 Zhuhai Shao
Zhuhai Shao Lili Xu
Lili Xu- Department of Endocrinology, The Affiliated Hospital of Qingdao University, Qingdao, China
This paper presents the diagnostic and therapeutic course of a 26-year-old male patient with maturity-onset diabetes of the young type 6 (MODY6) complicated by diabetic ketosis, resulting from a heterozygous neurogenic differentiation factor 1 (NEUROD1) c.-108G>C mutation. The patient was admitted due to “dry mouth, polydipsia, and polyuria lasting for 2 months.” The diagnosis of MODY6 was established based on blood glucose levels, glycosylated hemoglobin results, familial co-segregation of the variant (maternally inherited), and genetic sequencing data. This study analyzes the similarities and discrepancies between this case and classic MODY6, highlights the diagnostic significance of genetic testing in atypical cases, and puts forward the indications for genetic testing in clinically suspected MODY cases. Following individualized therapy with saxagliptin and acarbose, the patient achieved stable blood glucose control without insulin after 6 months, with partial recovery of islet function. This case supports that NEUROD1 mutations may retain incretin responsiveness, expanding therapeutic options for MODY6.
1 Introduction
Maturity-onset diabetes of the young (MODY) comprises a genetically heterogeneous group of monogenic forms of diabetes, characterized by autosomal dominant inheritance, early onset—typically before the age of 25—and primary β-cell dysfunction (1, 2). To date, 14 distinct MODY subtypes have been identified, each associated with specific genetic mutations and clinical phenotypes (3). Among these, MODY6, caused by heterozygous mutations in the neurogenic differentiation factor 1 (NEUROD1) gene, is a rare subtype that exhibits considerable phenotypic variability, ranging from mild hyperglycemia to ketosis-prone diabetes (4, 5).
The NEUROD1 gene, located on chromosome 2q32, encodes a basic helix-loop-helix (bHLH) transcription factor that plays a pivotal role in pancreatic β-cell development and the regulation of insulin secretion (6). Experimental studies have shown that NEUROD1 modulates the expression of SUR1 (sulfonylurea receptor 1), a critical subunit of β-cell ATP-sensitive K+ (KATP) channels (composed of SUR1 and Kir6.2). The SUR1-Kir6.2 complex mediates glucose-dependent insulin secretion (GSIS): increased glucose metabolism raises intracellular ATP levels, closing KATP channels and triggering Ca2+ influx to promote insulin release. Thus, NEUROD1 dysfunction links to impaired GSIS by downregulating SUR1 expression (6, 7). While most reported NEUROD1 mutations cluster within the transactivation domain and are typically associated with classic MODY6 phenotypes—characterized by non-ketotic, insulin-independent diabetes (4)—emerging evidence indicates that variants in the 5′ untranslated region (5′UTR) may disrupt transcriptional regulation, potentially resulting in atypical clinical presentations, including diabetic ketosis (8, 9).
Specifically, Su et al. (9) demonstrated that dinucleotide composition in the UTR region is a major determinant of RNA stability; variants altering this composition can reduce mRNA half-life and subsequent protein expression, which may explain why 5’UTR variants of NEUROD1 differ from transactivation domain mutations in phenotypic severity. Additionally, Horikawa et al. (5) reported that Japanese patients with NEUROD1-deficient MODY6 already exhibited heterogeneous β-cell dysfunction, with some cases showing near-normal insulin secretion and others presenting with severe impairment—hinting that NEUROD1 variant location (e.g., coding vs. non-coding regions) contributes to phenotypic variability. In our case, the NEUROD1 c.-108G>C variant in the 5’UTR may further expand this heterogeneity by disrupting transcriptional initiation or mRNA stability, ultimately leading to ketosis—a phenotype rarely reported in classic MODY6.
Building on the aforementioned evidence that 5’UTR variants of NEUROD1 may drive atypical MODY6 phenotypes, our study provides the first clinical evidence linking the NEUROD1 c.-108G>C 5’UTR variant to ketosis-prone MODY6—a presentation that challenges the traditional view of MODY6 as non-ketotic. Moreover, the patient’s positive response to DPP-4i therapy offers new insights into therapeutic options for NEUROD1-associated MODY6, which has previously been considered primarily insulin dependent. Here, we report a novel association between the NEUROD1 c.-108G>C variant and ketosis-prone MODY6, characterized by a favorable therapeutic response to dipeptidyl peptidase-4 inhibitor (DPP-4i) therapy. These findings highlight the importance of expanding genetic testing criteria to encompass atypical presentations of diabetes.
2 Case presentation
2.1 Clinical history
A 26-year-old unmarried male was admitted to the hospital with complaints of dry mouth, polydipsia, and polyuria persisting for two months. These symptoms began following a febrile episode two months prior to admission and were accompanied by foamy urine and an unintentional weight loss of 7kg. Outpatient evaluation revealed a fasting blood glucose level of 11.84 mmol/L. Regarding family history, his father was non-diabetic, whereas his mother, maternal aunt, and maternal grandfather were all diagnosed with diabetes, suggesting an autosomal dominant pattern of inheritance (Figure 1).
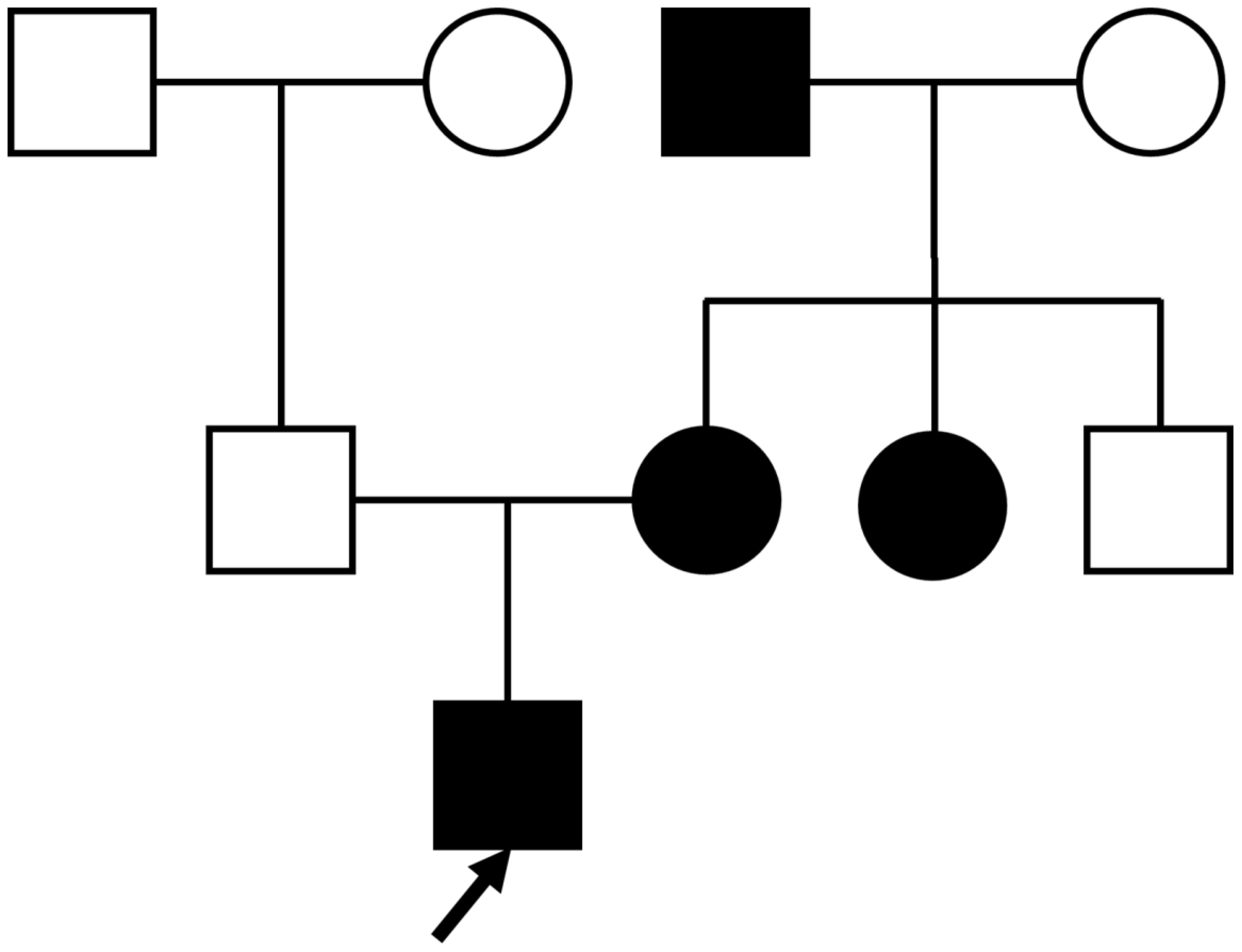
Figure 1. Proband’s family tree: Squares represent males, and circles represent females. The black symbol represents individuals with diabetes; the blank symbol represents normal individuals. The arrow indicates the proband.
2.2 Physical examination
On admission, the patient was afebrile (36.5°C) with a respiratory rate of 20 breaths per minute, a pulse of 111 beats per minute, and a blood pressure of 145/87 mmHg. Anthropometric assessment revealed a height of 186 cm, weight of 87.50 kg, body mass index (BMI) of 25.30 kg/m², waist circumference of 88 cm, and hip circumference of 106 cm. A comprehensive physical examination, including evaluation of the cardiovascular, respiratory, abdominal, and neurological systems, revealed no significant abnormalities.
2.3 Laboratory findings
The patient’s glycated hemoglobin (HbA1c) level was elevated at 12.80%. An insulin release test revealed a fasting insulin level of 0.98 μIU/ml (reference range: 2.60–24.90 μIU/ml), with 1h and 2h postprandial insulin levels of 3.90 μIU/ml and 6.82 μIU/ml, respectively. A C-peptide release test showed markedly reduced fasting C-peptide at 0.28 ng/ml (reference range: 1.10–4.40 ng/ml), with postprandial 1h and 2h C-peptide levels of 0.67 ng/ml and 0.92 ng/ml, respectively. Testing for islet autoantibodies, including glutamic acid decarboxylase autoantibodies, was negative. Blood lipid profile results were within normal limits: total cholesterol (TC) 4.63 mmol/L (reference value: 2.90–5.68 mmol/L), triglycerides (TG) 0.55 mmol/L (reference value: 0.00–1.70 mmol/L), low-density lipoprotein cholesterol (LDL-C) 3.00 mmol/L (reference value: 0.00–3.37 mmol/L), and high-density lipoprotein cholesterol (HDL-C) 0.95 mmol/L (reference value: 0.91–2.06 mmol/L). Liver function indicators were normal: alanine aminotransferase (ALT) 28 U/L (reference value: 9–50 U/L) and aspartate aminotransferase (AST) 25 U/L (reference value: 15–40 U/L). Renal function was also normal: serum creatinine (Scr) 53 μmol/L (reference value: 57–97 μmol/L), uric acid (UA) 257 μmol/L (reference value: 293–430 μmol/L), and blood urea nitrogen (BUN) 2.34 mmol/L (reference value: 3.10–8.00 mmol/L). The urinary albumin-to-creatinine ratio (UACR) was 4.61 mg/g. Urinalysis showed 3+ ketone bodies and 4+ glucose. Arterial blood gas (ABG) analysis revealed no metabolic acidosis, with pH 7.43 (reference value: 7.35–7.45), partial pressure of arterial oxygen (PaO2) 87 mmHg (reference value: 80–100 mmHg), partial pressure of arterial carbon dioxide (PCO2) 40 mmHg (reference value: 35–45 mmHg), standard bicarbonate (SB) 26.4 mmol/L (reference value: 22–27 mmol/L), and base excess (BE) 2.2 mmol/L (reference value: −3.0–3.0 mmol/L).
2.4 Genetic analysis
Whole-exome sequencing identified a heterozygous variant in the NEUROD1 gene (NM_002500.5): c.-108G>C, located in the 5′UTR (Figure 2). According to American College of Medical Genetics and Genomics (ACMG) guidelines (10), this variant was classified as of “uncertain significance” (PM2_Supporting: absent from population databases; PP1_Moderate: co-segregation with diabetes within the maternal lineage). Sanger sequencing confirmed the presence of the variant in both the proband and his diabetic mother, while it was absent in his non-diabetic father. No pathogenic variants were identified in other MODY-associated genes, including GCK and HNF1A.
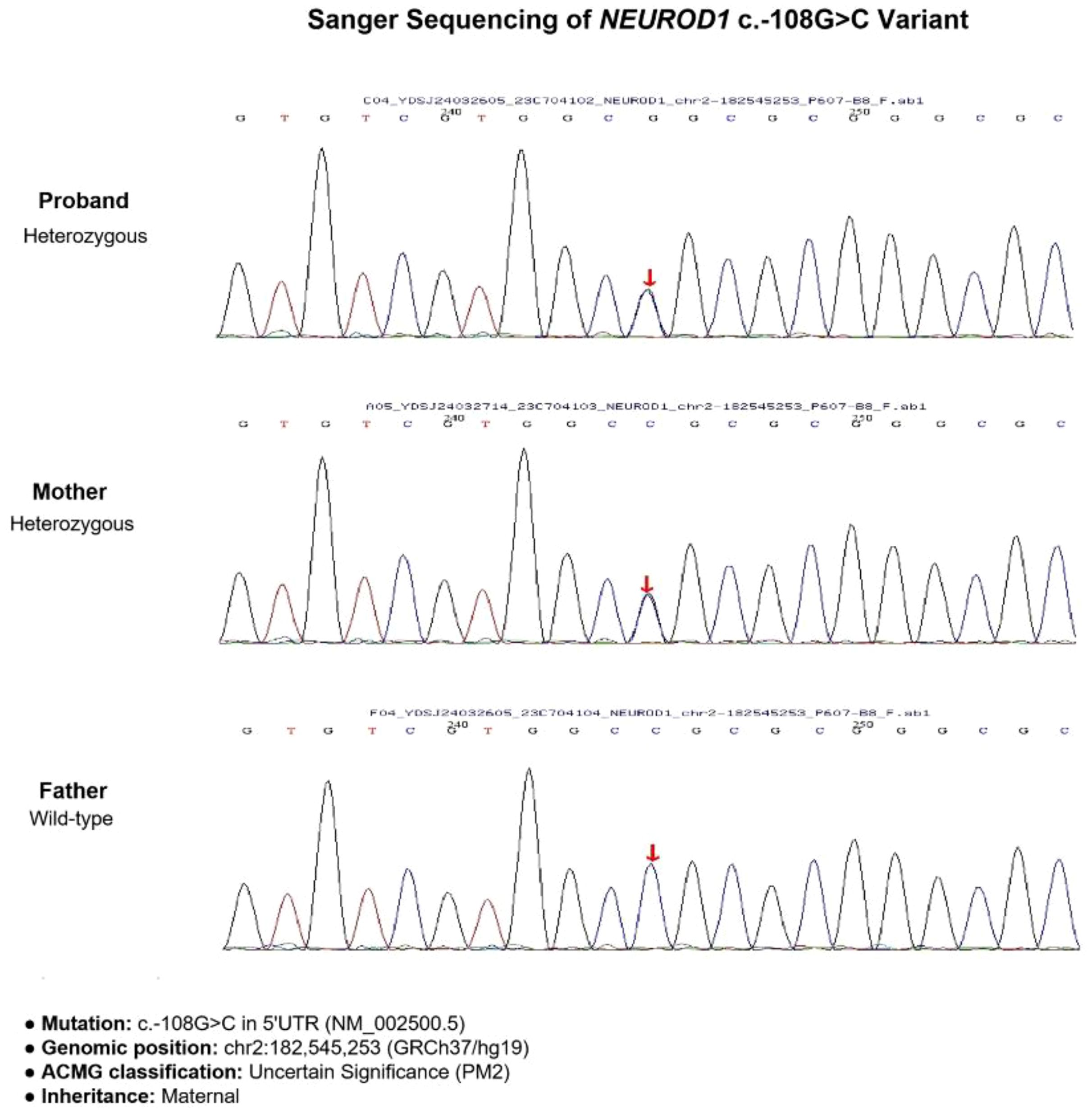
Figure 2. Gene sequencing results of the proband and his parents. The proband and his mother both carried the heterozygous variant.
2.5 Diagnosis and treatment
Based on the clinical presentation, autosomal dominant family history, and genetic findings, a diagnosis of MODY6 was established. After admission, the patient received intravenous fluid replacement and continuous subcutaneous insulin pump therapy with insulin lispro injection. The insulin regimen was set as follows: basal rate 16.7 U/day, with 5 U before breakfast, 4 U before lunch, and 5 U before dinner, totaling 30.7 U/day. The dosage was dynamically adjusted according to capillary blood glucose monitoring results (6–8 measurements daily). Concomitant medication included saxagliptin 5 mg once daily and acarbose 50 mg three times daily (to be chewed with meals). After urinary ketone bodies turned negative, the patient was discharged. One month post-discharge, partial recovery of islet function was observed, with a fasting C-peptide level of 0.58 ng/ml. At three months, fasting C-peptide had increased to 1.16 ng/ml, and HbA1c had decreased to 6.2%. By 6 months, insulin therapy was discontinued, and glycemic control remained stable without recurrence of ketosis. The detailed changes in the patient’s clinical indicators and treatment adjustments during the entire course are summarized in Table 1.
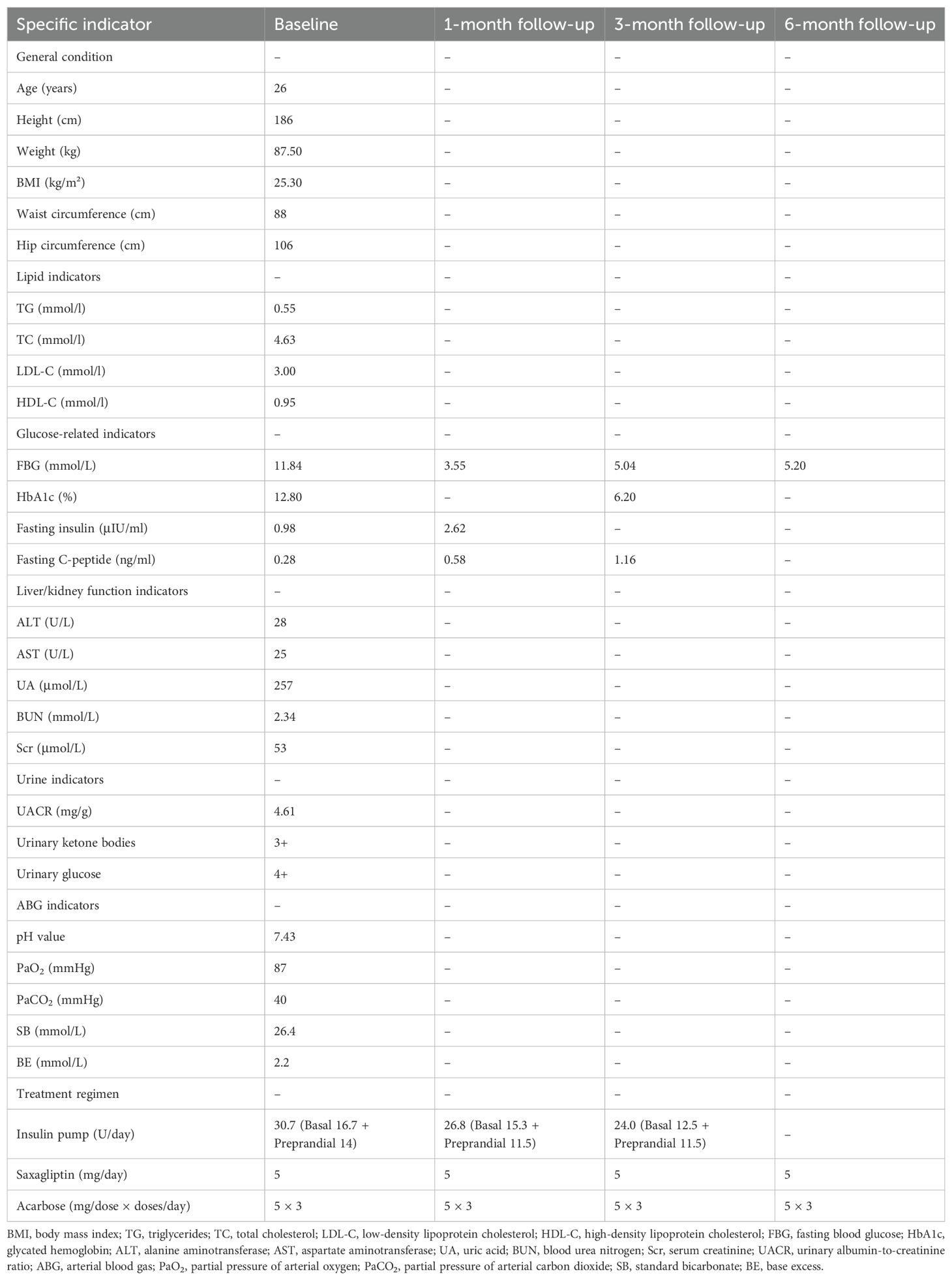
Table 1. Clinical indicators and treatment regimens of the patient at baseline and follow-up (1, 3, and 6 months).
3 Materials and methods
3.1 Study subjects
This study included one 26-year-old male patient diagnosed with MODY6 and his family members (father and mother). The patient was admitted to the Department of Endocrinology, Affiliated Hospital of Qingdao University, due to typical diabetic symptoms combined with a family history of diabetes. All participants provided written informed consent, and the study was approved by the Medical Ethics Committee of the Affiliated Hospital of Qingdao University (Ethical Approval No. QYFYWZLL30623).
3.2 Laboratory tests
Fasting blood glucose (FBG) was measured using the glucose oxidase method. HbA1c was determined via high-performance liquid chromatography. Serum samples (3 ml venous blood, centrifuged at 3000 rpm for 10min for separation) were used to detect insulin and C-peptide levels by chemiluminescent immunoassay. Glutamic acid decarboxylase autoantibodies were tested using enzyme-linked immunosorbent assay. Blood lipid indicators, liver function, and renal function were measured on an automatic biochemical analyzer with corresponding commercial kits. Urinalysis was performed using a urine chemistry analyzer to detect ketone bodies and glucose, and ABG analysis was conducted with a blood gas analyzer.
3.3 Genetic analysis
Peripheral venous blood (2 ml) was collected from the proband and his father and mother. Whole-exome sequencing (WES) was performed by MyGenostics (Beijing) Co., Ltd., with an average sequencing depth of 265.33× and a target region coverage of 99.79%. Sanger sequencing was used for family co-segregation verification of the identified variant. Variant pathogenicity was classified according to the ACMG guidelines.
4 Discussion
This study describes a patient with MODY6 presenting with diabetic ketosis, carrying a heterozygous NEUROD1 c.-108G>C variant. Although classified as “Uncertain Significance” by ACMG criteria (10, 11) (PM2_Supporting: absent in population databases; PP1_Moderate: maternal cosegregation), multiple lines of evidence support its pathogenicity. The proband exhibited classic MODY6 features (early-onset, non-autoimmune diabetes, β-cell dysfunction) along with atypical ketosis, consistent with previous reports associating NEUROD1 transactivation domain mutations with ketotic episodes (4, 5). Pedigree analysis confirmed the mutation was inherited from the diabetic mother while the non-diabetic father was wild-type (Figure 2), following an autosomal dominant pattern (3). Particularly, the c.-108G>C variant is located in the 5’UTR region, which contains key elements for transcriptional regulation, potentially affecting the expression regulation of NEUROD1 (12).
While functional studies are required to fully elucidate the molecular consequences, the observed clinical correlation in this family provides strong evidence supporting the disease association. These findings diverge from the classic MODY6 phenotype—characterized by the absence of ketosis and insulin independence for over five years after diagnosis—thereby underscoring the phenotypic heterogeneity of NEUROD1 mutations (3, 13). Based on the genotype-phenotype correlation and familial evidence, we propose re-evaluating the ACMG classification (PM1+PM2+PP1+PP4) to “Likely Pathogenic” for similar cases with strong phenotypic support.
4.1 Rationale for individualized treatment selection
The therapeutic strategy for this patient was formulated based on the integration of genetic background, clinical manifestations, and pharmacodynamic evidence, reflecting the principle of precision medicine in MODY management.
Selection of saxagliptin as the preferred DPP-4 inhibitor was supported by three key considerations. First, pharmacokinetic data indicate that saxagliptin has a favorable safety profile in Asian populations with minimal drug-drug interactions, which is critical for long-term maintenance therapy (14). The patient, a 26-year-old Chinese male with normal baseline liver and renal function, required a treatment option with low long-term organ toxicity, and saxagliptin’s predominantly renal excretion (with no need for dose adjustment in normal renal function) met this requirement. Second, the mechanism of DPP-4i, which involves upregulating anti-apoptotic factors to enhance β-cell survival and prolonging the half-life of endogenous incretins (e.g., GLP-1 and GIP) to stimulate insulin secretion, is highly congruent with the core objective of preserving residual β-cell function in NEUROD1-deficient MODY6 (15, 16). Given that NEUROD1 mutations impair β-cell development and insulin secretion rather than causing irreversible β-cell destruction, promoting β-cell survival is particularly relevant for this patient. Third, saxagliptin displays greater in vitro DPP-4 inhibitory potency than some other DPP-4 inhibitors (e.g., sitagliptin), rendering it more suitable for patients with severe baseline β-cell dysfunction, as illustrated by our patient (fasting C-peptide 0.28 ng/ml) (17).
Adjunctive use of acarbose was justified by the patient’s prominent postprandial hyperglycemia and the need for rapid glycemic control during ketosis resolution. Acarbose acts locally in the gastrointestinal tract to delay carbohydrate absorption (by inhibiting α-glucosidase) without systemic absorption, avoiding potential interference with saxagliptin’s mechanism of action (prolonging incretin half-life via DPP-4 inhibition). This combination addressed two key clinical needs simultaneously: acarbose rapidly reduced postprandial glucose spikes to prevent further ketosis exacerbation, while saxagliptin targeted the underlying β-cell dysfunction.
4.2 Clinical implications for MODY6 management
Current clinical guidelines for monogenic diabetes typically suggest insulin as the primary treatment for MODY6, based chiefly on observational data from cases with NEUROD1 coding region mutations (18, 19). However, this case demonstrates that a “short-term intensive insulin + long-term DPP-4 inhibitor” regimen can achieve insulin independence, with stable glycemic control and partial β-cell recovery. We propose that, for MODY6 patients with NEUROD1 variants (especially 5’UTR variants) and ketosis (without severe metabolic acidosis), DPP-4 inhibitors may be considered as maintenance therapy after short-term insulin to correct ketosis.
This case carries important implications for refining the indications for MODY genetic testing. Genetic evaluation should be considered in patients with antibody-negative diabetes who exhibit any of the following features (20): (1) unexplained ketosis, (2) early onset diabetes (even beyond age 25) accompanied by a family history suggestive of MODY, and (3) clinical features indicative of monogenic diabetes, even in the presence of variants classified as “uncertain significance” by ACMG criteria. Notably, the patient’s mother, who carried the same NEUROD1 mutation, exhibited a milder diabetic phenotype, suggesting variable expressivity. This highlights the importance of comprehensive genotypic and phenotypic evaluation of family members when assessing potential monogenic diabetes.
4.3 Study limitations
The limitations of this study include (1) the lack of functional validation and bioinformatics analysis (e.g., luciferase reporter assay) for the NEUROD1 c.-108G>C variant, which would help confirm its impact on transcriptional activity, and (2) short-term follow-up (6 months), which cannot confirm the long-term durability of glycemic control and β-cell recovery—future work will extend follow-up to 12 and 24 months to monitor for potential relapse of ketosis or β-cell function decline. Future research should incorporate in vitro experiments to assess the impact of the c.-108G>C variant on NEUROD1 transcriptional activity, as well as expand sample sizes to evaluate the long-term efficacy of DPP-4i in patients with NEUROD1-MODY. Furthermore, testing additional family members would enhance the robustness of the genotype-phenotype correlation analysis.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by The Ethics Committee of Qingdao University Affiliated Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
ZS: Writing – original draft. LX: Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
We thank the patient and his family for participating in this study, and the Department of Endocrinology, Affiliated Hospital of Qingdao University for technical support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Urakami T. Maturity-onset diabetes of the young (MODY): current perspectives on diagnosis and treatment. Diabetes Metab Syndr Obes. (2019) 12:1047–56. doi: 10.2147/DMSO.S179793
2. Nkonge KM, Nkonge DK, and Nkonge TN. The epidemiology, molecular pathogenesis, diagnosis, and treatment of maturity-onset diabetes of the young (MODY). Clin Diabetes Endocrinol. (2020) 6:20. doi: 10.1186/s40842-020-00112-5
3. El Nahas R, Missous G, Al-Tarakji M, Said-Ghali M, Hussain K, van Panhuys N, et al. Diagnosis and phenotypes of idiopathic diabetes: A systematic review. Endocr Pract. (2025) 31(10):1329–38. doi: 10.1016/j.eprac.2025.06.009
4. Li Y, Wen Q, Shao H, Hao M, Sun Y, and Liu T. A novel nonsense mutation c.747C>G in the NEUROD1 gene detected within a Chinese family affected by maturity-onset diabetes of the young type 6. J Diabetes. (2024) 16:e13607. doi: 10.1111/1753-0407.13607
5. Horikawa Y, Enya M, Mabe H, Fukushima K, Takubo N, Ohashi M, et al. NEUROD1-deficient diabetes (MODY6): Identification of the first cases in Japanese and the clinical features. Pediatr Diabetes. (2018) 19:236–42. doi: 10.1111/pedi.12553
6. Romer AI, Singer RA, Sui L, Egli D, and Sussel L. Murine perinatal β-cell proliferation and the differentiation of human stem cell-derived insulin-expressing cells require NEUROD1. Diabetes. (2019) 68:2259–71. doi: 10.2337/db19-0117
7. Pipatpolkai T, Usher S, Stansfeld PJ, and Ashcroft FM. New insights into K(ATP) channel gene mutations and neonatal diabetes mellitus. Nat Rev Endocrinol. (2020) 16:378–93. doi: 10.1038/s41574-020-0351-y
8. Sagen JV, Baumann ME, Salvesen HB, Molven A, Søvik O, and Njølstad PR. Diagnostic screening of NEUROD1 (MODY6) in subjects with MODY or gestational diabetes mellitus. Diabetes Med. (2005) 22:1012–5. doi: 10.1111/j.1464-5491.2005.01565.x
9. Su JY, Wang YL, Hsieh YT, Chang YC, Yang CH, Kang Y, et al. Multiplexed assays of human disease-relevant mutations reveal UTR dinucleotide composition as a major determinant of RNA stability. Elife. (2025) 13:RP97682. doi: 10.7554/eLife.97682.2
10. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
11. Wilcox EH, Webb RF, Tshering KC, Hughes MY, Cavé H, DiStefano MT, et al. Updated ACMG/AMP specifications for variant interpretation and gene curations from the ClinGen RASopathy expert panels. Genet Med Open. (2025) 3:103430. doi: 10.1016/j.gimo.2025.103430
12. Bohuslavova R, Fabriciova V, Smolik O, Lebrón-Mora L, Abaffy P, Benesova S, et al. NEUROD1 reinforces endocrine cell fate acquisition in pancreatic development. Nat Commun. (2023) 14:5554. doi: 10.1038/s41467-023-41306-6
13. Chua C, Tan CSH, Lim SC, and Vasanwala RF. A unique phenotype of maturity-onset diabetes of the young with a novel disease-causing insulin gene variant. JCEM Case Rep. (2025) 3:luae230. doi: 10.1210/jcemcr/luae230
14. Pan CY, Yang W, Tou C, Gause-Nilsson I, and Zhao J. Efficacy and safety of saxagliptin in drug-naïve Asian patients with type 2 diabetes mellitus: a randomized controlled trial. Diabetes Metab Res Rev. (2012) 28:268–75. doi: 10.1002/dmrr.1306
15. Yan X, Li X, Liu B, Huang J, Xiang Y, Hu Y, et al. Combination therapy with saxagliptin and vitamin D for the preservation of β-cell function in adult-onset type 1 diabetes: a multi-center, randomized, controlled trial. Signal Transduct Target Ther. (2023) 8:158. doi: 10.1038/s41392-023-01369-9
16. Nauck MA, Quast DR, Wefers J, and Pfeiffer AFH. The evolving story of incretins (GIP and GLP-1) in metabolic and cardiovascular disease: A pathophysiological update. Diabetes Obes Metab. (2021) 23 Suppl 3:5–29. doi: 10.1111/dom.14496
17. Wang A, Dorso C, Kopcho L, Locke G, Langish R, Harstad E, et al. Potency, selectivity and prolonged binding of saxagliptin to DPP4: maintenance of DPP4 inhibition by saxagliptin in vitro and ex vivo when compared to a rapidly-dissociating DPP4 inhibitor. BMC Pharmacol. (2012) 12:2. doi: 10.1186/1471-2210-12-2
18. Greeley SAW, Polak M, Njølstad PR, Barbetti F, Williams R, Castano L, et al. ISPAD Clinical Practice Consensus Guidelines 2022: The diagnosis and management of monogenic diabetes in children and adolescents. Pediatr Diabetes. (2022) 23:1188–211. doi: 10.1111/pedi.13426
19. Zečević K, Volčanšek Š, Katsiki N, Rizzo M, Milardović TM, Stoian AP, et al. Maturity-onset diabetes of the young (MODY) - in search of ideal diagnostic criteria and precise treatment. Prog Cardiovasc Dis. (2024) 85:14–25. doi: 10.1016/j.pcad.2024.03.004
Keywords: maturity-onset diabetes of the young, MODY6, NEUROD1 gene, diabetic ketosis, DPP-4 inhibitor
Citation: Shao Z and Xu L (2025) Case Report: NEUROD1 c.-108G>C mutation in a ketosis-prone MODY6 patient: implications for genetic testing and DPP-4 inhibitor therapy. Front. Endocrinol. 16:1673765. doi: 10.3389/fendo.2025.1673765
Received: 26 July 2025; Accepted: 20 October 2025;
Published: 03 November 2025.
Edited by:
Roma Patel, Albert Einstein College of Medicine, United StatesReviewed by:
Jayvadan Jayantilal Vaishnav, Parul University, IndiaNishant Parmar, Maharaja Sayajirao University of Baroda, India
Copyright © 2025 Shao and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lili Xu, cWRmeXhsbEBxZHUuZWR1LmNu