 Yinhong Zhang1†Shiyu Wang2†Aoyu Li2Wenjing Zhao1Fengyu Xia1Ying Chan1Junyue Lin1Xiaoyan Zhou1Suyun Li1Na Feng1
Yinhong Zhang1†Shiyu Wang2†Aoyu Li2Wenjing Zhao1Fengyu Xia1Ying Chan1Junyue Lin1Xiaoyan Zhou1Suyun Li1Na Feng1 Baosheng Zhu1*Li Li3*
Baosheng Zhu1*Li Li3*- 1Department of Medical Genetics, NHC Key Laboratory of Healthy Birth and Birth Defect Prevention in Western China, Yunnan Provincial Key Laboratory for Birth Defects and Genetic Diseases, The Afliated Hospital of Kunming University of Science and Technology, The First People’s Hospital of Yunnan Province, Kunming, China
- 2School of Medicine, Kunming University of Science and Technology, Kunming, China
- 3Department of Pediatrics, The Affiliated Hospital of Kunming University of Science and Technology, The First People’s Hospital of Yunnan Province, Kunming, China
Context: Congenital hypothyroidism (CH) is a congenital endocrine disorder with diverse clinical presentations. The genotype–phenotype relationship has recently become a focal point in genetic etiology research on CH.
Objective: To explore the correlation between genetic variants and the clinical and biochemical characteristics of patients with CH in Yunnan Province, Southwest China.
Methods: A retrospective analysis of 117 Yunnan-origin CH patients was conducted. Target regions capture next-generation sequencing (NGS) was used to screen for variations in all exons and their exon–intron boundaries in 27 CH-related genes. Patients were categorized into groups based on genetic variations; clinical outcomes were assessed through standardized follow-up.
Results: Among the 117 CH patients, 91 carried gene variations related to CH, yielding a detection rate of 77.8%. Notably, variations in DUOX2, DUOXA2, and TG was most prevalent. Specifically, DUOX2 gene variations were found in 67 CH patients; these mutations encompassed 47 variant types, with K530X, R885L, and R1110Q being the most common in the Chinese cohort. CH patients exhibiting goiter and thyroid dysgenesis required a higher initial levothyroxine (L-T4) dose. As the number of gene variants increased, thyroid morphology gradually shifted toward “goiter” and “dysgenesis”. No significant differences were observed in biochemical characteristics or clinical outcomes among genetic variant groups.
Conclusions: This study provides valuable insights into the genetic landscape of CH in Yunnan Province, highlighting the importance of genes associated with thyroid dyshormonogenesis. Genotype cannot effectively be used to predict CH phenotype and prognosis. Standardized treatment and follow-up are crucial for positive outcomes in CH children.
1 Introduction
Congenital hypothyroidism (CH) is a congenital endocrine disorder possibly caused by abnormalities in thyroid differentiation, migration, or development or insufficient synthesis of thyroid hormones (1). The estimated incidence of sporadic CH (outside iodine-deficient areas) is approximately 1/2000–1/3000 (2, 3). CH typically presents as prolonged physiological jaundice, abdominal distention, constipation, umbilical hernia, feeding difficulties, low-pitched crying, and distinctive facial features (4). However, due to the influence of maternal thyroid hormones during fetal development, affected individuals often do not exhibit evident symptoms at birth, potentially leading to underdiagnosis (5). Thyroid hormones not only impact a child’s physical growth and development but also play a critical role in intellectual development (6). Without timely treatment, newborns with CH may have irreversible physical and intellectual developmental deficits (7).
Based on the site at which the pathology causes thyroid dysfunction, CH can be classified into primary CH and central hypothyroidism. Central hypothyroidism may occur with isolated TSH deficiency but is more commonly associated with congenital hypopituitarism (8). Traditional newborn screening can detect only primary CH associated with elevated thyroid-stimulating hormone (TSH) levels (9). CH can also be categorized as permanent CH (PCH) and transient CH (TCH) according to its prognosis (10). PCH is mostly caused by thyroid absence or underdevelopment, leading to a continuous deficiency of thyroid hormones in affected individuals, who require lifelong levothyroxine (L-T4) replacement therapy. The etiology of TCH is often linked to maternal iodine deficiency, neonatal iodine exposure, maternal autoimmune thyroid disease, premature infant thyroid immaturity, etc., leading to temporary insufficient thyroid hormone secretion, which typically recovers by the age of 2–3 years, with discontinuation of L-T4 therapy (11, 12).
Despite considerable progress in the screening, diagnosis, and treatment of CH (13), the underlying mechanisms remain unclear. CH pathogenesis can be attributed to thyroid dysgenesis (TD) and dyshormonogenesis (DH). Recent studies have identified genetic defects associated with CH, with more than 40 associated genes, including TSHR, TITF2, NKX2 (TITF1), PAX8, NKX2-5, DUOX2, GLIS3, TUBB1, CDCA8, JAG1, HHEX, HES1, HOXA3, EYA1, TPO, SLC5A5, DUOX1, DUOXA2, SLC26A4, TG, TRHR, TSHB, IGSF1, HESX1, TBL1X, IYD, LEPR, PROP1, POU1F1, LHX3, andLHX4, among others (14).
Furthermore, research indicates strong population-specific differences in variants and phenotypes of CH. However, the relationships between genotype and thyroid development, clinical outcome, and L-T4 dose remain unclear. Therefore, further studies are needed such that the underlying genetic causes of CH can be fully understood. In this study, we comprehensively and systematically screened mutated genes through next-generation sequencing (NGS), aiming to investigate the correlation between the genetic mutation status of CH-related genes and phenotypes in Yunnan Province, Southwest China.
2 Methods
2.1 Patients
A total of 117 Yunnan-origin CH patients treated at the First People’s Hospital of Yunnan Province from January 2016 to January 2019 were retrospectively analyzed. These CH patients were diagnosed at an age ranging from 15 to 30 days; 54 males and 63 females were included. The inclusion criteria were as follows: (i) infants with negative thyroid autoantibody test results; (ii) lacking other congenital diseases or chromosomal abnormalities; and (iii) sporadic and non-consanguineous cases. The exclusion criteria were as follows: (i) central hypothyroidism or related syndromes and (ii) autoimmune diseases or family history thereof. The study area (Yunnan Province) has been classified as an iodine-sufficient region following implementation of the national universal salt iodization program.
This study was reviewed and approved by the Medical Ethics Committee of the First People’s Hospital of Yunnan Province (No. KHLL2023-KY037). Signed informed consent was obtained from the parents of all patients. The research protocol complied with the seventh revision of the Helsinki Declaration (2013).
2.2 Newborn screening and laboratory diagnostic criteria for CH
The newborns were all screened at the Yunnan Province Newborn Screening Center. Within 72 hours to 20 days after birth, dried blood spot samples were collected from the heel of the newborns. These samples were then transported to the screening center laboratory through a cold chain system, and testing was completed within 3 days. TSH values were measured using a time-resolved fluorescence immunoassay (Perkin Elmer), with a cutoff value set at ≥8 μmol/L. Newborns with TSH screening results exceeding this positive cutoff value were recalled for serum thyroid function tests. Thyroid function was assessed in the patient’s serum using five different parameters, and values were compared to established normal reference ranges (TSH: 0.87∼6.15 mIU/L, T4: 77.8∼170.0 nmol/L, FT4: 12.0∼18.6 pmol/L, T3: 1.80∼3.68 nmol/L, FT3: 5.1∼8.0 pmol/L). A diagnosis of CH was made when the patient’s serum FT4 level fell below the minimum threshold and the TSH level exceeded the maximum detectable threshold.
2.3 Ultrasound measurement of the thyroid in CH patients
The long diameter (L), wide diameter (W), and thick diameter (T) of the thyroid were measured using a SIEMENS-ACUSON-S2000 color Doppler ultrasound instrument. The probe frequency was set at 9~18 MHz, and the color scale parameter was conventionally set at 7 cm/s. The ultrasound examination procedure was as follows. After the child was calm or asleep, the child was placed in the supine position with appropriate neck elevation to fully expose the neck. The ultrasound probe was positioned in front of the neck, and during the examination, it was placed just below the thyroid cartilage. Transverse and longitudinal scans using two-dimensional ultrasound were initially conducted to carefully observe the thyroid’s size, shape, capsule, and internal echogenicity. The largest diameter of the right and left thyroid lobes and the isthmus was measured, and the formula V = 0.479LWT/1000 was used to calculate the volume of both lobes (15). Color Doppler flow imaging (CDFI) was used to assess the degree and distribution of blood flow within the thyroid. By comparing the thyroid volume of the CH patients with that of healthy infants of corresponding ages, the thyroids of the CH patients were categorized into normal, dysgenesis (agenesis or athyreosis, ectopy, orthotopic hypoplasia and hemiagenesis) and goiter groups.
2.4 Target regions capture, sequencing and pathogenicity analysis of gene variations
Five milliliters of peripheral blood was collected from the patients and their parents. Genomic DNA (gDNA) was extracted according to the kit manufacturer’s standard procedure (MagPure Buffy Coat DNA Midi KF Kit, MAGEN BIOTECH). The gDNA was broken into 100–500 bp fragments by Shearing Enzyme Premix Reagent (ENZYMATICS), and 200–300 bp fragments were subsequently collected by magnetic beads (Vazyme DNA Clean Beads, VAZYME). An “A” base was added at the 3’ overhang after the end of the repair process to ensure that the fragments could pair with the “T” base with a special adapter, after which a single individual DNA library was constructed after Linker-mediated-PCR and purification. The library was enriched for 16 h at 65 °C by array hybridization (KAPA Hyper Exome, ROCHE), followed by elution and post-capture amplification. The products were subsequently subjected to analysis with a CaliperGX_1000 Kit (Caliper Life Sciences) and BMG to estimate the magnitude of enrichment. The qualified products were pooled and quantified according to different library sizes. Then, single strands of the library products were prepared for circularization, and DNB was generated. Finally, the products were sequenced with PE100 + 10 using an MGISEQ-2000.
To detect potential variants in the patients, bioinformatics processing and data analysis were performed after receiving the primary sequencing data. Comprehensive analysis was conducted to screen for mutations in 27 genes associated with CH, including HESX1, LHX3, LHX4, SOX3, OTX2, PROP1, POU1F1, TRHR, TSHB, LEPR, TPO, TSHR, TBL1X, IYD, PAX8, NKX2-1, FOXE1, IGSF1, NKX2-5, JAG1, GLIS3, CDCA8, TG, DUOX2, DUOXA2, SLC5A5, and SLC26A4. Variants located in regions with <10× effective coverage were considered low confidence. Such sites were either re-evaluated by repeat sequencing or excluded from downstream analyses unless validated independently by Sanger sequencing. For cases with missing data at specific loci, these positions were treated as unavailable and not used for genotype–phenotype correlation analyses. Only high-confidence variants confirmed by both NGS and, when necessary, Sanger sequencing were included in the final dataset. The “clean reads” (with a length of 90 bp) were derived from targeted sequencing and filtering and subsequently aligned to the human genome reference (hg19) using the Burrows Wheeler Aligner (BWA) MultiVision software package. All SNVs and indels were filtered and estimated via multiple databases, including the 1000 Genomes Project (1KGP), Genome Aggregation Database (gnomAD), Human Gene Mutation Database (HGMD) and Clinvar. We used dbNSFP, which contains seven well-established in silico prediction programs (scale-invariant feature transform (SIFT), Polyphen2, LRT, Mutation Taster, and PhyloP), to predict the effect of missense variants. The pathogenicity of mutations was interpreted based on American College of Medical Genetics and Genomics (ACMG) guidelines (16). All mutations and potential pathogenic variants were validated using conventional Sanger sequencing methods.
2.5 Follow-up and management
Treatment, regular follow-up, and monitoring of confirmed cases were conducted by pediatric endocrinologists from the Newborn Screening Center. Several highly compliant patients received standardized outpatient follow-up and treatment, including assessments of height, weight, head circumference, Gesell development schedules, and L-T4 doses at 1, 2, and 3 years of age. All CH children temporarily stopped L-T4 treatment at the age of 2 years as part of the standardized treatment protocol. Thyroid function was tested after one month. If TSH and FT4 levels were abnormal, the original treatment dosage was resumed. These CH children were diagnosed with PCH and continued to receive regular treatment and follow-up. CH children who stopped L-T4 treatment and maintained normal thyroid function continuously for more than one year were diagnosed with TCH.
2.6 Statistical analyses
Statistical analysis was conducted using SPSS 21.0 software and GraphPad Prism 8 software. We summarized normally distributed quantitative variables using means and standard deviations (means ± SDs). For nonnormally distributed variables, we employed medians with interquartile intervals (Q1-Q3). Count data are expressed as the number of patients (percentages) [N (%)]. The normality of continuous data was assessed using the Shapiro–Wilk test. For normally distributed data with homogeneity of variance, the independent samples t-test or one-way analysis of variance (ANOVA) followed by Bonferroni post hoc correction was used for intergroup comparisons. The Kruskal–Walli’s test was used for nonnormally distributed continuous data. Comparison of count data was performed using the chi-square test or Fisher’s exact test. A P-value less than 0.05 was considered to indicate statistical significance. Graphs were generated using GraphPad Prism 8 software.
3 Results
3.1 Clinical and biochemical characteristics and phenotypes of CH
Our cohort comprised a total of 117 patients with CH, 91 of whom were identified as carriers of variations in CH-related genes; these patients included 46 males and 45 females. No statistically significant differences were detected between the male and female patients in terms of ethnic distribution, birth age, birth weight, birth height, age at diagnosis, maternal thyroid function, TSH levels at newborn screening (NBS), diagnostic serum TSH levels, diagnostic serum FT4 levels, initial L-T4 doses, thyroid morphology, or clinical outcome (Table 1).
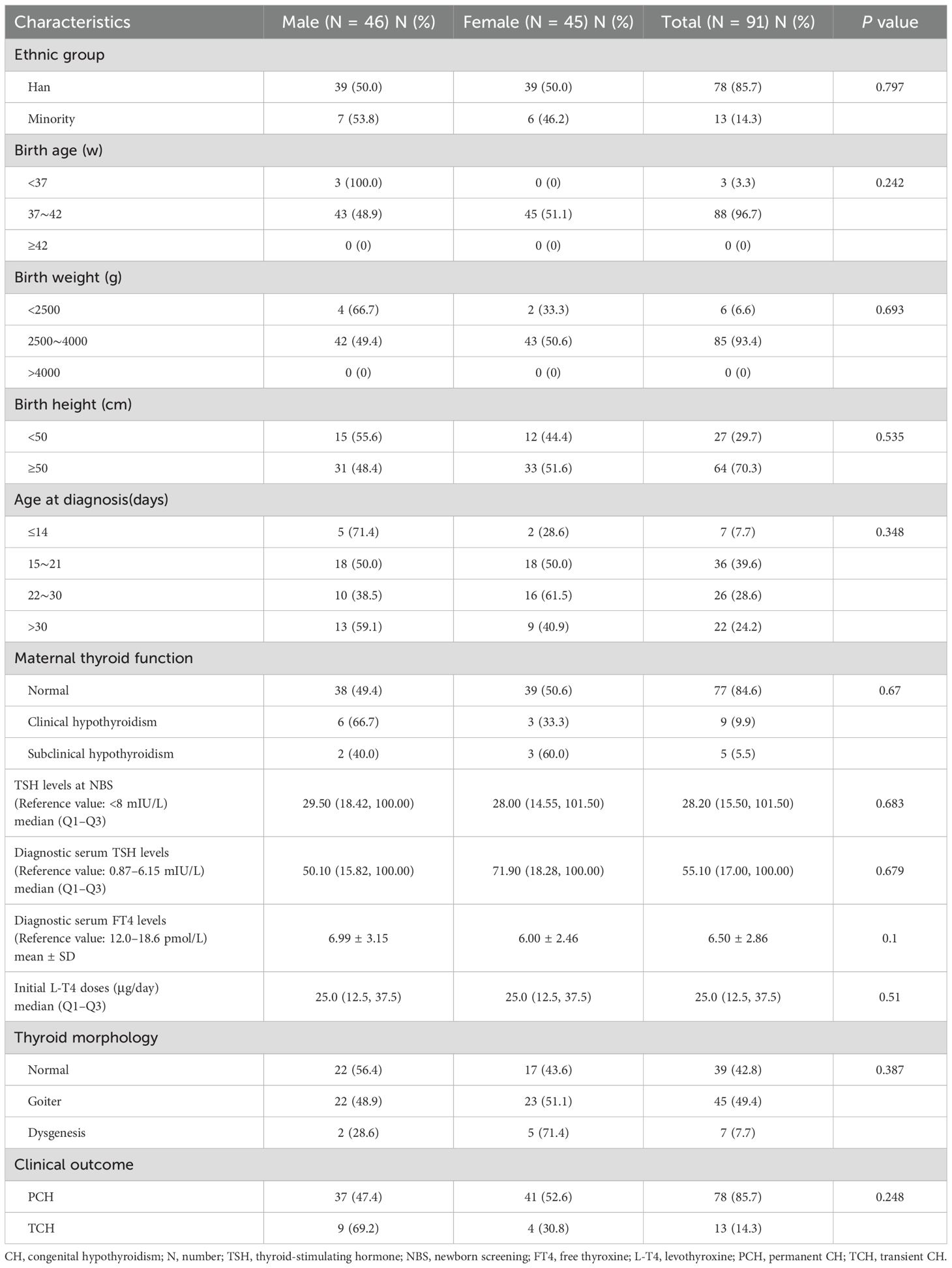
Table 1. Clinical and biochemical characteristics and clinical outcome of 91 children with CH.
Among the 91 patients, 39 had a normal thyroid, 45 had an enlarged thyroid, and 7 exhibited thyroid dysgenesis; 4 had thyroid agenesis, and 3 had orthotopic hypoplasia. After more than 2 years of treatment and follow-up, 78 cases were classified as PCH, whereas 13 cases were classified as TCH.
3.2 Genetic variation distribution in 91 CH patients
The detailed general information, biochemical test results, treatment results, clinical outcomes, and family molecular test results for the 91 patients are provided in Supplementary Table 1. Among them, 67 patients carried DUOX2 gene variations, 9 carried DUOXA2 gene variations, 6 carried TG gene variations, 6 carried TSHR gene variations, 5 carried TPO gene variations, and 2 each carried variations in the GLIS3, LHX3, PAX8, and TBL1X genes. Additionally, 1 patient each carried variations in the CDCA8, IGSF1, IYD, LEPR, POU1F1, SLC26A4, SLC5A5, and TRHR genes (Figure 1). Clearly, DUOX2 gene variations had the highest frequency in this cohort, accounting for 61.5% (67/109) of the total. The next most common variations involved DUOXA2, TG, TSHR, and TPO, which were associated with 8.3% (9/109), 5.5% (6/109), 5.5% (6/109), and 4.6% (5/109), respectively. A total of 47 variant types and 117 variant frequencies were identified in the DUOX2 gene, with c.1588A>T (p.K530X), c.2654G>T (p.R885L), c.3329G>A (p.R1110Q), c.2048G>T (p.R683L), c.3693 + 1G>T, c.2921G>A (p.R974H), and c.4027C>T (p.L1343F) accounting for 12.8%, 10.2%, 10.2%, 6.8%, 5.9%, 5.1%, and 4.2%, respectively. The most common DUOXA2 gene variant was c.738C>G (p.Y246X), accounting for 70.0% (7/10) of the total cases. Notably, digenic variants were found in 11 patients, and oligogenic variants were found in 3 patients. Among gene combinations, DUOX2+TG was the most common, accounting for 27.3% (3/11).
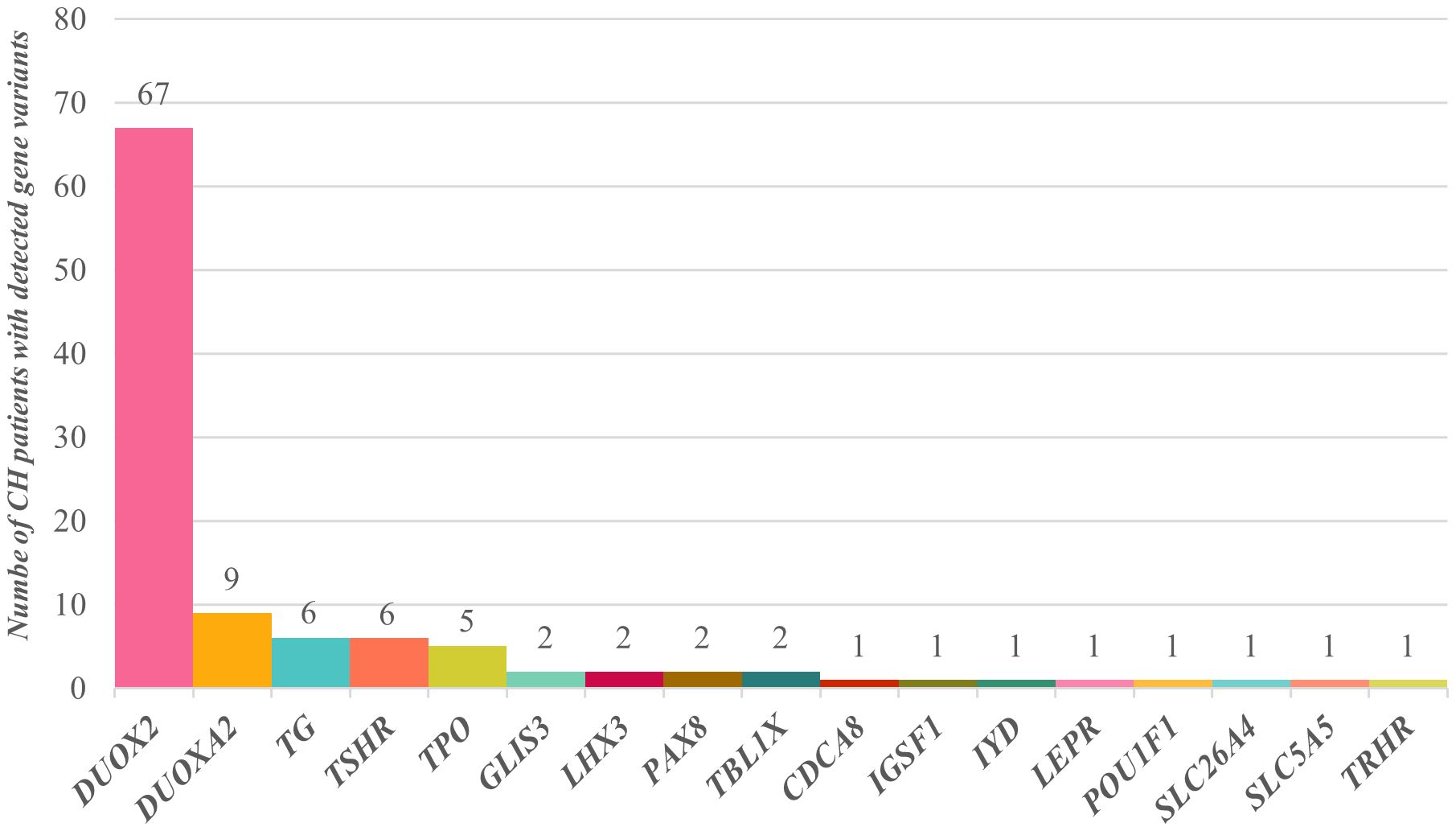
Figure 1. Distribution of CH-related gene variants in 91 children. Sixty-seven children had DUOX2 gene variants, 9 had DUOXA2 gene variants, 6 had TG gene variants, 6 had TSHR gene variants, and 5 had TPO gene variants. Variants in the GLIS3, LHX3, PAX8, and TBL1X genes were each detected in 2 children, and CDCA8, IGSF1, IYD, LEPR, POU1F1, SLC26A4, SLC5A5, and TRHR gene variants were each detected in a single child.
3.3 Analyses of relationships between thyroid morphology and biochemical/clinical characteristics
The CH patients were grouped according to thyroid morphology, and we conducted statistical analysis of differences in biochemical test results and clinical outcomes among the groups. We found statistically significant differences (P< 0.001) among the “normal”, “goiter”, and “dysgenesis” groups in terms of TSH levels at NBS, diagnostic serum TSH levels, and diagnostic serum FT4 levels. The “goiter” group and the “dysgenesis” group exhibited higher TSH levels at NBS and diagnostic serum TSH levels, corresponding to lower diagnostic serum FT4 levels. Moreover, the “goiter” group and the “dysgenesis” group required higher initial L-T4 doses than did the “normal” group. However, there was no statistically significant difference in clinical outcomes among the groups (P>0.05) (Table 2, Figure 2).
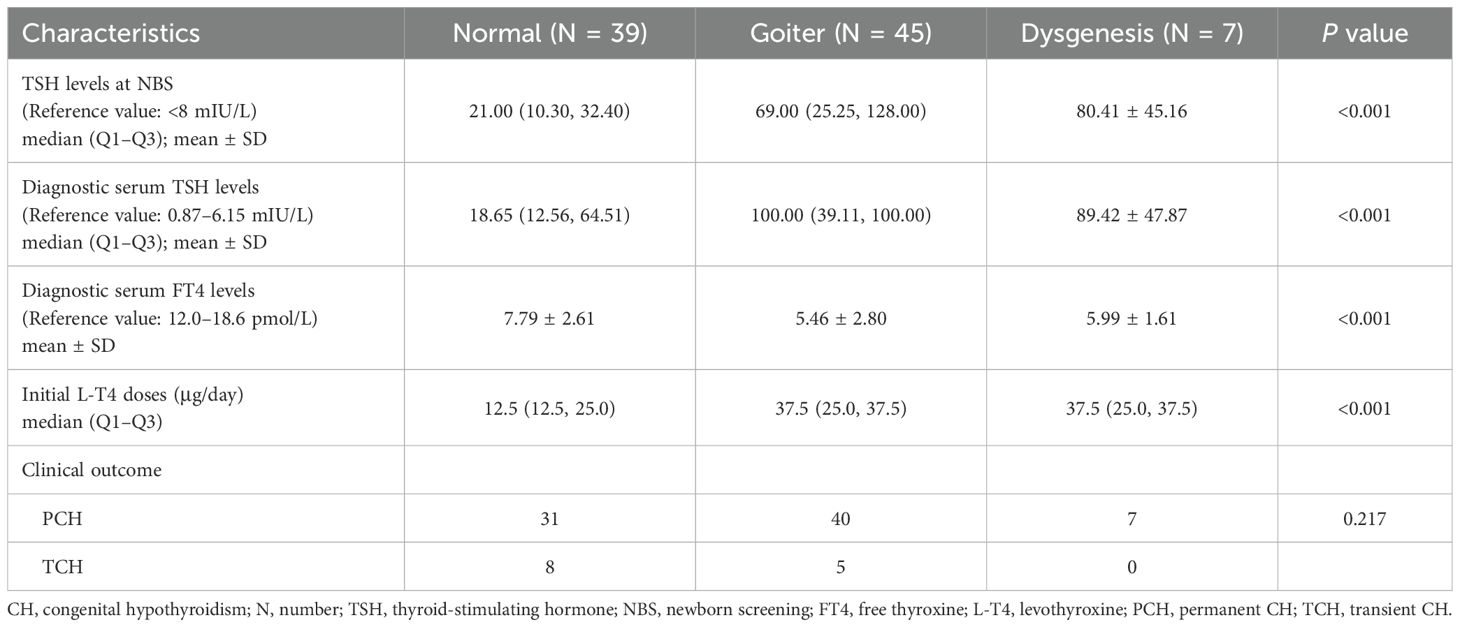
Table 2. Differences in biochemical and clinical characteristics of 91 children with CH among “normal”, “goiter”, and “dysgenesis” groups.
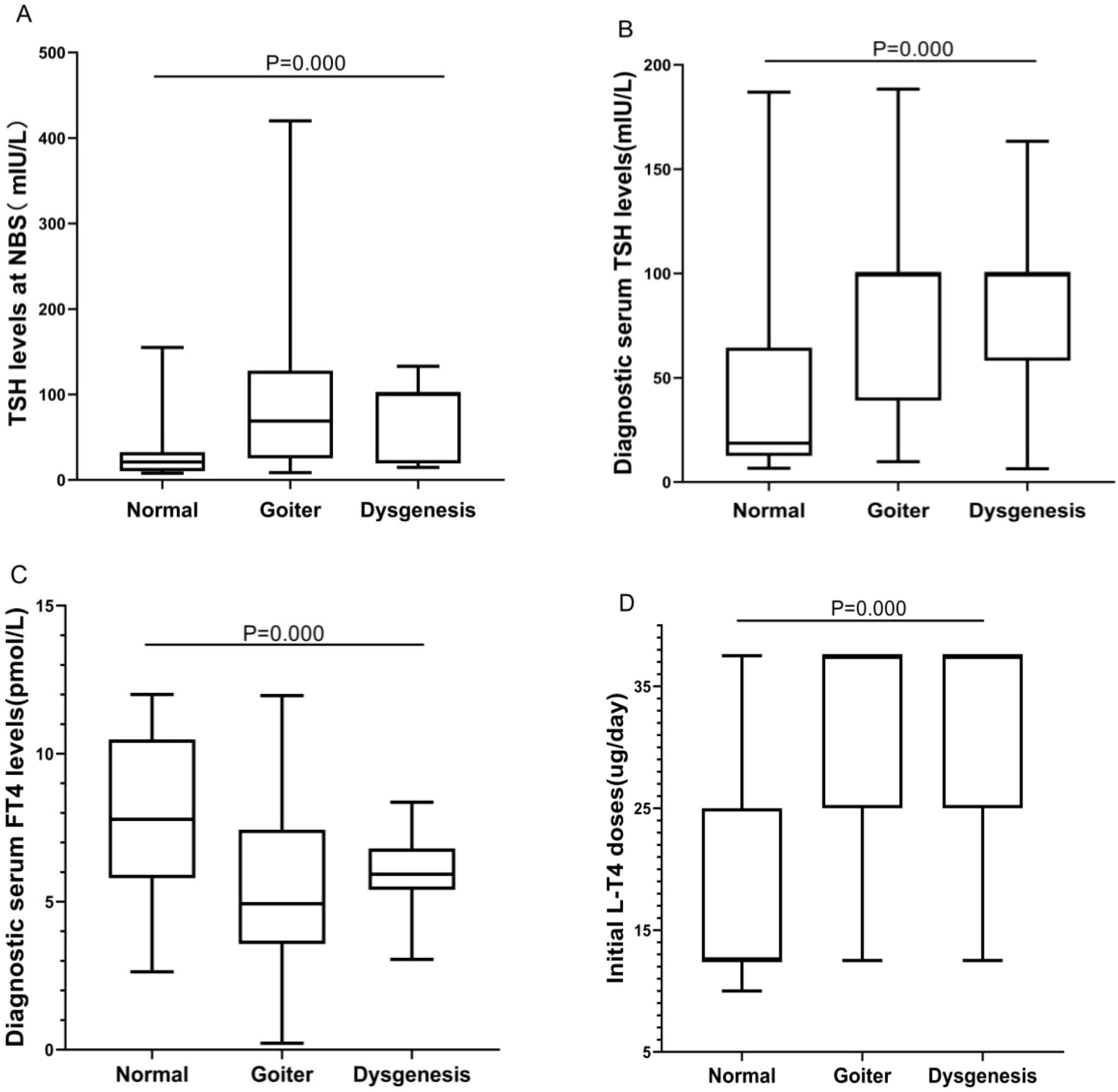
Figure 2. Differences in biochemical results and initial L-T4 doses among “normal”, “goiter”, and “dysgenesis” thyroid morphology groups. (A) The BOX plot chart shows the differences among the three groups regarding TSH levels at NBS. (B) The BOX plot chart shows the differences among the three groups regarding diagnostic serum TSH levels. (C) The BOX plot chart shows the differences among the three groups regarding diagnostic serum FT4 levels. (D) The BOX plot chart shows the differences among the three groups regarding the initial L-T4 doses. A P value <0.001 was considered to indicate an extremely significant difference.
3.4 Analyses of relationships between DUOX2 genotypes and biochemical/clinical characteristics
DUOX2 genotypes were divided into monoallelic variant and biallelic variant groups, and statistical analysis was conducted to assess differences between these two groups in terms of biochemical results, initial L-T4 doses, thyroid morphology, and clinical outcomes. There were no significant differences in TSH levels at NBS, diagnostic serum TSH levels, diagnostic serum FT4 levels, initial L-T4 doses, or clinical outcomes (all P> 0.05). However, there was a significant difference in thyroid morphology between the two groups (P = 0.05). A greater proportion of patients in the “DUOX2 monoallelic variant” group had normal thyroid morphology (50.00%, 10/20), whereas a greater proportion of patients in the “DUOX2 biallelic variant” group had goiter (59.57%, 28/47) (Table 3, Figure 3).
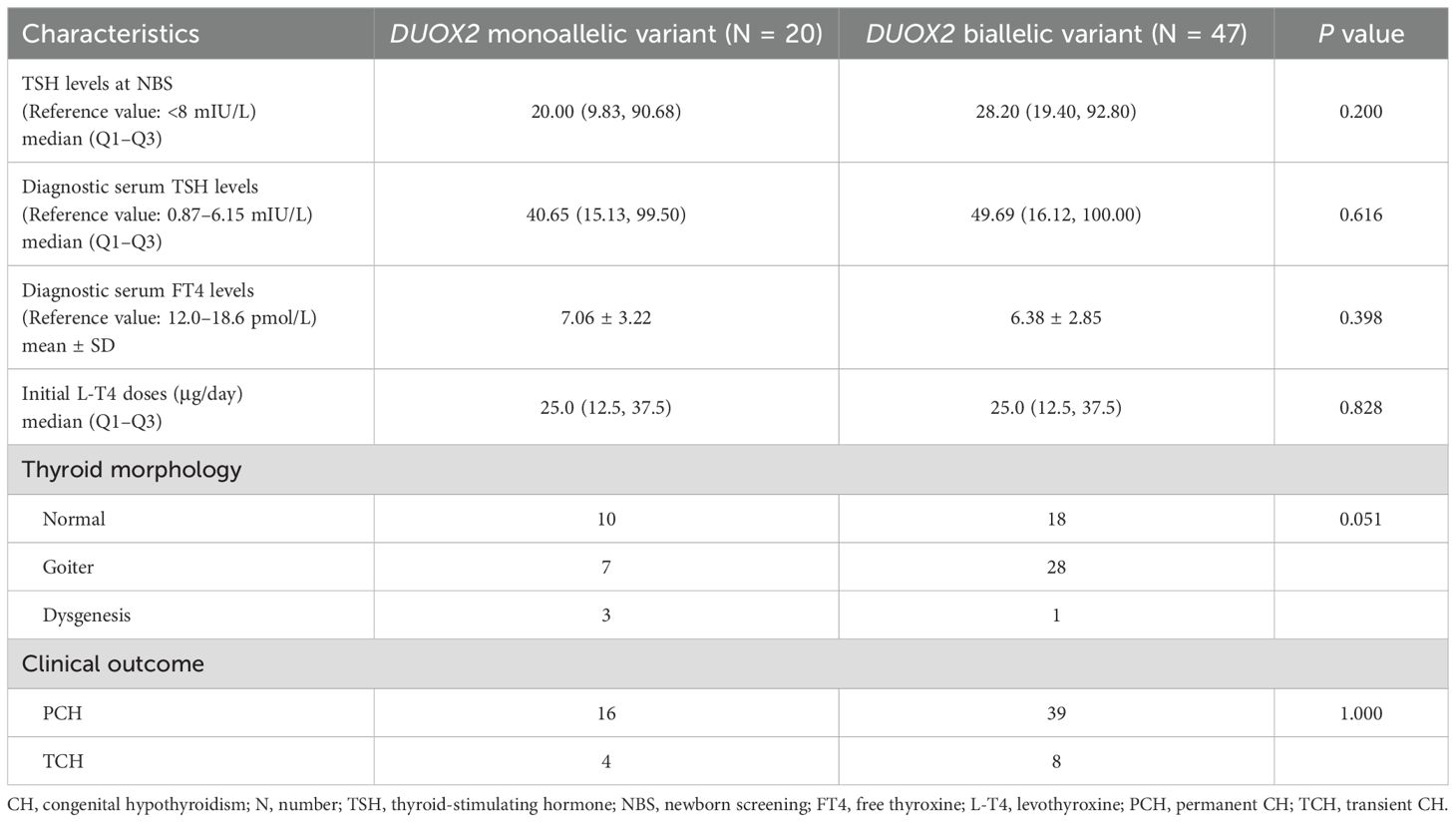
Table 3. Differences in biochemical and clinical characteristics of 67 children with CH between “DUOX2 monoallelic variant” and “DUOX2 biallelic variant” groups.
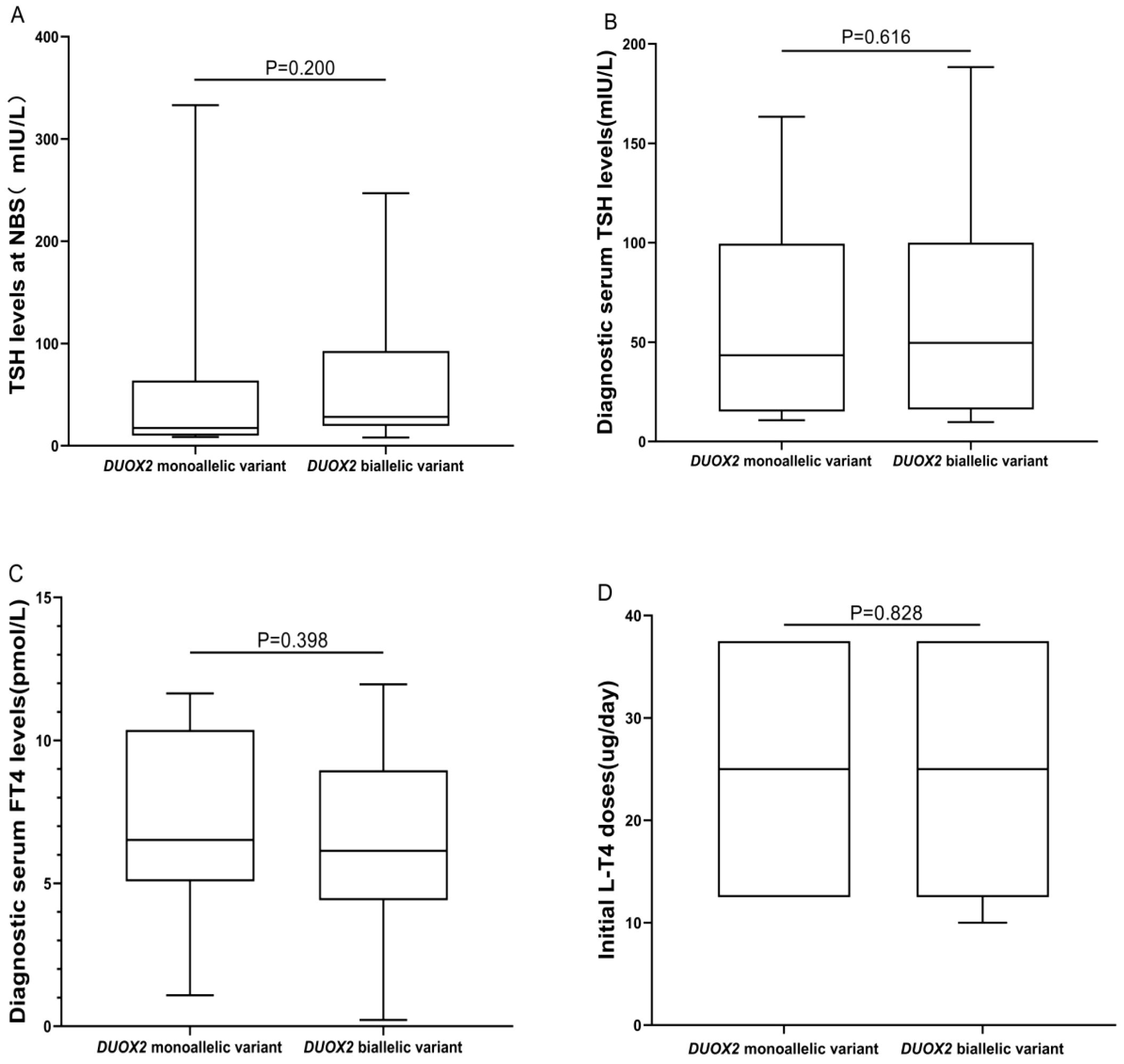
Figure 3. Differences in biochemical results and initial L-T4 doses between “DUOX2 monoallelic variant” and “DUOX2 biallelic variant” groups. (A) The BOX plot chart shows the differences between the DUOX2 monoallelic and biallelic variant groups regarding TSH levels at NBS. (B) The BOX plot chart shows the differences between the DUOX2 monoallelic and biallelic variant groups regarding diagnostic serum TSH levels. (C) The BOX plot chart shows the differences between the DUOX2 monoallelic and biallelic variant groups regarding diagnostic serum FT4 levels. (D) The BOX plot chart shows the differences between the DUOX2 monoallelic and biallelic variant groups regarding the initial L-T4 doses. P>0.05 was considered to indicate no statistical significance.
3.5 Analyses of relationships between genotypes and clinical phenotypes
To analyze relationships between genotypes and clinical phenotypes, the 91 CH patients were divided into four groups based on the type of gene variant: those carrying only one monoallelic variant were classified as the “monoallelic variant” group; those with two or more monoallelic variants were in the “more than one monoallelic variant” group; those with only one biallelic variant were in the “biallelic variant” group; and those with one or more monoallelic variants in addition to one biallelic variant were in the “biallelic variant + one or more monoallelic variant” group. The numbers of patients in these four groups were 32, 6, 45, and 8, respectively. There were no significant differences among the four groups in terms of TSH levels at NBS, diagnostic serum TSH levels, diagnostic serum FT4 levels, initial L-T4 doses, or clinical outcomes (P = 0.100, 0.528, 0.570, 0.348, 0.684, respectively). However, there was a significant difference in thyroid morphology among the four groups (P = 0.003). The “monoallelic variant” group had a greater proportion of individuals with “normal” thyroid morphology, accounting for 53.1% (17/32) of the population. With an increase in the number of gene variants, the thyroid morphology gradually shifted toward “goiter” and “dysgenesis” (Table 4, Figure 4).
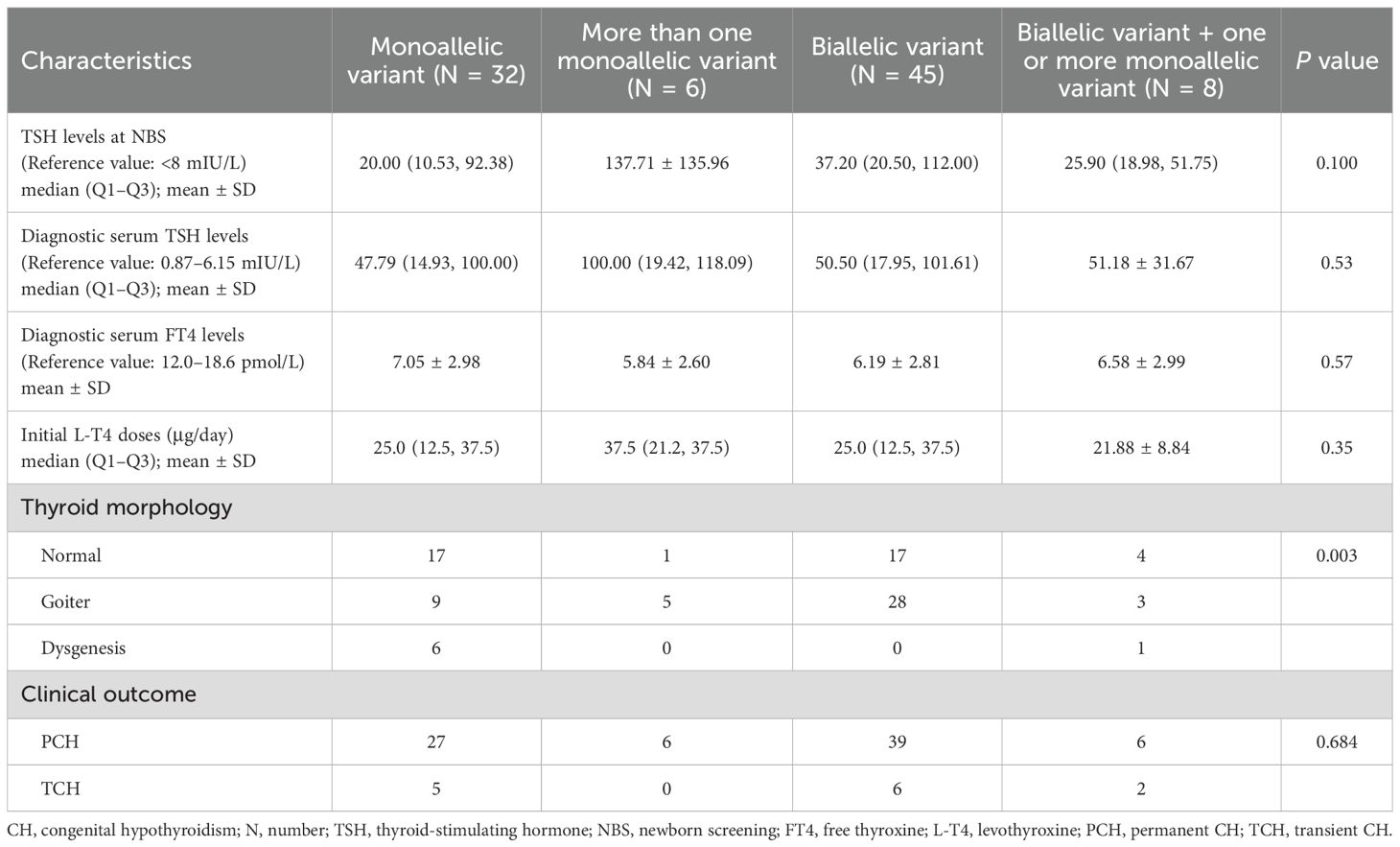
Table 4. Differences in biochemical and clinical characteristics of 91 children with CH based on gene variant combinations.
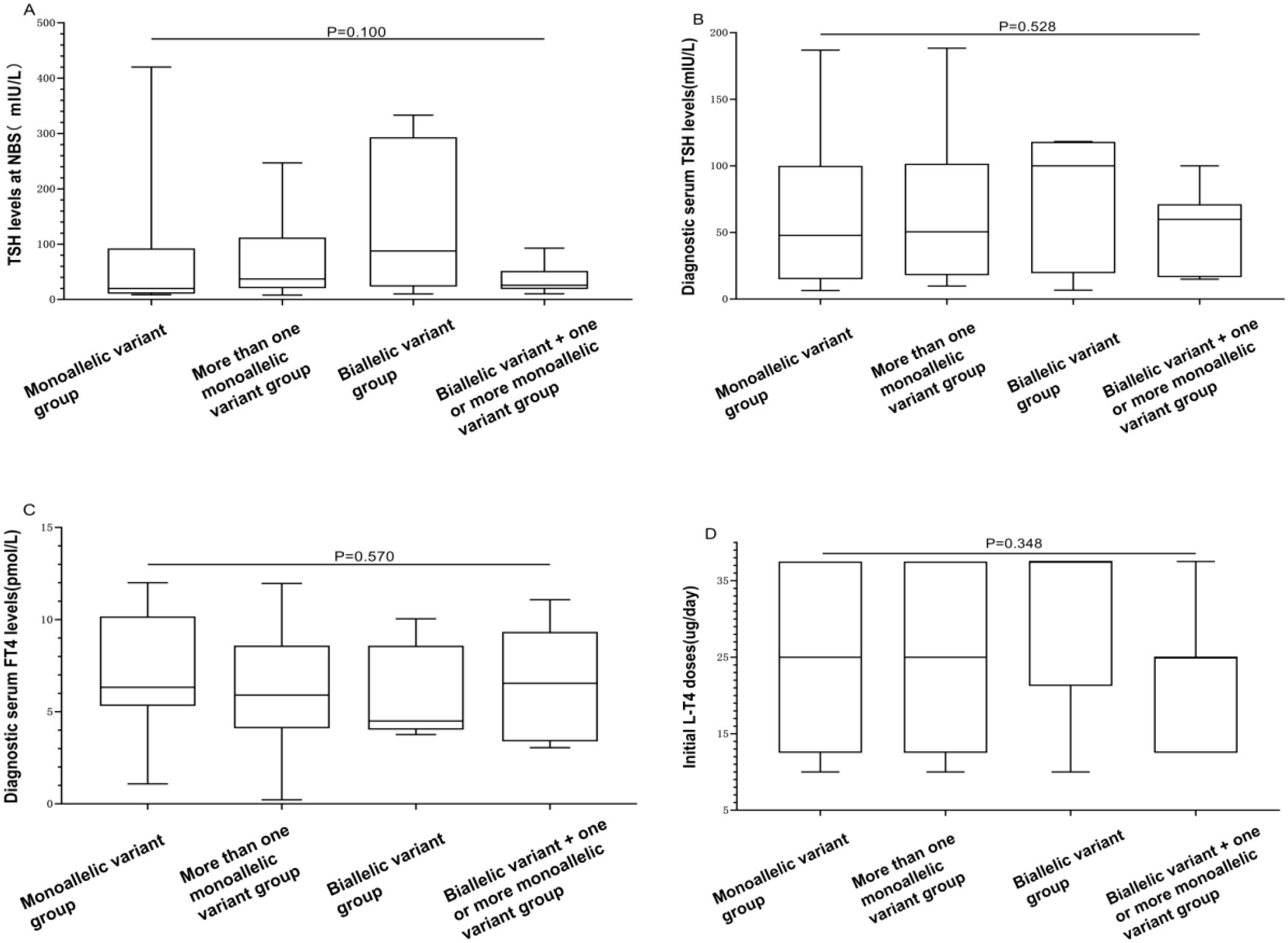
Figure 4. Differences in biochemical results and initial L-T4 doses among “monoallelic variant”, “more than one monoallelic variant”, “biallelic variant”, and “biallelic variant + one or more monoallelic variant” groups. (A) The BOX plot chart illustrates differences among the four groups concerning TSH levels at NBS. (B) The BOX plot chart displays differences among the four groups concerning diagnostic serum TSH levels. (C) The BOX plot chart shows differences among the four groups regarding diagnostic serum FT4 levels. (D) The BOX plot chart demonstrates differences among the four groups regarding the initial L-T4 doses. A P value > 0.05 was considered to indicate no statistically significant difference.
3.6 Growth and development of 25 children with CH of different genotypes
Among the 91 CH patients, 25 underwent standardized physical and intelligence assessments at the ages of 1, 2, and 3 years. The clinical outcomes included 23 cases of PCH and 2 cases of TCH. The physical development (height, weight, head circumference) and intellectual development (gross motor skills, fine motor skills, adaptive behavior, language, and personal-social behavior) of these patients were within the normal range, showing no significant differences compared to those of same-age children. The patients were divided into three groups based on the types of gene variants they carried. At the same monitoring time points, there were no significant differences in the above indicators among the patients with different gene variant types (all P> 0.05). Moreover, no significant differences in L-T4 doses at the maintenance level at the age of 3 years were detected among the three groups (all P> 0.05). A subset of 25 patients had complete standardized follow-up records at 1, 2, and 3 years; others were excluded due to incomplete data or drop-out (Table 5).
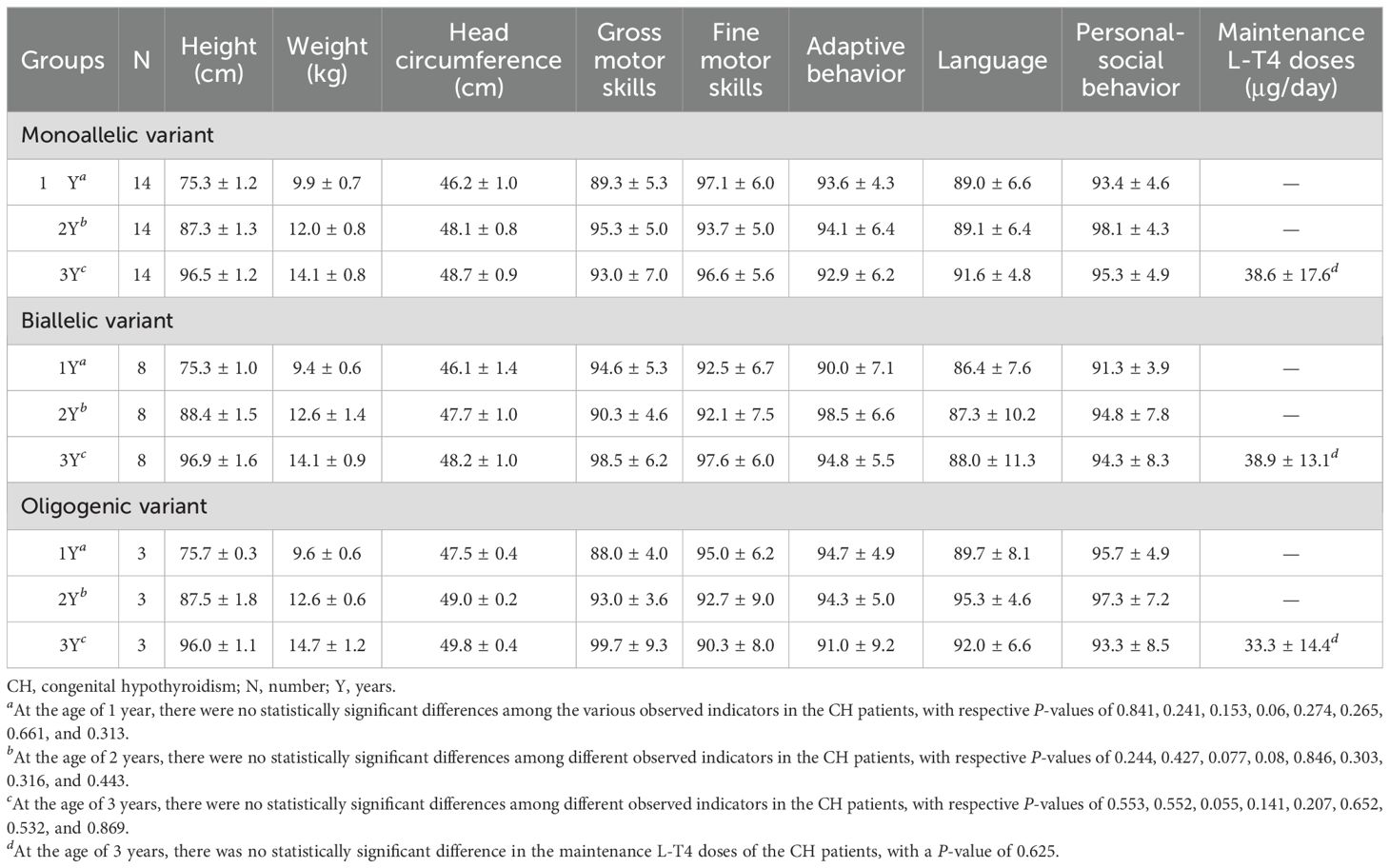
Table 5. Growth and development of 25 CH children with different gene variant combinations at 1 to 3 years of age.
4 Discussion
In this study, we employed targeted region capture NGS to analyze mutations in 27 CH-related genes in a comprehensive manner, aiming to unravel the molecular and clinical characteristics of 91 Chinese children with CH from Yunnan Province.
With widespread application of NGS, molecular research reports on CH have been increasing both in China and abroad. Application of genome sequencing technology for newborn screening is gradually becoming a future development trend (17–19). However, despite significant discoveries regarding CH-related genes, linking these genotypes to L-T4 treatment dosage and clinical outcomes poses a complex challenge. Our study detected relevant gene variations in 91 of 117 CH children from Yunnan, yielding a detection rate of 77.7% (91/117). No statistically significant differences were observed in the demographic or clinical characteristics between male and female patients, underscoring the uniform distribution of CH-related characteristics in this cohort. The five most commonly mutated genes were DUOX2, DUOXA2, TG, TSHR, and TPO. Notably, a subset of CH patients eventually exhibited TCH, constituting 14.3% (13/91) of the clinical outcomes. This finding is consistent with previous reports, as the clinical outcomes of CH detected through newborn screening continue to be primarily represented by PCH (11, 20).
CH is predominantly associated with TD and DH. DH accounts for approximately 35% of CHs, and a genetic cause is identified in 50% of patients; TD accounts for approximately 65% of CHs, and a genetic cause is identified in less than 5% of patients (21). Genes related to TD include PAX8, TSHR, NKX2-1, FOXE1, NKX2-5, TUBB1, and HHEX; these genes are often inherited in an autosomal dominant (AD) or autosomal recessive (AR) manner (22–25). On the other hand, genes associated with DH include DUOX2, DUOXA2, TPO, TG, SLC26A4, SLC5A5, and IYD, which are typically inherited in an AR-related manner (14, 26). In previous reports, the majority of CH cases were attributed to TD, as characterized by features such as agenesis or athyreosis, ectopy, orthotopic hypoplasia and hemiagenesis (27, 28). However, in our cohort, patients with a normal or enlarged thyroid constituted the overwhelming majority, accounting for 92.2% (84/91). Furthermore, among the five most common genetic variations in our cohort, DUOX2, DUOXA2, TG, and TPO mutations were associated with DH. Given the diverse ethnicities, geographical environments, dietary patterns and genetic backgrounds in China, existing research reports show slight variations in the ranking of gene variations (29, 30). Nevertheless, overall, genes related to DH dominate (31). In contrast, types of genetic variants associated with CH have been associated mainly with TD in Western populations (32, 33). It is undeniable that the mechanisms underlying CH occurrence display significant racial and ethnic variations.
In our cohort, 67 CH patients (61.5%) were identified to harbor DUOX2 variants, revealing a higher DUOX2 gene mutation rate, consistent with the findings of scholars in other regions of China, Korea, and Japan (34–37). This finding suggests that DUOX2 gene variants might be a primary genetic factor involved in the occurrence of CH in East Asian populations. Some studies from other countries report that variants in the TPO or TG gene are the most common cause of synthesis disorders, possibly linked to racial and regional variations (38–40). The variants c.1588A > T (p.K530X), c.3329G > A (p.R1110Q), and c.2654G>T (p.R885L) were most common in our cohort, with K530X previously reported to have a higher variant frequency in the Chinese population, emphasizing this significant characteristic. As a member of the NADPH oxidase family, DUOX2 plays a pivotal role in the generation of H2O2 in the thyroid, participating in a critical step in thyroid hormone synthesis. DUOX2 variations commonly impede iodination of thyroglobulin, consequently hampering synthesis of thyroid hormones (41). However, recent research has revealed that certain DUOX2 variants may also be associated with TD (15). In our study, three patients with either monoallelic or biallelic DUOX2 variants exhibited TD, suggesting a potential link between DUOX2 and thyroid dysgenesis in specific cases. A possible explanation is that the DUOX family exerts additional effects on thyroid development through interactions with other unidentified proteins. DUOXA2 resides on chromosome 15 in a head-to-head orientation with DUOX2 and plays a crucial role in the posttranslational processing and enzymatic activity of DUOX2 (42, 43). Both DUOXA2 and DUOX2 are vital components of the NADPH oxidase family (44). The main role of DUOXA2 is in assisting and facilitating the maturation and activation of DUOX2 (12, 44). Variations in DUOXA2 can impact the ability of the thyroid to generate H2O2, leading to disturbances in thyroid hormone synthesis (14). In our research, the DUOXA2 gene variant detection rate was 8.3% (9/109), suggesting that in the local population, DUOX2, in conjunction with DUOXA2, plays a major role in the occurrence of CH.
Several studies have proposed that CH patients with monoallelic DUOX2 variants tend to exhibit TCH and that those with biallelic DUOX2 variants are more likely to experience PCH (45). Our findings did not reveal significant differences in biochemical results, initial L-T4 doses, or clinical outcomes between patients with monoallelic and biallelic DUOX2 mutations. However, a significant association was found between DUOX2 biallelic variants and goiter, with monoallelic DUOX2 variants often associated with normal thyroid morphology. The above findings suggest a strong correlation between the quantity of DUOX2 variations and thyroid morphology. How to predict clinical outcomes based on these findings remains highly uncertain.
In general, the severity of the CH phenotype in relation to the quantity of CH gene variants remains unclear. Notably, this study revealed digenic variants in 11 patients and oligogenic variants in 3 patients. Among these gene combinations, DUOX2+TG was most common, constituting 27.3% (3/11). Wang et al. identified the most common digenic variant as DUOX2/DUOXA1 in 61 CH patients, in contrast to our findings (46). This discrepancy might be attributed to the smaller sample size in our study and variations in geographical and ethnic factors. Prior research suggests that the more gene variant sites or types there are, the more challenging it is for thyroid function to return to normal, requiring longer durations and larger doses of medication for maintenance (47, 48). In this study, we categorized CH patients into four groups based on the gene variant types and quantities. No statistically significant differences were observed among these groups regarding biochemical results, initial L-T4 doses or clinical outcomes. However, concerning thyroid morphology, the “monoallelic variant group” predominantly exhibited normal morphology, whereas goiters became more prevalent with an increase in gene variant types and quantities. These findings suggest that synergistic interactions between gene variants enhance the negative feedback loop of the hypothalamus-pituitary-thyroid axis, leading to elevated serum TSH levels, a gradual increase in thyroid volume, and eventual goiter. Despite these morphological differences, no significant differences were observed in clinical outcomes among the four groups, with PCH remaining the primary clinical outcome. A subset of 25 CH patients underwent three years of standardized follow-up, with no significant differences observed in physical or intellectual development assessments at 1, 2, and 3 years of age among patients with different gene types and quantities. Although the sample size is limited, these findings emphasize the paramount role of standardized L-T4 treatment and follow-up in influencing treatment outcomes for CH children.
Recent studies have increasingly highlighted the role of digenic and oligogenic inheritance patterns in CH, suggesting that combined variants in DUOX2, DUOXA2, TG, and other genes may exert synergistic effects on thyroid development and function. These findings reinforce our observation of multi-gene variants in a subset of patients and further underscore the complexity of genotype–phenotype correlations in CH (49). Integrating these recent insights, it becomes evident that comprehensive genetic screening and careful interpretation are essential, and that functional validation will be particularly important for variants of uncertain significance detected in oligogenic contexts (50).
Consistent with other East Asian cohorts, our Yunnan series showed a DUOX2-dominant genetic spectrum of primary CH. Population-based and hospital-based studies from mainland China similarly report dyshormonogenesis as the major etiology with DUOX2 being the most frequently mutated gene, often accompanied by DUOXA2/TG/TPO variants and occasional oligogenic patterns (30). In Korea, DUOX2 mutations are likewise a frequent cause with several recurrent alleles, supporting a shared East-Asian pattern (36). apanese data using targeted NGS panels also identify recurrent CH-associated variants detectable by screening panels, in line with an East-Asian mutation spectrum (37). In contrast, studies from consanguineous populations in the Middle East and from Sudan report a predominance of TPO and TG mutations among dyshormonogenesis cases, underscoring population-specific architectures likely shaped by founder effects and consanguinity (38). These comparisons suggest that our findings are generalizable within East Asia but may not extend to regions where TPO/TG defects prevail; this population contrast has been emphasized in recent reviews and consensus statements (14).
This study has several limitations. First, the relatively small sample size implies that the conclusions drawn may not comprehensively explain the genotype–phenotype correlation in CH patients. Future research should aim to expand the sample size for more robust analysis. Second, the CH patients in this study were exclusively from Yunnan Province. Given regional and ethnic differences, the findings of this study may not be representative of the entire country of mainland China, highlighting the need for multicenter prospective studies. Finally, further functional studies are warranted to validate the pathogenicity of these variants of uncertain significance (VUSs), enhancing the clarity of the genotype–phenotype relationship.
In conclusion, CH is a complex endocrine disorder, and although efforts have been made to decipher its genetic code, the relationship between genotype and phenotype is not straightforward. Possible genetic causes identified through NGS may not be possible for all CH patients, and unknown genes or environmental factors may play a role. Currently, supported by governmental initiatives, establishment of a standardized system for screening, recall, diagnosis, treatment and follow-up in newborn screening centers is crucial for ensuring the normal growth and development of CH children.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Ethics statement
This study was conducted in accordance with the guidelines of the Declaration of Helsinki and was approved by the Ethics Committee of The First People's Hospital of Yunnan Province (KHLL2023-KY037). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants' legal guardians/next of kin. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
YZ: Writing – original draft, Writing – review & editing. SW: Writing – original draft, Writing – review & editing. AL: Writing – original draft. WZ: Writing – original draft. FX: Writing – original draft. YC: Writing – original draft. JL: Writing – original draft. XZ: Writing – original draft. SL: Writing – review & editing. NF: Writing – original draft. BZ: Writing – original draft, Writing – review & editing. LL: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was supported by the “Famous Doctor” special project of the Ten Thousand People Program of Yunnan Province (YNWR-MY-2018-016), Co-operation Fund of Kunming Medical University and the Science and Technology Department of Yunnan Province (202101AY070001-262), Open Fund of Yunnan Provincial Key Laboratory for Birth Defects and Genetic Diseases (2020ZDKFKT001), Open Project of Yunnan Provincial Reproductive and Obstetrics and Gynecology Clinical Medicine Center (2022LCZXKF-SZ02), The Special Foundation for Basic Research Program of Yunnan Province (202101AT070233), The Xingdian Talent Support Project of Yunnan Province (XDYC-QNRC-2022-0267), Yunnan Provincial Special Project for Selecting High-level Scientific and Technological Talents and Innovation Teams -Technological Innovation Talent Training Project (202405AD350029).
Acknowledgments
We are grateful to the patients, their family members and all contributors for their participation.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2025.1640108/full#supplementary-material
Supplementary Table 1 | Clinical and molecular characteristics of 91 children with congenital hypothyroidism. M, male; F, female; SHT, subclinical hypothyroidism; CHT, clinical hypothyroidism; Y, years; M, months; NBS, newborn screening; TCH, transient congenital hypothyroidism; PCH, permanent congenital hypothyroidism; P, pathogenic; LP, likely pathogenic; VUS, variant of uncertain significance.
Abbreviations
CH, congenital hypothyroidism; TSH, thyroid-stimulating hormone; PCH, permanent CH; TCH, transient CH; L-T4, levothyroxine; TD, tothyroid dysgenesis; DH, dyshormonogenesis; NGS, next generation sequencing; T4, thyroxine; FT4, free thyroxine; T3, triiodothyronine; FT3, free triiodothyronine; NBS, newborn screening.
References
1. van Trotsenburg P, Stoupa A, Léger J, Rohrer T, Peters C, Fugazzola L, et al. Congenital hypothyroidism: A 2020–2021 consensus guidelines update-an ENDO-european reference network initiative endorsed by the european society for pediatric endocrinology and the european society for endocrinology. Thyroid. (2021) 31:387–419. doi: 10.1089/thy.2020.0333
2. Deng K, He C, Zhu J, Liang J, Li X, Xie X, et al. Incidence of congenital hypothyroidism in China: data from the national newborn screening program, 2013-2015. J Pediatr Endocrinol Metab. (2018) 31:601–8. doi: 10.1515/jpem-2017-0361
3. Kilberg MJ, Rasooly IR, LaFranchi SH, Bauer AJ, and Hawkes CP. Newborn screening in the US may miss mild persistent hypothyroidism. J Pediatr. (2018) 192:204–8. doi: 10.1016/j.jpeds.2017.09.003
4. Rose SR, Wassner AJ, Wintergerst KA, Yayah-Jones NH, Hopkin RJ, Chuang J, et al. Congenital hypothyroidism: screening and management. Pediatrics. (2023) 151(1):e2022060420. doi: 10.1542/peds.2022-060420
5. Wassner AJ. Congenital hypothyroidism. Clin Perinatol. (2018) 45:1–18. doi: 10.1016/j.clp.2017.10.004
6. He S, Ma X, Yang J, and Li L. Levothyroxine treatment for congenital hypothyroidism based on thyroid function: a 10-year clinical retrospective study. BMC EndocrDisord. (2022) 22:142. doi: 10.1186/s12902-022-01061-z
7. Bauer AJ and Wassner AJ. Thyroid hormone therapy in congenital hypothyroidism and pediatric hypothyroidism. Endocrine. (2019) 66:51–62. doi: 10.1007/s12020-019-02024-6
8. Libri DV, Trettene A, Bonomi M, Beck-Peccoz P, Persani L, and Salvatoni A. The unusual adequate development of a child with severe central hypothyroidsm negative at neonatal thyrotropin screening. J Endocrinol Invest. (2013) 36:788–9. doi: 10.3275/8963
9. Shapira SK, Hinton CF, Held PK, Jones E, Harry Hannon W, and Ojodu J. Single newborn screen or routine second screening for primary congenital hypothyroidism. Mol Genet Metab. (2015) 116:125–32. doi: 10.1016/j.ymgme.2015.08.003
10. Rastogi MV and LaFranchi SH. Congenital hypothyroidism. Orphanet J Rare Dis. (2010) 5:17. doi: 10.1186/1750-1172-5-17
11. Korzeniewski SJ, Grigorescu V, Kleyn M, Young WI, Birbeck G, Todem D, et al. Transient hypothyroidism at 3-year follow-up among cases of congenital hypothyroidism detected by newborn screening. J Pediatr. (2013) 162:177–82. doi: 10.1016/j.jpeds.2012.06.050
12. Peters C and Schoenmakers N. MECHANISMS IN ENDOCRINOLOGY: The pathophysiology of transient congenital hypothyroidism. Eur J Endocrinol. (2022) 187:R1–r16. doi: 10.1530/EJE-21-1278
13. Yao Y, Deng K, Zhu J, Xiang L, Yuan X, Li Q, et al. Increased incidence of congenital hypothyroidism in China: An analysis of 119 million screened newborns. Eur J Pediatr. (2023) 182:4477–86. doi: 10.1007/s00431-023-05108-8
14. Kostopoulou E, Miliordos K, and Spiliotis B. Genetics of primary congenital hypothyroidism-a review. Hormones (Athens). (2021) 20:225–36. doi: 10.1007/s42000-020-00267-x
15. Zhang T, Shen Y, Xu Y, Wu D, Chen C, and Yang R. Clinical, biochemical characteristics and genotype-phenotype analysis of congenital hypothyroidism diagnosed by newborn screening in China. Clin Chim Acta. (2023) 547:117459. doi: 10.1016/j.cca.2023.117459
16. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
17. Ceyhan-Birsoy O, Murry JB, Machini K, Lebo MS, Yu TW, Fayer S, et al. Interpretation of genomic sequencing results in healthy and ill newborns: results from the babySeq project. Am J Hum Genet. (2019) 104:76–93. doi: 10.1016/j.ajhg.2018.11.016
18. Huang X, Wu D, Zhu L, Wang W, Yang R, Yang J, et al. Application of a next-generation sequencing (NGS) panel in newborn screening efficiently identifies inborn disorders of neonates. Orphanet J Rare Dis. (2022) 17:66. doi: 10.1186/s13023-022-02231-x
19. Chen T, Fan C, Huang Y, Feng J, Zhang Y, Miao J, et al. Genomic sequencing as a first-tier screening test and outcomes of newborn screening. JAMA Netw Open. (2023) 6:e2331162. doi: 10.1001/jamanetworkopen.2023.31162
20. Alavi ER, Rafiei N, Rafiei R, and Farokhi E. Prevalence of transient congenital hypothyroidism among neonates. Ann Med Surg (Lond). (2021) 72:103083. doi: 10.1016/j.amsu.2021.103083
21. Stoupa A, Kariyawasam D, Polak M, and Carré A. Genetic of congenital hypothyroidism. Med Sci (Paris). (2022) 38:263–73. doi: 10.1051/medsci/2022028
22. Trueba SS, Augé J, Mattei G, Etchevers H, Martinovic J, Czernichow P, et al. PAX8, TITF1, and FOXE1 gene expression patterns during human development: new insights into human thyroid development and thyroid dysgenesis-associated malformations. J Clin Endocrinol Metab. (2005) 90:455–62. doi: 10.1210/jc.2004-1358
23. van Engelen K, Mommersteeg MT, Baars MJ, Lam J, Ilgun A, van Trotsenburg AS, et al. The ambiguous role of NKX2–5 mutations in thyroid dysgenesis. PloS One. (2012) 7:e52685. doi: 10.1371/journal.pone.0052685
24. Macchia PE, Lapi P, Krude H, Pirro MT, Missero C, Chiovato L, et al. PAX8 mutations associated with congenital hypothyroidism caused by thyroid dysgenesis. Nat Genet. (1998) 19:83–6. doi: 10.1038/ng0598-83
25. Stoupa A, Adam F, Kariyawasam D, Strassel C, Gawade S, Szinnai G, et al. TUBB1 mutations cause thyroid dysgenesis associated with abnormal platelet physiology. EMBO Mol Med. (2018) 10(17):3898. doi: 10.15252/emmm.201809569
26. Li M, Li X, Wang F, Ren Y, Zhang X, Wang J, et al. Genetic analysis of iodide transporter and recycling (NIS, PDS, SLC26A7, IYD) in patients with congenital hypothyroidism. Gene. (2022) 824:146402. doi: 10.1016/j.gene.2022.146402
27. Vassart G and Dumont JE. Thyroid dysgenesis: multigenic or epigenetic … or both? Endocrinology. (2005) 146:5035–7. doi: 10.1210/en.2005-1238
28. Mio C, Grani G, Durante C, and Damante G. Molecular defects in thyroid dysgenesis. Clin Genet. (2020) 97:222–31. doi: 10.1111/cge.13627
29. Xue P, Yang Y, Yun Q, Cui Y, Yu B, and Long W. Variant of TSHR is not a frequent cause of congenital hypothyroidism in chinese han patients. Int J Gen Med. (2021) 14:4135–43. doi: 10.2147/IJGM.S322726
30. Long W, Guo F, Yao R, Wang Y, Wang H, Yu B, et al. Genetic and phenotypic characteristics of congenital hypothyroidism in a chinese cohort. Front Endocrinol (Lausanne). (2021) 12:705773. doi: 10.3389/fendo.2021.705773
31. Zhang RJ, Yang GL, Cheng F, Sun F, Fang Y, Zhang CX, et al. The mutation screening in candidate genes related to thyroid dysgenesis by targeted next-generation sequencing panel in the Chinese congenital hypothyroidism. Clin Endocrinol (Oxf). (2022) 96:617–26. doi: 10.1111/cen.14577
32. Sun F, Zhang JX, Yang CY, Gao GQ, Zhu WB, Han B, et al. The genetic characteristics of congenital hypothyroidism in China by comprehensive screening of 21 candidate genes. Eur J Endocrinol. (2018) 178:623–33. doi: 10.1530/EJE-17-1017
33. Wang H, Kong X, Pei Y, Cui X, Zhu Y, He Z, et al. Mutation spectrum analysis of 29 causative genes in 43 Chinese patients with congenital hypothyroidism. Mol Med Rep. (2020) 22:297–309. doi: 10.3892/mmr.2020.11078
34. Tan M, Huang Y, Jiang X, Li P, Tang C, Jia X, et al. The prevalence, clinical, and molecular characteristics of congenital hypothyroidism caused by DUOX2 mutations: A population-based cohort study in guangzhou. Horm Metab Res. (2016) 48:581–8. doi: 10.1055/s-0042-112224
35. Ye L, Yin Y, Chen M, Gong N, Peng Y, Liu H, et al. Combined genetic screening and traditional newborn screening to improve the screening efficiency of congenital hypothyroidism. Front Pediatr. (2023) 11:1185802. doi: 10.3389/fped.2023.1185802
36. Park KJ, Park HK, Kim YJ, Lee KR, Park JH, Park JH, et al. DUOX2 mutations are frequently associated with congenital hypothyroidism in the korean population. Ann Lab Med. (2016) 36:145–53. doi: 10.3343/alm.2016.36.2.145
37. Watanabe D, Yagasaki H, Narusawa H, Saito T, Mitsui Y, Miyake K, et al. Screening of frequent variants associated with congenital hypothyroidism: a comparison with next generation sequencing. Endocr J. (2021) 68:1411–9. doi: 10.1507/endocrj.EJ21-0353
38. Cangul H, Aycan Z, Olivera-Nappa A, Saglam H, Schoenmakers NA, Boelaert K, et al. Thyroid dyshormonogenesis is mainly caused by TPO mutations in consanguineous community. Clin Endocrinol (Oxf). (2013) 79:275–81. doi: 10.1111/cen.12127
39. Bruellman RJ, Watanabe Y, Ebrhim RS, Creech MK, Abdullah MA, Dumitrescu AM, et al. Increased prevalence of TG and TPO mutations in Sudanese children with congenital hypothyroidism. J Clin Endocrinol Metab. (2020) 105:1564–72. doi: 10.1210/clinem/dgz297
40. Tobias L, Elias-Assad G, Khayat M, Admoni O, Almashanu S, and Tenenbaum-Rakover Y. Long-term outcome of patients with TPO mutations. J Clin Med. (2021) 10. doi: 10.3390/jcm10173898
41. Zamproni I, Grasberger H, Cortinovis F, Vigone MC, Chiumello G, Mora S, et al. Biallelic inactivation of the dual oxidase maturation factor 2 (DUOXA2) gene as a novel cause of congenital hypothyroidism. J Clin Endocrinol Metab. (2008) 93:605–10. doi: 10.1210/jc.2007-2020
42. Luxen S, Belinsky SA, and Knaus UG. Silencing of DUOX NADPH oxidases by promoter hypermethylation in lung cancer. Cancer Res. (2008) 68:1037–45. doi: 10.1158/0008-5472.CAN-07-5782
43. Hulur I, Hermanns P, Nestoris C, Heger S, Refetoff S, Pohlenz J, et al. A single copy of the recently identified dual oxidase maturation factor (DUOXA) 1 gene produces only mild transient hypothyroidism in a patient with a novel biallelic DUOXA2 mutation and monoallelic DUOXA1 deletion. J Clin Endocrinol Metab. (2011) 96:E841–845. doi: 10.1210/jc.2010-2321
44. Poncelet L, Dumont JE, Miot F, and De Deken X. The Dual Oxidase Duox2 stabilized with DuoxA2 in an enzymatic complex at the surface of the cell produces extracellular H(2)O(2) able to induce DNA damage in an inducible cellular model. Exp Cell Res. (2019) 384:111620. doi: 10.1016/j.yexcr.2019.111620
45. Fu C, Luo S, Zhang S, Wang J, Zheng H, Yang Q, et al. Next-generation sequencing analysis of DUOX2 in 192 Chinese subclinical congenital hypothyroidism (SCH) and CH patients. Clin Chim Acta. (2016) 458:30–4. doi: 10.1016/j.cca.2016.04.019
46. Wang F, Zang Y, Li M, Liu W, Wang Y, Yu X, et al. DUOX2 and DUOXA2 variants confer susceptibility to thyroid dysgenesis and gland-in-situ with congenital hypothyroidism. Front Endocrinol (Lausanne). (2020) 11:237. doi: 10.3389/fendo.2020.00237
47. Aycan Z, Cangul H, Muzza M, Bas VN, Fugazzola L, Chatterjee VK, et al. Digenic DUOX1 and DUOX2 mutations in cases with congenital hypothyroidism. J Clin Endocrinol Metab. (2017) 102:3085–90. doi: 10.1210/jc.2017-00529
48. Sun F, Zhang RJ, Cheng F, Fang Y, Yang RM, Ye XP, et al. Correlation of DUOX2 residual enzymatic activity with phenotype in congenital hypothyroidism caused by biallelic DUOX2 defects. Clin Genet. (2021) 100:713–21. doi: 10.1111/cge.14065
49. Stoupa A, Kariyawasam D, Jabot-Hanin F, Quoc AN, Hanein S, Rabeony T, et al. Digenic Inheritance Mode in Congenital Hypothyroidism due to Thyroid Dysgenesis: HYPOTYGEN translational cohort study. J Clin Endocrinol Metab. (2025) 9:dgaf004. doi: 10.1210/clinem/dgaf004
Keywords: congenital hypothyroidism, thyroid hormone, genotype-phenotype, next-generation sequencing, thyroid morphology, clinical outcome
Citation: Zhang Y, Wang S, Li A, Zhao W, Xia F, Chan Y, Lin J, Zhou X, Li S, Feng N, Zhu B and Li L (2025) Revealing the genotype-phenotype correlations of congenital hypothyroidism in Yunnan Province, Southwest China. Front. Endocrinol. 16:1640108. doi: 10.3389/fendo.2025.1640108
Received: 03 June 2025; Accepted: 30 September 2025;
Published: 17 November 2025.
Edited by:
Terry Francis Davies, Icahn School of Medicine at Mount Sinai, United StatesReviewed by:
Rossella Gaudino, University Hospital of Verona, ItalyJerzy Beltowski, Medical University of Lublin, Poland
Copyright © 2025 Zhang, Wang, Li, Zhao, Xia, Chan, Lin, Zhou, Li, Feng, Zhu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li Li, a2hlcmtsaWxpQHNpbmEuY29t; Baosheng Zhu, YnN6aHVAYWxpeXVuLmNvbQ==
†These authors share first authorship