 Ignacio Portales-Castillo
Ignacio Portales-Castillo Jakob Höppner
Jakob Höppner Harald Jüppner
Harald Jüppner Thomas J. Gardella2*
Thomas J. Gardella2*- 1Department of Medicine, Division of Nephrology, Washington University in St. Louis, St. Louis, MO, United States
- 2Endocrine Unit, Massachusetts General Hospital, and Harvard Medical School, Boston, MA, United States
- 3Pediatric Nephrology Unit, Massachusetts General Hospital, and Harvard Medical School, Boston, MA, United States
The parathyroid hormone receptor type 1 (PTH1R) is a G protein-coupled receptor that mediates the actions of parathyroid hormone (PTH) in the regulation of blood calcium levels, as well as PTH-related protein (PTHrP) in the regulation of skeletal development. Severe loss-of-function homozygous mutations in PTH1R are incompatible with life as in Blomstrand’s lethal chondrodysplasia, characterized by accelerated growth plate ossification. More recently, homozygous mutations located in the transmembrane helices, extracellular domains and C-tail of the PTH1R were identified in patients with milder conditions characterized by variable degrees of skeletal and mineral abnormalities. These include delayed ossification in Eiken syndrome, hypocalcemia in a pseudohypoparathyroidism-like disorder, and non-syndromic primary failure of tooth eruption; which is usually caused by heterozygous PTH1R mutations. Recent detailed pharmacologic characterization of these PTH1R mutants has revealed new insights into how even subtle perturbations in PTH1R function can result in disease.
Overview of human diseases caused by PTH1R mutations
The Parathyroid hormone/parathyroid hormone-related protein receptor (PTH/PTHrP type 1 receptor; or PTH1R) is a family B G protein-coupled receptor (GPCR) that transmits stimuli provided by two endogenous polypeptide ligands to thereby mediate distinct biological functions (1). PTH1R thus responds to PTH, which is secreted as an endocrine hormone from the parathyroid glands, to maintain homeostasis of calcium and phosphorus and it responds to PTHrP, which is secreted from diverse tissues as a paracrine factor, to play several regulatory roles, most prominently in endochondral bone formation, tooth eruption and mammary gland formation (1–4).
Intracellularly, the classic signaling pathway activated by PTH or PTHrP involves the stimulatory G protein alpha-subunit (GαS) – adenylyl cyclase (AC) – cAMP–protein kinase A (PKA) system. The activated PTH1R can also couple to the Gαq pathway to result in increased IP3/Ca2+ generation (5), as well as recruit βarrestin 1 and 2, key multifunctional effector proteins that classically mediate receptor internalization and desensitization, but can also promote signaling through non-canonical pathways (6).
Most of the inactivating PTH1R mutations found in humans are heterozygous and cause primary failure of tooth eruption (PFTE) (7–12). More than twenty such different mutations are spread throughout the different exons encoding the receptor. All reported PFTE mutations lead to a loss-of-function (LOF) effect, as assessed by direct measurement of signaling in vitro or molecular simulations (7–9, 11–13), while at least one intron variant is associated with PFTE (rs1575524795).
Five different heterozygous, gain-of-function PTH1R mutations (H223R, T410P, T410R, I458R, and I458K) have been identified and were shown to cause Jansen’s metaphyseal chondrodysplasia (JMC), a disease characterized by short stature, expanded growth plates and often severe hypercalcemia, hypophosphatemia and hypercalciuria (14–27). These clinical manifestations are consistent with excessive PTH1R signaling in growth plate chondrocytes, bone forming osteoblasts, and kidney target sites (28). All JMC mutations map to one of three amino acid residues in the PTH1R located at the cytosolic base of a transmembrane helices that are involved in a highly conserved network that, when perturbed by the JMC mutations, allows for increased interaction with G protein, and hence agonist-independent receptor activation (29).
The consistent impact of heterozygous activating PTH1R mutations in JMC has been well detailed (30, 31), and the heterozygous inactivating mutations in PFTE suggest that both PTH1R alleles are required for normal tooth development (13). In contrast, the twelve reported homozygous PTH1R mutations cause a wider spectrum of clinical abnormalities that are related to disruption of PTH- and/or PTHrP-dependent receptor functions (Table 1). Five of these mutations cause severe loss-of-function effects, as seen in Blomstrand’s lethal chondrodysplasia (BLC) (32–43). These mutations cause a profound acceleration in ossification of the entire skeleton, including the rib cage, thus leading impaired lung function and death soon after birth. Seven homozygous non-lethal PTH1R mutations were recently described in patients with various degrees of delayed ossification and PFTE, associated with PTH-resistant hypocalcemia in some cases (44–51) (Figure 1). However, these clinical and laboratory findings are distinct from those seen in patients with homozygous PTH mutations that reduce the peptide’s biological activity (52, 53) or heterozygous inactivating mutations in PTHrP that result in brachydactyly and short stature (54–57).
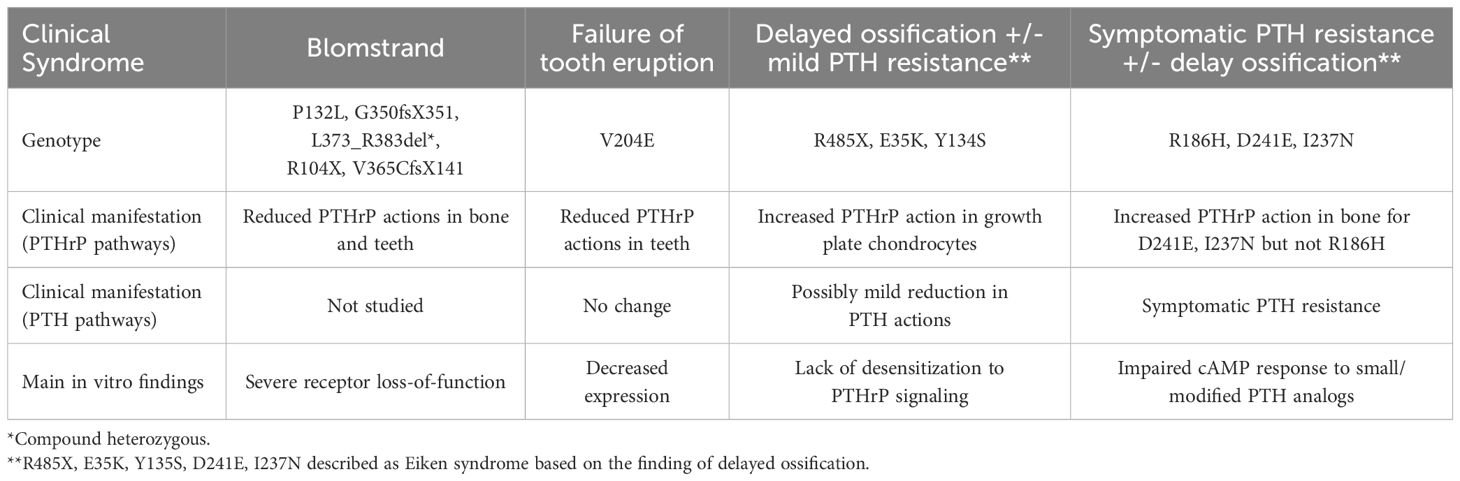
Table 1. Summary of clinical syndromes associated with homozygous PTH1R mutants.
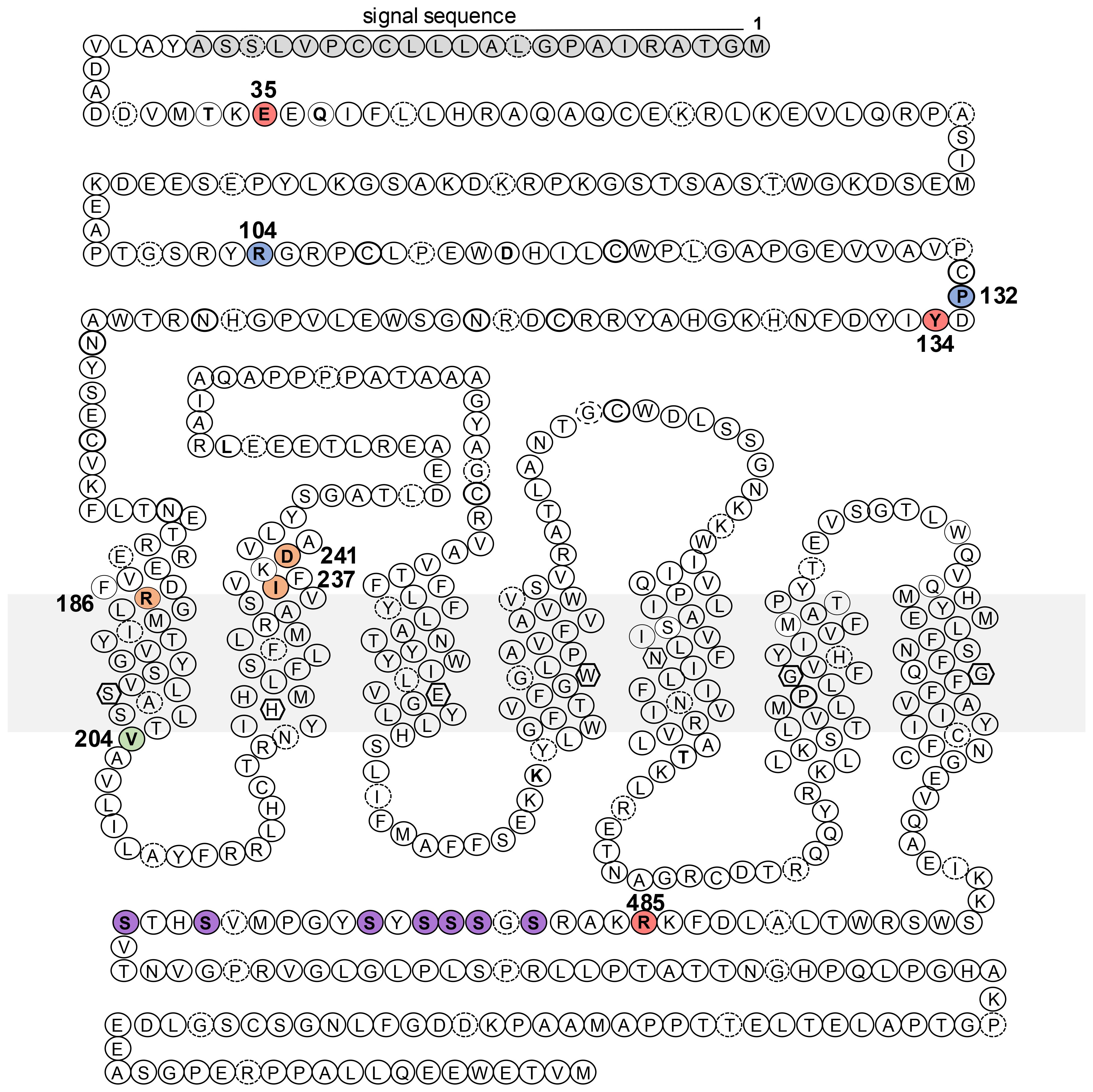
Figure 1. Schematic representation of the PTH1R amino acid sequence and sites of homozygous disease mutations. Indicated in the sequence are the locations of missense or nonsense mutations identified in patients with Blomstrand lethal chondrodysplasia (blue, P132, R104*), Eiken Skeletal dysplasia (salmon, R485, E35, Y132), pseudohypoparathyroidism-like disease (orange, R186, D241, I237) and primary failure of tooth eruption (green, V204). Also indicated are residues of the signal sequence (gray) and phosphorylatable serines (purple) in the C-tail. The gray rectangle represents the plasma membrane.
The effect of heterozygous disease-causing PTH1R mutants has been reviewed elsewhere (8, 10, 19, 58). We therefore focus our review on homozygous PTH1R mutations (Table 2). We first describe the clinical findings in BLC and contrast these findings with those of non-lethal diseases that are caused by homozygous PTH1R mutations. We follow with a pharmacologic characterization of the disease causing PTH1Rs mutants.
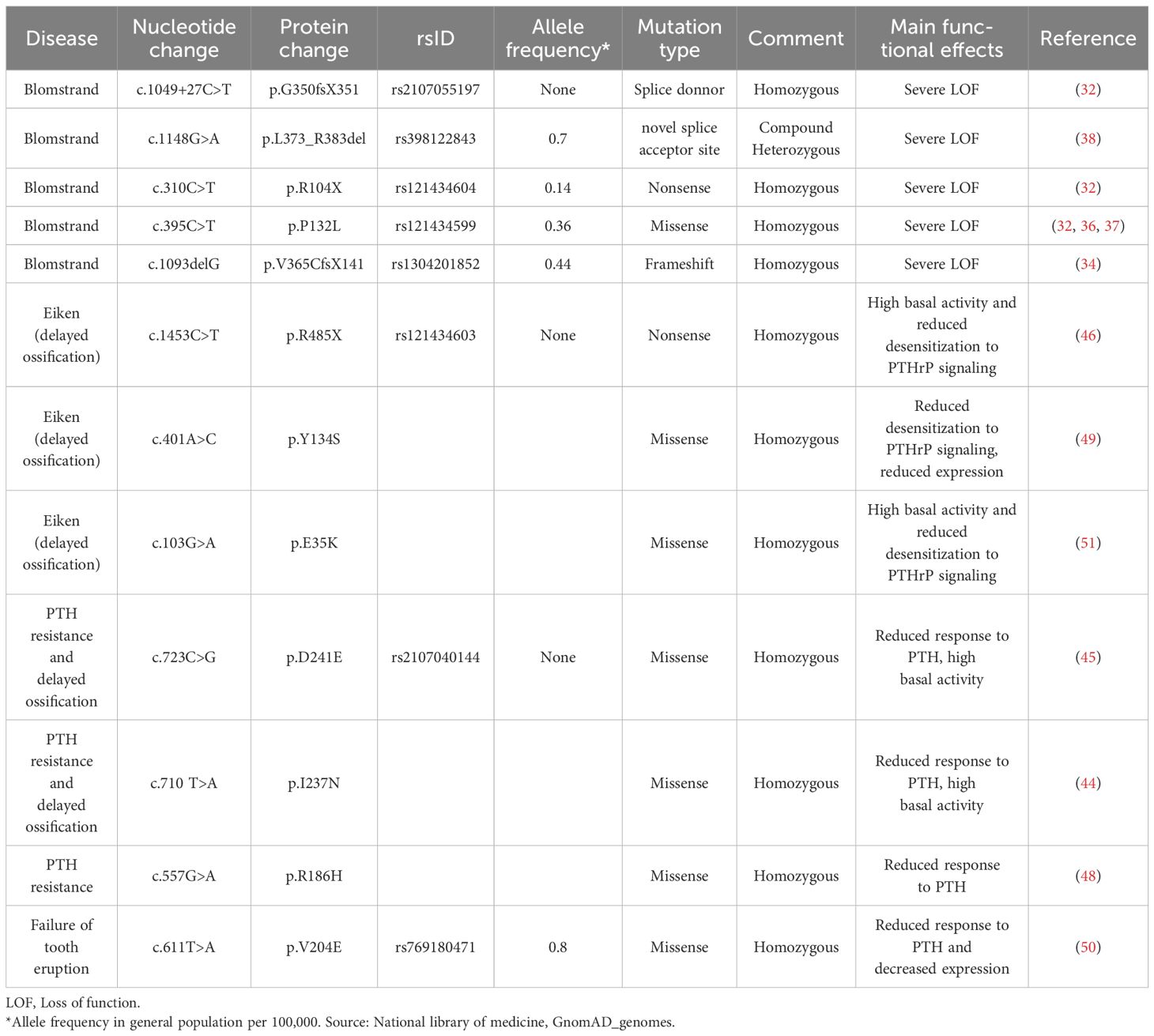
Table 2. Functional effects of PTH1R mutants.
Clinical syndromes associated with homozygous PTH1R mutations
Accelerated ossification in Blomstrand lethal chondrodysplasia
During endochondral bone formation, growth plate chondrocytes at the perichondrium synthesize PTHrP to maintain nearby cells expressing PTH1R in a proliferative state and hence delay their differentiation into hypertrophic chondrocytes and the subsequent process of bone mineralization (4, 59).
Mice that are null for PTHrP or the PTH1R die during the neonatal period (60). Their skeleton shows smaller bones in which histology of the growth plates reveals a shortness of the proliferative chondrocyte layer and an overall disruption of the otherwise highly organized zones of differentiating chondrocytes. Neonatal lethality of PTHrP-null mice can be prevented, at least partially, by overexpressing PTHrP transgenically in proliferating growth plate chondrocytes via the collagen type II promoter. These mice, however, show multiple other abnormalities, including altered skin formation, a failure of tooth eruption, and absent mammary glands (61), thus revealing broader roles of PTHrP during development.
The first report of Blomstrand lethal chondrodysplasia (BLC), was presented in 1985 (43). The affected infant was born prematurely at 22 weeks and died shortly after delivery. Postmortem exam showed generalized edema, tongue protrusion due to a short mandible and noticeably short limbs. The only obvious malformation of internal organs was aortic coarctation. Body radiographs showed markedly advanced ossification, which was corroborated histologically by nearly absent proliferating chondrocytes at the end of long bones. Over the next decade several reports followed that were similar in presentation, including fetal hydrops, flat nasal bridge, short limbs and reduced epiphyseal cartilage (32–43, 46). At least two fetuses had cataracts, suggesting hypocalcemia (39). The absence of mammary gland and impact on tooth eruption was documented only later (3).
Delayed ossification in Eiken skeletal dysplasia
Eiken syndrome was first described in 1984 in three siblings from consanguineous parents (47). In contrast to BLC, all patients appeared normal at birth. When one of the boys was 1 year old, it was noted that his hands and feet were short and broad. Skeletal survey showed delay ossification, which was prominent at the hands and feet. A sibling presented at age 9 years with short stature (3.4 standard deviations below WHO standard) and delayed ossification was noted in the second phalanx of the fifth finger and at the pubic bone, as well as in the epiphyses of other larger bones. The third child was initially evaluated at age 12. At that time, his physical examination revealed no obvious abnormalities, but radiographs revealed a delay in mineralization at the sacrum and metatarsals bones. Subsequent evaluations showed normal ossification in these three patients. Thus, the clinical syndrome described by Eiken et al. is that of delayed ossification.
It was nearly two decades later, in 2005, that Eiken syndrome was ascribed to PTH1R mutations (46). The family described by Eiken was thus found to have a homozygous non-sense mutation at residue arginine 485 (R485X) of the C-terminal tail of the PTH1R.
Two additional homozygous PTH1R mutations, E35K and Y134S, were subsequently described in patients, who presented with delayed ossification (49, 51). The patient with the E35K mutation exhibited delayed ossification as well as supernumerary epiphyses (pseudoepiphyses) and angle-shaped proximal phalanges (51). Interestingly, serum calcium and phosphate levels were within the normal range, but serum PTH was mildly elevated; similar findings of elevated PTH were reported for an additional patient with a homozygous R485X mutation, who was born after the initial description of this family (46). The patient with the Y134S mutation had primary failure of tooth eruption in addition to a delay in growth plate ossification, while serum PTH, calcium and phosphate levels were reported as normal (49).
The bone phenotype in most patients with Eiken syndrome (i.e., delayed ossification) points toward a gain-of-function effect on PTHrP/PTH1R-mediated signaling in the growth plate, as it is well established that this signaling response acts normally to sustain growth plate chondrocyte proliferation and to delay the differentiation of these cells toward the hypertrophic state. Moreover, the clinical syndrome described by Eiken is reminiscent of the findings in transgenic mice that overexpress PTHrP in chondrocytes (62). The other clinical manifestations of Eiken syndrome, however, such as the mild PTH resistance and PFTE, are more likely explained by a loss-of-function effect on PTH1R signaling responses in kidney and teeth, as mediated by PTH and PTHrP, respectively. The identified Eiken mutations thus appear to have pleiotropic effects on PTH1R function that are dependent on the context of cell-type and tissue.
PTH resistance with or without delayed ossification
Three other independent families have recently been described with delayed ossification, characteristically observed in Eiken syndrome, that was associated with significantly elevated PTH levels, as evidence for PTH-resistance, and symptomatic, in some cases severe hypocalcemia (44, 45). These laboratory abnormalities are reminiscent of pseudohypoparathyroidism, a disorder that is usually caused by impaired G protein expression or function (63). Thus, Demaret et al. reported a boy, who was born at term without obvious skeletal abnormalities. However, by age 1 year, he developed symptomatic hypocalcemia (ionized calcium: 0.68 mM; nl 1.2-1.4), requiring treatment with calcium carbonate and alfa calcidiol. Serum phosphate (3.39 mM; nl 1.36-1.74) and PTH (401 ng/L; nl 14-72) were markedly elevated, which in the context of normal renal function, is consistent with PTH-resistance (45). This patient was found to have a homozygous PTH1R mutation of D241E (c.723C>G). Because of delayed ossification as assessed by hand radiographs, he was diagnosed with Eiken syndrome (45).
More recently, Calder et al. reported two patients with severe hypocalcemia (44). Patient 1 presented at one week of age with hypocalcemic convulsions, while hypocalcemia was not discovered until age 12 years for patient number 2. Both patients had besides hypocalcemia, hyperphosphatemia and elevated PTH levels of 76.4 pmol/L (nl 1.6-7.2) and 36.5 ng/L (nl 1.1-6.9), respectively. In both cases, there were skeletal abnormalities characterized by short metacarpals, extensive metacarpal pseudo-epiphyses and deficient ossification of the sacrum. These patients were found to have a homozygous I237N-PTH1R mutation.
In addition to the above cases classified as Eiken syndrome, a single family from Portugal was identified, in which affected members exhibited PTH resistance but no skeletal abnormalities (48). The index case, a 68-female presented initially at age 22 with epilepsy. At age 49, she came to medical attention with muscle spasms, neurologic deterioration. A head computer tomography revealed calcification of the basal ganglia and cerebellum. During these episodes, she was found to have severe hypocalcemia and hyperphosphatemia, but with normal to mildly elevated serum PTH levels ranging from 45 pg/ml to 72 pg/ml (nl 9-72). The patient had also a blunted urinary cAMP response at 2 and 4 hours after PTH injection. Treatment with calcium and calcitriol improved her hypocalcemia but over the next twenty years her neurologic symptoms progressed, and she became bedridden. This patient was found to have a homozygous PTH1R mutation (R186H), which was also found in two of her siblings, who had elevated PTH levels. Their PTH1R mutation is located near the junction of the extracellular domain (ECD) and transmembrane domain helix-1 (TM1).
PFTE associated with a homozygous PTH1R mutation
A contrasting phenotype, i.e. skeletal but no biochemical abnormalities, was reported by Jelani et al, who identified 5 members of a five-generation family from Saudi Arabia who presented with PFTE (as in patients with heterozygous LOF PTH1R mutants) (50). While there were no apparent biochemical abnormalities or ossification abnormalities, other variable and minor deformities like palm hyperkeratosis, hypoplastic nasal bridges and clinodactyly were present. These patients were found to have a homozygous V204E missense PTH1R mutation, which maps to the intracellular end of the transmembrane domain helix-2 (TM2) in the PTH1R (64).
Functional properties of disease causing homozygous PTH1R mutations
Severe loss-of-function mutations associated with BLC
The first mutation found in BLC affected RNA splicing leading to an 11 amino acid deletion in the receptor (L373_R383del) (38). Despite this large deletion, the L373_R383del PTH1R mutant is well expressed in COS-7 cells but does not bind PTH ligands or mediate ligand-induced generation of cAMP or IP3 (38). Similar loss-of-function effects are seen for other mutants found in patients with BLC (34).
One missense mutation, (P132L) was also described in BLC (37). While cAMP generation is detectable when this mutant is transfected into HEK293 cells and treated with PTH (1–34) or PTHrP (1–36), the overall response is greatly diminished as compared to that seen with the wild-type receptor (65). These findings establish that the BLC phenotype in patients results from a profound reduction in PTH1R signaling.
Gain of PTHrP function in Eiken syndrome
The homozygous mutations identified in cases of BLC result in severe reductions in PTH1R function when assessed in vitro. In contrast, those mutations that are compatible with life have comparatively milder in vitro effects in cell-based assays. The R485X truncation, found in Eiken syndrome, results in the loss of most of the PTH1R C-tail, which is important for normal receptor desensitization and the translocation of the receptor from the plasma membrane to endosomes, as mediated by the binding of β-arrestin to the phosphorylated C-tail (66). Consistent with the classical model by which the PTH1R C-tail plays a critical role in recruiting β-arrestin, recent work in transfected HEK293 cells, demonstrated that the R485X mutation strongly impairs the capacity of the PTH1R to recruit β-arrestin2 in response to either PTH or PTHrP ligands (67). As a result, cells transfected with the R485X mutant exhibit increased cAMP generation in response to PTHrP(1–36). Interestingly, the R485X mutant also exhibits increased agonist-independent basal cAMP generation, which could be suppressed by overexpression of β-arrestin2. This result implies that β-arrestin interaction with the PTH1R core region plays a role in the tonic inhibition of signaling by the un-liganded PTH1R (67). These data are further consistent with recent cross-linking studies that identify specific proximity points between β-arrestin2 and both the TM core and C-tail regions of the PTH1R (68, 69).
In contrast to R485X, the E35K and Y134S PTH1R mutations, also found in Eiken syndrome, alter residues in the receptor’s N-terminal ECD. Based on the location of these mutations in the receptor’s ECD, each of the altered amino acids is predicted to affect interaction with the ligand, but not directly with βarrestin, as demonstrated by recent cryo-electron microscopy of the PTH1R in complex with PTH or PTHrP analogs (64, 70–72). In particular, residue 35 of PTH1R are within proximity of residue 19 in PTH and PTHrP (64, 73). Indeed, the two extracellular domain mutations were each found to moderately destabilize the receptor interaction with PTHrP(1–36) but had little if any effect on interaction with PTH(1–34). The decreased binding of PTHrP did not result in detectable reductions in the acute generation of cAMP. However, the mutations did lead to reduced PTHrP-induced βarrestin recruitment and impaired receptor desensitization, which resulted in an increase in cAMP accumulation after repeated stimulation with PTHrP, as compared to that observed on the wild-type receptor under the same conditions (67). The altered effects on PTH vs. PTHrP interaction can be explained by differences between the two PTH1R agonists at key amino acid positions, specifically at position 5 (histidine in PTHrP vs. isoleucine in PTH) and 19 (glutamate in PTH and arginine in PTHrP) (64).
The functional studies thus identified two distinct mechanisms by which Eiken syndrome PTH1R mutations destabilize interaction with βarrestin and hence lead to impaired desensitization of cAMP signaling. On the one hand, the R485X mutation directly removes the C-tail and thus destabilizes the fully engaged PTH1R-arrestin complex. The ECD mutants, E35K and Y134S, impair binding of PTHrP and, as a result, the weaker ligand-receptor complexes do not interact with βarrestin strongly enough to cause receptor desensitization (67). By impairing ligand-induced receptor desensitization, the two ECD mutations thus facilitate a gain-of-function effect on PTH1R signaling. In fact, in the growth plates where local PTHrP levels are expected to be abundant, inefficient PTH1R desensitization is likely to enhance activation of the down-stream signaling pathway resulting in the delayed chondrocyte differentiation that underlies the mineralization defect in Eiken syndrome. It likely would be revealing to generate and study mouse models of Eiken syndrome to test this working hypothesis.
Mild loss of PTH1R function associated with PTH resistance with or without skeletal abnormalities
Three PTH1R mutations found in patients with symptomatic hypocalcemia and PTH resistance map to the receptor TM1 (R186H) and TM2 (D241E and I237N) (64, 70). The Arginine-186 in TM1 interacts extensively with PTH and PTHrP (residues 3-14) and thus is likely to contribute importantly to ligand binding and activation processes (73, 74). The three PTH1R mutants exhibit surface expression levels that are comparable to those of the wild-type receptor, and their stimulation with either PTH(1–34) or PTHrP(1–36) results in similar cAMP as well as intracellular calcium signaling (44, 65). One important limitation of these in vitro assays is that overexpression of the PTH1R in the plasmid-transfected cells can mask subtle changes in ligand-affinity, which nevertheless may be relevant in vivo where quantities of the endogenous ligands and receptor are likely much lower. One strategy to probe for more subtle affinity changes involved the use of minimized N-terminal PTH; these peptides retain the capacity to activate the PTH1R but lack the stabilizing interactions which, for intact PTH(1–34)-based ligands, occur between the 15–34 region of the peptide and the N-terminal extracellular domain of the receptor (29, 71). Such N-terminal peptides can thus be more effective at detecting subtle affinity changes caused by mutations that impact the receptor’s ligand-binding pocket (74). Indeed, when probed with modified PTH(1–15) or PTH (1–11) peptide analogs, each of the mutants R186H, D241E and I237N showed a marked reduction in cAMP signaling potency vs. the wild-type receptor (44, 65). For the V204E mutation found in patients with PFTE, the in vitro assays revealed a 50% reduction in surface expression levels, which interestingly was also found for the Y134S mutant that also is associated with PFTE. These results with the Y134S and V204E mutants suggest that normal levels of PTH1R expression are required for proper tooth eruption.
Concluding remarks: unraveling the complex phenotypes of patients with homozygous PTH1R mutations
In contrast to Blomstrand’s lethal chondrodysplasia, several homozygous PTH1R mutations are compatible with life, yet lead to notable and complex phenotypes. Patients with delayed ossification (Eiken syndrome) can exhibit features of both gain-of-function effects of the mutant PTH1R toward PTHrP in developing bone but also loss-of-function effects toward PTH in the kidney (PTH-resistance) or toward PTHrP in developing teeth (PFTE). The functional characterization of these disease-causing mutations revealed varied effects on PTH1R function including diminished desensitization, expression, and subtle yet allele-specific alterations in ligand affinity. The generation of new mouse models should bring even deeper understanding of the cellular and molecular mechanisms causing these diseases, and, importantly, should help advance development of new therapeutic strategies that can be employed more broadly for diseases of bone and mineral metabolism.
Author contributions
IP-C: Conceptualization, Writing – review & editing, Writing – original draft. JH: Investigation, Writing – review & editing. HJ: Writing – review & editing, Conceptualization, Supervision. TG: Resources, Writing – original draft, Writing – review & editing, Supervision.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. IPC holds a Dean’s Scholars Award from the Washington University Division of Physician-Scientists, which is funded by a Burroughs Wellcome Fund Physician-Scientist Institutional Award. Funding for this article was provided by P01 DK11479 (TJG), R01 DK 11309 (TJG, HJ) and R01 DK046718 (HJ).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Martin TJ, Sims NA, and Seeman E. Physiological and pharmacological roles of PTH and PTHrP in bone using their shared receptor, PTH1R. Endocrine Rev. (2021) 42:383–406. doi: 10.1210/endrev/bnab005
2. Kobayashi T, Chung U-I, Schipani E, Starbuck M, Karsenty G, Katagiri T, et al. PTHrP and Indian hedgehog control differentiation of growth plate chondrocytes at multiple steps. Development. (2002) 129(12):2977–86. doi: 10.1242/dev.129.12.2977
3. Wysolmerski JJ, Cormier S, Philbrick W, Dann P, Zhang J, Roume J, et al. Absence of functional type 1 PTH/PTHrP receptors in humans is associated with abnormal breast development and tooth impactation. J Clin Endocrinol Metab. (2001) 86:1788–94. doi: 10.1210/jcem.86.4.7404
4. Lanske B, Karaplis AC, Lee K, Luz A, Vortkamp A, Pirro A, et al. PTH/PTHrP receptor in early development and Indian hedgehog-regulated bone growth. Science. (1996) 273:663–6. doi: 10.1126/science.273.5275.663
5. Sato T, Verma S, Khatri A, Dean T, Goransson O, Gardella TJ, et al. Comparable initial engagement of intracellular signaling pathways by parathyroid hormone receptor ligands teriparatide, abaloparatide, and long-acting PTH. JBMR plus. (2021) 5:e10441. doi: 10.1002/jbm4.10441
6. Ferrandon S, Feinstein TN, Castro M, Wang B, Bouley R, Potts JT, et al. Sustained cyclic AMP production by parathyroid hormone receptor endocytosis. Nat Chem Biol. (2009) 5:734–42. doi: 10.1038/nchembio.206
7. Decker E, Stellzig-Eisenhauer A, Fiebig BS, Rau C, Kress W, Saar K, et al. PTHR1 loss-of-function mutations in familial, nonsyndromic primary failure of tooth eruption. Am J Hum Genet. (2008) 83:781–6. doi: 10.1016/j.ajhg.2008.11.006
8. Frazier-Bowers SA, Hendricks HM, Wright JT, Lee J, Long K, Dibble CF, et al. Novel mutations in PTH1R associated with primary failure of eruption and osteoarthritis. J Dent Res. (2014) 93:134–9. doi: 10.1177/0022034513513588
9. Frazier-Bowers SA, Simmons D, Wright JT, Proffit WR, and Ackerman JL. Primary failure of eruption and PTH1R: the importance of a genetic diagnosis for orthodontic treatment planning. Am J Orthod Dentofacial Orthop. (2010) 137:160 e1–7; discussion -1. doi: 10.1016/j.ajodo.2009.10.019
10. Risom L, Christoffersen L, Daugaard-Jensen J, Hove HD, Andersen HS, Andresen BS, et al. Identification of six novel PTH1R mutations in families with a history of primary failure of tooth eruption. PloS One. (2013) 8:e74601. doi: 10.1371/journal.pone.0074601
11. Roth H, Fritsche LG, Meier C, Pilz P, Eigenthaler M, Meyer-Marcotty P, et al. Expanding the spectrum of PTH1R mutations in patients with primary failure of tooth eruption. Clin Oral Investig. (2014) 18:377–84. doi: 10.1007/s00784-013-1014-3
12. Yamaguchi T, Hosomichi K, Narita A, Shirota T, Tomoyasu Y, Maki K, et al. Exome resequencing combined with linkage analysis identifies novel PTH1R variants in primary failure of tooth eruption in Japanese. J Bone Miner Res. (2011) 26:1655–61. doi: 10.1002/jbmr.385
13. Modafferi C, Tabolacci E, Lo Vecchio F, Cassano I, Bertozzi R, Fargnoli A, et al. New Insight into the genotype-phenotype correlation of PTH1R variants and primary failure of tooth eruption on an Italian Cohort. Eur J Hum Genet. (2024) 32:1402–11. doi: 10.1038/s41431-024-01691-y
14. Bastepe M, Raas-Rothschild A, Silver J, Weissman I, Jüppner H, and Gillis D. A form of Jansen’s metaphyseal chondrodysplasia with limited metabolic and skeletal abnormalities is caused by a novel activating PTH/PTHrP receptor mutation. J Clin Endocrinol Metab. (2004) 89:3595–600. doi: 10.1210/jc.2004-0036
15. Charrow J and Poznanski AK. The Jansen type of metaphyseal chondrodysplasia: conformation of dominant inheritance and review of radiographic manifestations in the newborn and adult. J Med Genet. (1984) 18:321–7. doi: 10.1002/ajmg.1320180216
16. Gram PB, Fleming JL, Frame B, and Fine G. Metaphyseal chondrodysplasia of Jansen. J Bone Joint Surg. (1959) 41A:951–9. doi: 10.2106/00004623-195941050-00015
17. Holthusen W, Holt JF, and Stoeckenius M. The skull in metaphyseal chondrodysplasia type Jansen. Pediat Radiol. (1975) 3:137–44. doi: 10.1007/BF01006898
18. Jansen M. Über atypische Chondrodystrophie (Achondroplasie) und über eine noch nicht beschriebene angeborene Wachstumsstörung des Knochensystems: Metaphysäre Dysostosis. Zeitschr Orthop Chir. (1934) 61:253–86.
19. Jüppner H, Schipani E, and Silve C. Jansen’s metaphyseal chondrodysplasia and Blomstrand’s lethal chondrodysplasia: two genetic disorders caused by PTH/PTHrP receptor mutations. In: Bilezikian J, Raisz L, and Rodan G, editors. Principles of Bone Biology. Academic Press, San Diego, CA (2002). p. 1117–35.
20. Kruse K and Schütz C. Calcium metabolism in the Jansen type of metaphyseal dysplasia. Eur J Pediatr. (1993) 152:912–5. doi: 10.1007/BF01957529
21. Nampoothiri S, Fernandez-Rebollo E, Yesodharan D, Gardella TJ, Rush ET, Langman CB, et al. Jansen metaphyseal chondrodysplasia due to heterozygous H223R-PTH1R mutations with or without overt hypercalcemia. J Clin Endocrinol Metab. (2016) 101:4283–9. doi: 10.1210/jc.2016-2054
22. Savoldi G, Izzi C, Signorelli M, Bondioni MP, Romani C, Lanzi G, et al. Prenatal presentation and postnatal evolution of a patient with Jansen metaphyseal dysplasia with a novel missense mutation in PTH1R. Am J Med Genet A. (2013) 161:2614–9. doi: 10.1002/ajmg.a.v161a.10
23. Schipani E, Jensen GS, Pincus J, Nissenson RA, Gardella TJ, and Jüppner H. Constitutive activation of the cAMP signaling pathway by parathyroid hormone (PTH)/PTH-related peptide (PTHrP) receptors mutated at the two loci for Jansen's metaphyseal chondrodysplasia. Mol Endocrinol. (1997) 11:851–8. doi: 10.1210/mend.11.7.9934
24. Schipani E, Kruse K, and Jüppner H. A constitutively active mutant PTH-PTHrP receptor in Jansen-type metaphyseal chondrodysplasia. Science. (1995) 268:98–100. doi: 10.1126/science.7701349
25. Schipani E, Langman CB, Hunzelman J, LeMerrer M, Loke KY, Dillon MJ, et al. A novel PTH/PTHrP receptor mutation in Jansen's metaphyseal chondrodysplasia. J Clin Endocrinol Metab. (1999) 84:3052–7. doi: 10.1210/jcem.84.9.6000
26. Schipani E, Langman CB, Parfitt AM, Jensen GS, Kikuchi S, Kooh SW, et al. Constitutively activated receptors for parathyroid hormone and parathyroid hormone-related peptide in Jansen's metaphyseal chondrodysplasia. New Engl J Med. (1996) 335:708–14. doi: 10.1056/NEJM199609053351004
27. Silverthorn KG, Houston CS, and Duncan BP. Murk Jansen's metaphyseal chondrodysplasia with long-term followup. Pediatr Radiol. (1983) 17:119–23. doi: 10.1007/BF02388087
28. Saito H, Noda H, Gatault P, Bockenhauer D, Loke KY, Hiort O, et al. Progression of mineral ion abnormalities in patients with Jansen metaphyseal chondrodysplasia. J Clin Endocrinol Metab. (2018) 103:2660–9. doi: 10.1210/jc.2018-00332
29. Zhao L-H, Ma S, Sutkeviciute I, Shen D-D, Zhou XE, de Waal PW, et al. Structure and dynamics of the active human parathyroid hormone receptor-1. Science. (2019) 364:148–53. doi: 10.1126/science.aav7942
30. Höppner J, Firat D, Parvez-Khan M, Reyes M, Hanna P, Yadav PS, et al. A mouse model of Jansen’s metaphyseal chondrodysplasia for investigating disease mechanisms and candidate therapeutics. Proc Natl Acad Sci. (2025) 122:e2500176122. doi: 10.1073/pnas.2500176122
31. Reyes M, Firat D, Hanna P, Khan M, Bruce M, Shvedova M, et al. Substantially delayed maturation of growth plate chondrocytes in “Humanized” PTH1R mice with the H223R mutation of jansen's disease. JBMR plus. (2023) 7:e10802. doi: 10.1002/jbm4.10802
32. Hoogendam J, Farih-Sips H, Wynaendts LC, Lowik CW, Wit JM, and Karperien M. Novel mutations in the parathyroid hormone (PTH)/PTH-related peptide receptor type 1 causing Blomstrand osteochondrodysplasia types I and II. J Clin Endocrinol Metab. (2007) 92:1088–95. doi: 10.1210/jc.2006-0300
33. Oostra R, van der Harten J, Rijnders W, Scott R, Young M, and Trump D. Blomstrand osteochondrodysplasia: three novel cases and histological evidence for heterogeneity. Virchows Arch. (2000) 436:28–35. doi: 10.1007/PL00008195
34. Karperien MC, van der Harten HJ, van Schooten R, Farih-Sips H, den Hollander NS, Kneppers ALJ, et al. A frame-shift mutation in the type I parathyroid hormone/parathyroid hormone-related peptide receptor causing Blomstrand lethal osteochondrodysplasia. J Clin Endocrinol Metab. (1999) 84:3713–20. doi: 10.1210/jcem.84.10.6033
35. Galera M, de Silva Patricio F, Lederman H, Porciuncula C, Lopes Monlleo I, and Brunoni D. Blomstrand chondrodysplasia: a lethal sclerosing skeletal dysplasia. Case report and review. Pediatr Radiol. (1999) 29:842–5. doi: 10.1007/s002470050709
36. Zhang P, Jobert AS, Couvineau A, and Silve C. A homozygous inactivating mutation in the parathyroid hormone/parathyroid hormone-related peptide receptor causing Blomstrand chondrodysplasia. J Clin Endocrinol Metab. (1998) 83:3365–8. doi: 10.1210/jcem.83.9.5245
37. Karaplis AC, Bin He MT, Nguyen A, Young ID, Semeraro D, Ozawa H, et al. Inactivating mutation in the human parathyroid hormone receptor type 1 gene in Blomstrand chondrodysplasia. Endocrinology. (1998) 139:5255–8. doi: 10.1210/endo.139.12.6522
38. Jobert AS, Zhang P, Couvineau A, Bonaventure J, Roume J, LeMerrer M, et al. Absence of functional receptors parathyroid hormone and parathyroid hormone-related peptide in Blomstrand chondrodysplasia. J Clin Invest. (1998) 102:34–40. doi: 10.1172/JCI2918
39. Loshkajian A, Roume J, Stanescu V, Delezoide AL, Stampf F, and Maroteaux P. Familial Blomstrand Chondrodysplasia with advanced skeletal maturation: further delineation. Am J Med Genet. (1997) 71:283–8. doi: 10.1002/(SICI)1096-8628(19970822)71:3<283::AID-AJMG7>3.0.CO;2-V
40. den Hollander NS, van der Harten HJ, Vermeij-Keers C, Niermeijer MF, and Wladimiroff JW. First-trimester diagnosis of Blomstrand lethal osteochondrodysplasia. Am J Med Genet. (1997) 73:345–50. doi: 10.1002/(SICI)1096-8628(19971219)73:3<345::AID-AJMG22>3.0.CO;2-I
41. Leroy JG, Keersmaeckers G, Coppens M, Dumon JE, and Roels H. Blomstrand lethal chondrodysplasia. Am J Med Genet. (1996) 63:84–9. doi: 10.1002/(SICI)1096-8628(19960503)63:1<84::AID-AJMG17>3.0.CO;2-Q
42. Young ID, Zuccollo JM, and Broderick NJ. A lethal skeletal dysplasia with generalised sclerosis and advanced skeletal maturation: Blomstrand chondrodysplasia. J Med Genet. (1993) 30:155–7. doi: 10.1136/jmg.30.2.155
43. Blomstrand S, Claësson I, and Säve-Söderbergh J. A case of lethal congenital dwarfism with accelerated skeletal maturation. Pediatr Radiol. (1985) 15:141–3. doi: 10.1007/BF02388725
44. Calder AD, Allgrove J, Höppner J, Cheung M, Alexander S, Garagnani L, et al. Eiken syndrome with parathyroid hormone resistance due to a novel parathyroid hormone receptor type 1 mutation: clinical features and functional analysis. J Bone Mineral Res. (2024) 39:1596–605. doi: 10.1093/jbmr/zjae148
45. Demaret T, Wintjens R, Sana G, Docquir J, Bertin F, Ide C, et al. Case report: inactivating PTH/PTHrP signaling disorder type 1 presenting with PTH resistance. Front endocrinology. (2022) 13:928284. doi: 10.3389/fendo.2022.928284
46. Duchatelet S, Ostergaard E, Cortes D, Lemainque A, and Julier C. Recessive mutations in PTHR1 cause contrasting skeletal dysplasias in Eiken and Blomstrand syndromes. Hum Mol Genet. (2005) 14:1–5. doi: 10.1093/hmg/ddi001
47. Eiken M, Prag J, Petersen K, and Kaufmann H. A new familial skeletal dysplasia with severely retarded ossification and abnormal modeling of bones especially of the epiphyses, the hands, and feet. Eur J Pediatr. (1984) 141:231–5. doi: 10.1007/BF00572767
48. Guerreiro R, Brás J, Batista S, Pires P, Ribeiro H, Almeida R, et al. Pseudohypoparathyroidism Type I-b with neurological involvement is associated with a homozygous PTH1R mutation Genes. Brain Behav. (2016) 15:669–77. doi: 10.1111/gbb.12308
49. Jacob P, Soni JP, Mortier G, and Girisha KM. The third family with Eiken syndrome. Clin Genet. (2019) 96:378–9. doi: 10.1111/cge.13601
50. Jelani M, Kang C, Mohamoud HS, Al-Rehaili R, Almramhi MM, Serafi R, et al. A novel homozygous PTH1R variant identified through whole-exome sequencing further expands the clinical spectrum of primary failure of tooth eruption in a consanguineous Saudi family. Arch Oral Biol. (2016) 67:28–33. doi: 10.1016/j.archoralbio.2016.03.012
51. Moirangthem A, Narayanan D, Jacob P, Nishimura G, Mortier G, and Girisha K. Report of second case and clinical and molecular characterization of Eiken syndrome. Clin Genet. (2018) 94:457–60. doi: 10.1111/cge.2018.94.issue-5
52. Höppner J and Jüppner H. Rare genetic disorders that impair parathyroid hormone synthesis, secretion, or bioactivity provide insights into the diagnostic utility of different parathyroid hormone assays. Curr Opin Nephrol hypertension. (2024) 33:375–82. doi: 10.1097/MNH.0000000000000999
53. Hawkes CP, Al Jubeh JM, Li D, Tucker SE, Rajiyah T, and Levine MA. Novel PTH gene mutations causing isolated hypoparathyroidism. J Clin Endocrinol Metab. (2022) 107:e2449–e58. doi: 10.1210/clinem/dgac086
54. Bae J, Choi HS, Park SY, Lee D-E, and Lee S. Novel mutation in PTHLH related to brachydactyly type E2 initially confused with unclassical pseudopseudohypoparathyroidism. Endocrinol Metab. (2018) 33:252. doi: 10.3803/EnM.2018.33.2.252
55. Pereda A, Garzon-Lorenzo L, Garin I, Cruz-Rojo J, Sanchez del Pozo J, and Perez de Nanclares G. The p. R56* mutation in PTHLH causes variable brachydactyly type E. Am J Med Genet A. (2017) 173:816–9. doi: 10.1002/ajmg.a.v173.3
56. Sun J, Yang N, Xu Z, Cheng H, and Zhang X. A novel heterozygous mutation in PTHLH causing autosomal dominant brachydactyly type E complicated with short stature. Mol Genet Genomic Med. (2024) 12:e2393. doi: 10.1002/mgg3.v12.2
57. Wang J, Wang Z, An Y, Wu C, Xu Y, Fu Q, et al. Exome sequencing reveals a novel PTHLH mutation in a Chinese pedigree with brachydactyly type E and short stature. Clinica Chimica Acta. (2015) 446:9–14. doi: 10.1016/j.cca.2015.03.019
58. Silve C and Jüppner H. Genetic Disorders Caused by Mutations in the PTH/PTHrP Receptor, its Ligands, and Downstream Effector Molecules. In: Genetics of Bone Biology and Skeletal Disease. London, England: Elsevier (2018). p. 655–74.
59. Kronenberg H. Developmental regulation of the growth plate. Nature. (2003) 423:332–6. doi: 10.1038/nature01657
60. Günther T, Chen ZF, Kim J, Priemel M, Rueger JM, Amling M, et al. Genetic ablation of parathyroid glands reveals another source of parathyroid hormone. Nature. (2000) 406:199–203. doi: 10.1038/35018111
61. Wysolmerski J, Philbrick W, Dunbar M, Lanske B, Kronenberg H, and Broadus A. Rescue of the parathyroid hormone-related protein knockout mouse demonstrates that parathyroid hormone-related protein is essential for mammary gland development. Development. (1998) 125:1285–94. doi: 10.1242/dev.125.7.1285
62. Weir EC, Philbrick WM, Amling M, Neff LA, Baron R, and Broadus AE. Targeted overexpression of parathyroid hormone-related peptide in chondrocytes causes skeletal dysplasia and delayed endochondral bone formation. Proc Natl Acad Sci USA. (1996) 93:10240–5. doi: 10.1073/pnas.93.19.10240
63. Mantovani G, Bastepe M, Monk D, De Sanctis L, Thiele S, Usardi A, et al. Diagnosis and management of pseudohypoparathyroidism and related disorders: first international Consensus Statement. Nat Rev Endocrinology. (2018) 14:476–500. doi: 10.1038/s41574-018-0042-0
64. Kobayashi K, Kawakami K, Kusakizako T, Miyauchi H, Tomita A, Kobayashi K, et al. Endogenous ligand recognition and structural transition of a human PTH receptor. Mol Cell. (2022) 82:3468–83. e5. doi: 10.1016/j.molcel.2022.07.003
65. Portales-Castillo I, Dean T, Khatri A, Jüppner H, and Gardella TJ. Functional properties of two distinct PTH1R mutants associated with either skeletal defects or pseudohypoparathyroidism. JBMR plus. (2022) 6:e10604. doi: 10.1002/jbm4.10604
66. Vilardaga J-P, Krasel C, Chauvin S, Bambino T, Lohse MJ, and Nissenson RA. Internalization determinants of the parathyroid hormone receptor differentially regulate β-arrestin/receptor association. J Biol Chem. (2002) 277:8121–9. doi: 10.1074/jbc.M110433200
67. Portales-Castillo I, Dean T, Cheloha RW, Creemer BA, Vilardaga J-P, Savransky S, et al. Altered signaling and desensitization responses in PTH1R mutants associated with eiken syndrome. Commun Biol. (2023) 6:599. doi: 10.1038/s42003-023-04966-0
68. Aydin Y, Böttke T, Lam JH, Ernicke S, Fortmann A, Tretbar M, et al. Structural details of a Class B GPCR-arrestin complex revealed by genetically encoded crosslinkers in living cells. Nat Commun. (2023) 14:1151. doi: 10.1038/s41467-023-36797-2
69. Clark LJ, Krieger J, White AD, Bondarenko V, Lei S, Fang F, et al. Allosteric interactions in the parathyroid hormone GPCR–arrestin complex formation. Nat Chem Biol. (2020) 16:1096–104. doi: 10.1038/s41589-020-0567-0
70. Sano FK, Shimizume K, Kobayashi K, Awazu T, Kawakami K, Akasaka H, et al. Insights into G-protein coupling preference from cryo-EM structures of Gq-bound PTH1R. Nat Chem Biol. (2025), 1–9. doi: 10.1038/s41589-025-01957-6
71. Cary BP, Gerrard EJ, Belousoff MJ, Fletcher MM, Jiang Y, Russell IC, et al. Molecular insights into peptide agonist engagement with the PTH receptor. Structure. (2023) 31:668–76. e5. doi: 10.1016/j.str.2023.04.002
72. Zhai X, Mao C, Shen Q, Zang S, Shen D-D, Zhang H, et al. Molecular insights into the distinct signaling duration for the peptide-induced PTH1R activation. Nat Commun. (2022) 13:6276. doi: 10.1038/s41467-022-34009-x
73. Ehrenmann J, Schöppe J, Klenk C, Rappas M, Kummer L, Doré AS, et al. High-resolution crystal structure of parathyroid hormone 1 receptor in complex with a peptide agonist. Nat Struct Mol Biol. (2018) 25:1086–92. doi: 10.1038/s41594-018-0151-4
Keywords: PTH1R, Blomstrand, Eiken, tooth eruption, growth plate, delayed ossification, βarrestin, GPCR
Citation: Portales-Castillo I, Höppner J, Jüppner H and Gardella TJ (2025) Human diseases caused by homozygous PTH1R mutations. Front. Endocrinol. 16:1641292. doi: 10.3389/fendo.2025.1641292
Received: 04 June 2025; Accepted: 31 July 2025;
Published: 19 August 2025.
Edited by:
Michela Rossi, Bambino Gesù Children’s Hospital (IRCCS), ItalyReviewed by:
Christopher Stuart Walker, The University of Auckland, New ZealandCopyright © 2025 Portales-Castillo, Höppner, Jüppner and Gardella. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ignacio Portales-Castillo, aWduYWNpb0B3dXN0bC5lZHU=; Thomas J. Gardella, dGdhcmRlbGxhQG1naC5oYXJ2YXJkLmVkdQ==