 Olivia Pagliarosi
Olivia Pagliarosi Jessica Pepe
Jessica Pepe Andrea Del Fattore
Andrea Del Fattore Michela Rossi
Michela Rossi- 1Department of Clinical, Internal, Anesthesiology and Cardiovascular Sciences, Sapienza University, Rome, Italy
- 2Bone Physiopathology Research Unit, Translational Pediatric and Clinical Genetic Research Division, Bambino Gesù Children’s Hospital, IRCCS, Rome, Italy
The “vanishing bone disease” or Gorham-Stout disease (GSD) is a very rare disorder characterized by massive lymphatic and angiomatous proliferation accompanied by progressive osteolysis, without the deposition of new bone matrix. Because of its rare and complex clinical features, diagnosis is challenging and its etiopathogenesis is not completely known; the genetic basis of GSD has been hypothesized and different mutations have been reported in patients. Our review aims to describe all these genetic alterations found in GSD patients and their association with clinical features. The identification of a specific molecular pathway or genetic alteration in GSD could help in the diagnosis and possibly the treatment of this rare sporadic disease.
1 Introduction
Gorham-Stout disease (GSD), also known as “vanishing bone disease”, is a very rare disorder, to date not even 400 cases have been described, and is characterized by lymphatic and angiomatous proliferation accompanied by progressive osteolysis. Although a slight predilection for male, the disease does not show a clear sex bias or inheritance pattern and can occur at any age, with the majority of cases occurring in childhood (1), affecting one or multiple bones of either the axial or the appendicular skeleton; patients initially display a patchy osteoporosis condition, which progressively leads to skeletal deformity, shrinkage and eventual loss of the affected bone (2, 3). Moreover, both the medullary and cortical regions of affected bones present lymphatic vessels, which are not typically found in normal bones (4). The main reported symptoms are pain, weakness and impairment of the affected regions; however, some patients develop more severe complications such as chylothorax (5), which may cause respiratory distress, as well as vertebrae involvement leading to neurological deficits or paraplegia, bone infection and subsequent septic shock, and ultimately death (2).
GSD is frequently undiagnosed or misdiagnosed and is indeed often classified as a complex lymphatic abnormality (CLA) as some clinical features overlap with CLA diseases, such as generalized lymphatic anomaly (GLA), Kaposiform lymphangiomatosis (6) and channel-type lymphatic malformations (LM) (2). GLA is a rare and aggressive disease characterized by diffused lymphatic vessel proliferation with multi-organ involvement (mediastinum, lungs, bone, spleen, and soft tissues), and it primarily affects children and adolescents. Its symptoms include pleural and pericardial effusion, ascites, multiple cystic splenic lesions, gastrointestinal haemorrhage, multiple bone osteolysis (mostly skull and spine), lymphedema, and lymphorrhea (7) (Figure 1). GSD, in contrast, is distinguished by the progressive destruction and erosion of bone, particularly the cortical bone, often leading to its complete absorption with the presence of abnormal intraosseous LM, found also in regions adjacent to osteolytic lesions (Figure 1). Hu et al. carried out radiographic evaluations of 67 GSD cases, half of which present the disappearance of portions of bone; the initial stage of this process manifests as radiolucent foci in the intramedullary or subcortical regions (8). The femur was the most commonly involved site (8). In the same year, Lala et al. carried out another radiological study on a cohort of 51 patients, of which 19 met the criteria for GSD, and highlighted differences in osteolytic activity between GLA and GSD patients: GLA generally shows lytic areas confined to the medullary cavity, whereas GSD displays progressive osteolysis, with cortical bone loss (9). They also identified the ribs as the most commonly affected site in both groups, followed by the cranium, clavicle, and cervical spine in GSD, while the thoracic spine, humerus, and femur were most frequent in GLA. Additionally, GSD typically involves fewer bones than GLA (9) (Figure 1). Further investigation is necessary to fully differentiate these two lymphatic disorders.
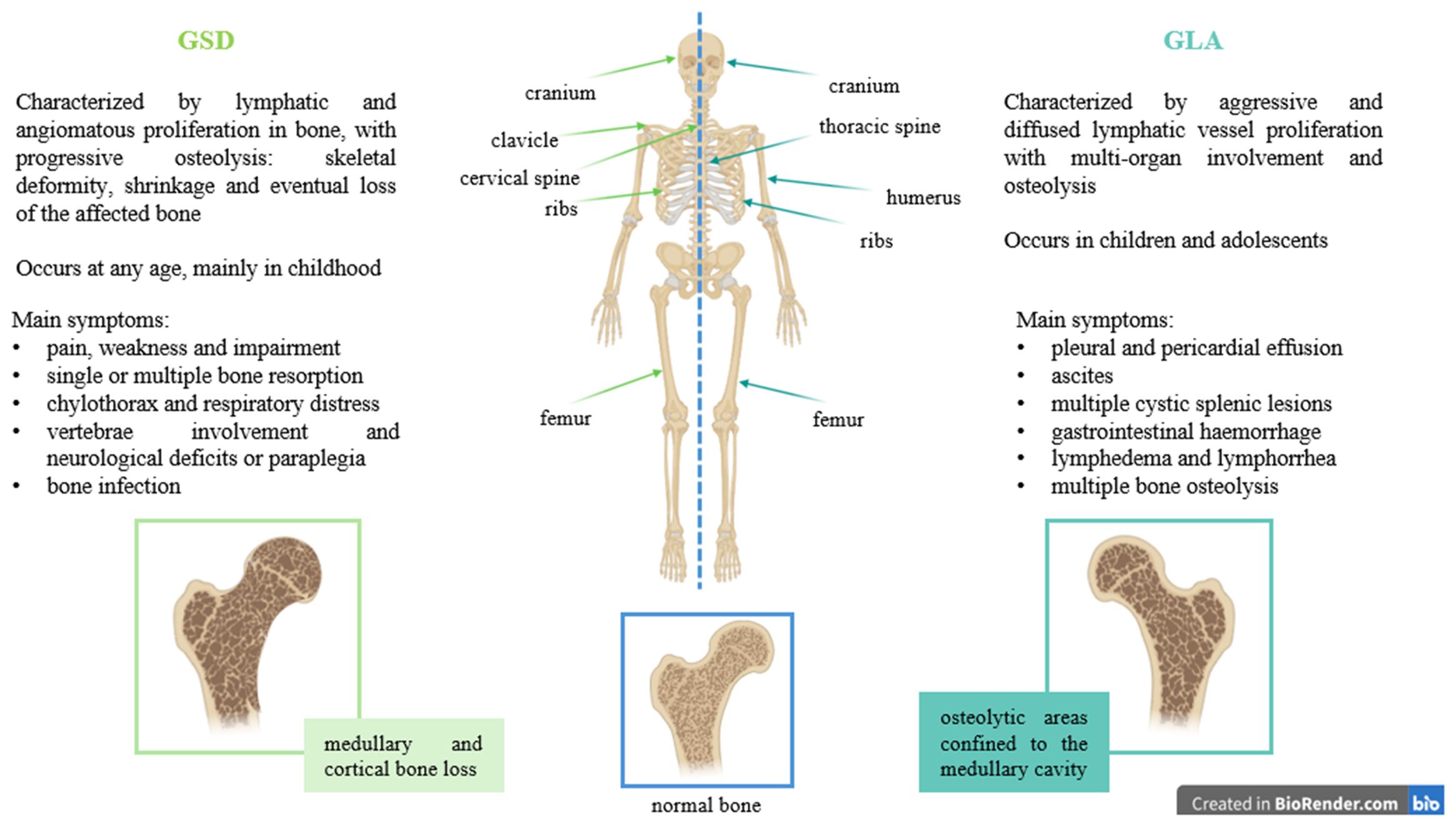
Figure 1. Differences between GSD and GLA, highlighting symptoms, bone involvement and type of osteolysis.
Therefore, the diagnosis of GSD remains challenging with no standardized guidelines; it is usually performed by exclusion criteria to rule out neoplastic processes, infections, as well as metabolic and endocrine disorders. Radiographs, bone scan and computed tomography are useful (10), but diagnosis must be confirmed by histopathological analysis of the bone lesion, which should reveal extensive bone resorption and angiomatous tissue, as well as excessive presence of fibrotic tissue without cellular atypia (11, 12). Indeed, the osteolytic process is characterized by the absence of increased osteoblast activity along the surfaces of the remaining bone fragments in sections of affected tissues; the disappearing bone is replaced by fibrovascular tissue rather than newly formed woven repair bone (2). Moreover, GSD osteoblastic cells exhibit ultrastructural alterations suggesting that they have either decreased synthetic activity or are undergoing degeneration (13). Furthermore, osteocytes within bone tissue close to the lesions have been reported to present enlarged lacuna, possibly related to osteocyte mediated-bone resorption activity (2, 12).
Due to its rarity and limited case studies, also the etiopathogenesis of GSD remains unclear. It has been proposed that the osteolytic process may result from excessive proliferation of endothelial and lymphatic vessels, accompanied by increased local blood flow, changes in pH or altered mechanical forces that affect bone remodeling (2). The secretion of cytokines and growth factors such as TNF (Tumor Necrosis Factor)-α, IL (Interleukin)-6 and VEGF (Vascular Endothelial Growth factor)-A/C has also been considered, given their stimulatory effect on lymphangiogenesis and osteoclast activity (7, 14–16), and their inhibitory effect on osteoblasts, particularly at high concentrations (2, 17–19). Recent studies have proposed that GSD may involve a primary imbalance in bone remodeling, characterized by increased osteoclast differentiation and resorption (20), along with an impairment of circulating bone cells (21). Moreover, immune dysregulation or inflammatory conditions could contribute to exacerbate the syndrome (22).
Depending on the severity of the disease and the extent of organ involvement, different strategies are used to treat GSD symptoms, including surgery, radiotherapy and drug treatment, which are only partially effective and none are curative, but patients experience relief and improved quality of life after treatment (1). Bone loss with functional impairment requires surgical procedures which consist of the resection of localized lesions and the reconstruction or stabilization of the bone by using bone grafts and/or prostheses (8, 23, 24); interventions are also carried out to prevent respiratory insufficiency and reduce or halt the fluid build-up in the pleural cavity (24). Without surgical intervention, morbidity and mortality rates are very high (2).
The use of radiotherapy has been described as successful and beneficial in several case reports, with a 75% overall success rate on local lesions with doses in the 30–45 Gy range (25–27). It has also been used to manage chylothorax in a GSD case (28). However, radiation could provoke serious problems, like secondary malignancy and growth restriction in children and adolescents who receive a high dose treatment (29).
Bisphosphonates and interferon alpha 2b are commonly prescribed. Bisphosphonates have been successfully used for the treatment of GSD patients for their anti-osteoclastic activity (30, 31). Hammer et al. reported a stabilization of the clinical and radiological picture in a patient treated with pamidronate every 3 months (31). In 2014, Liu et al. evaluated different bisphosphonate treatments (either zoledronic acid or pamidronate) with or without radiotherapy (40 Gy) on GSD patients, all of which stabilized disease progression as well as inhibited the enlargement of the osteolytic scope and increased bone mineral density (30). The authors also reviewed GSD bisphosphonate treatment investigations in literature, finding that the most commonly used was zoledronic acid, followed by alendronate, which often led to disease arrest (30). However, the use of anti-resorption drugs like bisphosphonates is often associated with side-effects, including atrial fibrillation, osteonecrosis of the jaw (1) and “frozen bone”, characterized by over-suppression of bone turnover (32). Interferon alpha 2b, an immunomodulatory and anti-angiogenic compound, has been used to stabilize GSD and can also be used in combination with bisphosphonates or after surgery (33–36). Other pharmaceuticals administered for clinical relief are the anti-angiogenic VEGF-neutralizing antibody Bevacizumab (37), propranolol (38), low molecular weight heparin (39), steroids, and vitamin D (2).
More recently, the repurposing of oncogenic treatments, such as Sirolimus or Alectinib, has become more frequent and will be further described in this review.
Over the past few years, the genetic basis of GSD has been largely hypothesized in case reports, although no definitive evidence has yet been established. The identification of specific germline mutations or somatic mosaicism in GSD could significantly aid in the diagnosis and possibly the treatment of this rare condition. This review aims to summarize all the reports investigating genetic alterations in GSD patients and explore their potential contribution to disease development, paving the way for the discovery of new therapeutic targets.
2 Genetic studies
In 2013, the first work suggesting that GLA and GSD patients could carry abnormal genomic copy number was presented at the first International Conference on Generalized Lymphatic Anomaly and Gorham-Stout Syndrome (11). Although no updates or reports have been released on that study, other genetic studies have emerged (Table 1).
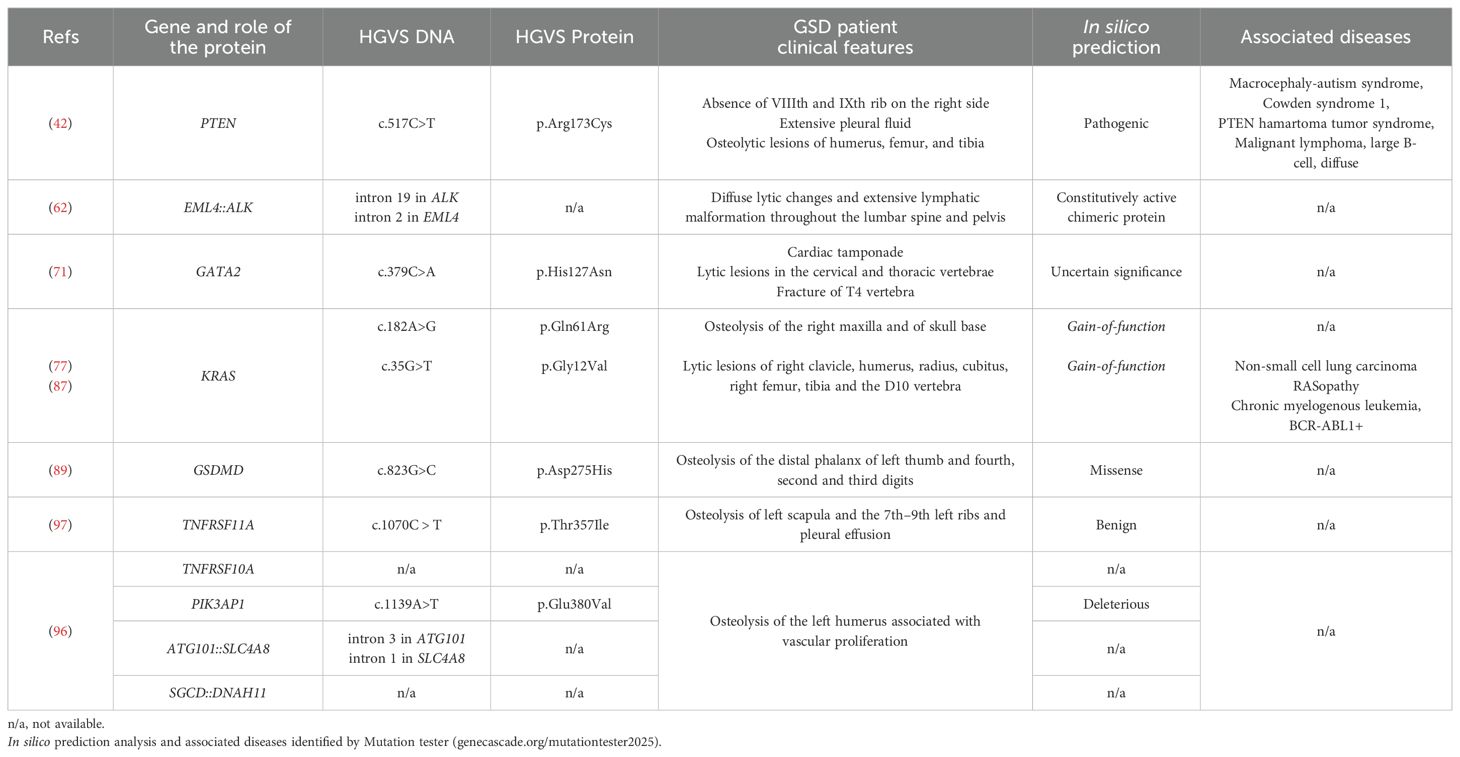
Table 1. Summary of the genetic variants identified in GSD patients.
2.1 PTEN
In 2012, Hopman et al. described a patient exhibiting absence of the 8th and 9th ribs on the right side, extensive pleural fluid, as well as osteolytic lesions in the humerus, femur and tibia. Additionally, he presented a vascular tumor affecting part of the right axilla and flank (42). Diagnosed with GSD and PTEN hamartoma tumor syndrome, the patient underwent genetic analysis which identified two variants in lymphocyte–derived DNA: a germline heterozygous mutation c.517C>T (p.Arg173Cys) of the PTEN (Phosphatase And Tensin Homolog) gene and a polymorphism c.649-26G>T in the TSC2 (TSC Complex Subunit 2) gene. Moreover, the analysis of DNA from the affected tissue revealed also the heterozygous variant c.2180C>T (p.Ala727Val) in the FLT4 (Fms Related Receptor Tyrosine Kinase 4, also known as VEGFR3) gene (42).
The TSC2 polymorphism was deemed non-pathogenic, while the PTEN mutation has been already described in patients with hamartoma (43). PTEN mutations have also been implicated in atypical endometrial hyperplasia, endometrial carcinoma (44), and glioblastoma (45, 46), bone metastases (47), multiple myeloma (48, 49), osteosarcoma (50–52), and other bone malignancies (53).
PTEN encodes a dual-specificity phosphatase protein that negatively regulates the PI3K/Akt/mTOR and MAPK (mitogen-activated protein kinase) signaling pathways. Stambolic et al. observed that Pten-deficient murine fibroblasts exhibited decreased sensitivity to cell death, as well as elevated protein kinase B or Akt (PKB/Akt) activity and phosphorylation (54), which promotes survival and oncogenesis (55). As a tumor suppressor, PTEN reduces intracellular phosphatidylinositol 3,4,5-trisphosphate levels, thereby inhibiting the PI3K/PKB/Akt axis (53, 54). Somatic changes in the PI3K/AKT/mTOR pathway have been observed in LM (7, 56). PTEN is further implicated in the regulation of osteoclast differentiation, survival and migration, as well as angiogenesis and lymphangiogenesis (57). The involvement of the phosphoinositide 3-kinase (PI3K) pathway in GSD is also supported by Rossi et al., who performed a transcriptomic analysis on mature osteoclasts differentiated from peripheral blood mononuclear cells of GSD patients, revealing an enrichment of this pathway (20). To define the role of PTEN in bone homeostasis and bone strength, Lorenz et al. generated a mouse model of Pten conditional Knock-out (Pten-cKO) in pre-osteoblasts and investigated osteoprogenitor cells; bone marrow stem cells isolated from Pten-cKO animals showed enhanced proliferation and osteogenic differentiation, resulting in increased trabecular bone volume and mechanical strength (58).
The mammalian target of rapamycin (mTOR) is a downstream kinase involved in the PI3K/Akt pathway, involved in metabolism, angiogenesis, cell motility and growth; its dysregulation has been documented in LM (59). Hence, the use of Sirolimus, an inhibitor of mTOR, has been tested on multiple vascular anomalies (60, 61) and on a cohort of 5 patients with GSD (41), demonstrating to be efficacious and well tolerated in these studies.
2.2 EML::ALK fusion
A recent paper reported a GSD patient with extensive LM in the lumbar spine and sacrum, with chronic cerebrospinal fluid leak and severe headaches. The patient underwent various unsuccessful treatments (62). Genetic testing using an oncology-focused next-generation sequencing panel on patient’s bone biopsy revealed an EML4::ALK (echinoderm microtubule-associated protein-like 4-anaplastic lymphoma kinase) fusion (62).
There are several types of EML4::ALK fusions, all containing the intracellular tyrosine kinase domain of ALK but differing in the truncation sites of EML4 (63). In vitro studies using NIH/3T3 mouse fibroblastic cells expressing different EML4::ALK variants demonstrated that these variants mainly activate the MAPK/ERK and STAT3 signaling pathways, promoting cell proliferation, survival, and invasion (63). The MAPK/ERK signaling pathway is associated with cell proliferation, and the mTOR and STAT3 pathways are associated with cell survival and apoptosis. The EML4::ALK fusion protein upregulates MAPK signaling and activates ERK; moreover, increased expression of STAT3 promotes the activation of mTOR thus inhibiting apoptosis of tumoral cells. Studies indicate that some regions of the EML4 gene induce tumorigenesis, in particular the HELP domain is necessary for the specific activation of RAS, which promotes the upregulation of RAS and the phosphorylation of ERK, inducing cell proliferation (64). These variants have been widely reported in tumors, particularly as the primary pathogenic driver in non-small cell lung cancer (65, 66), and have also been identified in a patient affected with GLA (62).
Anaplastic lymphoma kinase (ALK) is a tyrosine kinase receptor that plays a key role during development and is largely not expressed in most adult tissues; indeed, ALK becomes constitutively active when fused with EML4, driving oncogenic signaling; these fusion proteins are validated targets of tyrosine kinase inhibitors such as crizotinib, alectinib or lorlatinib (67–70). The aforementioned GSD patient was treated with alectinib, resulting in reduced swelling and pain, as well as decreased soft tissue edema in the LM (40).
An in-depth investigation into the influence of the chimeric mutation in GSD bone phenotype is still necessary, as well as the analysis of the effects of ALK inhibitors in the progression of the bone disease.
2.3 GATA2
In a very short case report, Oguz and colleagues investigated the genomic signature of a pediatric female diagnosed with GSD and presenting cardiac tamponade, who was successfully treated with Sirolimus (71). Genetic analysis was performed using a vascular anomaly panel which includes the NRAS, KRAS, FOXC2, FLT4, GJC2, VEGFC, PIEZO1 and GATA2 genes and heterozygous splicing mutation c.379C>A (p.His127Asn) in the GATA2 (Endothelial Transcription Factor GATA2) gene was detected (71).
GATA2 is a key factor in the generation and maintenance of hematopoietic stem and multipotent progenitor cells, and is involved in hematopoietic diseases, infections and cancer. Its altered expression is associated with immunodeficiency (so-called GATA2 deficiency) and acute myeloid leukemia (72). In hematopoietic stem progenitor cells, GATA2 activates the expression of diverse genes, including those encoding c-Kit receptor tyrosine kinase, erythroid and the megakaryocytic differentiation inducer GATA1 (72). Moreover, it is crucial for lymphatic vessel development (73) as demonstrated in Gata2 heterozygous deficient mice, which display delayed lymphatic recanalization after resection (74, 75). Furthermore, GATA2 modulates the expression of microRNA-126, regulator of lymphatic vessel development (73).
Tolkachov et al. analyzed the deletion of GATA2 in mesenchymal stem cells, revealing increased osteogenic differentiation and bone formation (76). Loss of GATA2 also reduced osteoprotegerin expression, enhancing osteoclastogenesis. In vivo, this resulted in enhanced bone formation accompanied by impaired trabecular bone and mechanical strength, confirming a role of GATA2 in bone turnover (76).
2.4 KRAS
So far, the most representative genetic study was performed by Nozawa and colleagues who analyzed a cohort of 6 GSD patients. Targeted sequencing analysis of cancer-related genes revealed the somatic KRAS c.182A>G (p.Gln61Arg) variant in frozen affected tissue of one patient (77). This gain-of-function mutation is well-documented in various human cancers (78) and congenital syndromes known as RASopathies (79). The mutation significantly promotes cell growth through the activation of the MAPK and PI3K/AKT signaling pathways (80). Indeed, the KRAS gene encodes K-Ras, a small GTPase that acts as an oncogenic molecular switch, regulating cell proliferation and survival (78, 81, 82). KRAS mutations in CLA patients impair GTP hydrolysis, resulting in hyperactive downstream signaling (78). Cells with oncogenic KRAS variants have an impact on the production of angiogenic and osteoclastogenic cytokines, including VEGF (83) and IL-6 (84). In addition, KRAS-mutant cancer cells modulate the inflammatory response, recruiting and activating immune cells, promoting pro-tumorigenic properties and cell evasion from immunosurveillance; moreover, these cells secrete molecules that promote the recruitment of activated macrophages, which also promote angiogenesis and osteoclastogenesis, contributing to the secretion of VEGF, IL-6 and TNF-α (85, 86). Indeed, Nozawa et al. suggested that macrophages harboring the variant may contribute to the production of large amounts of angiogenic and osteoclastogenic molecules (77).
In 2021, Homayun-Sepehr et al. identified another activating somatic mutation (c.35G>T, p.Gly12Val) of KRAS in the blood and affected tissue of a GSD patient using a comprehensive cancer panel containing 408 cancer-related genes (87). The mutation p.G12V was previously reported in a patient with malignant giant cell tumor of bone, a rare aggressive sarcoma characterized by the presence of multinucleated giant cells and poor clinical course (88). To assess the impact of the hyperactive KRAS variant in the lymphatic system, a genetically engineered mouse model that conditionally expressed the hyperactive form of Kras (p.Gly12Asp) in Lymphatic Endothelial Cells (LEC) was generated. The authors reported growth of lymphatic vessels in bone, impairment in lymphatic valve formation, and the development of chylothorax, resembling the vascular features presented by GSD patients (87). Moreover, a gene ontology analysis of the modulated genes identified in LEC derived from the hyperactive-KRAS mouse model showed increased expression of genes involved in angio/lymphangiogenesis, cell proliferation and migration, and metallopeptidase activity. The role of KRAS mutations in GSD bone still needs to be investigated.
2.5 Gasdermin D
More recently, Uehara et al. identified the biallelic missense variant c.823G>C (p.Asp275His) of the GSDMD (Gasdermin D) gene in a GSD patient with osteolysis of the distal phalanx of the left 4th, right 2nd, and 3rd digits, without lymphangiomatous proliferation observed in bone biopsy (89). As the variant is located at the exon-intron splice junction of exon 7, the authors hypothesized that the splicing process could be altered, but no alterations of GSDMD expression were found in a lymphoblastoid cell line derived from the patient’s PBMC (89).
GSDMD is a key regulator of pyroptosis, a type of programmed inflammatory cell death triggered by invasive infection and danger signals. Gasdermins mediate pore formation in the plasma membrane, leading to the loss of cell membrane integrity and leakage of cell cytosolic contents, inducing inflammation (90); GSDMD is transcriptionally regulated by NF-κB and the interferon regulatory factor 2 (91), and its activity is mediated by caspases and the NLRP3 inflammasome, already implied in osteoporosis (92, 93). Upon activation, caspases cleave GSDMD to generate an N-terminal cleavage product that triggers pyroptosis and the release of inflammatory cytokines such as IL-1β (94, 95).
Indeed, the connective tissue surrounding the bone of the aforementioned GSD patient had mild inflammatory cell infiltration, mainly characterized by macrophages. Moreover, investigation of GSDMD cleavage in monocytes of the GSD patient did not reveal the fragments generated with protein activation (89).
GSDMD has already been recognized as a critical player in bone metabolism by preventing bone loss. Indeed, an up-regulation of Gsdmd expression was detected during osteoclast differentiation of the murine macrophage RAW264.7 cell line and mouse bone marrow cells (89). In the late stage of osteoclast lineage commitment, Li et al. observed that the cleavage of Gsdmd also yielded a non-pyroptotic 20-kDa fragment that inhibits excessive osteoclastic resorption and bone loss by restricting the maturation and secretion of lysosomes (91). In fact, Gsdmd-deficient osteoclasts displayed enhanced lysosomal number, size, density and activity, and increased bone resorption activity in vitro. The Gsdmd-KO mice displayed an osteoporotic phenotype, with reduced trabecular bone and number, enhanced eroded surface and increased levels of serum bone resorption marker CTX-I (Carboxy-terminal type I collagen) compared to wild-type mice. Osteoblast parameters were minimally affected, indicating that the phenotype was not a result of impaired bone formation. In fact, gene expression analysis further confirmed that the most significant alterations occurred in osteoclasts rather than osteoblasts (91).
2.6 Multi-omics analysis
Yebenes Mayordomo et al. recently carried out a multi-omic analysis of data from whole-genome and RNA sequencing of the affected tissue and the surrounding normal tissue in a 45-year-old Gorham-Stout patient (96). A total of 643 mutations were identified across 233 genes, with a high frequency of insertions and deletions; among the most frequently mutated genes, TNFRSF10A, a tumor necrosis factor receptor involved in the mediation of apoptosis and the activation of NF-κB pathway, was identified (96). It belongs to the same receptor family as TNFRSF11A, found mutated (c.1070C > T, p.Thr357Ile) in a patient with GSD presenting osteolysis of the left scapula and the 7th–9th left ribs (97). Although the effect of this variant remains to be functionally characterized, it is well established that TNFRSF11A plays a key role in osteoclast differentiation and activity, and alterations in this gene have been associated with osteolytic diseases (98, 99). Additionally, Yebenes Mayordomo et al. identified a missense mutation (c.1139A>T) in the PIK3AP1 (Phosphoinositide-3-Kinase Adaptor Protein 1) gene (96), involved in the PTEN/PI3K/AKT signaling pathway and known for its role in regulating inflammation and the innate immune response.
The in-depth analysis discovered the presence of a substantial number of interchromosomal mutations, in particular chromosome translocations, suggesting that gene fusion variants could be frequent in GSD (96). The authors noted a gene variant found in chromosome 12 consisting of the fusion of ATG101 (Autophagy Related 101), involved in macroautophagy (100, 101), and SLC4A8 (Solute Carrier family 4 member 8), which could possibly affect the macrophage signaling pathway (96). Another gene fusion involved the SGCD (Sarcoglycan Delta) and DNAH11 (Dynein Axonemal Heavy Chain 11) genes (96); SGCD is associated with muscular dystrophy (102, 103), which could determine vascular malformations (104). These fusion variants still need to be fully described.
A high proportion of genes were either up- or down-regulated and the expression of gene families like VEGF or NOTCH drastically increased in GSD affected tissue compared to normal tissue (96). The expression of PI3K, as well as AKT and mTOR, was detected to be considerably decreased, while PTEN was increased (96), suggesting an altered stimulation of endothelial cell growth and angiogenesis through VEGFA and VEGFB and the VEGFR1-PI3K-AKT signaling pathway (105, 106).
3 Conclusions
Gorham-Stout disease is still an enigma. To date, only a few research studies have investigated the genetic alterations in a limited number of patients, employing heterogeneous methods and analyzing various types of tissues.
Many studies reported mutations that affect the PI3K/AKT signaling cascade as well as the MAPK pathway, with several collateral effects on macrophages, which impact osteoclastogenesis and bone turnover; moreover, for some of these pathways, drug treatment is still available (Table 2). A comprehensive analysis of these molecular mechanisms, combined with detailed phenotyping of bone involvement in a larger cohort of patients is essential to achieve a more complete understanding of GSD. Such insights are crucial to improve diagnosis and identify new therapeutic targets for this rare and debilitating syndrome.

Table 2. Pathways involved with the mutations identified in GSD patients and related available treatments.
Author contributions
OP: Writing – original draft. JP: Writing – original draft. AD: Funding acquisition, Writing – review & editing, Writing – original draft. MR: Funding acquisition, Writing – original draft.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This study was supported by grants from the Italian Ministry of Health with “Current Research funds” to A.D.F. and M.R.; M.R. was supported by Fondazione Umberto Veronesi.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
The handling editor FV declared a past co-authorship with the author JP.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Zhou Z, Qiu T, Zhou J, Zhang Z, Gong X, Zhang X, et al. Clinical features and current management experience in Gorham-Stout disease: a systematic review. Orphanet J Rare Dis. (2025) 20:134. doi: 10.1186/s13023-025-03649-9
2. Dellinger MT, Garg N, and Olsen BR. Viewpoints on vessels and vanishing bones in Gorham-Stout disease. Bone. (2014) 63:47–52. doi: 10.1016/j.bone.2014.02.011
3. Nikolaou VS, Chytas D, Korres D, and Efstathopoulos N. Vanishing bone disease (Gorham-Stout syndrome): A review of a rare entity. World J Orthop. (2014) 5:694–8. doi: 10.5312/wjo.v5.i5.694
4. Edwards JR, Williams K, Kindblom LG, Meis-Kindblom JM, Hogendoorn PC, Hughes D, et al. Lymphatics and bone. Hum Pathol. (2008) 39:49–55. doi: 10.1016/j.humpath.2007.04.022
5. Tie ML, Poland GA, and Rosenow EC 3rd. Chylothorax in Gorham’s syndrome. A common complication of a rare disease. Chest. (1994) 105:208–13. doi: 10.1378/chest.105.1.208
6. McDaniel CG, Adams DM, Steele KE, Hammill AM, Merrow AC, Crane JL, et al. Kaposiform lymphangiomatosis: Diagnosis, pathogenesis, and treatment. Pediatr Blood Cancer. (2023) 70:e30219. doi: 10.1002/pbc.30219
7. Ozeki M and Fukao T. Generalized lymphatic anomaly and gorham-stout disease: overview and recent insights. Adv Wound Care (New Rochelle). (2019) 8:230–45. doi: 10.1089/wound.2018.0850
8. Hu P, Yuan XG, Hu XY, Shen FR, and Wang JA. Gorham-Stout syndrome in mainland China: a case series of 67 patients and review of the literature. J Zhejiang Univ Sci B. (2013) 14:729–35. doi: 10.1631/jzus.B1200308
9. Lala S, Mulliken JB, Alomari AI, Fishman SJ, Kozakewich HP, and Chaudry G. Gorham-Stout disease and generalized lymphatic anomaly–clinical, radiologic, and histologic differentiation. Skeletal Radiol. (2013) 42:917–24. doi: 10.1007/s00256-012-1565-4
10. Zhang Y, Hao Q, Li X, Liu M, Sun X, and Wang R. Clinical and CT imaging features of different types of thoracic complex lymphatic anomaly. Quant Imaging Med Surg. (2025) 15:5567–81. doi: 10.21037/qims-24-1252
11. Dellinger MT GN, Ferry T, Kelly J, and Olsen BR. First international conference on generalized lymphatic anomaly and gorham-stout syndrome. IBMS BoneKEy. (2013). doi: 10.1038/bonekey.2013.210
12. Heyden G, Kindblom LG, and Nielsen JM. Disappearing bone disease. A Clin Histol Study J Bone Joint Surg Am. (1977) 59:57–61. doi: 10.2106/00004623-197759010-00009
13. Dickson GR, Hamilton A, Hayes D, Carr KE, Davis R, and Mollan RA. An investigation of vanishing bone disease. Bone. (1990) 11:205–10. doi: 10.1016/8756-3282(90)90215-K
14. Devlin RD, Bone HG 3rd, and Roodman GD. Interleukin-6: a potential mediator of the massive osteolysis in patients with Gorham-Stout disease. J Clin Endocrinol Metab. (1996) 81:1893–7. doi: 10.1210/jcem.81.5.8626854
15. Hominick D, Silva A, Khurana N, Liu Y, Dechow PC, Feng JQ, et al. VEGF-C promotes the development of lymphatics in bone and bone loss. Elife. (2018) 7. doi: 10.7554/eLife.34323
16. Yang Q, McHugh KP, Patntirapong S, Gu X, Wunderlich L, and Hauschka PV. VEGF enhancement of osteoclast survival and bone resorption involves VEGF receptor-2 signaling and beta3-integrin. Matrix Biol. (2008) 27:589–99. doi: 10.1016/j.matbio.2008.06.005
17. Glass GE, Chan JK, Freidin A, Feldmann M, Horwood NJ, and Nanchahal J. TNF-alpha promotes fracture repair by augmenting the recruitment and differentiation of muscle-derived stromal cells. Proc Natl Acad Sci U S A. (2011) 108:1585–90. doi: 10.1073/pnas.1018501108
18. Hu K and Olsen BR. Osteoblast-derived VEGF regulates osteoblast differentiation and bone formation during bone repair. J Clin Invest. (2016) 126:509–26. doi: 10.1172/JCI82585
19. McLaughlin KI, Milne TJ, Zafar S, Zanicotti DG, Cullinan MP, Seymour GJ, et al. The in vitro effect of VEGF receptor inhibition on primary alveolar osteoblast nodule formation. Aust Dent J. (2020) 65:196–204. doi: 10.1111/adj.12752
20. Rossi M, Buonuomo PS, Battafarano G, Conforti A, Mariani E, Algeri M, et al. Dissecting the mechanisms of bone loss in Gorham-Stout disease. Bone. (2020) 130:115068. doi: 10.1016/j.bone.2019.115068
21. Rossi M, Terreri S, Battafarano G, Rana I, Buonuomo PS, Di Giuseppe L, et al. Analysis of circulating osteoclast and osteogenic precursors in patients with Gorham-Stout disease. J Endocrinol Invest. (2024) 47:2775–84. doi: 10.1007/s40618-024-02365-8
22. Rossi M, Rana I, Buonuomo PS, Battafarano G, De Martino V, D’Agostini M, et al. Stimulation of treg cells to inhibit osteoclastogenesis in gorham-stout disease. Front Cell Dev Biol. (2021) 9:706596. doi: 10.3389/fcell.2021.706596
23. Pedroletti F, Rangarajan S, McCain JP, and Velez I. Conservative treatment of a pathologic fracture in a patient with Gorham-Stout disease. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. (2010) 109:e49–52. doi: 10.1016/j.tripleo.2009.08.045
24. Hagendoorn J, Yock TI, Borel Rinkes IH, Padera TP, and Ebb DH. Novel molecular pathways in Gorham disease: implications for treatment. Pediatr Blood Cancer. (2014) 61:401–6. doi: 10.1002/pbc.24832
25. Fontanesi J. Radiation therapy in the treatment of Gorham disease. J Pediatr Hematol Oncol. (2003) 25:816–7. doi: 10.1097/00043426-200310000-00016
26. Dunbar SF, Rosenberg A, Mankin H, Rosenthal D, and Suit HD. Gorham’s massive osteolysis: the role of radiation therapy and a review of the literature. Int J Radiat Oncol Biol Phys. (1993) 26:491–7. doi: 10.1016/0360-3016(93)90968-2
27. Heyd R, Micke O, Surholt C, Berger B, Martini C, Fuller J, et al. Radiation therapy for Gorham-Stout syndrome: results of a national patterns-of-care study and literature review. Int J Radiat Oncol Biol Phys. (2011) 81:e179–85. doi: 10.1016/j.ijrobp.2011.01.006
28. Duffy BM, Manon R, Patel RR, and Welsh JS. A case of Gorham’s disease with chylothorax treated curatively with radiation therapy. Clin Med Res. (2005) 3:83–6. doi: 10.3121/cmr.3.2.83
29. Bolling T, Willich N, and Ernst I. Late effects of abdominal irradiation in children: a review of the literature. Anticancer Res. (2010) 30:227–31.
30. Liu Y, Zhong DR, Zhou PR, Lv F, Ma DD, Xia WB, et al. Gorham-Stout disease: radiological, histological, and clinical features of 12 cases and review of literature. Clin Rheumatol. (2016) 35:813–23. doi: 10.1007/s10067-014-2780-2
31. Hammer F, Kenn W, Wesselmann U, Hofbauer LC, Delling G, Allolio B, et al. Gorham-Stout disease–stabilization during bisphosphonate treatment. J Bone Miner Res. (2005) 20:350–3. doi: 10.1359/JBMR.041113
32. Reid IR. Short-term and long-term effects of osteoporosis therapies. Nat Rev Endocrinol. (2015) 11:418–28. doi: 10.1038/nrendo.2015.71
33. Hagberg H, Lamberg K, and Astrom G. Alpha-2b interferon and oral clodronate for Gorham’s disease. Lancet. (1997) 350:1822–3. doi: 10.1016/S0140-6736(05)63639-2
34. Kose M, Pekcan S, Dogru D, Akyuz C, Ozcelik U, Ozsurekci Y, et al. Gorham-Stout Syndrome with chylothorax: successful remission by interferon alpha-2b. Pediatr Pulmonol. (2009) 44:613–5. doi: 10.1002/ppul.20849
35. Takahashi A, Ogawa C, Kanazawa T, Watanabe H, Suzuki M, Suzuki N, et al. Remission induced by interferon alfa in a patient with massive osteolysis and extension of lymph-hemangiomatosis: a severe case of Gorham-Stout syndrome. J Pediatr Surg. (2005) 40:E47–50. doi: 10.1016/j.jpedsurg.2004.11.015
36. Kuriyama DK, McElligott SC, Glaser DW, and Thompson KS. Treatment of Gorham-Stout disease with zoledronic acid and interferon-alpha: a case report and literature review. J Pediatr Hematol Oncol. (2010) 32:579–84. doi: 10.1097/MPH.0b013e3181edb464
37. Grunewald TGP, Damke L, Maschan M, Petrova U, Surianinova O, Esipenko A, et al. First report of effective and feasible treatment of multifocal lymphangiomatosis (Gorham-Stout) with bevacizumab in a child. Ann Oncol. (2010) 21:1733–4. doi: 10.1093/annonc/mdq331
38. Nir V, Guralnik L, Livnat G, Bar-Yoseph R, Hakim F, Ilivitzki A, et al. Propranolol as a treatment option in Gorham-Stout syndrome: a case report. Pediatr Pulmonol. (2014) 49:417–9. doi: 10.1002/ppul.22869
39. Brodszki N, Lansberg JK, Dictor M, Gyllstedt E, Ewers SB, Larsson MK, et al. A novel treatment approach for paediatric Gorham-Stout syndrome with chylothorax. Acta Paediatr. (2011) 100:1448–53. doi: 10.1111/j.1651-2227.2011.02361.x
40. Apsel Winger B, Dowd CF, Shimano KA, Devine WP, Mathes E, Frieden I, et al. Effective use of ALK inhibitors in EML4::ALK-positive lymphatic malformations. Pediatr Blood Cancer. (2025) 72:e31441. doi: 10.1002/pbc.31441
41. Ricci KW, Hammill AM, Mobberley-Schuman P, Nelson SC, Blatt J, Bender JLG, et al. Efficacy of systemic sirolimus in the treatment of generalized lymphatic anomaly and Gorham-Stout disease. Pediatr Blood Cancer. (2019) 66:e27614. doi: 10.1002/pbc.27614
42. Hopman SM, Van Rijn RR, Eng C, Bras J, Alders M, van der Horst CM, et al. PTEN hamartoma tumor syndrome and Gorham-Stout phenomenon. Am J Med Genet A. (2012) 158A:1719–23. doi: 10.1002/ajmg.a.35406
43. Tan MH, Mester J, Peterson C, Yang Y, Chen JL, Rybicki LA, et al. A clinical scoring system for selection of patients for PTEN mutation testing is proposed on the basis of a prospective study of 3042 probands. Am J Hum Genet. (2011) 88:42–56. doi: 10.1016/j.ajhg.2010.11.013
44. Tashiro H, Blazes MS, Wu R, Cho KR, Bose S, Wang SI, et al. Mutations in PTEN are frequent in endometrial carcinoma but rare in other common gynecological Malignancies. Cancer Res. (1997) 57:3935–40.
45. Bonneau D and Longy M. Mutations of the human PTEN gene. Hum Mutat. (2000) 16:109–22. doi: 10.1002/1098-1004(200008)16:2<109::AID-HUMU3>3.0.CO;2-0
46. Rasheed BK, Stenzel TT, McLendon RE, Parsons R, Friedman AH, Friedman HS, et al. PTEN gene mutations are seen in high-grade but not in low-grade gliomas. Cancer Res. (1997) 57:4187–90.
47. Gonzalez-Angulo AM, Ferrer-Lozano J, Stemke-Hale K, Sahin A, Liu S, Barrera JA, et al. PI3K pathway mutations and PTEN levels in primary and metastatic breast cancer. Mol Cancer Ther. (2011) 10:1093–101. doi: 10.1158/1535-7163.MCT-10-1089
48. Ge NL and Rudikoff S. Expression of PTEN in PTEN-deficient multiple myeloma cells abolishes tumor growth in vivo. Oncogene. (2000) 19:4091–5. doi: 10.1038/sj.onc.1203801
49. Hyun T, Yam A, Pece S, Xie X, Zhang J, Miki T, et al. Loss of PTEN expression leading to high Akt activation in human multiple myelomas. Blood. (2000) 96:3560–8.
50. Chen X, Bahrami A, Pappo A, Easton J, Dalton J, Hedlund E, et al. Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell Rep. (2014) 7:104–12. doi: 10.1016/j.celrep.2014.03.003
51. Freeman SS, Allen SW, Ganti R, Wu J, Ma J, Su X, et al. Copy number gains in EGFR and copy number losses in PTEN are common events in osteosarcoma tumors. Cancer. (2008) 113:1453–61. doi: 10.1002/cncr.23782
52. Suehara Y, Kitada R, Kamio S, Ogura K, Iwata S, Kobayashi E, et al. Analysis of cancer multigene panel testing for osteosarcoma in pediatric and adults using the center for cancer genomics and advanced therapeutics database in Japan. J Orthop Sci. (2024). doi: 10.1016/j.jos.2024.10.016
53. Xi Y and Chen Y. Oncogenic and therapeutic targeting of PTEN loss in bone Malignancies. J Cell Biochem. (2015) 116:1837–47. doi: 10.1002/jcb.25159
54. Stambolic V, Suzuki A, de la Pompa JL, Brothers GM, Mirtsos C, Sasaki T, et al. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell. (1998) 95:29–39. doi: 10.1016/S0092-8674(00)81780-8
55. Nicholson KM and Anderson NG. The protein kinase B/Akt signalling pathway in human Malignancy. Cell Signal. (2002) 14:381–95. doi: 10.1016/S0898-6568(01)00271-6
56. Rodriguez-Laguna L, Agra N, Ibanez K, Oliva-Molina G, Gordo G, Khurana N, et al. Somatic activating mutations in PIK3CA cause generalized lymphatic anomaly. J Exp Med. (2019) 216:407–18. doi: 10.1084/jem.20181353
57. Sugatani T, Alvarez U, and Hruska KA. PTEN regulates RANKL- and osteopontin-stimulated signal transduction during osteoclast differentiation and cell motility. J Biol Chem. (2003) 278:5001–8. doi: 10.1074/jbc.M209299200
58. Lorenz J, Richter S, Kirstein AS, Kolbig F, Nebe M, Schulze M, et al. Pten knockout in mouse preosteoblasts leads to changes in bone turnover and strength. JBMR Plus. (2024) 8:ziad016. doi: 10.1093/jbmrpl/ziad016
59. Kopec J, Salacinska-Los E, Orzechowska M, Sokolnicka M, Gawlowska-Marciniak A, and Przemyslaw P. mTOR pathway substrates present high activation in vascular malformations and significantly decrease with age. Diagn (Basel). (2023) 14. doi: 10.3390/diagnostics14010038
60. Adams DM, Trenor CC 3rd, Hammill AM, Vinks AA, Patel MN, Chaudry G, et al. Efficacy and safety of sirolimus in the treatment of complicated vascular anomalies. Pediatrics. (2016) 137:e20153257. doi: 10.1542/peds.2015-3257
61. Cavazos R, Patil MS, Gowda SH, Iacobas I, Rosenberg T, Fernandes CJ, et al. Sirolimus for vascular anomalies in the first year of life: a systematic review. J Perinatol. (2024) 44:1087–97. doi: 10.1038/s41372-024-01868-9
62. Apsel Winger B, Devine WP, Hsiao EC, Zapala M, Van Ziffle J, Gupta N, et al. EML4::ALK fusions in complex lymphatic malformations. Pediatr Blood Cancer. (2023) 70:e30516. doi: 10.22541/au.168076669.98356508/v1
63. Okamoto I and Nakagawa K. Echinoderm microtubule-associated protein-like 4-anaplastic lymphoma kinase-targeted therapy for advanced non-small cell lung cancer: molecular and clinical aspects. Cancer Sci. (2012) 103:1391–6. doi: 10.1111/j.1349-7006.2012.02327.x
64. Lei Y, Lei Y, Shi X, and Wang J. EML4-ALK fusion gene in non-small cell lung cancer. Oncol Lett. (2022) 24:277. doi: 10.3892/ol.2022.13397
65. Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. (2007) 448:561–6. doi: 10.1038/nature05945
66. Choi YL, Takeuchi K, Soda M, Inamura K, Togashi Y, Hatano S, et al. Identification of novel isoforms of the EML4-ALK transforming gene in non-small cell lung cancer. Cancer Res. (2008) 68:4971–6. doi: 10.1158/0008-5472.CAN-07-6158
67. Baba T, Inoue H, Matsuoka H, Kyakuno M, Yoshinaga Y, Takeguchi T, et al. A prompt diagnosis of ascites and dramatic effect of alectinib for advanced lung adenocarcinoma harboring EML4-ALK fusion: A case report. Intern Med. (2025). doi: 10.2169/internalmedicine.5397-25
68. Guo J, Zinner R, Zaorsky NG, Guo W, and Lu B. Systemic therapy for echinoderm microtubule-associated protein-like 4 anaplastic lymphoma kinase non-small cell lung cancer brain metastases. J Thorac Dis. (2016) 8:E1028–E31. doi: 10.21037/jtd.2016.09.09
69. Kijima T, Takeuchi K, Tetsumoto S, Shimada K, Takahashi R, Hirata H, et al. Favorable response to crizotinib in three patients with echinoderm microtubule-associated protein-like 4-anaplastic lymphoma kinase fusion-type oncogene-positive non-small cell lung cancer. Cancer Sci. (2011) 102:1602–4. doi: 10.1111/j.1349-7006.2011.01970.x
70. van der Wel JWT and de Langen AJ. Novel strategies for rare oncogenic drivers in non-small-cell lung cancer: An update from the 2024 Annual ESMO meeting. Lung Cancer. (2025) 204:108490. doi: 10.1016/j.lungcan.2025.108490
71. Oguz MM, Oguz B, Dogan V, Aydin B, Eyuboglu TS, Yesil S, et al. Cardiac tamponade in gorham-stout syndrome associated with GATA2 mutation. Indian J Pediatr. (2020) 87:239–40. doi: 10.1007/s12098-019-03174-1
72. Bresnick EH, Jung MM, and Katsumura KR. Human GATA2 mutations and hematologic disease: how many paths to pathogenesis? Blood Adv. (2020) 4:4584–92. doi: 10.1182/bloodadvances.2020002953
73. Mahamud MR, Geng X, Ho YC, Cha B, Kim Y, Ma J, et al. GATA2 controls lymphatic endothelial cell junctional integrity and lymphovenous valve morphogenesis through miR-126. Development. (2019) 146. doi: 10.1242/dev.184218
74. Watanabe-Asaka T, Hayashi M, Harada T, Uemura S, Takai J, Nakamura Y, et al. Perturbed collagen metabolism underlies lymphatic recanalization failure in Gata2 heterozygous deficient mice. J Biochem. (2024) 175:551–60. doi: 10.1093/jb/mvad122
75. Watanabe-Asaka T, Hayashi M, Uemura S, Takai J, Suzuki A, Moriguchi T, et al. GATA2 participates in the recanalization of lymphatic vessels after surgical lymph node extirpation. Genes Cells. (2021) 26:474–84. doi: 10.1111/gtc.12852
76. Tolkachov A, Fischer C, Ambrosi TH, Bothe M, Han CT, Muenzner M, et al. Loss of the hematopoietic stem cell factor GATA2 in the osteogenic lineage impairs trabecularization and mechanical strength of bone. Mol Cell Biol. (2018) 38. doi: 10.1128/MCB.00599-17
77. Nozawa A, Ozeki M, Niihori T, Suzui N, Miyazaki T, and Aoki Y. A somatic activating KRAS variant identified in an affected lesion of a patient with Gorham-Stout disease. J Hum Genet. (2020) 65:995–1001. doi: 10.1038/s10038-020-0794-y
78. Haigis KM. KRAS alleles: the devil is in the detail. Trends Cancer. (2017) 3:686–97. doi: 10.1016/j.trecan.2017.08.006
79. Aoki Y, Niihori T, Inoue S, and Matsubara Y. Recent advances in RASopathies. J Hum Genet. (2016) 61:33–9. doi: 10.1038/jhg.2015.114
80. Xiao S, Alshahrani M, Hu G, Tao P, and Verkhivker G. Accurate characterization of the allosteric energy landscapes, binding hotspots and long-range communications for KRAS complexes with effector proteins: integrative approach using microsecond molecular dynamics, deep mutational scanning of binding energetics and allosteric network modeling. bioRxiv. (2025). doi: 10.1101/2025.01.27.635141
81. Murugan AK, Grieco M, and Tsuchida N. RAS mutations in human cancers: Roles in precision medicine. Semin Cancer Biol. (2019) 59:23–35. doi: 10.1016/j.semcancer.2019.06.007
82. Prior IA, Lewis PD, and Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res. (2012) 72:2457–67. doi: 10.1158/0008-5472.CAN-11-2612
83. Rak J, Mitsuhashi Y, Bayko L, Filmus J, Shirasawa S, Sasazuki T, et al. Mutant ras oncogenes upregulate VEGF/VPF expression: implications for induction and inhibition of tumor angiogenesis. Cancer Res. (1995) 55:4575–80.
84. Ancrile B, Lim KH, and Counter CM. Oncogenic Ras-induced secretion of IL6 is required for tumorigenesis. Genes Dev. (2007) 21:1714–9. doi: 10.1101/gad.1549407
85. Dias Carvalho P, Guimaraes CF, Cardoso AP, Mendonca S, Costa AM, Oliveira MJ, et al. KRAS oncogenic signaling extends beyond cancer cells to orchestrate the microenvironment. Cancer Res. (2018) 78:7–14. doi: 10.1158/0008-5472.CAN-17-2084
86. Franco-Barrera MJ, Zavala-Cerna MG, Aguilar-Portillo G, Sanchez-Gomez DB, Torres-Bugarin O, Franco-Barrera MA, et al. Gorham-stout disease: a clinical case report and immunological mechanisms in bone erosion. Clin Rev Allergy Immunol. (2017) 52:125–32. doi: 10.1007/s12016-016-8594-z
87. Homayun-Sepehr N, McCarter AL, Helaers R, Galant C, Boon LM, Brouillard P, et al. KRAS-driven model of Gorham-Stout disease effectively treated with trametinib. JCI Insight. (2021) 6. doi: 10.1172/jci.insight.149831
88. Donigian S, Whiteway SL, Hipp SJ, Lybeck D, and Clark RO. Malignant giant cell tumor of bone with a KRAS G12V mutation. J Pediatr Hematol Oncol. (2022) 44:e268–e71. doi: 10.1097/MPH.0000000000002112
89. Uehara DT, Muramatsu T, Ishii S, Suzuki H, Fukushima K, Arasaki Y, et al. Identification of a biallelic missense variant in gasdermin D (c.823G > C, p.Asp275His) in a patient of atypical gorham-stout disease in a consanguineous family. JBMR Plus. (2023) 7:e10784. doi: 10.1002/jbm4.10784
90. Liu X, Xia S, Zhang Z, Wu H, and Lieberman J. Channelling inflammation: gasdermins in physiology and disease. Nat Rev Drug Discov. (2021) 20:384–405. doi: 10.1038/s41573-021-00154-z
91. Li Z, Ji S, Jiang ML, Xu Y, and Zhang CJ. The regulation and modification of GSDMD signaling in diseases. Front Immunol. (2022) 13:893912. doi: 10.3389/fimmu.2022.893912
92. Jiang N, An J, Yang K, Liu J, Guan C, Ma C, et al. NLRP3 inflammasome: A new target for prevention and control of osteoporosis? Front Endocrinol (Lausanne). (2021) 12:752546. doi: 10.3389/fendo.2021.752546
93. Tao Z, Wang J, Wen K, Yao R, Da W, Zhou S, et al. Pyroptosis in osteoblasts: A novel hypothesis underlying the pathogenesis of osteoporosis. Front Endocrinol (Lausanne). (2020) 11:548812. doi: 10.3389/fendo.2020.548812
94. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. (2016) 535:153–8. doi: 10.1038/nature18629
95. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. (2015) 526:660–5. doi: 10.1038/nature15514
96. Yebenes Mayordomo M, Al Shboul S, Gomez-Herranz M, Azfer A, Meynert A, Salter D, et al. Gorham-Stout case report: a multi-omic analysis reveals recurrent fusions as new potential drivers of the disease. BMC Med Genomics. (2022) 15:128. doi: 10.1186/s12920-022-01277-x
97. Li MH, Zhang HQ, Lu YJ, Gao P, Huang H, Hu YC, et al. Successful management of gorham-stout disease in scapula and ribs: A case report and literature review. Orthop Surg. (2018) 10:276–80. doi: 10.1111/os.12390
98. Gajewski D, Hennig AF, Grun R, Siggelkow H, Vishnolia S, Bastian L, et al. Paradoxical combination of osteosclerosis and osteopenia in an adult woman with biallelic TNFRSF11A loss-of-function variants escaping nonsense-mediated decay. JBMR Plus. (2025) 9:ziae179. doi: 10.1093/jbmrpl/ziae179
99. Hughes AE, Ralston SH, Marken J, Bell C, MacPherson H, Wallace RG, et al. Mutations in TNFRSF11A, affecting the signal peptide of RANK, cause familial expansile osteolysis. Nat Genet. (2000) 24:45–8. doi: 10.1038/71667
100. Hosokawa N, Sasaki T, Iemura S, Natsume T, Hara T, and Mizushima N. Atg101, a novel mammalian autophagy protein interacting with Atg13. Autophagy. (2009) 5:973–9. doi: 10.4161/auto.5.7.9296
101. Mercer CA, Kaliappan A, and Dennis PB. A novel, human Atg13 binding protein, Atg101, interacts with ULK1 and is essential for macroautophagy. Autophagy. (2009) 5:649–62. doi: 10.4161/auto.5.5.8249
102. Cox ML, Evans JM, Davis AG, Guo LT, Levy JR, Starr-Moss AN, et al. Exome sequencing reveals independent SGCD deletions causing limb girdle muscular dystrophy in Boston terriers. Skelet Muscle. (2017) 7:15. doi: 10.1186/s13395-017-0131-0
103. Younus M, Ahmad F, Malik E, Bilal M, Kausar M, Abbas S, et al. SGCD homozygous nonsense mutation (p.Arg97(*)) causing limb-girdle muscular dystrophy type 2F (LGMD2F) in a consanguineous family, a case report. Front Genet. (2018) 9:727. doi: 10.3389/fgene.2018.00727
104. Thapa S, Elhadidy S, and Asakura A. Vascular therapy for Duchenne muscular dystrophy (DMD). Fac Rev. (2023) 12:3. doi: 10.12703/r/12-3
105. Karar J and Maity A. PI3K/AKT/mTOR pathway in angiogenesis. Front Mol Neurosci. (2011) 4:51. doi: 10.3389/fnmol.2011.00051
Keywords: Gorham-Stout disease, rare disease, genetic characterization, genetic variants, molecular pathways
Citation: Pagliarosi O, Pepe J, Del Fattore A and Rossi M (2025) Exploring the genetic alterations of Gorham-Stout disease. Front. Endocrinol. 16:1654497. doi: 10.3389/fendo.2025.1654497
Received: 26 June 2025; Accepted: 01 August 2025;
Published: 19 August 2025.
Edited by:
Fabio Vescini, Azienda Sanitaria Universitaria Integrata di Udine, ItalyReviewed by:
Weiyang Zhong, First Affiliated Hospital of Chongqing Medical University, ChinaTaccyanna Mikulski Ali, Igenomix Brazil, Brazil
Copyright © 2025 Pagliarosi, Pepe, Del Fattore and Rossi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrea Del Fattore, YW5kcmVhLmRlbGZhdHRvcmVAb3BiZy5uZXQ=