prevent mortality and significantly improve the quality of life for affected individuals.
Data availability statement
The original data presented in the study are included in the article. Further inquiries can be directed to the corresponding authors.
Ethics statement
Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. The study was conducted in accordance with the local legislation and institutional requirements. Written informed consent was obtained from the individuals for the publication of any potentially identifiable data included in this article.
Author contributions
SM: Investigation, Conceptualization, Writing – review & editing, Visualization, Writing – original draft, Data curation. MA: Investigation, Writing – review & editing, Data curation. SH: Writing – review & editing. ES: Writing – review & editing. FL: Writing – review & editing. OB: Writing – review & editing. AI: Data curation, Investigation, Writing – review & editing. HE: Writing – review & editing, Investigation. AH: Funding acquisition, Writing – review & editing, Supervision. KK: Investigation, Supervision, Funding acquisition, Project administration, Visualization, Writing – review & editing. FQ: Visualization, Conceptualization, Validation, Writing – original draft, Funding acquisition, Investigation, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was supported by the Else Kröner-Fresenius-Stiftung & the Eva Luise und Horst Köhler-Stiftung within the Clinician Scientist program RISE to FQ and FL by the Deutsche Forschungsgemeinschaft project no. 314061271-TRR 205/2 to AH and KK and DFG grant KO 3588/2-1 to KK.
Acknowledgments
We thank the families for participating in and supporting this study, Pediatric endocrinologists, and fellows at GIA hospital for their great assistance in samples and data collection. The excellent technical assistance of Dana Landgraf is greatly appreciated. We particularly thank Wayne Lang for proofreading the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Allgrove J, Clayden GS, Grant DB, and Macaulay JC. Familial glucocorticoid deficiency with achalasia of the cardia and deficient tear production. Lancet. (1978) 1:1284–6. doi: 10.1016/S0140-6736(78)91268-0
2. Dumic M, Barišic N, Kusec V, Stingl K, Skegro M, and Stanimirovic A. Long-term clinical follow-up and molecular genetic findings in eight patients with triple A syndrome. Eur J Pediatr. (2012) 171:1453–9. doi: 10.1007/s00431-012-1745-1
3. Milenkovic T, Zdravkovic D, Savic N, Todorovic S, Mitrovic K, and Koehler K. Triple A syndrome: 32 years experience of a single centre (1977–2008). Eur J Pediatr November. (2010) 169:1323–8. doi: 10.1007/s00431-010-1222-7
4. Brooks B, Kleta R, Stuart C, Tuchman M, Jeong A, and Stergiopoulos S. Genotypic heterogeneity and clinical phenotype in triple A syndrome: a review of the NIH experience 2000–2005. Clin Genet. (2005) 68:215–21. doi: 10.1111/j.1399-0004.2005.00482.x
5. Prpic I, Huebner A, Persic M, Handschug K, and Pavletic M. Triple A syndrome: genotype–phenotype assessment. Clin Genet. (2003) 63:415–7. doi: 10.1034/j.1399-0004.2003.00070.x
6. Cehic M, Mitrovic K, Vukovic R, Milenkovic T, Kovacevic G, and Todorovic S. Very early and severe presentation of Triple A syndrome - case report and review of the literature. Front Endocrinol (Lausanne). (2024) 15:1431383. doi: 10.3389/fendo.2024.1431383
7. Cronshaw JM and Matunis MJ. The nuclear pore complex protein ALADIN is mislocalized in triple A syndrome. Proc Natl Acad Sci USA. (2003) 100:5823–7. doi: 10.1073/pnas.1031047100
8. Carvalhal S, Ribeiro SA, Arocena M, Kasciukovic T, Temme A, and Koehler K. The nucleoporin ALADIN regulates Aurora A localization to ensure robust mitotic spindle formation. Mol Biol Cell. (2015) 26:3424–38. doi: 10.1091/mbc.E15-02-0113
9. Hirano M, Furiya Y, Asai H, Yasui A, and Ueno S. ALADIN I482S causes selective failure of nuclear protein import and hypersensitivity to oxidative stress in triple A syndrome. Proc Natl Acad Sci USA. (2006) 103:2298–303. doi: 10.1073/pnas.0505598103
10. Koehler K, End K, Kind B, Landgraf D, Mitzscherling P, and Huebner A. Changes in differential gene expression in fibroblast cells from patients with triple A syndrome under oxidative stress. Horm Metab Res. (2013) 45:102–8. doi: 10.1055/s-0032-1331196
11. Kind B, Koehler K, Krumbholz M, Landgraf D, and Huebner A. Intracellular ROS level is increased in fibroblasts of triple A syndrome patients. J Mol Med. (2010) 88:1233–42. doi: 10.1007/s00109-010-0661-y
12. Tullio-Pelet A, Salomon R, Hadj-Rabia S, Mugnier C, De Laet MH, and Chaouachi B. Mutant WD-repeat protein in triple-A syndrome. Nat Genet. (2000) 26:332–5. doi: 10.1038/81642
13. Handschug K. Triple A syndrome is caused by mutations in AAAS, a new WD-repeat protein gene. Hum Mol Genet. (2001) 10:283–90. doi: 10.1093/hmg/10.3.283
14. Pogliaghi G, Cangiano B, Duminuco P, Vezzoli V, and Bonomi M. Triple-A syndrome (TAS): an in-depth overview on genetic and phenotype heterogeneity. Protein Pept Lett. (2020) 27:1192–203. doi: 10.2174/0929866527666200613215449
15. Patt H, Koehler K, Lodha S, Jadhav S, Yerawar C, and Huebner A. Phenotype–genotype spectrum of AAA syndrome from Western India and systematic review of literature. Endocrine Connections. (2017) 6:901–13. doi: 10.1530/EC-17-0255
16. Kallabi F, Belghuith N, Aloulou H, Kammoun T, Ghorbel S, and Hajji M. Clinical and genetic characterization of 26 Tunisian patients with allgrove syndrome. Arch Med Res. (2016) 47:105–10. doi: 10.1016/j.arcmed.2016.04.004
17. Kallabi F, Ben Rebeh I, Felhi R, Sellami D, Masmoudi S, and Keskes L. Molecular analysis of Libyan families with allgrove syndrome: geographic expansion of the ancestral mutation c.1331+1G>A in North Africa. Horm Res Paediatr. (2016) 85:18–21. doi: 10.1159/000441653
18. Charmandari E, Nicolaides NC, and Chrousos GP. Adrenal insufficiency. Lancet. (2014) 383:2152–67. doi: 10.1016/S0140-6736(13)61684-0
19. Qin K, Du X, and Rich BH. An Alu-mediated rearrangement causing a 3.2kb deletion and a novel two base pair deletion in AAAS gene as the cause of triple A syndrome. Mol Genet Metab. (2007) 92:359–63. doi: 10.1016/j.ymgme.2007.08.116
20. Schmittmann-Ohters K, Huebner A, Richter-Unruh A, and Hauffa BP. Clinical and novel molecular findings in a 6.8-year-old turkish boy with triple A syndrome. Horm Res Paediatr. (2001) 56:67–72. doi: 10.1159/000048093
21. Jaganathan K, Kyriazopoulou Panagiotopoulou S, McRae JF, Darbandi SF, Knowles D, and Li YI. Predicting splicing from primary sequence with deep learning. Cell. (2019) 176:535–548.e24. doi: 10.1016/j.cell.2018.12.015
22. Singh K, Puri RD, Bhai P, Arya AD, Chawla G, and Saxena R. Clinical heterogeneity and molecular profile of triple A syndrome: a study of seven cases. J Pediatr Endocrinol Metab. (2018) 31:799–807. doi: 10.1515/jpem-2018-0023
23. Musa SA, Hassan SS, Ahmed AI, Ngwiri T, Fadlalbari GF, and Ibrahim AA. Clinical profile, etiology, and diagnostic challenges of primary adrenal insufficiency in Sudanese children: 14-years’ experience from a resource limited setting. J Pediatr Endocrinol Metab. (2022) 35:231–7. doi: 10.1515/jpem-2021-0545
24. Derrar R, Boutimzine N, Laghmari A, Alouane A, and Daoudi R. Alacrymie congénitale révélant un syndrome d’allgrove: à propos de trois cas. Pan Afr Med J. (2015) 20:1–7. doi: 10.11604/pamj.2015.20.359.4717
25. Tibussek D, Ghosh S, Huebner A, Schaper J, Mayatepek E, and Koehler K. Crying without tears” as an early diagnostic sign-post of triple A (Allgrove) syndrome: two case reports. BMC Pediatr. (2018) 18:6. doi: 10.1186/s12887-017-0973-y
26. Belge Bilgin G, Bilgin C, Orscelik A, Musmar B, and Kandemirli SG. Isolated congenital lacrimal gland agenesis. Cureus. (2024) 16:e57732. doi: 10.7759/cureus.57732
27. Alwohaib M, Schellini SA, Elkhamary SM, and Al Shaikh O. Isolated bilateral congenital lacrimal gland agenesis – Report of two cases. Saudi J Ophthalmol. (2017) 31:257–9. doi: 10.1016/j.sjopt.2017.04.008
28. Flokas ME, Tomani M, Agdere L, and Brown B. Triple A syndrome (Allgrove syndrome): improving outcomes with a multidisciplinary approach. Pediatr Health Med Ther. (2019) 10:99–106. doi: 10.2147/PHMT.S173081
29. Grant DB, Barnes ND, Dumic M, Ginalska-Malinowska M, Milla PJ, and von Petrykowski W. Neurological and adrenal dysfunction in the adrenal insufficiency/alacrima/achalasia (3A) syndrome. Arch Dis Child. (1993) 68:779–82. doi: 10.1136/adc.68.6.779
30. Weiman DI, Gillespie MK, Hartley T, Osmond M, and Ito Y. Care4Rare Canada consortium, neurophysiological characteristics of allgrove (Triple A) syndrome: case report and literature review. Child Neurol Open. (2021) 8:2329048X211031059. doi: 10.1177/2329048X211031059
31. Aftab S, Manzoor J, Talat N, Khan HS, Subhanie M, and Khalid NA. Allgrove syndrome: adrenal insufficiency with hypertensive encephalopathy. J Coll Physicians Surg Pak. (2016) 26:790–2.
32. Houlden H. Clinical and genetic characterization of families with triple A (Allgrove) syndrome. Brain. (2002) 125:2681–90. doi: 10.1093/brain/awf270
33. Huebner A, Kaindl AM, Braun R, and Handschug K. NEW INSIGHTS INTO THE MOLECULAR BASIS OF THE TRIPLE A SYNDROME. Endocrine Res. (2002) 28:733–9. doi: 10.1081/ERC-120016998
34. Kallabi F, Ben Rhouma B, Baklouti S, Ghorbel R, Felhi R, and Keskes L. Splicing defects in the AAAS gene leading to both exon skipping and partial intron retention in a Tunisian patient with allgrove syndrome. Horm Res Paediatr. (2016) 86:90–3. doi: 10.1159/000446539
35. AlOmran HA, Busaleh F, Alhashim Z, AlHelal M, Alsaleh Y, and AlJabri A. Mineralocorticoid deficiency as an early presenting symptom of allgrove syndrome with novel mutation: A case report. Cureus. (2021) 13(11):e19316. doi: 10.7759/cureus.19316
36. Belmokhtar KY, Cherkaoui I, Lhousni S, Elidrissi Errahhali M, Elidrissi Errahhali M, and Charif M. Triple-A syndrome in Morocco: founder effect, age estimation of the AAAS c.1331 + 1G<A variant, and implications for genetic diagnosis. Mol Syndromol. (2024) 15:96–103. doi: 10.1159/000533894
37. Gaiani F, Gismondi P, Minelli R, Casadio G, de’Angelis N, and Fornaroli F. Case report of a familial triple: a syndrome and review of the literature. Medicine. (2020) 99:e20474. doi: 10.1097/MD.0000000000020474
Keywords: triple A syndrome, Allgrove syndrome, alacrima, achalasia, primary adrenal insufficiency, familial cases, Sub-Saharan Africa
Citation: Musa SA, Abdullah MA, Hassan SS, Streiff E, Lange F, Babiker OO, Ibrahim AA, Elshafie HA, Huebner A, Koehler K and Quitter F (2025) Insights into genetic and clinical profiles of triple A syndrome in Sudanese children. Front. Endocrinol. 16:1617552. doi: 10.3389/fendo.2025.1617552
Received: 24 April 2025; Accepted: 29 July 2025;
Published: 22 August 2025.
Edited by:
Henrik Falhammar, Karolinska Institutet (KI), SwedenReviewed by:
Lara Elizabeth Graves, Children’s Hospital at Westmead, AustraliaMarcellus Andre Walker Md, Johns Hopkins University, United States
Sami Bizzari, Hamdan bin Rashid Foundation for Medical and Educational Sciences, United Arab Emirates
Copyright © 2025 Musa, Abdullah, Hassan, Streiff, Lange, Babiker, Ibrahim, Elshafie, Huebner, Koehler and Quitter. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Friederike Quitter, RnJpZWRlcmlrZS5RdWl0dGVyQHVrZGQuZGU=; Salwa A. Musa, U2Fsd2FfMjFAaG90bWFpbC5jb20=