 Ming-Tai Chen1†
Ming-Tai Chen1† Rao-Qiong Wang2†
Rao-Qiong Wang2† Yu-Mei Qian2†Ting Peng2Xiao-Yu Lan2Ling-Ling Liang2Gang Luo2*
Yu-Mei Qian2†Ting Peng2Xiao-Yu Lan2Ling-Ling Liang2Gang Luo2* Qiu-Yu Liu3*
Qiu-Yu Liu3* Meng-Nan Liu2*
Meng-Nan Liu2*- 1Department of Cardiovascular Disease, Shenzhen Traditional Chinese Medicine Hospital, Shenzhen, China
- 2The Affiliated Traditional Chinese Medicine Hospital, Southwest Medical University, Luzhou, China
- 3School of Pharmacy, Southwest Medical University, Luzhou, China
Heart failure (HF), a serious stage of many cardiovascular illnesses associated with high morbidity and mortality, has emerged a major global public health concern worldwide. Its key pathophysiological mechanisms include cardiac remodeling, neurohormonal dysregulation and cardiac dysfunction. Despite established clinical treatments, adverse reactions and limited efficacy remain problems. In recent years, great emphasis has been paid to young blood, which is defined as blood from young people or in a youth associated physiological condition, and is abundant in various components that help maintain a youthful state of the organism. Currently, some progress has been made in the study of the anti-aging effects and mechanisms of young blood in older individuals, indicating therapeutic potential in the treatment of age-related diseases. In this paper, the major pathophysiological mechanisms of HF, the rejuvenating circulating factors in young blood and their positive effects on aging tissues are summarized. Moreover, the rejuvenating effects of young blood on the failing heart and the possible mechanisms of its action from multiple perspectives are investigated and discussed, aiming to provide theoretical foundation and potential therapeutic targets for the treatment of HF related diseases.
1 Introduction
Heart failure (HF) is a life-threatening clinical syndrome and a critical manifestation of multiple cardiovascular diseases. Marked by high incidence and mortality, it has become a major global public health concern. Epidemiological data reveal that the global prevalence of HF ranges from 1% to 10%, with an estimated incidence of 1 to 20 cases per 1000 person-years (1). Recognized risk factors for HF include hypertension, diabetes, coronary artery disease, obesity and smoking (2). Clinically, HF is primarily treated with diuretics, β-blockers and angiotensin-converting enzyme inhibitors, which have proven clinical efficacy (3). However, due to the high incidence of adverse reactions and recurrence rates associated with drug therapy, as well as the fact that some patients still fail to achieve ideal treatment outcomes after other standardized treatments, efforts are being actively made to explore and develop new drugs and alternative therapeutic approaches beyond pharmacotherapy.
Young blood refers to blood from young individuals or those exhibiting a physiologically youthful condition, whole blood including cells and plasma (cell-free, contains clotting factors), characterized by cellular and molecular compositions that facilitate the preservation of a youthful homeostasis. Recent studies employing heterochronic parabiosis-a technique that combines the circulatory systems of animals of varying ages to investigate systemic effects on aging and age-related diseases (4–6). They have shown that blood from young mice can enhance the functional capacity of aging tissues and organs, such as the liver, heart, brain and muscles (7, 8). The geroprotective qualities and specific mechanisms of young blood in aged organisms are progressively being clarified, demonstrating significant therapeutic potential for age-related illnesses (9–12). Given that the mechanisms underlying the association between young blood and HF have not yet been systematically integrated, this review aims to integrate the pathophysiological mechanisms of HF with the reparative effects and potential molecular mechanisms of young blood on the failing heart, so as to provide a systematic reference for clinical translation in this field.
2 Pathophysiological mechanisms of HF
HF is a clinical syndrome with diverse etiologies (13), characterized by diminished effective cardiac output (14), and manifested as symptoms such as dyspnea, fluid retention, nausea, vomiting and syncope (15). The primary causes are coronary artery disease, hypertension, valvular heart disease and other factors (16–18). The pathophysiological mechanisms of HF are illustrated in Figure 1.
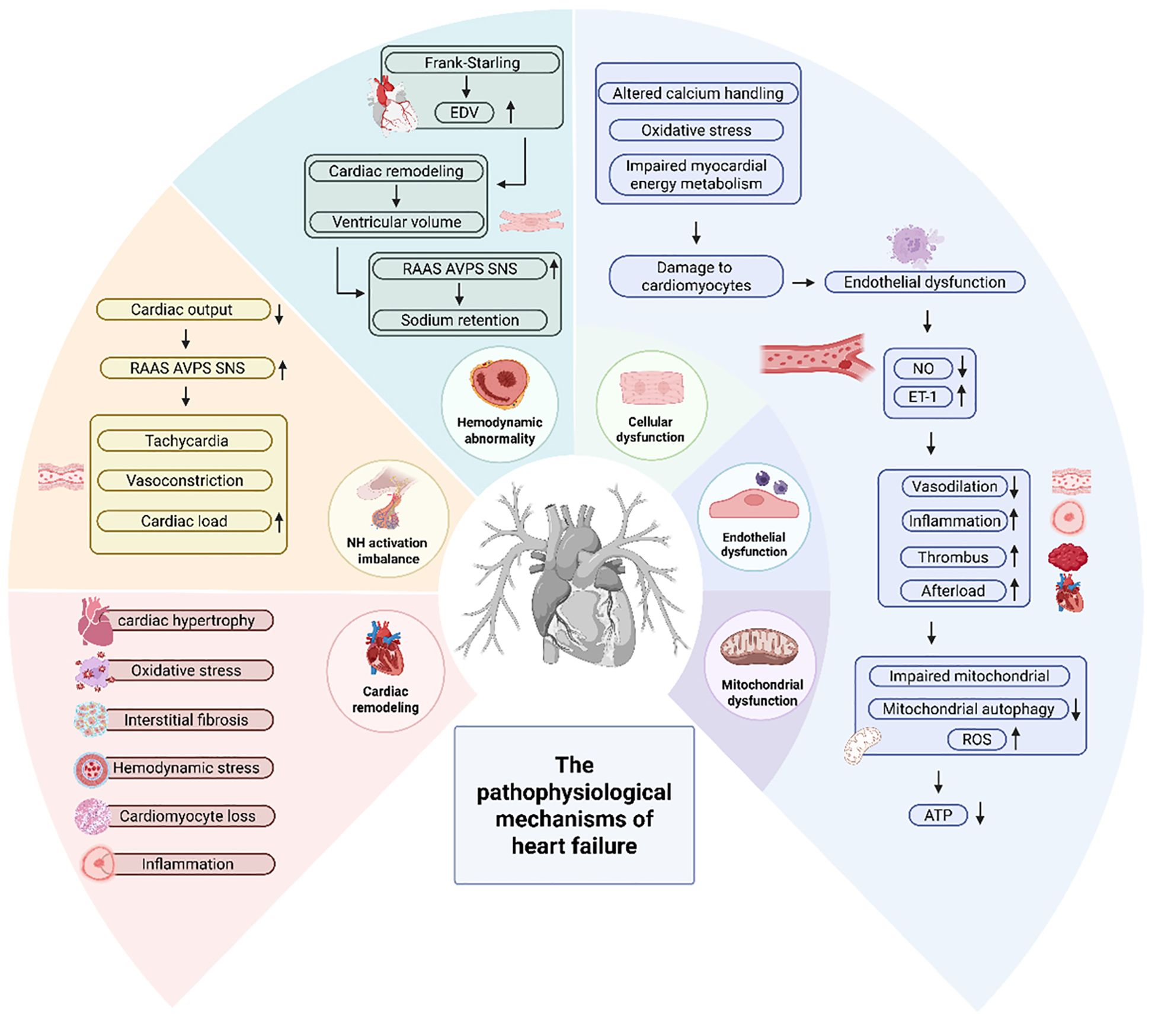
Figure 1. The pathophysiological mechanisms of HF.
2.1 Cardiac remodeling
Cardiac remodeling is frequently regarded as a pivotal factor influencing the clinical progression of HF. Cardiac remodeling is defined as changes in genomic expression leading to molecular, cellular and interstitial modifications, clinically evident as an increase in cardiac volume due to hemodynamic overload or injury, and a transformation in cardiac shape from elliptical to spherical, resulting in alterations in ventricular dimensions and both systolic and diastolic dysfunction (14, 19, 20). Cardiac remodeling is influenced by hemodynamic stress, neurohormonal activation and additional mechanisms now under investigation. The key steps leading to cardiac remodeling include cardiac hypertrophy, cardiomyocyte loss and interstitial fibrosis. During this remodeling, fibroblasts differentiate into myofibroblasts, activating their secretory functions and producing large amounts of extracellular matrix proteins. This results in scar tissue formation, which impedes the normal contractile function of cardiomyocytes, ultimately contributing to the development of HF in the long term (21, 22). Additional components involved include the interstitium, collagen and the coronary vascular system, with associated processes including ischaemia, cell necrosis and apoptosis. Patients with severe remodeling show progressive deterioration of cardiac function, with severe cases progressing to HF stages. Nonetheless, factors beyond remodeling can also influence the course of heart disease, and disease progression may occur in other ways in the absence of cardiac remodeling (19).
2.2 Neurohormonal activation imbalance
As a result of reduced cardiac output several neuroendocrine systems are activated, including the renin-angiotensin-aldosterone system (RAAS), arginine vasopressin system (AVPS) and the sympathetic nervous system (SNS). The persistent neurohormonal activation of the RAAS, AVPS and SNS plays a key role in the progression of HF. Extended activation of these systems leads to tachycardia, increased systemic vascular resistance, vasoconstriction, sodium and water retention, which in turn increases cardiac load and exacerbates HF. In addition, it leads to polyadrenergic receptor desensitization, cardiomyocyte hypertrophy, necrosis, cellular dysmorphism, myocardial fibrosis, renal arteriolar and venous vasoconstriction, diminished response to natriuretic peptides (NPs), peripheral vasoconstriction and vascular hypertrophy, and cardiac arrhythmia et al. (20, 23).
2.3 Hemodynamic abnormality
HF is characterized by pathological hemodynamic disturbances, including elevated cardiac filling pressures, which is associated with reduced cardiac output. The hemodynamic abnormalities in HF are not only influenced by cardiomyocytes, but also include important processes such as pericardial restriction, ventricular interactions and altered venous volumes (24). As the failing heart attempts to maintain adequate function, several compensatory mechanisms occur to ensure relatively normal cardiac output and maintain adequate cardiac function, including increased cardiac output and ventricular end-diastolic volume via the Frank-Starling mechanism, which increases cardiac output and cardiac workload, escalated ventricular volume and wall thickness via ventricular remodeling and maintained tissue perfusion by activating the neurohormonal system to increase mean arterial pressure, leading to increased myocardial contractility and sodium and water retention (25). The initial augmentation of ventricular wall thickness compensates for the heightened workload. However, it progressively diminishes cardiac diastolic function and leads to HF (16). These compensatory mechanisms, while initially beneficial in the early stages of HF, eventually lead to a vicious cycle of worsening HF (25). In addition, cardiac remodeling-induced cardiac changes can impair systolic function and reduce stroke volume output (26). Additionally, myocardial fibrosis, valve dysfunction and arrhythmias can also compromise systolic and diastolic function, thereby affecting cardiac output (16).
2.4 Cellular dysfunction
Decreased and impaired function of cardiomyocytes, including altered calcium handling and energy production (14), leads to impaired cardiac contractile function, cardiomyocyte apoptosis, myocardial ischemia and myocardial fibrosis. Among them, myocardial fibrosis destroys myocardial structure and leads to myocardial disorders, affecting the systolic and diastolic functions of the heart, leading to structural changes and inducing arrhythmias (27). Moreover, myocardial infarction significantly contributes to ventricular remodeling and the subsequent development of HF (28). Damage to cardiomyocytes involving inflammation, oxidative stress and impaired myocardial energy metabolism, they also are important causes of HF (14). Inflammation contributes to the pathogenesis and progression of HF through different mechanistic pathways. The HF state is characterized by an imbalance between pro-inflammatory and anti-inflammatory cytokines (29). The pro-inflammatory cytokines interleukin-1 (IL-1) and tumor necrosis factor-α (TNF-α) both induce systolic and diastolic dysfunction, and the latter also promotes adverse cardiac remodeling (30). Oxidative stress characterized by the generation of reactive oxygen species (ROS) and intrinsic energy metabolism. The generation of ROS and endogenous antioxidant defense systems are significant factors in the pathogenesis of cardiac remodeling and HF (31). When existing at low concentrations, ROS exert a pivotal function in maintaining cellular homeostasis. Nevertheless, an excess of ROS can lead to cellular dysregulation, lipid peroxidation, cellular injury and apoptosis. Ultimately, through the induction of cardiac fibroblast proliferation and the activation of matrix metalloproteinases, it triggers extracellular matrix remodeling, displaying profibrogenic properties. This process significantly influences the onset and progression of HF, contributing to the exacerbation of the pathological state (32). Van Bilsen et al. proposed the concept of myocardial metabolic remodeling in the context of myocardial energy metabolism disorders. They posited that HF induces a disturbance in the metabolism of carbohydrates and fats within myocardial cells, leading to alterations in the cardiac energy metabolic pathway and resulting in the abnormal structure and function of myocardial cells (33). There is increasing evidence that impaired myocardial energy metabolism can be one of the causes of HF, and HF can also exacerbate impaired myocardial energy metabolism. Therefore, reducing excessive myocardial energy expenditure and improving myocardial energy metabolism can improve the prognosis of HF (34).
2.5 Endothelial dysfunction
Endothelial cells are a monolayer of cells that cover the inner surface of blood vessels and act as a functional and structural barrier between the blood and the vessel wall, preventing platelets and leukocytes from adhering and aggregating, controlling permeability to plasma constituents, and regulating blood flow (35). Endothelial dysfunction, characterized by an imbalance in nitric oxide (NO, be defined in subsequent use) production and an increase in endothelin-1, leads to a shift in endothelial function toward reduced vasodilation, a pro-inflammatory state and pro-thrombotic properties. This is also one of the early mechanisms determining the decrease in organ perfusion, intolerance to physical exertion and the progression of HF. Additionally, it is associated with cardiovascular diseases such as coronary artery disease, hypertension and chronic HF (36–38). It leads to impaired vasodilation and compromised coronary blood flow and increased vascular resistance. This endothelial dysfunction leads to increased afterload, which puts additional stress on the heart. Some studies have shown that inflammation also induces endothelial dysfunction (16).
2.6 Mitochondrial dysfunction
Mitochondria are key double-membrane organelles for aerobic respiration in eukaryotic cells, mainly producing adenosine triphosphate (ATP) to provide energy for the organism, and participating in processes such as calcium homeostasis, lipid synthesis and regulation of apoptosis (39, 40). The heart, as the most metabolically active organ with the highest mitochondrial content, relies mainly on ATP generated by fatty acid oxidative metabolism to provide energy to maintain cardiac systolic and diastolic functions under physiological conditions. Bioenergetic homeostasis is accomplished almost exclusively through the “energy web” composed of the mitochondrial network and its associated phosphate transfer pairs (39). Common mitochondrial dysfunctions include decreased bioenergetics, increased oxidative stress, dysregulation of calcium homeostasis and altered mitochondrial dynamics (41). Mitochondrial dysfunction is thought to be one of the important pathogenetic mechanisms affecting cardiovascular diseases and is mainly associated with mitochondrial DNA (mtDNA) mutations, impaired mitochondrial autophagy, and elevated levels of mitochondria-derived ROS. Specifically, mitochondrial dysfunction has an important role in atherosclerosis, ischemic or reperfusion injury, calcium dyshomeostasis, neurodegenerative diseases, and cardiac systolic-diastolic dysfunction (41, 42).
3 Pharmacological treatment of HF
Clinical treatment options vary significantly depending on the type of HF patient, comorbidities and other clinical conditions (43). According to the 2022 Heart Failure Management Guidelines jointly issued by the American College of Cardiology/American Heart Association/American Heart Failure Society (AHA/ACC/HFSA), HF is classified based on left ventricular ejection fraction (LVEF), for HF with reduced ejection fraction (HFrEF, LVEF<40%, characterized by ventricular systolic dysfunction and progressive cardiac remodeling), renin-angiotensin system inhibitors (RASI), including angiotensin receptor–neprilysin inhibitor (ARNI), angiotensin-converting enzyme inhibitor (ACEI), angiotensin receptor blockers (ARB), mineralocorticoid receptor antagonist (MRA), sodium-dependent glucose transporters 2 inhibitors (SGLT2i) and β-receptor blocker. For HF with mildly reduced ejection fraction (HFmrEF, 40%≤LVEF ≤ 49%, possesses some characteristics of both HFrEF and HFpEF), SGLT2i is recommended in class 2a, ARNI, ACEI, ARB, MRA, and β-receptor blocker are recommended in class 2b. For HF with preserved ejection fraction (HFpEF, LVEF≥50%, mainly manifested by diastolic dysfunction, endothelial dysfunction, etc.), SGLT2i is recommended in class 2a, MRA and ARNI are recommended in class 2b. For HF with improved ejection fraction (HFiEF, previously HFrEF, with LVEF improved to ≥40% after treatment), ARNI, ACEI, ARB, MRA and β-receptor blockers (44). While guideline-directed pharmacological treatment can improve patient survival and quality of life, the burden of mortality in HF remains high. Pharmacological treatments are mainly symptom-directed, may lead to suboptimal stratification of treatment strategies, limited therapeutic efficacy, increased adverse events, and the emergence of drug resistance. These challenges collectively place patients at risk of progressive disease deterioration and poor clinical outcomes, so there is a need to look for more ideas and approaches to treating HF in order to achieve better outcomes (3, 45), and young blood offers a promising new avenue.
4 Therapeutic targets of young blood
Young blood contains a spectrum of circulating factors with rejuvenating properties, which play critical roles in sustaining physiological homeostasis, alleviating HF and other aging-related diseases, and promoting tissue and organ functional recovery. The therapeutic targets of young blood are shown in Table 1.
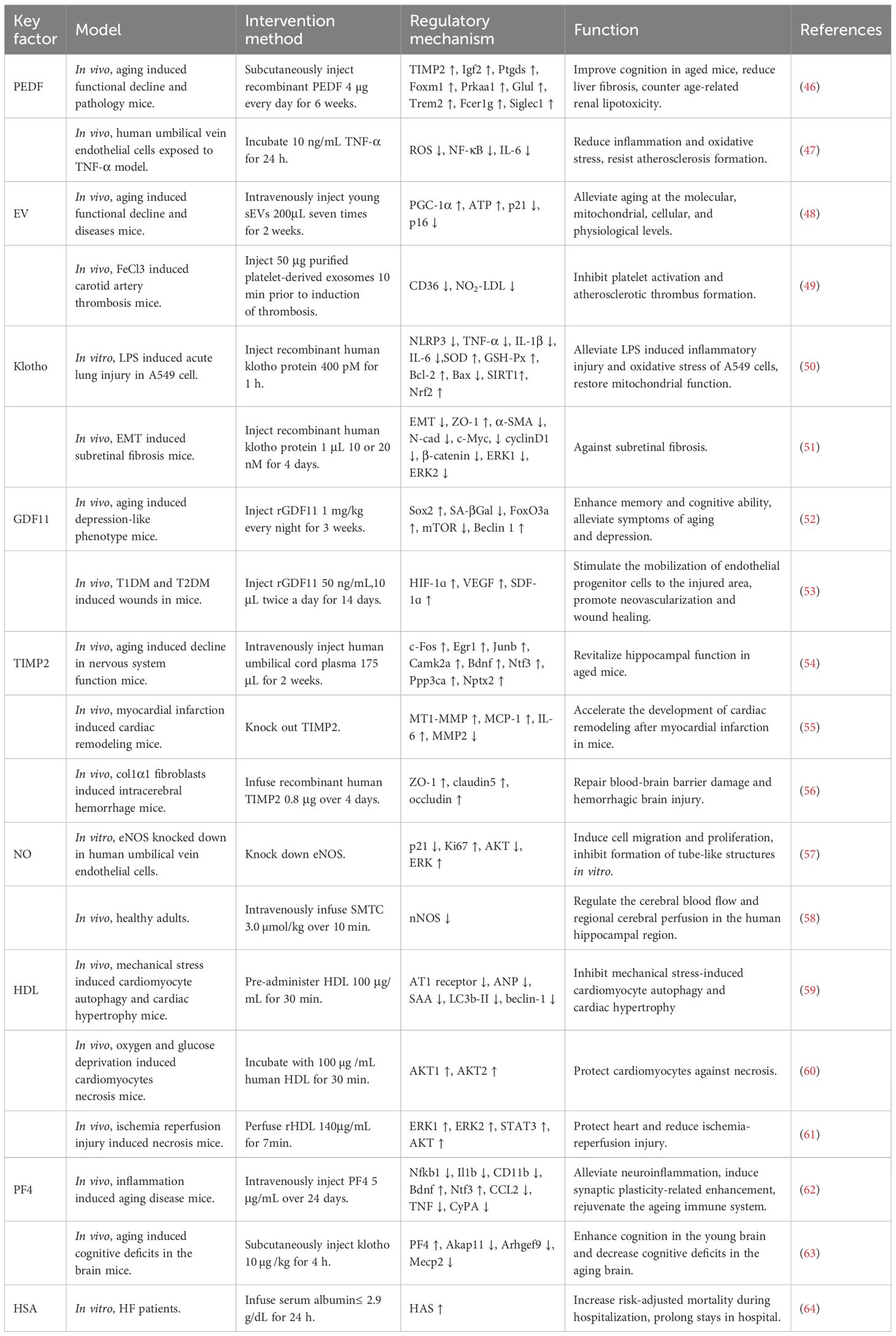
Table 1. The therapeutic targets of young blood.
4.1 Systemic mediators of young blood action
4.1.1 Pigment epithelium-derived factor
Among the serine protease inhibitor superfamily, PEDF has attracted attention as a 50-kDa secreted glycoprotein that exerts different physiological activities in various tissues, especially reducing myocardial fibrosis in HF while promoting cardiomyocyte apoptosis, thereby accelerating the progression of HF (65–67). Studies in animal models of injury and disease have shown that PEDF is involved in the maintenance of physiological homeostasis through its proven neurotrophic, antiangiogenic, immunomodulatory, tumor-killing and stem cell support functions. Meanwhile, PEDF induces a protective transcriptional profile in the hippocampus, liver and kidney of aged mice, improving cognitive function as well as liver and kidney integrity, suggesting that maintaining or supplementing PEDF levels in the elderly may have therapeutic implications for age-related diseases and chronic disorders of these tissues (46). Sho-Ichi Yamagishi et al. demonstrated that PEDF exerts anti-inflammatory, antioxidant and anti-atherosclerotic effects by inhibiting TNF-α-induced IL-6 expression through inhibition of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase-mediated ROS generation (47).
4.1.2 Extracellular vesicles
EVs are a heterogeneous group of nanoscale membrane vesicles composed of protein-rich lipid bilayers and closed proteins and RNAs originating from EV-producing cells that circulate throughout the body via the bloodstream and act as messengers in intercellular communication networks (48). Importantly, EVs plays critical therapeutic roles in HF through pro-angiogenic and cardiac remodeling-alleviating (68). EV-mediated communication is integral to the homeostasis of tissues and organs and significantly influences pathological processes (69). Xiaorui Chen et al. showed that young EVs have pleiotropic effects on systemic physiology and homeostasis, such as improving the morphology and function of aging tissues and organs, enhancing endurance and cognitive ability in aged mice, and improving mitochondrial dysfunction in aged cells, which may therefore explain the rejuvenating effects of young blood (48). Raghu Kalluri et al. observed that EVs can be classified into two main categories, extracellular vesicles and exosomes, among which exosomes are associated with immune responses, metabolic diseases and neurodegenerative diseases (70). Sowmya Srikanthan et al. demonstrated that platelet-derived exosomes inhibit atherosclerotic thrombosis by reducing CD36-dependent lipid loading in macrophages and suppressing platelet activation (49).
4.2 Key factors in young blood
4.2.1 Klotho
Klotho, an anti-aging gene, is expressed predominantly in the epithelial cells of the distal renal tubules of the kidney and in the choroid plexus of the brain (71). Yuechi Xu et al. showed that the human klotho gene encodes α-klotho, a multifunctional protein that includes three types: full-length transmembrane α-Klotho, truncated soluble α-Klotho, and secreted α-Klotho (72). Studies have shown that Klotho exerts a positive effect in HF by reducing inflammation, improving cardiac function and alleviating mitochondrial dysfunction (73, 74). Yanjun Zeng et al. demonstrated that Klotho inhibits lipopolysaccharide (LPS)-induced NOD-like receptor family pyrin domain containing 3 (NLRP3) inflammatory vesicle activation in A549 cells via the activation of the Sirtuin 1 (SIRT1)/nuclear factor erythroid 2-related factor 2 (Nrf2) signaling pathway. This process mitigates LPS-induced inflammatory injury in A549 cells and restores mitochondrial function (50). Yingle Jiang et al. showed that Klotho inhibits extracellular signal-regulated kinase (ERK)1/2 and wingless/integrated (Wnt)/β-catenin signaling pathway and attenuates the epithelial-mesenchymal transition of retinal pigment epithelial cells in subretinal fibrosis, thus exerting an anti-inflammatory effect on subretinal fibrosis (51).
4.2.2 Growth differentiation factor-11
GDF11 belongs to the transforming growth factor β superfamily, which plays a key role in embryonic development and is a critical regulator of a wide range of tissue patterning and formation (52). Carine Moigneu et al. demonstrated that GDF11 can act directly on hippocampal neurons in vitro to improve memory and alleviate aging and depression-like symptoms (52), while GDF11 deficient in cardiomyocytes leads to cardiac remodeling and eventually HF under pressure overload (75). Ying Zhang et al. showed that GDF11 promotes neovascularization and enhances diabetic wound healing by stimulating the mobilization of endothelial progenitor cells to injured areas mediated by the hypoxia-inducible factor (HIF)-1ɑ-vascular endothelial growth factor (VEGF)/stromal cell-derived factor (SDF)-1ɑ pathway (53). In addition, GDF11 has the ability to regulate muscle aging, reverse age-related decline in skeletal muscle function, improve mature adipocyte metabolism and anti-inflammatory properties (76–78).
4.2.3 TIMP2
TIMP2 holds a significant role within the family of tissue inhibitors of metalloproteinases, is highly expressed in the myocardium, and its deficiency results in exacerbate ventricular remodeling leading to severe HF (79). CASTELLANO et al. showed that human umbilical cord plasma and young rat plasma contain high levels of the anti-aging protein tissue TIMP2. Their study found that injection of human umbilical cord plasma into the hippocampus of aged mice activated this brain region, enhanced the cognitive function, promoted neuronal synapses formation, and improved the neuronal function in the hippocampus of the aged mice (54). Vijay Kandalam et al. demonstrate that TIMP2 defect promotes cardiac remodeling following myocardial infarction in mice by enhancing the activity of membrane type 1 matrix metalloproteinase (MT1-MMP) (6, 55). Lingling Xu et al. demonstrated that TIMP2 partially mediates fibroblasts to repair blood-brain barrier damage and hemorrhagic injury (56).
4.2.4 NO
NO is a highly reactive and diffusible, gaseous free radical, synthesized by three different isoforms of NO synthase (NOS): neuronal NOS (nNOS), inducible NOS (iNOS) and endothelial NOS (eNOS) (80, 81). The neurotransmitter function of NO depends on dynamic regulation of NOS (82). Shuhan Bu et al. demonstrated that knockdown of eNOS induced endothelial cell metastasis and proliferation through activation of mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) and inhibition of the phosphatidylinositol 3-kinase (PI3K)/AKT (protein kinase B, the key downstream target protein of PI3K) pathway, and inhibited the formation of tubular structures in vitro, confirming that eNOS regulates endothelial function by inversely controlling the proliferation and migration of endothelial cells as well as by directly regulating their tube-forming potential (57). Furthermore, Kevin O’Gallagher et al. demonstrated that nNOS modulates cerebral blood flow and local cerebral perfusion in the human hippocampal region through the administration of the nNOS inhibitor, S-methyl-L-thiotreponine (SMTC), which resulted in decreased cerebral blood flow (58). In addition, NO influences the progression of HF through alleviating vascular endothelial dysfunction, and influencing the nervous system and hemodynamics (83, 84).
4.2.5 High-density lipoprotein
HDL is a complex lipoprotein structure composed of lipids and proteins, along with the regulatory factors they transport, its level is related to the risk of HF (85). Lin Li et al. showed that HDL inhibits mechanical stress-induced cardiomyocyte autophagy and cardiac hypertrophy through the angiotensin II type 1 receptor-mediated PI3K/AKT pathway (59). Kristina K Durham et al. demonstrated that HDL protects cardiomyocytes from oxygen and glucose deprivation-induced necrosis via scavenger receptor class B, type 1 (SR-B1), PI3K, and AKT1 and AKT2 pathways (60). In addition, Marie-Claude Brulhart-Meynet et al. displayed that remodeled HDL mediated by sphingosine-1-phosphate (S1P) exerts a cardioprotective effect and mitigates ischemia-reperfusion injury through activation of ERK1/2, signal transducer and activator of transcription 3 (STAT3) and AKT (61).
4.2.6 Platelet factor 4
PF4 is a chemokine secreted by platelets, playing a role in coagulation and immunomodulation (86). PF4 indirectly influences the development of HF by impairing endothelial function and promoting atherosclerotic lesions (62). A study by Adam B Schroer et al. detected higher levels of PF4 in platelet fractions from young mice than from old mice by western blot analysis. They showed that PF4 has the ability to attenuate age-related neuroinflammation, induce synaptic plasticity-related enhancement, rejuvenate the aging immune system and rescue cognition in old age (86). Cana Park et al. showed that klotho activates platelet release of PF4 and restores the expression of aging and cognition-related factors in the hippocampus, thereby enhancing cognitive performance in young brains and reducing cognitive deficits in older brains (63).
4.2.7 Human serum albumin
HSA is the predominant serum (plasma without fibrinogen) protein and has been shown to have antioxidant properties (6, 87). Yali Wang et al. found that serum albumin levels are negatively correlated with the risk of atrial fibrillation, indicating that HSA may reduce the risk of atrial fibrillation (88). Hypoalbuminemia is prevalent in patients with HF and reduced albumin levels are associated with an increased prevalence of HFpEF, however, Tongqing Yao et al. found through data analysis that albumin therapy for HF carries certain risks (be showed in Table 1) (64). Lucia M Ruiz-Perera et al. experimentally found that human plasma and its component HSA play an important protective role against neuronal death induced by oxidative stress (6, 89).
5 Multidimensional analysis of cardiac repair mediated by young blood
The multiple circulating factors in young blood do not act in isolation, they exert positive influence on cardiac health through various mechanisms and play an important role in regulating cardiac physiological functions and improving pathological conditions. The multidimensional analysis of cardiac repair mediated by young blood is shown in Figure 2 and Table 2.
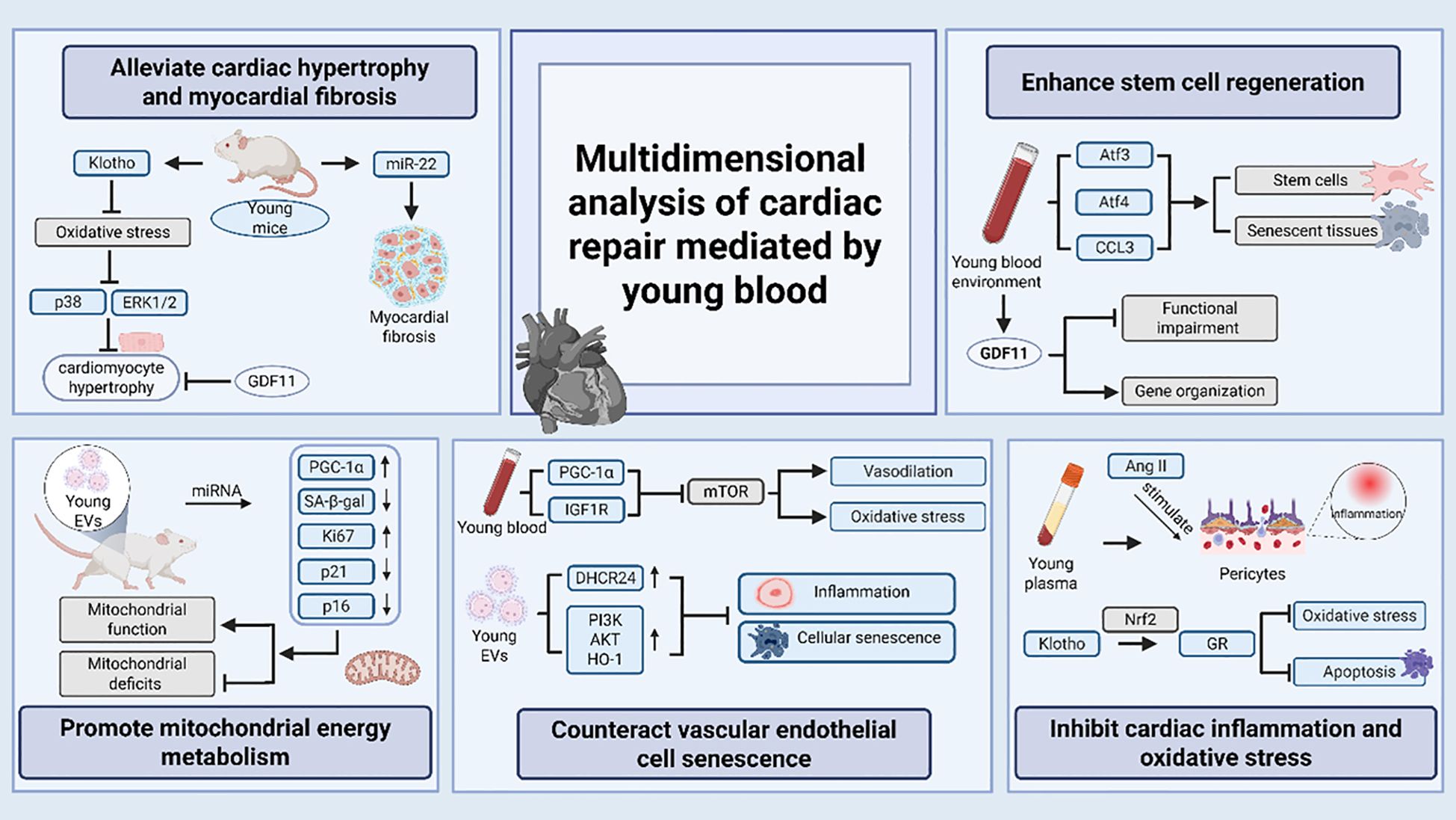
Figure 2. The specific mechanism of action of young blood.
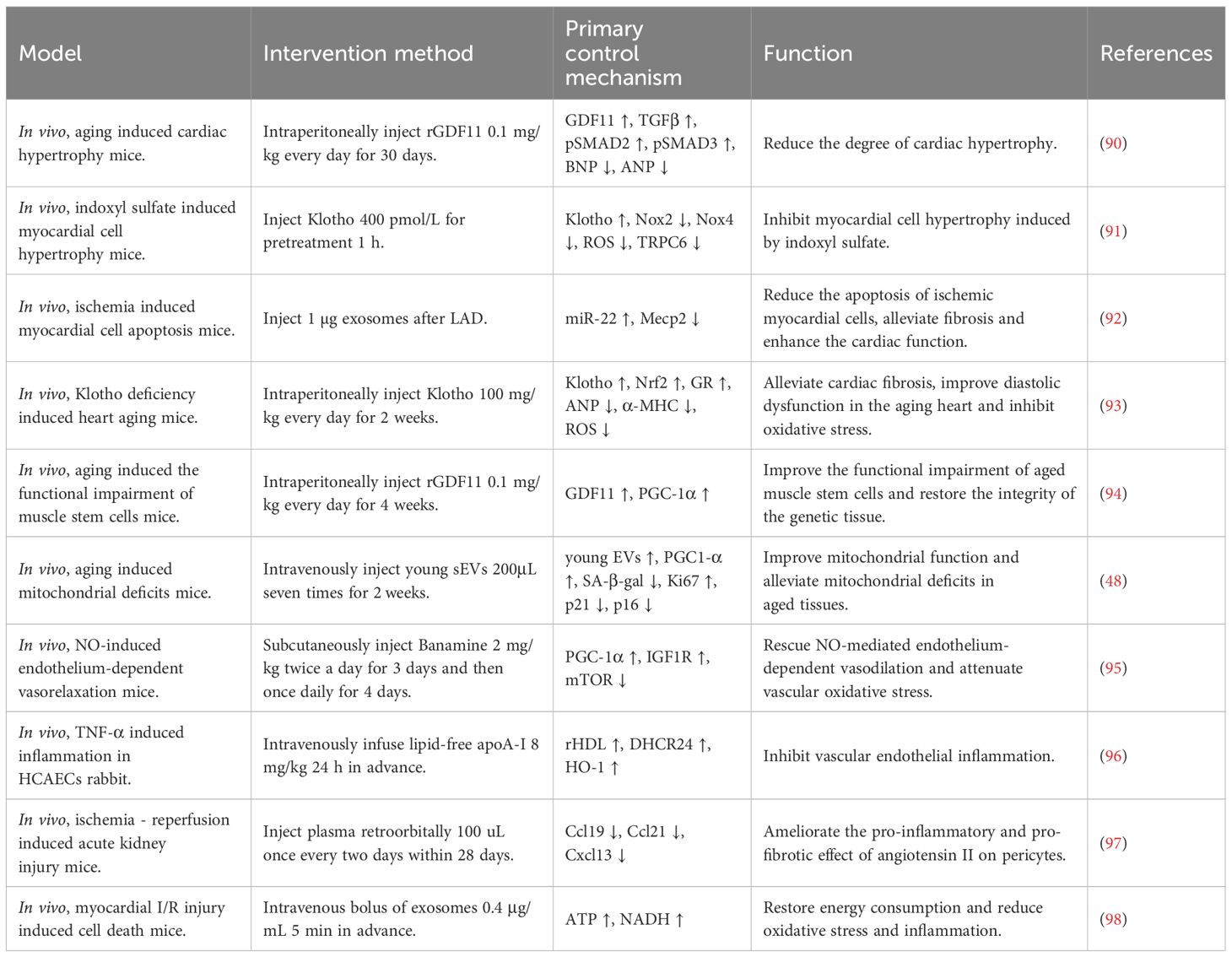
Table 2. The multidimensional analysis of cardiac repair mediated by young blood.
5.1 Alleviate cardiac hypertrophy and myocardial fibrosis
Cardiac hypertrophy is the result of compensatory hypertrophy of myocardial tissue, which is divided into physiological hypertrophy and pathological hypertrophy. In physiological hypertrophy, the heart has normal or enhanced contractile function, reversible hypertrophy, and intact energy metabolism without causing HF. In pathological hypertrophy, the heart is associated with cardiomyocyte death, myocardial fibrosis, incomplete reversibility of hypertrophy, and metabolic remodeling of the myocardium, which ultimately leads to systolic dysfunction and HF (99). Francesco S Loffredo et al. demonstrated that heterochronic parabiosis is able to reverse age-related cardiac hypertrophy. Further screening studies showed that GDF11 is a mediator of systemic anti-hypertrophic activity in young mice and has powerful anti-hypertrophic properties (90). Ke Yang et al. found that Klotho is predominantly synthesized in the kidneys and subsequently released into the serum. It has antioxidant activity and can inhibit the cardiomyocyte hypertrophy induced by indoxyl sulfate (an important uremic solute) by blocking oxidative stress and inhibiting the signaling pathways of p38 and ERK1/2 (91). Myocardial fibrosis is a pathological process in which excessive deposition of fibrous connective tissue in myocardial tissue leads to structural and functional abnormalities of myocardial fibers, affecting cardiac diastolic function, leading to arrhythmias and increasing the risk of cardiovascular events. Yuliang Feng et al. demonstrated that exosomes secreted by mesenchymal stem cells after ischemic preconditioning contain miR-22 that targets Mecp2. Through this mechanism, these exosomes can significantly reduce apoptosis of ischemic cardiomyocytes and myocardial fibrosis (92). Kai Chen et al. proved that cardiac-specific overexpression of glutathione reductase (a flavoprotein oxidoreductase, GR) attenuates cardiac fibrosis in aged mice, and Klotho secreted by young mice largely ameliorated Klotho deficiency-induced and aging-associated cardiac diastolic dysfunction (71).
5.2 Enhance stem cell regeneration
The decline in tissue regenerative capacity is a hallmark of aging and may be due to age-related changes in tissue-specific stem cells. Conboy et al. established paracrine pairs through heterochronic parabiosis and reveal that stem and progenitor cells retained their proliferative potential even in aging, and that serum from young animals reactivated the regeneration of senescent tissues (93). Shuai Ma et al. have found that hematopoietic stem and progenitor cells are sensitive to the environment of young blood by constructing an allogeneic symbiosis model and mapping the single-cell transcriptome, as well as the fact that young blood can induce changes in hematopoietic and immune systems by restoring the levels of key transcription factors, such as activating transcription factor (Atf)3, Atf4 and C-C motif chemokine ligand (CCL3), which promotes the activation of stem cells and systematic rejuvenation of senescent tissues (100). Manisha Sinha et al. demonstrated that supplementation of the body’s GDF11 levels through heterochronic symbiosis or systemic delivery of recombinant proteins reversed functional impairment and restored the integrity of gene organization in senescent muscle stem cells, suggesting an ameliorative effect of GDF11 on age-related skeletal muscle and stem cell dysfunction (76).
5.3 Promote mitochondrial energy metabolism
Declining mitochondrial bioenergetics is a characteristic feature of the aging process. Jenny L Gonzalez-Armenta et al. demonstrated that circulating factors themselves can mediate age-related structural and functional changes in mitochondria by using a heterochronic symbiosis model (101). Francisco Alejandro Lagunas-Rangely noted that heterochronic symbiosis of EV in young mice increased the expression of peroxisome proliferator-activated receptor gamma coactivator 1α (PGC-1α) in older mice through its miRNA carriers. These miRNAs respectively target amyloid β precursor protein, poly [ADP-ribose] polymerase 2, and HIF-1α inhibitor, which enhances mitochondrial function and attenuating mitochondrial defects in aging tissues (94). In addition, Xiaorui Chen et al. showed that young EVs stimulate PGC-1α expression both in vitro and in vivo via their miRNA cargo, improving mitochondrial function and enhancing mitochondrial energy metabolism to ameliorate degenerative changes and age-related dysfunctions, including cardiac (48).
5.4 Counteract vascular endothelial cell senescence
Vascular senescence is a degenerative change that occurs in the body with age and is manifested by thickening of the vascular wall, reduction of elastic fibers and dysfunction of endothelial cells. Endothelial cells are essential for vascular homeostasis, maintenance of blood flow, regulation of vascular tone, pro-inflammatory responses and neovascularization. Senescent endothelial cells display a flattened and enlarged morphology, reduced NO bioavailability, and increased secretion of various pro-inflammatory cytokines. The accumulation and deposition of which leads to vascular dysfunction and vascular senescence, and also promotes inflammation, thrombosis and atherosclerosis (102). Tamas Kiss et al. showed that, through a heterotopic symbiosis model, young blood can inhibit mammalian target of rapamycin (mTOR) signaling through activation of the PGC-1α and insulin-like growth factor 1 receptor (IGF1R) -mediated pathway and inhibiting mTOR signaling to promote vascular rejuvenation, improve NO-mediated endothelium-dependent vasodilation and attenuate vascular oxidative stress in aged mice (95). Plasma HDL content decreases with age. Ben J Wu et al. demonstrated that young plasma inhibits endothelial inflammation and decelerates vascular endothelial cell senescence through HDL-promoted 24-dehydrocholesterol reductase (DHCR24) expression, which in turn mediates PI3K/AKT/heme oxygenase-1 (HO-1) signaling pathway (96). In addition, Xiaorui Chen et al. demonstrated that young EVs not only improves cognitive function, counteracts mitochondrial defects, and enhances in vitro respiratory capacity by stimulating PGC-1α expression and enhancing mitochondrial energy metabolism, but also rejuvenates senescent tissues and inhibits cellular senescence, which is an indication of the ability of young EVs to ameliorate vascular endothelial cell aging (48).
5.5 Inhibit cardiac inflammation and oxidative stress
Blood is a fluid tissue composed of plasma and blood cells, and plasma is the liquid component of blood, accounting for about 55% of the blood volume. Shiyao Wei et al. found that young plasma by decreasing the expression of pro-inflammatory chemokines in angiotensin II-stimulated pericytes which are located between the endothelial cells of capillaries and the basement membranes, and which have physiological functions such as regulating vascular function, participating in angiogenesis and immunomodulation), the pro-inflammatory and pro-fibrotic effects of angiotensin II on pericytes can be ameliorated (97), thus contributing to attenuating the effects of inflammation on the development and progression of HF. Kai Chen et al. found that exogenously secreted Klotho promotes the expression of GR through Nrf2 activation, and verified that cardiac-specific expression of GR can diminish oxidative stress and apoptosis in the hearts of aged mice, with a protective effect against age-related cardiac injury (71). Shaina Ailawadi et al. found that multiple sources of stem cells, stress preconditioned cells, and genetically modified cells can generate cardioprotective exosomes to mitigate inflammation and oxidative stress in the heart (98, 103).
6 Discussion
HF remains one of the major challenges in modern medicine. In recent years, young blood has emerged as a promising research frontier with substantial potential for treating age-related diseases such as HF. Endi Xia et al. demonstrated that intravenous injection of young blood could alleviate hippocampus-dependent learning and memory deficits, and restore synapse formation and synaptic plasticity in mice with Alzheimer’s disease, which proved that young serum had a therapeutic effect on it (104). Anding Liu et al. demonstrated that infusing of young blood into aged mice alleviates aging-induced liver injury, liver fibrosis and reduces hepatocyte senescence (105). Shi-Yao Wei et al. also demonstrated that systemic administration of young plasma reduces the progression of renal fibrosis and the transition from acute injury to chronic renal disease et al. (97). Alkahest, a derivative of young blood, which was invested in and developed by Grifols, the world’s largest plasma manufacturer, has been used in preclinical studies of Alzheimer’s disease (106). Although Alkahest and similar young blood derivatives are still in the development and experimental stages, they still show great therapeutic promise and potential for young blood.
The great therapeutic potential and distinct advantages of young blood are reflected in the following aspects: (i) The multiple rejuvenating circulating factors in young blood offer the possibility of personalized treatment for HF. HF patients exhibit significant etiological heterogeneity and pathophysiological diversity, which traditional treatment protocols struggle to address uniformly. In contrast, young blood’s multi-component, multi-target synergistic effects enable tailored intervention across distinct pathological dimensions of HF. By leveraging biomarker-guided screening of active components and dose optimization, young blood therapies can be further integrated with other therapeutic modalities to establish precision-driven, personalized strategies aligned with each patient’s unique pathological profile. (ii) Circulating factors and mediators in young blood represent endogenous substances naturally produced by the body, functioning within normal physiological environments to ensure excellent biocompatibility and minimal immunogenicity. In contrast to synthetic therapeutic drugs or biologics, young blood-derived components carry a relatively lower risk of triggering immune rejection or other adverse reactions, enhancing patient tolerability. This inherent safety profile renders them an ideal candidate for long-term, safe management of HF, particularly for elderly patients or vulnerable populations with multiple comorbidities—while potentially mitigating treatment-related risks of dose dependency. (iii) The exploration of young blood transcends the limitations of traditional aging-related research and therapies, which typically focus on single symptoms or isolated pathological aspects of diseases. Unlike conventional approaches, young blood’s multi-component nature enables holistic, multi-target and multi-pathway improvements in cardiac and other tissue or organ functions. By modulating the organism’s overall aging trajectory, this strategy not only ameliorates age-related conditions like HF but also offers novel paradigms for preventing and treating aging-related liver and kidney diseases. This shift redirects research focus from merely treating diseases to proactively intervening in the organism’s systemic aging process, with the potential to redefine the research, prevention, and treatment paradigms for all aging-related disorders.
Although young blood shows great potential in improving the function of the failing heart, there are still some limitations in the current study. (i) The understanding of the role of young blood in cardiac regulation remains limited, primarily constrained by the interactions between various circulating factors and their specific mechanisms of action in regulating cardiac function. First, these circulating factors may regulate cardiac function through complex synergistic or antagonistic interactions among them, but the relative contribution of each factor and its dose-dependent effects still lack corresponding studies. Second, the current identification of key effectors is mainly based on a single factor research paradigm, and the relationship with young blood as a whole remains to be investigated. Third, directly infusing young blood for therapeutic purposes may carry risks such as immune rejection, allergic reactions and transmission of infections. Fourth, young blood’s components may instead exacerbate the progression of the disease, such as high levels of GDF11 cause severe cachexia, death, cardiac and skeletal muscle wasting (107), TNF-α and IL-6 promote cardiac remodeling and inflammation generation (108). Finally, current research on the subtype-specific effects of young blood and its circulating factors in HF is still in its preliminary stage. As noted, ARNI is recommended for HFrEF, while GDF11 and EVs synergistically improve ventricular systolic function via anti-myocardial hypertrophy and inhibiting myocardial fibrosis, respectively. SGLT2i is recommended for HFpEF, with Klotho reducing myocardial cell damage and NO alleviating endothelial vascular resistance elevation. (ii) The donor screening of young blood, the identification and isolation operation of active ingredients and regulatory issues regarding blood-derived therapies have not yet established unified standards. This makes it difficult to obtain circulating factors that meet the requirements of experimental research and clinical treatment, which affects the accuracy of the research results and the therapeutic effects in the clinic. Since most of the relevant studies focus on animal models, which have significant physiological differences from humans, leading to a scarcity of clinical data and insufficient evidence for the safety of young blood in clinical applications. (iii) The technology of artificially synthesizing active ingredients related to young blood has not yet achieved a breakthrough, and it is difficult to accurately construct their molecular structures with the existing biosynthesis technology, making it is impossible to satisfy the needs of a large number of patients relying on the blood of the donor alone. This may even give rise to ethical and social issues such as violations of donors’ health and damage to healthcare equity.
There is no dispute regarding the anti-aging effects of young blood, especially its therapeutic potential in HF. Future research should also focus on the following aspects: (i) In-depth mechanistic studies can help to clarify the relationship between the key effectors and young blood, and between young blood and HF subtypes. With the help of proteomics, metabolomics and other technologies to clarify the intrinsic relationship between the key effectors and young blood, analyze the specific effect mechanism of the key effectors on the heart and other aging tissues and organs. As well as explore the combined effects of young blood and existing therapeutic drugs and establish optimal dosage thresholds to avoid potential risks of young blood transfusion therapy. (ii) Unify the screening criteria for young blood donors, develop standardized procedures for the collection, storage and use of young blood, and strengthen the approval standards and supervision of blood therapies, while more in-depth techniques such as high-resolution proteomics and single-cell sequencing should be used for the identification and separation of active ingredients to accurately identify and extract the active ingredients in young blood. Furthermore, human clinical trials should be initiated through approaches such as organoid models and optimized administration dosages and methods to accumulate clinical data for therapeutic applications, validate the safety, efficacy and patient-specific responsiveness of young blood-based interventions, so as to accelerate the translation from existing animal experiments model to clinical practice. (iii) Strengthen the cooperation among multiple disciplines, such as biochemistry, cell biology, computer science, etc., and increase the funding and research investment on the artificial synthetic technology of active ingredients in young blood, focusing on the structural analysis and biosynthetic pathway of active ingredients, so as to make every effort to avoid the ethical and social problems caused by the direct infusion of young blood.
7 Conclusion
As a momentous breakthrough within the realm of anti-aging medicine, young blood therapy demonstrates unique advantages in enhancing cardiac function through the coordinated actions of multiple targets and pathways. The therapy transcends the limitations of traditional single-target therapy and offers an entirely novel intervention strategy and treatment paradigm for the management of HF and age-related maladies. However, there remain translational barriers from basic research to clinical application, as the complex interactions among various components of young blood, the lack of standardized systems, and ethical issues in clinical translation are urgently to be addressed. Future research should focus on current gaps. It is necessary to deeply analyze the dynamic interaction network of key circulating factors, clarify their synergistic or antagonistic effects and molecular regulatory pathways in different pathological microenvironments. Additionally, it is need to systematically evaluate the applicability of young blood therapy in special populations and improve risk early-warning mechanisms through dynamic monitoring of immunogenicity and long-term physiological impacts during treatment, thus gradually overcoming the core bottlenecks in translating basic research to clinical practice.
Author contributions
MC: Writing – original draft, Writing – review & editing. RW: Writing – original draft, Writing – review & editing. YQ: Writing – original draft, Writing – review & editing, Visualization. TP: Visualization, Writing – review & editing. XL: Data curation, Investigation, Writing – review & editing. LL: Data curation, Investigation, Writing – review & editing. GL: Supervision, Writing – review & editing. QL: Supervision, Writing – review & editing. ML: Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was supported by the National Natural Science Foundation of China (No. 82074378), the Project of Science & Technology Department of Sichuan Province (No. 2022YFS0618), Project of Office of Science & Technology and talent work of Luzhou (2023JYJ029), 2024 Traditional Chinese Medicine Guangdong Provincial Laboratory Project (HQCML-C-2024005), Shenzhen Science and Technology Program (JCYJ20230807094603007, JCYJ20240813152440051), Shenzhen Medical Research Fund (A2403028) and the Project of Southwest Medical University (2024ZKZ007, 2023ZYYQ04). The funder had no role in the study design, data analysis, or decision to publish.
Acknowledgments
All figures were produced by Biorender (https://app.biorender.com/) and obtained a publication license.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Groenewegen A, Rutten FH, Mosterd A, and Hoes AW. Epidemiology of heart failure. Eur J Heart Fail. (2020) 22:1342–56. doi: 10.1002/ejhf.1858
2. Dunlay SM and Roger VL. Understanding the epidemic of heart failure: past, present, and future. Curr Heart Fail Rep. (2014) 11:404–15. doi: 10.1007/s11897-014-0220-x
3. Golden L and Rogers JG. Heart failure drug therapy: new treatments, new guidelines. Tex Heart Inst J. (2022) 49:e227920. doi: 10.14503/THIJ-22-7920
4. Conboy MJ, Conboy IM, and Rando TA. Heterochronic parabiosis: historical perspective and methodological considerations for studies of aging and longevity. Aging Cell. (2013) 12:525–30. doi: 10.1111/acel.12065
5. Gulej R, Nyúl-Tóth Á, Csik B, Petersen B, Faakye J, Negri S, et al. Rejuvenation of cerebromicrovascular function in aged mice through heterochronic parabiosis: insights into neurovascular coupling and the impact of young blood factors. GeroScience. (2024) 46:327–47. doi: 10.1007/s11357-023-01039-2
6. Höving AL, Schmidt KE, Kaltschmidt B, Kaltschmidt C, and Knabbe C. The role of blood-derived factors in protection and regeneration of aged tissues. Int J Mol Sci. (2022) 23:9626. doi: 10.3390/ijms23179626
7. Carlson ME, Conboy MJ, Hsu M, Barchas L, Jeong J, Agrawal A, et al. Relative roles of TGF-beta1 and Wnt in the systemic regulation and aging of satellite cell responses. Aging Cell. (2009) 8:676–89. doi: 10.1111/j.1474-9726.2009.00517.x
8. Pan M, Wang P, Zheng C, Zhang H, Lin S, Shao B, et al. Aging systemic milieu impairs outcome after ischemic stroke in rats. Aging Dis. (2017) 8:519–30. doi: 10.14336/AD.2017.0710
9. Salpeter SJ, Khalaileh A, Weinberg-Corem N, Ziv O, Glaser B, and Dor Y. Systemic regulation of the age-related decline of pancreatic β-cell replication. Diabetes. (2013) 62:2843–8. doi: 10.2337/db13-0160
10. Horowitz AM and Villeda SA. Therapeutic potential of systemic brain rejuvenation strategies for neurodegenerative disease. F1000Research. (2017) 6:1291. doi: 10.12688/f1000research.11437.1
11. Baht GS, Silkstone D, Vi L, Nadesan P, Amani Y, Whetstone H, et al. Exposure to a youthful circulaton rejuvenates bone repair through modulation of β-catenin. Nat Commun. (2015) 6:7131. doi: 10.1038/ncomms8131
12. Jeon OH, Mehdipour M, Gil T-H, Kang M, Aguirre NW, Robinson ZR, et al. Systemic induction of senescence in young mice after single heterochronic blood exchange. Nat Metab. (2022) 4:995–1006. doi: 10.1038/s42255-022-00609-6
13. Feng J, Zhang Y, and Zhang J. Epidemiology and burden of heart failure in Asia. JACC Asia. (2024) 4:249–64. doi: 10.1016/j.jacasi.2024.01.013
14. Elendu C, Amaechi DC, Elendu TC, Fiemotonghan B-E, Okoye OK, Agu-Ben CM, et al. A comprehensive review of heart failure: Unraveling the etiology, decoding pathophysiological mechanisms, navigating diagnostic modalities, exploring pharmacological interventions, advocating lifestyle modifications, and charting the horizon of emerging therapies in the complex landscape of chronic cardiac dysfunction. Med (Baltimore). (2024) 103:e36895. doi: 10.1097/MD.0000000000036895
15. Ziaeian B and Fonarow GC. Epidemiology and aetiology of heart failure. Nat Rev Cardiol. (2016) 13:368–78. doi: 10.1038/nrcardio.2016.25
16. Sapna F, Raveena F, Chandio M, Bai K, Sayyar M, Varrassi G, et al. Advancements in heart failure management: A comprehensive narrative review of emerging therapies. Cureus. (2023) 15:e46486. doi: 10.7759/cureus.46486
17. Skrzypek A, Mostowik M, Szeliga M, Wilczyńska-Golonka M, Dębicka-Dąbrowska D, and Nessler J. Chronic heart failure in the elderly: still a current medical problem. Folia Med Cracov. (2018) 58:47–56. doi: 10.24425/fmc.2018.125703
18. Gallo G and Savoia C. Hypertension and heart failure: from pathophysiology to treatment. Int J Mol Sci. (2024) 25:6661. doi: 10.3390/ijms25126661
19. Cohn JN, Ferrari R, and Sharpe N. Cardiac remodeling–concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. Behalf of an International Forum on Cardiac Remodeling. J Am Coll Cardiol. (2000) 35:569–82. doi: 10.1016/s0735-1097(99)00630-0
20. Manolis AA, Manolis TA, and Manolis AS. Neurohumoral activation in heart failure. Int J Mol Sci. (2023) 24:15472. doi: 10.3390/ijms242015472
21. Gonciar D, Mocan T, and Agoston-Coldea L. Nanoparticles targeting the molecular pathways of heart remodeling and regeneration. Pharmaceutics. (2022) 14:711. doi: 10.3390/pharmaceutics14040711
22. Šalingová B, Červenák Z, Adamičková A, Chomanicová N, Valášková S, Gažová A, et al. Endothelial-mesenchymal transition or functional tissue regeneration - two outcomes of heart remodeling. Physiol Res. (2021) 70:S13–20. doi: 10.33549/physiolres.934780
23. Hartupee J and Mann DL. Neurohormonal activation in heart failure with reduced ejection fraction. Nat Rev Cardiol. (2017) 14:30–8. doi: 10.1038/nrcardio.2016.163
24. Verbrugge FH, Guazzi M, Testani JM, and Borlaug BA. Altered hemodynamics and end-organ damage in heart failure: impact on the lung and kidney. Circulation. (2020) 142:998–1012. doi: 10.1161/CIRCULATIONAHA.119.045409
25. Kemp CD and Conte JV. The pathophysiology of heart failure. Cardiovasc Pathol Off J Soc Cardiovasc Pathol. (2012) 21:365–71. doi: 10.1016/j.carpath.2011.11.007
26. Seferović PM, Polovina M, Bauersachs J, Arad M, Ben Gal T, Lund LH, et al. Heart failure in cardiomyopathies: a position paper from the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail. (2019) 21:553–76. doi: 10.1002/ejhf.1461
27. Gyöngyösi M, Winkler J, Ramos I, Do Q-T, Firat H, McDonald K, et al. Myocardial fibrosis: biomedical research from bench to bedside. Eur J Heart Fail. (2017) 19:177–91. doi: 10.1002/ejhf.696
28. Piątek-Matuszak P, Pasławski R, Pasławska U, Kiczak L, Płóciennik M, Janiszewski A, et al. Assessment of myocardial diastolic dysfunction as a result of myocardial infarction and extracellular matrix regulation disorders in the context of mesenchymal stem cell therapy. J Clin Med. (2022) 11:5430. doi: 10.3390/jcm11185430
29. Boulet J, Sridhar VS, Bouabdallaoui N, Tardif J-C, and White M. Inflammation in heart failure: pathophysiology and therapeutic strategies. Inflammation Res Off J Eur Histamine Res Soc Al. (2024) 73:709–23. doi: 10.1007/s00011-023-01845-6
30. Murphy SP, Kakkar R, McCarthy CP, and Januzzi JL. Inflammation in heart failure: JACC state-of-the-art review. J Am Coll Cardiol. (2020) 75:1324–40. doi: 10.1016/j.jacc.2020.01.014
31. Tsutsui H, Kinugawa S, and Matsushima S. Oxidative stress and heart failure. Am J Physiol Heart Circ Physiol. (2011) 301:H2181–2190. doi: 10.1152/ajpheart.00554.2011
32. van der Pol A, van Gilst WH, Voors AA, and van der Meer P. Treating oxidative stress in heart failure: past, present and future. Eur J Heart Fail. (2019) 21:425–35. doi: 10.1002/ejhf.1320
33. van Bilsen M, Smeets PJH, Gilde AJ, and van der Vusse GJ. Metabolic remodelling of the failing heart: the cardiac burn-out syndrome? Cardiovasc Res. (2004) 61:218–26. doi: 10.1016/j.cardiores.2003.11.014
34. Zhao XL and Yang JF. Research progress on the relationship between myocardial energetic metabolism and heart failure. Zhonghua Xin Xue Guan Bing Za Zhi. (2022) 50:404–9. doi: 10.3760/cma.j.cn112148-20210421-00363
35. Marti CN, Gheorghiade M, Kalogeropoulos AP, Georgiopoulou VV, Quyyumi AA, and Butler J. Endothelial dysfunction, arterial stiffness, and heart failure. J Am Coll Cardiol. (2012) 60:1455–69. doi: 10.1016/j.jacc.2011.11.082
36. Endemann DH and Schiffrin EL. Endothelial dysfunction. J Am Soc Nephrol JASN. (2004) 15:1983–92. doi: 10.1097/01.ASN.0000132474.50966.DA
37. Tsigkou V, Oikonomou E, Anastasiou A, Lampsas S, Zakynthinos GE, Kalogeras K, et al. Molecular mechanisms and therapeutic implications of endothelial dysfunction in patients with heart failure. Int J Mol Sci. (2023) 24:4321. doi: 10.3390/ijms24054321
38. De Luca M, Crisci G, Armentaro G, Cicco S, Talerico G, Bobbio E, et al. Endothelial dysfunction and heart failure with preserved ejection fraction-an updated review of the literature. Life Basel Switz. (2023) 14:30. doi: 10.3390/life14010030
39. Brown DA, Perry JB, Allen ME, Sabbah HN, Stauffer BL, Shaikh SR, et al. Expert consensus document: Mitochondrial function as a therapeutic target in heart failure. Nat Rev Cardiol. (2017) 14:238–50. doi: 10.1038/nrcardio.2016.203
40. Hinton A, Claypool SM, Neikirk K, Senoo N, Wanjalla CN, Kirabo A, et al. Mitochondrial structure and function in human heart failure. Circ Res. (2024) 135:372–96. doi: 10.1161/CIRCRESAHA.124.323800
41. Zong Y, Li H, Liao P, Chen L, Pan Y, Zheng Y, et al. Mitochondrial dysfunction: mechanisms and advances in therapy. Signal Transduct Target Ther. (2024) 9:124. doi: 10.1038/s41392-024-01839-8
42. Liu Y, Huang Y, Xu C, An P, Luo Y, Jiao L, et al. Mitochondrial dysfunction and therapeutic perspectives in cardiovascular diseases. Int J Mol Sci. (2022) 23:16053. doi: 10.3390/ijms232416053
43. Holst-Hansen A, Grimm D, and Wehland M. Finerenone in heart failure-A novel therapeutic approach. Int J Mol Sci. (2024) 25:13711. doi: 10.3390/ijms252413711
44. Heidenreich PA, Bozkurt B, Aguilar D, Allen LA, Byun JJ, Colvin MM, et al. 2022 AHA/ACC/HFSA guideline for the management of heart failure: A report of the American college of cardiology/American heart association joint committee on clinical practice guidelines. Circulation. (2022) 145:e895–e1032. doi: 10.1161/CIR.0000000000001063
45. Papadimitriou L, Moore CK, Butler J, and Long RC. The limitations of symptom-based heart failure management. Card Fail Rev. (2019) 5:74–7. doi: 10.15420/cfr.2019.3.2
46. Wang X, Tazearslan C, Kim S, Guo Q, Contreras D, Yang J, et al. In vitro heterochronic parabiosis identifies pigment epithelium-derived factor as a systemic mediator of rejuvenation by young blood. BioRxiv Prepr Serv Biol. (2024). 592258. doi: 10.1101/2024.05.02.592258
47. Yamagishi S-I, Inagaki Y, Nakamura K, Abe R, Shimizu T, Yoshimura A, et al. Pigment epithelium-derived factor inhibits TNF-alpha-induced interleukin-6 expression in endothelial cells by suppressing NADPH oxidase-mediated reactive oxygen species generation. J Mol Cell Cardiol. (2004) 37:497–506. doi: 10.1016/j.yjmcc.2004.04.007
48. Chen X, Luo Y, Zhu Q, Zhang J, Huang H, Kan Y, et al. Small extracellular vesicles from young plasma reverse age-related functional declines by improving mitochondrial energy metabolism. Nat Aging. (2024) 4:814–38. doi: 10.1038/s43587-024-00612-4
49. Srikanthan S, Li W, Silverstein RL, and McIntyre TM. Exosome poly-ubiquitin inhibits platelet activation, downregulates CD36 and inhibits pro-atherothombotic cellular functions. J Thromb Haemost JTH. (2014) 12:1906–17. doi: 10.1111/jth.12712
50. Zeng Y, Xu G, Feng C, Cai D, Wu S, Liu Y, et al. Klotho inhibits the activation of NLRP3 inflammasome to alleviate lipopolysaccharide-induced inflammatory injury in A549 cells and restore mitochondrial function through SIRT1/Nrf2 signaling pathway. Chin J Physiol. (2023) 66:335–44. doi: 10.4103/cjop.CJOP-D-23-00029
51. Jiang Y, Wen X, Jian X, Chen Q, and Li Y. Klotho attenuates epithelial−mesenchymal transition of retinal pigment epithelial cells in subretinal fibrosis by suppressing the ERK1/2 and Wnt/β−catenin signaling pathways. Int J Mol Med. (2025) 55:45. doi: 10.3892/ijmm.2025.5486
52. Moigneu C, Abdellaoui S, Ramos-Brossier M, Pfaffenseller B, Wollenhaupt-Aguiar B, de Azevedo Cardoso T, et al. Systemic GDF11 attenuates depression-like phenotype in aged mice via stimulation of neuronal autophagy. Nat Aging. (2023) 3:213–28. doi: 10.1038/s43587-022-00352-3
53. Zhang Y, Zhang Y-Y, Pan Z-W, Li Q-Q, Sun L-H, Li X, et al. GDF11 promotes wound healing in diabetic mice via stimulating HIF-1ɑ-VEGF/SDF-1ɑ-mediated endothelial progenitor cell mobilization and neovascularization. Acta Pharmacol Sin. (2023) 44:999–1013. doi: 10.1038/s41401-022-01013-2
54. Castellano JM, Mosher KI, Abbey RJ, McBride AA, James ML, Berdnik D, et al. Human umbilical cord plasma proteins revitalize hippocampal function in aged mice. Nature. (2017) 544:488–92. doi: 10.1038/nature22067
55. Kandalam V, Basu R, Abraham T, Wang X, Soloway PD, Jaworski DM, et al. TIMP2 deficiency accelerates adverse post-myocardial infarction remodeling because of enhanced MT1-MMP activity despite lack of MMP2 activation. Circ Res. (2010) 106:796–808. doi: 10.1161/CIRCRESAHA.109.209189
56. Xu L, Nirwane A, Xu T, Kang M, Devasani K, and Yao Y. Fibroblasts repair blood-brain barrier damage and hemorrhagic brain injury via TIMP2. Cell Rep. (2022) 41:111709. doi: 10.1016/j.celrep.2022.111709
57. Bu S, Nguyen HC, Nikfarjam S, Michels DCR, Rasheed B, Maheshkumar S, et al. Endothelial cell-specific loss of eNOS differentially affects endothelial function. PloS One. (2022) 17:e0274487. doi: 10.1371/journal.pone.0274487
58. O’Gallagher K, Puledda F, O’Daly O, Ryan M, Dancy L, Chowienczyk PJ, et al. Neuronal nitric oxide synthase regulates regional brain perfusion in healthy humans. Cardiovasc Res. (2022) 118:1321–9. doi: 10.1093/cvr/cvab155
59. Lin L, Liu X, Xu J, Weng L, Ren J, Ge J, et al. High-density lipoprotein inhibits mechanical stress-induced cardiomyocyte autophagy and cardiac hypertrophy through angiotensin II type 1 receptor-mediated PI3K/Akt pathway. J Cell Mol Med. (2015) 19:1929–38. doi: 10.1111/jcmm.12567
60. Durham KK, Chathely KM, and Trigatti BL. High-density lipoprotein protects cardiomyocytes against necrosis induced by oxygen and glucose deprivation through SR-B1, PI3K, and AKT1 and 2. Biochem J. (2018) 475:1253–65. doi: 10.1042/BCJ20170703
61. Brulhart-Meynet M-C, Braunersreuther V, Brinck J, Montecucco F, Prost J-C, Thomas A, et al. Improving reconstituted HDL composition for efficient post-ischemic reduction of ischemia reperfusion injury. PLoS One. (2015) 10:e0119664. doi: 10.1371/journal.pone.0119664
62. Sutter JD, de Veire NRV, Struyf S, Philippé J, Buyzere MD, and Damme JV. PF-4var/CXCL4L1 predicts outcome in stable coronary artery disease patients with preserved left ventricular function. PLoS One. (2012) 7:e31343. doi: 10.1371/journal.pone.0031343
63. Park C, Hahn O, Gupta S, Moreno AJ, Marino F, Kedir B, et al. Platelet factors are induced by longevity factor klotho and enhance cognition in young and aging mice. Nat Aging. (2023) 3:1067–78. doi: 10.1038/s43587-023-00468-0
64. Yao T, Xi Y, Chen F, Lin H, Qian J, and Liu X. Safety of human serum albumin infusion in heart failure patients with hypoproteinemia: a propensity score-matched analysis. Clin Sao Paulo Braz. (2025) 80:100659. doi: 10.1016/j.clinsp.2025.100659
65. Zhao Q, Liu Z, Huang B, Yuan Y, Liu X, Zhang H, et al. PEDF improves cardiac function in rats subjected to myocardial ischemia/reperfusion injury by inhibiting ROS generation via PEDF−R. Int J Mol Med. (2018) 41:3243–52. doi: 10.3892/ijmm.2018.3552
66. Zhang H, Hui H, Li Z, Pan J, Jiang X, Wei T, et al. Pigment epithelium-derived factor attenuates myocardial fibrosis via inhibiting Endothelial-to-Mesenchymal Transition in rats with acute myocardial infarction. Sci Rep. (2017) 7:41932. doi: 10.1038/srep41932
67. Rychli K, Niessner A, Hohensinner PJ, Ali KM, Kaun C, Neuhold S, et al. Prognostic value of pigment epithelium-derived factor in patients with advanced heart failure. Chest. (2010) 138:656–64. doi: 10.1378/chest.09-2739
68. Li H, Liu Y, Lin Y, Li S, Liu C, Cai A, et al. Cardiac repair using regenerating neonatal heart tissue-derived extracellular vesicles. Nat Commun. (2025) 16:1292. doi: 10.1038/s41467-025-56384-x
69. Lerner N, Chen I, Schreiber-Avissar S, and Beit-Yannai E. Extracellular vesicles mediate anti-oxidative response-in vitro study in the ocular drainage system. Int J Mol Sci. (2020) 21:6105. doi: 10.3390/ijms21176105
70. Kalluri R and LeBleu VS. The biology, function, and biomedical applications of exosomes. Science. (2020) 367:eaau6977. doi: 10.1126/science.aau6977
71. Chen K, Wang S, Sun QW, Zhang B, Ullah M, and Sun Z. Klotho deficiency causes heart aging via impairing the Nrf2-GR pathway. Circ Res. (2021) 128:492–507. doi: 10.1161/CIRCRESAHA.120.317348
72. Xu Y and Sun Z. Molecular basis of Klotho: from gene to function in aging. Endocr Rev. (2015) 36:174–93. doi: 10.1210/er.2013-1079
73. Chen K, Wang S, and Sun Z. In vivo cardiac-specific expression of adenylyl cyclase 4 gene protects against Klotho deficiency-induced heart failure. Transl Res. (2022) 244:101–13. doi: 10.1016/j.trsl.2022.01.006
74. Wang K, Li Z, Li Y, Liu X, Sun Y, Hong J, et al. Cardioprotection of Klotho against myocardial infarction-induced heart failure through inducing autophagy. Mech Ageing Dev. (2022) 207:111714. doi: 10.1016/j.mad.2022.111714
75. Zhu J, Zhang N, Zhao Y, Liu Q, Wang Y, Chen M, et al. Deficiency of GDF-11 accelerates TAC-induced heart failure by impairing cardiac angiogenesis. JACC Basic Transl Sci. (2023) 8:617–35. doi: 10.1016/j.jacbts.2022.11.004
76. Sinha M, Jang YC, Oh J, Khong D, Wu EY, Manohar R, et al. Restoring systemic GDF11 levels reverses age-related dysfunction in mouse skeletal muscle. Science. (2014) 344:649–52. doi: 10.1126/science.1251152
77. Frohlich J, Kovacovicova K, Raffaele M, Virglova T, Cizkova E, Kucera J, et al. GDF11 inhibits adipogenesis and improves mature adipocytes metabolic function via WNT/β-catenin and ALK5/SMAD2/3 pathways. Cell Prolif. (2022) 55:e13310. doi: 10.1111/cpr.13310
78. Zhang P, Zhai H, Zhang S, Ma X, Gong A, Xu Z, et al. GDF11 protects against mitochondrial-dysfunction-dependent NLRP3 inflammasome activation to attenuate osteoarthritis. J Adv Res. (2024). 73, S2090-1232(24)00323-0. doi: 10.1016/j.jare.2024.08.001
79. G S, K S, N N, A F, Q N, P S, et al. TIMP-2 mutant decreases MMP-2 activity and augments pressure overload induced LV dysfunction and heart failure. Arch Physiol Biochem. (2013) 119:65–74. doi: 10.3109/13813455.2012.755548
80. Król M and Kepinska M. Human nitric oxide synthase-its functions, polymorphisms, and inhibitors in the context of inflammation, diabetes and cardiovascular diseases. Int J Mol Sci. (2020) 22:56. doi: 10.3390/ijms22010056
81. Ally A, Powell I, Ally MM, Chaitoff K, and Nauli SM. Role of neuronal nitric oxide synthase on cardiovascular functions in physiological and pathophysiological states. Nitric Oxide Biol Chem. (2020) 102:52–73. doi: 10.1016/j.niox.2020.06.004
82. Zhou L and Zhu D-Y. Neuronal nitric oxide synthase: structure, subcellular localization, regulation, and clinical implications. Nitric Oxide Biol Chem. (2009) 20:223–30. doi: 10.1016/j.niox.2009.03.001
83. Y H, G Q, D M, C F, and X J. Exploring the role of iNOS in HFpEF-Related myocardial fibrosis: Involvement of PTEN-PI3K/AKT signaling pathway. Biochem Biophys Res Commun. (2024) 734. doi: 10.1016/j.bbrc.2024.150589
84. Roy R, Wilcox J, Webb AJ, and O’Gallagher K. Dysfunctional and dysregulated nitric oxide synthases in cardiovascular disease: mechanisms and therapeutic potential. Int J Mol Sci. (2023) 24:15200. doi: 10.3390/ijms242015200
85. P A, P Kv, S Mw, S Md, B Cm, V Ss, et al. Association of high-density lipoprotein parameters and risk of heart failure: A multicohort analysis. JACC Heart Fail. (2024) 12. doi: 10.1016/j.jchf.2024.03.007
86. Schroer AB, Ventura PB, Sucharov J, Misra R, Chui MKK, Bieri G, et al. Platelet factors attenuate inflammation and rescue cognition in ageing. Nature. (2023) 620:1071–9. doi: 10.1038/s41586-023-06436-3
87. Roche M, Rondeau P, Singh NR, Tarnus E, and Bourdon E. The antioxidant properties of serum albumin. FEBS Lett. (2008) 582:1783–7. doi: 10.1016/j.febslet.2008.04.057
88. Wang Y, Du P, Xiao Q, Li J, Liu X, Tan J, et al. Relationship between serum albumin and risk of atrial fibrillation: A dose-response meta-analysis. Front Nutr. (2021) 8:728353. doi: 10.3389/fnut.2021.728353
89. Ruiz-Perera LM, Höving AL, Schmidt KE, Cenan S, Wohllebe M, Greiner JFW, et al. Neuroprotection mediated by human blood plasma in mouse hippocampal slice cultures and in oxidatively stressed human neurons. Int J Mol Sci. (2021) 22:9567. doi: 10.3390/ijms22179567
90. Loffredo FS, Steinhauser ML, Jay SM, Gannon J, Pancoast JR, Yalamanchi P, et al. Growth differentiation factor 11 is a circulating factor that reverses age-related cardiac hypertrophy. Cell. (2013) 153:828–39. doi: 10.1016/j.cell.2013.04.015
91. Yang K, Wang C, Nie L, Zhao X, Gu J, Guan X, et al. Klotho protects against indoxyl sulphate-induced myocardial hypertrophy. J Am Soc Nephrol JASN. (2015) 26:2434–46. doi: 10.1681/ASN.2014060543
92. Feng Y, Huang W, Wani M, Yu X, and Ashraf M. Ischemic preconditioning potentiates the protective effect of stem cells through secretion of exosomes by targeting Mecp2 via miR-22. PloS One. (2014) 9:e88685. doi: 10.1371/journal.pone.0088685
93. Conboy IM, Conboy MJ, Wagers AJ, Girma ER, Weissman IL, and Rando TA. Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature. (2005) 433:760–4. doi: 10.1038/nature03260
94. Lagunas-Rangel FA. Aging insights from heterochronic parabiosis models. NPJ Aging. (2024) 10:38. doi: 10.1038/s41514-024-00166-0
95. Kiss T, Tarantini S, Csipo T, Balasubramanian P, Nyúl-Tóth Á, Yabluchanskiy A, et al. Circulating anti-geronic factors from heterochonic parabionts promote vascular rejuvenation in aged mice: transcriptional footprint of mitochondrial protection, attenuation of oxidative stress, and rescue of endothelial function by young blood. GeroScience. (2020) 42:727–48. doi: 10.1007/s11357-020-00180-6
96. Wu BJ, Chen K, Shrestha S, Ong KL, Barter PJ, and Rye K-A. High-density lipoproteins inhibit vascular endothelial inflammation by increasing 3β-hydroxysteroid-Δ24 reductase expression and inducing heme oxygenase-1. Circ Res. (2013) 112:278–88. doi: 10.1161/CIRCRESAHA.111.300104
97. Wei S-Y, Chou Y-H, Chang F-C, Huang S-Y, Lai C-F, and Lin S-L. Young plasma attenuated chronic kidney disease progression after acute kidney injury by inhibiting inflammation in mice. Aging Dis. (2023) 15:2786–98. doi: 10.14336/AD.2023.1230
98. Arslan F, Lai RC, Smeets MB, Akeroyd L, Choo A, Aguor ENE, et al. Mesenchymal stem cell-derived exosomes increase ATP levels, decrease oxidative stress and activate PI3K/Akt pathway to enhance myocardial viability and prevent adverse remodeling after myocardial ischemia/reperfusion injury. Stem Cell Res. (2013) 10:301–12. doi: 10.1016/j.scr.2013.01.002
99. Hou W, Luo W, Xi R, and Niu X. Research progress of myocardial energy metabolism remodeling in cardiac hypertrophy. Chin Heart J. (2025) 37:94–8. doi: 10.12125/j.chj.202308038
100. Ma S, Wang S, Ye Y, Ren J, Chen R, Li W, et al. Heterochronic parabiosis induces stem cell revitalization and systemic rejuvenation across aged tissues. Cell Stem Cell. (2022) 29:990–1005.e10. doi: 10.1016/j.stem.2022.04.017
101. Gonzalez-Armenta JL, Li N, Lee R-L, Lu B, and Molina AJA. Heterochronic parabiosis: old blood induces changes in mitochondrial structure and function of young mice. J Gerontol A Biol Sci Med Sci. (2021) 76:434–9. doi: 10.1093/gerona/glaa299
102. Hwang HJ, Kim N, Herman AB, Gorospe M, and Lee J-S. Factors and pathways modulating endothelial cell senescence in vascular aging. Int J Mol Sci. (2022) 23:10135. doi: 10.3390/ijms231710135
103. Ailawadi S, Wang X, Gu H, and Fan G-C. Pathologic function and therapeutic potential of exosomes in cardiovascular disease. Biochim Biophys Acta. (2015) 1852:1–11. doi: 10.1016/j.bbadis.2014.10.008
104. Xia E, Xu F, Hu C, Kumal JPP, Tang X, Mao D, et al. Young blood rescues the cognition of Alzheimer’s model mice by restoring the hippocampal cholinergic circuit. Neuroscience. (2019) 417:57–69. doi: 10.1016/j.neuroscience.2019.08.010
105. Liu A, Guo E, Yang J, Yang Y, Liu S, Jiang X, et al. Young plasma reverses age-dependent alterations in hepatic function through the restoration of autophagy. Aging Cell. (2018) 17:e12708. doi: 10.1111/acel.12708
106. Balistreri CR. Anti-inflamm-ageing and/or anti-age-related disease emerging treatments: A historical alchemy or revolutionary effective procedures? Mediators Inflammation. (2018) 2018:3705389. doi: 10.1155/2018/3705389
107. Harper SC, Johnson J, Borghetti G, Zhao H, Wang T, Wallner M, et al. GDF11 decreases pressure overload-induced hypertrophy, but can cause severe cachexia and premature death. Circ Res. (2018) 123:1220–31. doi: 10.1161/CIRCRESAHA.118.312955
Keywords: heart failure, young blood, circulating factors, cardiac repair, anti-aging pathways
Citation: Chen M-T, Wang R-Q, Qian Y-M, Peng T, Lan X-Y, Liang L-L, Luo G, Liu Q-Y and Liu M-N (2025) Rejuvenating the failing heart: multidimensional insights into young blood-mediated anti-aging pathways. Front. Endocrinol. 16:1653567. doi: 10.3389/fendo.2025.1653567
Received: 25 June 2025; Accepted: 12 August 2025;
Published: 03 September 2025.
Edited by:
Gaetano Santulli, Albert Einstein College of Medicine, United StatesReviewed by:
Tingting Zhao, University of Macau, Macao SAR, ChinaQida He, Xiamen University, China
Peymaneh Habibi, Tabriz University of Medical Sciences, Iran
Navya Katram, University of Oklahoma Health Sciences Center, United States
Copyright © 2025 Chen, Wang, Qian, Peng, Lan, Liang, Luo, Liu and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gang Luo, bHVvZ2FuZzE5ODJAMTYzLmNvbQ==; Qiu-Yu Liu, cS5saXVAc3dtdS5lZHUuY24=; Meng-Nan Liu, bGl1bWVuZ25hbkBzZW11LmVkdS5jbg==
†These authors have contributed equally to this work and share first authorship