 Jingyuan Ma
Jingyuan Ma Yanna Ma1†
Yanna Ma1† Yunshu Zhang
Yunshu Zhang Jifeng Liu
Jifeng Liu- 1The First Clinical Medical College, Liaoning University of Traditional Chinese Medicine, Shenyang, China
- 2The First Affiliated Hospital of Dalian Medical University, Dalian, China
Metabolic dysfunction-associated steatotic liver disease (MASLD) is now the most common chronic liver condition worldwide, closely linked to obesity, insulin resistance, and metabolic syndrome. It spans a spectrum from simple steatosis to metabolic dysfunction-associated steatohepatitis (MASH), fibrosis, and hepatocellular carcinoma. This review examines the core metabolic disruptions—particularly in lipid, glucose, bile acid, amino acid, and iron metabolism—that drive MASLD pathogenesis. It also explores how genetic variants such as PNPLA3, TM6SF2, GCKR, HSD17B13, and MBOAT7 contribute to disease susceptibility and variability in clinical outcomes. The interaction between genetic background and metabolic stress is central to the heterogeneity seen in disease progression and treatment response. We further discuss persistent clinical challenges and summarize recent advances in drugs, natural compounds, and microbiota-based strategies. Finally, we highlight the promise of multi-omics approaches to better stratify patients and personalize management. A clearer understanding of the molecular and clinical complexity of MASLD will be key to developing more effective and individualized strategies for diagnosis and treatment.
1 Introduction
Metabolic dysfunction-associated steatotic liver disease (MASLD) represents a spectrum of hepatic steatosis occurring in the absence of significant alcohol consumption or secondary causes of liver injury (1). Compared with the previous terminology, the current designation of MASLD is considered more appropriate, as it better emphasizes the central role of metabolic dysfunction in disease onset and progression (2, 3). A recent meta-analysis involving over 78 million participants from 38 countries estimated the global prevalence of MASLD at 30.2%, with higher rates in South America (34.0%) and Asia (30.9%) (4). Such a vast affected population underscores the urgent need for improved diagnostic and therapeutic strategies. As prevalence continues to rise globally, attention has shifted from its hepatic manifestations to its broader systemic impact. Beyond hepatic complications such as metabolic dysfunction-associated steatohepatitis (MASH), cirrhosis, and hepatocellular carcinoma (HCC), MASLD is now recognized as a multisystem disease (5). It is closely associated with a range of extrahepatic manifestations, including type 2 diabetes, cardiovascular disease, chronic kidney disease, and certain cancers (6, 7). These systemic consequences significantly contribute to the overall morbidity and mortality associated with MASLD.
In addition to lifestyle and metabolic contributors, accumulating evidence points to prenatal determinants as early-life factors influencing MASLD susceptibility (8). Maternal obesity, overnutrition, and metabolic disorders during pregnancy have been associated with increased hepatic steatosis in offspring, potentially via epigenetic programming of hepatic lipid metabolism and adipogenesis (9–11). These findings highlight a critical developmental window for future prevention and risk modification strategies.
The pathophysiology of MASLD involves complex interactions between metabolic dysregulation and genetic susceptibility. The key metabolic disturbances span lipid metabolism, glucose homeostasis, bile acid cycling, and iron handling (12–15), while common genetic variants in patatin-like phospholipase domain-containing protein 3 (PNPLA3), transmembrane 6 superfamily member 2 (TM6SF2), glucokinase regulatory protein (GCKR), hydroxysteroid 17-beta dehydrogenase 13 (HSD17B13), and membrane-bound O-acyltransferase domain-containing 7 (MBOAT7) significantly influence disease progression (16–19). This metabolic-genetic interplay creates substantial clinical heterogeneity, complicating the development of precision treatment approaches.
Persistent diagnostic and therapeutic challenges further hinder clinical management. Reliance on invasive liver biopsies persists due to limitations in non-invasive alternatives, while the absence of approved pharmacotherapeutics underscores unmet clinical needs (20). This review synthesizes current understanding of MASLD’s metabolic and genetic underpinnings, examines critical barriers to clinical management, and explores multi-omics strategies for advancing precision medicine in this complex disorder.
2 Metabolic dysregulation
MASLD arises from a multifaceted disruption of metabolic homeostasis involving several interrelated biochemical networks. Perturbations in lipid handling, glucose utilization, bile acid circulation, amino acid turnover, and iron metabolism collectively shape the metabolic landscape that underlies disease onset and progression. These metabolic imbalances not only contribute individually but also interact synergistically, amplifying hepatic dysfunction and fostering disease heterogeneity (Figure 1).
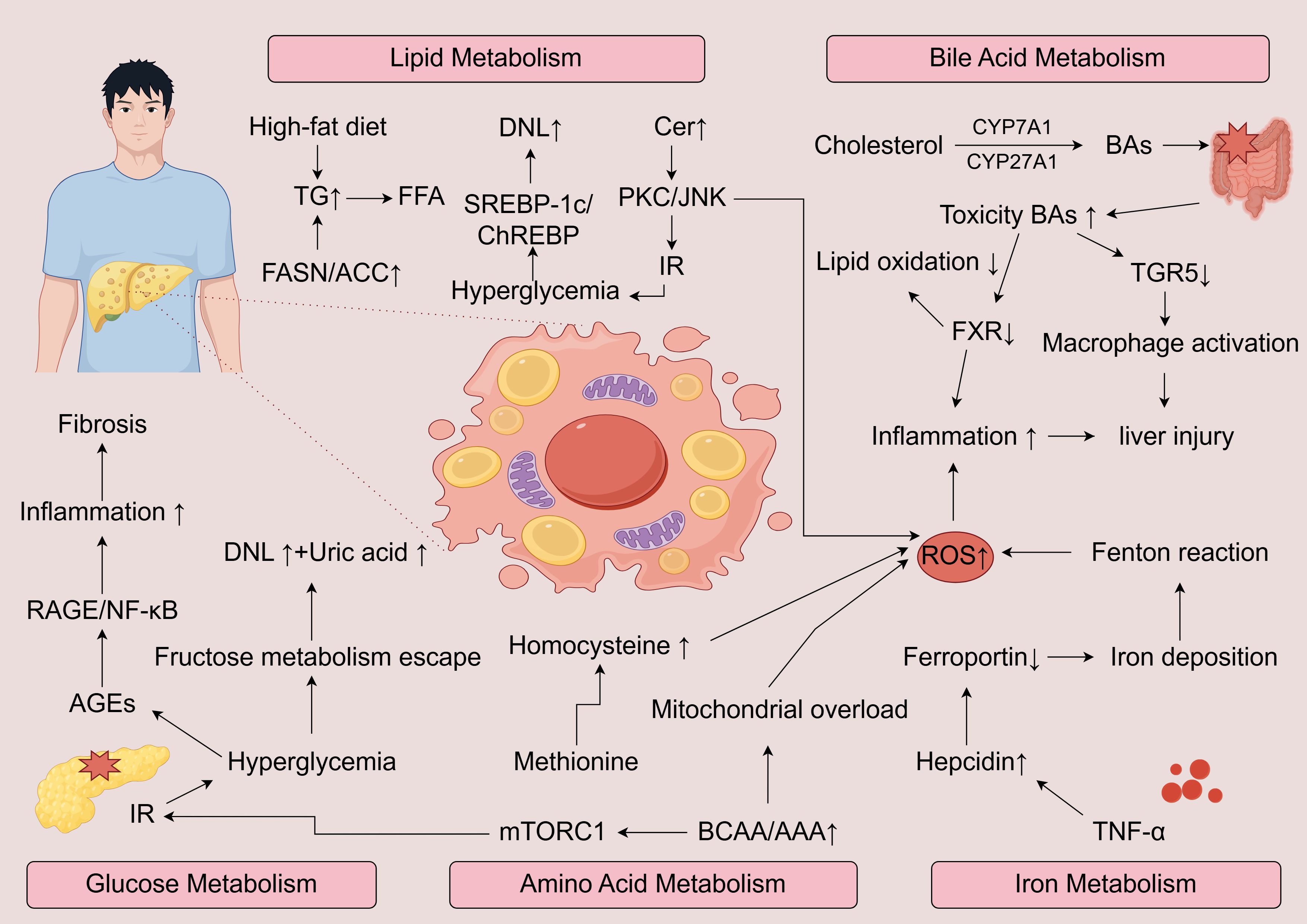
Figure 1. Schematic overview of the major metabolic pathways driving the progression of MASLD to MASH. Excess lipid intake enhances DNL, FFA accumulation, and ceramide synthesis, activating PKC/JNK signaling and insulin resistance. Disturbed glucose metabolism promotes AGEs-RAGE/NF-κB signaling, inflammation, and fibrosis. Altered bile acid metabolism with increased toxic bile acids and impaired FXR/TGR5 signaling exacerbates oxidative stress, macrophage activation, and hepatocellular injury. Dysregulated amino acid metabolism, including elevated BCAA/AAA and homocysteine, drives mTORC1 activation, mitochondrial overload, and ROS production. Iron overload due to hepcidin–ferroportin imbalance amplifies oxidative damage via the Fenton reaction. Together, these metabolic insults converge on excessive ROS generation and chronic inflammation, promoting hepatocyte injury, fibrosis, and disease progression.
2.1 Aberrant lipid metabolism
Among the various metabolic abnormalities, disordered lipid metabolism is widely regarded as a central driver of hepatic steatosis. Hepatic lipid overload stems from an imbalance between fatty acid influx, endogenous lipid synthesis, mitochondrial oxidation, and lipid export via very low-density lipoprotein (VLDL) secretion (21). In MASLD pathophysiology, hepatocyte lipid accumulation originates from three sources: dietary lipids, adipose-derived free fatty acids (FFAs), and enhanced de novo lipogenesis (DNL) (22). In the context of insulin resistance, enhanced lipolytic activity in peripheral adipose tissue leads to elevated plasma FFA levels, which are rapidly delivered to the liver through the portal circulation (23). Simultaneously, high-fat dietary patterns further augment lipid input. Hepatic DNL becomes hyperactive under these metabolic conditions, primarily driven by transcriptional activation of lipogenic regulators such as sterol regulatory element-binding protein-1c (SREBP-1c) and carbohydrate responsive element binding protein (ChREBP). These factors stimulate the expression of enzymes including fatty acid synthase (FASN) and acetyl-CoA carboxylase (ACC), leading to increased triglyceride production (24, 25). Carbohydrate metabolism intersects with lipogenesis via glucose-activated ChREBP and UDP-glucose-mediated SREBP-1c regulation, creating a metabolic crossroad between lipid synthesis and glycogen storage (26, 27). Chronic caloric excess disrupts this balance, overwhelming hepatocellular lipid-handling capacity.
Critically, MASLD progression reflects not triglyceride accumulation per se, but rather cytotoxic lipid species buildup. Saturated fatty acids, ceramides, and free cholesterol induce organelle stress through endoplasmic reticulum dysfunction, mitochondrial damage, and lysosomal permeabilization (28). These insults converge on cell death pathways—apoptosis, necroptosis, pyroptosis—that drive steatohepatitis development (29). Among these toxic lipids, ceramides (Cer) play a pivotal role by activating protein kinase C (PKC) and c-Jun N-terminal kinase (JNK) pathways, thereby impairing insulin signaling and inducing hepatocyte apoptosis (30–32). Cer accumulation further exacerbates mitochondrial dysfunction and reactive oxygen species (ROS) production, which activate inflammatory signaling and aggravate hepatic injury (33). In addition, excessive free cholesterol accumulation in hepatocytes can provoke ER stress and mitochondrial impairment, leading to activation of the NLRP3 inflammasome, release of pro-inflammatory cytokines, and polarization of Kupffer cells toward a pro-inflammatory phenotype (34–37). These stress and immune responses, initiated by lipid metabolic dysregulation, synergistically drive the transition from simple steatosis to non-alcoholic steatohepatitis (MASH).
2.2 Insulin resistance and glucose metabolic dysregulation
Insulin resistance (IR) is a pivotal pathological mechanism underlying the onset and progression of MASLD. It induces systemic metabolic disturbances primarily through dysregulated interactions among adipose tissue, liver, and skeletal muscle (38). In adipose tissue, IR results in aberrant activation of hormone-sensitive lipase (HSL), leading to enhanced lipolysis and increased release of FFAs into the circulation, which are subsequently taken up by the liver (39). This process significantly contributes to hepatic lipid overload and metabolic derangement. Simultaneously, elevated levels of pro-inflammatory adipokines (e.g., TNF-α, IL-6) and decreased adiponectin further impair AMP-activated protein kinase (AMPK)-mediated fatty acid oxidation, exacerbating lipid accumulation within hepatocytes (40).
In the liver, IR presents as “selective hepatic insulin resistance”: insulin’s ability to suppress gluconeogenesis is diminished, resulting in fasting hyperglycemia, whereas its stimulatory effect on DNL remains intact or even enhanced (41). This paradox is largely mediated by persistent activation of the mTORC1/SREBP-1c axis under hyperinsulinemic conditions, which drives fatty acid synthesis and triglyceride accumulation, thereby promoting hepatic steatosis and fibrosis (42). In addition, lipid intermediates such as diacylglycerol (DAG) activate protein kinase Cϵ (PKCϵ), impairing insulin signaling and perpetuating a vicious cycle of lipotoxicity and insulin resistance (43).
In skeletal muscle, IR impairs glucose clearance due to defective GLUT4 translocation, which forces the liver to compensate by redirecting excess glucose into lipogenesis via ChREBP-dependent metabolic reprogramming—constituting a “glucose-to-lipid” maladaptive feedback loop (44, 45). Chronic hyperglycemia further exacerbates hepatocellular injury through multiple mechanisms: excessive glucose activates the polyol pathway, depleting NADPH and inducing oxidative stress, while advanced glycation end-products (AGEs) trigger inflammatory cascades via the RAGE/NF-κB axis, thereby accelerating hepatic fibrogenesis (46–48).
At the metabolic level, hyperglycemia robustly promotes DNL through the ChREBP–FASN/ACC pathway. Fructose metabolism, owing to its “metabolic bypass” nature, circumvents key regulatory steps in glycolysis and rapidly enters the tricarboxylic acid (TCA) cycle, generating excess acetyl-CoA, which fuels lipogenesis and uric acid production while inducing ER stress and oxidative damage (49–52). Additionally, acetyl-CoA derived from glucose metabolism not only serves as a substrate for DNL but also epigenetically modulates lipogenic genes such as stearoyl-CoA desaturase 1 (SCD1) and diacylglycerol acyltransferase 2 (DGAT2) via histone acetylation, thereby establishing a long-lasting “metabolic memory” that predisposes hepatocytes to persistent lipid accumulation (53, 54).
Notably, hepatocellular lipotoxic mediators such as ceramides and DAGs activate PKCϵ, leading to serine phosphorylation of insulin receptor substrate (IRS), which disrupts PI3K/Akt signaling and aggravates hepatic insulin resistance (43, 55). Concurrently, IR-associated hyperinsulinemia enhances NADPH oxidase (NOX) activity, triggering ROS generation that further suppresses insulin signaling via the JNK/IKKβ pathway (56–58). Moreover, lipid peroxidation products such as 4-hydroxynonenal (4-HNE) directly impair mitochondrial function and modify key signaling proteins, amplifying inflammatory and fibrotic responses that facilitate the transition from simple steatosis to necroinflammatory injury and hepatic fibrosis (59, 60).
2.3 Bile acid metabolism dysregulation
Bile acids (BAs) are bioactive molecules synthesized in the liver from cholesterol and secreted into the intestine via the bile ducts, where they facilitate the emulsification, digestion, and absorption of dietary lipids (61). BA synthesis proceeds through two primary pathways: the classical (or neutral) pathway, in which cholesterol 7α-hydroxylase (CYP7A1) serves as the rate-limiting enzyme, and the alternative (or acidic) pathway, primarily mediated by sterol 27-hydroxylase (CYP27A1) (62). Once synthesized, BAs activate the farnesoid X receptor (FXR), which induces the expression of small heterodimer partner (SHP), thereby suppressing the transcription of CYP7A1 and CYP8B1, ultimately reducing BA synthesis through negative feedback regulation (63). In addition to biosynthetic control, FXR regulates BA transport by promoting hepatic BA efflux and limiting intrahepatic BA accumulation, thus protecting against cholestasis and hepatocellular injury (64). FXR also induces intestinal fibroblast growth factor 15/19 (FGF15/19), which further acts on the liver to inhibit BA synthesis and reinforce homeostatic regulation (65, 66).
Following their secretion and intestinal metabolism, BAs undergo reabsorption and enterohepatic circulation. A portion of BAs enters systemic circulation, where they interact with nuclear and membrane-bound receptors across multiple organs. This cross-organ communication enables BAs to function not only as digestive agents but also as metabolic signaling molecules that influence lipid and glucose metabolism, energy homeostasis, and immune responses (61).
In the context of MASLD, BA metabolic dysregulation has been identified as a critical contributor to disease pathogenesis. Alterations in BA composition and impaired FXR/TGR5 signaling may disrupt hepatic lipid and glucose homeostasis, promote hepatic fat accumulation, exacerbate inflammatory responses, and accelerate liver injury (67, 68). A study shows that several bile acids are significantly elevated in patients with MASLD, suggesting their potential utility as biomarkers for distinguishing MASH from simple steatosis (69).
2.4 Amino acid metabolism dysregulation
An expanding body of research underscores the significant contribution of amino acid metabolic disturbances to MASLD pathogenesis (70–73). As the liver serves as a central hub for amino acid synthesis, degradation, and interconversion, perturbations in systemic amino acid profiles are frequently observed in hepatic dysfunction (74). In MASLD, elevated circulating concentrations of branched-chain amino acids (BCAAs) and aromatic amino acids (AAAs) have been consistently identified (72, 75, 76). These elevations correlate strongly with insulin resistance and altered lipid handling, thereby facilitating hepatic fat accumulation and exacerbating inflammation (77). Beyond their metabolic roles, amino acid-derived intermediates can generate reactive oxygen species and impair mitochondrial function, further disrupting insulin signaling and accelerating disease progression (78).
Among individual amino acids, glycine levels are frequently depleted in MASLD. As a precursor for glutathione biosynthesis, reduced glycine availability compromises antioxidant defenses. Supplementation with glycine has demonstrated beneficial effects on liver steatosis and inflammation in both preclinical models and clinical trials (79). Disruptions in methionine metabolism are also prominent in MASLD, often resulting in hyperhomocysteinemia and diminished glutathione production—factors that collectively enhance oxidative stress and hepatic lipid deposition (80). In pediatric MASLD populations, metabolic alterations involving methionine, tyrosine, and tryptophan pathways have been observed, suggesting that such changes may manifest early in disease evolution (81). Additionally, aberrant expression of glutamine-metabolizing enzymes has been reported in MASLD livers, potentially affecting redox regulation and modulating immune responses (82).
2.5 Iron metabolism dysregulation
Iron, a vital micronutrient, participates in critical physiological processes such as oxygen transport, mitochondrial respiration, and DNA replication (83). Disruption of iron equilibrium can interfere with cellular function and has been implicated in the pathophysiology of diverse conditions, including anemia, neurodegenerative diseases, and malignancies (84). Increasing evidence also supports a critical role for iron metabolism dysregulation in the development and progression of MASLD (85, 86).
Approximately one-third of MASLD patients exhibit elevated serum ferritin levels, normal or mildly increased transferrin saturation, and mild hepatic iron deposition—a constellation referred to as “dysmetabolic iron overload syndrome (DIOS) “ (87). Excessive hepatic iron accumulation promotes the generation of hydroxyl radicals via the Fenton reaction, triggering lipid peroxidation, mitochondrial dysfunction, and apoptosis (88). These events exacerbate hepatic inflammation and fibrosis, ultimately accelerating the transition from simple steatosis to MASH.
Moreover, iron overload has been closely linked to insulin resistance, potentially by impairing insulin signaling and reducing insulin sensitivity, thereby facilitating hepatic fat accumulation (89). In individuals with MASLD, pro-inflammatory cytokines—particularly tumor necrosis factor-α (TNF-α)—can induce hepatic overexpression of the iron-regulatory hormone hepcidin. Elevated hepcidin suppresses ferroportin, the principal iron exporter, thereby promoting iron retention within hepatocytes and Kupffer cells. This accumulation enhances oxidative stress and pro-inflammatory signaling, exacerbating liver injury and fibrosis (90).
2.6 Metabolic-genetic crosstalk
MASLD results from the interplay between systemic metabolic stressors and genetic predisposition, forming a complex disease network. Metabolic disturbances—such as insulin resistance, lipotoxicity, and bile acid dysregulation—initiate and perpetuate hepatic injury, while genetic variants influence individual susceptibility, disease progression, and therapeutic response.
For instance, the PNPLA3 I148M variant impairs lipid droplet remodeling, promotes free cholesterol accumulation, and induces mitochondrial dysfunction in hepatic stellate cells (LX-2), thereby activating them and contributing to liver fibrosis (91, 92). The TM6SF2 E167K mutation reduces VLDL secretion, heightening hepatocellular lipid stress (93). In glucose metabolism, GCKR P446L enhances DNL in hyperinsulinemic states, especially under high-glucose dietary conditions (94). MBOAT7 loss impairs phospholipid remodeling, exacerbating lipid accumulation and inflammatory signaling under hyperinsulinemic or lipotoxic conditions, thereby promoting fibrosis (95, 96). In contrast, loss-of-function variants in HSD17B13 can attenuate hepatocellular injury and inflammatory responses under metabolic stresses such as lipotoxicity and oxidative stress, thereby conferring protection against MASH and fibrosis (19, 97).
These examples highlight that genetic risk is context-dependent, with phenotypic expression shaped by the metabolic milieu. Conversely, metabolic interventions may yield variable outcomes depending on genetic background. This metabolic-genetic crosstalk explains the clinical heterogeneity of MASLD and supports a shift toward genotype-informed, metabolism-guided precision medicine. Furthermore, gene-environment interactions play a pivotal role in modulating MASLD severity. Dietary composition (e.g., high sugar or saturated fat intake), physical inactivity, and even prenatal exposures may amplify or attenuate the phenotypic effects of genetic variants such as PNPLA3 I148M or TM6SF2 E167K. Understanding these interactions is crucial for translating genetic insights into effective lifestyle or pharmacological interventions.
3 Genetic polymorphisms
Genetic variation critically modulates MASLD susceptibility and progression (98, 99). Key single nucleotide polymorphisms (SNPs) in PNPLA3, TM6SF2, GCKR, HSD17B13, and MBOAT7 emerge as central regulators of hepatic lipid dynamics and fibrogenesis (100). Deciphering their molecular impacts provides critical insights for personalized therapeutic development. The genetic polymorphisms of MASLD are shown in Table 1. These associations have been validated across diverse ethnic groups using genome-wide association studies (GWAS) and candidate gene analyses. Sample sizes vary widely across studies—from a few hundred individuals to meta-analyses involving over one million participants. Techniques such as next-generation sequencing (NGS), TaqMan genotyping, and array-based GWAS platforms have been employed to establish robust links between these polymorphisms and MASLD phenotypes.
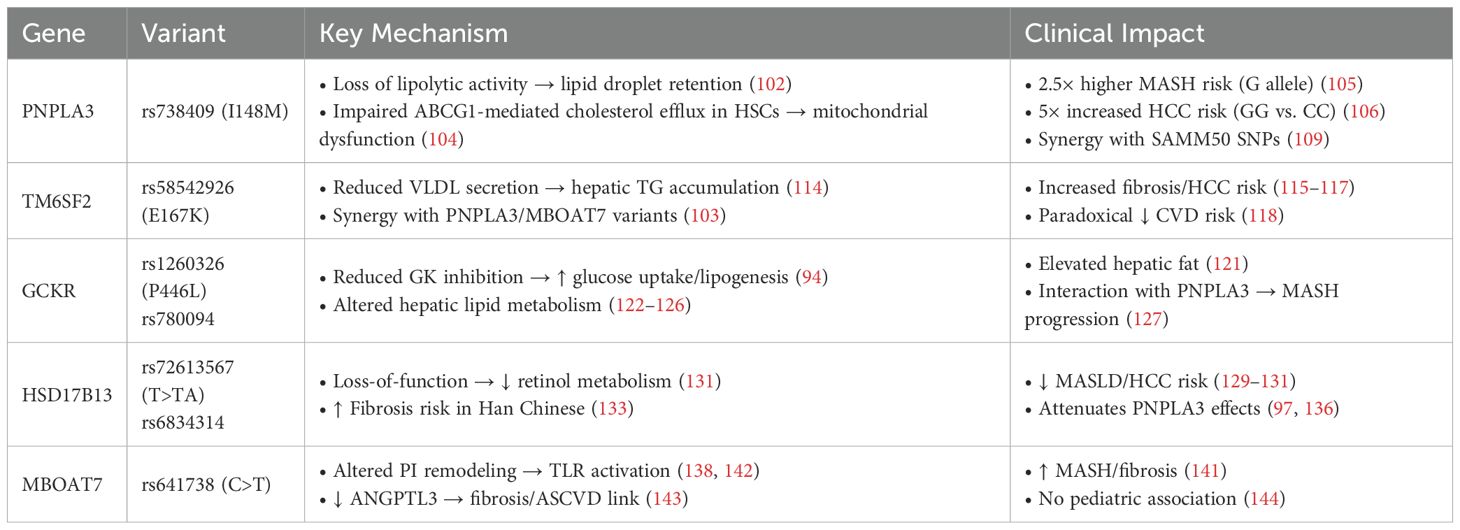
Table 1. Genetic polymorphisms in MASLD: functional impacts and clinical associations.
3.1 PNPLA3
The PNPLA3 rs738409 C>G variant (resulting in an isoleucine-to-methionine substitution at position 148, i.e., I148M) constitutes the strongest genetic risk determinant for MASLD (101). This substitution abolishes enzymatic activity, impairing lipid droplet hydrolysis and promoting intracellular retention of triglycerides and unesterified cholesterol (102). The pathogenic variant accumulates on lipid droplets, forming dysfunctional aggregates that prevent lipid release—a direct mechanistic link to hepatic steatosis (103).
Beyond steatosis, the I148M variant also impairs cholesterol handling in hepatic stellate cells (HSCs), specifically by reducing ABCG1-mediated cholesterol efflux. The resulting intracellular cholesterol buildup triggers mitochondrial dysfunction—evidenced by diminished ATP synthesis, elevated ROS, and compromised mitochondrial membrane potential—which collectively activate HSCs and promote fibrogenesis (104). Epidemiological analyses have linked the I148M variant to a significantly elevated risk of advanced liver disease. Carriers of the G allele exhibit an approximately 2.5-fold higher likelihood of developing MASH (105). Moreover, patients with the GG genotype show approximately five-fold increased HCC risk relative to CC individuals (106).
Importantly, the pathological influence of the PNPLA3 I148M variant extends beyond MASLD, suggesting broader implications across metabolic and fibrotic liver conditions. In patients with chronic hepatitis C, this polymorphism has also been associated with more severe steatosis and fibrosis, indicating its broader relevance across liver diseases (107, 108). Additionally, the combined polymorphisms in PNPLA3 and SAMM50—specifically four SNPs—have been linked to an increased risk of MASLD and elevated serum aspartate aminotransferase (AST)/alanine aminotransferase (ALT) levels, suggesting more severe hepatic injury (109). SAMM50 encodes a mitochondrial sorting and assembly machinery protein essential for maintaining mitochondrial structure and respiratory function (110). Variants in SAMM50 may disrupt mitochondrial integrity and enhance oxidative stress, thereby exacerbating hepatic steatosis and inflammation in synergy with PNPLA3 mutations. Collectively, the PNPLA3 I148M variant represents not only a major genetic driver of MASLD and its complications but also a potential biomarker for fibrosis and HCC risk, as well as a promising target for precision therapeutics (111, 112).
3.2 TM6SF2
TM6SF2 gene, located on chromosome 19p13.3, encodes a protein involved in hepatic lipid metabolism and has been strongly implicated in MASLD pathogenesis (113). A notable SNP, rs58542926, results in a glutamate-to-lysine substitution at position 167 (E167K) and is recognized as an independent risk factor for hepatic steatosis. This variant impairs the function of TM6SF2, reducing the efficiency of very low-density lipoprotein VLDL lipidation and secretion, which in turn causes triglyceride buildup within hepatocytes and promotes fatty liver developmen (114).
Carriers of the E167K variant not only face an elevated risk of MASLD but are also more prone to developing liver fibrosis and HCC (115–117). Additionally, emerging evidence suggests that TM6SF2 interacts synergistically with other genetic variants, including those in PNPLA3 and MBOAT7, further aggravating hepatic lipid accumulation, inflammation, and fibrotic progression (103).
Curiously, despite its association with liver disease, the E167K variant correlates with lower circulating triglyceride levels and a reduced risk of cardiovascular disease (CVD) (118). This paradox points to a potential metabolic trade-off, wherein hepatic fat retention occurs alongside decreased systemic lipid availability, possibly conferring some cardiovascular benefit. Nonetheless, the broader metabolic implications of this relationship remain incompletely understood and merit further investigation.
3.3 GCKR
GCKR gene encodes a critical regulator of glucose metabolism in hepatic and pancreatic tissues. By reversibly binding glucokinase (GK), GCKR modulates GK’s localization and enzymatic activity, serving as both a metabolic sensor and a safeguard against excessive glucose flux (94). Through this mechanism, GCKR contributes to the balance between glucose utilization and lipid synthesis, and its dysfunction can predispose the liver to metabolic stress and steatosis.
Genetic variants in GCKR have garnered attention due to their associations with diverse metabolic traits, including elevated fasting triglycerides, altered insulin sensitivity, and increased risk of MASLD (119–121). Among the most studied are rs1260326, which leads to a proline-to-leucine substitution at position 446 (P446L), and rs780094. Both variants have been robustly linked to hepatic fat accumulation and MASLD susceptibility (122). Functionally, the P446L variant reduces GCKR’s inhibitory effect on GK, enhancing hepatic glucose uptake and subsequent lipogenesis (94, 123). This, in turn, activates gluconeogenesis and de novo lipogenesis pathways, thereby promoting intracellular lipid accumulation and hepatic steatosis.
Another notable variant, rs780094, although located in an intronic region, modulates GCKR expression and has been linked to altered hepatic lipid metabolism (124–126). Carriers of this variant tend to exhibit elevated serum triglycerides and an increased risk of hepatic fat accumulation.
Interestingly, a population-based study in Turkey demonstrated that polymorphisms in both GCKR (rs1260326) and PNPLA3 (rs738409) are significantly associated with an elevated risk of MASH (127). Among them, the rs738409 variant appears to exert a stronger influence on disease progression.
3.4 HSD17B13
Hydroxysteroid 17-beta dehydrogenase 13 (HSD17B13) is a lipid droplet-associated hepatic enzyme with retinol dehydrogenase activity, involved in the metabolism of retinol (128). Genetic variants in HSD17B13, particularly the rs72613567 (T>TA) splice site insertion, have been extensively investigated in relation to MASLD (19). This variant results in a loss-of-function protein, which may mitigate hepatic inflammation and fibrosis by modulating retinoid metabolism, and has been associated with a reduced risk of both MASLD and HCC (129–131).
Studies have shown that both the rs72613567 and rs6834314 variants are negatively associated with MASLD and MASH, and correlate with a lower incidence of adverse hepatic outcomes in multiethnic Asian cohorts (132). However, in the Han Chinese population, the rs72613567:TA variant has been reported to be associated with an increased risk of liver fibrosis, suggesting a potential ethnic-specific effect (133). Moreover, a retrospective study found that the rs72613567:TA variant does not confer protection in advanced chronic liver disease and is associated with an increased risk of decompensation and mortality (134). Intriguingly, this variant may exert a protective effect against alcohol-induced liver damage, particularly in specific populations such as Han Chinese (135).
The interaction between HSD17B13 and other MASLD-associated genes, such as PNPLA3, has also attracted attention. Evidence suggests that the rs72613567:TA variant may attenuate the deleterious hepatic effects of the PNPLA3 I148M mutation, as reflected by lower serum transaminase levels and reduced hepatic inflammation (97). In a Japanese MASLD cohort, carriers of the HSD17B13 rs6834314 G allele exhibited a diminished impact of the PNPLA3 rs738409 GG genotype on the development of advanced fibrosis, further supporting a modifying role of HSD17B13 in genetic susceptibility to liver disease (136).
3.5 MBOAT7
MBOAT7 gene encodes a critical acyltransferase involved in the remodeling of phosphatidylinositol (PI), a key component of membrane phospholipids (137). MBOAT7 is highly expressed in hepatocytes and plays an essential role in maintaining membrane lipid composition and regulating intracellular signaling pathways (138). A functional variant, rs641738 C>T, has been strongly associated with MASLD susceptibility and progression in several large GWAS (139, 140).
The T allele of rs641738 is associated with decreased MBOAT7 expression, leading to reduced incorporation of arachidonic acid into PI and altering hepatic membrane lipid compositionn (96, 138). This promotes hepatic lipid accumulation and increases the risk of inflammation and fibrosis. A meta-analysis involving over one million participants found that this variant is significantly associated with increased liver fat content, elevated serum ALT levels, and a higher prevalence of MASH and advanced fibrosis—particularly among individuals of European descent (141).
In a multicenter liver biopsy cohort, the rs641738 T allele was positively correlated with fibrosis severity but showed no significant association with hepatic steatosis (104). In addition, loss of MBOAT7 function may activate the Toll-like receptor (TLR) signaling pathway and enhance the pro-inflammatory response of hepatic macrophages, further exacerbating liver injury and fibrogenesis (142). Among Han Chinese individuals, carriers of the rs641738 T allele exhibit reduced serum levels of angiopoietin-like protein 3 (ANGPTL3), which is associated with increased fibrosis severity and may mechanistically link MASLD with atherosclerotic cardiovascular disease (ASCVD) (143).
However, in contrast, another study reported no significant association between rs641738 and MASLD risk in overweight or obese children, suggesting that the genetic effects of MBOAT7 may vary by age and ethnicity (144). Collectively, current evidence supports a pivotal role for MBOAT7 in the pathogenesis of MASLD while underscoring the genetic heterogeneity of this variant across diverse populations.
4 Clinical challenges: from diagnosis to personalized treatment
Despite significant advances in understanding the pathophysiology of MASLD, numerous challenges persist in clinical practice (Table 2). From accurate diagnosis to individualized treatment, each step is hindered by technical limitations and implementation barriers, which impede early disease identification and restrict effective risk stratification and management of high-risk individuals.
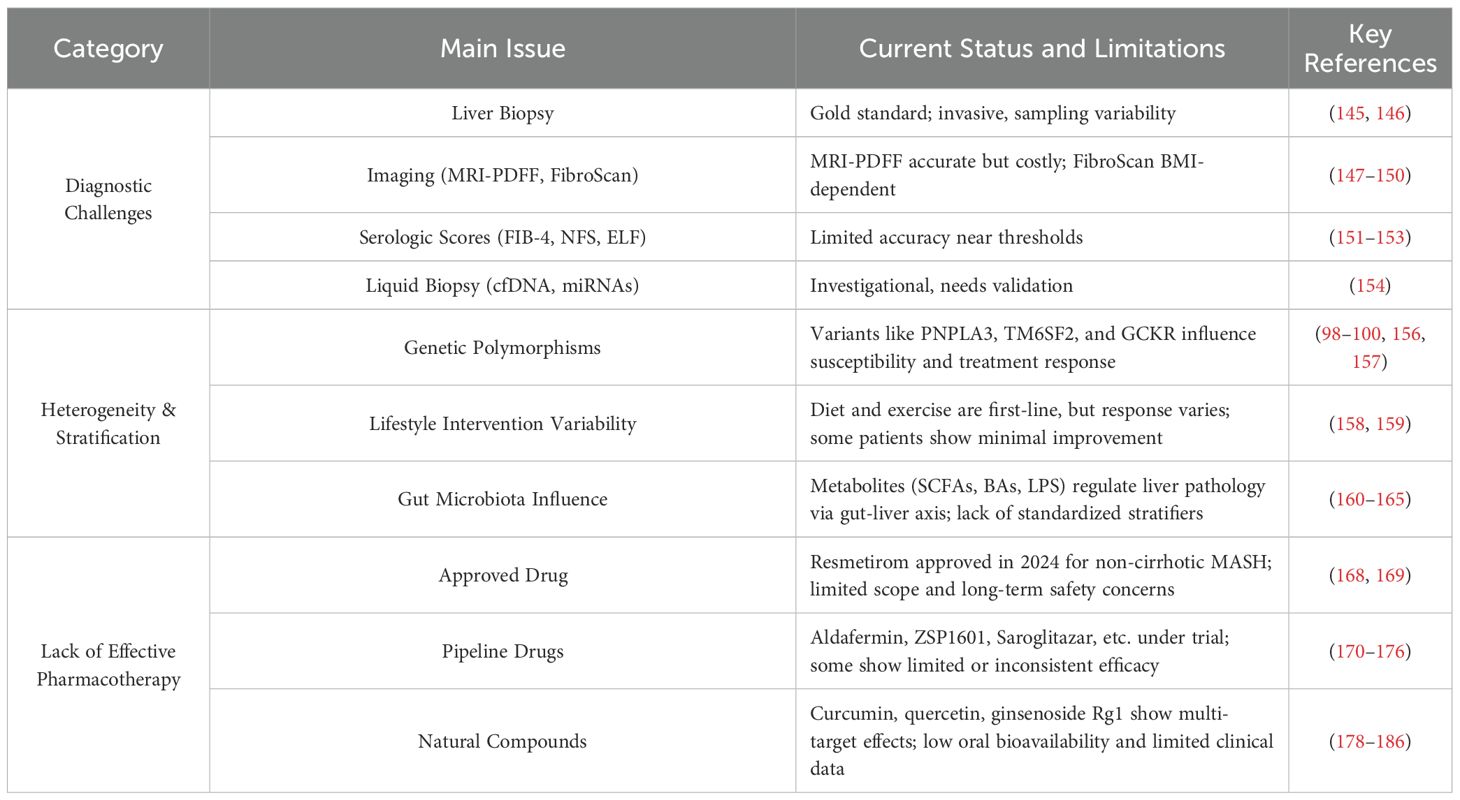
Table 2. Clinical challenges in MASLD: from diagnosis to personalized treatment.
4.1 Diagnostic challenges
Liver biopsy remains the gold standard for diagnosing MASLD and MASH due to its ability to directly assess steatosis, inflammation, and fibrosis (145). However, its clinical application is limited by invasiveness, risk of complications, sampling variability, and high cost (146). Consequently, non-invasive diagnostic approaches have gained traction. Imaging modalities such as MRI–proton density fat fraction (MRI-PDFF) and transient elastography (FibroScan) demonstrate good accuracy in evaluating hepatic steatosis and fibrosis (147, 148). While MRI-PDFF is constrained by limited accessibility and cost (149). FibroScan may yield less reliable results in obese individuals or those with hepatic inflammation (150). Serum-based scores including Fibrosis-4 (FIB-4) and the MASLD Fibrosis Score (NFS) are commonly used to estimate fibrosis, but their accuracy diminishes near threshold values, particularly in elderly patients or those with metabolic syndrome, leading to a risk of underdiagnosis (151–153). Emerging liquid biopsy tools, such as circulating free DNA and microRNAs, offer promise as minimally invasive biomarkers, yet remain in the exploratory stage due to limited sensitivity, specificity, and validation in large cohorts (154).
4.2 Challenges in addressing heterogeneity and stratification strategies
MASLD is a highly heterogeneous metabolic disorder, with a wide spectrum of histopathological phenotypes ranging from simple steatosis (SS) to MASH, cirrhosis, and HCC (155). This heterogeneity manifests not only in the rate and severity of disease progression but also in the variability of patient responses to treatment. As such, a “one-size-fits-all” therapeutic approach is insufficient to address the complexity of disease presentations in clinical practice.
Genetic background plays a pivotal role in driving this heterogeneity. As previously discussed, several genetic polymorphisms—including PNPLA3, TM6SF2, GCKR, and others—contribute to interindividual and interethnic differences in disease susceptibility and treatment response. For instance, patients carrying the PNPLA3 I148M variant exhibit greater reductions in ALT levels following semaglutide therapy (156). Similarly, rs738409 has been associated with variations in HbA1c response to dulaglutide, with potential sex-specific effects (157). These findings underscore the necessity of incorporating genetic profiling into personalized treatment strategies for MASLD.
Lifestyle interventions—including low-carbohydrate diets, Mediterranean-style diets, and structured physical activity—remain the cornerstone of MASLD management. While these approaches improve insulin sensitivity and reduce hepatic fat in many patients, responses are variable (158). Notably, some individuals fail to achieve significant reductions in hepatic steatosis despite strict adherence to lifestyle modification, suggesting that monotherapy may be insufficient in certain subgroups (159).
In recent years, the gut microbiota has emerged as a key contributor to MASLD heterogeneity. Significant interindividual differences exist in microbial composition, metabolite profiles, and microbiota-host interactions, all of which influence disease onset and progression (160). Microbial metabolites—including short-chain fatty acids (SCFAs), BAs, amino acid-derived compounds, trimethylamine-N-oxide (TMAO), and endogenous ethanol—modulate hepatic lipid metabolism, inflammation, and fibrosis via the gut-liver axis (161, 162). Disruption of the intestinal barrier can facilitate lipopolysaccharide (LPS) translocation, triggering hepatic inflammation and accelerating the transition from SS to MASH (163). Moreover, specific microbial genera such as Bacteroides and Prevotella produce SCFAs that regulate hepatic lipogenesis and energy homeostasis (164). The gut microbiota also modulates bile acid signaling through FXR and TGR5, thereby influencing hepatic inflammatory and fibrotic responses (165).
Despite growing interest, microbiota-based stratification strategies face several barriers, including the lack of highly specific microbial biomarkers, significant interethnic and geographic variation, and the complex nature of host-microbiota interactions (166, 167). These limitations currently hinder the clinical implementation of precision diagnostics and personalized interventions based on gut microbial profiling in MASLD.
4.3 Lack of effective pharmacotherapy
In March 2024, the U.S. Food and Drug Administration (FDA) granted accelerated approval to Resmetirom (brand name: Rezdiffra) for the treatment of adults with non-cirrhotic MASH, marking the first approved therapy for this indication and a significant milestone in the treatment of MASLD (168). However, its therapeutic scope is largely confined to patients with moderate to advanced fibrosis. Effective treatments for early-stage MASLD, decompensated cirrhosis, or patients with complex metabolic comorbidities remain lacking. Furthermore, long-term safety data are still limited, with concerns about potential cardiovascular risks and thyroid dysfunction requiring further investigation (169).
Beyond Resmetirom, several synthetic compounds are under clinical development targeting key MASLD pathways. Aldafermin (NGM282), an analog of fibroblast growth factor 19 (FGF19), has shown anti-steatotic and anti-fibrotic effects in early trials, but failed to produce significant dose-dependent improvements in fibrosis or MASH resolution in the phase IIb ALPINE 2/3 study (170–172). ZSP1601 (pan-phosphodiesterase inhibitor) reduced hepatic fat in Phase I/II, though long-term efficacy requires validation (173, 174). Other agents, such as the peroxisome proliferator-activated receptor (PPAR) agonist Saroglitazar, the apoptosis signal-regulating kinase 1 (ASK1) inhibitor Selonsertib, and the C-C chemokine receptor types 2 and 5 (CCR2/CCR5) antagonist Cenicriviroc, have shown limited or inconsistent efficacy in clinical trials, hindering broader application (175–177).
Natural compounds have attracted growing interest due to their multi-target mechanisms and lower toxicity profiles. Several phytochemicals—including curcumin, quercetin, and ginsenoside Rg1—have been shown to modulate lipid metabolism, reduce oxidative stress, and suppress inflammation through diverse molecular pathways (178–181). Curcumin inhibits the NF-κB signaling pathway, reducing pro-inflammatory cytokine expression, and has been shown in ApoE−/− mouse models to improve intestinal barrier integrity, lower endotoxin levels, and attenuate steatosis via the TLR4/NF-κB axis (182, 183). Quercetin activates the AMPK pathway, promoting fatty acid β-oxidation and reducing hepatic lipid accumulation (184). Moreover, natural products may beneficially modulate gut microbiota and restore gut-liver axis function, thereby contributing to systemic improvements in MASLD pathology (185).
Despite these advantages, clinical translation of natural products remains challenging, due primarily to poor oral bioavailability, unstable pharmacokinetics, and limited high-quality clinical data (186). Future studies should focus on enhancing delivery systems—such as nanoparticle carriers and targeted release technologies—to improve therapeutic efficacy and stability.
5 Future perspectives
As the complexity and heterogeneity of MASLD become increasingly apparent, precision medicine is emerging as a transformative approach to clinical management. The integration of multi-omics technologies—including genomics (e.g., PNPLA3, TM6SF2, HSD17B13), metabolomics (e.g., lipid and amino acid profiles), and microbiomics (e.g., gut–liver axis dynamics)—has enabled the construction of stratification frameworks that link molecular mechanisms to clinical phenotypes. These data-driven platforms lay the foundation for individualized risk prediction, disease monitoring, and therapeutic intervention, supporting a shift from uniform treatment paradigms to personalized care.
Building upon these insights, novel therapeutic strategies are being developed that leverage pharmacogenomic information (e.g., PNPLA3 I148M-guided therapy) and microbiome-based interventions. The latter, by reshaping bile acid signaling, immune responses, and metabolic pathways, holds promise in modulating key drivers of MASLD progression. In parallel, the therapeutic landscape is evolving toward integrated systems characterized by multi-target synergy, precision delivery technologies, and digital monitoring platforms—enabling dynamic regulation of disease trajectories and therapeutic responses.
The recent FDA approval of Resmetirom represents a therapeutic breakthrough in MASLD management, yet the disease’s multifactorial nature demands innovative strategies beyond single-target interventions. Critical areas for advancement include: (1) designing combination regimens that concurrently address lipid metabolism, inflammatory cascades, and fibrotic pathways; (2) validating non-invasive biomarkers for real-time disease monitoring; (3) developing integrative therapeutic platforms combining pharmacological agents with nutraceuticals; and (4) conducting multinational phase IV trials to establish longitudinal safety and efficacy profiles.Together, these efforts aim to bridge the gap between mechanistic discovery and clinical translation, ultimately fostering a paradigm shift toward predictive, preventive, and personalized hepatology.
6 Conclusion
MASLD is a multifactorial disease driven by a combination of metabolic dysfunction and genetic predisposition. Its pathogenesis involves lipid metabolic imbalance, insulin resistance, dysregulation of bile acid and amino acid metabolism, iron overload, and key genetic polymorphisms. Although advances have been made in diagnostic technologies and targeted therapeutic development, early detection and individualized treatment remain major clinical challenges. Moving forward, a precision medicine framework that integrates genomic, metabolic, and microbiome data will be essential for establishing comprehensive, stratified intervention models and achieving personalized management of MASLD—from risk prediction to mechanism-based therapy.
Author contributions
JM: Data curation, Investigation, Writing – original draft. YM: Data curation, Investigation, Writing – original draft. XW: Data curation, Investigation, Writing – original draft. JCL: Data curation, Investigation, Writing – original draft. YZ: Data curation, Investigation, Writing – original draft. JFL: Writing – review & editing. YG: Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Riazi K, Azhari H, Charette JH, Underwood FE, King JA, Afshar EE, et al. The prevalence and incidence of NAFLD worldwide: a systematic review and meta-analysis. Lancet Gastroenterol Hepatol. (2022) 7:851–61. doi: 10.1016/S2468-1253(22)00165-0
2. Huh Y, Cho YJ, and Nam GE. Recent epidemiology and risk factors of nonalcoholic fatty liver disease. J Obes Metab syndrome. (2022) 31:17–27. doi: 10.7570/jomes22021
3. Rinella ME, Lazarus JV, Ratziu V, Francque SM, Sanyal AJ, Kanwal F, et al. A multisociety Delphi consensus statement on new fatty liver disease nomenclature. J Hepatol. (2023) 79:1542–56. doi: 10.1016/j.jhep.2023.06.003
4. Amini-Salehi E, Letafatkar N, Norouzi N, Joukar F, Habibi A, Javid M, et al. Global Prevalence of Nonalcoholic Fatty Liver Disease: An Updated Review Meta-Analysis comprising a Population of 78 million from 38 Countries. Arch Med Res. (2024) 55:103043. doi: 10.1016/j.arcmed.2024.103043
5. Hagström H, Shang Y, Hegmar H, and Nasr P. Natural history and progression of metabolic dysfunction-associated steatotic liver disease. Lancet Gastroenterol Hepatol. (2024) 9:944–56. doi: 10.1016/S2468-1253(24)00193-6
6. Godoy-Matos AF, Silva Júnior WS, and Valerio CM. NAFLD as a continuum: from obesity to metabolic syndrome and diabetes. Diabetol Metab syndrome. (2020) 12:60. doi: 10.1186/s13098-020-00570-y
7. Llovet JM, Willoughby CE, Singal AG, Greten TF, Heikenwälder M, El-Serag HB, et al. Nonalcoholic steatohepatitis-related hepatocellular carcinoma: pathogenesis and treatment. Nat Rev Gastroenterol Hepatol. (2023) 20:487–503. doi: 10.1038/s41575-023-00754-7
8. Schoonejans JM and Ozanne SE. Developmental programming by maternal obesity: Lessons from animal models. Diabetic medicine: J Br Diabetic Assoc. (2021) 38:e14694. doi: 10.1111/dme.14694
9. Soderborg TK, Clark SE, Mulligan CE, Janssen RC, Babcock L, Ir D, et al. The gut microbiota in infants of obese mothers increases inflammation and susceptibility to NAFLD. Nat Commun. (2018) 9:4462. doi: 10.1038/s41467-018-06929-0
10. Godfrey KM, Sheppard A, Gluckman PD, Lillycrop KA, Burdge GC, McLean C, et al. Epigenetic gene promoter methylation at birth is associated with child’s later adiposity. Diabetes. (2011) 60:1528–34. doi: 10.2337/db10-0979
11. Bruce KD, Cagampang FR, Argenton M, Zhang J, Ethirajan PL, Burdge GC, et al. Maternal high-fat feeding primes steatohepatitis in adult mice offspring, involving mitochondrial dysfunction and altered lipogenesis gene expression. Hepatology. (2009) 50:1796–808. doi: 10.1002/hep.23205
12. Bhat N and Mani A. Dysregulation of lipid and glucose metabolism in nonalcoholic fatty liver disease. Nutrients. (2023) 15(10):2323. doi: 10.3390/nu15102323
13. Kakazu E, Mino M, and Kanto T. Role of amino acids in the regulation of hepatic gluconeogenesis and lipogenesis in metabolic dysfunction-associated steatotic liver disease. Clin Mol Hepatol. (2025) 31(3):771–95. doi: 10.3350/cmh.2025.0820
14. Chiang JYL and Ferrell JM. Bile acid metabolism in liver pathobiology. Gene Expression. (2018) 18:71–87. doi: 10.3727/105221618X15156018385515
15. Chen J, Li X, Ge C, Min J, and Wang F. The multifaceted role of ferroptosis in liver disease. Cell Death differentiation. (2022) 29:467–80. doi: 10.1038/s41418-022-00941-0
16. Ramandi A, Diehl AM, Sanyal AJ, and de Jong YP. Experimental models to investigate PNPLA3 in liver steatosis. Liver international: Off J Int Assoc Study Liver. (2025) 45:e70091. doi: 10.1111/liv.70091
17. Xue WY, Zhang L, Liu CM, Gao Y, Li SJ, Huai ZY, et al. Research progress on the relationship between TM6SF2 rs58542926 polymorphism and non-alcoholic fatty liver disease. Expert Rev Gastroenterol Hepatol. (2022) 16:97–107. doi: 10.1080/17474124.2022.2032661
18. Chavan SU, Rathi P, and Mandot A. Association of GCKR and MBOAT7 genetic polymorphisms with non-alcoholic fatty liver disease. Clin Exp Hepatol. (2024) 10:39–46. doi: 10.5114/ceh.2024.136326
19. Motomura T, Amirneni S, Diaz-Aragon R, Faccioli LAP, Malizio MR, Coard MC, et al. Is HSD17B13 genetic variant a protector for liver dysfunction? Future perspective as a potential therapeutic target. J personalized Med. (2021) 11(7):619. doi: 10.3390/jpm11070619
20. Sattar N, Forrest E, and Preiss D. Non-alcoholic fatty liver disease. BMJ (Clinical Res ed.). (2014) 349:g4596. doi: 10.1136/bmj.g4596
21. Bessone F, Razori MV, and Roma MG. Molecular pathways of nonalcoholic fatty liver disease development and progression. Cell Mol Life sciences: CMLS. (2019) 76:99–128. doi: 10.1007/s00018-018-2947-0
22. Shao M, Ye Z, Qin Y, and Wu T. Abnormal metabolic processes involved in the pathogenesis of non-alcoholic fatty liver disease (Review). Exp Ther Med. (2020) 20:26. doi: 10.3892/etm.2020.9154
23. Edgerton DS, Kraft G, Smith M, Farmer B, Williams PE, Coate KC, et al. Insulin’s direct hepatic effect explains the inhibition of glucose production caused by insulin secretion. JCI Insight. (2017) 2:e91863. doi: 10.1172/jci.insight.91863
24. Gnoni A, Siculella L, Paglialonga G, Damiano F, and Giudetti AM. 3,5-diiodo-L-thyronine increases de novo lipogenesis in liver from hypothyroid rats by SREBP-1 and ChREBP-mediated transcriptional mechanisms. IUBMB Life. (2019) 71:863–72. doi: 10.1002/iub.2014
25. Pengnet S, Sumarithum P, Phongnu N, Prommaouan S, Kantip N, Phoungpetchara I, et al. Naringin attenuates fructose-induced NAFLD progression in rats through reducing endogenous triglyceride synthesis and activating the Nrf2/HO-1 pathway. Front Pharmacol. (2022) 13:1049818. doi: 10.3389/fphar.2022.1049818
26. Régnier M, Carbinatti T, Parlati L, Benhamed F, and Postic C. The role of ChREBP in carbohydrate sensing and NAFLD development. Nat Rev Endocrinol. (2023) 19:336–49. doi: 10.1038/s41574-023-00809-4
27. Chen J, Zhou Y, Liu Z, Lu Y, Jiang Y, Cao K, et al. Hepatic glycogenesis antagonizes lipogenesis by blocking S1P via UDPG. Sci (New York N.Y.). (2024) 383:eadi3332. doi: 10.1126/science.adi3332
28. Inzaugarat ME, Wree A, and Feldstein AE. Hepatocyte mitochondrial DNA released in microparticles and toll-like receptor 9 activation: A link between lipotoxicity and inflammation during nonalcoholic steatohepatitis. Hepatology. (2016) 64:669–71. doi: 10.1002/hep.28666
29. Geng Y, Faber KN, de Meijer VE, Blokzijl H, and Moshage H. How does hepatic lipid accumulation lead to lipotoxicity in non-alcoholic fatty liver disease? Hepatol Int. (2021) 15:21–35. doi: 10.1007/s12072-020-10121-2
30. Summers SA. Ceramides in insulin resistance and lipotoxicity. Prog Lipid Res. (2006) 45:42–72. doi: 10.1016/j.plipres.2005.11.002
31. Hajduch E, Lachkar F, Ferré P, and Foufelle F. Roles of ceramides in non-alcoholic fatty liver disease. J Clin Med. (2021) 10(4):792. doi: 10.3390/jcm10040792
32. Thevissen K, François IE, Winderickx J, Pannecouque C, and Cammue BP. Ceramide involvement in apoptosis and apoptotic diseases. Mini Rev medicinal Chem. (2006) 6:699–709. doi: 10.2174/138955706777435643
33. Ziolkowska S, Binienda A, Jabłkowski M, Szemraj J, and Czarny P. The interplay between insulin resistance, inflammation, oxidative stress, base excision repair and metabolic syndrome in nonalcoholic fatty liver disease. Int J Mol Sci. (2021) 22(20):11128. doi: 10.3390/ijms222011128
34. de la Roche M, Hamilton C, Mortensen R, Jeyaprakash AA, Ghosh S, and Anand PK. Trafficking of cholesterol to the ER is required for NLRP3 inflammasome activation. J Cell Biol. (2018) 217:3560–76. doi: 10.1083/jcb.201709057
35. Lebeaupin C, Proics E, de Bieville CH, Rousseau D, Bonnafous S, Patouraux S, et al. ER stress induces NLRP3 inflammasome activation and hepatocyte death. Cell Death Dis. (2015) 6:e1879. doi: 10.1038/cddis.2015.248
36. Xu GX, Wei S, Yu C, Zhao SQ, Yang WJ, Feng YH, et al. Activation of Kupffer cells in NAFLD and NASH: mechanisms and therapeutic interventions. Front Cell Dev Biol. (2023) 11:1199519. doi: 10.3389/fcell.2023.1199519
37. Zhang J, Zhang K, Li Z, and Guo B. ER stress-induced inflammasome activation contributes to hepatic inflammation and steatosis. J Clin Cell Immunol. (2016) 7(5):457. doi: 10.4172/2155-9899.1000457
38. Pal SC and Méndez-Sánchez N. Insulin resistance and adipose tissue interactions as the cornerstone of metabolic (dysfunction)-associated fatty liver disease pathogenesis. World J Gastroenterol. (2023) 29:3999–4008. doi: 10.3748/wjg.v29.i25.3999
39. Shetty SS and Kumari S. Fatty acids and their role in type-2 diabetes (Review). Exp Ther Med. (2021) 22:706. doi: 10.3892/etm.2021.10138
40. Silva Figueiredo P, Carla Inada A, Marcelino G, Maiara Lopes Cardozo C, de Cássia Freitas K, de Cássia Avellaneda Guimarães R, et al. Fatty acids consumption: the role metabolic aspects involved in obesity and its associated disorders. Nutrients. (2017) 9(10):1158. doi: 10.3390/nu9101158
41. Bo T, Gao L, Yao Z, Shao S, Wang X, Proud CG, et al. Hepatic selective insulin resistance at the intersection of insulin signaling and metabolic dysfunction-associated steatotic liver disease. Cell Metab. (2024) 36:947–68. doi: 10.1016/j.cmet.2024.04.006
42. Laplante M and Sabatini DM. mTORC1 activates SREBP-1c and uncouples lipogenesis from gluconeogenesis. Proc Natl Acad Sci United States America. (2010) 107:3281–2. doi: 10.1073/pnas.1000323107
43. Jornayvaz FR and Shulman GI. Diacylglycerol activation of protein kinase Cϵ and hepatic insulin resistance. Cell Metab. (2012) 15:574–84. doi: 10.1016/j.cmet.2012.03.005
44. Park SS and Seo YK. Excess accumulation of lipid impairs insulin sensitivity in skeletal muscle. Int J Mol Sci. (2020) 21(6):1949. doi: 10.3390/ijms21061949
45. Philp A, Hargreaves M, and Baar K. More than a store: regulatory roles for glycogen in skeletal muscle adaptation to exercise. Am J Physiol Endocrinol Metab. (2012) 302:E1343–51. doi: 10.1152/ajpendo.00004.2012
46. Dey A and Chandrasekaran K. Hyperglycemia induced changes in liver: in vivo and in vitro studies. Curr Diabetes Rev. (2009) 5:67–78. doi: 10.2174/157339909788166864
47. Garza-Campos A, Prieto-Correa JR, Domínguez-Rosales JA, and Hernández-Nazará ZH. Implications of receptor for advanced glycation end products for progression from obesity to diabetes and from diabetes to cancer. World J Diabetes. (2023) 14:977–94. doi: 10.4239/wjd.v14.i7.977
48. Priken K, Tapia G, Cadagan C, Quezada N, Torres J, D’Espessailles A, et al. Higher hepatic advanced glycation end products and liver damage markers are associated with nonalcoholic steatohepatitis. Nutr Res (New York N.Y.). (2022) 104:71–81. doi: 10.1016/j.nutres.2022.04.005
49. Song Z, Xiaoli AM, and Yang F. Regulation and metabolic significance of de novo lipogenesis in adipose tissues. Nutrients. (2018) 10(10):1383. doi: 10.3390/nu10101383
50. Baharuddin B. The impact of fructose consumption on human health: effects on obesity, hyperglycemia, diabetes, uric acid, and oxidative stress with a focus on the liver. Cureus. (2024) 16:e70095. doi: 10.7759/cureus.70095
51. Choi YJ, Shin HS, Choi HS, Park JW, Jo I, Oh ES, et al. Uric acid induces fat accumulation via generation of endoplasmic reticulum stress and SREBP-1c activation in hepatocytes. Lab investigation; J Tech Methods Pathol. (2014) 94:1114–25. doi: 10.1038/labinvest.2014.98
52. Lanaspa MA, Sanchez-Lozada LG, Choi YJ, Cicerchi C, Kanbay M, Roncal-Jimenez CA, et al. Uric acid induces hepatic steatosis by generation of mitochondrial oxidative stress: potential role in fructose-dependent and -independent fatty liver. J Biol Chem. (2012) 287:40732–44. doi: 10.1074/jbc.M112.399899
53. Shi Y and Qi W. Histone modifications in NAFLD: mechanisms and potential therapy. Int J Mol Sci. (2023) 24(19):14653. doi: 10.3390/ijms241914653
54. Park G, Jung S, Wellen KE, and Jang C. The interaction between the gut microbiota and dietary carbohydrates in nonalcoholic fatty liver disease. Exp Mol Med. (2021) 53:809–22. doi: 10.1038/s12276-021-00614-x
55. Samuel VT, Liu ZX, Qu X, Elder BD, Bilz S, Befroy D, et al. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J Biol Chem. (2004) 279:32345–53. doi: 10.1074/jbc.M313478200
56. Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, et al. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest. (2004) 114:1752–61. doi: 10.1172/JCI21625
57. Houstis N, Rosen ED, and Lander ES. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature. (2006) 440:944–8. doi: 10.1038/nature04634
58. Solinas G and Karin M. JNK1 and IKKbeta: molecular links between obesity and metabolic dysfunction. FASEB journal: Off Publ Fed Am Societies Exp Biol. (2010) 24:2596–611. doi: 10.1096/fj.09-151340
59. Sutti S, Jindal A, Locatelli I, Vacchiano M, Gigliotti L, Bozzola C, et al. Adaptive immune responses triggered by oxidative stress contribute to hepatic inflammation in NASH. Hepatology. (2014) 59:886–97. doi: 10.1002/hep.26749
60. Albano E, Mottaran E, Vidali M, Reale E, Saksena S, Occhino G, et al. Immune response towards lipid peroxidation products as a predictor of progression of non-alcoholic fatty liver disease to advanced fibrosis. Gut. (2005) 54:987–93. doi: 10.1136/gut.2004.057968
61. Jia W, Li Y, Cheung KCP, and Zheng X. Bile acid signaling in the regulation of whole body metabolic and immunological homeostasis. Sci China. Life Sci. (2024) 67:865–78. doi: 10.1007/s11427-023-2353-0
62. Wei M, Tu W, and Huang G. Regulating bile acids signaling for NAFLD: molecular insights and novel therapeutic interventions. Front Microbiol. (2024) 15:1341938. doi: 10.3389/fmicb.2024.1341938
63. Bing H and Li YL. The role of bile acid metabolism in the occurrence and development of NAFLD. Front Mol Biosci. (2022) 9:1089359. doi: 10.3389/fmolb.2022.1089359
64. Chiang JYL and Ferrell JM. Bile acid receptors FXR and TGR5 signaling in fatty liver diseases and therapy. Am J Physiol Gastrointestinal liver Physiol. (2020) 318:G554–g573. doi: 10.1152/ajpgi.00223.2019
65. Kliewer SA and Mangelsdorf DJ. Bile acids as hormones: the FXR-FGF15/19 pathway. Digestive Dis (Basel Switzerland). (2015) 33:327–31. doi: 10.1159/000371670
66. Inagaki T, Choi M, Moschetta A, Peng L, Cummins CL, McDonald JG, et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. (2005) 2:217–25. doi: 10.1016/j.cmet.2005.09.001
67. Li Y, Jadhav K, and Zhang Y. Bile acid receptors in non-alcoholic fatty liver disease. Biochem Pharmacol. (2013) 86:1517–24. doi: 10.1016/j.bcp.2013.08.015
68. Long J, Xu Y, Zhang X, Wu B, and Wang C. Role of FXR in the development of NAFLD and intervention strategies of small molecules. Arch Biochem biophysics. (2024) 757:110024. doi: 10.1016/j.abb.2024.110024
69. Lai J, Luo L, Zhou T, Feng X, Ye J, and Zhong B. Alterations in circulating bile acids in metabolic dysfunction-associated steatotic liver disease: A systematic review and meta-analysis. Biomolecules. (2023) 13(9):1356. doi: 10.3390/biom13091356
70. Martínez-Montoro JI, Núñez-Sánchez M, Martinez-Sanchez MA, Balaguer-Román A, Fernández-Ruiz VE, Ferrer-Gómez M, et al. Hepatic and serum branched-chain fatty acid profile in patients with nonalcoholic fatty liver disease: A case-control study. Obes (Silver Spring Md.). (2023) 31:1064–74. doi: 10.1002/oby.23711
71. Amanatidou AI, Mikropoulou EV, Amerikanou C, Milanovic M, Stojanoski S, Bjelan M, et al. Plasma amino acids in NAFLD patients with obesity are associated with steatosis and fibrosis: results from the MAST4HEALTH study. Metabolites. (2023) 13(8):959. doi: 10.3390/metabo13080959
72. Liao Y, Chen Q, Liu L, Huang H, Sun J, Bai X, et al. Amino acid is a major carbon source for hepatic lipogenesis. Cell Metab. (2024) 36:2437–2448.e8. doi: 10.1016/j.cmet.2024.10.001
73. Deng Y, Hu M, Huang S, and Fu N. Molecular mechanism and therapeutic significance of essential amino acids in metabolically associated fatty liver disease. J Nutr Biochem. (2024) 126:109581. doi: 10.1016/j.jnutbio.2024.109581
74. Sano A, Kakazu E, Morosawa T, Inoue J, Kogure T, Ninomiya M, et al. The profiling of plasma free amino acids and the relationship between serum albumin and plasma-branched chain amino acids in chronic liver disease: a single-center retrospective study. J Gastroenterol. (2018) 53:978–88. doi: 10.1007/s00535-018-1435-5
75. Gaggini M, Carli F, Rosso C, Buzzigoli E, Marietti M, Della Latta V, et al. Altered amino acid concentrations in NAFLD: Impact of obesity and insulin resistance. Hepatology. (2018) 67:145–58. doi: 10.1002/hep.29465
76. Lake AD, Novak P, Shipkova P, Aranibar N, Robertson DG, Reily MD, et al. Branched chain amino acid metabolism profiles in progressive human nonalcoholic fatty liver disease. Amino Acids. (2015) 47:603–15. doi: 10.1007/s00726-014-1894-9
77. Sookoian S, Castaño GO, Scian R, Fernández Gianotti T, Dopazo H, Rohr C, et al. Serum aminotransferases in nonalcoholic fatty liver disease are a signature of liver metabolic perturbations at the amino acid and Krebs cycle level. Am J Clin Nutr. (2016) 103:422–34. doi: 10.3945/ajcn.115.118695
78. Sookoian S and Pirola CJ. The nonalcoholic steatohepatitis metabotype: Imbalance of circulating amino acids and transamination reactions reflect impaired mitochondrial function. Hepatology. (2018) 67:1177–8. doi: 10.1002/hep.29705
79. Rom O, Liu Y, Liu Z, Zhao Y, Wu J, Ghrayeb A, et al. Glycine-based treatment ameliorates NAFLD by modulating fatty acid oxidation, glutathione synthesis, and the gut microbiome. Sci Trans Med. (2020) 12(572):eaaz2841. doi: 10.1126/scitranslmed.aaz2841
80. Pacana T, Cazanave S, Verdianelli A, Patel V, Min HK, Mirshahi F, et al. Dysregulated hepatic methionine metabolism drives homocysteine elevation in diet-induced nonalcoholic fatty liver disease. PloS One. (2015) 10:e0136822. doi: 10.1371/journal.pone.0136822
81. Jin R, Banton S, Tran VT, Konomi JV, Li S, Jones DP, et al. Amino acid metabolism is altered in adolescents with nonalcoholic fatty liver disease-an untargeted, high resolution metabolomics study. J Pediatr. (2016) 172:14–19.e5. doi: 10.1016/j.jpeds.2016.01.026
82. Delgado TC, de Las Heras J, and Martínez-Chantar ML. Understanding gut-liver axis nitrogen metabolism in Fatty Liver Disease. Front Endocrinol. (2022) 13:1058101. doi: 10.3389/fendo.2022.1058101
83. Muckenthaler MU, Rivella S, Hentze MW, and Galy B. A red carpet for iron metabolism. Cell. (2017) 168:344–61. doi: 10.1016/j.cell.2016.12.034
84. Gozzelino R and Arosio P. Iron homeostasis in health and disease. Int J Mol Sci. (2016) 17(1):130. doi: 10.3390/ijms17010130
85. Shen X, Yu Z, Wei C, Hu C, and Chen J. Iron metabolism and ferroptosis in nonalcoholic fatty liver disease: what is our next step? Am J Physiol Endocrinol Metab. (2024) 326:E767–e775. doi: 10.1152/ajpendo.00260.2023
86. Sui Y, Geng X, Wang Z, Zhang J, Yang Y, and Meng Z. Targeting the regulation of iron homeostasis as a potential therapeutic strategy for nonalcoholic fatty liver disease. Metabolism: Clin Exp. (2024) 157:155953. doi: 10.1016/j.metabol.2024.155953
87. Qiu F, Wu L, Yang G, Zhang C, Liu X, Sun X, et al. The role of iron metabolism in chronic diseases related to obesity. Mol Med (Cambridge Mass.). (2022) 28:130. doi: 10.1186/s10020-022-00558-6
88. Chen H. Iron metabolism in non-alcoholic fatty liver disease: A promising therapeutic target. Liver Res (Beijing China). (2022) 6:203–13. doi: 10.1016/j.livres.2022.09.003
89. Sobieska K, Buczyńska A, Krętowski AJ, and Popławska-Kita A. Iron homeostasis and insulin sensitivity: unraveling the complex interactions. Rev endocrine Metab Disord. (2024) 25:925–39. doi: 10.1007/s11154-024-09908-7
90. Aigner E, Theurl I, Theurl M, Lederer D, Haufe H, Dietze O, et al. Pathways underlying iron accumulation in human nonalcoholic fatty liver disease. Am J Clin Nutr. (2008) 87:1374–83. doi: 10.1093/ajcn/87.5.1374
91. BasuRay S, Smagris E, Cohen JC, and Hobbs HH. The PNPLA3 variant associated with fatty liver disease (I148M) accumulates on lipid droplets by evading ubiquitylation. Hepatology. (2017) 66:1111–24. doi: 10.1002/hep.29273
92. Gou Y, Wang L, Zhao J, Xu X, Xu H, Xie F, et al. PNPLA3-I148M variant promotes the progression of liver fibrosis by inducing mitochondrial dysfunction. Int J Mol Sci. (2023) 24(11):9681. doi: 10.3390/ijms24119681
93. Faccioli LAP, Sun Y, Animasahun O, Motomura T, Liu Z, Kurihara T, et al. Human-induced pluripotent stem cell-based hepatic modeling of lipid metabolism-associated TM6SF2-E167K variant. Hepatology. (2024) 82(3):638–54. doi: 10.1097/HEP.0000000000000001065
94. Zhang Z, Ji G, and Li M. Glucokinase regulatory protein: a balancing act between glucose and lipid metabolism in NAFLD. Front Endocrinol. (2023) 14:1247611. doi: 10.3389/fendo.2023.1247611
95. Meroni M, Dongiovanni P, Longo M, Carli F, Baselli G, Rametta R, et al. Mboat7 down-regulation by hyper-insulinemia induces fat accumulation in hepatocytes. EBioMedicine. (2020) 52:102658. doi: 10.1016/j.ebiom.2020.102658
96. Thangapandi VR, Knittelfelder O, Brosch M, Patsenker E, Vvedenskaya O, Buch S, et al. Loss of hepatic Mboat7 leads to liver fibrosis. Gut. (2021) 70:940–50. doi: 10.1136/gutjnl-2020-320853
97. Abul-Husn NS, Cheng X, Li AH, Xin Y, Schurmann C, Stevis P, et al. A protein-truncating HSD17B13 variant and protection from chronic liver disease. New Engl J Med. (2018) 378:1096–106. doi: 10.1056/NEJMoa1712191
98. Sharma D and Mandal P. NAFLD: genetics and its clinical implications. Clinics Res Hepatol Gastroenterol. (2022) 46:102003. doi: 10.1016/j.clinre.2022.102003
99. Ioannou GN. Epidemiology and risk-stratification of NAFLD-associated HCC. J Hepatol. (2021) 75:1476–84. doi: 10.1016/j.jhep.2021.08.012
100. Trépo E and Valenti L. Update on NAFLD genetics: From new variants to the clinic. J Hepatol. (2020) 72:1196–209. doi: 10.1016/j.jhep.2020.02.020
101. Cherubini A, Casirati E, Tomasi M, and Valenti L. PNPLA3 as a therapeutic target for fatty liver disease: the evidence to date. Expert Opin Ther Targets. (2021) 25:1033–43. doi: 10.1080/14728222.2021.2018418
102. Danford CJ, Yao ZM, and Jiang ZG. Non-alcoholic fatty liver disease: a narrative review of genetics. J Biomed Res. (2018) 32:389–400. doi: 10.7555/JBR.32.20180045
103. Longo M, Meroni M, Paolini E, Erconi V, Carli F, Fortunato F, et al. TM6SF2/PNPLA3/MBOAT7 loss-of-function genetic variants impact on NAFLD development and progression both in patients and in in vitro models. Cell Mol Gastroenterol Hepatol. (2022) 13:759–88. doi: 10.1016/j.jcmgh.2021.11.007
104. Krawczyk M, Rau M, Schattenberg JM, Bantel H, Pathil A, Demir M, et al. Combined effects of the PNPLA3 rs738409, TM6SF2 rs58542926, and MBOAT7 rs641738 variants on NAFLD severity: a multicenter biopsy-based study. J Lipid Res. (2017) 58:247–55. doi: 10.1194/jlr.P067454
105. Salameh H, Hanayneh MA, Masadeh M, Naseemuddin M, Matin T, Erwin A, et al. PNPLA3 as a genetic determinant of risk for and severity of non-alcoholic fatty liver disease spectrum. J Clin Trans Hepatol. (2016) 4:175–91. doi: 10.14218/JCTH.2016.00009
106. Liu YL, Patman GL, Leathart JB, Piguet AC, Burt AD, Dufour JF, et al. Carriage of the PNPLA3 rs738409 C >G polymorphism confers an increased risk of non-alcoholic fatty liver disease associated hepatocellular carcinoma. J Hepatol. (2014) 61:75–81. doi: 10.1016/j.jhep.2014.02.030
107. Yasui K, Kawaguchi T, Shima T, Mitsuyoshi H, Seki K, Sendo R, et al. Effect of PNPLA3 rs738409 variant (I148 M) on hepatic steatosis, necroinflammation, and fibrosis in Japanese patients with chronic hepatitis C. J Gastroenterol. (2015) 50:887–93. doi: 10.1007/s00535-014-1018-z
108. Manchiero C, Nunes A, Magri MC, Dantas BP, Mazza CC, Barone AA, et al. The rs738409 polymorphism of the PNPLA3 gene is associated with hepatic steatosis and fibrosis in Brazilian patients with chronic hepatitis C. BMC Infect Dis. (2017) 17:780. doi: 10.1186/s12879-017-2887-6
109. Wang SW, Wang C, Cheng YM, Chen CY, Hsieh TH, Wang CC, et al. Genetic predisposition of metabolic dysfunction-associated steatotic liver disease: a population-based genome-wide association study. Hepatol Int. (2025) 19:415–27. doi: 10.1007/s12072-024-10769-0
110. Ott C, Ross K, Straub S, Thiede B, Götz M, Goosmann C, et al. Sam50 functions in mitochondrial intermembrane space bridging and biogenesis of respiratory complexes. Mol Cell Biol. (2012) 32:1173–88. doi: 10.1128/MCB.06388-11
111. Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. (2008) 40:1461–5. doi: 10.1038/ng.257
112. Sookoian S and Pirola CJ. Meta-analysis of the influence of I148M variant of patatin-like phospholipase domain containing 3 gene (PNPLA3) on the susceptibility and histological severity of nonalcoholic fatty liver disease. Hepatology. (2011) 53:1883–94. doi: 10.1002/hep.24283
113. Luo F, Oldoni F, and Das A. TM6SF2: A novel genetic player in nonalcoholic fatty liver and cardiovascular disease. Hepatol Commun. (2022) 6:448–60. doi: 10.1002/hep4.1822
114. Mahdessian H, Taxiarchis A, Popov S, Silveira A, Franco-Cereceda A, Hamsten A, et al. TM6SF2 is a regulator of liver fat metabolism influencing triglyceride secretion and hepatic lipid droplet content. Proc Natl Acad Sci United States America. (2014) 111:8913–8. doi: 10.1073/pnas.1323785111
115. Kozlitina J, Smagris E, Stender S, Nordestgaard BG, Zhou HH, Tybjærg-Hansen A, et al. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. (2014) 46:352–6. doi: 10.1038/ng.2901
116. Grandone A, Cozzolino D, Marzuillo P, Cirillo G, Di Sessa A, Ruggiero L, et al. TM6SF2 Glu167Lys polymorphism is associated with low levels of LDL-cholesterol and increased liver injury in obese children. Pediatr Obes. (2016) 11:115–9. doi: 10.1111/ijpo.12032
117. Liu YL, Reeves HL, Burt AD, Tiniakos D, McPherson S, Leathart JB, et al. TM6SF2 rs58542926 influences hepatic fibrosis progression in patients with non-alcoholic fatty liver disease. Nat Commun. (2014) 5:4309. doi: 10.1038/ncomms5309
118. Pirola CJ and Sookoian S. The dual and opposite role of the TM6SF2-rs58542926 variant in protecting against cardiovascular disease and conferring risk for nonalcoholic fatty liver: A meta-analysis. Hepatology. (2015) 62:1742–56. doi: 10.1002/hep.28142
119. Hu C, Zhang R, Wang C, Yu W, Lu J, Ma X, et al. Effects of GCK, GCKR, G6PC2 and MTNR1B variants on glucose metabolism and insulin secretion. PloS One. (2010) 5:e11761. doi: 10.1371/journal.pone.0011761
120. Perez-Martinez P, Delgado-Lista J, Garcia-Rios A, Mc Monagle J, Gulseth HL, Ordovas JM, et al. Glucokinase regulatory protein genetic variant interacts with omega-3 PUFA to influence insulin resistance and inflammation in metabolic syndrome. PloS One. (2011) 6:e20555. doi: 10.1371/journal.pone.0020555
121. Santoro N, Zhang CK, Zhao H, Pakstis AJ, Kim G, Kursawe R, et al. Variant in the glucokinase regulatory protein (GCKR) gene is associated with fatty liver in obese children and adolescents. Hepatology. (2012) 55:781–9. doi: 10.1002/hep.24806
122. Li J, Zhao Y, Zhang H, Hua W, Jiao W, Du X, et al. Contribution of rs780094 and rs1260326 polymorphisms in GCKR gene to non-alcoholic fatty liver disease: A meta-analysis involving 26,552 participants. Endocrine Metab Immune Disord Drug Targets. (2021) 21:1696–708. doi: 10.2174/1871530320999201126202706
123. Martin K, Hatab A, Athwal VS, Jokl E, and Piper Hanley K. Genetic contribution to non-alcoholic fatty liver disease and prognostic implications. Curr Diabetes Rep. (2021) 21:8. doi: 10.1007/s11892-021-01377-5
124. Fernandes Silva L, Vangipurapu J, Kuulasmaa T, and Laakso M. An intronic variant in the GCKR gene is associated with multiple lipids. Sci Rep. (2019) 9:10240. doi: 10.1038/s41598-019-46750-3
125. Zain SM, Mohamed Z, and Mohamed R. Common variant in the glucokinase regulatory gene rs780094 and risk of nonalcoholic fatty liver disease: a meta-analysis. J Gastroenterol Hepatol. (2015) 30:21–7. doi: 10.1111/jgh.12714
126. Petta S, Miele L, Bugianesi E, Cammà C, Rosso C, Boccia S, et al. Glucokinase regulatory protein gene polymorphism affects liver fibrosis in non-alcoholic fatty liver disease. PloS One. (2014) 9:e87523. doi: 10.1371/journal.pone.0087523
127. Husseini AA. Genotypic variation in CYP2E1, GCKR, and PNPLA3 among nonalcoholic steatohepatitis patients of Turkish origin. Mol Biol Rep. (2024) 51:845. doi: 10.1007/s11033-024-09787-w
128. Shi F, Zhao M, Zheng S, Zheng L, and Wang H. Advances in genetic variation in metabolism-related fatty liver disease. Front Genet. (2023) 14:1213916. doi: 10.3389/fgene.2023.1213916
129. Tang S, Zhang J, Mei TT, Zhang WY, Zheng SJ, and Yu HB. Association of HSD17B13 rs72613567: TA allelic variant with liver disease: review and meta-analysis. BMC Gastroenterol. (2021) 21:490. doi: 10.1186/s12876-021-02067-y
130. Zhang HB, Su W, Xu H, Zhang XY, and Guan YF. HSD17B13: A potential therapeutic target for NAFLD. Front Mol Biosci. (2021) 8:824776. doi: 10.3389/fmolb.2021.824776
131. Ma Y, Belyaeva OV, Brown PM, Fujita K, Valles K, Karki S, et al. 17-beta hydroxysteroid dehydrogenase 13 is a hepatic retinol dehydrogenase associated with histological features of nonalcoholic fatty liver disease. Hepatology. (2019) 69:1504–19. doi: 10.1002/hep.30350
132. Ting YW, Kong AS, Zain SM, Chan WK, Tan HL, Mohamed Z, et al. Loss-of-function HSD17B13 variants, non-alcoholic steatohepatitis and adverse liver outcomes: Results from a multi-ethnic Asian cohort. Clin Mol Hepatol. (2021) 27:486–98. doi: 10.3350/cmh.2020.0162
133. Liu WY, Eslam M, Zheng KI, Ma HL, Rios RS, Lv MZ, et al. Associations of hydroxysteroid 17-beta dehydrogenase 13 variants with liver histology in chinese patients with metabolic-associated fatty liver disease. J Clin Trans Hepatol. (2021) 9:194–202. doi: 10.14218/JCTH.2020.00151
134. Gil-Gómez A, Rojas Á, García-Lozano MR, Muñoz-Hernández R, Gallego-Durán R, Maya-Miles D, et al. Impact of a loss-of-function variant in HSD17B13 on hepatic decompensation and mortality in cirrhotic patients. Int J Mol Sci. (2022) 23(19):11840. doi: 10.3390/ijms231911840
135. Chen H, Zhang Y, Guo T, Yang F, Mao Y, Li L, et al. Genetic variant rs72613567 of HSD17B13 gene reduces alcohol-related liver disease risk in Chinese Han population. Liver international: Off J Int Assoc Study Liver. (2020) 40:2194–202. doi: 10.1111/liv.14616
136. Seko Y, Yamaguchi K, Tochiki N, Yano K, Takahashi A, Okishio S, et al. Attenuated effect of PNPLA3 on hepatic fibrosis by HSD17B13 in Japanese patients with non-alcoholic fatty liver disease. Liver international: Off J Int Assoc Study Liver. (2020) 40:1686–92. doi: 10.1111/liv.14495
137. Caddeo A, Spagnuolo R, and Maurotti S. MBOAT7 in liver and extrahepatic diseases. Liver international: Off J Int Assoc Study Liver. (2023) 43:2351–64. doi: 10.1111/liv.15706
138. Varadharajan V, Ramachandiran I, Massey WJ, Jain R, Banerjee R, Horak AJ, et al. Membrane-bound O-acyltransferase 7 (MBOAT7) shapes lysosomal lipid homeostasis and function to control alcohol-associated liver injury. eLife. (2024) 12:RP92243. doi: 10.7554/eLife.92243
139. Chen Y, Du X, Kuppa A, Feitosa MF, Bielak LF, O’Connell JR, et al. Genome-wide association meta-analysis identifies 17 loci associated with nonalcoholic fatty liver disease. Nat Genet. (2023) 55:1640–50. doi: 10.1038/s41588-023-01497-6
140. Mancina RM, Dongiovanni P, Petta S, Pingitore P, Meroni M, Rametta R, et al. The MBOAT7-TMC4 variant rs641738 increases risk of nonalcoholic fatty liver disease in individuals of european descent. Gastroenterology. (2016) 150:1219–1230.e6. doi: 10.1053/j.gastro.2016.01.032
141. Teo K, Abeysekera KWM, Adams L, Aigner E, Anstee QM, Banales JM, et al. rs641738C>T near MBOAT7 is associated with liver fat, ALT and fibrosis in NAFLD: A meta-analysis. J Hepatol. (2021) 74:20–30. doi: 10.1016/j.jhep.2020.08.027
142. Ren G, Bai C, Yi S, Cong Q, and Zhu Y. Mechanisms and therapeutic strategies for MAFLD targeting TLR4 signaling pathways. J innate Immun. (2024) 16:45–55. doi: 10.1159/000535524
143. Xu X, Xu H, Liu X, Zhang S, Cao Z, Qiu L, et al. MBOAT7 rs641738 (C>T) is associated with NAFLD progression in men and decreased ASCVD risk in elder Chinese population. Front Endocrinol. (2023) 14:1199429. doi: 10.3389/fendo.2023.1199429
144. Zusi C, Morandi A, Maguolo A, Corradi M, Costantini S, Mosca A, et al. Association between MBOAT7 rs641738 polymorphism and non-alcoholic fatty liver in overweight or obese children. Nutrition metabolism Cardiovasc diseases: NMCD. (2021) 31:1548–55. doi: 10.1016/j.numecd.2021.01.020
145. Chowdhury AB and Mehta KJ. Liver biopsy for assessment of chronic liver diseases: a synopsis. Clin Exp Med. (2023) 23:273–85. doi: 10.1007/s10238-022-00799-z
146. Thomaides-Brears HB, Alkhouri N, Allende D, Harisinghani M, Noureddin M, Reau NS, et al. Incidence of complications from percutaneous biopsy in chronic liver disease: A systematic review and meta-analysis. Digestive Dis Sci. (2022) 67:3366–94. doi: 10.1007/s10620-021-07089-w
147. Han MA, Saouaf R, Ayoub W, Todo T, Mena E, and Noureddin M. Magnetic resonance imaging and transient elastography in the management of Nonalcoholic Fatty Liver Disease (NAFLD). Expert Rev Clin Pharmacol. (2017) 10:379–90. doi: 10.1080/17512433.2017.1299573
148. Tang A, Tan J, Sun M, Hamilton G, Bydder M, Wolfson T, et al. Nonalcoholic fatty liver disease: MR imaging of liver proton density fat fraction to assess hepatic steatosis. Radiology. (2013) 267:422–31. doi: 10.1148/radiol.12120896
149. Boeriu A, Dobru D, and Fofiu C. Non-invasive diagnostic of NAFLD in type 2 diabetes mellitus and risk stratification: strengths and limitations. Life (Basel Switzerland). (2023) 13(12):2262. doi: 10.3390/life13122262
150. Garteiser P, Castera L, Coupaye M, Doblas S, Calabrese D, Dioguardi Burgio M, et al. Prospective comparison of transient elastography, MRI and serum scores for grading steatosis and detecting non-alcoholic steatohepatitis in bariatric surgery candidates. JHEP reports: Innovation Hepatol. (2021) 3:100381. doi: 10.1016/j.jhepr.2021.100381
151. Kang YW, Baek YH, and Moon SY. Sequential diagnostic approach using FIB-4 and ELF for predicting advanced fibrosis in metabolic dysfunction-associated steatotic liver disease. Diagnostics (Basel Switzerland). (2024) 14(22):2517. doi: 10.3390/diagnostics14222517
152. Sumida Y, Yoneda M, Tokushige K, Kawanaka M, Fujii H, Yoneda M, et al. FIB-4 first in the diagnostic algorithm of metabolic-dysfunction-associated fatty liver disease in the era of the global metabodemic. Life (Basel Switzerland). (2021) 11(2):143. doi: 10.3390/life11020143
153. Graupera I, Thiele M, Serra-Burriel M, Caballeria L, Roulot D, Wong GL, et al. Low accuracy of FIB-4 and NAFLD fibrosis scores for screening for liver fibrosis in the population. Clin Gastroenterol hepatology: Off Clin Pract J Am Gastroenterological Assoc. (2022) 20:2567–2576.e6. doi: 10.1016/j.cgh.2021.12.034
154. Turchinovich A, Baranova A, Drapkina O, and Tonevitsky A. Cell-free circulating nucleic acids as early biomarkers for NAFLD and NAFLD-associated disorders. Front Physiol. (2018) 9:1256. doi: 10.3389/fphys.2018.01256
155. Lonardo A, Leoni S, Alswat KA, and Fouad Y. History of nonalcoholic fatty liver disease. Int J Mol Sci. (2020) 21(16):5888. doi: 10.3390/ijms21165888
156. Urias E, Tedesco NR, Oliveri A, Raut C, Speliotes EK, and Chen VL. PNPLA3 risk allele association with ALT response to semaglutide treatment. Gastroenterology. (2024) 166:515–517.e2. doi: 10.1053/j.gastro.2023.11.018
157. Gavriilidis S, Andrianopoulou R, Ntenti C, Sarakapina A, Trakatelli C, Polyzos SA, et al. The effect of dulaglutide on glycated hemoglobin is associated with PNPLA3 I148M gene polymorphism in patients with type 2 diabetes mellitus. Endocrine. (2025) 87:73–8. doi: 10.1007/s12020-024-04007-8
158. Paris T, George ES, Roberts SK, and Tierney AC. The effects of diet and lifestyle interventions on insulin resistance in patients with nonalcoholic fatty liver disease: a systematic review. Eur J Gastroenterol Hepatol. (2017) 29:867–78. doi: 10.1097/MEG.0000000000000890
159. Liu Z, Jin P, Liu Y, Zhang Z, Wu X, Weng M, et al. A comprehensive approach to lifestyle intervention based on a calorie-restricted diet ameliorates liver fat in overweight/obese patients with NAFLD: a multicenter randomized controlled trial in China. Nutr J. (2024) 23:64. doi: 10.1186/s12937-024-00968-8
160. Jayakumar S and Loomba R. Review article: emerging role of the gut microbiome in the progression of nonalcoholic fatty liver disease and potential therapeutic implications. Alimentary Pharmacol Ther. (2019) 50:144–58. doi: 10.1111/apt.15314
161. Albillos A, de Gottardi A, and Rescigno M. The gut-liver axis in liver disease: Pathophysiological basis for therapy. J Hepatol. (2020) 72:558–77. doi: 10.1016/j.jhep.2019.10.003
162. Fang J, Yu CH, Li XJ, Yao JM, Fang ZY, Yoon SH, et al. Gut dysbiosis in nonalcoholic fatty liver disease: pathogenesis, diagnosis, and therapeutic implications. Front Cell infection Microbiol. (2022) 12:997018. doi: 10.3389/fcimb.2022.997018
163. Astbury S, Atallah E, Vijay A, Aithal GP, Grove JI, and Valdes AM. Lower gut microbiome diversity and higher abundance of proinflammatory genus Collinsella are associated with biopsy-proven nonalcoholic steatohepatitis. Gut Microbes. (2020) 11:569–80. doi: 10.1080/19490976.2019.1681861
164. Canfora EE, Meex RCR, Venema K, and Blaak EE. Gut microbial metabolites in obesity, NAFLD and T2DM. Nat Rev Endocrinol. (2019) 15:261–73. doi: 10.1038/s41574-019-0156-z
165. He LH, Yao DH, Wang LY, Zhang L, and Bai XL. Gut microbiome-mediated alteration of immunity, inflammation, and metabolism involved in the regulation of non-alcoholic fatty liver disease. Front Microbiol. (2021) 12:761836. doi: 10.3389/fmicb.2021.761836
166. Loomba R, Seguritan V, Li W, Long T, Klitgord N, Bhatt A, et al. Gut microbiome-based metagenomic signature for non-invasive detection of advanced fibrosis in human nonalcoholic fatty liver disease. Cell Metab. (2017) 25:1054–1062.e5. doi: 10.1016/j.cmet.2017.04.001
167. Tilg H, Zmora N, Adolph TE, and Elinav E. The intestinal microbiota fuelling metabolic inflammation. Nat Rev Immunol. (2020) 20:40–54. doi: 10.1038/s41577-019-0198-4
169. Noureddin M, Charlton MR, Harrison SA, Bansal MB, Alkhouri N, Loomba R, et al. Expert panel recommendations: practical clinical applications for initiating and monitoring resmetirom in patients with MASH/NASH and moderate to noncirrhotic advanced fibrosis. Clin Gastroenterol hepatology: Off Clin Pract J Am Gastroenterological Assoc. (2024) 22:2367–77. doi: 10.1016/j.cgh.2024.07.003
170. Harrison SA, Neff G, Guy CD, Bashir MR, Paredes AH, Frias JP, et al. Efficacy and safety of aldafermin, an engineered FGF19 analog, in a randomized, double-blind, placebo-controlled trial of patients with nonalcoholic steatohepatitis. Gastroenterology. (2021) 160:219–231.e1. doi: 10.1053/j.gastro.2020.08.004
171. Rinella ME, Lieu HD, Kowdley KV, Goodman ZD, Alkhouri N, Lawitz E, et al. A randomized, double-blind, placebo-controlled trial of aldafermin in patients with NASH and compensated cirrhosis. Hepatology. (2024) 79:674–89. doi: 10.1097/HEP.0000000000000607
172. Harrison SA, Abdelmalek MF, Neff G, Gunn N, Guy CD, Alkhouri N, et al. Aldafermin in patients with non-alcoholic steatohepatitis (ALPINE 2/3): a randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Gastroenterol Hepatol. (2022) 7:603–16. doi: 10.1016/S2468-1253(22)00017-6
173. Hu Y, Sun C, Chen Y, Liu YD, and Fan JG. Pipeline of new drug treatment for non-alcoholic fatty liver disease/metabolic dysfunction-associated steatotic liver disease. J Clin Trans Hepatol. (2024) 12:802–14. doi: 10.14218/JCTH.2024.00123
174. Hu Y, Li H, Zhang H, Chen X, Chen J, Xu Z, et al. ZSP1601, a novel pan-phosphodiesterase inhibitor for the treatment of NAFLD, A randomized, placebo-controlled phase Ib/IIa trial. Nat Commun. (2023) 14:6409. doi: 10.1038/s41467-023-42162-0
175. Gawrieh S, Noureddin M, Loo N, Mohseni R, Awasty V, Cusi K, et al. Saroglitazar, a PPAR-α/γ Agonist, for treatment of NAFLD: A randomized controlled double-blind phase 2 trial. Hepatology. (2021) 74:1809–24. doi: 10.1002/hep.31843
176. Harrison SA, Wong VW, Okanoue T, Bzowej N, Vuppalanchi R, Younes Z, et al. Selonsertib for patients with bridging fibrosis or compensated cirrhosis due to NASH: Results from randomized phase III STELLAR trials. J Hepatol. (2020) 73:26–39. doi: 10.1016/j.jhep.2020.02.027
177. Ratziu V, Sanyal A, Harrison SA, Wong VW, Francque S, Goodman Z, et al. Cenicriviroc treatment for adults with nonalcoholic steatohepatitis and fibrosis: final analysis of the phase 2b CENTAUR study. Hepatology. (2020) 72:892–905. doi: 10.1002/hep.31108
178. Li N, Cui C, Xu J, Mi M, Wang J, and Qin Y. Quercetin intervention reduced hepatic fat deposition in patients with nonalcoholic fatty liver disease: a randomized, double-blind, placebo-controlled crossover clinical trial. Am J Clin Nutr. (2024) 120:507–17. doi: 10.1016/j.ajcnut.2024.07.013
179. He Y, Chen X, Li Y, Liang Y, Hong T, Yang J, et al. Curcumin supplementation alleviates hepatic fat content associated with modulation of gut microbiota-dependent bile acid metabolism in patients with nonalcoholic simple fatty liver disease: a randomized controlled trial. Am J Clin Nutr. (2024) 120:66–79. doi: 10.1016/j.ajcnut.2024.05.017
180. Gao Y, Zhang S, Li J, Zhao J, Xiao Q, Zhu Y, et al. Effect and mechanism of ginsenoside Rg1-regulating hepatic steatosis in HepG2 cells induced by free fatty acid. Bioscience biotechnology Biochem. (2020) 84:2228–40. doi: 10.1080/09168451.2020.1793293
181. He B, Chen D, Zhang X, Yang R, Yang Y, Chen P, et al. Oxidative stress and ginsenosides: an update on the molecular mechanisms. Oxid Med Cell Longevity. (2022) 2022:9299574. doi: 10.1155/2022/9299574
182. Leclercq IA, Farrell GC, Sempoux C, dela Peña A, and Horsmans Y. Curcumin inhibits NF-kappaB activation and reduces the severity of experimental steatohepatitis in mice. J Hepatol. (2004) 41:926–34. doi: 10.1016/j.jhep.2004.08.010
183. Feng D, Zou J, Su D, Mai H, Zhang S, Li P, et al. Curcumin prevents high-fat diet-induced hepatic steatosis in ApoE(-/-) mice by improving intestinal barrier function and reducing endotoxin and liver TLR4/NF-κB inflammation. Nutr Metab. (2019) 16:79. doi: 10.1186/s12986-019-0410-3
184. Fukaya M, Sato Y, Kondo S, Adachi SI, Yoshizawa F, and Sato Y. Quercetin enhances fatty acid β-oxidation by inducing lipophagy in AML12 hepatocytes. Heliyon. (2021) 7:e07324. doi: 10.1016/j.heliyon.2021.e07324
185. Meng X, Li S, Li Y, Gan RY, and Li HB. Gut microbiota’s relationship with liver disease and role in hepatoprotection by dietary natural products and probiotics. Nutrients. (2018) 10(10):1457. doi: 10.3390/nu10101457
Keywords: MASLD, metabolic dysregulation, genetic polymorphisms, clinical management, precision medicine
Citation: Ma J, Ma Y, Wan X, Li J, Zhang Y, Liu J and Gao Y (2025) Metabolic and genetic mechanisms of metabolic dysfunction-associated steatotic liver disease: an integrative perspective from molecular pathways to clinical challenges. Front. Endocrinol. 16:1639064. doi: 10.3389/fendo.2025.1639064
Received: 05 June 2025; Accepted: 25 August 2025;
Published: 10 September 2025.
Edited by:
Antonio Marcus de Andrade Paes, Federal University of Maranhão, BrazilReviewed by:
Mohammed Abu El-Hamd, Sohag University, EgyptManuel Maliqueo, University of Chile, Chile
Copyright © 2025 Ma, Ma, Wan, Li, Zhang, Liu and Gao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jifeng Liu, MTE5MTQwNDYzOEBxcS5jb20=; Yunhai Gao, bG56eTEyM25AMTI2LmNvbQ==
†These authors have contributed equally to this work