 Nilda Gallardo
Nilda Gallardo Sara Artigas-Jerónimo
Sara Artigas-Jerónimo Lorena Mazuecos
Lorena Mazuecos Antonio Andrés
Antonio Andrés- 1Biochemistry Section, Faculty of Sciences and Chemical Technologies, University of Castilla-La Mancha, Ciudad Real, Spain
- 2DOE Research Group, Institute of Biomedicine, IDISCAM, Ciudad Real, Spain
Glucose is vital for brain physiological function, acting as the primary energy source that supports cognitive processes, neurotransmitter production, and overall health. The brain requires a constant supply of glucose, and the body has evolved protective mechanisms to maintain this supply during hypoglycemia. Increased appetite and food intake is a fundamental protective response. The precise network of brain regions, nerves, and connections responsible for initiating and coordinating these responses has not been fully identified or mapped. Neuroendocrine centers within the hypothalamus and brainstem monitor metabolic signals such as glucose, insulin, and leptin to regulate autonomic outflow, endocrine function, and behavior. Disruption of these central regulatory circuits contributes significantly to the pathogenesis of metabolic disorders, including obesity and type 2 diabetes mellitus (T2DM). Interestingly, incretin-based pharmacotherapies and bariatric surgery suppress food intake by acting on the brain, thereby enhancing the regulation of glucose homeostasis. This review summarizes current knowledge on the neural and hormonal pathways, including incretin signaling, involved in physiological glucose regulation, the mechanisms underlying their dysfunction in disease states, and the recent advances pointing to potential central targets for therapeutic intervention.
1 Introduction
Glucose is the primary energy substrate for most tissues and the almost exclusive fuel for the brain in both rodent and human. As noted by Mergenthaler et al. (2013) (1), the mammalian brain depends on glucose to sustain neuronal activity, neurotransmitter synthesis, and overall cognitive function. Thus, precise control of glucose metabolism is essential for brain and systemic health. To ensure a constant energy supply, plasma glucose concentrations are tightly regulated through highly coordinated mechanisms, a process known as glucose homeostasis (1, 2).
Traditionally, glucose homeostasis has been attributed to the balance between insulin and glucagon secretion by the pancreas (2). However, growing evidence highlights a pivotal role of the central nervous system (CNS) in sensing metabolic status and actively orchestrating systemic glucose regulation, shifting the paradigm from a passive to an integrative control system (3, 4). Key determinants of glucose homeostasis, such as insulin and glucagon secretion, peripheral insulin sensitivity, and glucose utilization, are under central modulation by the brain.
Within the brain, the hypothalamus and brainstem act as neuroendocrine hubs that integrate hormonal, nutritional, and neural signals to regulate feeding, autonomic tone, and endocrine outputs. Specialized hypothalamic glucose-sensing neurons detect changes in glucose availability through mechanisms shared with pancreatic β-cells and translate this information into adaptive autonomic and behavioral responses to maintain glucose stability. Through reciprocal connections with other brain regions, the hypothalamus and brainstem coordinate food intake, thermogenesis, and systemic energy balance.
Disruption of these regulatory circuits is increasingly recognized as a key contributor to metabolic disease. Central insulin and leptin resistance, hypothalamic inflammation, and impaired brainstem autonomic control are implicated in the pathogenesis of obesity and T2DM (4). Importantly, these alterations often precede overt hyperglycemia, suggesting that neuroendocrine dysfunction constitutes an early, causal step in metabolic deterioration (5) and may accelerate brain aging (6, 7) in both rodents and humans. Diet-induced hypothalamic stress, particularly from high-fat diets, further amplifies inflammatory and oxidative pathways, exacerbating both metabolic and cognitive decline.
Understanding how the hypothalamus and brainstem sense and integrate metabolic signals is therefore essential for elucidating the mechanisms underlying metabolic disease and identifying new therapeutic strategies. In this review, we summarize current insights into the neuroendocrine control of glucose homeostasis, discuss mechanisms of central dysfunction in metabolic disease, and highlight emerging therapies targeting brain pathways to restore systemic glucose balance. Much of the available evidence comes from rodent studies, as functional validation in humans remains limited.
2 Homeostatic control of glucose levels by the CNS
2.1 Hypothalamic regulation of glucose homeostasis
The core mechanisms of hypothalamic regulation of glucose homeostasis are conserved across mammals. The hypothalamus, particularly the arcuate nucleus (ARC), ventromedial (VMH), dorsomedial (DMH), and paraventricular (PVN) nuclei, plays a central role in regulating energy balance by sensing energy stores, metabolic signals, and nutrient availability. Adiposity signals such as insulin and leptin convey information about long-term energy sufficiency (8, 9), whereas gut-derived hormones including ghrelin, peptide YY (PYY), cholecystokinin (CKK), gastric inhibitory polypeptide (GIP), and glucagon-like peptides (GLP-1/GLP-2) reflect short-term changes in nutrient status and dietary intake across the life course (10–13).
Moreover, circulating nutrients (glucose, free fatty acids) and metabolites provide additional cues reflecting shifts in metabolic and environmental conditions. In addition, the biological clock within the hypothalamus also influences daily rhythms of glucose metabolism, with disrupted clock gene expression being closely associated with insulin resistance and type 2 diabetes (14).
Distinct hypothalamic neuronal populations in the ARC sense these signals and coordinate autonomic and neuroendocrine outputs to regulate feeding, energy expenditure, and glucose metabolism (4, 15). The proopiomelanocortin (POMC) and agouti-related peptide/neuropeptide Y (AgRP/NPY) neurons are two well-characterized populations that exert opposing influences on energy and glucose homeostasis. POMC neurons are anorexigenic and glucose-excited, promoting insulin sensitivity and glucose uptake when glucose and nutrients are abundant (16, 17). Conversely, AgRP/NPY neurons are orexigenic and glucose-inhibited; they become more active when blood glucose levels are low and ghrelin is high, stimulating hepatic glucose production to restore normoglycemia (18, 19).
Elegant studies using optogenetic and chemogenetic tools have confirmed, in mice, the dominant role of AgRP/NPY neurons in driving feeding behavior and acute metabolic adaptations during fasting and hypoglycemia (18, 20). In contrast, hypothalamic POMC neurons exert slower but more sustained effects, particularly in modulating sympathetic tone and energy expenditure (17). Nevertheless, the POMC-Melanocortin 4 Receptor (MC4R) circuit is essential for the body’s ability to counteract hypoglycemia, as demonstrated by studies showing that POMC or MC4R deficiency impairs glucagon secretion and hepatic glucose production in diabetic mice (21).
Building on this framework, recent evidence indicates that the functional interplay between orexigenic AgRP/NPY and anorexigenic POMC neurons extends beyond appetite control to encompass systemic glucose regulation. According to De Solis et al., simultaneous activation of AgRP/NPY neurons and inhibition of POMC neurons exert additive effects on feeding behavior yet produce distinct outcomes on systemic insulin sensitivity and hepatic gluconeogenesis compared with their independent modulation (22). Notably, this cooperative effect on feeding is not fully maintained in female mice. These observations suggest that hypothalamic circuits controlling energy balance operate through sex-dependent coordination between orexigenic and anorexigenic neurons. Such differences may contribute to the sexually dimorphic regulation of glucose homeostasis and the variable susceptibility to metabolic disorders observed across sexes.
Moreover, AgRP/NPY and POMC neurons in the ARC project to the VMH and DMH hypothalamic nuclei, as well as to second-order neurons in the PVN and extra-hypothalamic regions such as the brainstem. Through these connections, they influence both autonomic outflow and behavior, thereby coordinating adaptive responses in feeding, energy expenditure, and glucose homeostasis (20, 22).
The VMH and PVN contain specialized neurons that sense glucose, fatty acids, insulin, and leptin, integrating intrinsic detection with peripheral signals to monitor whole-body energy status. Within the VMH, glucose-excited (GE) neurons respond to postprandial hyperglycemia, while glucose-inhibited (GI) neurons detect hypoglycemia and initiate counterregulatory responses. Their activity is governed by mitochondrial ATP production and redox state, emphasizing the critical role of cellular energy metabolism in hypothalamic glucose sensing (23, 24).
Ultimately, PVN neurons modulate sympathetic and parasympathetic activity to regulate hepatic glucose production, glycogen storage, and systemic glucose availability, acting as a key effector that connects hypothalamic sensing to peripheral glucose homeostasis (25). Hypothalamic nuclei, including the PVH, LH, and ARC, connect extensively with brainstem and spinal autonomic centers, such as the nucleus of the solitary tract (NTS), dorsal motor nucleus of the vagus (DMV) and the intermediolateral nucleus of the spinal cord (IML), which relay hypothalamic signals to pancreatic islets, liver, and other metabolic tissues. Through these circuits, the hypothalamus regulates sympathetic and parasympathetic outputs, controlling pancreatic function, adipose storage, thermogenesis, and overall glucose homeostasis thereby closing the loop between central sensing and peripheral control of glucose.
2.2 Central signaling: insulin and leptin crosstalk
Insulin and leptin play a crucial role in the central regulation of glucose homeostasis, by informing the brain of the energy status of the body. In adulthood, both hormones act on hypothalamic neurons that control food intake, energy expenditure and glucose metabolism via shared intracellular pathways, including PI3K–Akt and STAT3 signaling cascades. These pathways suppress hepatic glucose production, enhance peripheral insulin sensitivity, and regulate food intake. Disruption of insulin–leptin crosstalk contributes to central insulin and leptin resistance, a hallmark of obesity and type 2 diabetes (15, 26).
In addition, leptin activates hypothalamic circuits that regulate glucose homeostasis via the autonomic nervous system, modulating pancreatic insulin and glucagon secretion and influencing liver, brown adipose tissue, and skeletal muscle metabolism. Remarkably, leptin signaling in POMC neurons can normalize glycemia, increase physical activity, and prevent diabetes, independently of food intake or body weight (17). Importantly, leptin can also improve glucose control even under insulin-deficient conditions (27).
Together, insulin and leptin act as complementary neuroendocrine signals that coordinate central control of energy balance and glucose metabolism, highlighting their synergistic role in maintaining systemic metabolic homeostasis. Consistent with animal studies, human and translational data indicate that perinatal perturbations in leptin and insulin signaling can reprogramming the hypothalamic control of glucose metabolism (28).
2.3 Developmental programming of the hypothalamus and its early-life sensitivity
Beyond their role in adulthood, AgRP and POMC neurons undergo a critical developmental window that shapes lifelong metabolic regulation, including the regulation of glucose homeostasis. This period is regulated by perineuronal nets (PNNs), specialized extracellular matrix structures that envelop leptin receptor-positive GABAergic neurons within the ARC, limiting plasticity but ensuring the stability of this inhibitory neuronal circuit throughout adulthood in humans and rodents (29). Although the timing of hypothalamic maturation differs between species, postnatal in rodents versus prenatal in humans, the concept of a developmentally restricted period of metabolic sensitivity appears conserved.
During this period, leptin acts as a neurotrophic factor, promoting the outgrowth of AgRP projections from the ARC to downstream targets, including the PVN and DMH (30, 31). This neurotrophic effect is time-limited: in leptin-deficient (ob/ob) mice, neonatal leptin replacement can largely restore AgRP projections, whereas treatment after postnatal day 28 fails to rescue circuit development. Moreover, the recovery of PVN projections is more robustly in females than in males. All of these indicate that leptin’s trophic effects are restricted to the developmental critical period for neuronal maturation (29, 32).
PNNs are also influenced by dietary interventions. Maternal high-fat diet during lactation (MHFD-L) disrupts PNNs maturation and enhances microglial activity in hypothalamic nuclei, leading to active engulfment of AgRP terminals within the PVN. This microglial activity reduces AgRP innervation density and remodels hypothalamic circuits, programming higher body weight and increased susceptibility to obesity and glucose dysregulation later in life (33, 34). Notably, microglial depletion during this postnatal period prevents both the reduction in PVN AgRP projections and the excessive weight gain observed in MHFD-L offspring, underscoring microglia as critical mediators of this developmental programming (29) (Figure 1).
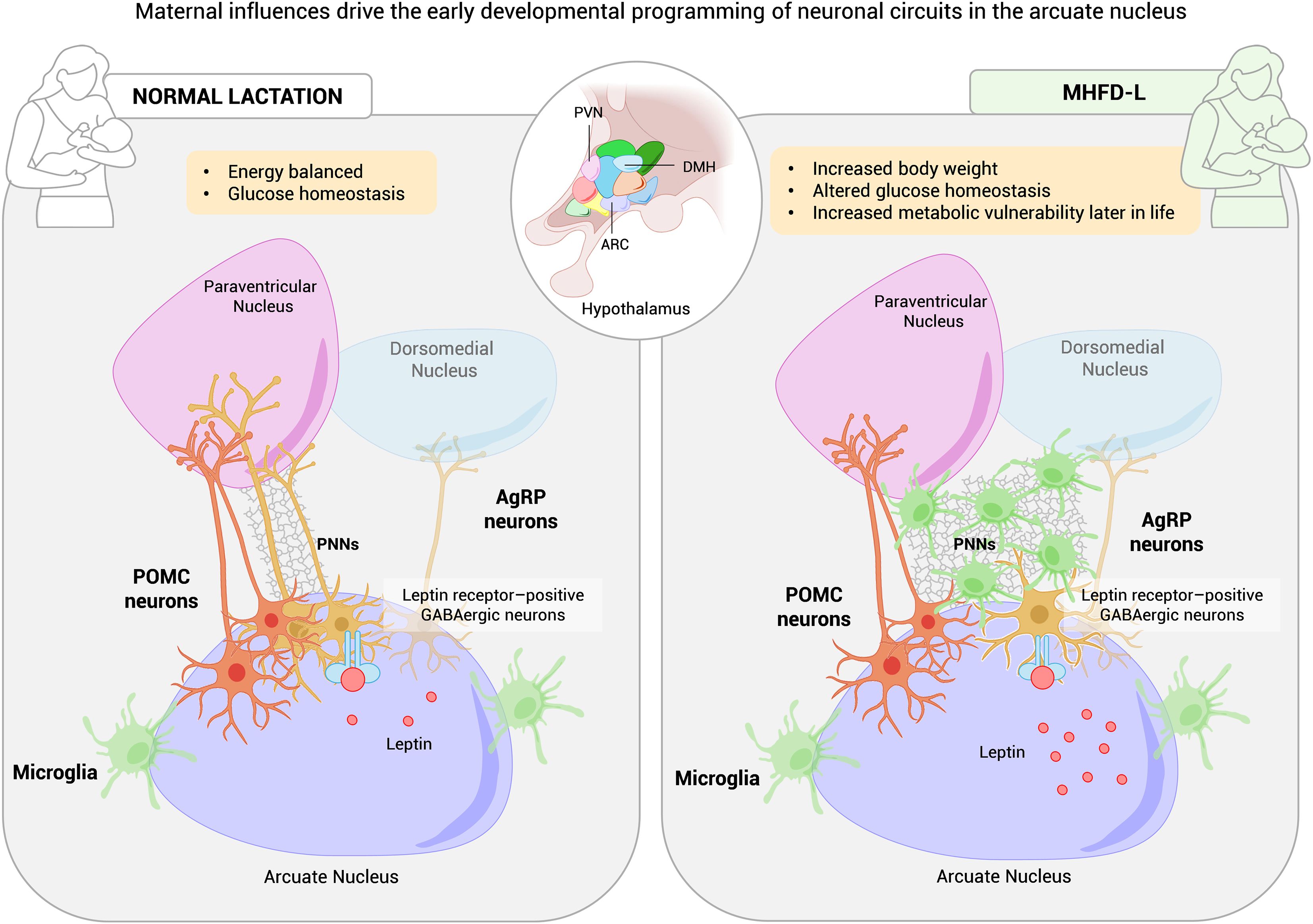
Figure 1. Impact of maternal high-fat diet on perineuronal nets (PNNs) and hypothalamic circuit plasticity controlling body weight and glucose metabolism. Schematic representation of how maternal exposure to a high-fat diet (HFD) alters the formation and remodeling of perineuronal nets (PNNs) in the arcuate nucleus (ARC) of offspring during the critical period of neuronal maturation. In both humans and rodents, maternal HFD during lactation (MHFD-L) increases perinatal leptin levels, disrupts PNN organization, and activates microglia, leading to degradation of AgRP neuronal terminals in the paraventricular nucleus (PVN). The resulting reduction in PVN-directed AgRP projections may predispose offspring to excessive weight gain and increased susceptibility to obesity and glucose dysregulation later in life. MHFD-L, maternal high-fat diet during lactation; Violet, ARC; Rose, PVN; Blue, DMH; Green, microglia; Orange, POMC neurons; Yellow, AgRP/NPY neurons.
Nutritional environment, and microglial activity converge to shape hypothalamic melanocortin circuits, with enduring effects on feeding behavior, energy expenditure, and glucose homeostasis. This developmental plasticity represents both a window of vulnerability, where adverse nutritional or hormonal cues can program future metabolic disease by impairing hypothalamic responses to insulin and leptin (33, 35), and a window of opportunity for intervention. Notably, this vulnerability appears to be more pronounced in human males. Although it remains unclear how PNNs themselves modulate blood glucose levels, establishing timely strategies aimed at normalizing circuit development during neuronal maturation could reduce the lifelong risk of obesity and type 2 diabetes.
2.4 Incretin mimetics in the central control of glycemia
Incretin mimetics such as GLP-1 receptor agonists (GLP-1RAs) are highly effective antidiabetic agents that enhance insulin secretion, improve glycemic control, and promote sustained weight loss. Recent findings show that both GLP-1R agonists, such as semaglutide, and bariatric surgery converge on central circuits controlling energy balance, although the neural mechanisms of surgery remain less defined. Beyond their potent anorectic actions, these interventions activate vagal afferents, brainstem nuclei like the NTS, and hypothalamic melanocortin pathways to modulate food intake and glucose homeostasis (36).
GLP-1 receptors are broadly distributed in the hypothalamus, brainstem, and mesolimbic system, where they exert region-specific effects on satiety, reward, and glycemic regulation. Preproglucagon (PPG) neurons in the NTS represent the main source of endogenous GLP-1; their activation suppresses feeding and modulates autonomic output. However, pharmacological GLP-1RAs act partly independently of these neurons, indicating that endogenous and therapeutic GLP-1 engage distinct yet overlapping gut-brain pathways (37, 38).
Recent studies identified glucose-sensing GLP-1R neurons in the DMH that lower blood glucose by enhancing vagal output to the pancreas through cAMP–PKA signaling (39). Optogenetic and imaging approaches further show that GLP-1R-expressing neurons in the ARC, PVN, and DMH nuclei inhibit AgRP/NPY neurons, providing a direct mechanism through which central GLP-1 signaling reduces hunger and improves glycemic control (40, 41). Together, these findings delineate a hierarchical GLP-1 network that integrates peripheral and central inputs to coordinate satiation, energy expenditure, and autonomic output.
Neuroimaging and preclinical data support a model where endogenous and pharmacological GLP-1 actions overlap but are not identical, involving parallel gut-brain loops. This dual signaling framework explains the remarkable efficacy of GLP-1RAs in producing durable metabolic improvements across species (42).
Building on these mechanisms, dual and triple incretin receptor agonists represent a major advance in metabolic therapy. Dual GLP-1/GIP agonists such as tirzepatide, and triple agonists targeting GLP-1, GIP, and glucagon receptors such as retatrutide, integrate complementary pathways to optimize glucose control, enhance fat oxidation and thermogenesis, and improve cardiovascular outcomes (43). These next-generation incretin agonists harness synergistic central and peripheral effects to achieve superior glycemic and weight benefits compared to single GLP-1 agonists.
Importantly, evidence from human and preclinical studies indicates sex-dependent differences in incretin responsiveness. Enhanced GLP-1R expression in female brain regions involved in aversive and reward processing, along with estrogen-dependent modulation of GLP-1 signaling, may underlie greater treatment efficacy and tolerability in females (40). These findings highlight the need to integrate sex as a biological variable in future research.
Altogether, the emerging picture places GLP-1 not only as a gut-derived hormone but as a central neuropeptide coordinating satiety, autonomic regulation, and glucose homeostasis. Understanding how endogenous and pharmacological incretin pathways interact, across neural circuits, metabolic organs, and sex-specific contexts, will be crucial to developing the next generation of personalized anti-obesity and antidiabetic therapies.
3 Brainstem and autonomic regulation of glucose homeostasis: insights into the gut–brain-liver axis
While the hypothalamus is a central hub for energy balance and glucose regulation, the brainstem provides essential autonomic control that maintains glucose homeostasis within narrow physiological limits (44, 45). Key regions in the brainstem include the dorsal vagal complex (DVC), comprising the nucleus of the solitary tract (NTS), dorsal motor nucleus of the vagus (DMV), and area postrema (AP), as well as the rostral ventrolateral medulla (RVLM), which together integrate visceral and hormonal signals and coordinate autonomic outputs to regulate pancreatic hormone secretion, hepatic glucose production, and peripheral glucose utilization (Figure 2).
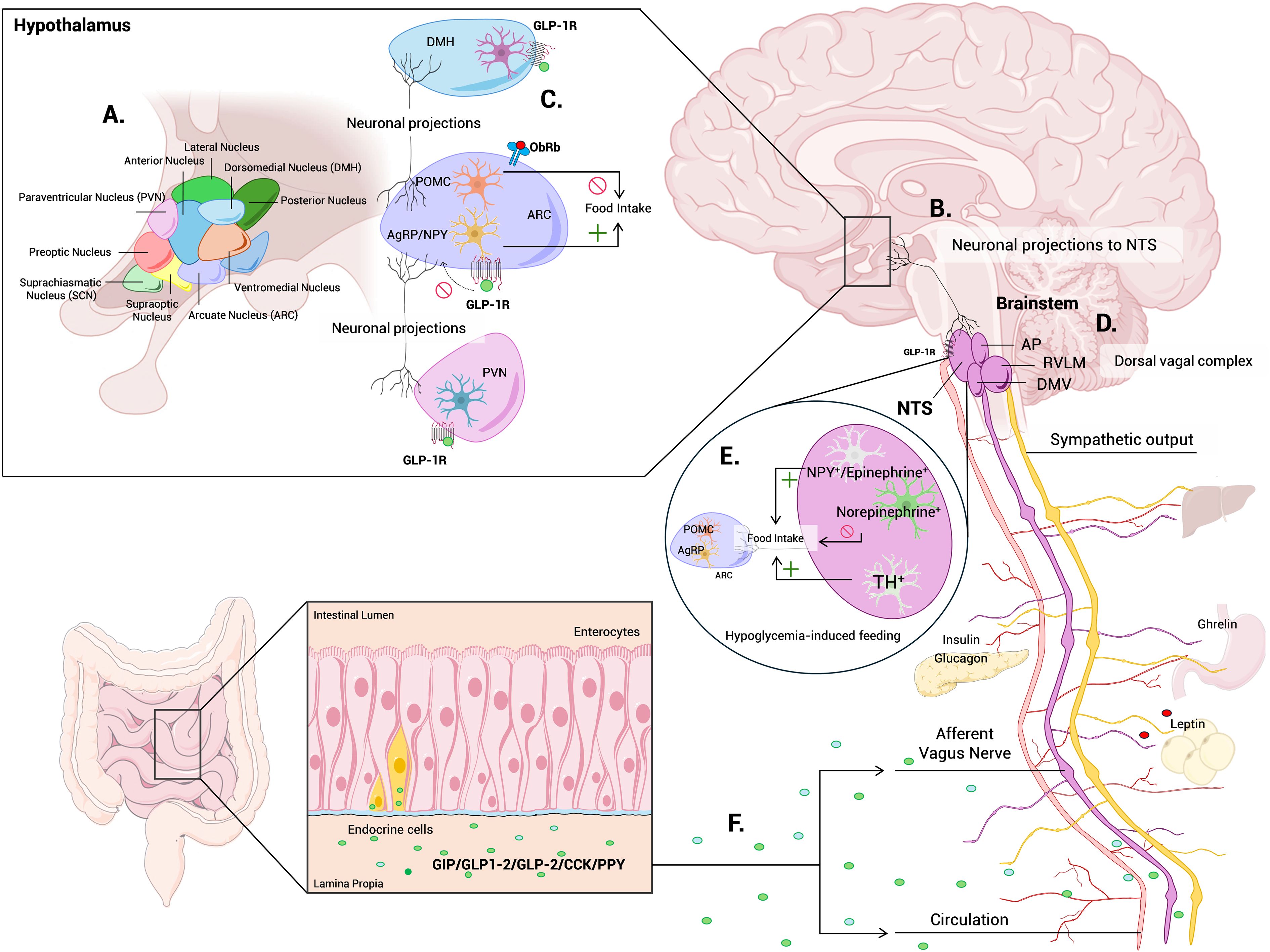
Figure 2. Neuroendocrine networks integrating hypothalamic and brainstem mechanisms of glucose homeostasis. Schematic overview summarizing the central circuits that coordinate glucose homeostasis through the hypothalamus-brainstem axis. (A) The ARC, DMH, VMH, LH, and PVN integrate hormonal and nutrient-derived signals via AgRP/NPY, POMC, and glucose-sensing neurons to regulate energy balance. (B) PVN projections to the NTS modulate autonomic activity controlling hepatic glucose output, glycogen storage and systemic glucose availability. (C) GLP-1 receptor-expressing neurons in the DMH and brainstem enhance hypothalamic-brainstem communication, promoting satiation and glucose control. (D) NTS, DMV, AP and RVLM areas in the brainstem provides essential autonomic control that maintains glucose homeostasis. (E) An ascending brainstem-hypothalamus pathway selectively drives appetite under energy deficit and hunger cues, in addition to its classical satiety function. (F) The gut-liver-brain axis contributes via vagal afferents, gut peptides (GLP-1, GIP, CCK, PYY), and microbiota-derived metabolites to modulate enteroendocrine signaling and neural pathways. ARC, arcuate nucleus; DMH; dorsomedial nucleus of hypothalamus; VMH; ventromedial nucleus of hypothalamus; LH, lateral hypothalamic area; PVN, paraventricular nucleus of hypothalamus; AgRP/NPY, agouti related peptide/neuropeptide Y neurons; POMC, pro-opiomelanocortin neurons; NTS, nucleus tractus solitaries; DMV, dorsal motor nucleus of the vagus; AP, area postrema; RVLM, rostral ventrolateral medulla; GLP-1, glucagon-like peptide 1; GIP, gastric inhibitory polypeptide; CCK, cholecystokinin; PYY, peptide YY; Violet, ARC; Rose, PVN; Blue, DMH; Green, microglia; Orange, POMC neurons; Yellow, AgRP/NPY neurons.
The vagus nerve contains the primary sensory neurons that monitor the gastrointestinal environment and transmit this information to the brainstem. The NTS serves as a major integrative center for visceral afferents from the gastrointestinal tract, liver, and pancreas. Vagal and sympathetic afferents from the gut synapse in the NTS, where neuronal activity modulates insulin secretion and hepatic glucose output (46, 47).
Recent advances have refined our understanding of gut-brain communication as a central regulator of metabolic homeostasis. Using intersectional genetic approaches, Borgmann et al., demonstrated that distinct populations of gut-innervating vagal afferents exert differential control over food intake and glucose metabolism by engaging separate neural circuits in the brain (48). Consistently, activation of intestinal mechanoreceptors during feeding generates satiety signals transmitted via vagal afferents to the nucleus tractus solitarius (NTS), where they inhibit hypothalamic AgRP/NPY neurons and activate anorexigenic pathways (49). By modulating autonomic balance-reducing sympathetic drive and enhancing parasympathetic signaling-gut-derived sensory inputs lower hepatic glucose output and improve insulin sensitivity. These findings highlight vagal sensory pathways as key integrators of intestinal, hepatic, and hypothalamic signals in the neurohormonal control of glucose homeostasis (Figure 2).
Within this framework, catecholaminergic circuits in the NTS integrate visceral afferent inputs to adapt feeding and glucose metabolism to changing energy states. Epinephrine/NPY-expressing neurons promote feeding, norepinephrine neurons suppress it, and a subset of tyrosine hydroxylase–expressing neurons drives glucoprivic feeding through hypothalamic AgRP and POMC neurons, revealing an orexigenic role for the vagal-NTS circuit beyond its classical satiety function (50–53) (Figure 2).
The NTS also receives descending projections from hypothalamic PVN, LH, and ARC neurons, allowing bidirectional communication that integrates circulating signals such as glucose, ghrelin, GLP-1, and leptin to coordinate autonomic and neuroendocrine regulation. Parallel brainstem centers, including the RVLM and the area postrema, contribute to sympathetic outflow that promotes hepatic gluconeogenesis, inhibits insulin secretion, and regulates feeding and lipolysis. Collectively, hypothalamic and brainstem neurons, together with sympathetic preganglionic circuits, form an integrated network that coordinates feeding behavior and glucose balance, whose dysfunction contributes to impaired hypoglycemia awareness, hyperglycemia in type 2 diabetes, and hormonal resistance (54–60).
The gut microbiota interfaces with the gut-liver-brain axis through bidirectional communication encompassing neural, immune, and endocrine pathways. This network involves vagal signaling, microbial metabolites such as short-chain fatty acids (SCFAs) and bile acids that influence host physiology, and immune responses elicited by microbial byproducts like lipopolysaccharides (LPS) reaching the liver and brain. While these interactions are essential for maintaining metabolic homeostasis, microbial dysbiosis can disrupt this equilibrium, promoting systemic inflammation, hepatic insulin resistance, and neuroendocrine dysfunction (61).
Future studies should further elucidate how the gut microbiota shapes neural, endocrine, and metabolic control within this interconnected system. Although SCFAs and bile acids have been identified as major mediators, the vast genetic and functional diversity of the microbiome likely conceals additional mechanisms influencing glucose homeostasis. Integrative approaches combining microbiomics, metabolomics, and neurophysiology will be crucial to bridge this gap (61).
Together, these insights into the gut-liver-brain network underscore how peripheral signals converge on central circuits to regulate glucose metabolism, an integrative framework that extends to the brain-pancreas axis, discussed next.
4 The brain-pancreas connection
Extending beyond incretin-mediated pathways, the brain-pancreas axis exemplifies how neuroendocrine circuits integrate peripheral metabolic cues with central control mechanisms to maintain glucose and energy homeostasis. The brain and peripheral nerves coordinate with pancreatic islets to regulate glucose. Glucose-sensing neurons in the hypothalamus, including AgRP/NPY, POMC and VMH, form the core of brain–pancreas connections. These neurons project to the PVN and the brainstem, ultimately communicating with the NTS and DMV. Through these pathways, brainstem structures control vagal and/or spinal outputs to the pancreatic islets, thereby regulating insulin and glucagon secretion (62–64).
Mammals have the capability to maintain blood glucose levels within strikingly narrow ranges under normal conditions. The maintenance of glucose homeostasis relies not only on the intrinsic sensing properties of pancreatic islet cells but also on complex neuroendocrine circuits and autonomic inputs from the sympathetic and parasympathetic nervous systems.
The vagus nerve, a major parasympathetic pathway, further modulates glucose metabolism by influencing both insulin release and hepatic glucose production (65). Direct evidence of autonomic control over islet function was provided by Rodriguez-Díaz et al., who demonstrated in mice with islets transplanted into the eye that autonomic axons innervating the islet directly regulate glucose homeostasis (65).
Pancreatic islets are thus under dual autonomic control. Sympathetic fibers, originating from thoracolumbar spinal segments, stimulate glucagon release from α-cells and suppress insulin secretion from β-cells, largely through modulation of islet blood flow (66).
Parasympathetic vagal input, under hypothalamic regulation, promotes insulin secretion during the cephalic phase of feeding by integrating sensory cues (taste, smell, vision) and responding to gut-derived hormones such as CCK and serotonin. While vagal signaling is dispensable for basal insulin secretion in fasting conditions, it becomes crucial for glucose-stimulated insulin release in the fed state. This dynamic interplay between sympathetic inhibition and parasympathetic stimulation coordinates islet hormone output according to metabolic state (67).
The gut microbiota also influences glucose homeostasis by communicating with the pancreas through various microbial metabolites like the SCFAs and branched-chain amino acids (BCAAs). In both type 1 and type 2 diabetes, microbial imbalance (dysbiosis) alters metabolite profiles, impacting pancreatic islet function. This can lead to effects like increased inflammation, insulin resistance, and the development or progression of diabetes (68). Understanding these complex gut-liver-pancreas-brain circuits could lead to new treatments to prevent or reverse metabolic dysfunction and improve insulin sensitivity.
5 Neuroendocrine dysfunction in disease
Neuroendocrine circuits that regulate glucose homeostasis are highly vulnerable to disruption in obesity, type 2 diabetes, and aging. Insulin resistance in the CNS impairs metabolic control, while autonomic dysregulation alters pancreatic function. Reduced vagal tone further limits insulin secretion and weakens counter-regulatory responses to hypoglycemia. Evidence from human and animal studies shows that these disturbances are accompanied by neuroinflammation, neurotransmitter imbalance, and structural brain changes, all of which contribute to progressive metabolic derangements and increased risk of cognitive decline (69).
Hypothalamic inflammation, characterized by gliosis and microglial activation in the ARC nucleus interferes with PI3K-Akt and STAT3 signaling and blunts POMC and AgRP neuronal responsiveness, thereby favoring orexigenic tone and systemic insulin resistance (70, 71).
In leptin-deficient models, chronic inflammatory signaling further exacerbates hyperphagia and metabolic dysfunction. Mechanistic studies implicate mitochondrial dysfunction and endoplasmic reticulum (ER) stress as key contributors, exemplified by POMC-specific Mitofusin 2 (Mfn2) deletion, which induces ER stress, leptin resistance, and obesity (72, 73).
Beyond inflammation, dysregulation of orexigenic neuropeptides such as neuropeptide Y (NPY) and melanin-concentrating hormone (MCH) worsens metabolic disease. In this regard, ablation of NPY Y2 receptors improves glucose and lipid profiles in ob/ob mice, whereas MCH deletion increases energy expenditure and glycemic control (74, 75).
Moreover, novel pituitary-brain circuits contribute to systemic glucose regulation, as a subset of AgRP-expressing neurons in the anterior pituitary, which responds to bile acid signals and regulates glucose tolerance independently of food intake; silencing these neurons improves glucose-stimulated insulin secretion (76).
However, the precise mechanisms linking brain activity to peripheral glucose handling in humans remain poorly defined, representing a critical frontier for translational research in metabolic regulation.
6 Emerging therapeutic perspectives
The identification of central mechanisms governing glucose homeostasis has opened new therapeutic avenues for obesity, insulin resistance, and diabetes. For instance, intranasal insulin bypasses the blood-brain barrier to enhance central insulin action, improving hepatic glucose production and peripheral glucose disposal without raising systemic insulin levels (77).
Dual and triple incretin receptor agonists, such as tirzepatide and retatrutide, combine peripheral and central actions that target mechanisms underlying sex differences in metabolic homeostasis and disease. These agents not only improve glycemic control and promote weight loss but also partially restore leptin sensitivity through microbiota-derived metabolites such as inosine (78, 79).
Systematically incorporating sex as a biological variable in both preclinical and clinical research will be essential to uncovering mechanistic differences, improving translational accuracy and ultimately guiding the development of sex-adapted therapies for diabetes and other metabolic disorders (80).
Neuromodulatory strategies, including vagus nerve stimulation, selective hepatic-celiac vagal modulation, optogenetics, and chemogenetics, also show promise in improving glycemic control and reducing inflammation (81, 82).
Lifestyle interventions, including intermittent fasting and time-restricted feeding, engage hypothalamic and autonomic pathways to reduce hepatic gluconeogenesis and enhance metabolic flexibility, while early-life nutritional modulation can shape hypothalamic plasticity and long-term glucose control (83).
Emerging therapies targeting mitochondrial quality control, inflammatory signaling, and post-translational modifications (e.g., neddylation) also hold potential to restore central metabolic function. Moreover, multi-omics approaches, integrating proteomics, metabolomics, epigenomics, and microbiomics, combined with artificial intelligence and circuit-level mapping, are accelerating biomarker discovery and precision therapeutic design (84).
Non-invasive neuroimaging tools, including positron emission tomography (PET) and functional magnetic resonance imaging (fMRI), complement these approaches by enabling longitudinal monitoring of brain metabolism and activity in humans (85–87).
7 Discussion
Central regulation of glucose homeostasis arises from the coordinated activity of neuroendocrine and autonomic circuits integrating hormonal and nutrient-derived signals from adipose tissue, gut, liver, and pancreas. Within this network, leptin, insulin, and incretin pathways converge on AgRP/NPY and POMC neurons in the ARC to control feeding, hepatic glucose output, and insulin sensitivity (4–14).
These hypothalamic circuits are developmentally programmed yet remain modifiable during early life. Maternal obesity, overnutrition, or inflammation can durably alter AgRP and POMC connectivity through microglial activation and impaired perineuronal net (PNN) maturation, establishing a “metabolic memory” that predisposes offspring to lifelong glucose dysregulation (29–35).
The brainstem complements hypothalamic control by integrating visceral feedback within the gut-liver-brain axis, central to metabolic homeostasis and a target for novel therapies (60–64). Chronic overnutrition and neuroinflammation exacerbate these impairments, promoting hypothalamic leptin and insulin resistance, hallmarks of metabolic disease and contributors to cognitive decline (70, 71).
Next-generation incretin mimetics and neuromodulatory interventions capitalize on these central-peripheral interactions to enhance metabolic flexibility and restore hormonal sensitivity (78, 79, 81–83). Integrating multi-omics and high-resolution neurocircuit mapping will refine biomarker discovery, improve translational fidelity, and inform precision strategies linking metabolic, neuroendocrine, and immune systems (84).
In summary, glucose homeostasis is governed by a distributed, plastic neuroendocrine network whose developmental and inflammatory plasticity shapes the trajectory of metabolic health. Leveraging this integrative understanding, bridging developmental neurobiology, incretin pharmacology, and neuromodulation, will be key to achieving durable metabolic restoration in obesity and diabetes.
8 Limitations and future directions
Despite major advances, translating mechanistic insights from animal models to humans remains a key challenge. Standardization across strains, diets, and experimental designs is needed to improve reproducibility and cross-study comparability. Longitudinal human cohorts combining neuroimaging, biofluid omics, and hormonal profiling will be instrumental to map central-peripheral interactions in metabolic control.
Future studies should incorporate sex as a biological variable, as estrogens acting through ERα signaling improve insulin sensitivity, β-cell survival, and central metabolic regulation, providing protection before menopause. Developing biomarkers to monitor central interventions will also accelerate translation into effective therapies.
Author contributions
NG: Conceptualization, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. SA-J: Formal Analysis, Investigation, Methodology, Visualization, Writing – review & editing. LM: Formal analysis, Investigation, Methodology, Visualization, Writing – review & editing. AA: Conceptualization, Formal analysis, Investigation, Methodology, Supervision, Validation, Visualization, Funding acquisition, Writing – original draft, Writing – review & editing.
Funding
The author(s) declared financial support was received for this work and/or its publication. This work was supported by Grants PID2021-128243OB-I00 (to NG) from Ministerio de Ciencia e Innovación, Spain, cofinanced by the European Regional Development Fund, (MCIN/AEI/FEDER/EU), and by Institutional Aids 2022-GRIN-34280 (to AA) from University of Castilla-La Mancha (UCLM).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Mergenthaler P, Lindauer U, Dienel GA, and Meisel A. Sugar for the brain: the role of glucose in physiological and pathological brain function. Trends Neurosci. (2013) 36:587–97. doi: 10.1016/j.tins.2013.07.001
2. Röder PV, Wu B, Liu Y, and Han W. Pancreatic regulation of glucose homeostasis. Exp Mol Med. (2016) 48:e219. doi: 10.1038/emm.2016.6
3. Güemes A and Georgiou P. Review of the role of the nervous system in glucose homoeostasis and future perspectives towards the management of diabetes. Bioelectron Med. (2018) 4:9. doi: 10.1186/s42234-018-0009-4
4. Jais A and Brüning JC. Arcuate nucleus-dependent regulation of metabolism-pathways to obesity and diabetes mellitus. Endocr Rev. (2022) 43:314–28. doi: 10.1210/endrev/bnab025
5. Myers MG, Leibel RL, Seeley RJ, and Schwartz MW. Obesity and leptin resistance: distinguishing cause from effect. Trends Endocrinol Metab. (2010) 21:643–51. doi: 10.1016/j.tem.2010.08.002
6. Wrighten SA, Piroli GG, Grillo CA, and Reagan LP. A look inside the diabetic brain: Contributors to diabetes-induced brain aging. Biochim Biophys Acta. (2009) 1792:444–53. doi: 10.1016/j.bbadis.2008.10.013
7. Biessels GJ and Despa F. Cognitive decline and dementia in diabetes mellitus: mechanisms and clinical implications. Nat Rev Endocrinol. (2018) 14:591–604. doi: 10.1038/s41574-018-0048-7
8. Elmquist JK, Bjørbaek C, Ahima RS, Flier JS, and Saper CB. Distributions of leptin receptor mRNA isoforms in the rat brain. J Comp Neurol. (1998) 395:535–47. doi: 10.1002/(SICI)1096-9861(19980615)395:4<535::AID-CNE9>3.0.CO;2-2
9. Cowley MA, Smart JL, Rubinstein M, Cerdán MG, Diano S, Horvath TL, et al. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature. (2001) 411:480–4. doi: 10.1038/35078085
10. Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, and Kangawa K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature. (1999) 402:656–60. doi: 10.1038/45230
11. Turton MD, O’Shea D, Gunn I, Beak SA, Edwards CM, Meeran K, et al. A role for glucagon-like peptide-1 in the central regulation of feeding. Nature. (1996) 379:69–72. doi: 10.1038/379069a0
12. Batterham RL, Cowley MA, Small CJ, Herzog H, Cohen MA, Dakin CL, et al. Gut hormone PYY(3-36) physiologically inhibits food intake. Nature. (2002) 418:650–4. doi: 10.1038/nature00887
13. Holliday A, Horner K, Johnson KO, Dagbasi A, and Crabtree DR. Appetite-related gut hormone responses to feeding across the life course. J Endocr Soc. (2025) 9:bvae223. doi: 10.1210/jendso/bvae223
14. Stenvers DJ, Scheer FAJL, Schrauwen P, la Fleur SE, and Kalsbeek A. Circadian clocks and insulin resistance. Nat Rev Endocrinol. (2019) 15:75–89. doi: 10.1038/s41574-018-0122-1
15. Timper K and Brüning JC. Hypothalamic circuits regulating appetite and energy homeostasis: pathways to obesity. Dis Model Mech. (2017) 10:679–89. doi: 10.1242/dmm.026609
16. Parton LE, Ye CP, Coppari R, Enriori PJ, Choi B, Zhang C-Y, et al. Glucose sensing by POMC neurons regulates glucose homeostasis and is impaired in obesity. Nature. (2007) 449:228–32. doi: 10.1038/nature06098
17. Berglund ED, Vianna CR, Donato J, Kim MH, Chuang J-C, Lee CE, et al. Direct leptin action on POMC neurons regulates glucose homeostasis and hepatic insulin sensitivity in mice. J Clin Invest. (2012) 122:1000–9. doi: 10.1172/JCI59816
18. Krashes MJ, Koda S, Ye C, Rogan SC, Adams AC, Cusher DS, et al. Rapid, reversible activation of AgRP neurons drives feeding behavior in mice. J Clin Invest. (2011) 121:1424–8. doi: 10.1172/JCI46229
19. Routh VH, Hao L, Santiago AM, Sheng Z, and Zhou C. Hypothalamic glucose sensing: making ends meet. Front Syst Neurosci. (2014) 8:236. doi: 10.3389/fnsys.2014.00236
20. Aponte Y, Atasoy D, and Sternson SM. AGRP neurons are sufficient to orchestrate feeding behavior rapidly and without training. Nat Neurosci. (2011) 14:351–5. doi: 10.1038/nn.2739
21. Tooke BP, Yu H, Adams JM, Jones GL, Sutton-Kennedy T, Mundada L, et al. Hypothalamic POMC or MC4R deficiency impairs counterregulatory responses to hypoglycemia in mice. Mol Metab. (2019) 20:194–204. doi: 10.1016/j.molmet.2018.11.004
22. De Solis AJ, Del Río-Martín A, Radermacher J, Chen W, Steuernagel L, Bauder CA, et al. Reciprocal activity of AgRP and POMC neurons governs coordinated control of feeding and metabolism. Nat Metab. (2024) 6:473–93. doi: 10.1038/s42255-024-00987-z
23. Fioramonti X, Contié S, Song Z, Routh VH, Lorsignol A, and Pénicaud L. Characterization of glucosensing neuron subpopulations in the arcuate nucleus: integration in neuropeptide Y and pro-opio melanocortin networks? Diabetes. (2007) 56:1219–27. doi: 10.2337/db06-0567
24. Stanley S, Moheet A, and Seaquist ER. Central mechanisms of glucose sensing and counterregulation in defense of hypoglycemia. Endocr Rev. (2019) 40:768–88. doi: 10.1210/er.2018-00226
25. Zsombok A, Desmoulins LD, and Derbenev AV. Sympathetic circuits regulating hepatic glucose metabolism: where we stand. Physiol Rev. (2024) 104:85–101. doi: 10.1152/physrev.00005.2023
26. Morton GJ, Meek TH, and Schwartz MW. Neurobiology of food intake in health and disease. Nat Rev Neurosci. (2014) 15:367–78. doi: 10.1038/nrn3745
27. Wang M, Chen L, Clark GO, Lee Y, Stevens RD, Ilkayeva OR, et al. Leptin therapy in insulin-deficient type I diabetes. Proc Natl Acad Sci U.S.A. (2010) 107:4813–9. doi: 10.1073/pnas.0909422107
28. Belgardt BF and Brüning JC. CNS leptin and insulin action in the control of energy homeostasis. Ann N Y Acad Sci. (2010) 1212:97–113. doi: 10.1111/j.1749-6632.2010.05799.x
29. Mirzadeh Z, Alonge KM, Cabrales E, Herranz-Pérez V, Scarlett JM, Brown JM, et al. Perineuronal net formation during the critical period for neuronal maturation in the hypothalamic arcuate nucleus. Nat Metab. (2019) 1:212–21. doi: 10.1038/s42255-018-0029-0
30. Bouret SG, Draper SJ, and Simerly RB. Trophic action of leptin on hypothalamic neurons that regulate feeding. Science. (2004) 304:108–10. doi: 10.1126/science.1095004
31. Pinto S, Roseberry AG, Liu H, Diano S, Shanabrough M, Cai X, et al. Rapid rewiring of arcuate nucleus feeding circuits by leptin. Science. (2004) 304:110–5. doi: 10.1126/science.1089459
32. Kamitakahara A, Bouyer K, Wang C-H, and Simerly R. A critical period for the trophic actions of leptin on AgRP neurons in the arcuate nucleus of the hypothalamus. J Comp Neurol. (2018) 526:133–45. doi: 10.1002/cne.24327
33. Vogt MC, Paeger L, Hess S, Steculorum SM, Awazawa M, Hampel B, et al. Neonatal insulin action impairs hypothalamic neurocircuit formation in response to maternal high-fat feeding. Cell. (2014) 156:495–509. doi: 10.1016/j.cell.2014.01.008
34. Mendoza-Romero HN, Biddinger JE, Bedenbaugh MN, and Simerly R. Microglia are required for developmental specification of AgRP innervation in the hypothalamus of offspring exposed to maternal high-fat diet during lactation. Elife. (2025) 13:RP101391. doi: 10.7554/eLife.101391
35. Chang G-Q, Gaysinskaya V, Karatayev O, and Leibowitz SF. Maternal high-fat diet and fetal programming: increased proliferation of hypothalamic peptide-producing neurons that increase risk for overeating and obesity. J Neurosci. (2008) 28:12107–19. doi: 10.1523/JNEUROSCI.2642-08.2008
36. Hankir MK and Lutz TA. Novel neural pathways targeted by GLP-1R agonists and bariatric surgery. Pflugers Arch. (2025) 477:171–85. doi: 10.1007/s00424-024-03047-3
37. Brierley DI, Holt MK, Singh A, de Araujo A, McDougle M, Vergara M, et al. Central and peripheral GLP-1 systems independently suppress eating. Nat Metab. (2021) 3:258–73. doi: 10.1038/s42255-021-00344-4
38. Gaykema RP, Newmyer BA, Ottolini M, Raje V, Warthen DM, Lambeth PS, et al. Activation of murine pre-proglucagon-producing neurons reduces food intake and body weight. J Clin Invest. (2017) 127:1031–45. doi: 10.1172/JCI81335
39. Huang Z, Liu L, Zhang J, Conde K, Phansalkar J, Li Z, et al. Glucose-sensing glucagon-like peptide-1 receptor neurons in the dorsomedial hypothalamus regulate glucose metabolism. Sci Adv. (2022) 8:eabn5345. doi: 10.1126/sciadv.abn5345
40. Kim KS, Park JS, Hwang E, Park MJ, Shin HY, Lee YH, et al. GLP-1 increases preingestive satiation via hypothalamic circuits in mice and humans. Science. (2024) 385:438–46. doi: 10.1126/science.adj2537
41. Singh I, Wang L, Xia B, Liu J, Tahiri A, El Ouaamari A, et al. Activation of arcuate nucleus glucagon-like peptide-1 receptor-expressing neurons suppresses food intake. Cell Biosci. (2022) 12:178. doi: 10.1186/s13578-022-00914-3
42. Hwang E, Portillo B, and Williams KW. Glucagon-like peptide 1 (GLP-1) action on hypothalamic feeding circuits. Endocrinology. (2025) 166:bqaf125. doi: 10.1210/endocr/bqaf125
43. Jastreboff AM, Kaplan LM, Frías JP, Wu Q, Du Y, Gurbuz S, et al. Triple-hormone-receptor agonist retatrutide for obesity - A phase 2 trial. N Engl J Med. (2023) 389:514–26. doi: 10.1056/NEJMoa2301972
44. Berthoud HR and Neuhuber WL. Functional and chemical anatomy of the afferent vagal system. Auton Neurosci. (2000) 85:1–17. doi: 10.1016/S1566-0702(00)00215-0
45. Grill HJ and Hayes MR. Hindbrain neurons as an essential hub in the neuroanatomically distributed control of energy balance. Cell Metab. (2012) 16:296–309. doi: 10.1016/j.cmet.2012.06.015
46. Rinaman L. Visceral sensory inputs to the endocrine hypothalamus. Front Neuroendocrinol. (2007) 28:50–60. doi: 10.1016/j.yfrne.2007.02.002
47. Appleyard SM, Marks D, Kobayashi K, Okano H, Low MJ, and Andresen MC. Visceral afferents directly activate catecholamine neurons in the solitary tract nucleus. J Neurosci. (2007) 27:13292–302. doi: 10.1523/JNEUROSCI.3502-07.2007
48. Borgmann D, Ciglieri E, Biglari N, Brandt C, Cremer AL, Backes H, et al. Gut-brain communication by distinct sensory neurons differently controls feeding and glucose metabolism. Cell Metab. (2021) 33:1466–1482.e7. doi: 10.1016/j.cmet.2021.05.002
49. Bai L, Mesgarzadeh S, Ramesh KS, Huey EL, Liu Y, Gray LA, et al. Genetic identification of vagal sensory neurons that control feeding. Cell. (2019) 179:1129–1143.e23. doi: 10.1016/j.cell.2019.10.031
50. Chen J, Cheng M, Wang L, Zhang L, Xu D, Cao P, et al. A vagal-NTS neural pathway that stimulates feeding. Curr Biol. (2020) 30:3986–3998.e5. doi: 10.1016/j.cub.2020.07.084
51. Cai H, Schnapp WI, Mann S, Miscevic M, Shcmit MB, Conteras M, et al. Neural circuits regulation of satiation. Appetite. (2024) 200:107512. doi: 10.1016/j.appet.2024.107512
52. Aklan I, Sayar Atasoy N, Yavuz Y, Ates T, Coban I, Koksalar F, et al. NTS catecholamine neurons mediate hypoglycemic hunger via medial hypothalamic feeding pathways. Cell Metab. (2020) 31:313–326.e5. doi: 10.1016/j.cmet.2019.11.016
53. Schwartz GJ. Roles for gut vagal sensory signals in determining energy availability and energy expenditure. Brain Res. (2018) 1693:151–3. doi: 10.1016/j.brainres.2018.04.004
54. Hayes MR, Bradley L, and Grill HJ. Endogenous hindbrain glucagon-like peptide-1 receptor activation contributes to the control of food intake by mediating gastric satiation signaling. Endocrinology. (2009) 150:2654–9. doi: 10.1210/en.2008-1479
55. Verberne AJM, Sabetghadam A, and Korim WS. Neural pathways that control the glucose counterregulatory response. Front Neurosci. (2014) 8:38. doi: 10.3389/fnins.2014.00038
56. Browning KN and Travagli RA. Central nervous system control of gastrointestinal motility and secretion and modulation of gastrointestinal functions. Compr Physiol. (2014) 4:1339–68. doi: 10.1002/cphy.c130055
57. Goehler LE, Erisir A, and Gaykema RPA. Neural-immune interface in the rat area postrema. Neuroscience. (2006) 140:1415–34. doi: 10.1016/j.neuroscience.2006.03.048
58. Riediger T. The receptive function of hypothalamic and brainstem centres to hormonal and nutrient signals affecting energy balance. Proc Nutr Soc. (2012) 71:463–77. doi: 10.1017/S0029665112000778
59. Koganezawa T, Shimomura Y, and Terui N. The role of the RVLM neurons in the viscero-sympathetic reflex: a mini review. Auton Neurosci. (2008) 142:17–9. doi: 10.1016/j.autneu.2008.03.007
60. Johansen VBI, Petersen J, Lund J, Mathiesen CV, Fenselau H, and Clemmensen C. Brain control of energy homeostasis: Implications for anti-obesity pharmacotherapy. Cell. (2025) 188:4178–212. doi: 10.1016/j.cell.2025.06.010
61. Wachsmuth HR, Weninger SN, and Duca FA. Role of the gut-brain axis in energy and glucose metabolism. Exp Mol Med. (2022) 54:377–92. doi: 10.1038/s12276-021-00677-w
62. Roh E, Song DK, and Kim M-S. Emerging role of the brain in the homeostatic regulation of energy and glucose metabolism. Exp Mol Med. (2016) 48:e216. doi: 10.1038/emm.2016.4
63. Yoon NA and Diano S. Hypothalamic glucose-sensing mechanisms. Diabetologia. (2021) 64:985–93. doi: 10.1007/s00125-021-05395-6
64. Rosario W, Singh I, Wautlet A, Patterson C, Flak J, Becker TC, et al. The brain-to-pancreatic islet neuronal map reveals differential glucose regulation from distinct hypothalamic regions. Diabetes. (2016) 65:2711–23. doi: 10.2337/db15-0629
65. Rodriguez-Diaz R, Speier S, Molano RD, Formoso A, Gans I, Abdulreda MH, et al. Noninvasive in vivo model demonstrating the effects of autonomic innervation on pancreatic islet function. Proc Natl Acad Sci U.S.A. (2012) 109:21456–61. doi: 10.1073/pnas.1211659110
66. Mateus Gonçalves L and Almaça J. Functional characterization of the human islet microvasculature using living pancreas slices. Front Endocrinol (Lausanne). (2020) 11:602519. doi: 10.3389/fendo.2020.602519
67. Owyang C and Heldsinger A. Vagal control of satiety and hormonal regulation of appetite. J Neurogastroenterol Motil. (2011) 17:338–48. doi: 10.5056/jnm.2011.17.4.338
68. Burcelin R, Serino M, Chabo C, Blasco-Baque V, and Amar J. Gut microbiota and diabetes: from pathogenesis to therapeutic perspective. Acta Diabetol. (2011) 48:257–73. doi: 10.1007/s00592-011-0333-6
69. Kullmann S, Heni M, Hallschmid M, Fritsche A, Preissl H, and Häring H-U. Brain insulin resistance at the crossroads of metabolic and cognitive disorders in humans. Physiol Rev. (2016) 96:1169–209. doi: 10.1152/physrev.00032.2015
70. Thaler JP and Schwartz MW. Minireview: Inflammation and obesity pathogenesis: the hypothalamus heats up. Endocrinology. (2010) 151:4109–15. doi: 10.1210/en.2010-0336
71. García-Cáceres C, Balland E, Prevot V, Luquet S, Woods SC, Koch M, et al. Role of astrocytes, microglia, and tanycytes in brain control of systemic metabolism. Nat Neurosci. (2019) 22:7–14. doi: 10.1038/s41593-018-0286-y
72. Zhang X, Zhang G, Zhang H, Karin M, Bai H, and Cai D. Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell. (2008) 135:61–73. doi: 10.1016/j.cell.2008.07.043
73. Schneeberger M, Dietrich MO, Sebastián D, Imbernón M, Castaño C, Garcia A, et al. Mitofusin 2 in POMC neurons connects ER stress with leptin resistance and energy imbalance. Cell. (2013) 155:172–87. doi: 10.1016/j.cell.2013.09.003
74. Naveilhan P, Svensson L, Nyström S, Ekstrand AJ, and Ernfors P. Attenuation of hypercholesterolemia and hyperglycemia in ob/ob mice by NPY Y2 receptor ablation. Peptides. (2002) 23:1087–91. doi: 10.1016/s0196-9781(02)00042-6
75. Segal-Lieberman G, Bradley RL, Kokkotou E, Carlson M, Trombly DJ, Wang X, et al. Melanin-concentrating hormone is a critical mediator of the leptin-deficient phenotype. Proc Natl Acad Sci U.S.A. (2003) 100:10085–90. doi: 10.1073/pnas.1633636100
76. Liu S-M, Ifebi B, Johnson F, Xu A, Ho J, Yang Y, et al. The gut signals to AGRP-expressing cells of the pituitary to control glucose homeostasis. J Clin Invest. (2023) 133:e164185. doi: 10.1172/JCI164185
77. Benedict C, Hallschmid M, Hatke A, Schultes B, Fehm HL, Born J, et al. Intranasal insulin improves memory in humans. Psychoneuroendocrinology. (2004) 29:1326–34. doi: 10.1016/j.psyneuen.2004.04.003
78. Polex-Wolf J, Deibler K, Hogendorf WFJ, Bau S, Glendorf T, Stidsen CE, et al. Glp1r-Lepr coexpressing neurons modulate the suppression of food intake and body weight by a GLP-1/leptin dual agonist. Sci Transl Med. (2024) 16:eadk4908. doi: 10.1126/scitranslmed.adk4908
79. Dong C, Zhou B, Zhao B, Lin K, Tian Y, Zhang R, et al. GLP-1RAs attenuated obesity and reversed leptin resistance partly via activating the microbiome-derived inosine/A2A pathway. Acta Pharm Sin B. (2025) 15:1023–38. doi: 10.1016/j.apsb.2024.12.006
80. Mauvais-Jarvis F, Arnold AP, and Reue K. A guide for the design of pre-clinical studies on sex differences in metabolism. Cell Metab. (2017) 25:1216–30. doi: 10.1016/j.cmet.2017.04.033
81. Pavlov VA and Tracey KJ. Bioelectronic medicine: Preclinical insights and clinical advances. Neuron. (2022) 110:3627–44. doi: 10.1016/j.neuron.2022.09.003
82. Waataja JJ, Asp AJ, and Billington CJ. Combining celiac and hepatic vagus nerve neuromodulation reverses glucose intolerance and improves glycemic control in pre- and overt-type 2 diabetes mellitus. Biomedicines. (2023) 11:2452. doi: 10.3390/biomedicines11092452
83. Zheng D, Hong X, He X, Lin J, Fan S, Wu J, et al. Intermittent fasting–improved glucose homeostasis is not entirely dependent on caloric restriction in db/db male mice. Diabetes. (2024) 73:864–78. doi: 10.2337/db23-0157
84. Song J, Wang C, Zhao T, Zhang Y, Xing J, Zhao X, et al. Multi-omics approaches for biomarker discovery and precision diagnosis of prediabetes. Front Endocrinol (Lausanne). (2025) 16:1520436. doi: 10.3389/fendo.2025.1520436
85. Iozzo P and Guzzardi MA. Imaging of brain glucose uptake by PET in obesity and cognitive dysfunction: life-course perspective. Endocr Connect. (2019) 8:R169–83. doi: 10.1530/EC-19-0348
86. Rebelos E, Latva-Rasku A, Koskensalo K, Pekkarinen L, Saukko E, Ihalainen J, et al. Insulin-stimulated brain glucose uptake correlates with brain metabolites in severe obesity: A combined neuroimaging study. J Cereb Blood Flow Metab. (2024) 44:407–18. doi: 10.1177/0271678X231207114
Keywords: hypothalamus, brainstem, glucose homeostasis, central regulatory circuits, vagus nerve, type 2 diabetes
Citation: Gallardo N, Artigas-Jerónimo S, Mazuecos L and Andrés A (2025) Neuroendocrine control of glucose homeostasis: integrative mechanisms from the hypothalamus to the brainstem. Front. Endocrinol. 16:1731725. doi: 10.3389/fendo.2025.1731725
Received: 24 October 2025; Accepted: 20 November 2025; Revised: 13 November 2025;
Published: 02 December 2025.
Edited by:
Ismael Valladolid-Acebes, Karolinska Institutet (KI), SwedenReviewed by:
Carolina Dalmasso, University of Kentucky, United StatesLuis Miguel Martínez Durán, Catholic University of Temuco, Chile
Leonardo Davier Martín Ríos, Ghent University, Belgium
Copyright © 2025 Gallardo, Artigas-Jerónimo, Mazuecos and Andrés. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nilda Gallardo, bmlsZGEuZ2FsbGFyZG9AdWNsbS5lcw==