 Hengpan Yao1†
Hengpan Yao1† Zhiyi Xia
Zhiyi Xia Fang Zhou
Fang Zhou- 1Department of Digestion, Children’s Hospital Affiliated to Zhengzhou University, Henan Children’s Hospital Zhengzhou Children’s Hospital, Zhengzhou, Henan, China
- 2Henan Provincial Key Laboratory of Children's Genetics and Metabolic Diseases, Henan Children’s Hospital, Zhengzhou, Henan, China
Background: With the rising global prevalence of childhood obesity, the incidence of metabolic dysfunction-associated steatotic liver disease (MASLD) in pediatric and adolescent populations is also increasing. The genetic mechanisms underlying MASLD remain incompletely elucidated. This study aimed to investigate the roles of eight genetic loci—PNPLA3-Ile148Met, PNPLA3-Lys434Glu, GCKR-Leu446Pro, TM6SF2-Glu167Lys, LEPR-Lys109Arg, LEPR-Lys656Asn, IRS1-Gly971Arg, and KLB-Arg728Gln—in the susceptibility to MASLD in Chinese children and adolescents, in order to provide scientific evidence for genetic research on MASLD. We hypothesized that PNPLA3-Ile148Met and TM6SF2-Glu167Lys variants confer susceptibility to MASLD in children.
Methods: A total of 350 children and adolescents aged 7–17 years were enrolled, including 196 with MASLD (case group) and 154 healthy controls. Demographics, medical history, anthropometric measurements, and hepatic B-ultrasound data were collected. Fasting morning blood samples were obtained for biochemical analysis and genotyping. Statistical analyses included chi-square tests, Fisher’s exact tests, and multivariate logistic regression to identify predictive factors for pediatric MASLD.
Results: The allele frequencies of PNPLA3-Ile148Met, PNPLA3-Lys434Glu, and TM6SF2-Glu167Lys were significantly higher in the MASLD group than in the control group (PNPLA3-Ile148Met and TM6SF2-Glu167Lys: both P_FDR < 0.001; PNPLA3-Lys434Glu: P_FDR=0.024), indicating an association with increased MASLD risk. In contrast, the allele frequencies of GCKR-Leu446Pro and LEPR-Lys109Arg were significantly lower in the MASLD group (P_FDR=0.009), suggesting potential protective effects. Multivariate logistic regression identified male sex, the PNPLA3-Ile148Met-GG genotype, and the TM6SF2-Glu167Lys-CT genotype as independent risk factors for MASLD. Additionally, carriers of the PNPLA3 rs738409 GG genotype exhibited significantly higher levels of AST, ALT, and TC compared to those with the GC genotype (all P_FDR < 0.001).
Conclusion: The PNPLA3-Ile148Met and TM6SF2-Glu167Lys gene polymorphisms are independent risk factors that significantly increase the risk of MASLD in Chinese children. Additionally, the PNPLA3-Lys434Glu variant is associated with increased risk at the allele level. In contrast, GCKR-Leu446Pro and LEPR-Lys109Arg may confer protective effects. This study provides new evidence for genetic susceptibility to MASLD in the pediatric population.
1 Introduction
The nomenclature of fatty liver disease has evolved through three phases: from “non-alcoholic fatty liver disease” (NAFLD) to “metabolically associated fatty liver disease” (MAFLD), and finally to the current standardized term MASLD. To better reflect its pathophysiological mechanisms, international consensus recommends renaming the former “NAFLD” to “MASLD”, removing the terms “non-alcoholic” and “fatty” to align with specific diagnostic criteria. For the convenience of future academic research and communication, this study adopts the revised MASLD designation (1–3). Pediatric MASLD is an important public health issue worldwide (4, 5). MASLD has become the most prevalent chronic liver disorder among children and adolescents worldwide (6), with its prevalence closely mirroring the obesity epidemic. Studies indicate a global prevalence rate of 13% in children and adolescents, while rates soar to 47.00% in a special population based on child obesity respectively (7). This health crisis is no longer confined to developed nations, as its prevalence in low-and middle-income countries is rising at an unprecedented rate. These patterns are directly linked to the global dietary shift toward Westernized diets (high-sugar, high-fructose beverages, and processed foods) and the widespread adoption of sedentary lifestyles. MASLD is increasingly being seen in children in China. Related analysis revealed that the total prevalence of MASLD in Chinese children is 6.30%, and the prevalence of overweight and obese children is 40.4%. MASLD is a clinicopathological syndrome characterized by hepatic steatosis, excluding ethanol and other definite liver damage factors (8). Recent studies have shown that the development of MASLD is associated with lipid accumulation, oxidative stress, gut microbiota imbalance and genetic factors (9, 10). Amid evolving lifestyles, dietary structures, and escalating obesity in younger populations, MASLD has emerged as the leading chronic liver disorder. The importance of MASLD in children goes far beyond the liver itself. It is a disease that affects multiple systems. Apart from liver-related morbidity, MASLD further associates with heightened incidence of cardiovascular disease, type 2 diabetes, and fatal outcomes (11). Genetic factors critically contribute to MASLD pathogenesis, given that not every obese child develops the condition (12). PNPLA3-Ile148Met (rs738409, patatin-like phospholipase domain-containing protein 3) is the most impactful genetic risk factor for steatotic liver disease (13, 14). Moreover, some meta-analyses have suggested that this variant is significantly associated with elevated serum alanine transaminase, aspartate transaminase, and gamma-glutamyltransferase concentrations and liver fat content (15, 16). TM6SF2 is involved in triglyceride secretion and the regulation of hepatic lipid metabolism (17, 18). When a mutation occurs at the TM6SF2-Glu167Lys site, it increases the risk of hepatic fat accumulation and liver injury.
However, despite growing evidence on genetic susceptibility to MASLD, most studies have focused on adult or non-East Asian populations, leaving a significant knowledge gap regarding genetic risk factors in Chinese children. To address this, we systematically selected eight single nucleotide polymorphisms (SNPs)—previously associated with MASLD in other populations but underexplored in Chinese pediatric cohorts—with minor allele frequency >0.10, aiming to identify susceptibility loci specific to this demographic. Our results demonstrate that PNPLA3-Ile148Met, PNPLA3-Lys434Glu, and TM6SF2-Glu167Lys significantly increase MASLD risk in Chinese children, thereby filling a critical research gap in this population. Notably, while GCKR-Leu446Pro has been widely reported as a risk locus in Western pediatric studies, it exhibited a protective effect in our cohort, highlighting the importance of population-specific genetic investigations.
2 Materials and methods
2.1 Data acquisition
A total of 350 children were enrolled in this study, including 196 children and adolescents with MASLD as the experimental group and 154 healthy children and adolescents as the control group. All participants and their legal guardians provided written informed consent. The study was approved by the Medical Ethics Committee of the Children’s Hospital Affiliated to Zhengzhou University, Henan Province(Approval No.: 2024-071-002). All participants were unrelated and of Han Chinese ethnicity.
2.2 Selection of 8 polymorphic loci
The 8 target SNPs were selected based on a comprehensive consideration of three key factors: (1) Sample size adaptability: Given the total sample size of 350 (196 in the experimental group and 154 in the control group), loci with a minor allele frequency > 0.10 were prioritized to avoid insufficient statistical power caused by rare variants; (2) Population frequency relevance: Loci with well-documented frequency data in Chinese populations (especially pediatric subgroups) were selected to ensure consistency with the study’s ethnic background; (3) MASLD association evidence: Only loci previously reported to be associated with MASLD or its pathological mechanisms (e.g., hepatic lipid metabolism, steatosis, and metabolic dysfunction) in international or Chinese adult studies were included, to focus on genetically meaningful targets for pediatric MASLD validation.
2.3 Sequencing and genotyping
Peripheral blood samples (200 μL) were collected from all participants, and genomic DNA was extracted using a commercial DNA extraction kit (Sangon Biotech, Cat. No.: B518253, Shanghai, China) following the manufacturer’s instructions; multiplex amplification primers were then designed based on the genomic positions of all 8 target SNPs to achieve targeted capture of the SNP regions. High-throughput sequencing was performed on the Illumina HiSeq X platform to generate 150 bp paired-end reads; raw sequencing data were filtered for quality (e.g., removing low-quality reads with Phred scores < 20), and the remaining high-quality reads were aligned to the human reference genome (GRCh37—hg19) using the bwa-mem2-v2.2.1 algorithm (19). Genotypes were finally identified using BCFtools-v1.9 (via the mpileup function) to generate genomic Variant Call Format (gVCF) files, which included both variant and wild-type genotype information for all target SNPs (20).
2.4 Liver ultrasound examination and MASLD diagnosis
2.4.1 Diagnostic criteria
MASLD was diagnosed per the Expert Consensus on the Diagnosis and Management of Metabolic Dysfunction-Associated Steatotic Liver Disease in Children (2025): (1) Ultrasonographic evidence of hepatic steatosis (e.g., liver-kidney echo discrepancy, attenuated echo penetration); (2) At least one cardiovascular/metabolic risk factor (e.g., BMI ≥ 85th percentile, abnormal blood glucose/lipids); (3) Exclusion of other liver injury causes (e.g., inherited metabolic disorders, drug-induced liver injury) (1).
2.4.2 Rationale for liver ultrasound
Liver ultrasound was used as it is non-invasive, safe for children, cost-effective, and widely accessible—consistent with international guidelines recommending it as the first-line tool for pediatric MASLD screening to identify clinically significant steatosis.
2.5 Statistical analysis and graphing
Statistical analyses were conducted using SPSS-v21.0, and graphs were generated via Python’s Matplotlib (v3.5). Hardy–Weinberg equilibrium (HWE) tests were performed for each SNP: Pearson χ² test was used if expected genotype counts ≥ 5, and Fisher’s two-tailed exact test was used if < 5. SNP allele/genotype frequency differences between groups were analyzed via chi-square test or Fisher’s exact test, as appropriate.
2.6 Genetic model selection for multivariate analysis
To ensure a robust and pre-specified approach for identifying independent genetic risk factors, we determined the most appropriate genetic model for each SNP prior to multivariate analysis. For all eight SNPs, we fitted univariate logistic regression models under additive, dominant, and recessive genetic models. The optimal model for each SNP was selected based on the lowest Akaike Information Criterion (AIC), indicating the best fit to the data. This pre-analysis led to the following model specifications for the final multivariate model:
1. PNPLA3-Ile148Met: A recessive model was used. This was represented by a dummy variable comparing the homozygous risk genotype (GG) against the combined group of heterozygotes and wild-type homozygotes (GC + CC).
2. TM6SF2-Glu167Lys: A dominant model was used. This was represented by a dummy variable comparing carriers of at least one risk allele (CT + TT) against wild-type homozygotes (CC).
Multivariable logistic regression (with MASLD status as the dependent variable) was then employed to identify independent predictive factors, incorporating the pre-specified genetic models for PNPLA3-Ile148Met and TM6SF2-Glu167Lys, alongside covariates including age, sex, and BMI. Variable selection was performed using a stepwise method.
Differences in biochemical indicators between PNPLA3-p.Ile148Met CG/GG genotypes were analyzed via Mann–Whitney U test (due to non-normal distribution). To control for type I errors in multiple testing, we applied false discovery rate (FDR) correction using the Benjamini–Hochberg procedure with the following specific implementations:
1. For genetic association analyses (Tables 1, 2, Figure 1): We performed separate FDR corrections for: The 8 SNP allele frequency comparisons (Table 1); The 13 genotype frequency comparisons (Table 2); Each set was corrected independently at α = 0.05 using the multipletests function in Python’s statsmodels library (method=‘fdr_bh’).
2. For biochemical marker comparisons (Table 3): Given the examination of 16 different biochemical parameters between genotype groups, we applied both FDR correction (primary method) and Bonferroni correction (secondary, more conservative method). The FDR threshold was set at q < 0.05.
3. For multivariate logistic regression (Table 4): P-values for the 5 predictor variables in the final model were subjected to FDR correction.
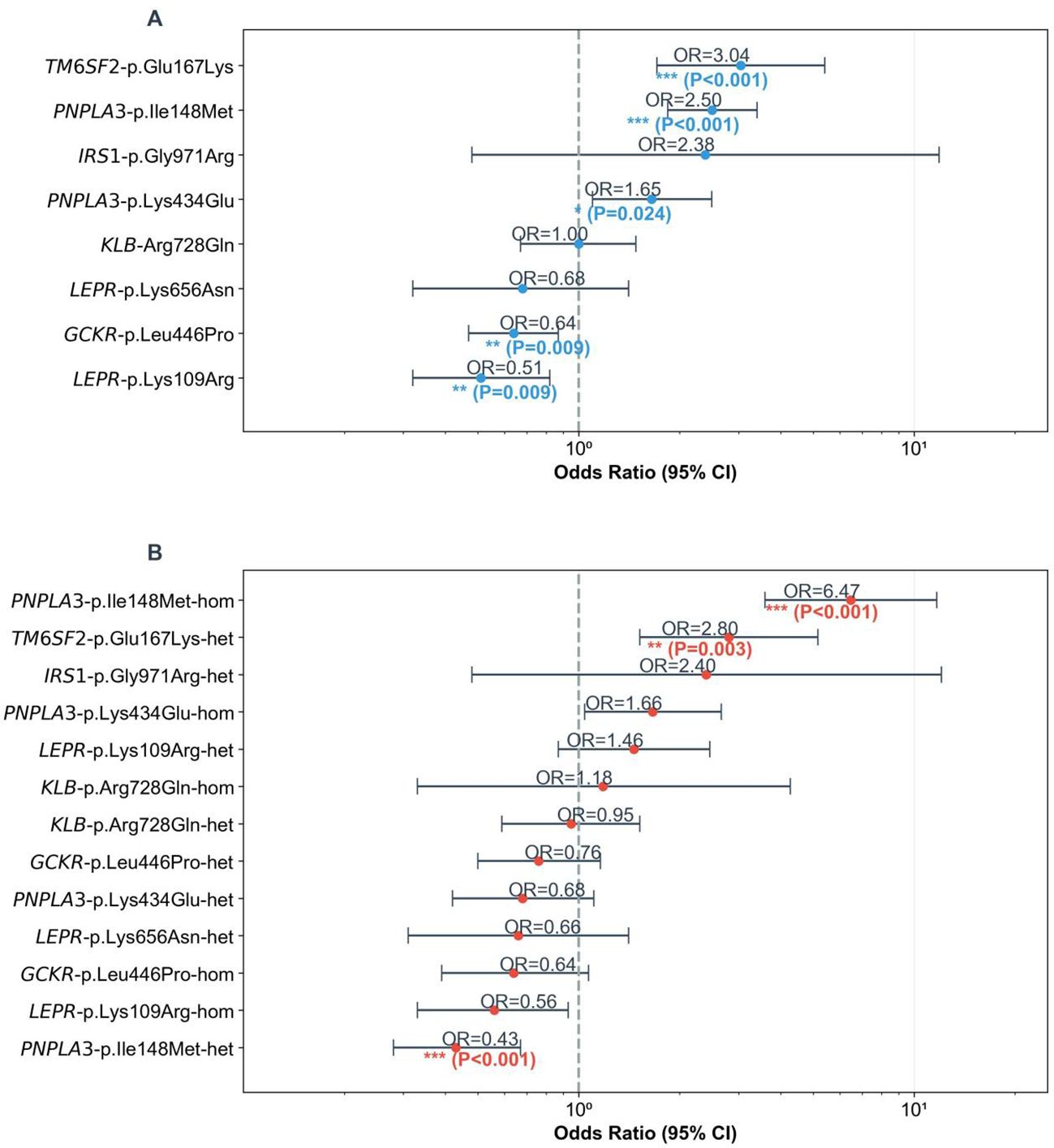
Figure 1. Forest plots of SNP associations with childhood MASLD. (A) ORs (MASLD/control) with 95% CIs for allele frequencies of 8 SNPs and MASLD. Dashed line = OR = 1; asterisks mark significance (***P<0.001, **P<0.010, *P<0.050). (B) Odds ratios (ORs, MASLD/control) with 95% CIs for homozygous/heterozygous genotypes of 8 SNPs and MASLD. Dashed line = OR = 1 (no association); asterisks mark significance. (***P<0.001, **P<0.010, *P<0.050).
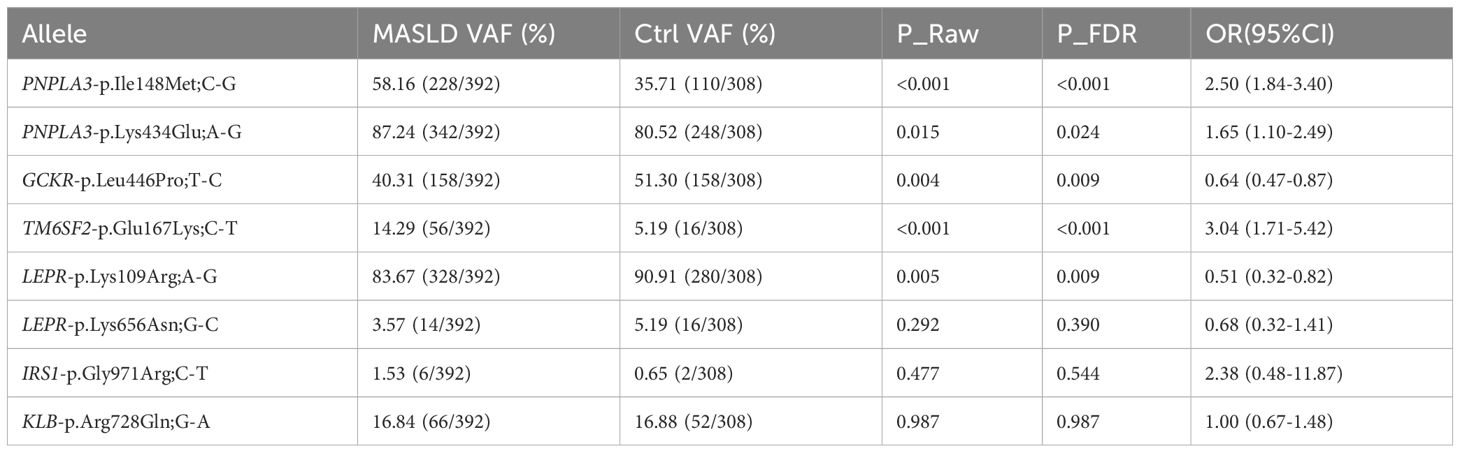
Table 1. Allele frequency distribution and association analysis results of 8 SNPs between MASLD and control groups in children.
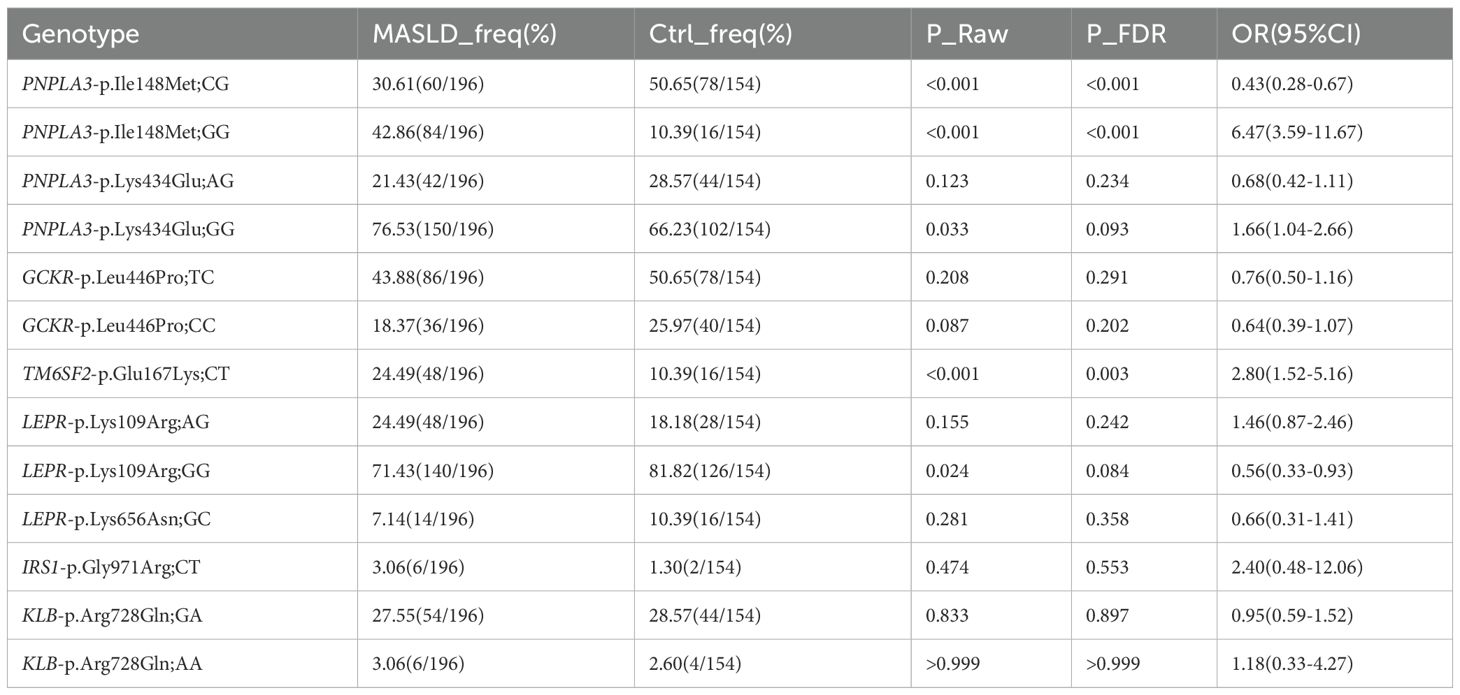
Table 2. Genotype frequency differences of SNPs between MASLD and control groups in children.
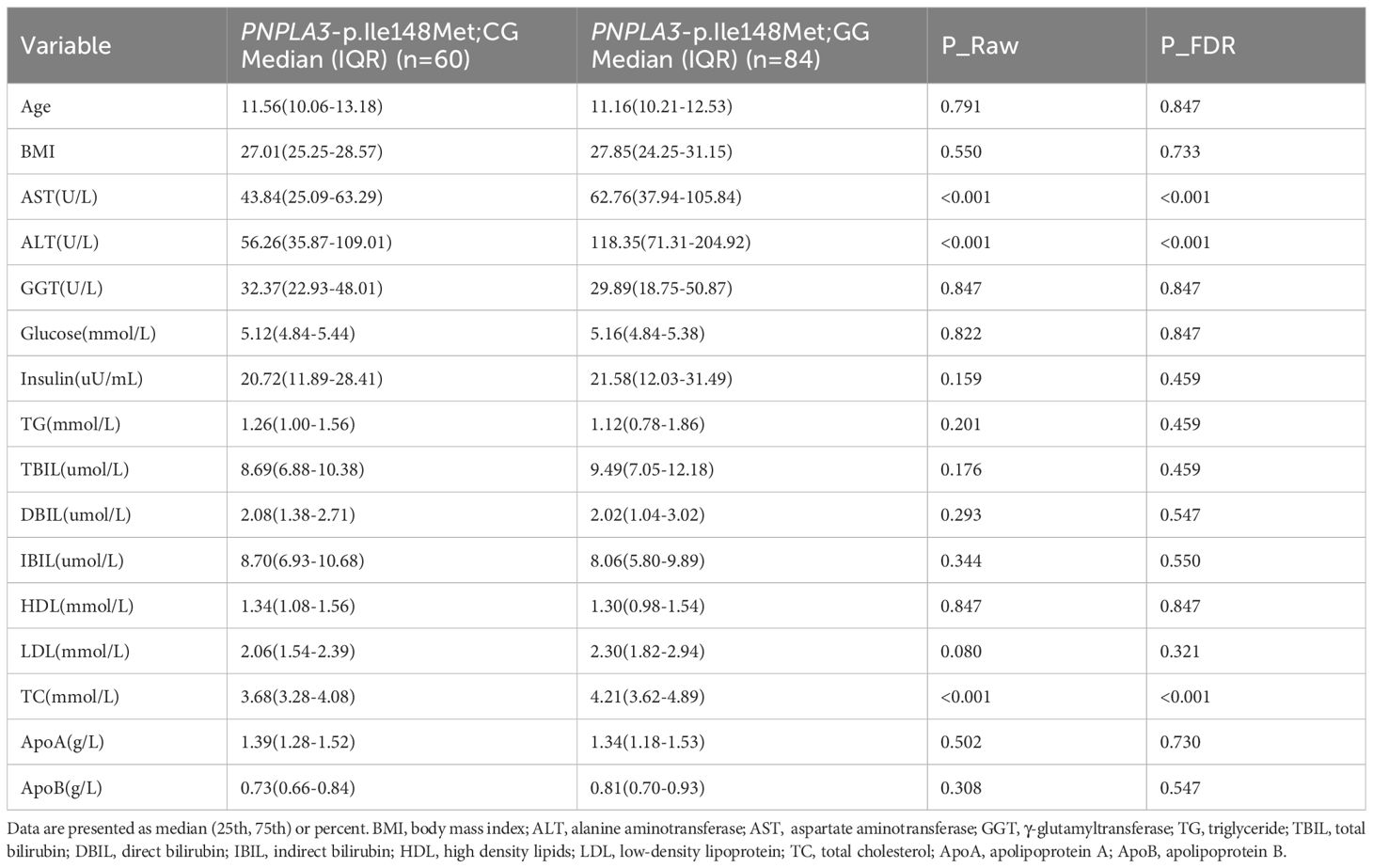
Table 3. Differential analysis of age, BMI and blood-related indicators between PNPLA3 - p.Ile148Met;CG and GG Genotype Groups.
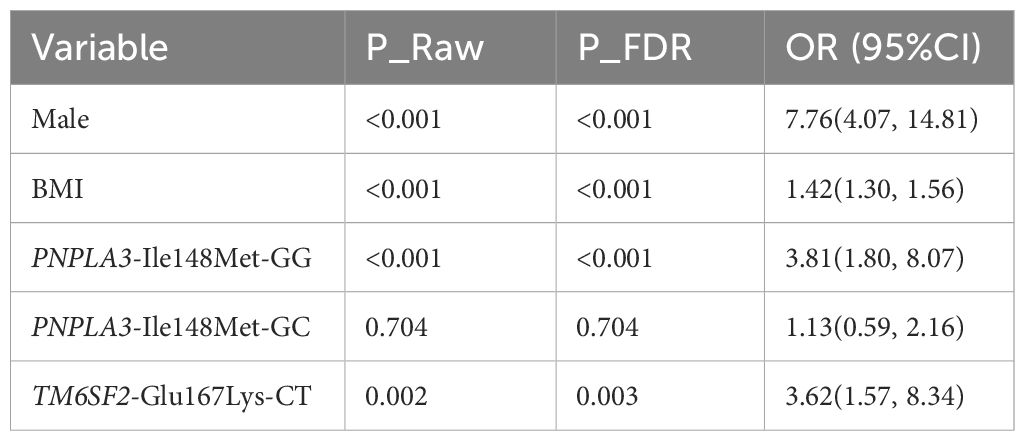
Table 4. Predictive factors for MASLD: multivariable logistic regression.
Associations with FDR-corrected P-values (denoted as P_FDR) < 0.050 were considered statistically significant. All statistical tests were two-sided.
2.7 In silico analysis of protein stability
To assess the potential structural impact of the identified non-synonymous variants, we performed in silico analysis on the PNPLA3 protein. The wild-type three-dimensional structure of human PNPLA3 was retrieved from the AlphaFold Database (https://alphafold.ebi.ac.uk). The DynaMut2 web server (https://biosig.lab.uq.edu.au/dynamut2/) was employed to predict the changes in protein stability upon mutation (21). This tool calculates the change in folding free energy (ΔΔG, kcal/mol) between the wild-type and mutant structures, where a negative ΔΔG value indicates a destabilizing effect.
3 Results
3.1 Hardy–Weinberg equilibrium validation of genotype distributions
To ensure the reliability of subsequent genetic association analyses, we first validated the genotype distributions of all target SNPs for deviation from Hardy–Weinberg equilibrium (HWE) in both the MASLD cohort (n=196) and the control cohort (n=154). Detailed HWE test results for all variants are presented in Supplementary Table 1.
In the control cohort, no deviation from HWE was observed for any of the tested variants (all P > 0.050), indicating that the control group had a genetically balanced genotype distribution and good population representativeness.
In the MASLD cohort, the PNPLA3-Ile148Met variant showed a significant deviation from HWE (P < 0.001), while all other variants conformed to HWE (all P > 0.050). This deviation is likely biologically driven rather than technical, as the PNPLA3 148M (G) allele is a well-established and potent risk factor for steatotic liver disease. The strong enrichment of the high-risk GG genotype in our case group—a hallmark of genuine association—naturally leads to a departure from HWE expectations within the affected population (13, 14, 22). This observation, coupled with the absence of HWE deviation for this SNP in our control group and for all other SNPs in both groups, supports the integrity of our genotyping data and the specific, robust association of PNPLA3-Ile148Met with pediatric MASLD.
3.2 Differences in allele frequencies of 8 SNPs between the MASLD group and the control group
The allele frequencies of three SNP loci (TM6SF2-Glu167Lys, PNPLA3-Ile148Met, and PNPLA3-Lys434Glu) in children and adolescents with MASLD were significantly higher than those in the healthy control group (TM6SF2-Glu167Lys: P_FDR<0.001, OR = 3.04; PNPLA3-Ile148Met: P_FDR<0.001, OR = 2.50; PNPLA3-Lys434Glu: P_FDR=0.024, OR = 1.65; Figure 1A, Table 1), suggesting an association between these three SNPs and an increased risk of MASLD. In silico stability analysis further predicted that both PNPLA3 missense variants (Ile148Met and Lys434Glu) have a destabilizing effect on the protein structure (ΔΔG = -0.55 and -0.23 kcal/mol, respectively).
In contrast, the variant allele frequencies of GCKR-Leu446Pro and LEPR-Lys109Arg were significantly lower in the MASLD group than in the control group (GCKR-p.Leu446Pro; T-C: P_FDR=0.009, OR = 0.64; LEPR-p.Lys109Arg; A-G: P_FDR=0.009, OR = 0.51). This implies that the variant sites of these two SNPs may serve as protective variants against MASLD, and individuals with the wild-type of these SNPs have a higher risk of developing MASLD.
3.3 Association between specific genotypes and MASLD susceptibility
Stratified analysis by genotype further confirmed the association of key variants with MASLD (Figure 1B, Table 2):
For PNPLA3-Ile148Met, the homozygous GG genotype and heterozygous GC genotype showed highly significant differences between the MASLD group and the control group (homozygous GG genotype: P_FDR<0.001, OR = 6.47; heterozygous GC genotype: P_FDR=0.007, OR = 0.43). This suggests that PNPLA3-Ile148Met exhibits a “recessive Mendelian-like inheritance pattern”—the homozygous GG genotype was significantly enriched in the MASLD group, and at the same time, it was observed that the heterozygous GC genotype had a significantly higher frequency in the control group. In addition, for TM6SF2-Glu167Lys, the CT genotype was significantly more frequent in the MASLD group than in the control group (P_FDR=0.003, OR = 2.80), a pattern similar to “dominant susceptibility”.
For other variants (e.g., GCKR-Leu446Pro, LEPR-Lys656Asn), although the ORs of their allele frequencies or genotypes deviated from 1 between the MASLD group and the control group, no statistically significant differences were observed in our sample (all P_FDR>0.050; Table 2). Notably, while the allele frequency of PNPLA3-Lys434Glu was significantly associated with MASLD risk (Table 1), its genotype frequencies did not reach statistical significance (Table 2), suggesting a potential dose-dependent effect that requires further validation.
3.4 Independent risk factors for pediatric MASLD
To identify independent risk factors for pediatric MASLD, a multivariate logistic regression model was constructed incorporating the pre-specified genetic models for PNPLA3-Ile148Met (recessive) and TM6SF2-Glu167Lys (dominant), with adjustment for potential confounding factors including age, sex, and body mass index (BMI). The final fitted model revealed that male sex, the PNPLA3-Ile148Met-GG genotype (under the recessive model), and the TM6SF2-Glu167Lys-CT genotype (under the dominant model, representing CT+TT carriers) were independent risk factors for MASLD (Table 4):
1. Male sex was associated with a 7.76-fold increased risk of MASLD (P_FDR<0.001);
2. Carriers of the PNPLA3 rs738409 GG genotype had a 3.81-fold higher risk of developing MASLD compared with the combined GC/CC genotype group (P_FDR<0.001);
3. Carriers of the TM6SF2-Glu167Lys risk allele (CT/TT genotypes) had a 3.62-fold higher risk of MASLD compared to CC homozygotes (P_FDR=0.003).
Notably, after adjusting for confounding factors and under the recessive model, the PNPLA3-Ile148Met-GC genotype, when grouped with the CC genotype as the reference, showed no significant association with MASLD risk (P_FDR=0.704).
3.5 Differences in biochemical indicators among children with MASLD carrying different PNPLA3 rs738409 genotypes
When comparing biochemical indicators between children with MASLD carrying different PNPLA3 rs738409 genotypes (GG vs. GC), significant differences were observed in three key markers (Table 3): the levels of aspartate aminotransferase (AST), alanine aminotransferase (ALT), and total cholesterol (TC) were all significantly higher in the GG genotype group than in the GC genotype group (all P_FDR<0.001). No significant differences were found between the two genotypes in other indicators, such as body mass index (BMI), glucose, triglycerides (TG), and high-density lipoprotein (HDL) (all P_FDR>0.050).
Collectively, the results of this study confirm the following key findings: First, PNPLA3-Ile148Met (especially the GG genotype) and TM6SF2-Glu167Lys (especially the CT genotype) are significantly associated with an increased risk of MASLD in children, and both were identified as independent risk factors for pediatric MASLD after adjusting for confounding factors like age, sex, and BMI. Second, two SNPs were found to potentially act as protective variants against pediatric MASLD: GCKR-Leu446Pro and LEPR-Lys109Arg, as their allele frequencies were significantly lower in the MASLD group than in the control group (GCKR-Leu446Pro: P_FDR=0.009, OR = 0.64; LEPR-Lys109Arg: P_FDR=0.009, OR = 0.51), suggesting that carriers of these variant sites may have a reduced risk of MASLD.
4 Discussion
This case-control study provides robust evidence for the significant association between specific genetic variants and the risk of MASLD in Chinese children. Our principal findings confirm that PNPLA3-Ile148Met and TM6SF2-Glu167Lys are independent genetic risk factors in this population, thereby validating their central role in pediatric MASLD pathogenesis across ethnicities. Notably, we report the novel and paradoxical finding that the GCKR-Leu446Pro variant, widely recognized as a risk allele in Western adult cohorts, appears to confer a protective effect in our Chinese pediatric cohort. Furthermore, we identified a significant association of the PNPLA3-Lys434Glu variant with increased MASLD risk at the allele level, highlighting the potential contribution of additional variation within this pivotal gene. These results not only validate the roles of established genes but also underscore critical, population-specific characteristics in the genetic architecture of pediatric MASLD.
Our findings robustly reinforce the pivotal role of PNPLA3 in MASLD susceptibility. The PNPLA3-Ile148Met variant (rs738409 G allele) was significantly enriched in our MASLD cohort and was identified as an independent risk factor, consistent with a vast body of international literature (22, 23). As previously reported (24–26), this variant reduces the TG hydrolase activity of PNPLA3 while enhancing its lysophosphatidic acid acyltransferase (LPAAT) activity, thereby inhibiting lipid breakdown and promoting intrahepatic steatosis. Additionally, the significantly higher levels of aspartate aminotransferase (AST), alanine aminotransferase (ALT), and total cholesterol (TC) in children with the GG genotype further validate that this homozygous variant exacerbates liver injury—consistent with Stefano Romeo et al.’s observation that 148M allele carriers have elevated liver enzymes (27), and A. Kotronen et al.’s finding that rs738409 correlates with serum AST levels (28). This suggests PNPLA3-Ile148Met not only increases MASLD susceptibility but also associates with more severe hepatic dysfunction in pediatric patients. The observed deviation from Hardy-Weinberg equilibrium specifically in the MASLD case group for this variant is a recognized hallmark of a genuine, high-effect risk allele, as the over-representation of risk homozygotes disrupts genetic equilibrium in the affected population.
To further elucidate the potential structural and functional impacts of the identified PNPLA3 variants, we performed in silico analysis. Using the DynaMut2 online tool, we compared the wild-type protein structure (obtained from the AlphaFold database, https://alphafold.ebi.ac.uk) with the mutant structures. The analysis predicted that both the Ile148Met and Lys434Glu substitutions are destabilizing to the PNPLA3 protein structure. The Ile148Met variant showed a predicted stability change (ΔΔG) of -0.55 kcal/mol, while the Lys434Glu variant had a ΔΔG of -0.23 kcal/mol. A negative ΔΔG value indicates a decrease in folding stability, which may lead to protein misfolding, accelerated degradation, or a loss of proper enzymatic function (25, 29, 30). For the well-characterized Ile148Met variant, this computational prediction aligns with established experimental evidence showing that the mutation impairs triglyceride hydrolase activity and promotes aberrant lipid accumulation. Although the Lys434Glu variant has been less studied, its predicted destabilizing effect provides a plausible mechanistic basis for its association with MASLD risk observed at the allele level in our cohort (31). This suggests that, similar to the Ile148Met variant, the Lys434Glu substitution may compromise protein integrity and contribute to dysregulated hepatic lipid metabolism.
Similarly, our data confirm the importance of TM6SF2-Glu167Lys (rs58542926 T allele) as an independent risk factor for pediatric MASLD, supporting the conclusion of Goffredo et al (32). The dominant model of susceptibility observed in our cohort aligns with the proposed mechanism whereby the E167K variant causes misfolding and degradation of the TM6SF2 protein (33), impairing very low-density lipoprotein (VLDL) assembly and triglyceride secretion from the liver. It is noteworthy that the frequency of the risk CT genotype in our Han Chinese MASLD group (24.49%) was substantially higher than that reported in some other ethnicities (e.g., 12.19% in Caucasians) (22), highlighting prominent population-specific genetic characteristics and underscoring the necessity of ethnic-specific genetic studies.
A pivotal and novel finding of our study is the protective association of the GCKR-Leu446Pro variant (OR = 0.64) in Chinese children with MASLD. This stands in stark contrast to its well-established role as a risk allele for hepatic steatosis in Western adult cohorts, highlighting the profound context-dependency of genetic risk. Mechanistically, the Leu446Pro variant causes a loss-of-function in the glucokinase regulator, leading to enhanced hepatic glycolysis and de novo lipogenesis (DNL)—the conventional explanation for its steatotic effect. We propose that in the specific context of childhood obesity—where dynamic growth and severe insulin resistance may not yet be fully entrenched—the resulting persistent glucokinase activity may paradoxically offer an advantage by improving hepatic glucose disposal. This potential benefit for systemic glucose homeostasis might temporarily outweigh the pro-steatotic DNL effect, yielding a net protective phenotype in our young cohort. This hypothesis is supported by functional evidence suggesting the variant’s pathogenicity is modulated by metabolic background, such as the presence of diabetes, which was rare in our population (34).
At the allele level, we also observed a significant association between the PNPLA3-Lys434Glu variant and increased MASLD risk. Although this variant did not exhibit significant genotype-level associations, its allele frequency enrichment in MASLD patients and its predicted destabilizing effect on protein structure (ΔΔG = -0.23 kcal/mol) suggest a potential role in disease susceptibility that warrants further investigation in larger cohorts.
From a clinical perspective, our identification of PNPLA3-Ile148Met and TM6SF2-Glu167Lys as independent risk factors, combined with the powerful non-genetic risk factor of male sex (OR = 7.76), provides a practical framework for refining early MASLD screening strategies in Chinese children. For instance, male children carrying the PNPLA3 GG genotype or the TM6SF2 CT/TT genotypes could be prioritized for more intensive lifestyle counseling and liver ultrasound monitoring, enabling timely intervention to prevent disease progression.
5 Limitations and future directions
While this study identifies significant genetic associations with pediatric MASLD, several limitations must be acknowledged. First, the cross-sectional nature of our design precludes the inference of causal relationships between the genetic variants and the development of MASLD. Second, the diagnosis of MASLD was based on ultrasonography rather than liver histology. Although ultrasound is a widely recommended, non-invasive first-line tool for population screening, it has limited sensitivity for detecting mild steatosis (<20% fat infiltration) and cannot assess liver fibrosis (35), a key determinant of disease progression. Third, our cohort was composed exclusively of Han Chinese children, which, while homogenous for initial discovery, limits the generalizability of our findings to other ethnic populations. Finally, we lacked detailed data on environmental and lifestyle factors, such as dietary habits (e.g., fructose intake) and physical activity levels, which are known to modulate genetic risk and are critical components of MASLD pathogenesis.
To address these limitations and build upon our findings, future research should prioritize several avenues. Prospective, longitudinal multiethnic cohorts are needed to establish the temporal sequence of genetic risk leading to MASLD and to validate the population-specific effects we observed, particularly for GCKR-Leu446Pro. The integration of multi-omics data—including genomics, gut microbiome profiling, and metabolomics—will be essential to elucidate the functional pathways through which these genetic variants operate and to uncover how they interact with environmental exposures. Ultimately, such integrated approaches are crucial for transitioning from genetic association to a mechanistic understanding of pediatric MASLD, paving the way for personalized risk prediction and targeted preventive strategies.
6 Conclusion
In summary, we show that the PNPLA3-Ile148Met and TM6SF2-Glu167Lys gene polymorphisms are independent risk factors that significantly increase the risk of MASLD in Chinese children. Additionally, the PNPLA3-Lys434Glu variant is associated with increased risk at the allele level. In contrast, GCKR-Leu446Pro and LEPR-Lys109Arg may confer protective effects. This study provides new evidence for genetic susceptibility to MASLD in the pediatric population.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statement
The studies involving humans were approved by Medical Ethics Committee of Children’s Hospital Affiliated Zhengzhou University, Henan Province (Approval No. 2024-071-002). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
HY: Conceptualization, Methodology, Writing – original draft, Project administration. ZX: Conceptualization, Methodology, Formal analysis, Investigation, Writing – review & editing, Supervision, Funding acquisition. YL: Investigation, Data curation, Validation, Writing – review & editing. MD: Investigation, Resources, Data curation, Writing – review & editing. KY: Software, Formal analysis, Visualization, Writing – review & editing. FZ: Conceptualization, Writing – review & editing, Supervision, Project administration.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was supported by Henan Provincial Science and Technology Plan Project (NO.232102311124).
Acknowledgments
First and foremost, I would like to express my sincere gratitude to FZ (first corresponding author) for their invaluable guidance throughout this study. Their profound expertise in pediatric hepatology and genetics not only shaped the study design, data analysis, and manuscript revision but also provided consistent encouragement that kept this research on track. I am equally thankful to YL, MD, and KY for their dedicated efforts in sample collection, clinical data curation, and high-throughput sequencing data processing—their meticulous work laid a solid foundation for the completion of this study. Additionally, this research was financially supported by the Henan Provincial Science and Technology Plan Project (No. 232102311124), and I would like to acknowledge the support from the Medical Ethics Committee of Zhengzhou Children’s Hospital for approving the study protocol (Approval No.: 2024-071-002), which ensured the ethical conduct of our research involving pediatric participants. I also appreciate the help from colleagues at the Zhengzhou Children’s Hospital and Henan Institute of Medical Genetics for their assistance in participant recruitment and laboratory technical support. Finally, my heartfelt thanks go to my family for their unwavering love and support, which gave me the strength to overcome challenges during the research process.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2025.1689656/full#supplementary-material.
Abbreviations
NAFLD, non-alcoholic fatty liver disease; MAFLD, metabolically associated fatty liver disease; MASLD, metabolic dysfunction-associated steatotic liver disease; PNPLA3, patatin-like phospholipase domain-containing protein 3; LPAAT, lysophosphatidic acid acyltransferase; ATGL, adipose triglyceride lipase; ABHD5, α/β-hydrolase domain containing-5; TAG, triacylglycerol; STAT3, signal transducer and activator of transcription 3; HSCs, hepatic stellate cells; PPAR γ, peroxisome proliferator-activated receptor γ; AP-1, activator protein-1; PROTAC, proteolysis-targeting chimera; SREBPlC, sterol regulator element binding protein 1C; LDs, lipid droplets; LDL, low-density lipoprotein;LPA, lipoprotein a; ROS: reactive oxygen species; VAF, Variant Allele Frequency.
References
1. Subspecialty Group of Gastroenterology, the Society of Pediatrics, Chinese Medical Association, Chinese Children′s Chronic Liver Disease Collaborative Group, and Editorial Board, Chinese Journal of Pediatrics. Expert consensus on the diagnosis and management of metabolic dysfunction-associated steatotic liver disease in children (2025). Zhonghua Er Ke Za Zhi. (2025) 63:960–6. doi: 10.3760/cma.j.cn112140-20250411-00323
2. Lee HY and Yoon EL. Metabolic dysfunction-associated steatotic liver disease, recent revision of terminology and its implications. Korean J Gastroenterol. (2025) 85:126–30. doi: 10.4166/kjg.2025.027
3. European Society for Pediatric Gastroenterology, Hepatology and Nutrition (ESPGHAN), European Association for the Study of the Liver (EASL), North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition (NASPGHAN), Latin-American Society for Pediatric Gastroenterology, Hepatology, and Nutrition (LASPGHAN), Asian Pan-Pacific Society for Pediatric Gastroenterology, Hepatology and Nutrition (APPSPGHAN), and Pan Arab Society for Pediatric Gastroenterology and Nutrition (PASPGHAN). Paediatric steatotic liver disease has unique characteristics: A multisociety statement endorsing the new nomenclature. J Pediatr Gastroenterol Nutr. (2024) 78:1190–6. doi: 10.1002/jpn3.12156
4. Thomas JA, Kendall BJ, El-Serag HB, Thrift AP, and Macdonald GA. Hepatocellular and extrahepatic cancer risk in people with non-alcoholic fatty liver disease. Lancet Gastroenterol Hepatol. (2024) 9:159–69. doi: 10.1016/S2468-1253(23)00275-3
5. Mitrovic B, Gluvic ZM, Obradovic M, Radunovic M, Rizzo M, and Banach M. Non-alcoholic fatty liver disease, metabolic syndrome, and type 2 diabetes mellitus: where do we stand today? Arch Med Sci. (2023) 19(4):884–894. doi: 10.5114/aoms/150639
6. Paik JM, Kabbara K, Eberly KE, Younossi Y, Henry L, and Younossi ZM. Global burden of NAFLD and chronic liver disease among adolescents and young adults. Hepatology. (2022) 75:1204–17. doi: 10.1002/hep.32228
7. Lee EJ, Choi M, Ahn SB, Yoo JJ, Kang SH, and Cho Y. Prevalence of nonalcoholic fatty liver disease in pediatrics and adolescents: a systematic review and meta-analysis. World J Pediatr. (2024) 20:569–80. doi: 10.1007/s12519-024-00814-1
8. Takeichi Y, Miyazawa T, Sakamoto S, Hanada Y, Wang L, and Gotoh K. Non-alcoholic fatty liver disease in mice with hepatocyte-specific deletion of mitochondrial fission factor. Diabetologia. (2021) 64:2092–107. doi: 10.1007/s00125-021-05488-2
9. Guo X, Yin X, Liu Z, and Wang J. Non-alcoholic fatty liver disease (NAFLD) pathogenesis and natural products for prevention and treatment. Int J Mol Sci. (2022) 23:15489. doi: 10.3390/ijms232415489
10. Njei B, Al-Ajlouni YA, Ugwendum D, Abdu M, Forjindam A, and Mohamed MF. Genetic and epigenetic determinants of non-alcoholic fatty liver disease (NAFLD) in lean individuals: a systematic review. Transl Gastroenterol Hepatol. (2024) 9:11. doi: 10.21037/tgh-23-31
11. Yoo JJ, Kim W, Kim MY, Jun DW, Kim SG, and Yeon JE. Recent research trends and updates on nonalcoholic fatty liver disease. Clin Mol Hepatol. (2019) 25:1–11. doi: 10.3350/cmh.2018.0037
12. Draijer L, Benninga M, and Koot B. Pediatric NAFLD: an overview and recent developments in diagnostics and treatment. Expert Rev Gastroenterol Hepatol. (2019) 13:447–61. doi: 10.1080/17474124.2019.1595589
13. Riccio S, Melone R, Vitulano C, Guida P, Maddaluno I, and Guarino S. Advances in pediatric non-alcoholic fatty liver disease: From genetics to lipidomics. World J Clin Pediatr. (2022) 11:221–38. doi: 10.5409/wjcp.v11.i3.221
14. Ericson E, Bergenholm L, Andréasson AC, Dix CI, Knöchel J, and Hansson SF. Hepatic patatin-like phospholipase domain-containing 3 levels are increased in I148M risk allele carriers and correlate with NAFLD in humans. Hepatol Commun. (2022) 6:2689–701. doi: 10.1002/hep4.2032
15. Li J, Hua W, Ji C, Rui J, Zhao Y, and Xie C. Effect of the patatin-like phospholipase domain containing 3 gene (PNPLA3) I148M polymorphism on the risk and severity of nonalcoholic fatty liver disease and metabolic syndromes: A meta-analysis of paediatric and adolescent individuals. Pediatr Obes. (2020) 15:e12615. doi: 10.1111/ijpo.12615
16. Ramandi A, Diehl AM, Sanyal AJ, and de Jong YP. Experimental models to investigate PNPLA3 in liver steatosis. Liver Int. (2025) 45:e70091. doi: 10.1111/liv.70091
17. Li XY, Liu Z, Li L, Wang HJ, and Wang H. TM6SF2 rs58542926 is related to hepatic steatosis, fibrosis and serum lipids both in adults and children: A meta-analysis. Front Endocrinol (Lausanne). (2022) 13:1026901. doi: 10.3389/fendo.2022.1026901
18. Faccioli L, Sun Y, Animasahun O, Motomura T, Liu Z, and Kurihara T. Human-induced pluripotent stem cell-based hepatic modeling of lipid metabolism-associated TM6SF2-E167K variant. Hepatology. (2025) 82(3):638–54. doi: 10.1097/HEP.0000000000001065
19. Li H and Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. (2009) 25:1754–60. doi: 10.1093/bioinformatics/btp324
20. Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. (2011) 27:2987–93. doi: 10.1093/bioinformatics/btr509
21. Rodrigues C, Pires D, and Ascher DB. DynaMut2: Assessing changes in stability and flexibility upon single and multiple point missense mutations. Protein Sci. (2021) 30:60–9. doi: 10.1002/pro.3942
22. Hudert CA, Selinski S, Rudolph B, Bläker H, Loddenkemper C, and Thielhorn R. Genetic determinants of steatosis and fibrosis progression in paediatric non-alcoholic fatty liver disease. Liver Int. (2019) 39:540–56. doi: 10.1111/liv.14006
23. Lin YC, Chang PF, Chang MH, and Ni YH. Genetic variants in GCKR and PNPLA3 confer susceptibility to nonalcoholic fatty liver disease in obese individuals. Am J Clin Nutr. (2014) 99:869–74. doi: 10.3945/ajcn.113.079749
24. Kumari M, Schoiswohl G, Chitraju C, Paar M, Cornaciu I, and Rangrez AY. Adiponutrin functions as a nutritionally regulated lysophosphatidic acid acyltransferase. Cell Metab. (2012) 15:691–702. doi: 10.1016/j.cmet.2012.04.008
25. Mikaeeli S and Cohen DE. Loss or gain of function: The functional complexity of the PNPLA3 I148M variant. J Hepatol. (2025) 82:778–80. doi: 10.1016/j.jhep.2025.01.019
26. BasuRay S, Smagris E, Cohen JC, and Hobbs HH. The PNPLA3 variant associated with fatty liver disease (I148M) accumulates on lipid droplets by evading ubiquitylation. Hepatology. (2017) 66:1111–24. doi: 10.1002/hep.29273
27. Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, and Pennacchio LA. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. (2008) 40:1461–5. doi: 10.1038/ng.257
28. Kotronen A, Johansson LE, Johansson LM, Roos C, Westerbacka J, and Hamsten A. A common variant in PNPLA3, which encodes adiponutrin, is associated with liver fat content in humans. Diabetologia. (2009) 52:1056–60. doi: 10.1007/s00125-009-1285-z
29. Teskey G, Tiwari N, Butcko AJ, Kumar A, Yadav A, and Huang YMM. Lipid droplet targeting of the lipase coactivator ABHD5 and the fatty liver disease-causing variant PNPLA3 I148M is required to promote liver steatosis. J Biol Chem. (2025) 301:108186. doi: 10.1016/j.jbc.2025.108186
30. Wang Y, Hong S, Hudson H, Kory N, Kinch LN, and Kozlitina J. PNPLA3(148M) is a gain-of-function mutation that promotes hepatic steatosis by inhibiting ATGL-mediated triglyceride hydrolysis. J Hepatol. (2025) 82:871–81. doi: 10.1016/j.jhep.2024.10.048
31. Roy A, Paul I, Chakraborty P, Saha A, and Ray S. Unlocking the influence of PNPLA3 mutations on lipolysis: Insights into lipid droplet formation and metabolic dynamics in metabolic dysfunction-associated steatotic liver disease. Biochim Biophys Acta Gen Subj. (2025) 1869:130766. doi: 10.1016/j.bbagen.2025.130766
32. Goffredo M, Caprio S, Feldstein AE, D'Adamo E, Shaw MM, and Pierpont B. Role of TM6SF2 rs58542926 in the pathogenesis of nonalcoholic pediatric fatty liver disease: A multiethnic study. Hepatology. (2016) 63:117–25. doi: 10.1002/hep.28283
33. Kozlitina J, Smagris E, Stender S, Nordestgaard BG, Zhou HH, and Tybjærg-Hansen A. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. (2014) 46:352–6. doi: 10.1038/ng.2901
34. Kimura M, Iguchi T, Iwasawa K, Dunn A, Thompson WL, and Yoneyama Y. En masse organoid phenotyping informs metabolic-associated genetic susceptibility to NASH. Cell. (2022) 185:4216–4232.e16. doi: 10.1016/j.cell.2022.09.031
Keywords: MASLD, PNPLA3, TM6SF2, pediatric hepatology, genetics, polymorphism, genetic susceptibility
Citation: Yao H, Xia Z, Liu Y, Dong M, Yang K and Zhou F (2025) PNPLA3-Ile148Met and TM6SF2-Glu167Lys increase susceptibility to metabolic dysfunction-associated steatotic liver disease in children. Front. Endocrinol. 16:1689656. doi: 10.3389/fendo.2025.1689656
Received: 20 August 2025; Accepted: 20 October 2025;
Published: 31 October 2025.
Edited by:
Kapil Upadhyay, University of Michigan, United StatesReviewed by:
Peerzada Tajamul Mumtaz, University of Nebraska-Lincoln, United StatesHadla Hariri, University of Michigan, United States
Sathyabaarathi Ravichandran, Jackson Laboratory for Genomic Medicine, United States
Copyright © 2025 Yao, Xia, Liu, Dong, Yang and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fang Zhou, emhvdWZhbmc3ODA2QDE2My5jb20=
†These authors share first authorship