 Daniel Bikle
Daniel Bikle- Departments of Medicine and Dermatology, University of California San Francisco and Veterans Administration Medical Center, San Francisco, CA, United States
Fractures engender a multimillion dollar medical cost to society with substantial morbidity and mortality for the patients. In long bones fracture repair takes place at 3 distinct sites: intramembranous bone formation along the outer surface of the periosteum, endochondral bone formation bridging the fracture site, and intramedullary bone formation within the marrow at the ends of the fractured bone. Fracture repair occurs in 4 overlapping phases. 1. Hematoma formation and a proinflammatory response that activates the stem cells, 2. initiation of chondrogenesis, osteogenesis, and angiogenesis, 3. mineralization of the soft callus to form the hard callus, 4. remodeling of the callus to regenerate an intact bone. Stem cells in the periosteum, marrow, and overlying muscle supply the cells for the repair process. Parathyroid hormone (PTH) in its 1–34 form (teriparatide) and its analog abaloparatide are promising drugs to promote fracture repair. PTH acts via its receptor (PTHR) which is expressed in essentially all skeletal cells involved in fracture repair. Its anabolic actions are mediated by a number of interacting pathways including cAMP/PKA, Wnt, BMP, IGF1 and the bidirectional signaling of Ephrin B2/Eph B4. Progress in this field will lead to better treatment of fractures especially those slow or fail to heal.
Highlights
1. Fractures result in major medical costs and substantial morbidity and mortality for the patients.
2. In long bones there are 3 distinct sites of fracture repair: intramembranous on outer side of the periosteum, endochondral bridging the fracture site, and intramedullary within the marrow at the ends of the fractured bones.
3. Fracture repair occurs in 4 phases starting with hematoma formation and inflammation activating the stem cell population to supply the cells needed to generate the callus, followed by mineralization of the callus which is then remodeled to the pre fracture state.
4. The stem cells responding to the fracture originate primarily from the periosteum, marrow, and overlying muscle although transdifferentiation of osteocytes at the fracture site contributes to intramedullary bone formation.
5. Parathyroid hormone and its analog abaloparatide are promising drugs to expedite fracture repair.
Introduction: the clinical problem
In the United States there are an estimated two million fractures each year resulting in nearly a half million hospital admissions, 2.5 million office visits, and 180,000 nursing home admissions (1). Hip fractures account for only about 14% of all fractures, but they account for most of the costs and morbidity, estimated around $35,000 to $54,000 per fracture, with a 20-30% mortality within one year (2, 3). Although most fractures heal within 6–8 weeks, a significant percentage do not with delayed and even nonunion prolonging both the costs and morbidity (4). Bone turnover markers such as C-terminal telopeptide of collagen I (CTX), N-terminal propeptide of type 1 procollagen (P1NP) (5) or periostin (6) may provide markers for fracture healing. In the decades between 1970 and 2010, the rate of hip fractures in the Framingham Heart Study population decreased by 4.4%/year. This was attributed to better treatment of osteoporosis as well as a decrease in smoking and heavy drinking (7). However, this appears to have plateaued according to an analysis performed in 2015 (1). This likely relates to a decline in testing for osteoporosis and thus its treatment (8). Thus, fractures continue to be a major health problem. This review attempts to summarize what we know of the cellular mechanisms that are involved in fracture repair with a subsequent focus on how parathyroid hormone and its analogs impact those cellular mechanisms to promote the repair process. The discussion will be restricted to long bone fractures in which both intramembranous and endochondral bone formation occur as well as the less well studied intramedullary bone formation within the marrow at the ends of the fractured bone (9, 10).
The three sites of fracture repair
The repair of fractures of long bones occurs in 3 sites of new bone formation (BF) that are distinct in both location and process as shown in Figure 1 (9, 11). Site 1 marks the location of intramembranous BF along the periosteal surface of the intact bone. Bone formation at site 1 does not require an intermediate chondrocyte step. Site 3 marks the location of intramedullary BF forming within the marrow adjacent to the fracture site. BF at this site has been overlooked in studies using an intramedullary pin to stabilize the Fx as this obliterates the marrow space precluding new BF. As will be discussed BF at this site is quite distinct from BF at the other sites in the origin of the progenitor cells, although like that at site 1 a chondrogenic intermediary step is not involved. Site 2 is the bridging region between the broken ends of bone, and its development proceeds via endochondral BF that in many ways recapitulates the early development of bone (12).
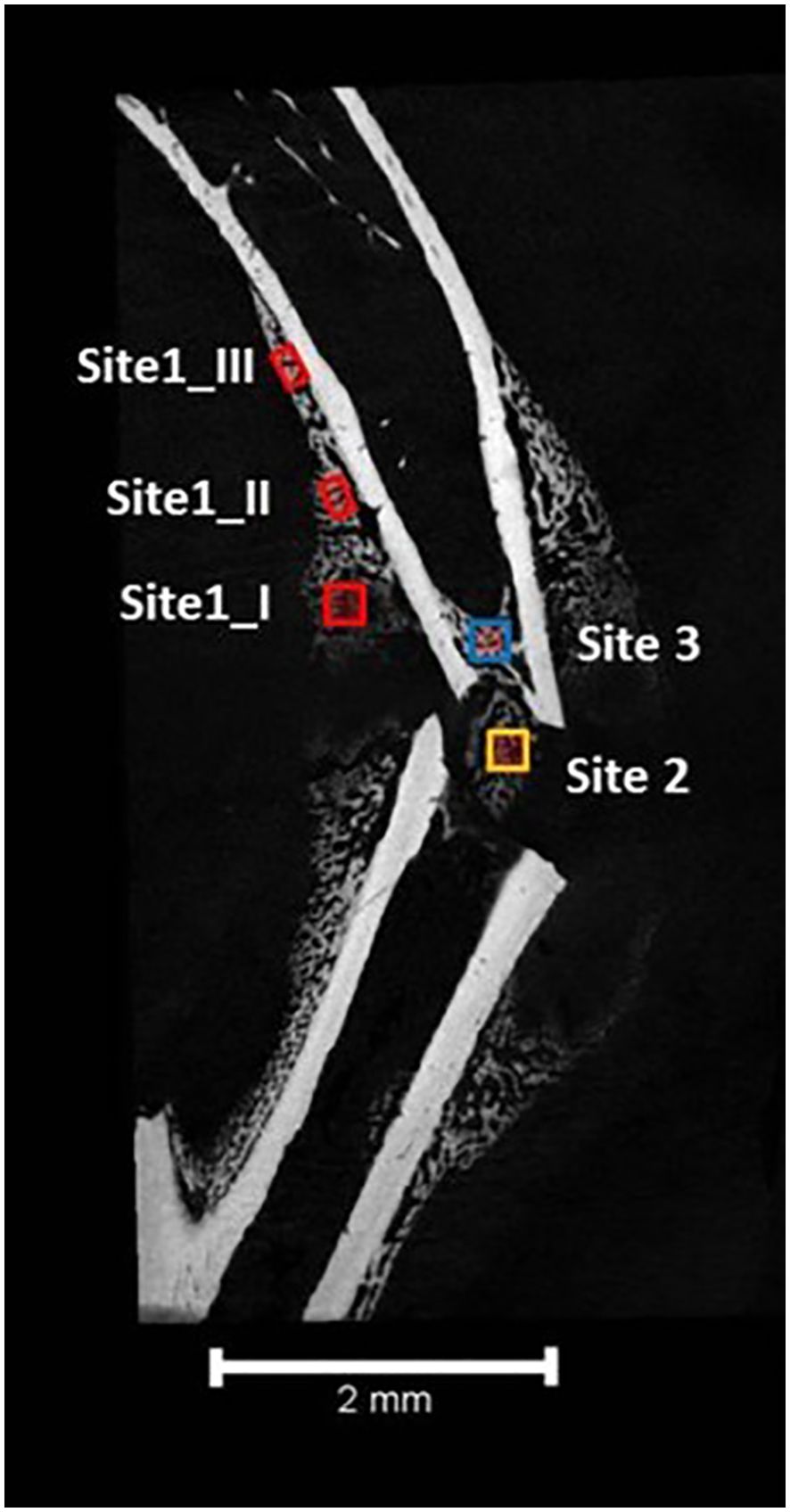
Figure 1. The three sites of long bone fracture repair. Site 1 involves intramembranous bone formation along the periosteum a short distance away from the fracture site. Stem cells from the periosteum as they differentiate into osteoblasts provide the source of progenitors for this site. Site 2 involves endochondral bone formation bridging the ends of the broken bone. Stem cells from the periosteum also provide the cells for this site as they differentiate first into chondrocytes. Site 3 occurs within the marrow at the ends of the broken bone. Stem cells from the marrow and the osteocytes within the cortical bone at the site of fracture differentiate or transdifferentiate into the osteoblasts for repair at this site.
The four phases of fracture repair
The four phases of fracture repair have been well described in previous reports (13) and are summarized here and depicted in Figure 2. The repair process described is that of long bone fractures. This is a multifaceted process in which inflammation plays a key role. Factors such as age, comorbidities such as diabetes mellitus, obesity, and autoimmune diseases affect healing and contribute to the risk of dysunion or nonunion. That said in general phase 1 lasts about 5 days, phase 2 likewise about 5 days, phase 3 several weeks, and the final phase 4 months to even years (14) with overlap from one phase to the next.

Figure 2. Four phases of fracture repair. The first phase involves formation of the hematoma into which inflammatory cells invade, activating the stem cells. The second phase includes angiogenesis and vascular invasion with the initiation of both endochondral bone formation to start forming the soft callus and osteogenesis in both the intramedullary and intramembranous sites. The third phase involves the mineralization of the soft callus with formation of the hard callus. The fourth phase marks the remodeling of the callus into lamellar bone to regenerate the bone to its pre fractured form.
Phase 1
This phase is marked by the formation of a hematoma at the fracture site by erythrocytes and platelets infiltrated by immune cells from the marrow including neutrophils and monocytes (15, 16). The monocytes differentiate into macrophages that in the early pro-inflammatory environment develop the M1 phenotype that prefer the hypoxic environment of the initially avascular hematoma. They secrete a variety of inflammatory cytokines including chemokine (C-C motif) ligand 2 (CCL2) for neutrophil recruitment and granulocyte-colony stimulating factor (G-CSF) and granulocyte macrophage-stimulating factor (GM-CSF) for neutrophil maintenance as well as cytokines like the interleukins IL-1α and β, IL-2, IL-6, interferon gamma (IFNγ) and tumor necrosis factor alpha (TNFα) that activate T cells while recruiting mesenchymal skeletal progenitor cells (MSC) (17). (Skeletal stem cells, SSC, and mesenchymal stem cells, MSC will be used interchangeably as they are generally treated as such in the literature reviewed in this report). Moreover, the M1 macrophages induce receptor activator of nuclear factor kappa-b ligand (RANKL) expression in osteoblasts and T (in particular, Th17) cells to promote osteoclastogenesis necessary to initiate the remodeling of the callus (17). The apoptosis of the osteocytes at the fracture site further promotes the recruitment of osteoclasts (18). Following this initial proinflammatory response M2 macrophages appear by recruitment of macrophages from the bone marrow and transition of M1 to M2 macrophages. These cells secrete IL-10 to reduce the pro-inflammatory response (19), and transforming growth factor beta (TGFβ, vascular epithelial growth factor (VEGF), insulin like growth factor1 (IGF-1), and bone morphogenic protein 2 (BMP2) to promote vascularization and MSC differentiation into osteoblasts (20, 21). MSCs can likewise suppress the proinflammatory response by killing the inflammatory cells by direct contact and secreting cytokines such as TGFβ that induce Treg and inhibit neutrophil migration (22). Platelet derived growth factor (PDGF) secretion from macrophages stimulates periostin enabling the response of the periosteal stem cells to injury (23). Similarly platelets within the fracture callus also secrete PDGF, VEGF, TGFβ, and IGF-1 facilitating the increase in vascularization, proliferation of MSCs, osteoblast differentiation and chondrogenesis (24).
Phase 2
This phase is marked by angiogenesis, cartilage formation, and development of the soft callus. Endochondral bone formation occurs in the region between the fractured ends of the bone, the bridge region, while intramembranous bone formation occurs along the periosteum at some distance from the fracture site. A third site, that within the medullary regions at the ends of the fractured bone, is best seen in non-stabilized fractures or in drill hole models. Intramedullary bone begins somewhat later (day 5–10 in mice) and is transient (9–11). At the first two sites stem cells from the periosteum proliferate and differentiate into osteoblasts or chondrocytes (25, 26), although stem cells from muscle also contribute (27–29). Runx2 regulates osteoblastogenesis, whereas osterix suppresses runx2 facilitating the development of chondrocytes (30). Preceding the expression of runx2 is upregulation of the glucose transporter GLUT 1, indicating the high energy demands for osteoblastogenesis (31). Hedgehog signaling interacting with both the wnt and BMP pathways regulates these early events in osteoblast differentiation and endochondral ossification (32). Notch signaling inhibits wnt signaling leading to proliferation of osteoprogenitors but inhibition of their differentiation (33), whereas BMP (in particular BMP2) signaling promotes osteoblast differentiation (34). IGF-1 promotes both chondrocyte and osteoblast proliferation and differentiation (35) and is a key mediator of the role of parathyroid hormone (PTH) in this regard (36). Ephrin B2 plays a critical role in coordinating these events (11, 37) as does calcium via its calcium sensing receptor (38). Intramedullary bone formation appears to be initiated following transdifferentiation of osteocytes from the cortex at the fracture site (9, 39) and differentiation of leptin receptor (LepR+) expressing stem cells in the marrow (40). The cartilage collar forms the soft callus starting at day 3 in mice, but it subsequently mineralizes, with the chondrocytes initially becoming hypertrophic, switching from producing collagen II to collagen X and eventually transdifferentiating into osteoblasts, a process reminiscent of that in the growth plate (41). This resemblance to bone formation at the growth plate is further supported by a genetic analysis of the genes involved showing similarity between fracture healing and embryonic development of bone (42). IL-6 plays an important role in the mineralization process (43). Vascularization of the soft callus is driven primarily by VEGF, fibroblast growth factor 1 (FGF1), and TGFβ coming from the M2 macrophages and platelets as noted above as well as osteoblasts (44). The type of vessel may also matter. Endomycin expressing endothelial cells (H cells) are more associated with osteoprogenitors and osteogenesis than endomycin low endothelium (45). Intramembranous bone formation along the periosteum develops without going through a chondrocyte phase to form the hard callus. Intramedullary bone formation likewise does not proceed through a chondrocyte intermediate step. Unlike the development of the soft and hard callus, it begins between 5–10 days, and is essentially gone by 28 days (9, 10).
Phase 3
This phase is marked by cartilage removal and its calcification with more complete formation of the hard callus. The chondrocytes mineralize and are either removed by osteoclasts or transition into osteoblasts (9, 46, 47). This process is accompanied by and perhaps induced by vascularization as the hypertrophic chondrocytes lose their sox9 and collagen X expression while increasing runx2 and β-catenin expression followed by that of osteoblast markers such as alkaline phosphatase, osterix, osteopontin, and osteocalcin (13, 48). Tumor necrosis factor alpha (TNFα) plays a role in stimulating osteoclast formation, and likely plays a role in the mineralization process (49). The induction of matrix metalloproteinases such as MMP13 facilitates the degradation of the matrix (47). The down regulation of noggin, an inhibitor of BMP stimulation of osteoblast differentiation, is also required for the mineralization process to proceed (50). Woven bone is initially formed that with the recruitment of osteoclasts is remodeled into lamellar bone during the fourth phase.
Phase 4
This phase involves the remodeling of the woven bone into lamellar bone to restore the bone to its pre fracture condition including restoration of the hematopoietic and trabecular structures. This process begins after 3–4 weeks but extends for months. Cytokines such as IL-1, IL-6 and TNFα are major drivers (28, 47, 51). BMPs promote cartilage resorption and recruitment of osteoblasts (51, 52) with the wnt signaling pathway playing a major role (18). The cells involved include the osteoclasts that resorb the cartilage and help remodel the woven bone with subsequent rebuilding of the lamellar bone by osteoblasts (47, 53, 54). With time the number and role of osteoclasts diminishes as osteoblasts and osteocytes become the primary bone cells, and in human bone the Haversian system with its central vascular canal and canalicular network are reestablished (55). Human osteoblasts from fractures that fail to heal show marked changes in these pathways of growth and differentiation (56).
Skeletal stem/progenitor cells during fracture repair
In an initial quest to identify the skeletal stem cell from which all skeletal progenitor cells were derived Chan et al (57) identified cells from the mouse growth plate that they purified by fluorescence activated cell sorting (FACS) that were capable of differentiating into bone, cartilage, and stromal cell lineages. These cells, called osteochondral skeletal stem cells (ocSSC), were identified with the markers CD45-,Ter119-,Tie2-,αυ+, Thy-,6C3-,CD105-,CD200+. The authors hypothesized that this cell type was the source of all the osteochondroprogenitors similar to the single stem cell in the hematopoietic system leading to the subsequent cell lines. Similar cells were found in the fracture callus, which the authors called fracture induced bone, cartilage, stromal progenitors (f-BCSP) (58). Several years later Chan et al. identified what they considered the human skeletal stem cell in this case marked as PDPN+,CD145-,CD73+,CD164+ which like the mouse skeletal stem cell could be induced to produce bone, cartilage, and stromal cell lineages. These cells were not progenitors for fat or muscle. However, this group subsequently identified a second group of stem cells called perivascular skeletal stem cells (pvSSC) identified with markers CD45-,CD31-,Pdgfrα+,Sca1+,CD24+. These cells are the source of adipogenic progenitors. but also osteochondrostromal progenitors (59). These cells met the stem cell criteria of being self-renewing and multipotent. Furthermore, these cells were induced to proliferate in response to injury such as fracture, and produce the more differentiated cell lines required to heal the fracture. The concept of a single skeletal stem cell was further challenged by the discovery of other niches of skeletal stem cells in different regions of the skeleton and under different regulation (25, 40, 60–62). Moreover other tissues like fat and muscle could provide stem cells that contributed to fracture repair (40). The main niches for skeletal stem cells involved in fracture repair of long bones are the periosteum, marrow, and muscle (Figure 3).
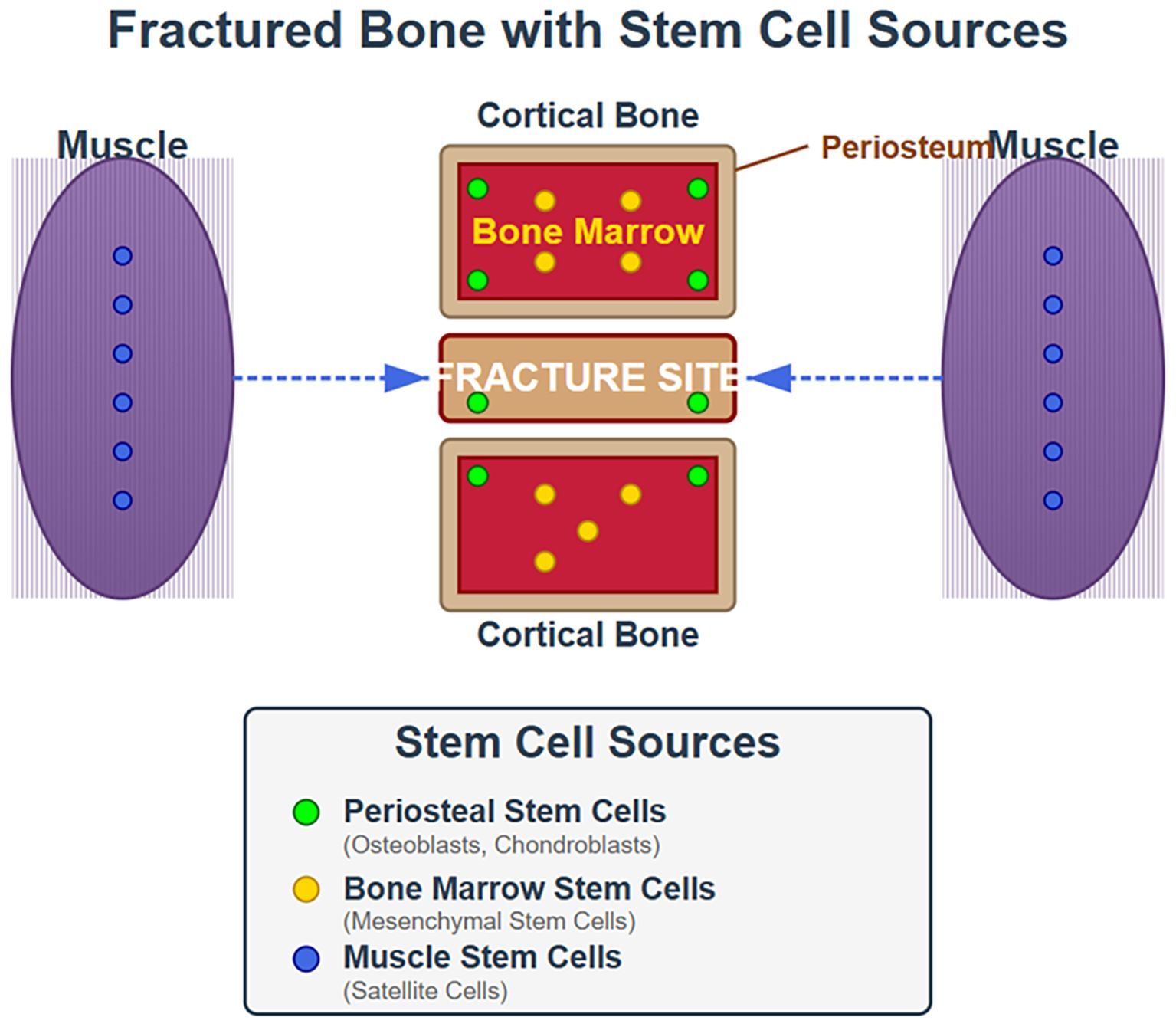
Figure 3. Origin of stem cells involved in fracture repair. This cartoon illustrates that during fracture repair stem cells from the periosteum, bone marrow and muscle all contribute albeit to different extents.
Periosteal stem cells
These cells reside in the inner cambium layer of the periosteum (63). They are considered the main source of cells involved in fracture repair of long bones (27) and are superior to bone marrow stem cells when used in therapy for fracture healing (64). In the mouse they express markers such as CD90, CD105, CD51, and CD29, Sca1 and in humans CD90, CD105, and CD73 (65). In response to injury (fracture) these stem cells lose their markers and change their cell fate, for example from generating only osteoblasts within the kidney capsule to generating both chondrocytes and osteoblasts in response to fracture (66). To trace the fate of these cells and their progeny in response to fracture various cre recombinases (cre) have been used to label the cells in combination with a fluorescent probe activated by the cre. These cres have not proved to be highly specific. However, gli 1 cre is a useful means of labeling periosteal stem cells and their response to injury (67) in contradistinction to adiponectin cre which preferentially labels Leptin receptor + (LepR+) cells in the bone marrow. The glioma associated protein (gli1+) expressing stem cells give rise to periostin (postn) + cells (68), many of which express the markers of the stem cells identified by Chan et al (57). Cathepsin K (ctsk) cre, alpha smooth muscle actin (αSMA) cre, and paired homeobox 1 (prrx1) cre have also been used to label periosteal stem cells, but also label bone marrow stromal cells and other tissues (67, 69). These cres do not necessarily label the same cells, as cells from the periosteum with characteristics of skeletal stem cells have been shown by scRNA-seq to be heterogeneous (63, 70). The review by Salhotra et al (32) provides a good overview of the various transcription factors and pathways involved in the fracture repair process as the stem cells differentiate into their different fates.
Bone marrow stem cells
In the adult LepR+ expressing SSC are a major source of osteoblasts (59). These reside as perivascular stromal cells in the bone marrow. These cells largely overlap with CXCL12+ abundant reticular cells (CAR cells) in the marrow, and both contribute to either osteogenesis (osteo-CAR) or adipogenesis (adipo-CAR) subsets (71, 72).The osteo-CAR cells are associated the endomucin high expressing H type vessels mentioned earlier, whereas the adipo-CAR cells are associated with endomucin low expressing L type vessels (sinusoid) or non vasculature (40). Zinc finger protein 467 appears to regulate the differentiation of these stem cells promoting the adipogenic fate (73, 74). The osteo-CAR cells contribute to fracture repair (75), but the role of the adipo-CAR cells is less clear. However, LepR+ Adiponectin+ cells, likely overlapping if not the same as adipo-CAR cells, were the major contributors to bone formation within drill hole injuries (67) and may be a source of cells forming the intramedullary bone during bicortical fractures (9). Similarly marrow adipogenic lineage precursors also likely overlap adipo-CAR cells, and have been shown to promote osteoclastogenesis and secrete BMP receptor inhibitors to inhibit bone formation during skeletal remodeling (76, 77). Their role during fracture repair is not clear. The interferon-induced GTP binding protein Mx1 marks an osteoprogenitor pool of cells on the endosteal surface and bone marrow that contributes to fracture repair (78). Other markers have labeled various cells within the bone marrow, but they also label periosteal stem cells so determining their distinct contribution to fracture repair is unclear.
Muscle stem cells
Although not strictly speaking skeletal stem cells, the juxtaposition of muscle to bone, and the injury to both during fractures has generated interest in the contribution of muscle to fracture repair. Separating muscle from bone over fracture sites with a cell impermeable membrane disrupts the repair process (79). Moreover, muscle secretes a number of myokines that promote fracture repair (80). Fibro-adipogenic progenitors (FAP) cells in muscle are key cells involved in muscle regeneration (81). But they also have osteogenic potential (80). Julien et al (27) labeled muscle stem cells with prrx1 cre and demonstrated their incorporation into the skeletal repair process. The Pax7 or Pax3 muscle satellite cells do not appear to play a role (82). He et al. (83) labeled Prg (lubricin) expressing FAP cells and likewise showed their contribution to chondrocytes and osteoblasts during fracture repair. Similar results were observed when muscle cells were labeled with αSMA cre, that labeled both perivascular and satellite cells, although Pax7 labeled satellite cells did not contribute (84).
Role of parathyroid hormone and its analogs in fracture repair
Clinical and animal studies
Parathyroid hormone (PTH), in particular the 1–34 portion of PTH (teriparatide), is effective in promoting fracture repair in a number of animal and human studies (85–87), although it is not yet FDA approved for this purpose in humans. Gorter et al. (88) summarized 41 animal studies in which various drugs, antiresorptive and anabolic, were used to treat fractures in either osteoporotic or non-osteoporotic animal models. PTH proved superior to the antiresorptives such as alendronate in showing an increase in bone mineral content, callus formation, biomechanical strength, improved radiologic and histologic healing, and improved union rate (89–91). Clinical studies have been less consistent. Retrospective studies (92, 93) and some randomized controlled trials (RCT) (94–96) but not all RCT (95, 97) have shown acceleration of union, although the incidence of non unions does not appear to be reduced.
Abaloparatide (Abl) is a synthetic 36aa analog of parathyroid hormone related peptide (PTHrP) to which it shares 76% homology and 42% homology with teriparatide (98). It signals through the same receptor as PTH and PTHrP and also promotes fracture repair (89). Although both teriparatide and abaloparatide act via the same receptor, their effects on the receptor differ (99). The parathyroid receptor has two conformations: R° and RG. Abaloparatide has enhanced selectivity for the RG form of the receptor relative to the R° form compared to teriparatide. The RG conformation mediates a faster and less sustained cell signaling response associated with a greater anabolic and less catabolic effect on bone metabolism (100). The molecular basis for this is well described in a review by Vilardaga et al (101). These differences also lead to substantial differences in their effects on the transcriptome (102). Several animal studies comparing abaloparatide to teriparatide on the balance between bone formation and bone resorption have demonstrated a greater increase in the expression of genes associated with anabolism and bone mineral density with abaloparatide (103, 104). For example, abaloparatide and teriparatide differ in their modulation of RANKL expression (abaloparatide has a lesser effect) perhaps explaining part of their differences in their effects on fracture repair (100, 105). In animal studies lower doses of abaloparatide have a greater effect on trabecular and cortical bone than equivalent doses of teriparatide (106, 107), although at higher doses the effects on bone of the two drugs appear to be equivalent (103). However, in a study in ovariectomized monkeys abaloparatide did not increase cortical porosity or elevate serum calcium (108), unlike earlier studies with teriparatide or full length PTH in this animal model (109, 110), indicating a lesser effect on bone resorption. With respect to clinical studies the Dose-finding Study of Abaloparatide (ACTIVE trial) demonstrated that 40 and 80ug abaloparatide increased bone mineral density at the lumbar spine, femoral neck, and total hip vs placebo and more than a 20ug teriparatide dose at the hip. This occurred with less increase in the bone resorption marker CTX-1 and fewer incidences of hypercalcemia (111, 112). Moreover, in a meta-analysis abaloparatide showed a greater reduction in both vertebral and non-vertebral fractures compared to teriparatide (113).
Cellular mechanisms of action during fracture repair
PTH originates only from the parathyroid gland, whereas PTHrP is expressed in a number of tissues including the periosteum of bone, where its expression is increased following fracture. Mice deficient in either PTH or PTHrP have delayed fracture repair (114, 115) including deletion of PTHrP specifically from the periosteum (116).The PTH/PTHrP receptor is found in a number of bone cells that participate in fracture repair including mesenchymal stem cells (MSC) in the marrow and periosteum, chondrocytes, osteoblasts, and osteocytes (117), and thus would be expected to participate in all sites of bone formation during fracture repair. In the marrow PTH/PTHrP likely promotes intramedullary bone in part by downregulation of the transcription factor Zfp467, that otherwise promotes adipogenesis while blocking osteogenesis (74, 118). The ability of PTH and PTHrP to stimulate the proliferation of osteochondral progenitors (119), and act on both chondrocytes and osteoblasts to promote chondrogenesis and osteogenesis (85, 120, 121), is critical to fracture repair. Runx 1 appears to play a major role in PTH stimulated chondrogenesis (122), whereas a number of transcription factors including runx2 play a role in osteogenesis. PTH also promotes angiogenesis (123), critical for remodeling during fracture repair (124). This includes not only the vascular invasion during the endochondral phase of bone formation, but the increase in blood vessels within the marrow and the bone cortex itself (123–125). In these actions PTH promotes the proliferation of the αSMA+ perivascular stromal cells (126). As will be discussed subsequently, PTH not only increases the cross talk between chondrocytes, osteoblasts and osteoclasts via ephrinB2/EphB4 signaling (37, 127) but also the cross talk between ephrinB2 expressing osteoblasts and the EphB4 expressing endomucin+ blood vessels in bone (123). The increase in intracortical and intramedullary blood vessels could provide perivascular osteoprogenitors for intramedullary bone formation during fracture repair as noted above.
PTH acts through a number of different pathways
These have been well reviewed by Liu et al (128). The PTH receptor activation of the cAMP/PKA and the Ca2+/PKC pathways occurs via distinct G proteins, Gs and Gq. G12/13 mediates the phospholipase D-transforming protein RhoA pathway that has been little studied in fracture repair. In general the anabolic pro osteogenic actions are mediated through the cAMP/PKA pathway (129, 130), whereas the Ca2+/PKC pathway underlies the catabolic actions (131). In skeletal stem cells (SSC) the increase in cAMP leading to the increase in PKA activity and the cAMP response element binding protein (p-CREB) promotes their proliferation and osteogenic differentiation (130). Although the Ca2+/PKC pathway has been associated with catabolic actions, at least one report indicates that PKCδ may promote osteogenesis in MSC (132). The PKA-SIK (salt inducible kinase) pathway plays an important role downstream of PTH activation of the cAMP/PKA pathway. PTH by activating PKA phosphorylates and inactivates SIK2 which in the non-phosphorylated form would phosphorylate the histone deacetylases HDAC4 and 5 keeping them out of the nucleus. However, with SIK2 inactivated, HDAC 4 and 5 can enter the nucleus where they inhibit the function of MEF2, a transcription factor that otherwise drives Sost expression, an inhibitor of bone formation (133). Inactivation of SIK2 also enables the CREB regulated transcription factor (CRTC2) to enter the nucleus promoting bone resorption (133). The transcription factor nascent polypeptide-associated complex and coactivator alpha (αNAC) is also a substrate for PTH activated PKA. Its phosphorylation enables its entrance into the nucleus where it promotes expression of genes involved with bone formation (134). αNAC also colocalizes with and facilitates junD stimulation of LRP6 expression (135). As described below LRP6 serves as a coreceptor for the PTH receptor (PTHR) enhancing its function.
Wnt pathway. PTH stimulates the Wnt pathway thus increasing active β-catenin levels in periosteal SSC expanding this population to provide the cells for fracture repair (136). PTH increases the levels of other members of the Wnt signaling pathway including LRP5, Wnt7b and Wnt10b (137) while reducing Wnt inhibitors such as Dkk1 (138), Wasf2 (139), and in osteocytes Sost (140). At least some of the Wnt10b comes from PTH stimulation of T cells (141). The coreceptor for the Wnt receptor frizzled (FZD), LRP6, also serves as a coreceptor for the PTH receptor (PTHR) facilitating PTH stimulation of cAMP production (142). The enhanced PTHR/LRP6 signaling in turn can inhibit sclerostin production via the PKA/SIK2 pathway feeding back positively on Wnt signaling including PTHR/LRP6 signaling, a feed forward loop. However, this interaction is blocked by N-cadherin providing some negative control (143). The PTHR/LRP6 signaling pathway in osteocytes could contribute to PTH promotion of intramedullary bone formation during fracture repair but has not been tested.
BMP/SMAD pathway. PTH activates the SMAD1 pathway in the periosteum, mediating downstream targets of BMP2 and increasing osteogenesis (144). This is activated by the PTH induced endocytosis of the PTHR/LRP6 complex leading to phosphorylation of SMAD1 and stimulation of αSMA+ stem cell differentiation into osteoblasts (145).
IGF1 signaling. PTH stimulates the expression of IGF1 and its receptor (IGF1R) in bone (146, 147). IGF1 (148) and its receptor (149) are required for the anabolic actions of PTH on bone. The chondrocytes in the growth plates (GP) of global (glo)IGF1KO mice show decreased proliferation and increased apoptosis. Differentiation of the proliferating chondrocytes to hypertrophic chondrocytes is delayed with decreased vascular invasion and mineralization (150). The delay in vascular invasion is consistent with the induction by IGF1 of hypoxia inducible factor (HIF)1α and vascular endothelial growth factor (VEGF) (151). Bone formation is reduced in the gloIGF1KO mice that survive postnatally. Although bone formation is reduced, trabecular bone volume (BV/TV) in the proximal tibia is increased (152), a result reflecting the dual effect of IGF1 on osteoblast and osteoclast activity (153, 154). IGF1 stimulates RANKL expression by osteoblasts thus promoting osteoclastogenesis (154–156). IGF1 signaling plays a comparable critical role during fracture repair. IGF1 and IGF1R are expressed in chondrocytes, osteoblasts, and osteoclasts, and their levels increase throughout fracture repair (157, 158). This is associated with a marked increase in proliferation of cells within the cambial layer of the periosteum, increase in Sox2 labeled stem cells, increase in VEGF expression, and invasion into the callus. Shi et al (159) demonstrated that PTH stimulated gli1+ stem cell progenitor differentiation in this case to chondrocytes via IGF signaling, and it is likely that IGF signaling underlies other PTH actions during fracture repair.
Ephrinb2/EphB4 signaling. Among the means by which PTH and IGF1 promote the intercellular communication required for fracture repair involves the bidirectional ephrinB2 (EfnB2)/EphB4 signaling, which when disrupted impairs fracture repair (11). This bidirectional signaling of EfnB2/EphB4 occurs between all cells in bone (127) including the blood vessels (123, 160). Of particular relevance to fracture repair is that this bidirectional signaling promotes the motility of mesenchymal stem and endothelial cells, promotes osteochondral differentiation, and stimulates VEGF induced angiogenesis (161–164). In fracture studies with col2EfnB2KO mice (11) vascular invasion was markedly reduced as were VEGF expression and numbers of stem cells in the invasion front. IGF1 is essential for the expression of EfnB2 and EphB4 in those cells engaged in fracture repair (37). Stimulation of EfnB2 and EphB4 expression by PTH (165) requires IGF1/IGF1R. Blocking the EfnB2/EphB4 interactions with specific inhibitors or gene deletions blocks basal and IGF1 stimulated chondrocyte, osteoblast, and osteoclast differentiation, and blocks IGF1 stimulated fracture repair (11). Thus, like PTH and IGF1, EfnB2/EphB4 signaling is found in and between essentially all the cells involved with fracture repair and is required for the fracture repair process.
Conclusions
Fractures are a major medical concern, and especially if the repair process is prolonged, leading to huge morbidity, mortality, and costs. Thus understanding the mechanisms by which fracture repair takes place will likely lead to better therapy improving the outcomes for the patients involved. Repair of the fracture rallies all cells in the bone and bone marrow to alter their cell fate with the goal of regenerating bone to its pre fracture condition. Within bone and bone marrow exist different stem cell niches. It is these cells that respond to the challenge to alter their cell fate to repair the fracture. Indeed, these skeletal stem cells are being used clinically to expedite fracture repair especially in conditions of delayed repair or nonunion. One class of drugs, PTH and its analogs also shows promise in facilitating the repair process. As is appropriate for the complexity of fracture repair, a number of signaling pathways are activated by PTH to promote fracture repair. Many of the anabolic actions of PTH are mediated by the cAMP/PKA pathway. However, Wnt signaling, IGF1 signaling and the bidirectional signaling of EfnB2/EphB4 play major and interacting roles as well. With progress in this field we can anticipate that the burden of fractures, especially those with delayed or non-unions will be lessened.
Author contributions
DB: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. US Department of Veterans Affairs provided salary and facilities. The financial support via grant 1I01 BX005854 from the Veterans Health Administration.
Acknowledgments
The author acknowledges the investigative skills of Dr. Yongmei Wang into mechanisms of bone fracture repair and the role of PTH in this process within our laboratory.
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that Generative AI was used in the creation of this manuscript. I used claude.ai to help generate figures 2 and 3.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Lewiecki EM, Wright NC, Curtis JR, Siris E, Gagel RF, Saag KG, et al. Hip fracture trends in the United States, 2002 to 2015. Osteoporos Int. (2018) 29:717–22. doi: 10.1007/s00198-017-4345-0
2. Gu Q, Koenig L, Mather RC 3rd, and Tongue J. Surgery for hip fracture yields societal benefits that exceed the direct medical costs. Clin Orthop Relat Res. (2014) 472:3536–46. doi: 10.1007/s11999-014-3820-6
3. Johnell O, Kanis JA, Jonsson B, Oden A, Johansson H, De Laet C, et al. The burden of hospitalised fractures in Sweden. Osteoporos Int. (2005) 16:222–8. doi: 10.1007/s00198-004-1686-2
4. Hak DJ, Fitzpatrick D, Bishop JA, Marsh JL, Tilp S, Schnettler R, et al. Delayed union and nonunions: epidemiology, clinical issues, and financial aspects. Injury. (2014) 45 Suppl 2:S3–7. doi: 10.1016/j.injury.2014.04.002
5. Stewart CC, O'Hara NN, Bzovsky S, Bahney CS, Sprague S, Slobogean GP, et al. Bone turnover markers as surrogates of fracture healing after intramedullary fixation of tibia and femur fractures. Bone Joint Res. (2022) 11:239–50. doi: 10.1302/2046-3758.114.BJR-2021-0226.R1
6. Yan J, Liu HJ, Li H, Chen L, Bian YQ, Zhao B, et al. Circulating periostin levels increase in association with bone density loss and healing progression during the early phase of hip fracture in Chinese older women. Osteoporos Int. (2017) 28:2335–41. doi: 10.1007/s00198-017-4034-z
7. Swayambunathan J, Dasgupta A, Rosenberg PS, Hannan MT, Kiel DP, Bhattacharyya T, et al. Incidence of hip fracture over 4 decades in the framingham heart study. JAMA Intern Med. (2020) 180:1225–31. doi: 10.1001/jamainternmed.2020.2975
8. Miller PD. Underdiagnosis and undertreatment of osteoporosis: the battle to be won. J Clin Endocrinol Metab. (2016) 101:852–9. doi: 10.1210/jc.2015-3156
9. Wang Y, Chen L, Kang M, Ling L, Tian F, Won-Kim SH, et al. The fracture callus is formed by progenitors of different skeletal origins in a site-specific manner. JBMR Plus. (2019) 3:e10193. doi: 10.1002/jbm4.10193
10. Inoue S, Takito J, and Nakamura M. Site-specific fracture healing: comparison between diaphysis and metaphysis in the mouse long bone. Int J Mol Sci. (2021) 22. doi: 10.3390/ijms22179299
11. Wang Y, Ling L, Tian F, Won Kim SH, Ho S, Bikle DD, et al. Ablation of ephrin B2 in col2 expressing cells delays fracture repair. Endocrinology. (2020) 161. doi: 10.1210/endocr/bqaa179
12. Einhorn TA and Gerstenfeld LC. Fracture healing: mechanisms and interventions. Nat Rev Rheumatol. (2015) 11:45–54. doi: 10.1038/nrrheum.2014.164
13. Saul D and Khosla S. Fracture healing in the setting of endocrine diseases, aging, and cellular senescence. Endocr Rev. (2022) 43:984–1002. doi: 10.1210/endrev/bnac008
14. ElHawary H, Baradaran A, Abi-Rafeh J, Vorstenbosch J, Xu L, Efanov JI, et al. Bone healing and inflammation: principles of fracture and repair. Semin Plast Surg. (2021) 35:198–203. doi: 10.1055/s-0041-1732334
15. Kovtun A, Bergdolt S, Wiegner R, Radermacher P, Huber-Lang M, Ignatius A, et al. The crucial role of neutrophil granulocytes in bone fracture healing. Eur Cell Mater. (2016) 32:152–62. doi: 10.22203/ecm.v032a10
16. Maruyama M, Rhee C, Utsunomiya T, Zhang N, Ueno M, Yao Z, et al. Modulation of the inflammatory response and bone healing. Front Endocrinol (Lausanne). (2020) 11:386. doi: 10.3389/fendo.2020.00386
17. Molitoris KH, Huang M, and Baht GS. Osteoimmunology of fracture healing. Curr Osteoporos Rep. (2024) 22:330–9. doi: 10.1007/s11914-024-00869-z
18. Choy MHV, Wong RMY, Chow SKH, Li MC, Chim YN, Li TK, et al. How much do we know about the role of osteocytes in different phases of fracture healing? A systematic review. J Orthop Translat. (2020) 21:111–21. doi: 10.1016/j.jot.2019.07.005
19. Vi L, Baht GS, Soderblom EJ, Whetstone H, Wei Q, Furman B, et al. Macrophage cells secrete factors including LRP1 that orchestrate the rejuvenation of bone repair in mice. Nat Commun. (2018) 9:5191. doi: 10.1038/s41467-018-07666-0
20. Gong L, Zhao Y, Zhang Y, and Ruan Z. The macrophage polarization regulates MSC osteoblast differentiation in vitro. Ann Clin Lab Sci. (2016) 46:65–71.
21. Xu W, Yang Y, Li N, and Hua J. Interaction between mesenchymal stem cells and immune cells during bone injury repair. Int J Mol Sci. (2023) 24. doi: 10.3390/ijms241914484
22. Medhat D, Rodriguez CI, and Infante A. Immunomodulatory effects of MSCs in bone healing. Int J Mol Sci. (2019) 20. doi: 10.3390/ijms20215467
23. Gao B, Deng R, Chai Y, Chen H, Hu B, Wang X, et al. Macrophage-lineage TRAP+ cells recruit periosteum-derived cells for periosteal osteogenesis and regeneration. J Clin Invest. (2019) 129:2578–94. doi: 10.1172/JCI98857
24. Wildemann B, Schmidmaier G, Ordel S, Stange R, Haas NP, Raschke M, et al. Cell proliferation and differentiation during fracture healing are influenced by locally applied IGF-I and TGF-beta1: comparison of two proliferation markers, PCNA and BrdU. J BioMed Mater Res B Appl Biomater. (2003) 65:150–6. doi: 10.1002/jbm.b.10512
25. Perrin S and Colnot C. Periosteal skeletal stem and progenitor cells in bone regeneration. Curr Osteoporos Rep. (2022) 20:334–43. doi: 10.1007/s11914-022-00737-8
26. Wang T, Zhang X, and Bikle DD. Osteogenic differentiation of periosteal cells during fracture healing. J Cell Physiol. (2017) 232:913–21. doi: 10.1002/jcp.25641
27. Julien A, Perrin S, Martinez-Sarra E, Kanagalingam A, Carvalho C, Luka M, et al. Skeletal stem/progenitor cells in periosteum and skeletal muscle share a common molecular response to bone injury. J Bone Miner Res. (2022) 37:1545–61. doi: 10.1002/jbmr.4616
28. Glass GE, Chan JK, Freidin A, Feldmann M, Horwood NJ, Nanchahal J, et al. TNF-alpha promotes fracture repair by augmenting the recruitment and differentiation of muscle-derived stromal cells. Proc Natl Acad Sci U.S.A. (2011) 108:1585–90. doi: 10.1073/pnas.1018501108
29. Shah K, Majeed Z, Jonason J, and O’Keefe RJ. The role of muscle in bone repair: the cells, signals, and tissue responses to injury. Curr Osteoporos Rep. (2013) 11:130–5. doi: 10.1007/s11914-013-0146-3
30. Yoshida CA, Komori H, Maruyama Z, Miyazaki T, Kawasaki K, Furuichi T, et al. SP7 inhibits osteoblast differentiation at a late stage in mice. PloS One. (2012) 7:e32364. doi: 10.1371/journal.pone.0032364
31. Wei J, Shimazu J, Makinistoglu MP, Maurizi A, Kajimura D, Zong H, et al. Glucose uptake and runx2 synergize to orchestrate osteoblast differentiation and bone formation. Cell. (2015) 161:1576–91. doi: 10.1016/j.cell.2015.05.029
32. Salhotra A, Shah HN, Levi B, and Longaker MT. Mechanisms of bone development and repair. Nat Rev Mol Cell Biol. (2020) 21:696–711. doi: 10.1038/s41580-020-00279-w
33. Novak S, Roeder E, Sinder BP, Adams DJ, Siebel CW, Grcevic D, et al. Modulation of Notch1 signaling regulates bone fracture healing. J Orthop Res. (2020) 38:2350–61. doi: 10.1002/jor.24650
34. Lin GL and Hankenson KD. Integration of BMP, Wnt, and notch signaling pathways in osteoblast differentiation. J Cell Biochem. (2011) 112:3491–501. doi: 10.1002/jcb.23287
35. Wang Y, Cheng Z, Elalieh HZ, Nakamura E, Nguyen MT, Mackem S, et al. IGF-1R signaling in chondrocytes modulates growth plate development by interacting with the PTHrP/Ihh pathway. J Bone Miner Res. (2011) 26:1437–46. doi: 10.1002/jbmr.359
36. Bikle DD and Wang Y. Insulin like growth factor-I: a critical mediator of the skeletal response to parathyroid hormone. Curr Mol Pharmacol. (2012) 5:135–42. doi: 10.2174/1874467211205020135
37. Wang Y, Menendez A, Fong C, ElAlieh HZ, Chang W, Bikle DD, et al. Ephrin B2/EphB4 mediates the actions of IGF-I signaling in regulating endochondral bone formation. J Bone Miner Res. (2014) 29:1900–13. doi: 10.1002/jbmr.2196
38. Cheng Z, Li A, Tu CL, Maria CS, Szeto N, Herberger A, et al. Calcium-sensing receptors in chondrocytes and osteoblasts are required for callus maturation and fracture healing in mice. J Bone Miner Res. (2020) 35:143–54. doi: 10.1002/jbmr.3864
39. Torreggiani E, Matthews BG, Pejda S, Matic I, Horowitz MC, Grcevic D, et al. Preosteocytes/osteocytes have the potential to dedifferentiate becoming a source of osteoblasts. PloS One. (2013) 8:e75204. doi: 10.1371/journal.pone.0075204
40. Trompet D, Melis S, Chagin AS, and Maes C. Skeletal stem and progenitor cells in bone development and repair. J Bone Miner Res. (2024) 39:633–54. doi: 10.1093/jbmr/zjae069
41. Zhou X, von der Mark K, Henry S, Norton W, Adams H, de Crombrugghe B, et al. Chondrocytes transdifferentiate into osteoblasts in endochondral bone during development, postnatal growth and fracture healing in mice. PloS Genet. (2014) 10:e1004820. doi: 10.1371/journal.pgen.1004820
42. Bais M, McLean J, Sebastiani P, Young M, Wigner N, Smith T, et al. Transcriptional analysis of fracture healing and the induction of embryonic stem cell-related genes. PloS One. (2009) 4:e5393. doi: 10.1371/journal.pone.0005393
43. Prystaz K, Kaiser K, Kovtun A, Haffner-Luntzer M, Fischer V, Rapp AE, et al. Distinct effects of IL-6 classic and trans-signaling in bone fracture healing. Am J Pathol. (2018) 188:474–90. doi: 10.1016/j.ajpath.2017.10.011
44. Hu K and Olsen BR. Osteoblast-derived VEGF regulates osteoblast differentiation and bone formation during bone repair. J Clin Invest. (2016) 126:509–26. doi: 10.1172/JCI82585
45. Kusumbe AP, Ramasamy SK, and Adams RH. Coupling of angiogenesis and osteogenesis by a specific vessel subtype in bone. Nature. (2014) 507:323–8. doi: 10.1038/nature13145
46. Hu DP, Ferro F, Yang F, Taylor AJ, Chang W, Miclau T, et al. Cartilage to bone transformation during fracture healing is coordinated by the invading vasculature and induction of the core pluripotency genes. Development. (2017) 144:221–34. doi: 10.1242/dev.130807
47. Bahney CS, Zondervan RL, Allison P, Theologis A, Ashley JW, Ahn J, et al. Cellular biology of fracture healing. J Orthop Res. (2019) 37:35–50. doi: 10.1002/jor.24170
48. Duan P and Bonewald LF. The role of the wnt/beta-catenin signaling pathway in formation and maintenance of bone and teeth. Int J Biochem Cell Biol. (2016) 77:23–9. doi: 10.1016/j.biocel.2016.05.015
49. Valles G, Bensiamar F, Maestro-Paramio L, Garcia-Rey E, Vilaboa N, Saldana L, et al. Influence of inflammatory conditions provided by macrophages on osteogenic ability of mesenchymal stem cells. Stem Cell Res Ther. (2020) 11:57. doi: 10.1186/s13287-020-1578-1
50. Aspenberg P, Jeppsson C, and Economides AN. The bone morphogenetic proteins antagonist Noggin inhibits membranous ossification. J Bone Miner Res. (2001) 16:497–500. doi: 10.1359/jbmr.2001.16.3.497
51. Tsiridis E, Upadhyay N, and Giannoudis P. Molecular aspects of fracture healing: which are the important molecules? Injury. (2007) 38 Suppl:1, S11–25. doi: 10.1016/j.injury.2007.02.006
52. Lissenberg-Thunnissen SN, de Gorter DJ, Sier CF, and Schipper IB. Use and efficacy of bone morphogenetic proteins in fracture healing. Int Orthop. (2011) 35:1271–80. doi: 10.1007/s00264-011-1301-z
53. Marsell R and Einhorn TA. The biology of fracture healing. Injury. (2011) 42:551–5. doi: 10.1016/j.injury.2011.03.031
54. Casanova M, Schindeler A, Peacock L, Lee L, Schneider P, Little DG, et al. Characterization of the developing lacunocanalicular network during fracture repair. JBMR Plus. (2021) 5:e10525. doi: 10.1002/jbm4.10525
55. Yu B, Pacureanu A, Olivier C, Cloetens P, and Peyrin F. Assessment of the human bone lacuno-canalicular network at the nanoscale and impact of spatial resolution. Sci Rep. (2020) 10:4567. doi: 10.1038/s41598-020-61269-8
56. Hofmann A, Ritz U, Hessmann MH, Schmid C, Tresch A, Rompe JD, et al. Cell viability, osteoblast differentiation, and gene expression are altered in human osteoblasts from hypertrophic fracture non-unions. Bone. (2008) 42:894–906. doi: 10.1016/j.bone.2008.01.013
57. Chan CK, Seo EY, Chen JY, Lo D, McArdle A, Sinha R, et al. Identification and specification of the mouse skeletal stem cell. Cell. (2015) 160:285–98. doi: 10.1016/j.cell.2014.12.002
58. Marecic O, Tevlin R, McArdle A, Seo EY, Wearda T, Duldulao C, et al. Identification and characterization of an injury-induced skeletal progenitor. Proc Natl Acad Sci U.S.A. (2015) 112:9920–5. doi: 10.1073/pnas.1513066112
59. Ambrosi TH, Sinha R, Steininger HM, Hoover MY, Murphy MP, Koepke LS, et al. Distinct skeletal stem cell types orchestrate long bone skeletogenesis. Elife. (2021) 10. doi: 10.7554/eLife.66063
60. Solidum JGN, Jeong Y, and Heralde F. 3rd & Park, D. Differential regulation of skeletal stem/progenitor cells in distinct skeletal compartments. Front Physiol. (2023) 14:1137063. doi: 10.3389/fphys.2023.1137063
61. Sun J, Hu L, Bok S, Yallowitz AR, Cung M, McCormick J, et al. A vertebral skeletal stem cell lineage driving metastasis. Nature. (2023) 621:602–9. doi: 10.1038/s41586-023-06519-1
62. Mizuhashi K, Ono W, Matsushita Y, Sakagami N, Takahashi A, Saunders TL, et al. Resting zone of the growth plate houses a unique class of skeletal stem cells. Nature. (2018) 563:254–8. doi: 10.1038/s41586-018-0662-5
63. Matsushita Y, Ono W, and Ono N. Skeletal stem cells for bone development and repair: diversity matters. Curr Osteoporos Rep. (2020) 18:189–98. doi: 10.1007/s11914-020-00572-9
64. Gao X, Ruzbarsky JJ, Layne JE, Xiao X, and Huard J. Stem cells and bone tissue engineering. Life (Basel). (2024) 14. doi: 10.3390/life14030287
65. Duchamp de Lageneste O, Julien A, Abou-Khalil R, Frangi G, Carvalho C, Cagnard N, et al. Periosteum contains skeletal stem cells with high bone regenerative potential controlled by Periostin. Nat Commun. (2018) 9:773. doi: 10.1038/s41467-018-03124-z
66. Debnath S, Yallowitz AR, McCormick J, Lalani S, Zhang T, Xu R, et al. Discovery of a periosteal stem cell mediating intramembranous bone formation. Nature. (2018) 562:133–9. doi: 10.1038/s41586-018-0554-8
67. Jeffery EC, Mann TLA, Pool JA, Zhao Z, and Morrison SJ. Bone marrow and periosteal skeletal stem/progenitor cells make distinct contributions to bone maintenance and repair. Cell Stem Cell. (2022) 29:1547–1561 e1546. doi: 10.1016/j.stem.2022.10.002
68. Yin B, Shen F, Ma Q, Liu Y, Han X, Cai X, et al. Identification of Postn+ periosteal progenitor cells with bone regenerative potential. JCI Insight. (2024) 9. doi: 10.1172/jci.insight.182524
69. Liu H, Li P, Zhang S, Xiang J, Yang R, Liu J, et al. Prrx1 marks stem cells for bone, white adipose tissue and dermis in adult mice. Nat Genet. (2022) 54:1946–58. doi: 10.1038/s41588-022-01227-4
70. Matthews BG, Novak S, Sbrana FV, Funnell JL, Cao Y, Buckels EJ, et al. Heterogeneity of murine periosteum progenitors involved in fracture healing. Elife. (2021) 10. doi: 10.7554/eLife.58534
71. Baccin C, Al-Sabah J, Velten L, Helbling PM, Grunschlager F, Hernandez-Malmierca P, et al. Combined single-cell and spatial transcriptomics reveal the molecular, cellular and spatial bone marrow niche organization. Nat Cell Biol. (2020) 22:38–48. doi: 10.1038/s41556-019-0439-6
72. Zhou BO, Yue R, Murphy MM, Peyer JG, and Morrison SJ. Leptin-receptor-expressing mesenchymal stromal cells represent the main source of bone formed by adult bone marrow. Cell Stem Cell. (2014) 15:154–68. doi: 10.1016/j.stem.2014.06.008
73. You L, Pan L, Chen L, Chen JY, Zhang X, Lv Z, et al. Suppression of zinc finger protein 467 alleviates osteoporosis through promoting differentiation of adipose derived stem cells to osteoblasts. J Transl Med. (2012) 10:11. doi: 10.1186/1479-5876-10-11
74. Quach JM, Walker EC, Allan E, Solano M, Yokoyama A, Kato S, et al. Zinc finger protein 467 is a novel regulator of osteoblast and adipocyte commitment. J Biol Chem. (2011) 286:4186–98. doi: 10.1074/jbc.M110.178251
75. Matsushita Y, Nagata M, Kozloff KM, Welch JD, Mizuhashi K, Tokavanich N, et al. A Wnt-mediated transformation of the bone marrow stromal cell identity orchestrates skeletal regeneration. Nat Commun. (2020) 11:332. doi: 10.1038/s41467-019-14029-w
76. Yu W, Zhong L, Yao L, Wei Y, Gui T, Li Z, et al. Bone marrow adipogenic lineage precursors promote osteoclastogenesis in bone remodeling and pathologic bone loss. J Clin Invest. (2021) 131. doi: 10.1172/JCI140214
77. Zou W, Rohatgi N, Brestoff JR, Li Y, Barve RA, Tycksen E, et al. Ablation of fat cells in adult mice induces massive bone gain. Cell Metab. (2020) 32:801–813 e806. doi: 10.1016/j.cmet.2020.09.011
78. Park D, Spencer JA, Koh BI, Kobayashi T, Fujisaki J, Clemens TL, et al. Endogenous bone marrow MSCs are dynamic, fate-restricted participants in bone maintenance and regeneration. Cell Stem Cell. (2012) 10:259–72. doi: 10.1016/j.stem.2012.02.003
79. Harry LE, Sandison A, Paleolog EM, Hansen U, Pearse MF, Nanchahal J, et al. Comparison of the healing of open tibial fractures covered with either muscle or fasciocutaneous tissue in a murine model. J Orthop Res. (2008) 26:1238–44. doi: 10.1002/jor.20649
80. Sui H, Dou J, Shi B, and Cheng X. The reciprocity of skeletal muscle and bone: an evolving view from mechanical coupling, secretory crosstalk to stem cell exchange. Front Physiol. (2024) 15:1349253. doi: 10.3389/fphys.2024.1349253
81. Molina T, Fabre P, and Dumont NA. Fibro-adipogenic progenitors in skeletal muscle homeostasis, regeneration and diseases. Open Biol. (2021) 11:210110. doi: 10.1098/rsob.210110
82. Julien A, Kanagalingam A, Martinez-Sarra E, Megret J, Luka M, Menager M, et al. Direct contribution of skeletal muscle mesenchymal progenitors to bone repair. Nat Commun. (2021) 12:2860. doi: 10.1038/s41467-021-22842-5
83. He Q, Lu J, Liang Q, Yao L, Sun T, Wang H, et al. Prg4+ fibro-adipogenic progenitors in muscle are crucial for bone fracture repair. bioRxiv. (2025). doi: 10.1101/2025.05.14.654160
84. Matthews BG, Torreggiani E, Roeder E, Matic I, Grcevic D, Kalajzic I, et al. Osteogenic potential of alpha smooth muscle actin expressing muscle resident progenitor cells. Bone. (2016) 84:69–77. doi: 10.1016/j.bone.2015.12.010
85. Nakazawa T, Nakajima A, Shiomi K, Moriya H, Einhorn TA, Yamazaki M, et al. Effects of low-dose, intermittent treatment with recombinant human parathyroid hormone (1-34) on chondrogenesis in a model of experimental fracture healing. Bone. (2005) 37:711–9. doi: 10.1016/j.bone.2005.06.013
86. Alkhiary YM, Gerstenfeld LC, Krall E, Westmore M, Sato M, Mitlak BH, et al. Enhancement of experimental fracture-healing by systemic administration of recombinant human parathyroid hormone (PTH 1-34). J Bone Joint Surg Am. (2005) 87:731–41. doi: 10.2106/JBJS.D.02115
87. Lou S, Lv H, Wang G, Zhang L, Li M, Li Z, et al. The effect of teriparatide on fracture healing of osteoporotic patients: A meta-analysis of randomized controlled trials. BioMed Res Int. (2016) 2016:6040379. doi: 10.1155/2016/6040379
88. Gorter EA, Reinders CR, Krijnen P, Appelman-Dijkstra NM, and Schipper IB. The effect of osteoporosis and its treatment on fracture healing a systematic review of animal and clinical studies. Bone Rep. (2021) 15:101117. doi: 10.1016/j.bonr.2021.101117
89. Lanske B, Chandler H, Pierce A, Brown J, Ominsky M, Kostenuik P, et al. Abaloparatide, a PTH receptor agonist with homology to PTHrP, enhances callus bridging and biomechanical properties in rats with femoral fracture. J Orthop Res. (2019) 37:812–20. doi: 10.1002/jor.24254
90. Milstrey A, Wieskoetter B, Hinze D, Grueneweller N, Stange R, Pap T, et al. Dose-dependent effect of parathyroid hormone on fracture healing and bone formation in mice. J Surg Res. (2017) 220:327–35. doi: 10.1016/j.jss.2017.07.019
91. Lin EA, Liu CJ, Monroy A, Khurana S, and Egol KA. Prevention of atrophic nonunion by the systemic administration of parathyroid hormone (PTH 1-34) in an experimental animal model. J Orthop Trauma. (2012) 26:719–23. doi: 10.1097/BOT.0b013e31826f5b9e
92. Huang TW, Chuang PY, Lin SJ, Lee CY, Huang KC, Shih HN, et al. Teriparatide improves fracture healing and early functional recovery in treatment of osteoporotic intertrochanteric fractures. Med (Baltimore). (2016) 95:e3626. doi: 10.1097/MD.0000000000003626
93. Kim SJ, Park HS, Lee DW, and Lee JW. Short-term daily teriparatide improve postoperative functional outcome and fracture healing in unstable intertrochanteric fractures. Injury. (2019) 50:1364–70. doi: 10.1016/j.injury.2019.06.002
94. Peichl P, Holzer LA, Maier R, and Holzer G. Parathyroid hormone 1–84 accelerates fracture-healing in pubic bones of elderly osteoporotic women. J Bone Joint Surg Am. (2011) 93:1583–7. doi: 10.2106/JBJS.J.01379
95. Aspenberg P, Genant HK, Johansson T, Nino AJ, See K, Krohn K, et al. Teriparatide for acceleration of fracture repair in humans: a prospective, randomized, double-blind study of 102 postmenopausal women with distal radial fractures. J Bone Miner Res. (2010) 25:404–14. doi: 10.1359/jbmr.090731
96. Almirol EA, Chi LY, Khurana B, Hurwitz S, Bluman EM, Chiodo C, et al. Short-term effects of teriparatide versus placebo on bone biomarkers, structure, and fracture healing in women with lower-extremity stress fractures: A pilot study. J Clin Transl Endocrinol. (2016) 5:7–14. doi: 10.1016/j.jcte.2016.05.004
97. Chesser TJ, Fox R, Harding K, Halliday R, Barnfield S, Willett K, et al. The administration of intermittent parathyroid hormone affects functional recovery from trochanteric fractured neck of femur: a randomised prospective mixed method pilot study. Bone Joint J. (2016) 98-B:840–5. doi: 10.1302/0301-620X.98B6.36794
98. Mann R, Wigglesworth MJ, and Donnelly D. Ligand-receptor interactions at the parathyroid hormone receptors: subtype binding selectivity is mediated via an interaction between residue 23 on the ligand and residue 41 on the receptor. Mol Pharmacol. (2008) 74:605–13. doi: 10.1124/mol.108.048017
99. Hattersley G, Dean T, Corbin BA, Bahar H, and Gardella TJ. Binding selectivity of abaloparatide for PTH-type-1-receptor conformations and effects on downstream signaling. Endocrinology. (2016) 157:141–9. doi: 10.1210/en.2015-1726
100. Ricarte FR, Le Henaff C, Kolupaeva VG, Gardella TJ, and Partridge NC. Parathyroid hormone(1-34) and its analogs differentially modulate osteoblastic Rankl expression via PKA/SIK2/SIK3 and PP1/PP2A-CRTC3 signaling. J Biol Chem. (2018) 293:20200–13. doi: 10.1074/jbc.RA118.004751
101. Vilardaga JP, Clark LJ, White AD, Sutkeviciute I, Lee JY, Bahar I, et al. Molecular mechanisms of PTH/PTHrP class B GPCR signaling and pharmacological implications. Endocr Rev. (2023) 44:474–91. doi: 10.1210/endrev/bnac032
102. Mosca MJ, He Z, Ricarte FR, Le Henaff C, and Partridge NC. Differential effects of PTH (1-34), PTHrP (1-36), and abaloparatide on the murine osteoblast transcriptome. J Endocr Soc. (2023) 8:bvad156. doi: 10.1210/jendso/bvad156
103. Le Henaff C, Ricarte F, Finnie B, He Z, Johnson J, Warshaw J, et al. Abaloparatide at the same dose has the same effects on bone as PTH (1-34) in mice. J Bone mineral Res. (2020) 35:714–24. doi: 10.1002/jbmr.3930
104. Makino A, Hasegawa T, Takagi H, Takahashi Y, Hase N, Amizuka N, et al. Frequent administration of abaloparatide shows greater gains in bone anabolic window and bone mineral density in mice: A comparison with teriparatide. Bone. (2021) 142:115651. doi: 10.1016/j.bone.2020.115651
105. Bernhardsson M and Aspenberg P. Abaloparatide versus teriparatide: a head to head comparison of effects on fracture healing in mouse models. Acta Orthop. (2018) 89:674–7. doi: 10.1080/17453674.2018.1523771
106. Sahbani K, Cardozo CP, Bauman WA, and Tawfeek HA. Abaloparatide exhibits greater osteoanabolic response and higher cAMP stimulation and beta-arrestin recruitment than teriparatide. Physiol Rep. (2019) 7:e14225. doi: 10.14814/phy2.14225
107. Makino A, Takagi H, Takahashi Y, Hase N, Sugiyama H, Yamana K, et al. Abaloparatide exerts bone anabolic effects with less stimulation of bone resorption-related factors: A comparison with teriparatide. Calcif Tissue Int. (2018) 103:289–97. doi: 10.1007/s00223-018-0422-4
108. Doyle N, Varela A, Haile S, Guldberg R, Kostenuik PJ, Ominsky MS, et al. Abaloparatide, a novel PTH receptor agonist, increased bone mass and strength in ovariectomized cynomolgus monkeys by increasing bone formation without increasing bone resorption. Osteoporos Int. (2018) 29:685–97. doi: 10.1007/s00198-017-4323-6
109. Sato M, Westmore M, Ma YL, Schmidt A, Zeng QQ, Glass EV, et al. Teriparatide [PTH(1-34)] strengthens the proximal femur of ovariectomized nonhuman primates despite increasing porosity. J Bone mineral Res. (2004) 19:623–9. doi: 10.1359/JBMR.040112
110. Fox J, Miller MA, Recker RR, Turner CH, and Smith SY. Effects of treatment of ovariectomized adult rhesus monkeys with parathyroid hormone 1–84 for 16 months on trabecular and cortical bone structure and biomechanical properties of the proximal femur. Calcif Tissue Int. (2007) 81:53–63. doi: 10.1007/s00223-007-9036-y
111. Leder BZ, O'Dea LS, Zanchetta JR, Kumar P, Banks K, McKay K, et al. Effects of abaloparatide, a human parathyroid hormone-related peptide analog, on bone mineral density in postmenopausal women with osteoporosis. J Clin Endocrinol Metab. (2015) 100:697–706. doi: 10.1210/jc.2014-3718
112. Bilezikian JP, Hattersley G, Fitzpatrick LA, Harris AG, Shevroja E, Banks K, et al. Abaloparatide-SC improves trabecular microarchitecture as assessed by trabecular bone score (TBS): a 24-week randomized clinical trial. Osteoporos Int. (2018) 29:323–8. doi: 10.1007/s00198-017-4304-9
113. Hernandez AV, Perez-Lopez FR, Piscoya A, Pasupuleti V, Roman YM, Thota P, et al. Comparative efficacy of bone anabolic therapies in women with postmenopausal osteoporosis: A systematic review and network meta-analysis of randomized controlled trials. Maturitas. (2019) 129:12–22. doi: 10.1016/j.maturitas.2019.08.003
114. Zhou W, Yu L, Fan J, Wan B, Jiang T, Yin J, et al. Endogenous Parathyroid Hormone Promotes Fracture Healing by Increasing Expression of BMPR2 through cAMP/PKA/CREB Pathway in Mice. Cell Physiol Biochem. (2017) 42:551–63. doi: 10.1159/000477605
115. Wang YH, Qiu Y, Han XD, Xiong J, Chen YX, Shi HF, et al. Haploinsufficiency of endogenous parathyroid hormone-related peptide impairs bone fracture healing. Clin Exp Pharmacol Physiol. (2013) 40:715–23. doi: 10.1111/1440-1681.12161
116. Wang M, Nasiri AR, Broadus AE, and Tommasini SM. Periosteal PTHrP regulates cortical bone remodeling during fracture healing. Bone. (2015) 81:104–11. doi: 10.1016/j.bone.2015.07.008
117. Yang M, Arai A, Udagawa N, Zhao L, Nishida D, Murakami K, et al. Parathyroid hormone shifts cell fate of a leptin receptor-marked stromal population from adipogenic to osteoblastic lineage. J Bone Miner Res. (2019) 34:1952–63. doi: 10.1002/jbmr.3811
118. Le PT, Liu H, Alabdulaaly L, Vegting Y, Calle IL, Gori F, et al. The role of Zfp467 in mediating the pro-osteogenic and anti-adipogenic effects on bone and bone marrow niche. Bone. (2021) 144:115832. doi: 10.1016/j.bone.2020.115832
119. Nishida S, Yamaguchi A, Tanizawa T, Endo N, Mashiba T, Uchiyama Y, et al. Increased bone formation by intermittent parathyroid hormone administration is due to the stimulation of proliferation and differentiation of osteoprogenitor cells in bone marrow. Bone. (1994) 15:717–23. doi: 10.1016/8756-3282(94)90322-0
120. Kulkarni NH, Wei T, Kumar A, Dow ER, Stewart TR, Shou J, et al. Changes in osteoblast, chondrocyte, and adipocyte lineages mediate the bone anabolic actions of PTH and small molecule GSK-3 inhibitor. J Cell Biochem. (2007) 102:1504–18. doi: 10.1002/jcb.21374
121. Casado-Diaz A, Dorado G, Giner M, Montoya MJ, Navarro-Valverde C, Diez-Perez A, et al. Proof of concept on functionality improvement of mesenchymal stem-cells, in postmenopausal osteoporotic women treated with teriparatide (PTH1-34), after suffering atypical fractures. Calcif Tissue Int. (2019) 104:631–40. doi: 10.1007/s00223-019-00533-0
122. Wang J, Wang X, Holz JD, Rutkowski T, Wang Y, Zhu Z, et al. Runx1 is critical for PTH-induced onset of mesenchymal progenitor cell chondrogenic differentiation. PloS One. (2013) 8:e74255. doi: 10.1371/journal.pone.0074255
123. Zhao S, Hasegawa T, Hongo H, Yamamoto T, Abe M, Yoshida T, et al. Intermittent PTH administration increases bone-specific blood vessels and surrounding stromal cells in murine long bones. Calcif Tissue Int. (2020). doi: 10.1007/s00223-020-00776-2
124. Jiang X, Xu C, Shi H, and Cheng Q. PTH1–34 improves bone healing by promoting angiogenesis and facilitating MSCs migration and differentiation in a stabilized fracture mouse model. PloS One. (2019) 14:e0226163. doi: 10.1371/journal.pone.0226163
125. Root SH, Wee NKY, Novak S, Rosen CJ, Baron R, Matthews BG, et al. Perivascular osteoprogenitors are associated with transcortical channels of long bones. Stem Cells. (2020) 38:769–81. doi: 10.1002/stem.3159
126. Zhao S, Hasegawa T, Hongo H, Yamamoto T, Abe M, Yoshida T, et al. Intermittent PTH administration increases bone-specific blood vessels and surrounding stromal cells in murine long bones. Calcif Tissue Int. (2021) 108:391–406. doi: 10.1007/s00223-020-00776-2
127. Zhao C, Irie N, Takada Y, Shimoda K, Miyamoto T, Nishiwaki T, et al. Bidirectional ephrinB2-EphB4 signaling controls bone homeostasis. Cell Metab. (2006) 4:111–21. doi: 10.1016/j.cmet.2006.05.012
128. Liu H, Liu L, and Rosen CJ. PTH and the regulation of mesenchymal cells within the bone marrow niche. Cells. (2024) 13. doi: 10.3390/cells13050406
129. Kao R, Lu W, Louie A, and Nissenson R. Cyclic AMP signaling in bone marrow stromal cells has reciprocal effects on the ability of mesenchymal stem cells to differentiate into mature osteoblasts versus mature adipocytes. Endocrine. (2012) 42:622–36. doi: 10.1007/s12020-012-9717-9
130. Chen B, Lin T, Yang X, Li Y, Xie D, Cui H, et al. Intermittent parathyroid hormone (1-34) application regulates cAMP-response element binding protein activity to promote the proliferation and osteogenic differentiation of bone mesenchymal stromal cells, via the cAMP/PKA signaling pathway. Exp Ther Med. (2016) 11:2399–406. doi: 10.3892/etm.2016.3177
131. Kulebyakin K, Tyurin-Kuzmin P, Sozaeva L, Voloshin N, Nikolaev M, Chechekhin V, et al. Dynamic balance between PTH1R-dependent signal cascades determines its pro- or anti-osteogenic effects on MSC. Cells. (2022) 11. doi: 10.3390/cells11213519
132. Kuo SW, Rimando MG, Liu YS, and Lee OK. Intermittent administration of parathyroid hormone 1–34 enhances osteogenesis of human mesenchymal stem cells by regulating protein kinase cdelta. Int J Mol Sci. (2017) 18. doi: 10.3390/ijms18102221
133. Wein MN, Foretz M, Fisher DE, Xavier RJ, and Kronenberg HM. Salt-inducible kinases: physiology, regulation by cAMP, and therapeutic potential. Trends Endocrinol Metab. (2018) 29:723–35. doi: 10.1016/j.tem.2018.08.004
134. Pellicelli M, Miller JA, Arabian A, Gauthier C, Akhouayri O, Wu JY, et al. The PTH-Galphas-protein kinase A cascade controls alphaNAC localization to regulate bone mass. Mol Cell Biol. (2014) 34:1622–33. doi: 10.1128/MCB.01434-13
135. Pellicelli M, Hariri H, Miller JA, and St-Arnaud R. Lrp6 is a target of the PTH-activated alphaNAC transcriptional coregulator. Biochim Biophys Acta Gene Regul Mech. (2018) 1861:61–71. doi: 10.1016/j.bbagrm.2018.01.008
136. Yukata K, Xie C, Li TF, Takahata M, Hoak D, Kondabolu S, et al. Aging periosteal progenitor cells have reduced regenerative responsiveness to bone injury and to the anabolic actions of PTH 1–34 treatment. Bone. (2014) 62:79–89. doi: 10.1016/j.bone.2014.02.002
137. Ono N, Nakashima K, Schipani E, Hayata T, Ezura Y, Soma K, et al. Constitutively active PTH/PTHrP receptor specifically expressed in osteoblasts enhances bone formation induced by bone marrow ablation. J Cell Physiol. (2012) 227:408–15. doi: 10.1002/jcp.22986
138. Guo J, Liu M, Yang D, Bouxsein ML, Saito H, Galvin RJ, et al. Suppression of Wnt signaling by Dkk1 attenuates PTH-mediated stromal cell response and new bone formation. Cell Metab. (2010) 11:161–71. doi: 10.1016/j.cmet.2009.12.007
139. Uyama M, Kawanami M, and Tamura M. Wasf2: a novel target of intermittent parathyroid hormone administration. Int J Mol Med. (2013) 31:1243–7. doi: 10.3892/ijmm.2013.1315
140. Kiryaman G, Enabulele I, Banville ML, and Divieti Pajevic P. The evolving role of PTH signaling in osteocytes. Endocrinology. (2025) 166. doi: 10.1210/endocr/bqaf034
141. Li JY, Walker LD, Tyagi AM, Adams J, Weitzmann MN, Pacifici R, et al. The sclerostin-independent bone anabolic activity of intermittent PTH treatment is mediated by T-cell-produced Wnt10b. J Bone Miner Res. (2014) 29:43–54. doi: 10.1002/jbmr.2044
142. Li C, Xing Q, Yu B, Xie H, Wang W, Shi C, et al. Disruption of LRP6 in osteoblasts blunts the bone anabolic activity of PTH. J Bone Miner Res. (2013) 28:2094–108. doi: 10.1002/jbmr.1962
143. Revollo L, Kading J, Jeong SY, Li J, Salazar V, Mbalaviele G, et al. N-cadherin restrains PTH activation of Lrp6/beta-catenin signaling and osteoanabolic action. J Bone Miner Res. (2015) 30:274–85. doi: 10.1002/jbmr.2323
144. Ogita M, Rached MT, Dworakowski E, Bilezikian JP, and Kousteni S. Differentiation and proliferation of periosteal osteoblast progenitors are differentially regulated by estrogens and intermittent parathyroid hormone administration. Endocrinology. (2008) 149:5713–23. doi: 10.1210/en.2008-0369
145. Yu B, Zhao X, Yang C, Crane J, Xian L, Lu W, et al. Parathyroid hormone induces differentiation of mesenchymal stromal/stem cells by enhancing bone morphogenetic protein signaling. J Bone Miner Res. (2012) 27:2001–14. doi: 10.1002/jbmr.1663
146. Pfeilschifter J, Laukhuf F, Muller-Beckmann B, Blum WF, Pfister T, Ziegler R, et al. Parathyroid hormone increases the concentration of insulin-like growth factor-I and transforming growth factor beta 1 in rat bone. J Clin Invest. (1995) 96:767–74. doi: 10.1172/JCI118121
147. Canalis E, Centrella M, Burch W, and McCarthy TL. Insulin-like growth factor I mediates selective anabolic effects of parathyroid hormone in bone cultures. J Clin Invest. (1989) 83:60–5. doi: 10.1172/JCI113885
148. Bikle DD, Sakata T, Leary C, Elalieh H, Ginzinger D, Rosen CJ, et al. Insulin-like growth factor I is required for the anabolic actions of parathyroid hormone on mouse bone. J Bone mineral Res. (2002) 17:1570–8. doi: 10.1359/jbmr.2002.17.9.1570
149. Wang Y, Nishida S, Boudignon BM, Burghardt A, Elalieh HZ, Hamilton MM, et al. IGF-I receptor is required for the anabolic actions of parathyroid hormone on bone. J Bone mineral Res. (2007) 22:1329–37. doi: 10.1359/jbmr.070517
150. Nixon AJ, Goodrich LR, Scimeca MS, Witte TH, Schnabel LV, Watts AE, et al. Gene therapy in musculoskeletal repair. Ann N Y Acad Sci. (2007) 1117:310–27. doi: 10.1196/annals.1402.065
151. Schipani E, Maes C, Carmeliet G, and Semenza GL. Regulation of osteogenesis-angiogenesis coupling by HIFs and VEGF. J Bone mineral Res. (2009) 24:1347–53. doi: 10.1359/jbmr.090602
152. Bikle D, Majumdar S, Laib A, Powell-Braxton L, Rosen C, Beamer W, et al. The skeletal structure of insulin-like growth factor I-deficient mice. J Bone mineral Res. (2001) 16:2320–9. doi: 10.1359/jbmr.2001.16.12.2320
153. Wang Y, Nishida S, Elalieh HZ, Long RK, Halloran BP, Bikle DD, et al. Role of IGF-I signaling in regulating osteoclastogenesis. J Bone mineral Res. (2006) 21:1350–8. doi: 10.1359/jbmr.060610
154. Wang Y, Nishida S, Sakata T, Elalieh HZ, Chang W, Halloran BP, et al. Insulin-like growth factor-I is essential for embryonic bone development. Endocrinology. (2006) 147:4753–61. doi: 10.1210/en.2006-0196
155. Hill PA, Reynolds JJ, and Meikle MC. Osteoblasts mediate insulin-like growth factor-I and -II stimulation of osteoclast formation and function. Endocrinology. (1995) 136:124–31. doi: 10.1210/endo.136.1.7828521
156. Rubin J, Ackert-Bicknell CL, Zhu L, Fan X, Murphy TC, Nanes MS, et al. IGF-I regulates osteoprotegerin (OPG) and receptor activator of nuclear factor-kappaB ligand in vitro and OPG in vivo. J Clin Endocrinol Metab. (2002) 87:4273–9. doi: 10.1210/jc.2002-020656
157. Dimitriou R, Tsiridis E, and Giannoudis PV. Current concepts of molecular aspects of bone healing. Injury. (2005) 36:1392–404. doi: 10.1016/j.injury.2005.07.019
158. Wildemann B, Schmidmaier G, Brenner N, Huning M, Stange R, Haas NP, et al. Quantification, localization, and expression of IGF-I and TGF-beta1 during growth factor-stimulated fracture healing. Calcif Tissue Int. (2004) 74:388–97. doi: 10.1007/s00223-003-0117-2
159. Shi Y, Liao X, Long JY, Yao L, Chen J, Yin B, et al. Gli1(+) progenitors mediate bone anabolic function of teriparatide via Hh and Igf signaling. Cell Rep. (2021) 36:109542. doi: 10.1016/j.celrep.2021.109542
160. Wang HU, Chen ZF, and Anderson DJ. Molecular distinction and angiogenic interaction between embryonic arteries and veins revealed by ephrin-B2 and its receptor Eph-B4. Cell. (1998) 93:741–53. doi: 10.1016/s0092-8674(00)81436-1
161. Sawamiphak S, Seidel S, Essmann CL, Wilkinson GA, Pitulescu ME, Acker T, et al. Ephrin-B2 regulates VEGFR2 function in developmental and tumour angiogenesis. Nature. (2010) 465:487–91. doi: 10.1038/nature08995
162. Wang Y, Nakayama M, Pitulescu ME, Schmidt TS, Bochenek ML, Sakakibara A, et al. Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature. (2010) 465:483–6. doi: 10.1038/nature09002
163. Arthur A, Zannettino A, Panagopoulos R, Koblar SA, Sims NA, Stylianou C, et al. EphB/ephrin-B interactions mediate human MSC attachment, migration and osteochondral differentiation. Bone. (2011) 48:533–42. doi: 10.1016/j.bone.2010.10.180
164. Tonna S, Takyar FM, Vrahnas C, Crimeen-Irwin B, Ho PW, Poulton IJ, et al. EphrinB2 signaling in osteoblasts promotes bone mineralization by preventing apoptosis. FASEB J. (2014) 28:4482–96. doi: 10.1096/fj.14-254300
Keywords: parathyroid hormone, fracture, skeletal stem cells, cellular signaling, abaloparatide, muscle stem cells
Citation: Bikle D (2025) Fracture healing: cellular mechanisms and impact of parathyroid hormone and its analogs. Front. Endocrinol. 16:1703129. doi: 10.3389/fendo.2025.1703129
Received: 10 September 2025; Accepted: 20 October 2025;
Published: 17 November 2025.
Edited by:
Antonio Desmond McCarthy, National University of La Plata, ArgentinaReviewed by:
Maria Angelica Rivoira, Universidad Nacional de Córdoba, ArgentinaSilvina Mastaglia, University of Buenos Aires, Argentina
Copyright © 2025 Bikle. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniel Bikle, RGFuaWVsLmJpa2xlQHVjc2YuZWR1