 Andrea Di Credico1,2,3†
Andrea Di Credico1,2,3† Giulia Gaggi1,2,3†
Giulia Gaggi1,2,3† Sandra Bibbò1,2,3
Sandra Bibbò1,2,3 Timothy A. Blenkinsop4
Timothy A. Blenkinsop4 Angela Di Baldassarre1,2,3*
Angela Di Baldassarre1,2,3* Barbara Ghinassi2,3,5
Barbara Ghinassi2,3,5- 1Department of Medicine and Aging Sciences, G. d'Annunzio University of Chieti-Pescara, Chieti, Italy
- 2Cell Reprogramming and Differentiation Lab, Center of Advanced Studies and Technologies (CAST), G. d'Annunzio University of Chieti-Pescara, Chieti, Italy
- 3UdA-Tech Lab, G. D’Annunzio University of Chieti-Pescara, Chieti, Italy
- 4Department of Ophthalmology, Cell Development and Regenerative Biology, Black Family Stem Cell Institute, Icahn School of Medicine at Mount Sinai, New York, NY, United States
- 5Department of Innovative Technologies in Medicine and Dentistry, G. D’Annunzio University of Chieti-Pescara, Chieti, Italy
Objective: Prenatal exposure to environmental endocrine-disrupting chemicals (EDCs) has been increasingly linked to neurodevelopmental impairment. Bisphenol S (BPS) and perfluoro-octane sulfonate (PFOS), two widely distributed EDCs detected in maternal and fetal tissues, raise concern due to their potential to interfere with brain development even at low environmental doses.
Methods: a phenotypic screening on human iPSC-derived cerebral organoids was performed to explore whether chronic exposure to BPS and PFOS could affect key neurodevelopmental processes.
Results: Both compounds affected key neurodevelopmental processes, including neuronal proliferation, cortical specification, synaptogenesis, glutamatergic differentiation, mitochondrial function, and choroid plexus formation. Importantly, TUNEL assay confirmed the absence of significant cytotoxicity. BPS exposure was associated with reduced ERβ, GPER, and phosphorylated Akt expression, suggesting a possible involvement of estrogen-related pathways. PFOS exposure coincided with decreased transthyretin expression, suggesting a potential influence on thyroid hormone availability.
Conclusions: Exposure to multiple EDCs may disrupt distinct endocrine axes, producing cumulative impacts on human brain development. These findings underscore the value of human-relevant models for identifying endocrine-mediated neurodevelopmental hazards. While the observed molecular changes suggest distinct hormonal pathways may be involved, future mechanistic studies, including co-exposures with receptor modulators, will be required to establish causal relationships.
Introduction
Human brain development is a tightly regulated process that begins in early embryogenesis and extends into young adulthood (1). It involves sequential phases of neurogenesis, neuronal migration, and synaptogenesis, which are essential for the formation of functional neural circuits underlying cognition, sensory processing, and behavior (1–3).
Disruptions during these critical stages can have long-lasting consequences, contributing to neurodevelopmental disorders such as autism spectrum disorder (ASD), attention-deficit/hyperactivity disorder (ADHD), and intellectual disabilities (4).
Among the environmental factors implicated in altered neurodevelopment, endocrine-disrupting chemicals (EDCs) have emerged as compounds of concern. Bisphenol S (BPS) and perfluoro-octane sulfonate (PFOS) are two widespread EDCs found in consumer products including plastics, thermal receipts, non-stick coatings, and personal care items (5–8).Human biomonitoring studies have documented measurable PFOS levels in Italian populations (9, 10) and detectable urinary BPS in a European adult cohort (11). Although these data derive from separate studies, together they support a plausible real-life scenario in which individuals are chronically exposed to both compounds. Investigating BPS and PFOS in combination at environmentally relevant, low-dose levels reflects this realistic exposure pattern, while also considering their mechanistic complementarity, BPS modulating estrogen-receptor pathways (12–14) and PFOS disrupting thyroid-hormone signaling (15) which may yield additive or synergistic neurodevelopmental effects.
From a regulatory perspective, several frameworks, including the EU REACH Regulation and international guidance documents, have incorporated assays addressing EDC mechanisms, particularly those linked to estrogen receptor (ER) or thyroid hormone receptor signaling (16). However, most validated in vitro and in vivo tests still focus on receptor binding, hormone synthesis, or systemic hormone levels, without directly assessing neurodevelopmental endpoints (17). Consequently, human-relevant models that capture hormone-mediated effects on brain development remain scarce, despite the increasing recognition of these pathways as critical determinants of developmental neurotoxicity (18).
Estrogen and thyroid hormone signaling play pivotal roles in early brain development. Estrogen receptors (ERα, ERβ, and the G-protein–coupled estrogen receptor, GPER) influence neural stem cell proliferation, differentiation, and synaptogenesis (19). Similarly, thyroid hormones act through nuclear receptors to regulate neural progenitor proliferation, neuronal migration, synaptic maturation, and oligodendrocyte differentiation (20, 21).
Disruption of these hormone-regulated pathways during sensitive developmental windows has been associated with cognitive impairment, neurobehavioral abnormalities, and defective myelination (20, 22).
Notably, both chemicals can cross the human placenta during pregnancy (23), a period characterized by high sensitivity to hormonal cues. Yet, despite growing concern (24–26), the specific mechanisms by which these EDCs may impact the human brain remain poorly understood. Most studies to date rely on animal models or high-dose, short-term exposures that do not reflect real-life scenarios (27, 28), and often fail to capture the complexity of human neurodevelopment (29).
The ability of stem cells to generate diverse cell types offers great potential to model several diseases (30–35), and the development of brain organoids derived from human induced pluripotent stem cells (hiPSCs) offers a powerful platform to model early brain formation in a human-relevant context (36, 37). These three-dimensional structures mimic the architecture and functionality of the human brain on a miniature scale (38), offering an accurate representation of the complex cellular interactions and developmental processes that occur in vivo (39, 40).In this study, we employed brain organoids to investigate the effects of chronic exposure to environmental doses of BPS and PFOS, individually and in combination, on human neurodevelopment. We examined a range of morphological and molecular parameters, including progenitor proliferation, cortical specification, synaptogenesis, glutamatergic differentiation, mitochondrial function, and hormone receptor–related signaling. Our findings aim to provide mechanistic insights into how real-life exposures to persistent pollutants may compromise brain development and inform ongoing efforts in environmental health risk assessment.
Materials and methods
Human iPSCs maintenance and culture
Two lines of hiPSCs from male donors (41), and (30HU-002, iXcell Biotechnology) were cultured on a monolayer of irradiated embryonic murine fibroblasts (A24903, Thermo Fischer Scientific, Waltham, MA, USA) in Dulbecco’s Modified Eagle’s Medium/Nutrient Mixture F-12 (DMEM/F12, 11320033, Gibco) supplemented with 20% Knockout Serum Replacement (KO), 1% penicillin/streptomycin, 2 mM l-glutamine, 1% MEM nonessential amino acid, 55μM β-Mercaptoethanol (all Life Technologies) and 5 ng/mL basic fibroblast growth factor (bFGF) (R&D Systems) or in Matrigel (CLS354263, Corning, Sommerville, MA, USA)-coated plates using mTesR plus medium (Stemcell Technologies). hiPSCs were maintained at 37 °C, 5% CO2. iPSCs were split every 5 days using 1x EDTA, and 10 µM ROCK Inhibitor Y-27632.
The hiPSC lines underwent standard banking and quality control prior to use. Banking was performed at early passages under controlled cryopreservation conditions using KO serum in 10%DMSO. Quality control included verification of pluripotency marker expression by immunofluorescence and mycoplasma testing. Only hiPSC batches passing all quality control criteria were expanded and used for subsequent differentiation experiments.
Cerebral organoids differentiation protocol and EDCs treatment
Cerebral organoids were obtained using the STEMdiff Cerebral Organoid kit (STEMCELL TECHNOLOGIES, Vancouver, Canada) based on the protocol published by Lancaster et al. (36). Briefly, on Day 0, hiPSCs were seeded in 96-well round-bottom ultra-low attachment microplate (Corning, Sommerville, MA, USA) in Seeding Medium (Basal Medium 1 + Supplement A + 10 µM ROCK Inhibitor Y-27632). Cells were cultured in Organoid Formation Medium (Basal Medium 1 + Supplement A), and then from Day 5 in a Neuronal induction medium (Basal Medium 1 + Supplement B) in 24-well ultra-low attachment plate (Corning 3473). On day 7, organoids were embedded in Matrigel drop (CLS354263, Corning, Sommerville, MA, USA) and incubated in Expansion Medium (Basal Medium 2 + Supplement C and D) in 6-Well Ultra-Low Adherent (100-0083). From day 10 onwards, organoids were cultured in Maturation Medium (Basal Medium 2 + supplement E) and shaken continuously at 65 rpm using Celltron (INFORS HT, I69222). To study the EDCs effects, organoids were chronically treated with 50nM BPS, and/or 100nM of PFOS (Wellington Laboratories Inc., Canada) and vehicle (CTRL, methanol 0.129%, a concentration that has been shown to be non-toxic in previous studies (24, 42)) from day 10 onward. Medium change with EDCs was performed every 3 days. On days 20 and 40, cerebral organoids were collected and analyzed. Any organoids that showed leakage from Matrigel or that had fused together were excluded for subsequent analysis.
Organoids imaging: morphological, morphometrical and immunofluorescent analyses
For morphometrical analysis, brain organoid brightfield images were acquired using EVOS M7000 during the maturation phase at different timepoints. The area was measured by Fiji (ImageJ) software version 1.54k using the “Threshold” function, while the shape analysis was performed by measuring the organoid circularity (circularity index: 4 *area/perimeter).
For the other analyses, brain organoids were embedded in optimal cutting temperature (OCT) compound (Leica, Wetzlar, Germany) then frozen at -80 °C, and 10 μm thick sections were obtained using the CM1950 cryostat (Leica, Wetzlar, Germany).
For hematoxylin and eosin staining, slides were fixed in acetone for 5 min, incubated in Mayer’s hemalum solution (TC62200QQ, Titolchimica) for 1 min and, after washings, with eosin for 45 sec. Slides were mounted with Histo mount medium and the ventricular-like zone (VLZ) area was measured using Fiji (ImageJ) software version 1.54k: the outer edges of VLZ were identified; then the internal empty area was subtracted to obtain the actual VLZ area expressed in µm2.
For immunofluorescent staining, cryosections were fixed in acetone, permeabilized with 0.5% Triton for 15 min and blocked in 5% BSA for 30 min. The following primary antibodies were used for immunofluorescence staining: anti-Ki-67 (53-5698-82, Thermo Fisher Scientific, MA, US), anti-synaptophysin (SAB4502906, Sigma-Aldrich, St. Louis, Missouri, US), anti-TBR1 (WH0010716M1, Sigma-Aldrich, St. Louis, Missouri, US), anti-Ctip2 (ab233713, Abcam, Cambridge, UK), anti-SATB2 (ab34735, Abcam, Cambridge, UK), anti-transthyretin (MA5-32634, Thermo Fisher Scientific, MA, US), anti-β-Tubulin III (T8578, Sigma-Aldrich, St. Louis, Missouri, US), anti-NMDAR2B (ab65783, Abcam, Cambridge, UK), anti-VGlutT1(ab180188, Abcam, Cambridge, UK), and anti-PSD-95 (ab18258, Abcam, Cambridge, UK). Immunofluorescent images were acquired using Operetta CLS (Revvity). Due to limitations in nuclear segmentation accuracy within densely proliferative zones, Ki-67 expression was quantified as the ratio of Ki-67+ area to DAPI+ area using constant thresholding parameters across samples, using Fiji (ImageJ) version 1.54K. PSD95 results were obtained as the ratio of PSD95+ area to DAPI+ area. This approach provided a robust and reproducible measure of proliferative activity while minimizing segmentation bias.
Nuclear CTIP2 and SATB2 translocation levels were evaluated using Harmony Software (Revvity). Briefly, nuclear segmentation was performed to identify the nuclei population based on DAPI-guided masks, and an adjacent outer region was selected to delimit the cytoplasmic compartment. Protein fluorescence intensity was quantified in pixels for both nuclear and cytoplasmic regions. Nuclear translocation levels were then calculated by dividing the nuclear fluorescence by the cytoplasmic fluorescence, and results were expressed as arbitrary units. Image acquisition was performed with a PerkinElmer Operetta CLS high-content imaging system, optimized for broad-field coverage to ensure consistency and reproducibility across multiple fields and biological replicates. This approach prioritized sampling depth and quantitative robustness over subnuclear resolution, allowing objective assessment of CP-specific expression patterns across large sample areas.
TUNEL assay
Cell apoptosis was evaluated on day-40 cerebral organoid cryosections using the In Situ Cell Death Detection Kit, TMR red (Roche, cat. no. 12156792910), following the manufacturer’s instructions with minor adaptations.
Organoids cryosections were fixed in acetone, permeabilized with 0.5% Triton for 15 min then incubated with the TUNEL reaction mixture (Enzyme Solution + Label Solution) for 1 h at 37 °C in a humidified chamber. After washing in PBS, nuclei were counterstained with DAPI and mounted with antifade medium.
Immunoblotting
Brain organoid protein contents were determined by Western blotting. Organoids were lysed in ice-cold RIPA lysis buffer (89900, Thermo Fisher Scientific, MA, US) adding protease and phosphatase inhibitors. Samples were then sonicated and centrifuged (14,000 × g) for 20 min at 4˚C. The protein concentration of each sample was determined in triplicate with a Pierce™ BCA Protein Assay Kits (23225, Thermo Fisher Scientific, MA, US). Equal amounts of protein (15 ug) were separated by pre-cast 4-15% gels (Bio-Rad Laboratories, CA, US) and transferred to a PVDF membrane (Millipore A/S, Copenhagen, Denmark). Afterward, No-Stain™ Protein Labeling Reagent (A44449, Thermo Fisher Scientific, MA, US) was used to check for total protein loading and transfer efficiency. After blocking in TBS-Tween 3% BSA, membranes were incubated overnight with the primary antibody at 4 °C, and then with the appropriate HRP-conjugated secondary antibody for 1 h. Bands were visualized with ECL (Millipore) and recorded with a digital camera (iBright, Thermo Fisher Scientific, MA, US). Bands were quantified using Image Lab version 6.0 (Bio-Rad Laboratories) and determined as the total band intensity adjusted for background intensity and normalized for beta-actin.
Primary antibodies used were anti-total OXPHOS (ab110411, Abcam, Cambridge, UK), anti-beta actin (MA1-140, Thermo Fisher Scientific, MA, US), anti-synaptophysin (SAB4502906, Sigma-Aldrich, St. Louis, Missouri, US), anti-TBR1 (WH0010716M1, Sigma-Aldrich, St. Louis, Missouri, US), anti-Ctip2 (ab233713, Abcam, Cambridge, UK), anti-SATB2 (ab34735, Abcam, Cambridge, UK), anti-transthyretin (MA5-32634, Thermo Fisher Scientific, MA, US), anti-ER alpha (ab76228, Abcam, Cambridge, UK), anti-ER beta (ab187291, Abcam, Cambridge, UK), anti GPER (ab39742, Abcam, Cambridge, UK), anti-pAkt (4058S, Cell Signaling, Danvers, MA, US), anti-Akt (4691S, Cell Signaling, Danvers, MA, US), anti-pERK1-2 (9101, Cell Signaling, Danvers, MA, US), anti-ERK1-2 (9102, Cell Signaling, Danvers, MA, US), anti-pmTOR (ab109268, Abcam, Cambridge, UK), and anti-mTOR (PA534663, Thermo Fisher Scientific, MA, US).
The secondary antibodies used were HRP-conjugated goat anti-rabbit IgG (31460, Thermo Fisher Scientific, MA, US), and goat anti-mouse IgG (31430, Thermo Fisher Scientific, MA, US).
Statistical analysis
The one-way analysis of variance (ANOVA) was used to check for significant differences when the only independent variable was represented by the treatments (organoids circularity, VLZ area, all immunofluorescence, and western blot data). When ANOVA showed significant results, Tukey’s post-hoc analysis was used to check for differences between the control and treatments. For organoids area and proliferation index, a mixed-effect analysis was used. Time and treatment were considered as independent variables and area or proliferation index as outcome measures. In this case, Fisher’s Least Significant Difference (LSD) test was used as a post-hoc analysis. Results were considered significant when p<0.05. All statistical analyses were performed using GraphPad PRISM version 10.2.1 (GraphPad Software, LLC).
Results
Chronic exposure to BPS affects size but not shape of developing brain organoids
To analyze the effects of environmental xenobiotics on the early and critical phases of human neurodevelopment, brain organoids were chronically exposed to BPS (50 nM), PFOS (100 nM), or a combination of both. These doses fall within the range of concentrations found in various human fetal tissues and organs, and are therefore representative of the actual exposure of the developing fetus (43–45). The treatment phase started on day 10 of culture, when neuronal induction was completed, and organoids began maturation, continuing up to day 40. It has already been reported, indeed, that 40 to 60-day-old organoids recapitulate the mid-fetal brain (10–16 post-conception weeks) (46, 47). A scheme of the cell culture protocol is reported in Figure 1A.
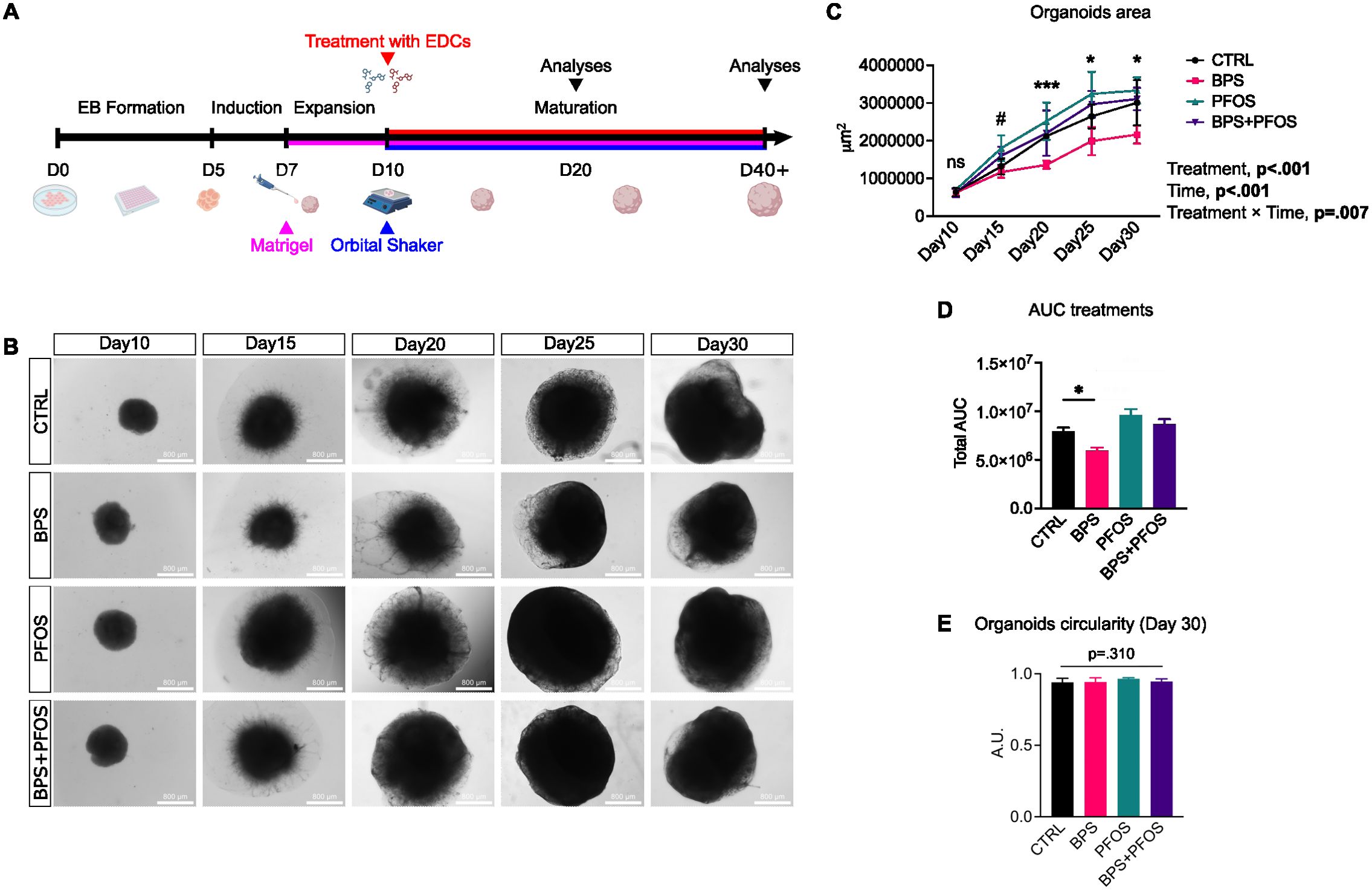
Figure 1. Effect of BPS and/or PFOS on the growth of brain organoids. (A) Schematic of the cerebral organoids differentiation timeline. (B) Representative brightfield microscopy images of the organoids from day 10 to day 30. Scale bar, 800 µm, 4x magnification. (C) Measures of the organoid area during the cell culture expressed in μm2, and mixed-effect analysis results. * p<.05, *** p<.001CTRL vs. BPS, and # p<.05 CTRL vs. PFOS; ns, non-significant. (D) Quantification of the AUC of different treatments over time. (E) Measures of organoid circularity at day 30. Organoid area and circularity results are based on n = 4 independent (2 for each cell line) organoid batches (biological replicates), each derived from independent differentiation runs, and that for each batch, at least n = 3 organoids were analyzed. Data are reported as mean and standard deviation.
The effects of the treatments on the external morphology (size and shape) of the brain organoids were analyzed. The organoid area was monitored during the first phases of the maturation process using brightfield images (Figure 1B). Mixed-effect analysis showed that both time and treatments had significant effects on organoid growth (Figure 1C): indeed, the organoids kept on growing during the culture in all the experimental conditions, but the BPS-treated remained constantly smaller than the other samples, as confirmed by the area under the curve (AUC) analysis (Figure 1D).
Then, the organoid circularity was measured; this parameter is used in organoid morphological studies because its changes often accompany alterations in the maturation process (48). Treatments did not affect the external shape of the organoids, as no changes were detected in the circularity values. (Figure 1E).
BPS and PFOS influence VLZ and neural stem cell proliferation inside the brain organoid
The developing human brain is characterized by ventricle-like cavities (ventricular-like zone – VLZ), proliferative regions of neural stem cells that, over time, move outward, producing the multilayered cortical-like structure. We then analyzed whether antenatal exposure to BPS and/or PFOS can induce abnormalities in the VLZ of the developing brain. Based on morphological features and spatial marker distribution, these VLZ structures were identified as round, densely nucleated regions located beneath the cortical-like plate. Supplementary Figure S1 shows that Ki-67+ proliferative progenitors are predominantly localized within these internal areas, whereas βIII-tubulin (Tuj1) expression is enriched in the surrounding cortical regions, suggesting a spatial segregation between proliferating and post-mitotic cells consistent with early cortical development. At the end of the maturation period (day 40), brain organoids underwent hematoxylin and eosin staining, and the VLZ were analyzed. Data evidenced that the average VLZ area was smaller in BPS+PFOS samples compared to the control (Figures 2A, B).
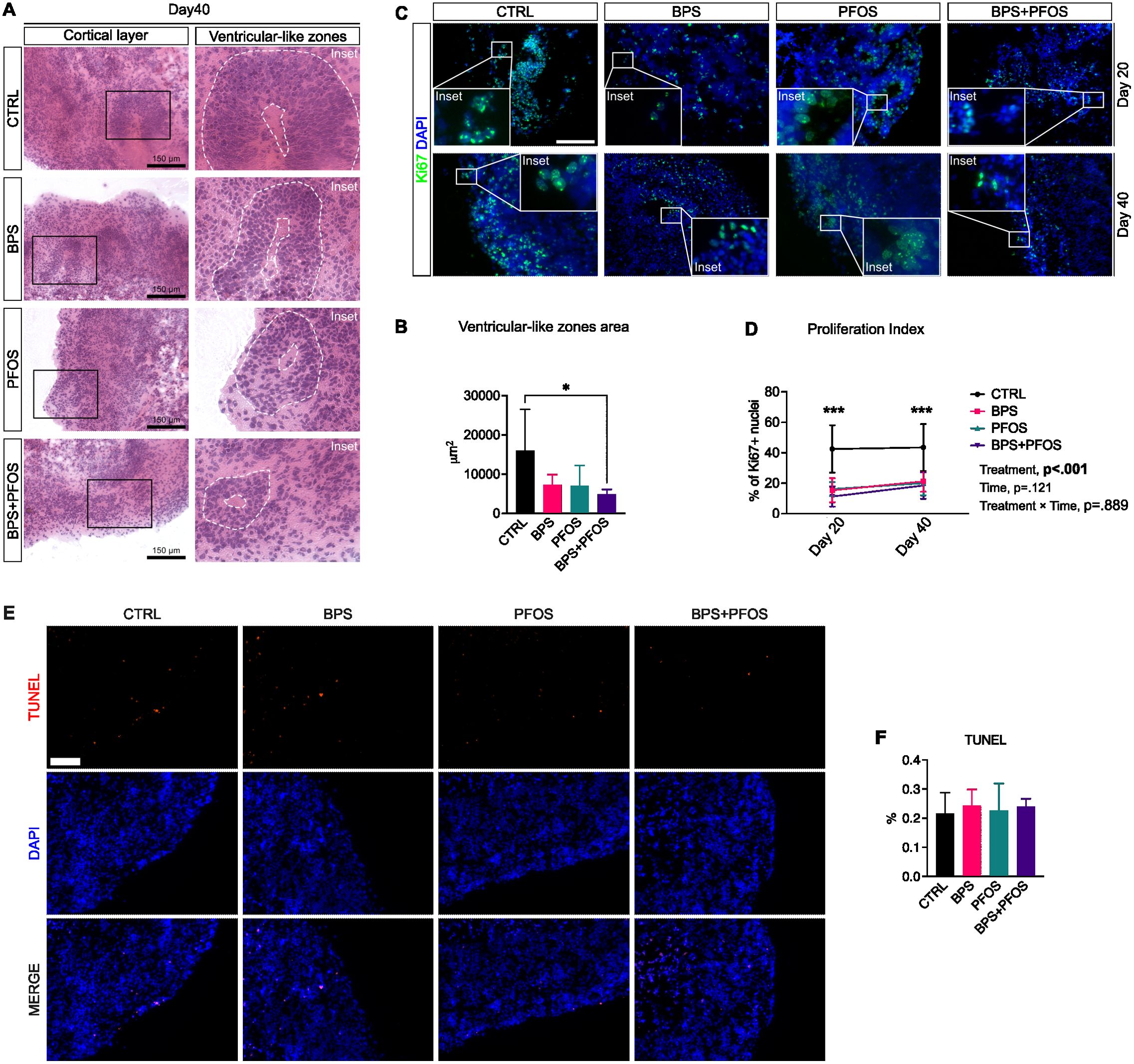
Figure 2. (A) Hematoxylin/eosin staining of cerebral organoids at day 40. Scale bar, 150 µm, original magnification 20 ; the insets highlight representative VLZ structures. (B) Area of the VLZ expressed in μm2 at day 40. * p<.05. (C) Immunostaining of cerebral organoids for Ki67 (green fluorescence) at day 20 (upper row) and 40 (lower row). Nuclei are counterstained with DAPI (blue fluorescence). Scale bar, 150 µm, original magnification 20 . (D) Percentage of Ki67+ nuclei at days 20 and 40; the main mixed-effects analysis results are also reported. (E) Immunostaining of cerebral organoids for TUNEL (red fluorescence). Nuclei are counterstained with DAPI (blue fluorescence). Scale bar, 100 µm, original magnification 20 (F) Percentage of TUNEL+ nuclei at day 40 in the different condition analyzed by one-way ANOVA. VLZ area, proliferation index and TUNEL+ nuclei results are based on n = 4 independent (2 for each cell line) organoid batches (biological replicates), each derived from independent differentiation runs, and that for each batch, at least n = 3 organoids were analyzed with at least n = 5 fields per organoid. *** p<.001 CTRL vs treatments. In all graphs, data are reported as mean and standard deviation.
Then, to quantify the proliferation index of the neuronal progenitors, we examined the expression of Ki-67, a typical cell proliferation marker, by immunofluorescence. Analyses were performed on 20-day and 40-day organoids to monitor the effect of the treatments during the maturation period. As expected, proliferative zones were predominantly detected at the cortical level and within the VLZ (Figure 2C).
The quantification of Ki-67+ nuclei revealed that, under all experimental conditions, the proliferation rate remained constant throughout the maturation phase, with no significant change observed between day 20 and day 40. However, control organoids showed a significantly higher percentage of Ki-67+ nuclei compared to those exposed to BPS, PFOS, and BPS+PFOS, both on day 20 and 40 (Figure 2D).
Since reductions in Ki-67+ cells could reflect either impaired proliferation or increased cell death, we performed a TUNEL assay at day 40 to assess whether the observed effects might result from unspecific cytotoxicity (Figure 2E). The analysis did not reveal any statistically significant increase in apoptotic cells across treatment groups compared to controls (Figure 2F), indicating that chronic low-dose exposure to BPS, PFOS, or their combination did not induce measurable apoptosis in this system.
Taken together, these findings suggest that the decrease in proliferating progenitors is unlikely due to overt cytotoxicity but reflects specific alterations in neurodevelopmental programs induced by chronic exposure to environmentally relevant concentrations of BPS and PFOS.
BPS and/or PFOS interfere with the development of cerebral cortical layers and choroid plexus formation
We then analyzed the effect of BPS and/or PFOS exposure on the internal architecture of brain organoids, focusing on the development of the cortical plate (CP) and the formation of the choroid plexus (ChP). To assess cortical development, we first examined T-box brain 1 (TBR1) and synaptophysin (SYP) expression in 40-day-old brain organoids (Figure 3B). TBR1 is a transcription factor of the T-box family, expressed soon after cortical progenitors begin differentiating, and it is highly detected in early-born neurons of the preplate (49). Immunofluorescence analysis showed that TBR1 expression was reduced by BPS and BPS+PFOS treatment compared to the control, while no differences were observed when cells were exposed to PFOS alone (Figure 3C). Western blot analysis confirmed a decrease in TBR1 expression following BPS exposure compared to control (Figures 3J, K).
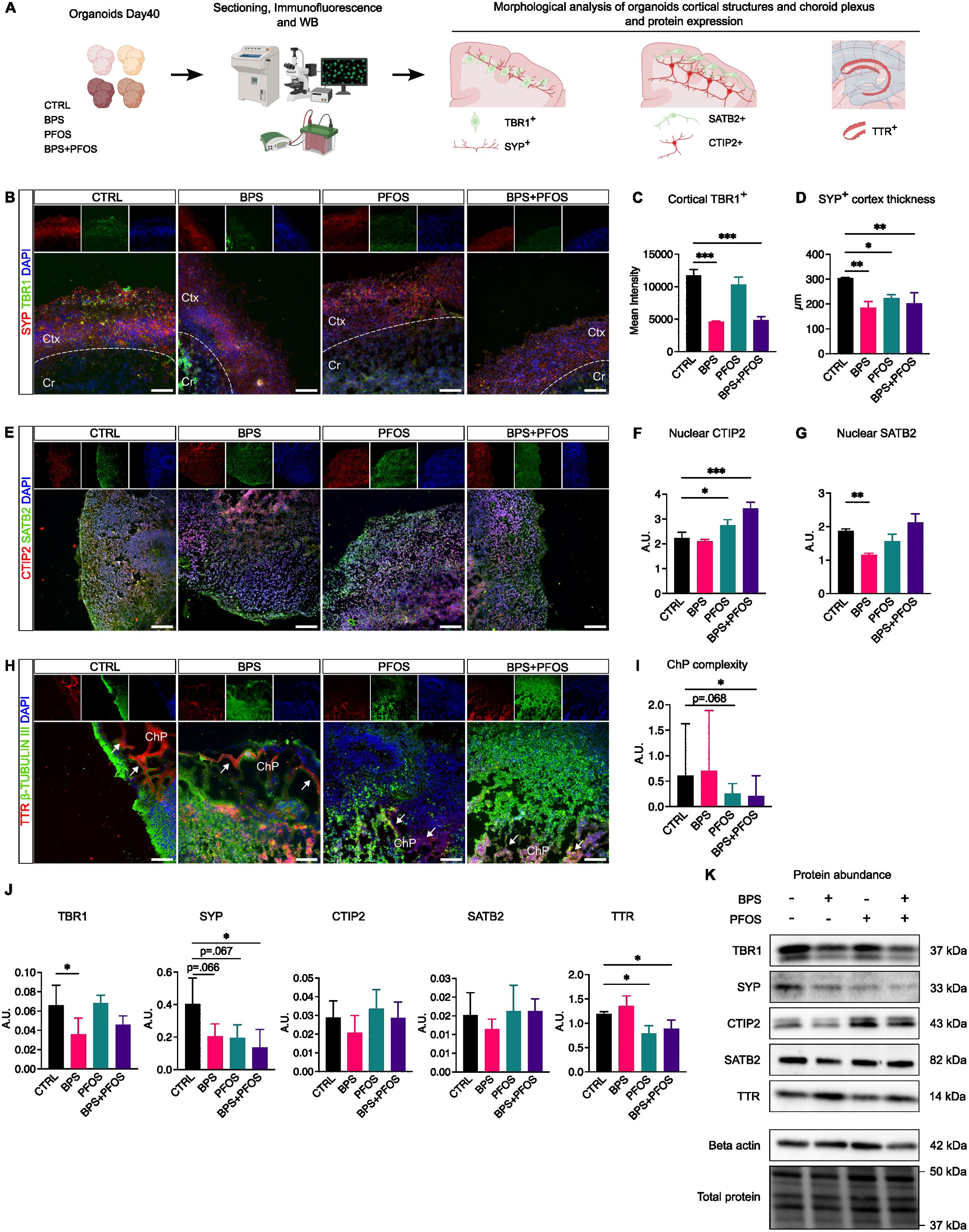
Figure 3. Effect of BPS and/or PFOS on brain organoid architectures. (A) Experimental procedures and workflow. (B) SYP (red fluorescence) and TBR1 (green fluorescence) localization in control and treated brain organoids; the cortical and central zones are divided by the dashed line. (C) Quantification of TBR1 expression at cortical level. (D) Quantification of SYN positive cortical thickness. (E) Representative images of CTIP and SATB2 localization. (F, G) Quantification of nuclear levels of CTIP2 and SATB2. (H) TTR (red fluorescence) and -Tubulin III (green fluorescence) expression. White arrows indicate the TTR+ zone surrounding the cavities. (I) Measures of choroid plexus complexity. (J, K) Western blot images and band quantification of TBR1, SYP, SATB2, CTIP2, and TTR expression in the different experimental conditions. Microscopic images results are based on n = 4 independent (2 for each cell line) organoid batches (biological replicates), each derived from independent differentiation runs, and that for each batch, at least n = 3 organoids were analyzed with at least n = 5 fields per organoid. Western blot results are based on n = 4 independent (2 for each cell line) organoid batches (biological replicates), each derived from independent differentiation runs, and for each batch, n = 3 organoids were pooled. In the immunofluorescence, nuclei are counterstained with Dapi (blue fluorescence). Scale bar 100 µm, original magnification: 20 ; Ctx, cortex; Cr, central region; ChP, choroid plexus. In all graphs, data are reported as mean and standard deviation. *p<.05, **p<.01, ***p<.001.
We then focused on SYP, a synaptic vesicle membrane protein expressed throughout the brain and widely used as a marker of synaptogenesis (50). In 40-day organoids, SYP signal was detected primarily in the cortical region, where newly differentiated neurons begin to establish synaptic connections (Figure 3B). Consistent with its known expression pattern during early development, SYP immunoreactivity was not restricted to discrete synaptic puncta but showed a diffuse distribution along neuronal process. This pattern is typical of immature neurons and reflects ongoing synaptic assembly and vesicle transport, as reported in previous studies (51, 52). The thickness of the SYP-positive zone, corresponding to the CP, was significantly reduced in all treated groups, with the most pronounced decrease observed following BPS and BPS+PFOS exposure (Figure 3D). Western blot quantification of SYP confirmed the downregulation of this protein, particularly in the BPS+PFOS group (Figures 3J, K). To strengthen these findings, we performed an additional co-staining for PSD95, a canonical postsynaptic marker, together with Tuj1, which labels the neuronal cytoplasm. The PSD95 signal was predominantly localized in the cortical regions and showed a substantial reduction in treated organoids, consistent with the observed SYP pattern (Supplementary Figure S2). These findings indicate that both presynaptic (SYP) and postsynaptic (PSD95) compartments are impaired by chronic exposure to BPS and PFOS, supporting the hypothesis of altered synaptogenesis and delayed cortical maturation.
We then analyzed the effects of BPS and/or PFOS on neuronal specification within the CP. In the developing mammalian neocortex, the thickness of CP increases, allowing the six specialized cortical layers to develop, with a specific layer-dependent expression of neuronal subtype markers (53). Specifically, we focused on COUP TF1-interacting protein 2 (CTIP2) and SATB homeobox 2 (SATB2): initially co-expressed by early born cortical neurons, the expression of these molecules becomes more selective during CP maturation. SATB2 predominantly marks intracortical projection neurons in the upper layers (II-IV), while CTIP2 identifies subcortical projection neurons in deeper layers (V-VI). Immunofluorescence analysis showed that in day-40 brain organoids, SATB2 was mainly localized in the outermost layers of the CP, whereas CTIP2 labeling was more diffuse, mainly staining the inner zones. This maturation pattern was altered by the treatments, as in all treated samples, the separation between SATB2+ and CTIP2+ populations was less clear, showing a mixed positivity without the layered localization seen in controls (Figure 4E). Being transcription factors, CTIP2 and SATB2 translocate into the nucleus to interact with DNA. Thus, we quantified the nuclear expression of these markers: results showed that organoids treated with PFOS and the BPS+PFOS combination had significantly increased nuclear CTIP2 compared to controls (Figure 4F). Conversely, BPS treatment reduced nuclear SATB2 content relative to controls (Figure 4G). No differences in total CTIP2 and SATB2 expression were detected via Western blot (Figures 4J, K). These findings suggest that BPS and PFOS may impair the normal processes of CP formation and neuronal specification, affecting TRB1 expression and disrupting the nuclear translocation of SATB2 and CTIP2.
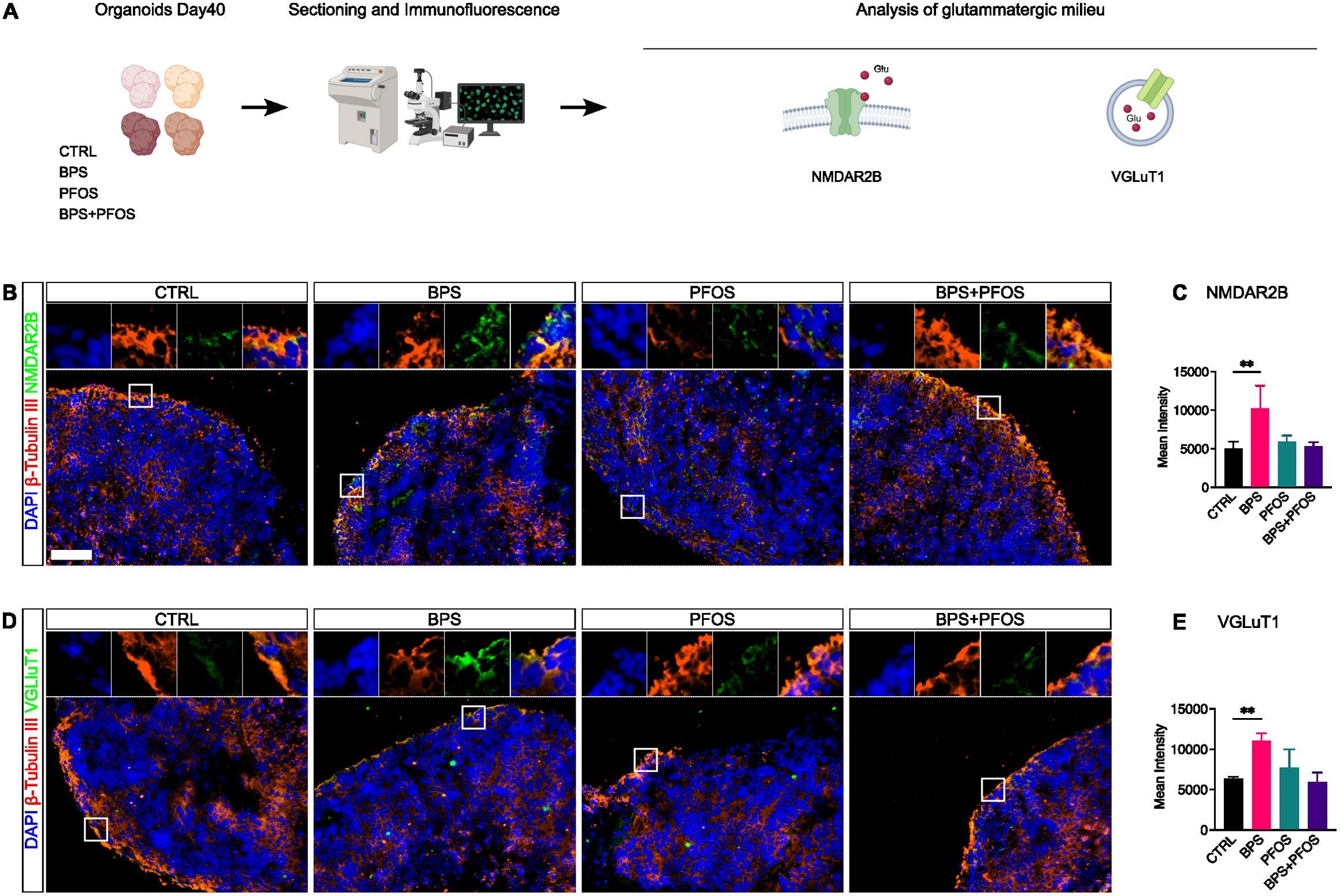
Figure 4. Effect of BPS and/or PFOS on NMDAR2B and VGluT1 expression in 40-days organoids. (A) Experimental procedures and workflow. (B-E) Expression and quantification of NMDAR2B and VGluT1 by immunofluorescence. In all graphs, data are reported as mean and standard deviation. *p<.05, **p<.01, ***p<.001. Microscopic images results are based on n = 4 independent (2 for each cell line) organoid batches (biological replicates), each derived from independent differentiation runs, and that for each batch, at least n = 3 organoids were analyzed with at least n = 5 fields per organoid. Nuclei are counterstained with DAPI (blue fluorescence) while neural cytoskeleton with β-Tubulin III (red fluorescence). Scale bar, 100 µm, original magnification 20 . White square indicates the region magnified in the inset.
We finally analyzed the effects of BPS and/or PFOS exposure on the ChP formation. It has already been reported that day-40 brain organoids display internal areas of columnar or cuboidal cells enriched in transthyretin (TTR) that delimit small cavities in which the cerebrospinal fluid is secreted (54). Thus, to analyze ChP development, immunofluorescent analysis was performed by labeling brain organoid slices for β-Tubulin III, an early marker of neural differentiation, and for TTR, which identifies the ChP. In our models, the EDCs dramatically interfere with the ChP development, as evidenced by a weaker TTR fluorescence and cavities of minor dimensions in the treated samples (Figure 3H). To confirm this observation, we measured the “complexity” of ChP architecture; ChP complexity was calculated as luminal area divided by total ChP area (i.e., the area delineated by TTR+ staining). This parameter reflects the degree of folding and branching of the ChP epithelium, which is a morphological hallmark of its maturation and secretory specialization. Results evidenced BPS+PFOS treatment significantly affected the development of the ChP, changing its architecture (Figure 3I). Finally, Western blot analysis confirmed the downregulation of TTR protein expression after PFOS and BPS+PFOS treatments compared to control (Figures 3J, K).
BPS exposure affects glutamatergic neurogenesis
The formation of the human cerebral cortex in vivo involves the assembly of canonical circuits, with glutamate being the principal excitatory neurotransmitter (53). To investigate the effect of EDCs on glutamatergic neurogenesis, we examined NMDAR2B and VGluT1 expression through immunofluorescence analysis. NMDAR2B is a subunit of NMDA-type glutamate receptors that localizes at both postsynaptic and extra-synaptic sites, including neurites and soma, and plays a central role in neuronal excitability, calcium signaling, and synaptic plasticity (55, 56). These markers were co-stained with βIII-tubulin (Tuj1) to better visualize their localization and distribution.
Both NMDAR2B and VGluT1 immunoreactivity were detected primarily within the CP (Figures 5B, D), where newly generated glutamatergic neurons begin to form synaptic connections. This cortical localization is biologically consistent with the developmental role of these markers in excitatory circuit assembly.
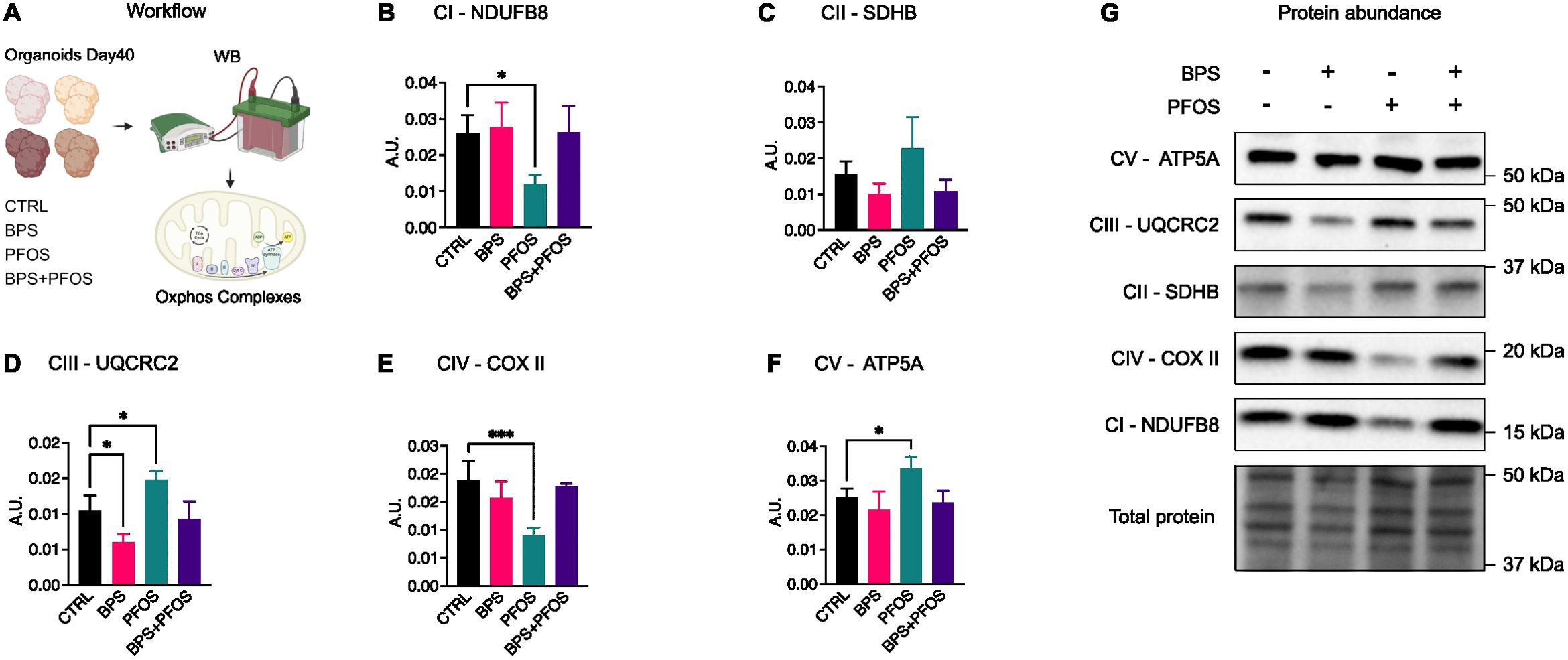
Figure 5. Effect of BPS and/or PFOS on the ETC complexes proteins. (A) Schematics of the experimental workflow: on day 40, brain organoids were processed for Western blot analysis and the expression of the five ETC complexes was evaluated, (B-G) Western blot images and band quantification of Complex I (NDUFB8), Complex II (SDHB), Complex III (UQCRC2), Complex IV (COXII), Complex V (ATP5A). Western blot results are based on n = 4 independent (2 for each cell line) organoid batches (biological replicates), each derived from independent differentiation runs, and for each batch, n = 3 organoids were pooled. In all graphs, data are reported as mean and standard deviation. *p<.05, ***p<.001.
Both antigens exhibited a partially clustered positivity, consistent with their expression pattern during early synaptic development (57, 58). Quantitative analysis of fluorescence intensity revealed that BPS exposure, unlike PFOS, significantly increased NMDAR2B and VGluT1 expression, suggesting an EDC-driven modulation of glutamatergic maturation (Figures 5C, E).
BPS and PFOS differentially regulate the expression of the mitochondrial oxidative phosphorylation complexes
To determine whether the morphological and molecular modifications observed upon xenobiotic treatment were also accompanied by metabolic alterations in developing neurons, an analysis of mitochondrial oxidative phosphorylation (OXPHOS) complexes was performed. OXPHOS represents the primary energy source necessary for cellular functions, and mitochondrial activity plays a key role in neuronal development, axiogenesis, and synaptogenesis (59).
Day-40 brain organoids were lysed, and the expression of electron transport chain (ETC) complexes was assessed by Western blot analysis (Figure 5A). Results indicated that exposure to BPS and/or PFOS modulated the expression of several OXPHOS components, although with distinct compound- and complex-specific profiles (Figure 5G). Specifically, PFOS exposure reduced Complex I (NADH dehydrogenase ubiquinone 1 beta subcomplex subunit 8 – NDUFB8, Figure 5B) and Complex IV (cytochrome c oxidase subunit 2 – COX II, Figure 5E) protein levels, while increasing Complex III (ubiquinol-cytochrome c reductase core protein 2 - UQCRC2, Figure 5D) and Complex V (ATP synthase lipid-binding protein - ATP5A, Figure 5F) expression. In contrast, BPS treatment led to a reduction in Complex III compared to control (Figure 5D). The expression of Complex II (succinate dehydrogenase complex II, subunit B - SDHB) was not significantly regulated by treatments (Figure 5C).
Chronic exposure to BPS, but not PFOS, disrupts genomic and non-genomic estrogen signaling pathways
To explore potential mechanisms underlying the morphological and metabolic alterations previously observed, we investigated estrogen signaling pathways, because a growing body of evidence suggests that EDCs including bisphenols and perfluoroalkyl substances, may exert their effects by interfering with estrogen-mediated signal transduction. Both genomic and non-genomic estrogen signaling routes have been implicated in neurodevelopment and may be considered susceptible targets for xenoestrogenic compounds. The genomic pathway involves classical nuclear estrogen receptors (ERα and ERβ), which, upon ligand binding, act as transcription factors to regulate gene expression programs essential for neurodevelopment. In contrast, the non-genomic pathway is initiated by membrane-bound receptors, notably G protein-coupled estrogen receptor 1 (GPER), and leads to rapid intracellular responses through activation of signaling cascades such as ERK1/2 and Akt–mTOR.
Results showed that only BPS treatment interfered with the genomic signaling pathway, resulting in a significant decrease in ERβ expression (Figures 6A, B). Conversely, analysis of the non-genomic pathway revealed a downregulation of GPER expression following exposure to BPS and the BPS+PFOS combination (Figures 6C, D). As the key downstream effectors of this pathway ERK1/2 and Akt–mTOR signaling cascades, rely on phosphorylation for activation, we analyzed their phosphorylation levels. Results evidenced that pERK1/2 remained unaffected by any treatment, while a reduction in pAkt levels was observed after exposure to BPS and BPS+PFOS. In contrast, mTOR phosphorylation showed no significant change (Figures 6C, D).
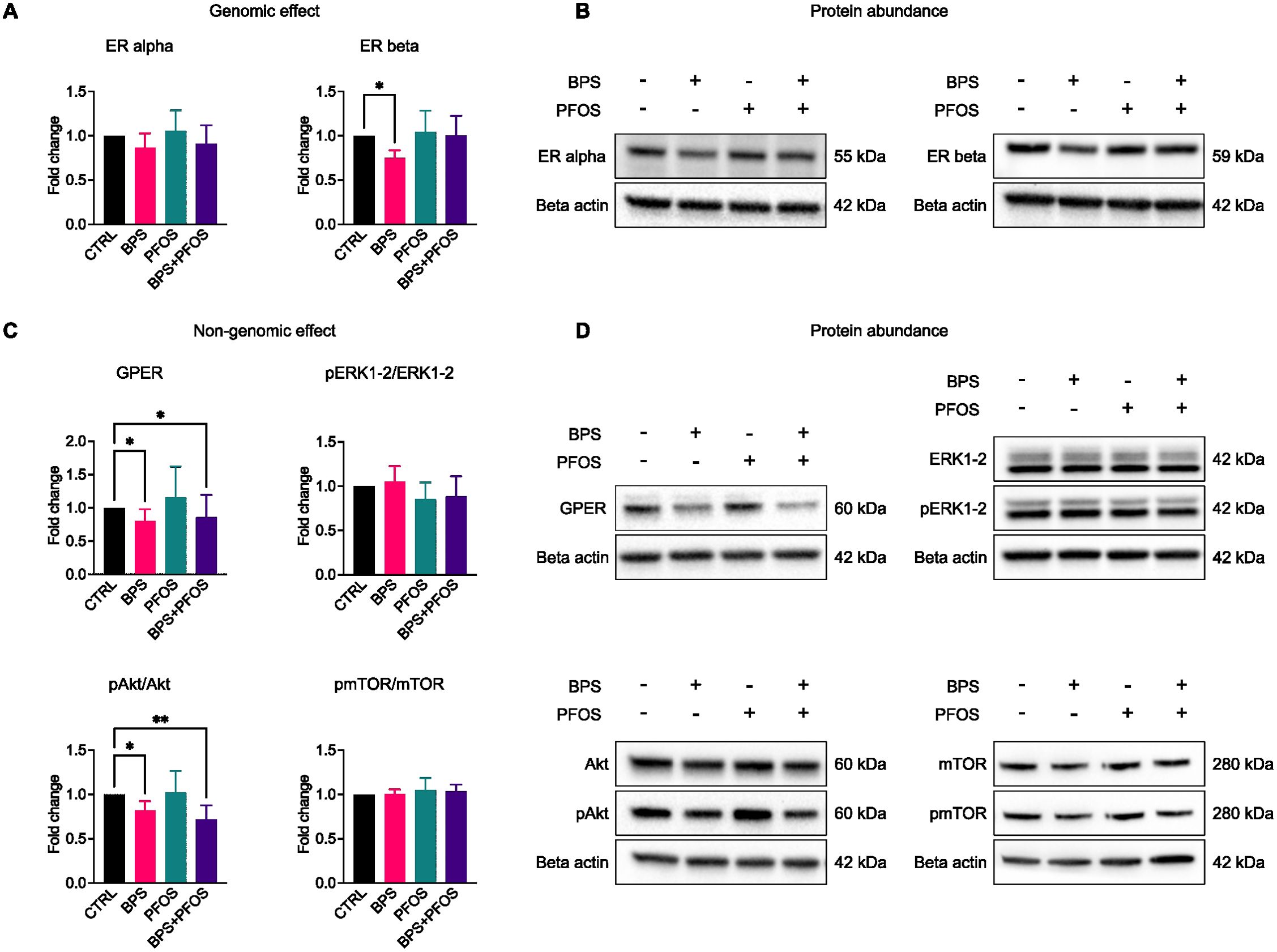
Figure 6. Effects of BPS and/or PFOS on genomic and non-genomic cascades. Western blot images and band quantification of ER alpha, ER beta (A, B), GPER, ERK1-2, Akt, and mTOR (C, D). Western blot results are based on n = 8 independent (4 for each cell line) organoid batches (biological replicates), each derived from independent differentiation runs, and for each batch, n = 3 organoids were pooled. Results were obtained from 8 independent experiments (4 for each cell line), 3 organoids were pooled for each condition. * p<.05, ** p<.01.
Discussion
Concerns are mounting about the immediate and long-term consequences of human exposure to EDCs such as BPS and PFOS. These compounds interfere with hormonal signaling and may compromise developmental processes and long-term health. Pregnancy is a particularly vulnerable period, as both chemicals cross the placental barrier and reach fetal tissues, potentially causing lasting alterations.
Several prospective and epidemiological studies have linked prenatal EDC exposure to disruptions in neurodevelopmental trajectories, with potential consequences for cognition, emotional regulation, and social behavior (60, 61). Evidence from animal models supports these associations (62), though interspecies differences in neuroendocrine development limit their predictive value (29). By evaluating BPS and PFOS both individually and in combination at concentrations within the range detected in pregnant women’s serum (63, 64), this study leverages a human brain organoid model to replicate realistic exposure scenarios. This approach not only differentiates compound-specific from interaction-driven effects but also provides a biologically relevant framework to elucidate whether chronic co-exposure can intensify neurodevelopmental risk.
This study investigated the impact of chronic low-dose exposure to BPS and PFOS on selected aspects of early human brain development, using a cerebral organoid model. By examining structural, cellular, and molecular parameters, we identified several exposure-related changes suggestive of altered neurodevelopmental trajectories.
Our data show that both BPS and PFOS affect brain organoid growth during maturation, without altering overall morphology but causing a slight reduction in size, particularly in BPS-treated samples. This reduction appears to reflect decreased proliferative activity within the VLZ, the neuroepithelial region containing precursors for cortical excitatory neurons. Organoids exposed to BPS and/or PFOS exhibited lower expression of the proliferative marker Ki67, suggesting disruption of the neurogenic timeline. Importantly, TUNEL assay did not reveal any increase in apoptosis across experimental conditions, indicating that the reduction in Ki67 expression is not attributable to cytotoxicity but rather to altered proliferation. These findings confirm and expand our previous data (5–7) highlighting that even low-dose exposure to bisphenols or perfluoroalkyls can alter the proliferative rate and biological characteristics of stem cells present in both the developing fetus and fetal annexes. Since these progenitors must tightly regulate the balance between self-renewal and differentiation, early interference may induce long-lasting alterations in brain development.
Starting from the VLZ, proliferative neural stem cells migrate outward to generate the multilayered CP, composed of deep- and superficial-layer neurons (65). In BPS-exposed organoids, we observed a reduction in TBR1 expression, a transcription factor critical for early-born neuron identity and cortical layer organization (66). Proper regulation of TBR1 is essential for cortical development, and its downregulation suggests altered CP maturation. Whether due to fewer neurons populating the cortical plate or to lower TBR1 expression per cell, both interpretations converge on a biologically relevant impairment in CP formation. In parallel, we found reduced expression and thickness of SYP-positive areas across all treated samples, indicating impaired synaptogenesis. Since SYP marks synaptic density (67), its reduction reinforces the idea that BPS and PFOS interfere with neuronal connectivity. This observation was further supported by the decreased expression of PSD95, a canonical postsynaptic marker, whose distribution pattern and intensity closely mirrored that of Synaptophysin. Together, these two markers point to a converging effect on both presynaptic and postsynaptic compartments, suggesting a delay or impairment in the formation of functional synapses. These findings align with prior studies showing that BPA, structurally similar to BPS, disrupts synaptogenesis and induces behavioral alterations in animal models (68). Despite BPS being marketed as a safer alternative to BPA, it is often detected at higher concentrations in both maternal and fetal samples (69, 70) raising concerns about its safety profile. Recent work in rodents further supports our findings, demonstrating that BPS impairs hippocampal neurogenesis and newborn neuron maturation (71).
The effects of BPS and PFOS on cortical plate development extend beyond proliferation and early neurogenesis, involving alterations in neuronal subtype specification. SATB2 and CTIP2 are transcription factors essential for the development of intracortical and subcortical projection neurons, respectively (72). While Western blot analysis did not reveal changes in their expression levels, morphological analysis showed a disrupted laminar organization in treated organoids. In controls, SATB2 and CTIP2 displayed distinct layer-specific localization, with SATB2 labeling upper CP layers and CTIP2 marking deeper strata. In contrast, EDC-treated samples exhibited a more diffuse distribution, indicating impaired neuron positioning and specification.
To further explore transcription factor activity, we employed high-content imaging and broad-field acquisitions, which enabled quantitative evaluation of nuclear versus cytoplasmic signal across thousands of cells. Although this method does not capture fine subnuclear details, it provides an effective readout of transcription factor localization dynamics and supports robust population-level comparisons. PFOS, alone or in combination with BPS, increased nuclear translocation of CTIP2, suggesting enhanced subcortical neuron fate at the expense of callosal projections. Conversely, BPS exposure was associated with reduced nuclear SATB2 levels, hinting at compromised development of commissural neurons. These shifts in cortical identity may contribute to aberrant brain connectivity, a hallmark of several neurodevelopmental disorders (73).
The ChP, responsible for producing cerebrospinal fluid (CSF), plays a crucial role in maintaining brain homeostasis during development (74). The main protein synthesized and secreted by the ChP is transthyretin (TTR), which is essential for the transport of thyroid hormones from the blood into the CSF (75). Reduced TTR expression and the morphological alterations of the TTR+ regions, as observed in PFOS-exposed organoids, may contribute to impaired thyroid hormone delivery to the developing CNS. Since thyroid hormones are essential for neural progenitor proliferation, cortical layering, and synapse formation, this decrease in TTR may help explain the concurrent deficits observed in neurogenesis, cortical plate maturation, and synaptic development within our model. Disruption of ChP development may thus have widespread consequences, including impaired CSF production, altered barrier integrity, and disrupted signaling between the brain and peripheral organs. These findings are consistent with prior evidence showing that EDCs can affect the blood–brain and blood–CSF barriers and impair CSF production, potentially contributing to neurodevelopmental disorders (76). Given the essential role of thyroid hormones in normal brain development, even subtle alterations in TTR expression may influence thyroid hormone availability to the CNS during fetal growth, with long-term consequences. While this association is biologically plausible, further studies are needed to determine whether and how such changes functionally impact thyroid hormone–dependent neurodevelopmental processes.
Glutamate is the main excitatory neurotransmitter in the central nervous system, and glutamatergic signaling is fundamental for synaptic plasticity, learning, and memory (77, 78). In our model, organoids exposed to BPS showed increased expression of NMDAR2B and VGluT1, suggesting an association with enhanced glutamatergic marker expression during early differentiation. While this may suggest an accelerated excitatory differentiation program, whether these molecular alterations translate into functional changes in glutamatergic connectivity remains to be investigated. Our findings align with previous studies reporting that EDCs, such as BPA, a structural analog of BPS, can enhance glutamatergic activity and disrupt synaptic dynamics in vivo (79). Likewise, BPA exposure was associated with increased VGluT1 expression in rodent models, possibly contributing to aberrant synaptic function (80). These results support the idea that BPS, though introduced as a safer alternative to BPA, may exert comparable effects on excitatory neuronal pathways. Interestingly, while BPS alone increased expression of glutamatergic markers, co-exposure with PFOS attenuated this upregulation. Although the mechanistic basis of this modulation remains to be clarified, it highlights the importance of considering combined chemical effects in neurodevelopmental risk assessment. Indeed, mixtures of EDCs may lead to additive, synergistic, or antagonistic outcomes (5–7, 24, 81), complicating the prediction of their actual toxicological impact in real-life scenarios.
Mitochondria are central regulators of energy metabolism in the developing brain, supporting neurogenesis, synaptic formation, and plasticity. Alterations in mitochondrial protein expression have been implicated in various neurodevelopmental and neurodegenerative conditions. In our study, BPS and PFOS exposure resulted in distinct alterations in the expression of proteins from the mitochondrial electron transport chain (ETC), suggesting that these compounds may modulate mitochondrial regulatory pathways during neurodevelopment. Specifically, PFOS-treated organoids showed downregulation of Complex I and Complex IV. While we did not assess functional respiration, this expression pattern is consistent with previous studies reporting PFOS-related effects on mitochondrial biology and redox homeostasis (82, 83). Concurrently, PFOS induced an upregulation of Complex III and Complex V components, which could reflect a compensatory shift in mitochondrial protein regulation. BPS-treated organoids exhibited a different profile, with decreased expression of Complex III subunits. Similar findings have been described in studies showing BPS-associated alterations in mitochondrial protein expression and oxidative stress marker (84). Notably, in the combined BPS+PFOS condition, the ETC expression pattern more closely resembled that of BPS alone, suggesting potential dominance of BPS-mediated effects under co-exposure conditions. Overall, these findings highlight that BPS and PFOS can differentially influence the expression of mitochondrial ETC proteins. While these observations point to a potential role for mitochondrial signaling in EDC-induced neurodevelopmental effects, further functional studies are required to determine whether these molecular changes impact energy metabolism or oxidative balance in developing neurons.
Estrogens play a crucial role in brain development, regulating key processes such as neuronal proliferation, migration, and synapse formation. Given the hormone-sensitive nature of the developing brain, prenatal exposure to EDCs may interfere with critical neurodevelopmental pathways. Based on previous evidence indicating that bisphenols and perfluoroalkyls can act as xenoestrogens (85, 86), we investigated whether the morphological and metabolic alterations observed in our model might be associated with disruptions in estrogen signaling. In our study, BPS exposure was associated with decreased expression of ERβ, a nuclear receptor involved in neurodevelopmental gene regulation, and with altered levels of GPER, the membrane-bound estrogen receptor known to mediate non-genomic signaling. In parallel, a reduction in Akt phosphorylation was observed in BPS-treated organoids, in the absence of significant changes in mTOR or ERK1/2 phosphorylation. These observations are consistent with a possible modulation of the GPER–PI3K/Akt axis. Notably, this pattern aligns with our recent findings in a different cellular model, where BPS and PFOS were shown to alter both genomic and non-genomic estrogen signaling in stem cells (12).
While these data suggest a potential involvement of estrogen receptors in mediating the effects of BPS, it is important to note that the PI3K/Akt and ERK1/2 pathways can also be regulated by ER-independent mechanisms, which may themselves be modulated by xenobiotics such as BPS or PFOS. Furthermore, the tissue-specific nature of GPER downstream signaling must be considered: in developing neural contexts, GPER may preferentially engage the Akt cascade while bypassing other canonical targets such as ERK1/2 or mTOR (87). These findings align with previous reports of EDC-mediated impairment of estrogen receptor activity in neuronal models (88), and reinforce the view of BPS as xenoestrogen with specific effects on non-genomic pathways, which might be particularly relevant in the context of early neurodevelopment, where even subtle hormonal imbalances may result in long-term structural and functional deficits (89). It is worth noting that activation of estrogen receptors can enhance mitochondrial biogenesis and regulate expression of oxidative phosphorylation complexes in neuronal and non-neuronal tissues (90, 91).
In our model, the observed reduction of OXPHOS protein expression after BPS exposure may therefore reflect, at least in part, disruption of estrogen receptor–mediated mitochondrial regulation. Overall, our results support a hypothesis-generating association between BPS exposure and altered estrogen receptor–linked signaling in human brain organoids. However, the lack of direct functional manipulation (e.g., use of receptor agonists/antagonists) precludes definitive mechanistic conclusions. In addition, an involvement of the Akt signaling axis independently of estrogen receptor activation cannot be excluded. Several environmental stimuli, including xenobiotics, are known to modulate Akt and ERK1/2 through alternative, estrogen-independent mechanisms, which may contribute to the observed changes in our model.
Although PFOS did not alter classical estrogen signaling markers in our model, its inclusion in the estrogen pathway assessment was necessary to verify potential crosstalk with estrogenic mechanisms, given previous reports of receptor-mediated activities for some PFAS. In our study, PFOS exposure did not produce direct changes in ER-related markers, suggesting a limited contribution of classical estrogen signaling to PFOS neurotoxicity in this system. Nonetheless, PFOS exposure profoundly affected brain organoid development, disrupting choroid plexus (ChP) formation and reducing transthyretin (TTR1) expression, alterations that may indirectly impact thyroid hormone availability during critical stages of neurodevelopment. This hypothesis aligns with the literature evidence indicating that PFOS can interfere with the thyroid hormone axis at multiple levels (92–94), potentially contributing to deficits in neurogenesis, synaptogenesis, and cognitive functions. The concordance between our findings and existing reports reinforces the plausibility of thyroid-related mechanisms in mediating PFOS effects. These observations also highlight that simultaneous exposure to multiple EDCs, each potentially targeting distinct hormonal axes such as estrogens and thyroid hormones, may result in complex neurodevelopmental disturbances. Such effects cannot be easily extrapolated from single-compound studies, reinforcing the relevance of mixture-based toxicological approaches.
Study limitations
This study presents some limitations that should be acknowledged:
- the experiments were conducted exclusively using hiPSCs derived from male donors, which may limit the generalizability of our findings. Given the well-established influence of sex hormones on brain development, future studies should include female-derived hiPSCs to evaluate potential sex-specific responses to BPS and PFOS exposure.
- although the concentrations of BPS and PFOS used in our model are in the range of those detected in human biological fluids, the absence of a dose–response assessment limits our ability to define threshold levels for toxicity and to explore potential non-monotonic effects, which are common among EDCs.
- our analysis focused primarily on morphological and molecular endpoints, with particular attention to glutamatergic differentiation due to its known sensitivity to environmental toxicants and relevance in excitatory–inhibitory balance. However, functional consequences—such as altered electrophysiological activity, synaptic integration, or long-term neuronal network maturation—were not addressed in this study and require dedicated follow-up investigations.
- While our data support the involvement of estrogen signaling—based on the observed changes in ERβ and GPER expression and in Akt phosphorylation—we did not assess downstream transcriptional activity or specific gene targets, which would be necessary to fully characterize the genomic consequences of this pathway. In addition, the observed modulation of Akt and ERK signaling may also reflect activation of estrogen-independent mechanisms, which were not explored in the present study and should be considered in future investigations. Finally, the potential contribution of other endocrine axes, such as thyroid hormone signaling in PFOS-exposed samples, remains hypothetical and warrants direct functional validation to confirm its role in the observed neurodevelopmental phenotypes.
- Another limitation of our study is the absence of glial cell characterization. Although the genesis of glial cells in brain organoids typically emerges at later stages (>80–100 days) (95), we did not assess the presence or maturation of glial populations in our 40-day model. As a result, our findings are limited to neuronal development and do not capture potential EDC-related effects on astrocytes or other glial lineages.
Conclusion
Our findings demonstrate that chronic exposure to environmental doses of BPS and PFOS interferes with key stages of human brain development in cerebral organoids, including progenitor proliferation, neuronal identity specification, synaptogenesis, and mitochondrial protein expression. BPS acted as a xenoestrogen, disrupting both genomic (ERβ) and non-genomic (GPER-Akt) estrogen signaling. PFOS, although not altering the estrogenic pathway, induced changes consistent with reduced availability of thyroid hormones to the developing CNS, as suggested by decreased TTR1 expression.
These data highlight that distinct EDCs can target different hormonal systems, with potential for converging or synergistic effects on neurodevelopment in the real-life mixed exposure. Low-dose exposure to EDCs can therefore produce biologically significant outcomes with possible long-term consequences, underscoring the need for precautionary approaches in their risk assessment. Our data provide mechanistic and dose-relevant evidence to inform regulatory strategies aimed at reducing early-life exposure to persistent and bio-accumulative pollutants.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
Ethical approval was not required for the studies on humans in accordance with the local legislation and institutional requirements because only commercially available established cell lines were used.
Author contributions
AC: Writing – original draft, Methodology, Investigation, Formal Analysis, Conceptualization. GG: Formal Analysis, Writing – review & editing, Investigation, Validation, Conceptualization, Methodology. SB: Validation, Formal Analysis, Writing – review & editing. TB: Validation, Writing – review & editing. AB: Funding acquisition, Writing – review & editing, Project administration, Methodology, Conceptualization, Supervision. BG: Project administration, Methodology, Writing – review & editing, Supervision, Funding acquisition.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the European Union - Next Generation EU, under the National Recovery and Resilience Plan (ECS00000041) VITALITY.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2025.1692333/full#supplementary-material
References
1. Stiles J and Jernigan TL. The basics of brain development. Neuropsychol Rev. (2010) 20:327–48. doi: 10.1007/s11065-010-9148-4
2. Gaggi G, Di Credico A, Izzicupo P, Iannetti G, Di Baldassarre A, and Ghinassi B. Chemical and biological molecules involved in differentiation, maturation, and survival of dopaminergic neurons in health and parkinson’s disease: physiological aspects and clinical implications. Biomedicines. (2021) 9:754. doi: 10.3390/biomedicines9070754
3. Lenroot RK and Giedd JN. The changing impact of genes and environment on brain development during childhood and adolescence: Initial findings from a neuroimaging study of pediatric twins. Dev Psychopathol. (2008) 20:1161–75. doi: 10.1017/S0954579408000552
4. Short AK and Baram TZ. Early-life adversity and neurological disease: age-old questions and novel answers. Nat Rev Neurol. (2019) 15:657–69. doi: 10.1038/s41582-019-0246-5
5. Di Credico A, Gaggi G, Bucci I, Ghinassi B, and Di Baldassarre A. The effects of combined exposure to bisphenols and perfluoroalkyls on human perinatal stem cells and the potential implications for health outcomes. IJMS. (2023) 24:15018. doi: 10.3390/ijms241915018
6. Gaggi G, Di Credico A, Barbagallo F, Ballerini P, Ghinassi B, and Di Baldassarre A. Antenatal exposure to plastic pollutants: study of the bisphenols and perfluoroalkyls effects on human stem cell models. Expo Health. (2024) 16:693–714. doi: 10.1007/s12403-023-00586-5
7. Gaggi G, Di Credico A, Barbagallo F, Ghinassi B, and Di Baldassarre A. Bisphenols and perfluoroalkyls alter human stem cells integrity: A possible link with infertility. Environ Res. (2023) 235:116487. doi: 10.1016/j.envres.2023.116487
8. Ramadan M, Cooper B, and Posnack NG. Bisphenols and phthalates: Plastic chemical exposures can contribute to adverse cardiovascular health outcomes. Birth Defects Res. (2020) 112:1362–85. doi: 10.1002/bdr2.1752
9. De Felip E, Abballe A, Albano FL, Battista T, Carraro V, Conversano M, et al. Current exposure of Italian women of reproductive age to PFOS and PFOA: A human biomonitoring study. Chemosphere. (2015) 137:1–8. doi: 10.1016/j.chemosphere.2015.03.046
10. Ingelido AM, Abballe A, Dellatte E, Ferri F, Iacovella N, Marra V, et al. Ten years of PFOS and PFOA human biomonitoring in Italy: Exposure levels and determinants of exposure. Chemosphere. (2025) 376:144297. doi: 10.1016/j.chemosphere.2025.144297
11. Husøy T, Andreassen M, Hjertholm H, Carlsen MH, Norberg N, Sprong C, et al. The Norwegian biomonitoring study from the EU project EuroMix: Levels of phenols and phthalates in 24-hour urine samples and exposure sources from food and personal care products. Environ Int. (2019) 132:105103. doi: 10.1016/j.envint.2019.105103
12. Gaggi G, Di Credico A, Bibbò S, Corneo B, Ferlin A, Di Baldassarre A, et al. Interference of estrogen signaling by endocrine disruptors in male and female cells: Potential implications of BPS and PFOS in human development. Eur J Cell Biol. (2025) 104:151509. doi: 10.1016/j.ejcb.2025.151509
13. Rosenfeld CS and Cooke PS. Endocrine disruption through membrane estrogen receptors and novel pathways leading to rapid toxicological and epigenetic effects. J Steroid Biochem Mol Biol. (2019) 187:106–17. doi: 10.1016/j.jsbmb.2018.11.007
14. Viñas R and Watson CS. Bisphenol S disrupts estradiol-induced nongenomic signaling in a rat pituitary cell line: effects on cell functions. Environ Health Perspect. (2013) 121:352–8. doi: 10.1289/ehp.1205826
15. Coperchini F, Croce L, Ricci G, Magri F, Rotondi M, Imbriani M, et al. Thyroid disrupting effects of old and new generation PFAS. Front Endocrinol. (2021) 11:612320. doi: 10.3389/fendo.2020.612320
16. European Chemical Agency (ECHA) and European Food Safety Authority (EFSA) with the technical support of the Joint Research Centre (JRC), Andersson N, Arena M, Auteri D, Barmaz S, Grignard E, et al. Guidance for the identification of endocrine disruptors in the context of Regulations (EU) No 528/2012 and (EC) No 1107/2009. EFS2. (2018) 16. doi: 10.2903/j.efsa.2018.5311
17. Crofton KM, Mundy WR, and Shafer TJ. Developmental neurotoxicity testing: A path forward. Congenital Anomalies. (2012) 52:140–6. doi: 10.1111/j.1741-4520.2012.00377.x
18. Patisaul HB. REPRODUCTIVE TOXICOLOGY: Endocrine disruption and reproductive disorders: impacts on sexually dimorphic neuroendocrine pathways. Reproduction. (2021) 162:F111–30. doi: 10.1530/REP-20-0596
19. Arevalo M-A, Azcoitia I, and Garcia-Segura LM. The neuroprotective actions of oestradiol and oestrogen receptors. Nat Rev Neurosci. (2015) 16:17–29. doi: 10.1038/nrn3856
20. Bernal J. “Thyroid hormones in brain development and function.,”. In: Feingold KR, Ahmed SF, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Dungan K, et al, editors. Endotext. MDText.com, Inc, South Dartmouth (MA (2000). Available online at: http://www.ncbi.nlm.nih.gov/books/NBK285549/.
21. Vujovic F and Farahani RM. Thyroid hormones and brain development: A focus on the role of mitochondria as regulators of developmental time. Cells. (2025) 14:150. doi: 10.3390/cells14030150
22. Kajta M and Wójtowicz AK. Impact of endocrine-disrupting chemicals on neural development and the onset of neurological disorders. Pharmacol Rep. (2013) 65:1632–9. doi: 10.1016/S1734-1140(13)71524-X
23. Mallozzi M, Bordi G, Garo C, and Caserta D. The effect of maternal exposure to endocrine disrupting chemicals on fetal and neonatal development: A review on the major concerns. Birth Defects Res Pt C. (2016) 108:224–42. doi: 10.1002/bdrc.21137
24. Di Credico A, Weiss A, Corsini M, Gaggi G, Ghinassi B, Wilbertz JH, et al. Machine learning identifies phenotypic profile alterations of human dopaminergic neurons exposed to bisphenols and perfluoroalkyls. Sci Rep. (2023) 13:21907. doi: 10.1038/s41598-023-49364-y
25. Naderi M and Kwong RWM. A comprehensive review of the neurobehavioral effects of bisphenol S and the mechanisms of action: New insights from in vitro and in vivo models. Environ Int. (2020) 145:106078. doi: 10.1016/j.envint.2020.106078
26. Xu Y, Nie J, Lu C, Hu C, Chen Y, Ma Y, et al. Effects and mechanisms of bisphenols exposure on neurodegenerative diseases risk: A systemic review. Sci Total Environ. (2024) 919:170670. doi: 10.1016/j.scitotenv.2024.170670
27. Nakagawa Y and Tayama S. Metabolism and cytotoxicity of bisphenol A and other bisphenols in isolated rat hepatocytes. Arch Toxicol. (2000) 74:99–105. doi: 10.1007/s002040050659
28. Qian Y, Ducatman A, Ward R, Leonard S, Bukowski V, Lan Guo N, et al. Perfluorooctane sulfonate (PFOS) induces reactive oxygen species (ROS) production in human microvascular endothelial cells: role in endothelial permeability. J Toxicol Environ Health Part A. (2010) 73:819–36. doi: 10.1080/15287391003689317
29. Watson RE, DeSesso JM, Hurtt ME, and Cappon GD. Postnatal growth and morphological development of the brain: a species comparison. Birth Defect Res B. (2006) 77:471–84. doi: 10.1002/bdrb.20090
30. Di Baldassarre A, Cimetta E, Bollini S, Gaggi G, and Ghinassi B. Human-induced pluripotent stem cell technology and cardiomyocyte generation: progress and clinical applications. Cells. (2018) 7:48. doi: 10.3390/cells7060048
31. Di Credico A, Gaggi G, Izzicupo P, Ferri L, Bonanni L, Iannetti G, et al. Real-time monitoring of levetiracetam effect on the electrophysiology of an heterogenous human iPSC-derived neuronal cell culture using microelectrode array technology. Biosensors. (2021) 11:450. doi: 10.3390/bios11110450
32. Gaggi G, Di Credico A, Guarnieri S, Mariggiò MA, Ballerini P, Di Baldassarre A, et al. Human fetal membrane-mesenchymal stromal cells generate functional spinal motor neurons in vitro. iScience. (2022) 25:105197. doi: 10.1016/j.isci.2022.105197
33. Gaggi G, Di Credico A, Guarnieri S, Mariggiò MA, Di Baldassarre A, and Ghinassi B. Human mesenchymal amniotic fluid stem cells reveal an unexpected neuronal potential differentiating into functional spinal motor neurons. Front Cell Dev Biol. (2022) 10:936990. doi: 10.3389/fcell.2022.936990
34. Gaggi G, Di Credico A, Izzicupo P, Alviano F, Di Mauro M, Di Baldassarre A, et al. Human mesenchymal stromal cells unveil an unexpected differentiation potential toward the dopaminergic neuronal lineage. IJMS. (2020) 21:6589. doi: 10.3390/ijms21186589
35. Gaggi G, Izzicupo P, Di Credico A, Sancilio S, Di Baldassarre A, and Ghinassi B. Spare parts from discarded materials: fetal annexes in regenerative medicine. IJMS. (2019) 20:1573. doi: 10.3390/ijms20071573
36. Lancaster MA and Knoblich JA. Generation of cerebral organoids from human pluripotent stem cells. Nat Protoc. (2014) 9:2329–40. doi: 10.1038/nprot.2014.158
37. Qian X, Song H, and Ming G. Brain organoids: advances, applications and challenges. Development. (2019) 146:dev166074. doi: 10.1242/dev.166074
38. Lee C-T, Bendriem RM, Wu WW, and Shen R-F. 3D brain Organoids derived from pluripotent stem cells: promising experimental models for brain development and neurodegenerative disorders. J BioMed Sci. (2017) 24:59. doi: 10.1186/s12929-017-0362-8
39. Guy B, Zhang JS, Duncan LH, and Johnston RJ. Human neural organoids: Models for developmental neurobiology and disease. Dev Biol. (2021) 478:102–21. doi: 10.1016/j.ydbio.2021.06.012
40. Kim J, Koo B-K, and Knoblich JA. Human organoids: model systems for human biology and medicine. Nat Rev Mol Cell Biol. (2020) 21:571–84. doi: 10.1038/s41580-020-0259-3
41. Amabile G, Welner RS, Nombela-Arrieta C, D’Alise AM, Di Ruscio A, Ebralidze AK, et al. In vivo generation of transplantable human hematopoietic cells from induced pluripotent stem cells. Blood. (2013) 121:1255–64. doi: 10.1182/blood-2012-06-434407
42. Nguyen ST, Nguyen HT-L, and Truong KD. Comparative cytotoxic effects of methanol, ethanol and DMSO on human cancer cell lines. BioMed Res Ther. (2020) 7:3855–9. doi: 10.15419/bmrat.v7i7.614
43. Jin H, Xie J, Mao L, Zhao M, Bai X, Wen J, et al. Bisphenol analogue concentrations in human breast milk and their associations with postnatal infant growth. Environ pollut. (2020) 259:113779. doi: 10.1016/j.envpol.2019.113779
44. Lorigo M and Cairrao E. Fetoplacental vasculature as a model to study human cardiovascular endocrine disruption. Mol Aspects Med. (2022) 87:101054. doi: 10.1016/j.mam.2021.101054
45. Rivera-Núñez Z, Kinkade CW, Khoury L, Brunner J, Murphy H, Wang C, et al. Prenatal perfluoroalkyl substances exposure and maternal sex steroid hormones across pregnancy. Environ Res. (2023) 220:115233. doi: 10.1016/j.envres.2023.115233
46. Luo C, Lancaster MA, Castanon R, Nery JR, Knoblich JA, and Ecker JR. Cerebral organoids recapitulate epigenomic signatures of the human fetal brain. Cell Rep. (2016) 17:3369–84. doi: 10.1016/j.celrep.2016.12.001
47. Trevino AE, Sinnott-Armstrong N, Andersen J, Yoon S-J, Huber N, Pritchard JK, et al. Chromatin accessibility dynamics in a model of human forebrain development. Science. (2020) 367:eaay1645. doi: 10.1126/science.aay1645
48. Pagliaro A, Finger R, Zoutendijk I, Bunschuh S, Clevers H, Hendriks D, et al. Temporal morphogen gradient-driven neural induction shapes single expanded neuroepithelium brain organoids with enhanced cortical identity. Nat Commun. (2023) 14:7361. doi: 10.1038/s41467-023-43141-1
49. Hevner RF, Shi L, Justice N, Hsueh Y-P, Sheng M, Smiga S, et al. Tbr1 regulates differentiation of the preplate and layer 6. Neuron. (2001) 29:353–66. doi: 10.1016/S0896-6273(01)00211-2
50. Wiedenmann B and Franke WW. Identification and localization of synaptophysin, an integral membrane glycoprotein of Mr 38,000 characteristic of presynaptic vesicles. Cell. (1985) 41:1017–28. doi: 10.1016/S0092-8674(85)80082-9
51. Li L, Tasic B, Micheva KD, Ivanov VM, Spletter ML, Smith SJ, et al. Visualizing the distribution of synapses from individual neurons in the mouse brain. PloS One. (2010) 5:e11503. doi: 10.1371/journal.pone.0011503
52. Sarnat HB, Flores-Sarnat L, and Trevenen CL. Synaptophysin immunoreactivity in the human hippocampus and neocortex from 6 to 41 weeks of gestation. J Neuropathol Exp Neurol. (2010) 69:234–45. doi: 10.1097/NEN.0b013e3181d0151f
53. Lodato S and Arlotta P. Generating neuronal diversity in the mammalian cerebral cortex. Annu Rev Cell Dev Biol. (2015) 31:699–720. doi: 10.1146/annurev-cellbio-100814-125353
54. Pellegrini L, Bonfio C, Chadwick J, Begum F, Skehel M, and Lancaster MA. Human CNS barrier-forming organoids with cerebrospinal fluid production. Science. (2020) 369:eaaz5626. doi: 10.1126/science.aaz5626
55. Newpher TM and Ehlers MD. Glutamate receptor dynamics in dendritic microdomains. Neuron. (2008) 58:472–97. doi: 10.1016/j.neuron.2008.04.030
56. Xu X, Li T, Luo Q, Hong X, Xie L, and Tian D. Bisphenol-A rapidly enhanced passive avoidance memory and phosphorylation of NMDA receptor subunits in hippocampus of young rats. Toxicol Appl Pharmacol. (2011) 255:221–8. doi: 10.1016/j.taap.2011.06.022
57. Gambrill AC and Barria A. NMDA receptor subunit composition controls synaptogenesis and synapse stabilization. Proc Natl Acad Sci USA. (2011) 108:5855–60. doi: 10.1073/pnas.1012676108
58. Woelfle S and Boeckers TM. Layer-specific vesicular glutamate transporter 1 immunofluorescence levels delineate all layers of the human hippocampus including the stratum lucidum. Front Cell Neurosci. (2021) 15:789903. doi: 10.3389/fncel.2021.789903
59. Hroudová J and Fišar Z. Control mechanisms in mitochondrial oxidative phosphorylation. Neural Regener Res. (2013) 8:363–75. doi: 10.3969/j.issn.1673-5374.2013.04.009
60. Braun JM, Yolton K, Stacy SL, Erar B, Papandonatos GD, Bellinger DC, et al. Prenatal environmental chemical exposures and longitudinal patterns of child neurobehavior. NeuroToxicology. (2017) 62:192–9. doi: 10.1016/j.neuro.2017.07.027
61. Grandjean P and Landrigan PJ. Neurobehavioural effects of developmental toxicity. Lancet Neurol. (2014) 13:330–8. doi: 10.1016/S1474-4422(13)70278-3
62. Masuo Y and Ishido M. Neurotoxicity of endocrine disruptors: possible involvement in brain development and neurodegeneration. J Toxicol Environ Health Part B. (2011) 14:346–69. doi: 10.1080/10937404.2011.578557
63. Høyer BB, Ramlau-Hansen CH, Obel C, Pedersen HS, Hernik A, Ogniev V, et al. Pregnancy serum concentrations of perfluorinated alkyl substances and offspring behaviour and motor development at age 5–9 years – a prospective study. Environ Health. (2015) 14. doi: 10.1186/1476-069x-14-2
64. Li A, Zhuang T, Shi W, Liang Y, Liao C, Song M, et al. Serum concentration of bisphenol analogues in pregnant women in China. Sci Total Environ. (2020) 707:136100. doi: 10.1016/j.scitotenv.2019.136100
65. Di Lullo E and Kriegstein AR. The use of brain organoids to investigate neural development and disease. Nat Rev Neurosci. (2017) 18:573–84. doi: 10.1038/nrn.2017.107
66. Crespo I, Pignatelli J, Kinare V, Méndez-Gómez HR, Esgleas M, Román MJ, et al. Tbr1 misexpression alters neuronal development in the cerebral cortex. Mol Neurobiol. (2022) 59:5750–65. doi: 10.1007/s12035-022-02936-x
67. Tarsa L and Goda Y. Synaptophysin regulates activity-dependent synapse formation in cultured hippocampal neurons. Proc Natl Acad Sci USA. (2002) 99:1012–6. doi: 10.1073/pnas.022575999
68. Welch C and Mulligan K. Does bisphenol A confer risk of neurodevelopmental disorders? What we have learned from developmental neurotoxicity studies in animal models. IJMS. (2022) 23:2894. doi: 10.3390/ijms23052894
69. Pan Y, Deng M, Li J, Du B, Lan S, Liang X, et al. Occurrence and maternal transfer of multiple bisphenols, including an emerging derivative with unexpectedly high concentrations, in the human maternal–fetal–placental unit. Environ Sci Technol. (2020) 54:3476–86. doi: 10.1021/acs.est.0c00206
70. Zhao H, Li J, Ma X, Huo W, Xu S, and Cai Z. Simultaneous determination of bisphenols, benzophenones and parabens in human urine by using UHPLC-TQMS. Chin Chem Lett. (2018) 29:102–6. doi: 10.1016/j.cclet.2017.06.013
71. Tiwari S, Phoolmala, Goyal S, Yadav RK, and Chaturvedi RK. Bisphenol-F and bisphenol-S (BPF and BPS) impair the stemness of neural stem cells and neuronal fate decision in the hippocampus leading to cognitive dysfunctions. Mol Neurobiol. (2024) 61:9347–68. doi: 10.1007/s12035-024-04160-1
72. Leone DP, Heavner WE, Ferenczi EA, Dobreva G, Huguenard JR, Grosschedl R, et al. Satb2 regulates the differentiation of both callosal and subcerebral projection neurons in the developing cerebral cortex. Cereb Cortex. (2015) 25:3406–19. doi: 10.1093/cercor/bhu156
73. Markicevic M, Fulcher BD, Lewis C, Helmchen F, Rudin M, Zerbi V, et al. Cortical excitation: inhibition imbalance causes abnormal brain network dynamics as observed in neurodevelopmental disorders. Cereb Cortex. (2020) 30:4922–37. doi: 10.1093/cercor/bhaa084
74. Lun MP, Monuki ES, and Lehtinen MK. Development and functions of the choroid plexus–cerebrospinal fluid system. Nat Rev Neurosci. (2015) 16:445–57. doi: 10.1038/nrn3921
75. Richardson SJ, Wijayagunaratne RC, D’Souza DG, Darras VM, and Van Herck SLJ. Transport of thyroid hormones via the choroid plexus into the brain: the roles of transthyretin and thyroid hormone transmembrane transporters. Front Neurosci. (2015) 9:66. doi: 10.3389/fnins.2015.00066
76. Denuzière A and Ghersi-Egea J-F. Cerebral concentration and toxicity of endocrine disrupting chemicals: The implication of blood-brain interfaces. NeuroToxicology. (2022) 91:100–18. doi: 10.1016/j.neuro.2022.04.004
77. Hunt DL and Castillo PE. Synaptic plasticity of NMDA receptors: mechanisms and functional implications. Curr Opin Neurobiol. (2012) 22:496–508. doi: 10.1016/j.conb.2012.01.007
78. Reiner A and Levitz J. Glutamatergic signaling in the central nervous system: ionotropic and metabotropic receptors in concert. Neuron. (2018) 98:1080–98. doi: 10.1016/j.neuron.2018.05.018
79. Jardim NS, Sartori G, Sari MHM, Müller SG, and Nogueira CW. Bisphenol A impairs the memory function and glutamatergic homeostasis in a sex-dependent manner in mice: Beneficial effects of diphenyl diselenide. Toxicol Appl Pharmacol. (2017) 329:75–84. doi: 10.1016/j.taap.2017.05.035
80. Li D, Ai S, Huang C, Liu Z-H, and Wang H-L. Icariin rescues developmental BPA exposure induced spatial memory deficits in rats. Toxicol Appl Pharmacol. (2024) 482:116776. doi: 10.1016/j.taap.2023.116776
81. Hamid N, Junaid M, and Pei D-S. Combined toxicity of endocrine-disrupting chemicals: A review. Ecotoxicol Environ Saf. (2021) 215:112136. doi: 10.1016/j.ecoenv.2021.112136
82. Han R, Hu M, Zhong Q, Wan C, Liu L, Li F, et al. Perfluorooctane sulphonate induces oxidative hepatic damage via mitochondria-dependent and NF-κB/TNF-α-mediated pathway. Chemosphere. (2018) 191:1056–64. doi: 10.1016/j.chemosphere.2017.08.070
83. Hofmann A, Mishra JS, Yadav P, Dangudubiyyam SV, Blesson CS, and Kumar S. PFOS impairs mitochondrial biogenesis and dynamics and reduces oxygen consumption in human trophoblasts. J Environ Sci Public Health. (2023) 7:164–75. doi: 10.26502/jesph.96120197
84. Zhang X, Gong H, Zhao Y, Wu Y, Cheng J, Song Y, et al. Bisphenol S impairs mitochondrial function by targeting Myo19/oxidative phosphorylation pathway contributing to axonal and dendritic injury. Environ Int. (2024) 186:108643. doi: 10.1016/j.envint.2024.108643
85. Cimmino I, Fiory F, Perruolo G, Miele C, Beguinot F, Formisano P, et al. Potential mechanisms of bisphenol A (BPA) contributing to human disease. IJMS. (2020) 21:5761. doi: 10.3390/ijms21165761
86. Moon MK. Concern about the safety of bisphenol A substitutes. Diabetes Metab J. (2019) 43:46. doi: 10.4093/dmj.2019.0027
87. Upadhayay S, Gupta R, Singh S, Mundkar M, Singh G, and Kumar P. Involvement of the G-protein-coupled estrogen receptor-1 (GPER) signaling pathway in neurodegenerative disorders: A review. Cell Mol Neurobiol. (2023) 43:1833–47. doi: 10.1007/s10571-022-01301-9
88. Le HH and Belcher SM. Rapid signaling actions of environmental estrogens in developing granule cell neurons are mediated by estrogen receptor β. Endocrinology. (2010) 151:5689–99. doi: 10.1210/en.2010-0710
89. Sellers KJ, Denley MCS, Saito A, Foster EM, Salgarella I, Delogu A, et al. Brain-synthesized oestrogens regulate cortical migration in a sexually divergent manner. Eur J Neurosci. (2020) 52:2646–63. doi: 10.1111/ejn.14755
90. Klinge CM. Estrogens regulate life and death in mitochondria. J Bioenerg Biomembr. (2017) 49:307–24. doi: 10.1007/s10863-017-9704-1
91. Guajardo-Correa E, Silva-Agüero JF, Calle X, Chiong M, Henríquez M, García-Rivas G, et al. Estrogen signaling as a bridge between the nucleus and mitochondria in cardiovascular diseases. Front Cell Dev Biol. (2022) 10:968373. doi: 10.3389/fcell.2022.968373
92. Davidsen N, Ramhøj L, Ballegaard A-SR, Rosenmai AK, Henriksen CS, and Svingen T. Perfluorooctanesulfonic acid (PFOS) disrupts cadherin-16 in the developing rat thyroid gland. Curr Res Toxicol. (2024) 6:100154. doi: 10.1016/j.crtox.2024.100154
93. Davidsen N, Ramhøj L, Lykkebo CA, Kugathas I, Poulsen R, Rosenmai AK, et al. PFOS-induced thyroid hormone system disrupted rats display organ-specific changes in their transcriptomes. Environ pollut. (2022) 305:119340. doi: 10.1016/j.envpol.2022.119340
94. Du Y, Chen C, Zhou G, Cai Z, Man Q, Liu B, et al. Perfluorooctanoic acid disrupts thyroid-specific genes expression and regulation via the TSH-TSHR signaling pathway in thyroid cells. Environ Res. (2023) 239:117372. doi: 10.1016/j.envres.2023.117372
Keywords: brain organoids, neurodevelopment, neural differentiation, endocrine disrupting chemicals, BPS, PFOS
Citation: Di Credico A, Gaggi G, Bibbò S, Blenkinsop TA, Di Baldassarre A and Ghinassi B (2025) Endocrine-disrupting effects of environmental BPS and PFOS on human brain organoid development. Front. Endocrinol. 16:1692333. doi: 10.3389/fendo.2025.1692333
Received: 25 August 2025; Accepted: 21 November 2025; Revised: 13 November 2025;
Published: 04 December 2025.
Edited by:
Marta Lombó, University of León, SpainReviewed by:
Zhengliang Gao, Tongji University, ChinaKatharina Koch, Leibniz-Institut für Umweltmedizinische Forschung (IUF), Germany
Copyright © 2025 Di Credico, Gaggi, Bibbò, Blenkinsop, Di Baldassarre and Ghinassi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Angela Di Baldassarre, YS5kaWJhbGRhc3NhcnJlQHVuaWNoLml0
†These authors have contributed equally to this work