 Angel A. Lee
Angel A. Lee Laura J. Den Hartigh
Laura J. Den Hartigh- 1Department of Medicine: Metabolism, Endocrinology, and Nutrition, University of Washington, Seattle, WA, United States
- 2UW Medicine Diabetes Institute, Seattle, WA, United States
The aged population, expected to double by 2050, makes up a large proportion of people living with metabolic disease. Obesity rates in the elderly are rapidly increasing, with estimates that nearly 40% of men and women over the age of 60 are classified as obese. White adipose tissue (WAT) is a highly metabolically active organ that undergoes significant changes during both obesity and aging, and metabolic dysfunction in WAT is a major cause for elevated diabetes risk. A marked difference in fat distribution is often reported between men and women. Many studies suggest that pre-menopausal women are protected from the accumulation of visceral adiposity due to gonadal estrogen, which exerts cardiometabolic benefits. Men with obesity harbor a disproportionately higher volume of intra-abdominal fat than premenopausal age-matched women with obesity, an effect that is negated by menopause as women begin to gain intra-abdominal fat. Post-menopausal women are at increased risk of developing diabetes, which can be mitigated by estrogen replacement therapy, suggesting an important role for sex steroids in diabetes risk. In addition to being highly responsive to gonadal estrogens, WAT has the capacity to convert androgens into estrogens, which may similarly impact WAT distribution and metabolism. Estrogens, comprised primarily of estrone (E1) and estradiol (E2) within WAT, are biosynthesized from circulating androgens androstenedione (A4) and testosterone (T) by aromatase (CYP19A1), which is highly expressed in human and mouse adipose tissue. In post-menopausal women, WAT becomes the predominant source of estrogen production, with age-associated increases in WAT aromatase expression that are mirrored by obesity. In contrast to ovarian estrogen production, in which E2 is the predominant estrogen type, E1 tends to be the predominant estrogen post-menopause. To date, little is known about WAT-derived estrogens and their impact on metabolic health, but emerging evidence suggests that increased E1 levels may contribute to metabolic dysfunction in aging. This review will introduce known sex differences in adipose metabolism associated with aging, obesity, and diabetes, and discuss the impact of WAT-derived sex hormones on local and systemic metabolism.
1 Introduction
The global rise in obesity has become one of the most pressing health challenges of our time as a leading contributor to global morbidity and mortality. Between 1975 and 2014, the average body weight of adults worldwide dramatically increased, with women gaining an equivalent of an additional 1.5 kg per decade (1). The striking rise in body mass index (BMI) is a comorbidity to other metabolic diseases, including type 2 diabetes (T2D), which is driven in part by dysregulated white adipose tissue (WAT) function. WAT is a highly metabolically active organ that undergoes significant physiological changes during obesity as well as aging. WAT can expand via increased recruitment of pre-adipocytes, hence increasing the total number of adipocytes (hyperplasia), or via the enlargement of existing adipocytes (hypertrophy). WAT exists as many depots distributed throughout the body, and in general are categorized as either subcutaneous (sWAT) or visceral/omental (vWAT). sWAT is the most abundant depot in healthy people and contributes to metabolic health (2, 3). By contrast, excess vWAT is associated with metabolic syndrome, which includes the constellation of type 2 diabetes, hypertension, insulin resistance, dyslipidemia, systemic inflammation, and atherosclerosis (4, 5). During periods of nutrient excess (i.e. obesity), WAT may become severely dysfunctional due to maximization of WAT expansion potential, resulting in ectopic fat accumulation and lipotoxicity in other organs (2). In women, the transition to menopause significantly increases metabolic risk, with postmenopausal women facing a five times greater risk of central obesity compared to premenopausal women (6–8). This significant shift in fat distribution from subcutaneous regions in the hips and thighs to visceral depots is attributed to the loss of ovarian sex steroid production, in which case, adipose tissue then becomes the primary site of estrogen production (7, 9).
The gonads are classically thought to contribute to the majority of circulating sex hormone levels, and by extension are assumed to primarily determine sex steroid exposure to other tissues in the body. This largely ignores the contribution of local tissues to extra-gonadal sex hormone effects (10). The metabolic impact of estrogens produced in adipose tissue, for example, remains poorly understood, and will be a major focus of this review. During reproductive years, when gonadal estradiol (E2) is the dominant estrogen, premenopausal women tend to accumulate more subcutaneous fat, particularly in the hips and thighs (10). This pattern presents metabolic benefits, as subcutaneous fat is associated with protective effects, while visceral fat promotes a greater metabolic risk (10). However, in postmenopausal women, circulating estrogen levels decline, and the remaining levels of circulating estrogens reflect what is produced in extragonadal sites like adipose tissue (11). Studies that support circulating levels of estrogen being a secondary outcome of estrogen production for postmenopausal women and men clarify what may occur in extragonadal tissues but does not capture the local or tissue-specific actions of estrogen itself. The mechanisms and consequences underlying the production of estrogen in different adipose tissue depots remain unclear. Additionally, the metabolic impact on adipose tissue by endogenously produced estrogens appears to be influenced by age, sex, and metabolic disease. These variations in estrogen regulation and its implications for metabolic health warrant additional investigation.
This review will investigate the impact of age, sex, and metabolic disease on endogenous adipose tissue estrogen metabolism, including differences between subcutaneous and visceral fat depots. Observational studies in humans across the age span and preclinical studies in rodent models that have contributed to our knowledge regarding the relationships between adipose and sex steroids will be discussed. Firstly, adipocyte metabolism in healthy, aging, obese, and diabetic individuals will be presented, followed by the response of adipose tissue to estrogen. Next, we will explore the regional differences of estrogen activity in different depots of fat, including estrogen conversion mechanisms in adipose tissue and the changes that occur with aging. Finally, the impact of these endogenously produced estrogens on metabolic function will be discussed. This review thus offers a unique adipocentric perspective on estrogen metabolism in health and disease across the lifespan.
2 Adipocyte metabolism
In a healthy state, WAT importantly contributes to the maintenance of lipid and glucose homeostasis. Adipocytes store triglycerides and release free fatty acids (FFA), as well as synthesize and secrete adipokines, to maintain metabolic homeostasis. The WAT secretome is composed of cytokines, adipokines, and other factors, which can be reviewed here (12), and adipose depot location greatly influences metabolic health (13, 14). However, the differences in depot specific WAT sites and their molecular properties are much less understood (15). Adipocytes within WAT can expand in both number (hyperplasia) and in size (hypertrophy) which has been shown to be regulated by nutrient availability and sex steroids (10, 16). Healthy expansion of WAT includes increased vascularization and anti-inflammatory signals (17, 18). Thus, adipose tissue can be considered as an energy balance “hub” that integrates the body’s requirements for energy storage and utilization by other organ systems.
To understand the role of WAT on metabolic function and dysfunction, it must first be understood that while obesity is commonly defined using BMI, it is not a reliable indicator of metabolic health on its own. Subsets of individuals who classify as having obesity can have “metabolically healthy obesity’ (MHO) or “metabolically unhealthy obesity” (MUHO), and it is also possible to be “Metabolically unhealthy with a normal weight” (MUHNW) (19), suggesting there are clear exceptions that may be misclassified if one were to rely on BMI alone. Several studies have been conducted on these subsets of patients to clarify the root connection between obesity and metabolic decline. Demographics ranging between men, women, and postmenopausal women show that there is a correlation between a beneficial phenotype for metabolic outcomes and subcutaneous fat accumulation, whereas visceral fat accumulation, including omental adipocyte hypertrophy, leads to a decline in metabolic health (20–25). Adipose tissue distribution can contribute to metabolic dysfunction associated with aging, obesity, and diabetes, to be discussed in more detail in the next sections.
2.1 Aging
The aged population, herein pertaining to adults over the age of 60, is expected to double by 2050 (26). Aging is a major contributor to the growing global prevalence of metabolic diseases, which include the constellation of obesity, diabetes, and cardiovascular disease (CVD). Obesity rates among the elderly are rapidly increasing, with estimates that 37.5% of men and 39.4% of women over age 60 are classified as obese in the United States (27). With aging, fat mass and tissue distribution go through significant changes. Fat depot sizes reach a peak in middle to advanced age (28), following a substantial decline, which appears to be a result of decreased hypertrophy rather than hyperplasia, as the capacity for WAT to continue preadipocyte differentiation declines with age (29–31). Included in this decline is a redistribution of fat from subcutaneous to visceral depots, particularly in post-menopausal women, which is associated with an increased risk for metabolic dysfunction, including insulin resistance (IR), as adipose tissue releases excess FFA and inflammatory mediators that impair insulin signaling (32, 33). Additionally, with age, the decline in WAT mass is associated with accumulation of fat in ectopic regions in non-adipose tissue organs like skeletal muscle (34), liver (35), and bone marrow (36), which exacerbates metabolic dysfunction (37). Aging-related changes in WAT distribution in women and men are depicted in Figure 1.
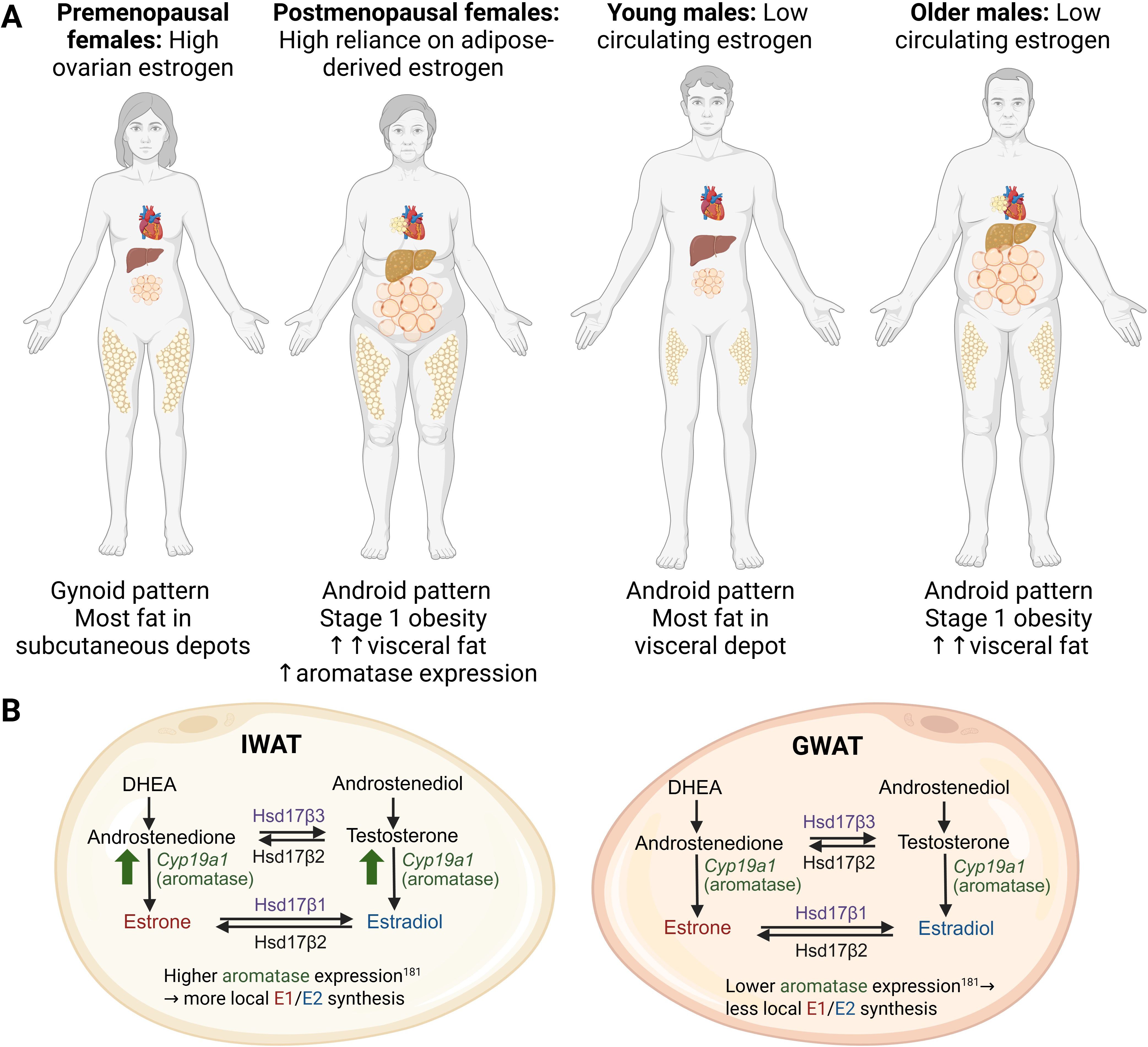
Figure 1. (A) Fat distribution changes across the age span in women and men. Premenopausal women tend to store most fat in subcutaneous depots. After menopause, more fat accumulates in the visceral compartment, including ectopic fat surrounding the heart and in the liver. Young and older men tend to accumulate most of their fat in the intra-abdominal region, with more visceral and ectopic fat accumulation as they age. (B) Estrogen synthesis pathways in subcutaneous inguinal white adipose tissue (IWAT) and visceral gonadal white adipose tissue (GWAT). In both IWAT and GWAT, estrogens can be converted from androstenedione and/or testosterone due to the presence of aromatase (Cyp19a1). DHEA, androstenedione, androstenediol, and testosterone enter WAT from the circulation. Estrone (E1) results from androstenedione conversion, while estradiol (E2) is a product of testosterone conversion by aromatase. E1 and E2 can interconvert due to the action of 17-beta hydroxysteroid dehydrogenases 1 and 2 (Hsd17β1 and Hsd17β2). Aromatase expression and activity tends to be higher in IWAT. Created in BioRender. Lee, (A) (2025) https://BioRender.com/62mlzmg.
A relationship between aging and chronic, low-grade inflammation exists, in which a term called “inflammaging” arose, coined by Dr. Franceschi in the year 2000 (38). With this, proinflammatory cytokines are notably increased, including immune cells infiltrating WAT (38). Aged WAT is characterized by a proinflammatory microenvironment with elevated expression of inflammatory genes associated with metabolic disease (39). Aging is also associated with macrophage infiltration into primarily visceral adipose depots which drives a pro-inflammatory state and further increases adipose dysfunction (40). In terms of function and metabolism, older WAT exhibits reduced lipolysis and lipid storage capacity, driven by increased fibrosis and reduced plasticity (41). Additionally, WAT from older individuals exhibits a decline in anti-inflammatory adipokines like adiponectin, and an upregulation in pro-inflammatory cytokines that include tumor necrosis factor alpha (TNF-α) and interleukin-6 (IL-6) which can be reviewed here. In post-menopausal women, a similar pattern of decreased circulating estrogen levels has been correlated to decreased adiponectin secretion and elevated levels of TNF-α and IL-6 (42, 43). This chronic inflammation in aging WAT contributes to hepatic insulin resistance and systemic metabolic decline (44). The association between decreased adiponectin levels in postmenopausal women exacerbating metabolic dysfunction is supported by a cross-sectional study by Karim et al. which suggests an inverse association with endogenous concentrations of estrogen and adiponectin and ghrelin and a positive association with leptin and endogenous estrogens (45). While cross-sectional studies may be limited in design, a longitudinal study by Tai et al. in Taiwanese postmenopausal women showed that higher serum adiponectin levels were associated with lower BMI and decreased risk of hyperlipidemia. Similar results were seen in another cross-sectional study in which menopausal age category was accounted for, but with no influence (46). Associations between decreased adiponectin and enhanced insulin resistance and other metabolic diseases is supported by several other studies (47–49).
2.2 Obesity and diabetes
Obesity, which is characterized by an expansion of WAT, occurs through two primary mechanisms: hyperplasia and hypertrophy. Hyperplasia, referring to an increase in the number of adipocytes, is associated with an increase in subcutaneous fat volume and protective signals, and is associated with MHO (50). A greater number of small adipocytes has been associated with improved insulin sensitivity, reduced inflammation, and less ectopic lipid accumulation, as the production of new cells leads to a greater capacity for nutrient storage (51). On the contrary, hypertrophic adipocytes, in which existing adipocytes enlarge, is associated with increased dysfunction and MUHO (50), which contributes to IR and subsequent T2D (52, 53). The molecular and genetic mechanisms underlying obesity-driven hyperplasia and hypertrophy are reviewed here (54). Hypertrophic adipocytes are associated with an interference in lipolysis and adipokine secretion. This reduces cellular stability, increasing the risk of cell death resulting in the chronic, low-grade inflammation that we see in tandem with metabolic syndrome (25). Hypertrophy leads to multiple fold increase in infiltration of macrophages in WAT (55, 56), which promotes a pro-inflammatory and insulin resistant environment. There are several mechanisms that lead to this state, including the promotion of cell death. As WAT expands beyond its capacity, hypoxia is induced, leading to cellular stress and necrosis (57). This exacerbates macrophage inflammation, and thus the chronic pro-inflammatory state that impairs adipocyte function and insulin sensitivity which contributes to systemic dysregulation (57, 58). Furthermore, dysfunctional adipocytes caused by obesity have impaired storage capabilities, promoting the release of FFA into circulation and ectopic fat accumulation which interfere with insulin signaling pathways and contribute to systemic insulin resistance and eventual T2D (44). The pathophysiology of obesity-related diabetes has several nuances related to the disruption of metabolic homeostasis caused by increased adiposity. Obesity-induced insulin resistance is driven by adipocyte dysfunction, coupled with chronic inflammation, and ectopic lipid deposition in non-adipose tissues, similar to the effects seen in aging, which other reviews describe as a compounding effect (59, 60).
3 Sex steroids and metabolic disease
Sex steroid synthesis pathways have been well described (61, 62). Briefly, as shown in Figure 1, gonadal sex steroids are synthesized from cholesterol by the enzymatic action of steroidogenic acute regulatory protein (StAR) and CYP11A1. Cholesterol-derived pregnenolone (Preg) can be further metabolized into progesterone or dehydroepiandrosterone (DHEA) by specific enzymes, followed by androstenedione (A4) and/or testosterone (T), which can be terminally converted into estrogens. A4 is primarily converted into E1, while T is converted into E2, both directed by CYP19A1 (aromatase) activity. E1 and E2 can interconvert due to the activity of several hydroxysteroid dehydrogenase (HSD) enzymes, HSD17β subtypes 1, 7, and 12 (62). E2 derived from testosterone is the primary estrogen produced from the ovaries, while DHEA-derived A4 leads primarily to E1 in adipose tissue (63). Thus, E2 tends to dominate in people with high gonadal function, while E1 becomes more prevalent as gonadal function declines, as occurs in aging, obesity, PCOS, and post-menopause (64–66). The impact of circulating sex steroids on metabolic health and aging is summarized in Table 1.
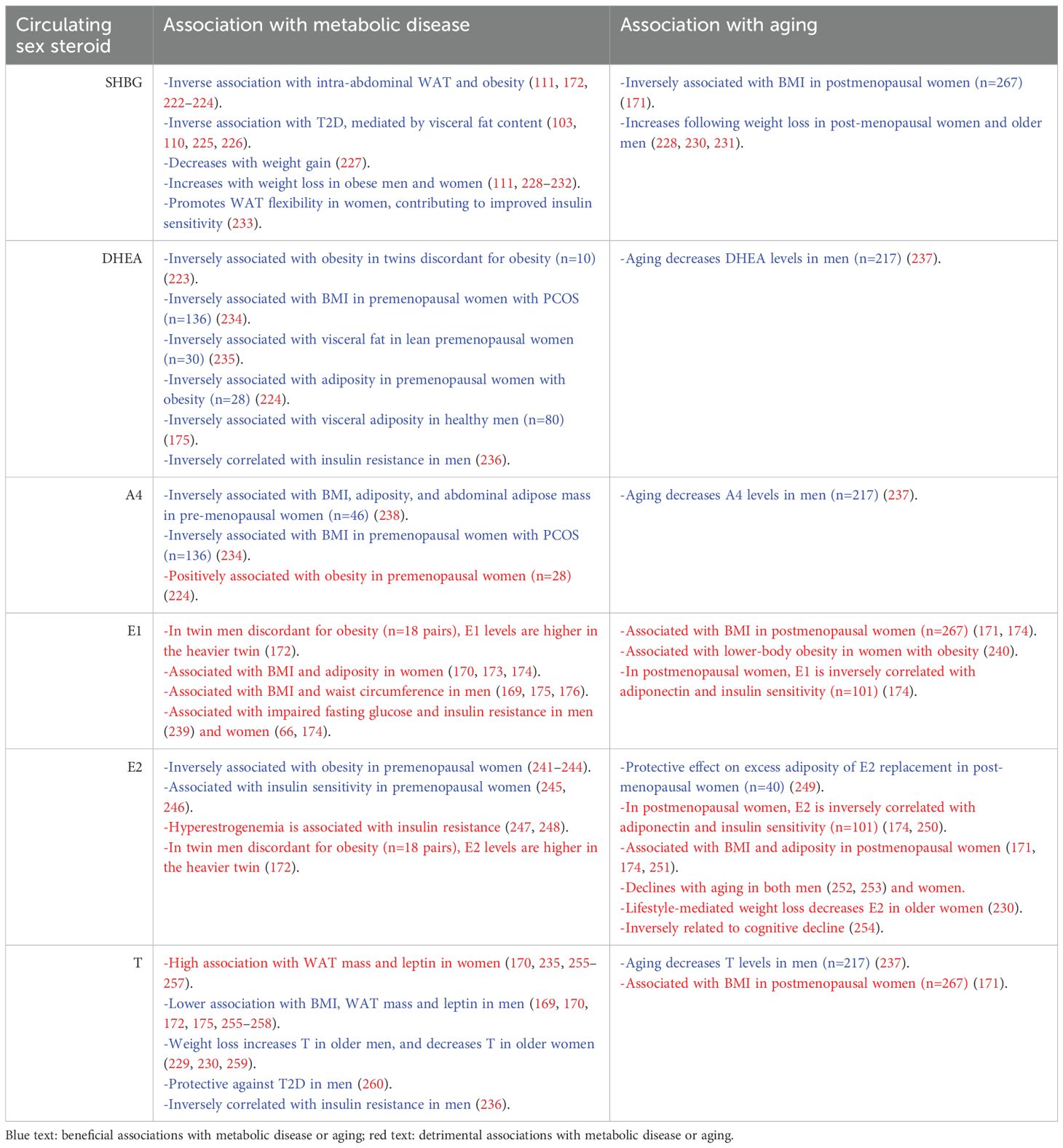
Table 1. Summary of the impact of circulating sex steroids on metabolic disease and aging in humans.
A notable sexual dimorphism exists in body fat composition and adipocyte metabolism, which is influenced by sex steroids that include estrogens and androgens (51) (Figure 1). Women generally have higher subcutaneous fat storage capacity than men, which provides some protection against metabolic dysfunction, while men tend to store more visceral fat, putting men at greater risk for developing IR (67, 68). After menopause, women tend to store a similar amount of adipose tissue in the visceral compartment as men, implicating a lack of gonadal estrogen signaling in visceral fat accumulation and subsequent increased risk for IR (69). Indeed, subcutaneous adipose tissue expresses high levels of estrogen receptors (37). Hyperplasia and hypertrophy can occur in a depot-specific manner, as intra-abdominal fat depots primarily expand through hypertrophy, while subcutaneous WAT has been seen to expand through both methods (54). Moreover, visceral and subcutaneous WAT expansion occurs in a sexually dimorphic manner. Male mice develop diet-induced obesity primarily though visceral fat hyperplasia, while female mice do so via both visceral and subcutaneous fat hyperplasia (70, 71) with sex steroids playing a major role (70, 72). The direct impact of gonadal sex steroids on WAT function is not well understood, which prompts further investigation into the sexual dimorphism of adipocyte function and the mechanisms of gonadal steroids in these respective tissues. Moreover, T2D is more prevalent in men than women, with an estimated 13% of men and 11% of women between the ages of 20–79 classified as diabetic in 2016 (67, 73). Post-menopausal women are also at increased risk of developing diabetes, an effect that can be mitigated by estrogen replacement therapy (74), suggesting an important role for estrogens in diabetes risk (Table 1).
Animal studies have supported the protective impact of estrogens on metabolic disease risk (summarized in Table 2). Female mice and rats that have undergone ovariectomy (OVX), effectively removing all gonadal estrogens, have increased adiposity and are more prone to diet-induced obesity than sham-operated mice, with elevated adipose tissue inflammation (75–79). These effects of OVX appear to be consistent across rodent species (mice and rats), strain (rats: Sprague-Dawley, Wistar), and with age at OVX (4–10 weeks of age in mice). Estrogen treatment promotes anti-inflammatory and insulin sensitizing effects in both male and female mice (80, 81), and estrogen replacement reverses some of the detrimental metabolic effects of ovariectomy (80, 82–84). By contrast, castrated male rodents have been shown to exhibit improved glucose and insulin tolerance with reduced adiposity in some studies (85, 86), and in others display worsened adiposity, WAT inflammation, and glucose tolerance (87, 88). Perturbation of estrogen signaling in mice has primarily been achieved by genetic manipulation of estrogen receptors (ERα, ERβ) or aromatase in mice. Global deletion of ERα has been shown to increase adiposity, systemic and adipose tissue inflammation, and insulin resistance in mice (89–92). Similarly, adipocyte-specific deletion of ERα increases inflammation concurrently with adipocyte hypertrophy (93). Studies of mice with global aromatase deficiency consistently show an increased propensity towards obesity and insulin resistance (94–96), and mice with adipocyte-specific aromatase overexpression exhibit improved insulin sensitivity and reduced inflammation (97). Collectively, studies in mice highlight the distinct metabolic impacts of the loss of gonadal androgens and estrogens on systemic metabolism.

Table 2. Summary of the impact of perturbing circulating and WAT-derived sex steroids on metabolic disease and aging in mouse models.
In addition to being a major target of gonadal sex hormones, adipose tissue can also synthesize and store them (Figure 1). The decline in gonadal sex hormone production that occurs in post-menopausal women and in aged men coincides with increased sex hormone synthesis within adipose tissue (76, 98). Increased synthesis of adipose tissue-derived sex hormones has also been reported in the setting of obesity (99). The mechanisms by which adipose tissue can become a source for sex hormones will be discussed in Section 3.2.4., and emerging knowledge regarding the metabolic impact of WAT-derived sex hormones is summarized in Table 3.
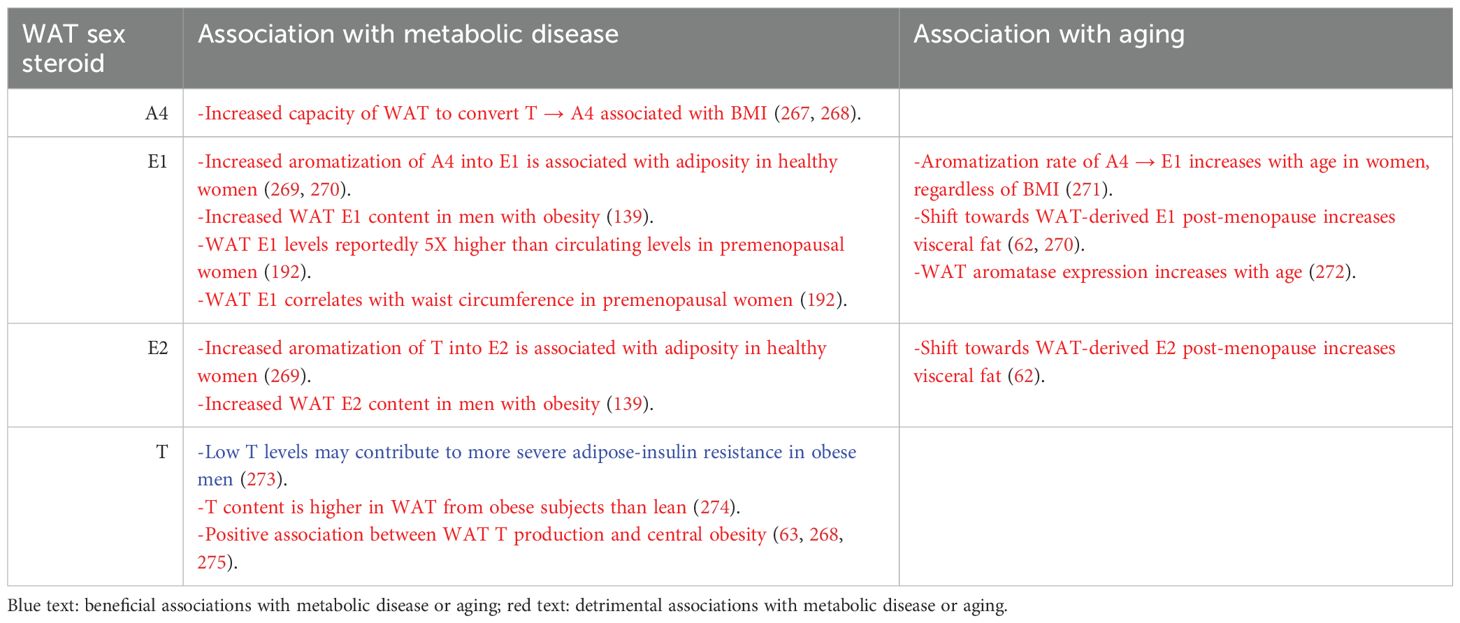
Table 3. Summary of the impact of WAT-derived sex steroids on metabolic disease and aging in humans.
3.1 Sex hormone-binding globulin and adipose tissue
Sex hormone-binding globulin (SHBG) is a glycoprotein primarily synthesized in the liver. SHBG regulates the bioavailability of sex steroids in the bloodstream, including testosterone (T) and estradiol (E2). There is compelling evidence that supports SHBG’s involvement in glucose and lipid metabolism and its role as a biomarker for obesity-related disorders including T2D (100–102). The mechanisms of SHBG’s regulation in the context of sex differences and adiposity are incompletely understood. This section will discuss the current knowledge and gaps thereof.
The most established link between SHBG and metabolic health is its inverse association with insulin resistance. Low circulating SHBG levels are predictive of T2D development, independent of BMI (103). Particular single nucleotide polymorphisms (SNPs) for SHBG confer increased risk for T2D (102, 104). This brings to question if SHBG may mediate a relationship between adiposity and impaired glucose metabolism (100). Mechanistically, this seems to involve insulin’s inhibitory effect on hepatic SHBG production. Hyperinsulinemia, which is a hallmark of insulin resistance, suppresses SHBG synthesis, creating a cycle of increasing free sex steroid concentrations and worsening metabolic outcomes (105–107). Interestingly, recent studies suggest that SHBG may partially explain sex differences in glucose regulation, reporting that SHBG mediates a proportion of the association between sex and fasting glucose levels, as well as T2D incidence (101). The degree to which SHBG independently influences metabolic health, or rather if it acts as a marker of other underlying processes, is not well agreed upon in current literature. A study by Sofer et al. (2018) tested the hypothesis that SHBG provides metabolic protection by feeding transgenic mice expressing human SHBG a high-fat diet (HFD) for 4.5 months. Their results revealed no protective effects from SHBG expression on obesity or dysglycemia in either male or female mice (108). SHBG transgenic mice appeared to gain weight similarly to wild-type (WT) controls. Furthermore, fasting glucose, insulin, and insulin resistance measured using HOMA-IR found no significant differences (108). It was also observed that female SHBG transgenic mice showed higher fasting glucose levels than WT controls, suggesting that SHBG may play a detrimental role in certain contexts (108). The authors speculate that the absence of metabolic protection may result from the longer duration of the HFD in their model, which may have allowed for compensatory mechanisms to override an early protective benefit that SHBG may provide. They also acknowledge that SHBG may not be causally protective but rather serve as a biomarker for metabolic health. Contrastingly, a study by Saez-Lopez et al. (2020) reported that SHBG overexpression protected male, transgenic mice against HFD-induced obesity as well as metabolic disease. Over the course of 8 weeks, compared to WT controls on the same diet, SHBG transgenic mice demonstrated significantly less weight gain, smaller epididymal white adipose tissue (EWAT) depots, and a healthier metabolic profile including lower insulin, leptin, and resistin while also demonstrating higher adiponectin levels (109). The authors proposed that this protective effect was mediated through enhanced lipolysis in WAT, as SHBG transgenic mice had elevated expression of lipolytic genes including beta-3- adrenergic receptor (Adrb3), interferon regulatory factor-4 (Irf4), and perilipin-1 (Plin) and increased phosphorylation of protein kinase A (PKA), extracellular signal-regulated kinases 1/2 (ERK-1/2), and hormone sensitive lipase (HSL), which may suggest that SHBG plays a more active role in adipocyte metabolism than previously thought. A notable difference between the two studies is their duration of HFD exposure. Sofer et al. used a longer, 4.5-month model which may have allowed for metabolic adaptations, or saturation effects of any protective role SHBG may play. The shorter 8-week model by Saez-Lopez et al. may reflect a more acute metabolic response. Further support for SHBG’s protective role comes from a longitudinal human study observing changes in SHBG and diabetes risk over the menopause transition (110). This study found that increasing levels of SHBG were associated with a decreased risk of T2D after adjusting for covariates. Furthermore, stable or increasing rates of change in SHBG were independently associated with a lower risk of diabetes compared to decreasing rates of change (110). This may suggest that SHBG exerts effects on glucose regulation beyond its known role as a regulator of sex steroids. Comparisons between its role with circulating sex hormones and those that are endogenously produced are also not fully understood, especially in the context of obesity related adipocyte dysfunction.
A critical question remains: Is SHBG simply a biomarker of obesity and metabolic health, or does it act as an active contributor to metabolic regulation? Cross-sectional human studies show that low SHBG levels predict T2D and metabolic syndrome incidence, even after adjusting for adiposity (111). However, these associations may not imply causation. Studies have demonstrated that SHBG levels are inversely correlated with markers of adiposity that include BMI and WHR (111, 112). In post-menopausal women, it appears that lower SHBG concentrations are associated with higher central adiposity (113). A separate study found that SHBG levels tend to increase with age linearly in healthy post-menopausal women (114). while another suggests this increase relates to the increase of circulating free testosterone in late-postmenopausal women (115). Further studies could be beneficial to clarify the interaction between hormonal aging and metabolic health in postmenopausal women with and without metabolic syndrome. Despite numerous studies that show a relationship between SHBG and adiposity, the findings are not uniform across all populations. A 1997 study found that SHBG negatively correlated with BMI, WHR, insulin, and testosterone levels in both premenopausal and postmenopausal women, however, E2 levels correlated positively with SHBG only in the premenopausal group (116). The relationship between SHBG and IAAT similarly does not reach a consensus across studies. Several cross-sectional studies have shown an inverse relationship between SHBG and IAAT, suggesting that visceral fat may actively suppress SHBG synthesis, potentially through inflammatory pathways or hepatic fat accumulation (111, 117). Contrasting to these findings, the first of a kind to publish longitudinal data regarding the relationship of IAAT gain and SHBG from postmenopausal women indicate that higher baseline SHBG levels may predict greater IAAT gain over time, a finding that challenges previous cross-sectional studies (118). These inconsistencies call for additional investigation into the temporality and causality of the SHBG-adiposity relationship.
3.2 Estradiol and estrone in metabolism
Estrogens influence many physiological processes including lipid metabolism and adipose tissue distribution. Estrogen has three primary forms, estrone (E1), estradiol (E2), and estriol (E3). Three estrogen receptors have been described: ERα, ERβ, and G-protein coupled estrogen receptor 1 (GPER1), also known as G-protein coupled receptor 30 (GPR30). Upon activation, estrogen receptors translocate to the nucleus where they dimerize and bind to specific DNA sequences, termed estrogen response elements, to initiate estrogen-dependent gene transcription. Estrogens can also exert receptor-mediated non-genomic effects via GPER1, including interactions with cell membrane-associated complexes such as caveolin-1, other G-proteins, and receptor tyrosine kinases such as EGFR, IGF-1, and MAPK (119, 120). E2 has the highest affinity for ERα and ERβ and is thus considered the strongest estrogen (121). E1 and E3 are weaker estrogens, with higher affinity for ERα and ERβ, respectively (121). Only E1 and E2 have reported agonism for GPER1 (122). ERα and ERβ are widely distributed throughout the body, with major and roughly equivalent expression in the brain and liver (123). Notably, metabolic tissues such as WAT, skeletal muscle, bone, and the heart have higher expression of ERα, while other tissues such as bone, prostate, testes, and ovaries have higher expression of ERβ. To date, GPER1 expression has been reported in reproductive tissues (testes, prostate, and endometrium), immune cells, metabolic tissues (adipose, liver, pancreas, and skeletal muscle), and in certain cancers (prostate, ovarian, cervical, breast, and lung) (122, 124–126). E2, or 17β-estradiol, is the predominant estrogen in premenopausal women, and it is the more biologically active form as it has a higher affinity for estrogen receptors than other estrogen forms during reproductive years (121). However, post-menopause, E1 becomes the dominat estrogen, where it is synthesized in WAT via aromatase activity (62, 127, 128). Many studies have shown that E2 beneficially influences metabolism through mechanisms involving reduced food intake and increased energy expenditure (75, 129–131). Many of the beneficial effects of E2 on energy metabolism are increasingly being attributed to its interaction with GPER1, including its rapid effects on insulin sensitivity, hepatic glucose and lipid metabolism (132, 133). However, much less is known regarding how the prevalence of E1 in postmenopausal women may influence metabolic health. Emerging evidence suggests that E1 is not associated with the same beneficial effects on insulin sensitivity reported for E2 and may promote inflammation (99, 134). Obese mice supplemented with E1 display a robust pro-inflammatory phenotype, while E2 treatment resulted in an anti-inflammatory phenotype, an effect dependent on NFκB activation by E1 that was replicated in cultured adipocytes and breast cancer cell lines (134). In addition, obesity is associated with higher circulating and WAT-derived E1 levels (64, 135–138), which may be due to the capacity for higher fat content to convert androgens into more estrogens (139, 140), and in particular E1 (62, 141). Rats fed an E1-enriched diet gained twice as much body weight as control rats (142), suggesting that E1 could promote obesity. More research is needed to improve our understanding of the metabolic impact of changing estrogens concentration and type on metabolism post-menopause.
The metabolic benefits of E2 have been well studied, with evidence that it promotes insulin-sensitivity and improved lipid metabolism in tandem with estrogen receptors, primarily ERα (90) and GPER1 (133). In mice, estrogens act via ERα and GPER1 to regulate insulin sensitivity (143). Studies have shown that ERα knockout (KO) mice in both males and females have worse metabolic profiles including developing obesity and worsened glucose homeostasis (90, 143–145). Recent research has also suggested that estrogen’s metabolic effects are not only mediated through direct action on adipocytes, but also through endothelial mechanisms of ERα that improve insulin transport to skeletal muscle (146). ERα appears to be required to induce the positive effect that E2 has on insulin sensitivity in mice (89, 90). Recent mouse models of global GPER1 deficiency report similar phenotypes. GPER1 KO mice are more prone to obesity, insulin resistance, inflammation, and dyslipidemia than control mice (147–149). Furthermore, in humans, individuals with T2D and poorly managed glucose control showed a significant decrease in ERα mRNA expression (145). The systemic effects of estrogens are well documented, especially regarding the role of E2 in promoting insulin sensitivity and reducing lipid accumulation, but a major limitation in our current understanding of how estrogens impact metabolism is limited by a lack of understanding the tissue-specific mechanisms that drive metabolic regulation and WAT distribution (130). Tissue-specific estrogen actions will be discussed in the next sections.
3.2.1 Skeletal muscle
Current literature supports that the expression of glucose transporter-4 (GLUT4) in myocytes may be influenced by ERα in mouse models, suggesting ERα positively regulates GLUT4 expression, contributing to the systemic differences in insulin resistance seen in ERα KO mice (150). An in vivo experiment similarly showed that increased ERα expression increased skeletal muscle glucose uptake in ovariectomized female mice given E2 (151). Another study found that the expression of ERα in skeletal muscle had a significant, inverse relationship with adiposity and fasting insulin levels (152). However, a separate study showed that ERα is sufficient, but is not required, to protect mice from metabolic dysfunction in skeletal muscle and in women with insulin resistance and obesity (153). This necessitates future studies to consider the role of estrogens in skeletal muscle, and what mechanisms compensate for individuals with low to no ERα expression resulting from changes like aging and obesity (153). The expression of ERα is not uniform across all tissues in the body but is highly expressed in female reproductive tissues including ovaries and breast, WAT, liver, and other tissues which can be reviewed here (154). ERβ, which was not experimentally shown to regulate GLUT4 expression (150), is primarily expressed in male reproductive organs and other tissues (154). The sexual dimorphism of estrogen synthesis and estrogen receptors would benefit from additional investigations that consider the tissue-specific mechanisms in males and females and how this differs regarding our understanding of how E2 maintains a protective effect.
3.2.2 Liver
There is strong evidence that estrogens regulate hepatic glucose and lipid metabolism through ERs, particularly ERα (155–157). Loss of ERα in hepatocytes appears to impair glucose tolerance and increase lipid accumulation in the liver, worsening adiposity and IR in both male and female mice, with one study reporting a greater effect in female mice (158, 159). Orally ingested estrogens have also been reported to induce acute cholestasis, causing a decrease in bile flux which is detrimental to cholesterol homeostasis (159). The role of E2 has been well established to play a regulatory metabolic role via ERα in peripheral tissues such as skeletal muscle and WAT (130, 160–162) (further discussed in Section 3.2.3). There is evidence that ERα plays a similar role in the liver, with lower, but similar expression patterns to WAT (130, 162). E2/ERα signaling appears to increase lipid and glucose metabolism in the liver by influencing transcriptional factors that increase lipolysis while decreasing lipogenesis and gluconeogenesis (162). The role of E1 in the liver is much less clear. Some studies report that elevated E1 levels may contribute to hepatic IR and increased inflammation (134). An interesting direction from studies on male patients with hepatic cirrhosis states that E1, not E2, may play a larger role on sustaining increased circulating levels of estrogen for patients with liver cirrhosis through the peripheral conversion of androgens to E1 (163, 164). This specific hormonal profile may have clinical relevance on the progression of cirrhosis and altered fat distribution. Further research regarding the conversion mechanisms of androgens to estrogens and the use of aromatase inhibitors could improve our current understanding of E1 function in the liver and how it may impact metabolic homeostasis.
3.2.3 Adipose tissue
In premenopausal women, E2 promotes lipid storage predominantly in subcutaneous adipose tissue (SAT) and inhibits excessive lipid accumulation in visceral adipose tissue (VAT) (165). E2 contributes to metabolic homeostasis in adipose tissue specifically by decreasing the activity of lipoprotein lipase and subsequent lipogenesis, and has proven to influence hyperplasia of subcutaneous adipocytes (166). E2 also increases preadipocyte proliferation, suggesting an adipogenic effect (167). This protective effect appears to diminish in postmenopausal women, where a decrease in circulating E2 by the gonads leads to a deficiency, which has been associated with increased VAT deposition (168).
E1 has consistently been found to positively associate with BMI, waist circumference, and adiposity in men and women across the life span (169–176). The dominant estrogen post-menopause is E1, and while there is compelling speculation regarding its metabolic impact, there is not yet sufficient evidence for conclusive roles for E1 in energy metabolism. E2 has been studied for its endogenous metabolic benefits, yet the role of E1 in WAT function and metabolism remains much less understood. As the predominant estrogen circulating post-menopause, E1 is primarily synthesized in adipose tissue through the aromatization of androgens via aromatase (CYP19A1) (62, 177). A seminal publication suggested that E1 may exert metabolic effects distinct from those of E2, indicating that elevated E1 levels correlate with increased adiposity and insulin resistance in postmenopausal women (134). This supports the notion that E1 may contribute to metabolic dysfunction in WAT, while also supporting that E2 does not appear to have the same negative effects. It is not clear if the metabolic impact of E1 is dependent on local conversion mechanisms to E2, and if estrogen receptors and aromatase activity differ in varying depots and how this may impact the effects of E1 on metabolism. Whether E1 possesses direct metabolic effects remains a topic of interest.
The protective role attributed to E2 may be depot-specific, as ERα must be available for E2 to have a protective effect (178). Similar to what has been observed in skeletal muscle, it appears that the metabolic benefit of E2 is regulated by receptor signaling, which may vary in different fat depots. In adipose tissue, deletion of ERα (but not ERβ) is associated with metabolic decline (93, 179). In overweight pre-menopausal women, SAT in the abdominal region contains more ERα relative to ERβ than in gluteal SAT (180). Another study showed that E2/ER signaling plays a significant role in mediating sex differences of VAT accumulation, where males express less ERα in VAT than females (181).
Much has been learned about peripheral estrogen receptor-mediated effects using genetically perturbed mice. Male and female mice deficient in ERα have a higher degree of adiposity, insulin resistance, and glucose intolerance than WT mice (90, 92), a phenotype that closely resembles humans lacking either ERα or aromatase. Additional approaches to silencing ERα, including adeno-associated viral techniques and ERαlox/lox/Adiponectin-Cre (Adipo-ERα) mice, revealed that disrupting adipose tissue ERα recapitulated this phenotype (93), suggesting that the adipocyte is a major target of estrogens to impart beneficial effects on systemic metabolism.
3.2.4 Aromatase activity and estrogen conversion in adipose tissue
In addition to its capacity to respond to gonadal estrogens, WAT has the capacity to convert circulating androgens into estrogens (127, 128, 182) (Figures 1 and 2). Such endogenously converted estrogens can then function in an autocrine or paracrine manner (183, 184). Estrogen biosynthesis is catalyzed by aromatase P450, encoded by the CYP19A1 gene, which is highly expressed in human and mouse WAT (185, 186). In ovaries, aromatase is largely driven by the cyclic AMP response element binding protein (CREB) promoter (98). In contrast to the ovaries, in which the major substrate for estrogen aromatization is testosterone, the major substrates for aromatase-mediated estrogenesis in WAT are DHEA and androstenedione, and may be driven by inflammatory cytokines (62, 98). Another potential direction with promise lies in exploring the less understood pathways of aromatase activity and estrogen conversion between E2 and E1, and how it may modulate a tissue-specific outcome. WAT converts androgens to E1 via CYP19A1 (62, 141), an enzyme that appears to increase with adiposity in males due to the decline in available testosterone (187). Indeed, people with obesity have been reported to have increased CYP19A1 expression in WAT (99, 141, 188, 189). In another study, aromatase gene expression in SAT positively correlated with increased adiposity and IR, but interestingly not with circulating E2 (190). SAT tends to express aromatase in higher levels compared to VAT, whereas VAT appears to express more of 17β-hydroxysteroid dehydrogenase (17β-HSD), an enzyme which interconverts E1 and E2 (191). Another study found that, in women, aromatase levels in VAT positively correlated with adipocyte hypertrophy, suggesting that aromatase activity may be associated with VAT gain and overall metabolic dysfunction (189). These findings suggest that aromatase activity appears to regionally differ between fat depots (Figure 1B). Future analysis into aromatase expression levels in different depots to analyze its metabolic impact would benefit from an added component of comparing levels of E1 and E2 produced in these sites. A separate study found that E1 is the dominating form of estrogen in WAT for premenopausal women, showing a 5–10 times greater concentration of E1 in SAT and VAT compared to serum levels (192). They also found that the expression of aromatase positively correlated with the E1 concentration in VAT (192). In another study, women with obesity appear to express much higher levels of aromatase in SAT compared to VAT (193). The observed correlation between aromatase expression and its effect on adiposity and IR implicates E1 in metabolic dysfunction. Further research should seek to distinguish if E1 has direct metabolic effects or if its metabolic impact is mediated through the local conversion to E2.
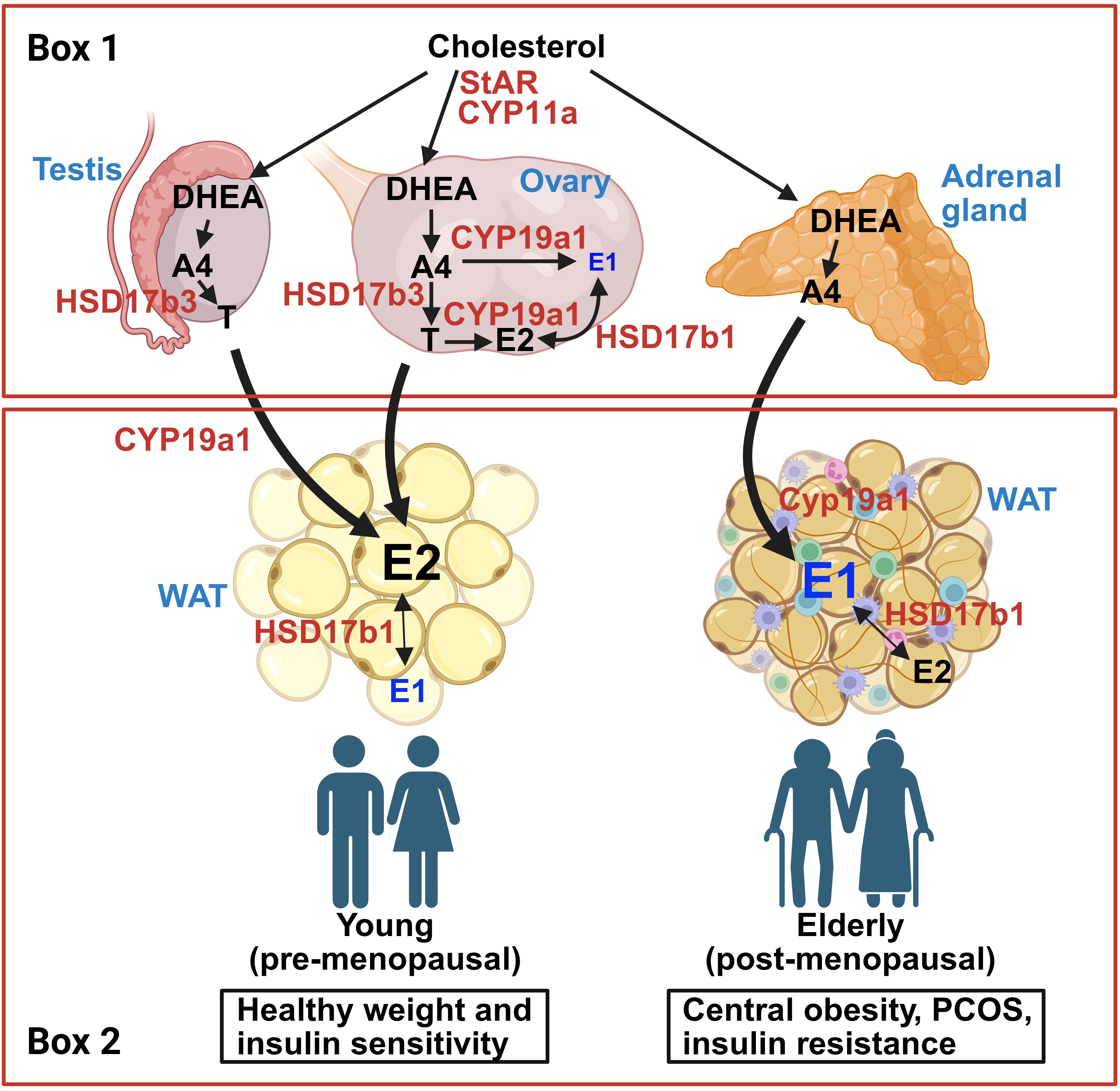
Figure 2. Schematic for estrogen synthesis pathways derived from cholesterol involving gonads (testes and ovaries), adrenal gland, and adipose tissue. Box 1: Gonadal and adrenal sex hormone synthesis pathways. Circulating cholesterol is transported across the mitochondrial membrane by steroidogenic acute regulatory protein (StAR) and CYP11A1 and is eventually converted to dehydroepiandrosterone (DHEA) via CYP17a1. In both testes and ovaries, DHEA is converted to androstenedione (A4) and testosterone (T) by HSD3b1 and HSD17b3, respectively. When aromatase (CYP19a1) is present, A4 and T are converted into estrone (E1) and estradiol (E2), respectively. Functional gonads release high levels of T from testes, and E2 from ovaries. Like the gonads, the adrenal gland can convert circulating cholesterol to androgens, primarily A4. Box 2: Adipose tissue androgen conversion pathways from healthy young people (left) and those with obesity, insulin resistance, PCOS, and/or advanced age (right). In people with robust gonadal function, circulating androgens (A4, T) are converted to E2 due to aromatase activity in white adipose tissue (WAT). Some E2 will convert to E1 due to HSD17b1 activity. In people with reduced gonadal function (i.e. advanced age, obesity, PCOS, and/or insulin resistance), the major circulating androgen is A4, which predominantly converts to E1 in WAT. Created in BioRender. Den Hartigh, L. (2025) https://BioRender.com/oqv7exy.
Mechanistic studies in mice provide clues regarding the metabolic effects of local estrogens. Mice globally deficient in aromatase (Cyp19a1 KO) are more prone to aging-associated increase in abdominal obesity and insulin resistance (94–96). Providing exogenous E2 rescues the obesogenic phenotype in Cyp19a1 KO mice, suggesting the lack of E2 drives the increased adiposity. While rare, aromatase deficiency in humans also leads to insulin resistance and T2D (194), suggesting a beneficial effect of aromatase. In support of this, a single study has shown improved insulin sensitivity in male mice with transgenic aromatase overexpression specifically from WAT, driven largely by increased E2 (97). While these genetic studies offer a starting point in modulating estrogen production capacity in general terms, only one study has addressed the potentially divergent effects of perturbing particular estrogens. Qureshi et al. found that in obese mice, E1 promoted a pro-inflammatory phenotype, while E2 dampened inflammation, an effect supported by transcriptomics (134). Further, higher E1:E2 ratios were predictive of ER-positive tumor growth (134), suggesting differential impact of E1 and E2 on obesity-related breast cancer incidence. The impact of particular estrogens on breast tissue and tumor burden will be discussed in the next section.
3.2.5 Breast tissue
Breast tissue appears to have a distinct estrogenic profile than other fat depots, despite breast tissue having a significant portion of WAT. Similar to WAT, the expression of aromatase in breast tissue is highly regulated and involved in estrogen synthesis. E2 in breast tissue is not associated with protective metabolic effects, but rather has been linked to proliferative tumor growth (195). Elevated aromatase expression in breast WAT increases local E2 production, which may contribute to estrogen receptor-positive (ER+) breast cancer pathogenesis (135, 195, 196). This localized estrogen synthesis differs from systemic estrogen metabolism as breast WAT appears to maintain high estrogenic activity, especially for postmenopausal women where WAT is the key source of estrogen synthesis. Moreover, tumor-bearing breast tissue has been shown to have higher aromatase expression within adipose in close proximity to the tumor than in distal tissue within the same breast (197), suggesting an adverse role for endogenously produced estrogens in breast cancer. Inflammatory cytokines secreted by hypertrophic breast tissue in individuals with obesity appear to exacerbate aromatase production and thus local estrogen production (198). In addition, the impact of post-menopausal estrogens derived from WAT on breast cancer incidence has been studied extensively, with evidence that ER-positive breast cancer incidence increases with age (199). Multivariate analyses suggest that estrogens are the most important factors associated with the elevated breast cancer risk in postmenopausal women with obesity (200). Finally, it has recently been reported that E1 and E2 derived from breast WAT have opposing pro- and anti-inflammatory transcriptional profiles, respectively (134).
3.4 Menopausal hormone therapy
The use of menopausal hormone therapy (MHT) to reduce metabolic dysfunction associated with menopause has been studied. However a consensus regarding its efficacy is far from established. Studies generally show that estrogen replacement therapy in postmenopausal women can promote weight loss and improve metabolic markers including fat distribution, notably by increasing subcutaneous fat and reducing visceral fat mass (201–203). The majority of MHT trials have been done in women of normal weight (BMI<30); as such, the reported impact of MHT on body weight and adiposity has been modest (74, 204–206). After adjusting for age and BMI, the Nurse’s Health Study (n=21,028) showed that MHT (estrogen alone, progesterone alone, or the combination) users had a 20% reduced risk for diabetes than non-users (207). Similarly, MHT users in the postmenopausal estrogen/progestin interventions (PEPI) trial showed improved fasting glucose and insulin levels (204). MHT has also been shown to reduce homeostatic model assessment of insulin resistance (HOMA-IR) in postmenopausal women, and with a nearly 3-fold greater benefit in postmenopausal women with T2D (204, 208). All the MHT studies listed to this point used either conjugated estrogen (CE) alone or in combination with a progestin, delivered orally. More recently, bioidentical E2 with or without a progestin has become a more common method of MHT. To date, there is very little known about the impact of other MHT delivery methods, including transdermal patches, vaginal rings, gels, creams, or suppositories, on general metabolic health. No randomized controlled trials have directly examined the metabolic impact of oral vs. transdermal HRT, but observational studies reveal the potential for reduced risk for thromboembolism and dyslipidemia with transdermal delivery when compared to oral (209, 210). This could be due to the first-pass through the liver with oral MHT, enabling increased triglycerides, coagulation factors, and C-reactive protein, which is minimized with transdermal therapy (211, 212). By contrast, while both transdermal and oral MHT delivery methods have been shown to reduce diabetes risk, oral MHT led to a greater risk reduction (213).
Conflicting data suggest that estrogen’s metabolic effects may depend on context. Initial results from the Women’s Health Initiative (WHI) showed that conjugated equine estrogen with or without medroxyprogesterone acetate let to worse cardiometabolic outcomes in postmenopausal women, dampening enthusiasm for MHT (214). It has since been suggested that, since the MHT study population in the WHI was on average more than 10 years post-menopause, metabolic benefits of MHT may be greater in women within 10 years of menopause, introducing the importance of MHT timing into the equation (215). Indeed, subsequent re-analyses of the WHI data revealed cardiometabolic benefits of MHT in women aged 50-59 (216–218). Some studies report that high circulating E2 levels are associated with increased inflammation and adipocyte dysfunction, particularly in postmenopausal individuals with obesity (174, 219). This discrepancy may be attributed to differences in estrogen metabolism, receptor expression, or interactions with other hormones such as androgens and insulin which can be reviewed here (220). Other studies support that E2’s protective effects on WAT expansion may be depot-specific, as the expression of estrogen receptor ERα, which has been shown to be necessary for E2 to maintain its protective effect of inflammation in both males and females, may be higher in subcutaneous fat (181, 221). Moreover, subcutaneous WAT has been shown to have a higher E2 conversion rate than visceral WAT (64, 192). By contrast, visceral WAT has been reported to produce more E1 than subcutaneous WAT (64). To date, it is not known how MHT impacts WAT responsiveness or estrogen conversion capacity in postmenopausal women.
4 Conclusions
Endogenously produced estrogens in WAT appear to impact energy homeostasis, fat distribution, and inflammation. The patterns which emerge in studies to date show that the impact of estrogens synthesized locally in WAT depots are dependent on factors like age, sex, and depot, with subcutaneous fat generally presenting higher estrogenic activity than visceral fat.
As women enter menopause, the shift in estrogen production localized to estrogens produced in WAT is primarily driven by aromatase activity. The interconversion between estradiol to estrone in WAT may play a significant role in estrone dominance over estradiol in circulation post-menopause. How estrone may influence visceral adiposity, inflammation, and insulin resistance is a topic of growing interest that requires investigation that separate the longitudinal and systemic impact presented in covariates like sex, age, depot, species, and tissue type. Examples include the difference in bioavailability and expression of key enzymes involved in estrogen production, conversion, transport, and signaling, which include but are not limited to aromatase, HSD17β1, SHBG, TNF-α, IL-6 and ERα. Further investigation to clarify how WAT responds to estrogenic signaling across the life span will lead to a more comprehensive understanding of metabolic decline with age, especially for women post-menopause. Developing therapies require the precision of understanding the interplay between the human hormonal milieu and metabolic health to manage metabolic dysfunction in diverse populations.
Author contributions
AL: Conceptualization, Visualization, Writing – review & editing, Writing – original draft. LD: Conceptualization, Supervision, Writing – review & editing, Funding acquisition, Visualization.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. National Institutes of Health R01-DK135756 (PI: LD).
Acknowledgments
BioRender was used to generate figures.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary
ADRB3: beta-3- adrenergic receptor
BMI: body mass index
CE: conjugated estrogen
CVD: cardiovascular disease
CYP19A1: aromatase
DHEA: dehydroepiandrosterone
E1: estrone
E2: estradiol
E3: estriol
ERα: estrogen receptor alpha
ERβ: estrogen receptor beta
ERK1/2: extracellular signal-regulated kinases 1/2
EWAT: epididymal white adipose tissue
FFA: free fatty acids
GLUT4: glucose transporter 4
GPER1: G-protein coupled estrogen receptor
GPR30: G-protein coupled receptor 30
HFD: high fat diet
HOMA-IR: homeostatic model assessment of insulin resistance
HSD: hydroxysteroid dehydrogenase
HSL: hormone sensitive lipase
IAAT: intra-abdominal adipose tissue
IL-6: interleukin-6
IR: insulin resistance
IRF4: interferon regulatory factor-4
KO: knock out
MHO: metabolically healthy obesity
MHT: menopausal hormone therapy
MUHNW: metabolically unhealthy normal weight
MUHO: metabolically unhealthy obesity
OVX: ovariectomy
Preg: pregnenolone
PKA: protein kinase A
PLIN: perilipin
SAT: subcutaneous adipose tissue
SHBG: sex hormone binding globulin
StAR: steroidogenic acute regulatory protein
sWAT: subcutaneous white adipose tissue
T: testosterone
T2D: type 2 diabetes
TNFα: tumor necrosis factor alpha
VAT: visceral adipose tissue
vWAT: visceral white adipose tissue
WAT: white adipose tissue
WHI: Women’s Health Initiative
WHR: waist-to-hip ratio
WT: wild type.
References
1. (NCD-RisC) NRFC. Trends in adult body-mass index in 200 countries from 1975 to 2014: a pooled analysis of 1698 population-based measurement studies with 19·2 million participants. Lancet. (2016) 387:1377–96. doi: 10.1016/S0140-6736(16)30054-X
2. Chait A and den Hartigh LJ. Adipose tissue distribution, inflammation and its metabolic consequences, including diabetes and cardiovascular disease. Front Cardiovasc Med. (2020) 7:22. doi: 10.3389/fcvm.2020.00022
3. Reddy P, Lent-Schochet D, Ramakrishnan N, McLaughlin M, and Jialal I. Metabolic syndrome is an inflammatory disorder: A conspiracy between adipose tissue and phagocytes. Clin Chim Acta. (2019) 496:35–44. doi: 10.1016/j.cca.2019.06.019
4. Wajchenberg BL. Subcutaneous and visceral adipose tissue: their relation to the metabolic syndrome. Endocr Rev. (2000) 21:697–738. doi: 10.1210/edrv.21.6.0415
5. Kwon H, Kim D, and Kim JS. Body fat distribution and the risk of incident metabolic syndrome: A longitudinal cohort study. Sci Rep. (2017) 7:10955. doi: 10.1038/s41598-017-09723-y
6. Donato GB, Fuchs SC, Oppermann K, Bastos C, and Spritzer PM. Association between menopause status and central adiposity measured at different cutoffs of waist circumference and waist-to-hip ratio. Menopause. (2006) 13:280–5. doi: 10.1097/01.gme.0000177907.32634.ae
7. Hill JH, Solt C, and Foster MT. Obesity associated disease risk: the role of inherent differences and location of adipose depots. Horm Mol Biol Clin Investig. (2018) 33:1–16. doi: 10.1515/hmbci-2018-0012
8. Kranendonk ME, van Herwaarden JA, Stupkova T, de Jager W, Vink A, Moll FL, et al. Inflammatory characteristics of distinct abdominal adipose tissue depots relate differently to metabolic risk factors for cardiovascular disease: distinct fat depots and vascular risk factors. Atherosclerosis. (2015) 239:419–27. doi: 10.1016/j.atherosclerosis.2015.01.035
9. Qureshi R, Picon-Ruiz M, Sho M, Van Booven D, Nunes de Paiva V, Diaz-Ruano AB, et al. Estrone, the major postmenopausal estrogen, binds ERa to induce SNAI2, epithelial-to-mesenchymal transition, and ER+ breast cancer metastasis. Cell Rep. (2022) 41:111672. doi: 10.1016/j.celrep.2022.111672
10. Steiner BM and Berry DC. The regulation of adipose tissue health by estrogens. Front Endocrinol (Lausanne). (2022) 13:889923. doi: 10.3389/fendo.2022.889923
11. Simpson ER. Sources of estrogen and their importance. J Steroid Biochem Mol Biol. (2003) 86:225–30. doi: 10.1016/s0960-0760(03)00360-1
12. Pogodziński D, Ostrowska L, Smarkusz-Zarzecka J, and Zyśk B. Secretome of adipose tissue as the key to understanding the endocrine function of adipose tissue. Int J Mol Sci. (2022) 23:1–14. doi: 10.3390/ijms23042309
13. Kahn D, Macias E, Zarini S, Garfield A, Zemski Berry K, MacLean P, et al. Exploring visceral and subcutaneous adipose tissue secretomes in human obesity: implications for metabolic disease. Endocrinology. (2022) 163:1–11. doi: 10.1210/endocr/bqac140
14. Walker GE, Marzullo P, Ricotti R, Bona G, and Prodam F. The pathophysiology of abdominal adipose tissue depots in health and disease. Horm Mol Biol Clin Investig. (2014) 19:57–74. doi: 10.1515/hmbci-2014-0023
15. Tchkonia T, Thomou T, Zhu Y, Karagiannides I, Pothoulakis C, Jensen MD, et al. Mechanisms and metabolic implications of regional differences among fat depots. Cell Metab. (2013) 17:644–56. doi: 10.1016/j.cmet.2013.03.008
16. Lee MJ and Fried SK. Sex-dependent depot differences in adipose tissue development and function; role of sex steroids. J Obes Metab Syndr. (2017) 26:172–80. doi: 10.7570/jomes.2017.26.3.172
17. Clemente-Suárez VJ, Redondo-Flórez L, Beltrán-Velasco AI, Martín-Rodríguez A, Martínez-Guardado I, Navarro-Jiménez E, et al. The role of adipokines in health and disease. Biomedicines. (2023) 11:1–41. doi: 10.3390/biomedicines11051290
18. Kawai T, Autieri MV, and Scalia R. Adipose tissue inflammation and metabolic dysfunction in obesity. Am J Physiol Cell Physiol. (2021) 320:C375–91. doi: 10.1152/ajpcell.00379.2020
19. Mathew H, Farr OM, and Mantzoros CS. Metabolic health and weight: Understanding metabolically unhealthy normal weight or metabolically healthy obese patients. Metabolism. (2016) 65:73–80. doi: 10.1016/j.metabol.2015.10.019
20. Stefan N, Kantartzis K, Machann J, Schick F, Thamer C, Rittig K, et al. Identification and characterization of metabolically benign obesity in humans. Arch Intern Med. (2008) 168:1609–16. doi: 10.1001/archinte.168.15.1609
21. Karelis AD, Faraj M, Bastard JP, St-Pierre DH, Brochu M, Prud’homme D, et al. The metabolically healthy but obese individual presents a favorable inflammation profile. J Clin Endocrinol Metab. (2005) 90:4145–50. doi: 10.1210/jc.2005-0482
22. Brochu M, Tchernof A, Dionne IJ, Sites CK, Eltabbakh GH, Sims EA, et al. What are the physical characteristics associated with a normal metabolic profile despite a high level of obesity in postmenopausal women? J Clin Endocrinol Metab. (2001) 86:1020–5. doi: 10.1210/jcem.86.3.7365
23. O’Connell J, Lynch L, Cawood TJ, Kwasnik A, Nolan N, Geoghegan J, et al. The relationship of omental and subcutaneous adipocyte size to metabolic disease in severe obesity. PloS One. (2010) 5:e9997. doi: 10.1371/journal.pone.0009997
24. Liu F, He J, Liu B, Zhang P, Wang H, Sun X, et al. Association of omental adipocyte hypertrophy and fibrosis with human obesity and type 2 diabetes. Obes (Silver Spring). (2021) 29:976–84. doi: 10.1002/oby.23155
25. Veilleux A, Caron-Jobin M, Noël S, Laberge PY, and Tchernof A. Visceral adipocyte hypertrophy is associated with dyslipidemia independent of body composition and fat distribution in women. Diabetes. (2011) 60:1504–11. doi: 10.2337/db10-1039
26. Fakhouri TH, Ogden CL, Carroll MD, Kit BK, and Flegal KM. Prevalence of obesity among older adults in the United States, 2007-2010. In: NCHS Data Brief. Hyattsville, MD, USA: US Department of Health and Human Services, Center for Disease Control and Prevention (2012). p. 1–8.
27. Batsis JA and Zagaria AB. Addressing obesity in aging patients. Med Clin North Am. (2018) 102:65–85. doi: 10.1016/j.mcna.2017.08.007
28. Cartwright MJ, Tchkonia T, and Kirkland JL. Aging in adipocytes: potential impact of inherent, depot-specific mechanisms. Exp Gerontol. (2007) 42:463–71. doi: 10.1016/j.exger.2007.03.003
29. Kirkland JL, Tchkonia T, Pirtskhalava T, Han J, and Karagiannides I. Adipogenesis and aging: does aging make fat go MAD? Exp Gerontol. (2002) 37:757–67. doi: 10.1016/s0531-5565(02)00014-1
30. Bertrand HA, Lynd FT, Masoro EJ, and Yu BP. Changes in adipose mass and cellularity through the adult life of rats fed ad libitum or a life-prolonging restricted diet. J Gerontol. (1980) 35:827–35. doi: 10.1093/geronj/35.6.827
31. Spalding KL, Arner E, Westermark PO, Bernard S, Buchholz BA, Bergmann O, et al. Dynamics of fat cell turnover in humans. Nature. (2008) 453:783–7. doi: 10.1038/nature06902
32. Tchernof A and Després JP. Pathophysiology of human visceral obesity: an update. Physiol Rev. (2013) 93:359–404. doi: 10.1152/physrev.00033.2011
33. Kahn SE, Hull RL, and Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. (2006) 444:840–6. doi: 10.1038/nature05482
34. Goodpaster BH, He J, Watkins S, and Kelley DE. Skeletal muscle lipid content and insulin resistance: evidence for a paradox in endurance-trained athletes. J Clin Endocrinol Metab. (2001) 86:5755–61. doi: 10.1210/jcem.86.12.8075
35. Gan L, Chitturi S, and Farrell GC. Mechanisms and implications of age-related changes in the liver: nonalcoholic Fatty liver disease in the elderly. Curr Gerontol Geriatr Res. (2011) 2011:831536. doi: 10.1155/2011/831536
36. Aaron N, Costa S, Rosen CJ, and Qiang L. The implications of bone marrow adipose tissue on inflammaging. Front Endocrinol (Lausanne). (2022) 13:853765. doi: 10.3389/fendo.2022.853765
37. Mancuso P and Bouchard B. The impact of aging on adipose function and adipokine synthesis. Front Endocrinol (Lausanne). (2019) 10:137. doi: 10.3389/fendo.2019.00137
38. Fulop T, Larbi A, Pawelec G, Khalil A, Cohen AA, Hirokawa K, et al. Immunology of aging: the birth of inflammaging. Clin Rev Allergy Immunol. (2023) 64:109–22. doi: 10.1007/s12016-021-08899-6
39. Lumeng CN and Saltiel AR. Inflammatory links between obesity and metabolic disease. J Clin Invest. (2011) 121:2111–7. doi: 10.1172/JCI57132
40. Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, and Ferrante AW. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. (2003) 112:1796–808. doi: 10.1172/JCI19246
41. Zhang YX, Ou MY, Yang ZH, Sun Y, Li QF, and Zhou SB. Adipose tissue aging is regulated by an altered immune system. Front Immunol. (2023) 14:1125395. doi: 10.3389/fimmu.2023.1125395
42. Su HI and Freeman EW. Hormone changes associated with the menopausal transition. Minerva Ginecol. (2009) 61:483–9.
43. Cioffi M, Esposito K, Vietri MT, Gazzerro P, D’Auria A, Ardovino I, et al. Cytokine pattern in postmenopause. Maturitas. (2002) 41:187–92. doi: 10.1016/s0378-5122(01)00286-9
44. Hotamisligil GS. Inflammation and metabolic disorders. Nature. (2006) 444:860–7. doi: 10.1038/nature05485
45. Karim R, Stanczyk FZ, Brinton RD, Rettberg J, Hodis HN, and Mack WJ. Association of endogenous sex hormones with adipokines and ghrelin in postmenopausal women. J Clin Endocrinol Metab. (2015) 100:508–15. doi: 10.1210/jc.2014-2834
46. Ebong IA, Michos ED, Wilson M, Appiah D, Schreiner PJ, Racette SB, et al. Adipokines and adiposity among postmenopausal women of the Multi-Ethnic Study of Atherosclerosis. Menopause. (2024) 31:209–17. doi: 10.1097/GME.0000000000002261
47. Chu MC, Cosper P, Orio F, Carmina E, and Lobo RA. Insulin resistance in postmenopausal women with metabolic syndrome and the measurements of adiponectin, leptin, resistin, and ghrelin. Am J Obstet Gynecol. (2006) 194:100–4. doi: 10.1016/j.ajog.2005.06.073
48. Störk S, Bots ML, Angerer P, von Schacky C, Grobbee DE, Angermann CE, et al. Low levels of adiponectin predict worsening of arterial morphology and function. Atherosclerosis. (2007) 194:e147–153. doi: 10.1016/j.atherosclerosis.2006.11.044
49. Huang WY, Chang CC, Chen DR, Kor CT, Chen TY, and Wu HM. Circulating leptin and adiponectin are associated with insulin resistance in healthy postmenopausal women with hot flashes. PloS One. (2017) 12:e0176430. doi: 10.1371/journal.pone.0176430
50. Blüher M. Metabolically healthy obesity. Endocr Rev. (2020) 41:1–16. doi: 10.1210/endrev/bnaa004
51. Palmer BF and Clegg DJ. The sexual dimorphism of obesity. Mol Cell Endocrinol. (2015) 402:113–9. doi: 10.1016/j.mce.2014.11.029
52. Grundy SM. Adipose tissue and metabolic syndrome: too much, too little or neither. Eur J Clin Invest. (2015) 45:1209–17. doi: 10.1111/eci.12519
53. Goossens GH. The role of adipose tissue dysfunction in the pathogenesis of obesity-related insulin resistance. Physiol Behav. (2008) 94:206–18. doi: 10.1016/j.physbeh.2007.10.010
54. Horwitz A and Birk R. Adipose tissue hyperplasia and hypertrophy in common and syndromic obesity-the case of BBS obesity. Nutrients. (2023) 15:1–22. doi: 10.3390/nu15153445
55. Weisberg SP, Hunter D, Huber R, Lemieux J, Slaymaker S, Vaddi K, et al. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J Clin Invest. (2006) 116:115–24. doi: 10.1172/JCI24335
56. Johnson AR, Milner JJ, and Makowski L. The inflammation highway: metabolism accelerates inflammatory traffic in obesity. Immunol Rev. (2012) 249:218–38. doi: 10.1111/j.1600-065X.2012.01151.x
57. Cinti S, Mitchell G, Barbatelli G, Murano I, Ceresi E, Faloia E, et al. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J Lipid Res. (2005) 46:2347–55. doi: 10.1194/jlr.M500294-JLR200
58. Ellulu MS, Patimah I, Khaza’ai H, Rahmat A, and Abed Y. Obesity and inflammation: the linking mechanism and the complications. Arch Med Sci. (2017) 13:851–63. doi: 10.5114/aoms.2016.58928
59. Frasca D, Blomberg BB, and Paganelli R. Aging, obesity, and inflammatory age-related diseases. Front Immunol. (2017) 8:1745. doi: 10.3389/fimmu.2017.01745
60. Thomas AL, Alarcon PC, Divanovic S, Chougnet CA, Hildeman DA, and Moreno-Fernandez ME. Implications of inflammatory states on dysfunctional immune responses in aging and obesity. Front Aging. (2021) 2:732414. doi: 10.3389/fragi.2021.732414
61. Payne AH and Hales DB. Overview of steroidogenic enzymes in the pathway from cholesterol to active steroid hormones. Endocr Rev. (2004) 25:947–70. doi: 10.1210/er.2003-0030
62. Kuryłowicz A. Estrogens in adipose tissue physiology and obesity-related dysfunction. Biomedicines. (2023) 11:1–23. doi: 10.3390/biomedicines11030690
63. Li J, Papadopoulos V, and Vihma V. Steroid biosynthesis in adipose tissue. Steroids. (2015) 103:89–104. doi: 10.1016/j.steroids.2015.03.016
64. Hetemäki N, Savolainen-Peltonen H, Tikkanen MJ, Wang F, Paatela H, Hämäläinen E, et al. Estrogen metabolism in abdominal subcutaneous and visceral adipose tissue in postmenopausal women. J Clin Endocrinol Metab. (2017) 102:4588–95. doi: 10.1210/jc.2017-01474
65. Dumitrescu R, Mehedintu C, Briceag I, Purcarea VL, and Hudita D. The polycystic ovary syndrome: an update on metabolic and hormonal mechanisms. J Med Life. (2015) 8:142–5.
66. Kim N and Chun S. Association between the serum estrone-to-estradiol ratio and parameters related to glucose metabolism and insulin resistance in women with polycystic ovary syndrome. Clin Exp Reprod Med. (2021) 48:374–9. doi: 10.5653/cerm.2021.04553
67. Kautzky-Willer A and Handisurya A. Metabolic diseases and associated complications: sex and gender matter! Eur J Clin Invest. (2009) 39:631–48. doi: 10.1111/j.1365-2362.2009.02161.x
68. Kotani K, Tokunaga K, Fujioka S, Kobatake T, Keno Y, Yoshida S, et al. Sexual dimorphism of age-related changes in whole-body fat distribution in the obese. Int J Obes Relat Metab Disord. (1994) 18:207–2.
69. Rubin R. Postmenopausal women with a “Normal” BMI might be overweight or even obese. JAMA. (2018) 319:1185–7. doi: 10.1001/jama.2018.0423
70. Jeffery E, Wing A, Holtrup B, Sebo Z, Kaplan JL, Saavedra-Peña R, et al. The adipose tissue microenvironment regulates depot-specific adipogenesis in obesity. Cell Metab. (2016) 24(1):142–50. doi: 10.1016/j.cmet.2016.05.012
71. Jeffery E, Church CD, Holtrup B, Colman L, and Rodeheffer MS. Rapid depot-specific activation of adipocyte precursor cells at the onset of obesity. Nat Cell Biol. (2015) 17:376–85. doi: 10.1038/ncb3122
72. Sebo ZL and Rodeheffer MS. Testosterone metabolites differentially regulate obesogenesis and fat distribution. Mol Metab. (2021) 44:101141. doi: 10.1016/j.molmet.2020.101141
73. Tramunt B, Smati S, Grandgeorge N, Lenfant F, Arnal JF, Montagner A, et al. Sex differences in metabolic regulation and diabetes susceptibility. Diabetologia. (2020) 63:453–61. doi: 10.1007/s00125-019-05040-3
74. Margolis KL, Bonds DE, Rodabough RJ, Tinker L, Phillips LS, Allen C, et al. Effect of oestrogen plus progestin on the incidence of diabetes in postmenopausal women: results from the Women’s Health Initiative Hormone Trial. Diabetologia. (2004) 47:1175–87. doi: 10.1007/s00125-004-1448-x
75. Riant E, Waget A, Cogo H, Arnal JF, Burcelin R, and Gourdy P. Estrogens protect against high-fat diet-induced insulin resistance and glucose intolerance in mice. Endocrinology. (2009) 150:2109–17. doi: 10.1210/en.2008-0971
76. Vieira Potter VJ, Strissel KJ, Xie C, Chang E, Bennett G, Defuria J, et al. Adipose tissue inflammation and reduced insulin sensitivity in ovariectomized mice occurs in the absence of increased adiposity. Endocrinology. (2012) 153:4266–77. doi: 10.1210/en.2011-2006
77. Bruun JM, Nielsen CB, Pedersen SB, Flyvbjerg A, and Richelsen B. Estrogen reduces pro-inflammatory cytokines in rodent adipose tissue: studies in vivo and in vitro. Horm Metab Res. (2003) 35:142–6. doi: 10.1055/s-2003-39074
78. Blaustein JD and Wade GN. Ovarian influences on the meal patterns of female rats. Physiol Behav. (1976) 17:201–8. doi: 10.1016/0031-9384(76)90064-0
79. Wallen WJ, Belanger MP, and Wittnich C. Sex hormones and the selective estrogen receptor modulator tamoxifen modulate weekly body weights and food intakes in adolescent and adult rats. J Nutr. (2001) 131:2351–7. doi: 10.1093/jn/131.9.2351
80. Camporez JP, Lyu K, Goldberg EL, Zhang D, Cline GW, Jurczak MJ, et al. Anti-inflammatory effects of oestrogen mediate the sexual dimorphic response to lipid-induced insulin resistance. J Physiol. (2019) 597:3885–903. doi: 10.1113/JP277270
81. Litwak SA, Wilson JL, Chen W, Garcia-Rudaz C, Khaksari M, Cowley MA, et al. Estradiol prevents fat accumulation and overcomes leptin resistance in female high-fat diet mice. Endocrinology. (2014) 155:4447–60. doi: 10.1210/en.2014-1342
82. Cruz AGD, Santos JDMD, Alves EDS, Santos ARMD, Trinca BF, Camargo FN, et al. Metabolic effects of late-onset estradiol replacement in high-fat-fed ovariectomized mice. Curr Res Physiol. (2025) 8:100144. doi: 10.1016/j.crphys.2025.100144
83. Zhu L, Brown WC, Cai Q, Krust A, Chambon P, McGuinness OP, et al. Estrogen treatment after ovariectomy protects against fatty liver and may improve pathway-selective insulin resistance. Diabetes. (2013) 62:424–34. doi: 10.2337/db11-1718
84. Bhardwaj P, Du B, Zhou XK, Sue E, Giri D, Harbus MD, et al. Estrogen protects against obesity-induced mammary gland inflammation in mice. Cancer Prev Res (Phila). (2015) 8:751–9. doi: 10.1158/1940-6207.CAPR-15-0082
85. Macotela Y, Boucher J, Tran TT, and Kahn CR. Sex and depot differences in adipocyte insulin sensitivity and glucose metabolism. Diabetes. (2009) 58:803–12. doi: 10.2337/db08-1054
86. Fraenkel M, Caloyeras J, Ren SG, and Melmed S. Sex-steroid milieu determines diabetes rescue in pttg-null mice. J Endocrinol. (2006) 189:519–28. doi: 10.1677/joe.1.06656
87. Aoki A, Fujitani K, Takagi K, Kimura T, Nagase H, and Nakanishi T. Male hypogonadism causes obesity associated with impairment of hepatic gluconeogenesis in mice. Biol Pharm Bull. (2016) 39:587–92. doi: 10.1248/bpb.b15-00942
88. Baik M, Jeong JY, Park SJ, Yoo SP, Lee JO, Lee JS, et al. Testosterone deficiency caused by castration increases adiposity in male rats in a tissue-specific and diet-dependent manner. Genes Nutr. (2020) 15:14. doi: 10.1186/s12263-020-00673-1
89. Ribas V, Nguyen MT, Henstridge DC, Nguyen AK, Beaven SW, Watt MJ, et al. Impaired oxidative metabolism and inflammation are associated with insulin resistance in ERalpha-deficient mice. Am J Physiol Endocrinol Metab. (2010) 298:E304–319. doi: 10.1152/ajpendo.00504.2009
90. Heine PA, Taylor JA, Iwamoto GA, Lubahn DB, and Cooke PS. Increased adipose tissue in male and female estrogen receptor-alpha knockout mice. Proc Natl Acad Sci U S A. (2000) 97:12729–34. doi: 10.1073/pnas.97.23.12729
91. Naaz A, Zakroczymski M, Heine P, Taylor J, Saunders P, Lubahn D, et al. Effect of ovariectomy on adipose tissue of mice in the absence of estrogen receptor alpha (ERalpha): a potential role for estrogen receptor beta (ERbeta). Horm Metab Res. (2002) 34:758–63. doi: 10.1055/s-2002-38259
92. Ohlsson C, Hellberg N, Parini P, Vidal O, Bohlooly-Y M, Rudling M, et al. Obesity and disturbed lipoprotein profile in estrogen receptor-alpha-deficient male mice. Biochem Biophys Res Commun. (2000) 278:640–5. doi: 10.1006/bbrc.2000.3827
93. Davis KE, Neinast MD, Sun K, Skiles WM, Bills JD, Zehr JA, et al. The sexually dimorphic role of adipose and adipocyte estrogen receptors in modulating adipose tissue expansion, inflammation, and fibrosis. Mol Metab. (2013) 2:227–42. doi: 10.1016/j.molmet.2013.05.006
94. Jones ME, Thorburn AW, Britt KL, Hewitt KN, Misso ML, Wreford NG, et al. Aromatase-deficient (ArKO) mice accumulate excess adipose tissue. J Steroid Biochem Mol Biol. (2001) 79:3–9. doi: 10.1016/s0960-0760(01)00136-4
95. Takeda K, Toda K, Saibara T, Nakagawa M, Saika K, Onishi T, et al. Progressive development of insulin resistance phenotype in male mice with complete aromatase (CYP19) deficiency. J Endocrinol. (2003) 176:237–46. doi: 10.1677/joe.0.1760237
96. Jones ME, Thorburn AW, Britt KL, Hewitt KN, Wreford NG, Proietto J, et al. Aromatase-deficient (ArKO) mice have a phenotype of increased adiposity. Proc Natl Acad Sci U S A. (2000) 97:12735–40. doi: 10.1073/pnas.97.23.12735
97. Ohlsson C, Hammarstedt A, Vandenput L, Saarinen N, Ryberg H, Windahl SH, et al. Increased adipose tissue aromatase activity improves insulin sensitivity and reduces adipose tissue inflammation in male mice. Am J Physiol Endocrinol Metab. (2017) 313:E450–62. doi: 10.1152/ajpendo.00093.2017
98. Simpson ER, Zhao Y, Agarwal VR, Michael MD, Bulun SE, Hinshelwood MM, et al. Aromatase expression in health and disease. Recent Prog Horm Res. (1997) 52:185–213; discussion 213-184.
99. Kozakowski J, Gietka-Czernel M, Leszczyńska D, and Majos A. Obesity in menopause - our negligence or an unfortunate inevitability? Prz Menopauzalny. (2017) 16:61–5. doi: 10.5114/pm.2017.68594
100. Daka B, Rosen T, Jansson PA, Råstam L, Larsson CA, and Lindblad U. Inverse association between serum insulin and sex hormone-binding globulin in a population survey in Sweden. Endocr Connect. (2013) 2:18–22. doi: 10.1530/EC-12-0057
101. Raeisi-Dehkordi H, Amiri M, Rathmann W, Zeller T, Adamski J, Bano A, et al. Sex hormone-binding globulin may explain sex differences for glucose homeostasis and incidence of type 2 diabetes: the KORA study. Eur J Epidemiol. (2024) 39:915–24. doi: 10.1007/s10654-024-01136-2
102. Ding EL, Song Y, Manson JE, Hunter DJ, Lee CC, Rifai N, et al. Sex hormone-binding globulin and risk of type 2 diabetes in women and men. N Engl J Med. (2009) 361:1152–63. doi: 10.1056/NEJMoa0804381
103. Le TN, Nestler JE, Strauss JF, and Wickham EP. Sex hormone-binding globulin and type 2 diabetes mellitus. Trends Endocrinol Metab. (2012) 23:32–40. doi: 10.1016/j.tem.2011.09.005
104. Perry JR, Weedon MN, Langenberg C, Jackson AU, Lyssenko V, Sparsø T, et al. Genetic evidence that raised sex hormone binding globulin (SHBG) levels reduce the risk of type 2 diabetes. Hum Mol Genet. (2010) 19:535–44. doi: 10.1093/hmg/ddp522
105. Li M, Chi X, Wang Y, Setrerrahmane S, Xie W, and Xu H. Trends in insulin resistance: insights into mechanisms and therapeutic strategy. Signal Transduct Target Ther. (2022) 7:216. doi: 10.1038/s41392-022-01073-0
106. Pasquali R, Casimirri F, De Iasio R, Mesini P, Boschi S, Chierici R, et al. Insulin regulates testosterone and sex hormone-binding globulin concentrations in adult normal weight and obese men. J Clin Endocrinol Metab. (1995) 80:654–8. doi: 10.1210/jcem.80.2.7852532
107. Strain G, Zumoff B, Rosner W, and Pi-Sunyer X. The relationship between serum levels of insulin and sex hormone-binding globulin in men: the effect of weight loss. J Clin Endocrinol Metab. (1994) 79:1173–6. doi: 10.1210/jcem.79.4.7962291
108. Sofer Y, Nevo N, Vechoropoulos M, Shefer G, Osher E, Landis N, et al. Human sex hormone-binding globulin does not provide metabolic protection against diet-induced obesity and dysglycemia in mice. Endocr Connect. (2018) 7:91–6. doi: 10.1530/EC-17-0240
109. Saez-Lopez C, Villena JA, Simó R, and Selva DM. Sex hormone-binding globulin overexpression protects against high-fat diet-induced obesity in transgenic male mice. J Nutr Biochem. (2020) 85:108480. doi: 10.1016/j.jnutbio.2020.108480
110. Hedderson MM, Capra A, Lee C, Habel LA, Lee J, Gold EB, et al. Longitudinal changes in sex hormone binding globulin (SHBG) and risk of incident diabetes: the study of women’s health across the nation (SWAN). Diabetes Care. (2024) 47:676–82. doi: 10.2337/dc23-1630
111. Azrad M, Gower BA, Hunter GR, and Nagy TR. Intra-abdominal adipose tissue is independently associated with sex-hormone binding globulin in premenopausal women. Obes (Silver Spring). (2012) 20:1012–5. doi: 10.1038/oby.2011.375
112. Weinberg ME, Manson JE, Buring JE, Cook NR, Seely EW, Ridker PM, et al. Low sex hormone-binding globulin is associated with the metabolic syndrome in postmenopausal women. Metabolism. (2006) 55:1473–80. doi: 10.1016/j.metabol.2006.06.017
113. Maggio M, Lauretani F, Basaria S, Ceda GP, Bandinelli S, Metter EJ, et al. Sex hormone binding globulin levels across the adult lifespan in women–the role of body mass index and fasting insulin. J Endocrinol Invest. (2008) 31:597–601. doi: 10.1007/BF03345608
114. Stanczyk FZ, Sriprasert I, Karim R, Hwang-Levine J, Mack WJ, and Hodis HN. Concentrations of endogenous sex steroid hormones and SHBG in healthy postmenopausal women. J Steroid Biochem Mol Biol. (2022) 223:106080. doi: 10.1016/j.jsbmb.2022.106080
115. Skałba P, Wójtowicz M, and Sikora J. Androgen and SHBG serum concentrations in late post-menopause women. Med Sci Monit. (2003) 9:CR152–156.
116. Pasquali R, Vicennati V, Bertazzo D, Casimirri F, Pascal G, Tortelli O, et al. Determinants of sex hormone-binding globulin blood concentrations in premenopausal and postmenopausal women with different estrogen status. Virgilio-Menopause-Health Group. Metabolism. (1997) 46:5–9. doi: 10.1016/s0026-0495(97)90159-1
117. Bonnet F, Velayoudom Cephise FL, Gautier A, Dubois S, Massart C, Camara A, et al. Role of sex steroids, intrahepatic fat and liver enzymes in the association between SHBG and metabolic features. Clin Endocrinol (Oxf). (2013) 79:517–22. doi: 10.1111/cen.12089
118. Goss AM, Darnell BE, Brown MA, Oster RA, and Gower BA. Longitudinal associations of the endocrine environment on fat partitioning in postmenopausal women. Obes (Silver Spring). (2012) 20:939–44. doi: 10.1038/oby.2011.362
119. Levin ER. Integration of the extranuclear and nuclear actions of estrogen. Mol Endocrinol. (2005) 19:1951–9. doi: 10.1210/me.2004-0390
120. Kato S, Endoh H, Masuhiro Y, Kitamoto T, Uchiyama S, Sasaki H, et al. Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science. (1995) 270:1491–4. doi: 10.1126/science.270.5241.1491
121. Zhu BT, Han GZ, Shim JY, Wen Y, and Jiang XR. Quantitative structure-activity relationship of various endogenous estrogen metabolites for human estrogen receptor alpha and beta subtypes: Insights into the structural determinants favoring a differential subtype binding. Endocrinology. (2006) 147:4132–50. doi: 10.1210/en.2006-0113
122. Hernández-Silva CD, Villegas-Pineda JC, and Pereira-Suárez AL. Expression and role of the G protein-coupled estrogen receptor (GPR30/GPER) in the development and immune response in female reproductive cancers. Front Endocrinol (Lausanne). (2020) 11:544. doi: 10.3389/fendo.2020.00544
123. Biason-Lauber A and Lang-Muritano M. Estrogens: Two nuclear receptors, multiple possibilities. Mol Cell Endocrinol. (2022) 554:111710. doi: 10.1016/j.mce.2022.111710
124. Samartzis N, Samartzis EP, Noske A, Fedier A, Dedes KJ, Caduff R, et al. Expression of the G protein-coupled estrogen receptor (GPER) in endometriosis: a tissue microarray study. Reprod Biol Endocrinol. (2012) 10:30. doi: 10.1186/1477-7827-10-30
125. Notas G, Kampa M, and Castanas E. G protein-coupled estrogen receptor in immune cells and its role in immune-related diseases. Front Endocrinol (Lausanne). (2020) 11:579420. doi: 10.3389/fendo.2020.579420
126. Prossnitz ER and Barton M. The G-protein-coupled estrogen receptor GPER in health and disease. Nat Rev Endocrinol. (2011) 7:715–26. doi: 10.1038/nrendo.2011.122
127. Nimrod A and Ryan KJ. Aromatization of androgens by human abdominal and breast fat tissue. J Clin Endocrinol Metab. (1975) 40:367–72. doi: 10.1210/jcem-40-3-367
128. Cleland WH, Mendelson CR, and Simpson ER. Aromatase activity of membrane fractions of human adipose tissue stromal cells and adipocytes. Endocrinology. (1983) 113:2155–60. doi: 10.1210/endo-113-6-2155
129. Kim M, Neinast MD, Frank AP, Sun K, Park J, Zehr JA, et al. ERα upregulates Phd3 to ameliorate HIF-1 induced fibrosis and inflammation in adipose tissue. Mol Metab. (2014) 3:642–51. doi: 10.1016/j.molmet.2014.05.007
130. Mauvais-Jarvis F, Clegg DJ, and Hevener AL. The role of estrogens in control of energy balance and glucose homeostasis. Endocr Rev. (2013) 34:309–38. doi: 10.1210/er.2012-1055
131. Pereira RI, Casey BA, Swibas TA, Erickson CB, Wolfe P, and Van Pelt RE. Timing of estradiol treatment after menopause may determine benefit or harm to insulin action. J Clin Endocrinol Metab. (2015) 100:4456–62. doi: 10.1210/jc.2015-3084
132. Sharma G and Prossnitz ER. Targeting the G protein-coupled estrogen receptor (GPER) in obesity and diabetes. Endocr Metab Sci. (2021) 2:1–8. doi: 10.1016/j.endmts.2021.100080
133. Liu L, Zhou Y, Liu J, Zhang X, He C, Zeng X, et al. GPER in metabolic homeostasis and disease: molecular mechanisms, nutritional regulation, and therapeutic potential. J Transl Med. (2025) 23:960. doi: 10.1186/s12967-025-07005-0
134. Qureshi R, Picon-Ruiz M, Aurrekoetxea-Rodriguez I, Nunes de Paiva V, D’Amico M, Yoon H, et al. The major pre- and postmenopausal estrogens play opposing roles in obesity-driven mammary inflammation and breast cancer development. Cell Metab. (2020) 31:1154–1172.e1159. doi: 10.1016/j.cmet.2020.05.008
135. Bhardwaj P, Au CC, Benito-Martin A, Ladumor H, Oshchepkova S, Moges R, et al. Estrogens and breast cancer: Mechanisms involved in obesity-related development, growth and progression. J Steroid Biochem Mol Biol. (2019) 189:161–70. doi: 10.1016/j.jsbmb.2019.03.002
136. Baglietto L, English DR, Hopper JL, MacInnis RJ, Morris HA, Tilley WD, et al. Circulating steroid hormone concentrations in postmenopausal women in relation to body size and composition. Breast Cancer Res Treat. (2009) 115:171–9. doi: 10.1007/s10549-008-0069-3
137. Lukanova A, Lundin E, Zeleniuch-Jacquotte A, Muti P, Mure A, Rinaldi S, et al. Body mass index, circulating levels of sex-steroid hormones, IGF-I and IGF-binding protein-3: a cross-sectional study in healthy women. Eur J Endocrinol. (2004) 150:161–71. doi: 10.1530/eje.0.1500161
138. Key TJ, Appleby PN, Reeves GK, Roddam A, Dorgan JF, Longcope C, et al. Body mass index, serum sex hormones, and breast cancer risk in postmenopausal women. J Natl Cancer Inst. (2003) 95:1218–26. doi: 10.1093/jnci/djg022
139. Kley HK, Deselaers T, Peerenboom H, and Krüskemper HL. Enhanced conversion of androstenedione to estrogens in obese males. J Clin Endocrinol Metab. (1980) 51:1128–32. doi: 10.1210/jcem-51-5-1128
140. Kley HK, Edelmann P, and Krüskemper HL. Relationship of plasma sex hormones to different parameters of obesity in male subjects. Metabolism. (1980) 29:1041–5. doi: 10.1016/0026-0495(80)90214-0
141. MacDonald PC, Edman CD, Hemsell DL, Porter JC, and Siiteri PK. Effect of obesity on conversion of plasma androstenedione to estrone in postmenopausal women with and without endometrial cancer. Am J Obstet Gynecol. (1978) 130:448–55. doi: 10.1016/0002-9378(78)90287-9
142. Remesar X, Tang V, Ferrer E, Torregrosa C, Virgili J, Masanés RM, et al. Estrone in food: a factor influencing the development of obesity? Eur J Nutr. (1999) 38:247–53. doi: 10.1007/s003940050068
143. Bryzgalova G, Gao H, Ahren B, Zierath JR, Galuska D, Steiler TL, et al. Evidence that oestrogen receptor-alpha plays an important role in the regulation of glucose homeostasis in mice: insulin sensitivity in the liver. Diabetologia. (2006) 49:588–97. doi: 10.1007/s00125-005-0105-3
144. Park CJ, Zhao Z, Glidewell-Kenney C, Lazic M, Chambon P, Krust A, et al. Genetic rescue of nonclassical ERα signaling normalizes energy balance in obese Erα-null mutant mice. J Clin Invest. (2011) 121:604–12. doi: 10.1172/JCI41702
145. Yang W, Jiang W, Liao W, Yan H, Ai W, Pan Q, et al. An estrogen receptor α-derived peptide improves glucose homeostasis during obesity. Nat Commun. (2024) 15:3410. doi: 10.1038/s41467-024-47687-6
146. Sacharidou A, Chambliss K, Peng J, Barrera J, Tanigaki K, Luby-Phelps K, et al. Endothelial ERα promotes glucose tolerance by enhancing endothelial insulin transport to skeletal muscle. Nat Commun. (2023) 14:4989. doi: 10.1038/s41467-023-40562-w
147. Haas E, Bhattacharya I, Brailoiu E, Damjanović M, Brailoiu GC, Gao X, et al. Regulatory role of G protein-coupled estrogen receptor for vascular function and obesity. Circ Res. (2009) 104:288–91. doi: 10.1161/CIRCRESAHA.108.190892
148. Sharma G, Hu C, Brigman JL, Zhu G, Hathaway HJ, and Prossnitz ER. GPER deficiency in male mice results in insulin resistance, dyslipidemia, and a proinflammatory state. Endocrinology. (2013) 154:4136–45. doi: 10.1210/en.2013-1357
149. Prossnitz ER and Hathaway HJ. What have we learned about GPER function in physiology and disease from knockout mice? J Steroid Biochem Mol Biol. (2015) 153:114–26. doi: 10.1016/j.jsbmb.2015.06.014
150. Barros RP, MaChado UF, Warner M, and Gustafsson JA. Muscle GLUT4 regulation by estrogen receptors ERbeta and ERalpha. Proc Natl Acad Sci U S A. (2006) 103:1605–8. doi: 10.1073/pnas.0510391103
151. Gorres BK, Bomhoff GL, Morris JK, and Geiger PC. In vivo stimulation of oestrogen receptor α increases insulin-stimulated skeletal muscle glucose uptake. J Physiol. (2011) 589:2041–54. doi: 10.1113/jphysiol.2010.199018
152. Ribas V, Drew BG, Zhou Z, Phun J, Kalajian NY, Soleymani T, et al. Skeletal muscle action of estrogen receptor α is critical for the maintenance of mitochondrial function and metabolic homeostasis in females. Sci Transl Med. (2016) 8:334ra354. doi: 10.1126/scitranslmed.aad3815
153. Iñigo MR, Amorese AJ, Tarpey MD, Balestrieri NP, Jones KG, Patteson DJ, et al. Estrogen receptor-α in female skeletal muscle is not required for regulation of muscle insulin sensitivity and mitochondrial regulation. Mol Metab. (2020) 34:1–15. doi: 10.1016/j.molmet.2019.12.010
154. Chen P, Li B, and Ou-Yang L. Role of estrogen receptors in health and disease. Front Endocrinol (Lausanne). (2022) 13:839005. doi: 10.3389/fendo.2022.839005
155. Gao H, Bryzgalova G, Hedman E, Khan A, Efendic S, Gustafsson JA, et al. Long-term administration of estradiol decreases expression of hepatic lipogenic genes and improves insulin sensitivity in ob/ob mice: a possible mechanism is through direct regulation of signal transducer and activator of transcription 3. Mol Endocrinol. (2006) 20:1287–99. doi: 10.1210/me.2006-0012
156. Shen M and Shi H. Sex hormones and their receptors regulate liver energy homeostasis. Int J Endocrinol. (2015) 2015:294278. doi: 10.1155/2015/294278
157. Parthasarathy C, Renuka VN, and Balasubramanian K. Sex steroids enhance insulin receptors and glucose oxidation in Chang liver cells. Clin Chim Acta. (2009) 399:49–53. doi: 10.1016/j.cca.2008.09.011
158. Zhu L, Shi J, Luu TN, Neuman JC, Trefts E, Yu S, et al. Hepatocyte estrogen receptor alpha mediates estrogen action to promote reverse cholesterol transport during Western-type diet feeding. Mol Metab. (2018) 8:106–16. doi: 10.1016/j.molmet.2017.12.012
159. Qiu S, Vazquez JT, Boulger E, Liu H, Xue P, Hussain MA, et al. Hepatic estrogen receptor α is critical for regulation of gluconeogenesis and lipid metabolism in males. Sci Rep. (2017) 7:1661. doi: 10.1038/s41598-017-01937-4
160. Iorga A, Cunningham CM, Moazeni S, Ruffenach G, Umar S, and Eghbali M. The protective role of estrogen and estrogen receptors in cardiovascular disease and the controversial use of estrogen therapy. Biol Sex Differ. (2017) 8:33. doi: 10.1186/s13293-017-0152-8
161. Yasrebi A, Rivera JA, Krumm EA, Yang JA, and Roepke TA. Activation of estrogen response element-independent ERα Signaling protects female mice from diet-induced obesity. Endocrinology. (2017) 158:319–34. doi: 10.1210/en.2016-1535
162. Mahboobifard F, Pourgholami MH, Jorjani M, Dargahi L, Amiri M, Sadeghi S, et al. Estrogen as a key regulator of energy homeostasis and metabolic health. BioMed Pharmacother. (2022) 156:113808. doi: 10.1016/j.biopha.2022.113808
163. Gordon GG, Olivo J, Rafil F, and Southren AL. Conversion of androgens to estrogens in cirrhosis of the liver. J Clin Endocrinol Metab. (1975) 40:1018–26. doi: 10.1210/jcem-40-6-1018
164. Kley HK, Krüskemper HL, and Keck E. Estrone and estradiol in patients with cirrhosis of the liver: effects of ACTH and dexamethasone. J Clin Endocrinol Metab. (1976) 43:557–60. doi: 10.1210/jcem-43-3-557
165. Brown LM and Clegg DJ. Central effects of estradiol in the regulation of food intake, body weight, and adiposity. J Steroid Biochem Mol Biol. (2010) 122:65–73. doi: 10.1016/j.jsbmb.2009.12.005
166. Cooke PS and Naaz A. Role of estrogens in adipocyte development and function. Exp Biol Med (Maywood). (2004) 229:1127–35. doi: 10.1177/153537020422901107
167. Roncari DA and Van RL. Promotion of human adipocyte precursor replication by 17beta-estradiol in culture. J Clin Invest. (1978) 62:503–8. doi: 10.1172/JCI109153
168. Lizcano F and Guzmán G. Estrogen deficiency and the origin of obesity during menopause. BioMed Res Int. (2014) 2014:757461. doi: 10.1155/2014/757461
169. Bélanger C, Hould FS, Lebel S, Biron S, Brochu G, and Tchernof A. Omental and subcutaneous adipose tissue steroid levels in obese men. Steroids. (2006) 71:674–82. doi: 10.1016/j.steroids.2006.04.008
170. Seyfart T, Friedrich N, Kische H, Bülow R, Wallaschofski H, Völzke H, et al. Association of sex hormones with physical, laboratory, and imaging markers of anthropometry in men and women from the general population. PloS One. (2018) 13:e0189042. doi: 10.1371/journal.pone.0189042
171. McTiernan A, Wu L, Chen C, Chlebowski R, Mossavar-Rahmani Y, Modugno F, et al. Relation of BMI and physical activity to sex hormones in postmenopausal women. Obes (Silver Spring). (2006) 14:1662–77. doi: 10.1038/oby.2006.191
172. Vihma V, Naukkarinen J, Turpeinen U, Hämäläinen E, Kaprio J, Rissanen A, et al. Metabolism of sex steroids is influenced by acquired adiposity-A study of young adult male monozygotic twin pairs. J Steroid Biochem Mol Biol. (2017) 172:98–105. doi: 10.1016/j.jsbmb.2017.06.007
173. Kleerekoper M, Nelson DA, Peterson EL, Wilson PS, Jacobsen G, and Longcope C. Body composition and gonadal steroids in older white and black women. J Clin Endocrinol Metab. (1994) 79:775–9. doi: 10.1210/jcem.79.3.8077360
174. Marchand GB, Carreau AM, Weisnagel SJ, Bergeron J, Labrie F, Lemieux S, et al. Increased body fat mass explains the positive association between circulating estradiol and insulin resistance in postmenopausal women. Am J Physiol Endocrinol Metab. (2018) 314:E448–56. doi: 10.1152/ajpendo.00293.2017
175. Tchernof A, Després JP, Bélanger A, Dupont A, Prud’homme D, Moorjani S, et al. Reduced testosterone and adrenal C19 steroid levels in obese men. Metabolism. (1995) 44:513–9. doi: 10.1016/0026-0495(95)90060-8
176. Brind J, Strain G, Miller L, Zumoff B, Vogelman J, and Orentreich N. Obese men have elevated plasma levels of estrone sulfate. Int J Obes. (1990) 14:483–6.
177. Grodin JM, Siiteri PK, and MacDonald PC. Source of estrogen production in postmenopausal women. J Clin Endocrinol Metab. (1973) 36:207–14. doi: 10.1210/jcem-36-2-207
178. Björntorp P. The regulation of adipose tissue distribution in humans. Int J Obes Relat Metab Disord. (1996) 20:291–302.
179. Blüher M. Importance of estrogen receptors in adipose tissue function. Mol Metab. (2013) 2:130–2. doi: 10.1016/j.molmet.2013.07.001
180. Gavin KM, Cooper EE, and Hickner RC. Estrogen receptor protein content is different in abdominal than gluteal subcutaneous adipose tissue of overweight-to-obese premenopausal women. Metabolism. (2013) 62:1180–8. doi: 10.1016/j.metabol.2013.02.010
181. Tao Z, Zheng LD, Smith C, Luo J, Robinson A, Almeida FA, et al. Estradiol signaling mediates gender difference in visceral adiposity via autophagy. Cell Death Dis. (2018) 9:309. doi: 10.1038/s41419-018-0372-9
182. Hemsell DL, Grodin JM, Brenner PF, Siiteri PK, and MacDonald PC. Plasma precursors of estrogen. II. Correlation of the extent of conversion of plasma androstenedione to estrone with age. J Clin Endocrinol Metab. (1974) 38:476–9. doi: 10.1210/jcem-38-3-476
183. Rubinow KB. An intracrine view of sex steroids, immunity, and metabolic regulation. Mol Metab. (2018) 15:92–103. doi: 10.1016/j.molmet.2018.03.001
184. Labrie F, Bélanger A, Luu-The V, Labrie C, Simard J, Cusan L, et al. DHEA and the intracrine formation of androgens and estrogens in peripheral target tissues: its role during aging. Steroids. (1998) 63:322–8. doi: 10.1016/s0039-128x(98)00007-5
185. Simpson ER. Aromatase: biologic relevance of tissue-specific expression. Semin Reprod Med. (2004) 22:11–23. doi: 10.1055/s-2004-823023
186. Bolt HM and Göbel P. Formation of estrogens from androgens by human subcutaneous adipose tissue in vitro. Horm Metab Res. (1972) 4:312–3. doi: 10.1055/s-0028-1097099
187. Cohen PG. Aromatase, adiposity, aging and disease. The hypogonadal-metabolic-atherogenic-disease and aging connection. Med Hypotheses. (2001) 56:702–8. doi: 10.1054/mehy.2000.1169
188. Schindler AE, Ebert A, and Friedrich E. Conversion of androstenedione to estrone by human tissue. J Clin Endocrinol Metab. (1972) 35:627–30. doi: 10.1210/jcem-35-4-627
189. Ostinelli G, Laforest S, Denham SG, Gauthier MF, Drolet-Labelle V, Scott E, et al. Increased adipose tissue indices of androgen catabolism and aromatization in women with metabolic dysfunction. J Clin Endocrinol Metab. (2022) 107:e3330–42. doi: 10.1210/clinem/dgac261
190. Ahmed F, Hetty S, Laterveer R, Surucu EB, Mathioudaki A, Hornbrinck E, et al. Altered expression of aromatase and estrogen receptors in adipose tissue from men with obesity or type 2 diabetes. J Clin Endocrinol Metab. (2025) 110(10):e3410–e3424. doi: 10.1210/clinem/dgaf038
191. Geer EB and Shen W. Gender differences in insulin resistance, body composition, and energy balance. Gend Med. (2009) 6 Suppl 1:60–75. doi: 10.1016/j.genm.2009.02.002
192. Hetemäki N, Mikkola TS, Tikkanen MJ, Wang F, Hämäläinen E, Turpeinen U, et al. Adipose tissue estrogen production and metabolism in premenopausal women. J Steroid Biochem Mol Biol. (2021) 209:105849. doi: 10.1016/j.jsbmb.2021.105849
193. Rice S, Patel B, Bano G, Ugwumadu A, and Whitehead SA. Aromatase expression in abdominal omental/visceral and subcutaneous fat depots: a comparison of pregnant and obese women. Fertil Steril. (2012) 97:1460–1466.e1461. doi: 10.1016/j.fertnstert.2012.03.008
194. Jones ME, Boon WC, Proietto J, and Simpson ER. Of mice and men: the evolving phenotype of aromatase deficiency. Trends Endocrinol Metab. (2006) 17:55–64. doi: 10.1016/j.tem.2006.01.004
195. Yue W, Wang JP, Li Y, Fan P, Liu G, Zhang N, et al. Effects of estrogen on breast cancer development: Role of estrogen receptor independent mechanisms. Int J Cancer. (2010) 127:1748–57. doi: 10.1002/ijc.25207
196. Bulun SE, Chen D, Moy I, Brooks DC, and Zhao H. Aromatase, breast cancer and obesity: a complex interaction. Trends Endocrinol Metab. (2012) 23:83–9. doi: 10.1016/j.tem.2011.10.003
197. Barakat R, Oakley O, Kim H, Jin J, and Ko CJ. Extra-gonadal sites of estrogen biosynthesis and function. BMB Rep. (2016) 49:488–96. doi: 10.5483/bmbrep.2016.49.9.141
198. Kolb R and Zhang W. Obesity and breast cancer: A case of inflamed adipose tissue. Cancers (Basel). (2020) 12:1–18. doi: 10.3390/cancers12061686
199. Jenkins EO, Deal AM, Anders CK, Prat A, Perou CM, Carey LA, et al. Age-specific changes in intrinsic breast cancer subtypes: a focus on older women. Oncologist. (2014) 19:1076–83. doi: 10.1634/theoncologist.2014-0184
200. Key TJ, Appleby PN, Reeves GK, Travis RC, Brinton LA, Helzlsouer KJ, et al. Steroid hormone measurements from different types of assays in relation to body mass index and breast cancer risk in postmenopausal women: Reanalysis of eighteen prospective studies. Steroids. (2015) 99:49–55. doi: 10.1016/j.steroids.2014.09.001
201. Mauvais-Jarvis F, Manson JE, Stevenson JC, and Fonseca VA. Menopausal hormone therapy and type 2 diabetes prevention: evidence, mechanisms, and clinical implications. Endocr Rev. (2017) 38:173–88. doi: 10.1210/er.2016-1146
202. Costa GBC, Carneiro G, Umeda L, Pardini D, and Zanella MT. Influence of menopausal hormone therapy on body composition and metabolic parameters. Biores Open Access. (2020) 9:80–5. doi: 10.1089/biores.2019.0050
203. Gambacciani M, Ciaponi M, Cappagli B, Piaggesi L, De Simone L, Orlandi R, et al. Body weight, body fat distribution, and hormonal replacement therapy in early postmenopausal women. J Clin Endocrinol Metab. (1997) 82:414–7. doi: 10.1210/jcem.82.2.3735
204. Valery T, Miller M, John LaRosa M, Vanessa Barnabei M, Craig Kessler MD, Levin G, et al. Effects of estrogen or estrogen/progestin regimens on heart disease risk factors in postmenopausal women. The Postmenopausal Estrogen/Progestin Interventions (PEPI) Trial. The Writing Group for the PEPI Trial. JAMA. (1995) 273:199–208. doi: 10.1001/jama.1995.03520270033028
205. Kanaya AM, Herrington D, Vittinghoff E, Lin F, Grady D, Bittner V, et al. Glycemic effects of postmenopausal hormone therapy: the Heart and Estrogen/progestin Replacement Study. A randomized, double-blind, placebo-controlled trial. Ann Intern Med. (2003) 138:1–9. doi: 10.7326/0003-4819-138-1-200301070-00005
206. Jensen LB, Vestergaard P, Hermann AP, Gram J, Eiken P, Abrahamsen B, et al. Hormone replacement therapy dissociates fat mass and bone mass, and tends to reduce weight gain in early postmenopausal women: a randomized controlled 5-year clinical trial of the Danish Osteoporosis Prevention Study. J Bone Miner Res. (2003) 18:333–42. doi: 10.1359/jbmr.2003.18.2.333
207. Manson JE, Rimm EB, Colditz GA, Willett WC, Nathan DM, Arky RA, et al. A prospective study of postmenopausal estrogen therapy and subsequent incidence of non-insulin-dependent diabetes mellitus. Ann Epidemiol. (1992) 2:665–73. doi: 10.1016/1047-2797(92)90011-e
208. Lovre D, Lindsey SH, and Mauvais-Jarvis F. Effect of menopausal hormone therapy on components of the metabolic syndrome. Ther Adv Cardiovasc Dis. (2016) 11:33–43. doi: 10.1177/1753944716649358
209. Bergendal A, Kieler H, Sundström A, Hirschberg AL, and Kocoska-Maras L. Risk of venous thromboembolism associated with local and systemic use of hormone therapy in peri- and postmenopausal women and in relation to type and route of administration. Menopause. (2016) 23:593–9. doi: 10.1097/GME.0000000000000611
210. Goldštajn M, Mikuš M, Ferrari FA, Bosco M, Uccella S, Noventa M, et al. Effects of transdermal versus oral hormone replacement therapy in postmenopause: a systematic review. Arch Gynecol Obstet. (2023) 307:1727–45. doi: 10.1007/s00404-022-06647-5
211. Pinkerton JV. Hormone therapy for postmenopausal women. N Engl J Med. (2020) 382:446–55. doi: 10.1056/NEJMcp1714787
212. Kaunitz AM. Transdermal and vaginal estradiol for the treatment of menopausal symptoms: the nuts and bolts. Menopause. (2012) 19:602–3. doi: 10.1097/gme.0b013e31824c8a5a
213. de Lauzon-Guillain B, Fournier A, Fabre A, Simon N, Mesrine S, Boutron-Ruault MC, et al. Menopausal hormone therapy and new-onset diabetes in the French Etude Epidemiologique de Femmes de la Mutuelle Générale de l’Education Nationale (E3N) cohort. Diabetologia. (2009) 52:2092–100. doi: 10.1007/s00125-009-1456-y
214. Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML, et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results From the Women’s Health Initiative randomized controlled trial. JAMA. (2002) 288:321–33. doi: 10.1001/jama.288.3.321
215. Hodis HN and Mack WJ. A “window of opportunity:” the reduction of coronary heart disease and total mortality with menopausal therapies is age- and time-dependent. Brain Res. (2011) 1379:244–52. doi: 10.1016/j.brainres.2010.10.076
216. Manson JE, Chlebowski RT, Stefanick ML, Aragaki AK, Rossouw JE, Prentice RL, et al. Menopausal hormone therapy and health outcomes during the intervention and extended poststopping phases of the Women’s Health Initiative randomized trials. JAMA. (2013) 310:1353–68. doi: 10.1001/jama.2013.278040
217. Boardman HM, Hartley L, Eisinga A, Main C, Roqué i Figuls M, Bonfill Cosp X, et al. Hormone therapy for preventing cardiovascular disease in post-menopausal women. Cochrane Database Syst Rev. (2015) 2015:CD002229. doi: 10.1002/14651858.CD002229.pub4
218. Rossouw JE, Prentice RL, Manson JE, Wu L, Barad D, Barnabei VM, et al. Postmenopausal hormone therapy and risk of cardiovascular disease by age and years since menopause. JAMA. (2007) 297:1465–77. doi: 10.1001/jama.297.13.1465
219. Mair KM, Gaw R, and MacLean MR. Obesity, estrogens and adipose tissue dysfunction - implications for pulmonary arterial hypertension. Pulm Circ. (2020) 10:2045894020952019. doi: 10.1177/2045894020952023
220. Straub RH. The complex role of estrogens in inflammation. Endocr Rev. (2007) 28:521–74. doi: 10.1210/er.2007-0001
221. Bracht JR, Vieira-Potter VJ, De Souza Santos R, Öz OK, Palmer BF, and Clegg DJ. The role of estrogens in the adipose tissue milieu. Ann N Y Acad Sci. (2020) 1461:127–43. doi: 10.1111/nyas.14281
222. Hynes MC, Watling CZ, Dunneram Y, Key TJ, and Perez-Cornago A. Associations of body composition measures with circulating insulin-like growth factor-I, testosterone, and sex hormone-binding globulin concentrations in 16,000 men. Int J Obes (Lond). (2024) 48:1809–17. doi: 10.1038/s41366-024-01633-0
223. Vihma V, Heinonen S, Naukkarinen J, Kaprio J, Rissanen A, Turpeinen U, et al. Increased body fat mass and androgen metabolism - A twin study in healthy young women. Steroids. (2018) 140:24–31. doi: 10.1016/j.steroids.2018.08.006
224. De Pergola G, Zamboni M, Sciaraffia M, Turcato E, Pannacciulli N, Armellini F, et al. Body fat accumulation is possibly responsible for lower dehydroepiandrosterone circulating levels in premenopausal obese women. Int J Obes Relat Metab Disord. (1996) 20:1105–10.
225. Stangl TA, Wiepjes CM, Smit RAJ, van Hylckama Vlieg A, Lamb HJ, van der Velde JHPM, et al. Association between low sex hormone-binding globulin and increased risk of type 2 diabetes is mediated by increased visceral and liver fat: results from observational and mendelian randomization analyses. Diabetes. (2024) 73:1793–804. doi: 10.2337/db23-0982
226. Bray GA. Obesity and reproduction. Hum Reprod. (1997) 12 Suppl 1:26–32. doi: 10.1093/humrep/12.suppl_1.26
227. Singh P, Covassin N, Sert-Kuniyoshi FH, Marlatt KL, Romero-Corral A, Davison DE, et al. Overfeeding-induced weight gain elicits decreases in sex hormone-binding globulin in healthy males-Implications for body fat distribution. Physiol Rep. (2021) 9:e15127. doi: 10.14814/phy2.15127
228. Aroda VR, Christophi CA, Edelstein SL, Perreault L, Kim C, Golden SH, et al. Circulating sex hormone binding globulin levels are modified with intensive lifestyle intervention, but their changes did not independently predict diabetes risk in the Diabetes Prevention Program. BMJ Open Diabetes Res Care. (2020) 8:1–10. doi: 10.1136/bmjdrc-2020-001841
229. Kim C, Dabelea D, Kalyani RR, Christophi CA, Bray GA, Pi-Sunyer X, et al. Changes in visceral adiposity, subcutaneous adiposity, and sex hormones in the diabetes prevention program. J Clin Endocrinol Metab. (2017) 102:3381–9. doi: 10.1210/jc.2017-00967
230. Bennett WL, He JH, Michos ED, Kalyani RR, Clark JM, Woodward M, et al. Weight loss differentially impacts sex hormones in women and men with type 2 diabetes: look AHEAD sex hormone study. J Clin Endocrinol Metab. (2025) 110:e2001–7. doi: 10.1210/clinem/dgae584
231. Mucinski JM, Kelley DE, Winters SJ, and Goodpaster BH. Effects of weight loss on testosterone, sex hormone-binding globulin, adiposity, and insulin sensitivity in women and men. Obes (Silver Spring). (2025) 33:962–73. doi: 10.1002/oby.24269
232. Leenen R, van der Kooy K, Seidell JC, Deurenberg P, and Koppeschaar HP. Visceral fat accumulation in relation to sex hormones in obese men and women undergoing weight loss therapy. J Clin Endocrinol Metab. (1994) 78:1515–20. doi: 10.1210/jcem.78.6.8200956
233. Abildgaard J, Aleliunaite A, Horvath C, Palani N, Henriksen TI, Zhong J, et al. Sex hormone-binding globulin controls sex-specific lipolytic activity in human abdominal subcutaneous adipocytes. Mol Metab. (2025) 98:102189. doi: 10.1016/j.molmet.2025.102189
234. Moran C, Arriaga M, Arechavaleta-Velasco F, and Moran S. Adrenal androgen excess and body mass index in polycystic ovary syndrome. J Clin Endocrinol Metab. (2015) 100:942–50. doi: 10.1210/jc.2014-2569
235. Keller JL, Casson PR, and Toth MJ. Relationship of androgens to body composition, energy and substrate metabolism and aerobic capacity in healthy, young women. Steroids. (2011) 76:1247–51. doi: 10.1016/j.steroids.2011.06.001
236. Tchernof A, Després JP, Dupont A, Bélanger A, Nadeau A, Prud’homme D, et al. Relation of steroid hormones to glucose tolerance and plasma insulin levels in men. Importance of visceral adipose tissue. Diabetes Care. (1995) 18:292–9. doi: 10.2337/diacare.18.3.292
237. Couillard C, Gagnon J, Bergeron J, Leon AS, Rao DC, Skinner JS, et al. Contribution of body fatness and adipose tissue distribution to the age variation in plasma steroid hormone concentrations in men: the HERITAGE Family Study. J Clin Endocrinol Metab. (2000) 85:1026–31. doi: 10.1210/jcem.85.3.6427
238. Marchand GB, Carreau AM, Laforest S, Côté JA, Daris M, Cianflone K, et al. Circulating steroid levels as correlates of adipose tissue phenotype in premenopausal women. Horm Mol Biol Clin Investig. (2018) 34:1–10. doi: 10.1515/hmbci-2017-0082
239. Jasuja GK, Travison TG, Davda M, Rose AJ, Zhang A, Kushnir MM, et al. Circulating estrone levels are associated prospectively with diabetes risk in men of the Framingham Heart Study. Diabetes Care. (2013) 36:2591–6. doi: 10.2337/dc12-2477
240. Kirschner MA and Samojlik E. Sex hormone metabolism in upper and lower body obesity. Int J Obes. (1991) 15 Suppl 2:101–8.
241. Freeman EW, Sammel MD, Lin H, and Gracia CR. Obesity and reproductive hormone levels in the transition to menopause. Menopause. (2010) 17:718–26. doi: 10.1097/gme.0b013e3181cec85d
242. Santoro N, Crawford SL, Lasley WL, Luborsky JL, Matthews KA, McConnell D, et al. Factors related to declining luteal function in women during the menopausal transition. J Clin Endocrinol Metab. (2008) 93:1711–21. doi: 10.1210/jc.2007-2165
243. Randolph JF, Sowers M, Gold EB, Mohr BA, Luborsky J, Santoro N, et al. Reproductive hormones in the early menopausal transition: relationship to ethnicity, body size, and menopausal status. J Clin Endocrinol Metab. (2003) 88:1516–22. doi: 10.1210/jc.2002-020777
244. Su HI, Sammel MD, Freeman EW, Lin H, DeBlasis T, and Gracia CR. Body size affects measures of ovarian reserve in late reproductive age women. Menopause. (2008) 15:857–61. doi: 10.1097/gme.0b013e318165981e
245. Yan H, Yang W, Zhou F, Li X, Pan Q, Shen Z, et al. Estrogen improves insulin sensitivity and suppresses gluconeogenesis via the transcription factor foxo1. Diabetes. (2019) 68:291–304. doi: 10.2337/db18-0638
246. De Paoli M, Zakharia A, and Werstuck GH. The role of estrogen in insulin resistance: A review of clinical and preclinical data. Am J Pathol. (2021) 191:1490–8. doi: 10.1016/j.ajpath.2021.05.011
247. Masuyama H and Hiramatsu Y. Potential role of estradiol and progesterone in insulin resistance through constitutive androstane receptor. J Mol Endocrinol. (2011) 47:229–39. doi: 10.1530/JME-11-0046
248. Vejrazkova D, Vcelak J, Vankova M, Lukasova P, Bradnova O, Halkova T, et al. Steroids and insulin resistance in pregnancy. J Steroid Biochem Mol Biol. (2014) 139:122–9. doi: 10.1016/j.jsbmb.2012.11.007
249. Brussaard HE, Gevers Leuven JA, Frölich M, Kluft C, and Krans HM. Short-term oestrogen replacement therapy improves insulin resistance, lipids and fibrinolysis in postmenopausal women with NIDDM. Diabetologia. (1997) 40:843–9. doi: 10.1007/s001250050758
250. Muka T, Nano J, Jaspers L, Meun C, Bramer WM, Hofman A, et al. Associations of steroid sex hormones and sex hormone-binding globulin with the risk of type 2 diabetes in women: A population-based cohort study and meta-analysis. Diabetes. (2017) 66:577–86. doi: 10.2337/db16-0473
251. Li Y, Streicher SA, Franke AA, Tome AN, White KK, Shvetsov Y, et al. Association of adiposity phenotypes with 27-hydroxycholesterol and sex hormones: the multiethnic cohort study. J Clin Endocrinol Metab. (2025) 110:e1488–98. doi: 10.1210/clinem/dgae549
252. Vermeulen A, Kaufman JM, Goemaere S, and van Pottelberg I. Estradiol in elderly men. Aging Male. (2002) 5:98–102. doi: 10.1080/tam.5.2.98.102
253. Wu A, Shi Z, Martin S, Vincent A, Heilbronn L, and Wittert G. Age-related changes in estradiol and longitudinal associations with fat mass in men. PloS One. (2018) 13:e0201912. doi: 10.1371/journal.pone.0201912
254. Xu Q, Ji M, Huang S, and Guo W. Association between serum estradiol levels and cognitive function in older women: a cross-sectional analysis. Front Aging Neurosci. (2024) 16:1356791. doi: 10.3389/fnagi.2024.1356791
255. Varma B, Ogunmoroti O, Ndumele CE, Kazzi B, Rodriquez CP, Osibogun O, et al. Associations between endogenous sex hormone levels and adipokine levels in the Multi-Ethnic Study of Atherosclerosis. Front Cardiovasc Med. (2022) 9:1062460. doi: 10.3389/fcvm.2022.1062460
256. Loh NY, Humphreys E, Karpe F, Tomlinson JW, Noordam R, and Christodoulides C. Sex hormones, adiposity, and metabolic traits in men and women: a Mendelian randomisation study. Eur J Endocrinol. (2022) 186:407–16. doi: 10.1530/EJE-21-0703
257. Söderberg S, Olsson T, Eliasson M, Johnson O, Brismar K, Carlström K, et al. A strong association between biologically active testosterone and leptin in non-obese men and women is lost with increasing (central) adiposity. Int J Obes Relat Metab Disord. (2001) 25:98–105. doi: 10.1038/sj.ijo.0801467
258. De Pergola G, Triggiani V, Giorgino F, Cospite MR, Garruti G, Cignarelli M, et al. The free testosterone to dehydroepiandrosterone sulphate molar ratio as a marker of visceral fat accumulation in premenopausal obese women. Int J Obes Relat Metab Disord. (1994) 18:659–64.
259. Van de Velde F, Deventer K, Van Gansbeke W, Van Eenoo P, Van Renterghem P, Fiers T, et al. Metabolism of testosterone during weight loss in men with obesity. J Steroid Biochem Mol Biol. (2021) 209:105851. doi: 10.1016/j.jsbmb.2021.105851
260. Wittert G and Grossmann M. Obesity, type 2 diabetes, and testosterone in ageing men. Rev Endocr Metab Disord. (2022) 23:1233–42. doi: 10.1007/s11154-022-09746-5
261. Gao J, He J, Shi X, Stefanovic-Racic M, Xu M, O’Doherty RM, et al. Sex-specific effect of estrogen sulfotransferase on mouse models of type 2 diabetes. Diabetes. (2012) 61:1543–51. doi: 10.2337/db11-1152
262. Rogers NH, Perfield JW, Strissel KJ, Obin MS, and Greenberg AS. Reduced energy expenditure and increased inflammation are early events in the development of ovariectomy-induced obesity. Endocrinology. (2009) 150:2161–8. doi: 10.1210/en.2008-1405
263. Mayes JS and Watson GH. Direct effects of sex steroid hormones on adipose tissues and obesity. Obes Rev. (2004) 5:197–216. doi: 10.1111/j.1467-789X.2004.00152.x
264. Cahyadi DD, Warita K, and Hosaka YZ. Depot-specific adiposity changes in ovariectomized mice on high-fat diet. J Vet Med Sci. (2025) 87:241–7. doi: 10.1292/jvms.24-0442
265. Dakin RS, Walker BR, Seckl JR, Hadoke PW, and Drake AJ. Estrogens protect male mice from obesity complications and influence glucocorticoid metabolism. Int J Obes (Lond). (2015) 39:1539–47. doi: 10.1038/ijo.2015.102
266. Mann SN, Hadad N, Nelson Holte M, Rothman AR, Sathiaseelan R, Ali Mondal S, et al. Health benefits attributed to 17α-estradiol, a lifespan-extending compound, are mediated through estrogen receptor α. Elife. (2020) 9:1–30. doi: 10.7554/eLife.59616
267. Fouad Mansour M, Pelletier M, Boulet MM, Mayrand D, Brochu G, Lebel S, et al. Oxidative activity of 17β-hydroxysteroid dehydrogenase on testosterone in male abdominal adipose tissues and cellular localization of 17β-HSD type 2. Mol Cell Endocrinol. (2015) 414:168–76. doi: 10.1016/j.mce.2015.06.016
268. Quinkler M, Sinha B, Tomlinson JW, Bujalska IJ, Stewart PM, and Arlt W. Androgen generation in adipose tissue in women with simple obesity–a site-specific role for 17beta-hydroxysteroid dehydrogenase type 5. J Endocrinol. (2004) 183:331–42. doi: 10.1677/joe.1.05762
269. Longcope C, Baker R, and Johnston CC. Androgen and estrogen metabolism: relationship to obesity. Metabolism. (1986) 35:235–7. doi: 10.1016/0026-0495(86)90206-4
270. Edman CD and MacDonald PC. Effect of obesity on conversion of plasma androstenedione to estrone in ovulatory and anovulator young women. Am J Obstet Gynecol. (1978) 130:456–61. doi: 10.1016/0002-9378(78)90288-0
271. Cleland WH, Mendelson CR, and Simpson ER. Effects of aging and obesity on aromatase activity of human adipose cells. J Clin Endocrinol Metab. (1985) 60:174–7. doi: 10.1210/jcem-60-1-174
272. Bulun SE and Simpson ER. Competitive reverse transcription-polymerase chain reaction analysis indicates that levels of aromatase cytochrome P450 transcripts in adipose tissue of buttocks, thighs, and abdomen of women increase with advancing age. J Clin Endocrinol Metab. (1994) 78:428–32. doi: 10.1210/jcem.78.2.8106632
273. Li X, Liu J, Zhou B, Li Y, Wu Z, Meng H, et al. Sex differences in the effect of testosterone on adipose tissue insulin resistance from overweight to obese adults. J Clin Endocrinol Metab. (2021) 106:2252–63. doi: 10.1210/clinem/dgab325
274. Wagner IV, Sahlin L, Savchuk I, Klöting N, Svechnikov K, and Söder O. Adipose tissue is a potential source of hyperandrogenism in obese female rats. Obes (Silver Spring). (2018) 26:1161–7. doi: 10.1002/oby.22198
275. Corbould AM, Bawden MJ, Lavranos TC, Rodgers RJ, and Judd SJ. The effect of obesity on the ratio of type 3 17beta-hydroxysteroid dehydrogenase mRNA to cytochrome P450 aromatase mRNA in subcutaneous abdominal and intra-abdominal adipose tissue of women. Int J Obes Relat Metab Disord. (2002) 26:165–75. doi: 10.1038/sj.ijo.0801886
Keywords: sex steroids, sex hormones, adipocyte, aromatase, estradiol, estrone, obesity, menopause
Citation: Lee AA and Den Hartigh LJ (2025) Metabolic impact of endogenously produced estrogens by adipose tissue in females and males across the lifespan. Front. Endocrinol. 16:1682231. doi: 10.3389/fendo.2025.1682231
Received: 08 August 2025; Accepted: 06 October 2025;
Published: 17 October 2025.
Edited by:
Luís Pedro Rato, University of Évora, PortugalReviewed by:
Akira Takamata, Nara Women’s University, JapanVittorio Emanuele Bianchi, University of the Republic of San Marino, San Marino
Copyright © 2025 Lee and Den Hartigh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laura J. Den Hartigh, bGF1cmFkaEB1dy5lZHU=