
- Department of Pathology and Cell Biology, University of Montreal and Research Center, Maisonneuve-Rosemont Hospital, Montreal, QC, Canada
Precise control of the neutrophil death program provides a balance between their defense functions and safe clearance, whereas impaired regulation of neutrophil death is thought to contribute to a wide range of inflammatory pathologies. Apoptosis is essential for neutrophil functional shutdown, removal of emigrated neutrophils, and timely resolution of inflammation. Neutrophils receive survival and pro-apoptosis cues from the inflammatory microenvironment and integrate these signals through surface receptors and common downstream mechanisms. Among these receptors are the leukocyte-specific membrane receptors β2 integrins that are best known for regulating adhesion and phagocytosis. Accumulating evidence indicate that outside-in signaling through the β2 integrin Mac-1 can generate contrasting cues in neutrophils, leading to promotion of their survival or apoptosis. Binding of Mac-1 to its ligands ICAM-1, fibrinogen, or the azurophilic granule enzyme myeloperoxidase suppresses apoptosis, whereas Mac-1-mediated phagocytosis of bacteria evokes apoptotic cell death. Mac-1 signaling is also target for the anti-inflammatory, pro-resolving mediators, including lipoxin A4, aspirin-triggered lipoxin A4, and resolvin E1. This review focuses on molecular mechanisms underlying Mac-1 regulation of neutrophil apoptosis and highlights recent advances how hierarchy of survival and pro-apoptosis signals can be harnessed to facilitate neutrophil apoptosis and the resolution of inflammation.
Introduction
Neutrophils form the first line of defense against invading pathogens or tissue injury. They are rapidly recruited to the sites of infection/injury and play a prominent role in the initiation and progression of the inflammatory response. Their many defense mechanisms that destroy invading pathogens are also capable of inflicting damage to the surrounding tissue (Nathan, 2006). Under certain conditions, these harmful consequences became dominant and prolong inflammation. Once the pathogens are cleared, neutrophils are thought to undergo constitutive apoptosis (Savill et al., 2002). This process renders neutrophils unresponsive to extracellular stimuli, allows their recognition and removal by macrophages (Savill et al., 2002; Gilroy et al., 2004; Nathan and Ding, 2010) and limits their potentially harmful actions. Neutrophil accumulation in inflamed tissues is a balance of their recruitment and removal. Conversely, effective resolution of inflammation requires cessation of neutrophil recruitment as well as timely removal of emigrated neutrophils from the site of inflammation. Apoptosis, which ensures that neutrophils are securely marked for disposal, emerged as a control point in resolving inflammation (Filep and El Kebir, 2009; Fox et al., 2010; Perretti, 2012). Pro-survival and pro-apoptosis signals from the inflammatory milieu can, however, influence the execution of the constitutive death program, thereby profoundly affecting the fate of neutrophils and the outcome of the inflammatory response (reviewed in Filep and El Kebir, 2010; Fox et al., 2010; Geering and Simon, 2011).
β2 integrins are leukocyte-specific adhesion molecules that govern neutrophil adhesion and transmigration across the activated endothelium and phagocytosis of pathogens (Ross, 2000; Ley et al., 2007). Growing evidence demonstrates that outside-in signaling through β2 integrins can generate contrasting cues in neutrophils, leading to promotion of their survival or apoptosis. This review will focus on the hierarchy of these signaling circuits and the underlying molecular mechanisms, and will discuss how interference with β2 integrin signaling could be harnessed for promoting neutrophil apoptosis to enhance the resolution of inflammation.
Neutrophil Apoptosis: A Control Point for the Resolution of Inflammation
Neutrophil Apoptosis during Homeostasis and Inflammation
Mature neutrophils are terminally differentiated cells that have the shortest lifespan among leukocytes in the circulation. Neutrophil lifespan is generally thought to be in the range of 8–20 h, though recent data with in vivo labeling suggest a lifespan of 5.4 days under physiological conditions in humans (Pillay et al., 2010). Aged neutrophils die by constitutive (or spontaneous) apoptosis. This mechanism is essential to maintain the balance of cellular homeostasis under physiological conditions (Cartwright et al., 1964; Coxon et al., 1996). Apoptosis renders neutrophils unresponsive to extracellular stimuli and leads to expression of “eat-me” signals, so that neutrophils can be recognized and removed by macrophages in the spleen and bone marrow and Kupffer cells in the liver (Savill et al., 1989, 2002). In mice, these three organs contribute equally to removal of senescent neutrophils (Furze and Rankin, 2008).
During inflammation, extending the lifespan of neutrophils during transendothelial migration and at the sites of infection is critical for efficient destruction of pathogens (Watson et al., 1997; Savill et al., 2002; Nathan, 2006). Once this is accomplished, neutrophils may undergo necrosis, apoptosis, NETosis (neutrophil extracellular trap cell death) (Brinkmann et al., 2004; Fuchs et al., 2007), or autophagy (Remijsen et al., 2011) with the type of death profoundly affecting the outcome of the inflammatory response.
Apoptotic neutrophil death in situ has multiple pro-resolution actions. In addition to becoming unresponsive to agonists and stopping production of inflammatory mediators, apoptotic neutrophils can sequester cytokines (Ariel et al., 2006; Ren et al., 2008) and their phagocytosis by macrophages induces macrophage polarization from a pro-inflammatory (M1) to a pro-resolution (M2) phenotype (Fadok et al., 1998). M2 macrophages secrete mediators, such as IL-10 and TGFβ, which mediate resolution and tissue repair (Ariel and Serhan, 2012; Sica and Mantovani, 2012). Interestingly, injection of large quantities of apoptotic neutrophils protected mice against endotoxin shock (Ren et al., 2008).
In non-resolving inflammation, neutrophils persist at the inflamed site and are liable to cause tissue destruction (Nathan and Ding, 2010; Soehnlein, 2012). Neutrophil recruitment may occur normally or may become excessive, but neutrophils persist as a result of delayed apoptosis or decreased clearance by macrophages (Haslett, 1999; Savill et al., 2002). The abnormal host response creates a persistent inflammatory microenvironment with ongoing release of inflammatory mediators and damage-associated molecular patterns (Nathan and Ding, 2010; Serhan, 2011).
Neutrophil Apoptosis in Human Disease
The tight regulation of neutrophil death is also evident under pathological conditions, though it is often difficult to decide whether prolonged survival or apoptosis is most favorable from the host’s perspective. Consistently, both accelerated and delayed neutrophil apoptosis could have severe pathological consequences. For example, infections with the opportunistic pathogen Pseudomonas aeruginosa (Allen et al., 2005), influenza virus A (Colamussi et al., 1999), or HIV (Elbim et al., 2009) as well as autoimmune diseases, such as systemic lupus erythematosus (Courtney et al., 1999) shorten neutrophil lifespan by accelerating apoptosis, leading to impaired antimicrobial defenses and increased susceptibility to recurrent infections. Intracellular pathogens may use apoptotic neutrophils as a Trojan horse to infect and propagate in macrophages (Laskay et al., 2008; Rupp et al., 2009). On the other hand, delayed neutrophil apoptosis appears to be a component of the pathophysiology in patients with inflammatory diseases, including acute respiratory distress syndrome (ARDS) (Matute-Bello et al., 1997; chronic pulmonary obstructive disease (COPD) (Brown et al., 2009), viral pneumonia (Lindemans et al., 2006), sepsis (Ertel et al., 1998), burn (Chitnis et al., 1996), acute coronary artery disease (Garlichs et al., 2004), rheumatoid arthritis (Wong et al., 2009), and cystic fibrosis (McKeon et al., 2008), and frequently correlates with disease severity and outcome.
Distinct Molecular Features of Neutrophil Apoptosis
A complex network of intracellular death and survival pathways regulates neutrophil apoptosis and the balance of these circuits would ultimately determine the fate of neutrophils. Since neutrophils undergo apoptosis even in the absence of any extracellular stimuli, this type of death is called spontaneous or constitutive programed cell death. However, under most conditions, neutrophils receive both pro-survival and pro-apoptosis cues, and the net effect is likely determined by the balance of these signals. Neutrophil apoptosis shares many morphological features with apoptosis in other cell types; however, it involves distinct molecular mechanisms in executing the cell death program. Predominant expression of the anti-apoptotic protein myeloid cell leukemia-1 (Mcl-1), restricted function of mitochondria to apoptosis, ROS production, release of proteases from azurophilic granules, and unusual roles for nuclear proteins are hallmarks of regulation of apoptosis in neutrophils.
Mcl-1 Regulation of Neutrophil Survival and Apoptosis
Spontaneous neutrophil apoptosis rely upon the balance of pro- and anti-apoptotic members of the Bcl-2 family. Mature human neutrophils constitutively express the pro-apoptotic Bcl-2-associated X protein (Bax), Bcl-2-associated death promoter (Bad), Bcl-2 homologous antagonist/killer (Bak), Bcl-2 homology-3 (BH-3)-interacting domain death agonist (Bid), Bcl-2 interacting protein (Bim), and Bcl-2 interacting killer (Bik) as well as the anti-apoptotic Bcl-2 homolog Mcl-1, and to a much lesser extent A1 and Bcl-XL, but not Bcl-2 (Akgul et al., 2001; Moulding et al., 2001). The pro-apoptotic Bcl-2 homologs have relatively long half-lives and their cellular levels change very little during exposure of neutrophils to agents that either accelerate or delay apoptosis. Genetic deletion of bim or bax/bak results in increased neutrophil numbers in mice (Lindsten et al., 2000). Mcl-1 contains a PEST domain [rich in proline (P), glutamic acid (E), serine (S), and threonine (T)], which facilitates its proteasomal degradation, resulting in a very short half-life (Edwards et al., 2004). Mcl-1 levels closely correlate with neutrophil survival kinetics (Moulding et al., 1989; Hamasaki et al., 1998; Leuenroth et al., 2000; Kato et al., 2006; Dzhagalov et al., 2007). Survival of myeloid cells decreases following treatment with antisense oligonucleotides against Mcl-1 (Moulding et al., 2000). Myeloid lineage-specific knockout of mcl-1 reduces neutrophil numbers by accelerating apoptosis (Dzhagalov et al., 2007; Steimer et al., 2009). Recent data indicate that Mcl-1 levels drop in advance of apoptosis, even in the presence of caspase inhibitors (Wardle et al., 2011), indicating that Mcl-1 functions as a regulator and a downstream target of caspase activation. On the other hand, unchanged or even increased Mcl-1 expression has been detected in neutrophils of patients with Crohn’s disease (Catarzi et al., 2008) or severe sepsis (Fotouhi-Ardakani et al., 2010). Mcl-1 promotion of neutrophil survival is thought to involve heterodimerization with and neutralization of Bim or Bak in the mitochondrial outer membrane (Reed, 2006; Brenner and Mak, 2009), resulting in maintenance of the mitochondrial transmembrane potential (ΔΨm) and prevention of release of pro-apoptotic proteins.
Mature neutrophils contain a low number of mitochondria that may have a role restricted to apoptosis (Maianski et al., 2004). Thus, mitochondrial respiration in mature neutrophils is low and mitochondria generate only small amounts of ATP by oxidative phosphorylation (Maianski et al., 2004). The mitochondrial poison cyanide does not affect neutrophil function. Nevertheless, neutrophil mitochondria maintain a transmembrane potential, forms a complex network that plays a role in chemotaxis, phagocytosis, and triggering apoptosis (Fossati et al., 2003). Mitochondria contains pro-apoptotic proteins cytochrome c, second mitochondria-derived activator of caspases (Smac)/DIABLO (direct IAP-binding protein with low pl), apoptosis-inducing factor (AIF), and endonuclease G (Saelens et al., 2004). Loss of ΔΨm evokes release of these proteins. Cytochrome c and Smac appear to be required for optimal caspase-3 activation (Altznauer et al., 2004). Loss of ΔΨm precedes development of apoptotic morphology in neutrophils undergoing constitutive (Maianski et al., 2004) and induced apoptosis.
Role of Reactive Oxygen Species and Redox Balance
Neutrophils generate high amounts of ROS by NADPH oxidase in response to soluble stimuli as well as following phagocytosis of bacteria in order to destroy invading pathogens (Nauseef, 2007). High amounts ROS can inflict damage to the surrounding cell and evoke necrosis. ROS also function as intracellular signaling molecules. The intrinsic or mitochondrial pathway of apoptosis is likely initiated through ROS generation (Kasahara et al., 1997; Maianski et al., 2004; Xu et al., 2010), though the source(s) of ROS in aging non-activated neutrophils is still unknown. Ligation of the cell surface death receptors, TNF, Fas, or TRAIL (TNF-related apoptosis-inducing ligand) receptors triggers the formation of the death-inducing signaling complex (DISC), which through downstream adaptor proteins, such as Fas-associated death domain (FADD) leads to activation of NADPH oxidase and cleavage of caspase-8 (Green, 2000). Likewise, phagocytosis of opsonized microorganisms through complement and/or Fcγ receptors evokes ROS-mediated activation of caspase-8 and subsequently caspase-3, leading to neutrophil apoptosis (Watson et al., 1996; Perskvist et al., 2002; DeLeo, 2004). Consistent with these findings, patients with chronic granulomatous diseases that lacks functional NADPH oxidase exhibit increased neutrophil viability (Fadeel et al., 1998) and reduced neutrophil apoptosis following ingestion of bacteria (Coxon et al., 1996). The effects of ROS are balanced by neutrophil antioxidant defenses, including catalase, superoxide dismutase, and glutathione. These proteins become depleted during ex vivo culture of neutrophils parallel with development of apoptotic morphology (Watson, 2002; Melley et al., 2005). Loss of GSH by chloramines or GSH depletion during activation of the respiratory burst predisposes to apoptosis (Melley et al., 2005).
Regulation of Apoptosis by Granular and Nuclear Proteins
Certain granular and nuclear proteins have also been implicated in the modulation of the cell death program. For example, cathepsin D is released from the azurophilic granules during apoptosis and may contribute to activation of caspase-3 through processing of caspase-8 and Bid (Conus et al., 2008). Consistently, pharmacological or genetic inhibition of cathepsin D results in delayed neutrophil apoptosis. Mature neutrophils constitutively express the cyclin-dependent kinases CDK1, CDK2, and CDK5 (Rosales et al., 2004; Rossi et al., 2006). Culture of neutrophils with R-roscovitine, a non-selective inhibitor of cyclin-dependent kinases enhances apoptosis likely through down-regulation of Mcl-1 expression (Rossi et al., 2006; Leitch et al., 2010). Unlike in other cell types, proliferating cell nuclear antigen (PCNA) is expressed in the cytoplasm of mature neutrophils and is bound to pro-caspases, resulting in suppression of neutrophil apoptosis (Witko-Sarsat et al., 2010). Conversely, decreased cytoplasmic PCNA expression resulted in augmented neutrophil apoptosis. In contrast, during constitutive apoptosis, another nuclear protein myeloid nuclear differentiation antigen (MNDA) is cleaved by caspases and accumulates in the cytoplasm, where it promotes proteasomal degradation of Mcl-1 and subsequently collapse of mitochondrial transmembrane potential (Fotouhi-Ardakani et al., 2010). Bacterial constituents and platelet-activating factor prevent cytoplasmic MNDA accumulation parallel with preservation of Mcl-1 and suppression of apoptosis (Fotouhi-Ardakani et al., 2010).
Suppression of Apoptosis: Intracellular Survival Pathways
Although apoptosis is a default fate of neutrophils, in the inflammatory microenvironment, neutrophils are likely exposed to various pro-survival signals, including granulocyte macrophage colony stimulating factor (GM-CSF) (Colotta et al., 1992; Lee et al., 1993), leukotriene B4 (Lee et al., 1999), C5a (Lee et al., 1993), the acute-phase reactants C-reactive protein (Khreiss et al., 2002), and serum amyloid A (El Kebir et al., 2007; Christenson et al., 2008) as well as bacterial constituents LPS (Colotta et al., 1992; Lee et al., 1993) and bacterial DNA (József et al., 2004). Multiple kinase pathways are involved in determining the fate of neutrophils. For example, GM-CSF activates the Jak2/STAT and phosphoinositide-3-kinase (PI3K)/Akt pathways, leading to preservation of Mcl-1 expression and retardation of apoptosis (Klein et al., 2000; Epling-Burnette et al., 2001). PI3K generates PtdIns(3,4,5)P3, which also influences NF-κB and cAMP-response-element-binding protein (CREB) and thus may generate additional pro-survival signals (Ward et al., 2004). Many inflammatory mediators also activate the MAPK/ERK pathway that, in turn, inhibits the intrinsic pathway of apoptosis (Filep and El Kebir, 2010; Geering and Simon, 2011). ERK 1/2 and Akt phosphorylate Bad and Bax, leading to dissociation of phosphorylated Bad and Bax from the anti-apoptotic protein Mcl-1 (Akgul et al., 2001; El Kebir et al., 2007). Concomitant activation of Akt and ERK appears to be required for suppression of neutrophil apoptosis, and transient activation of Akt without ERK activation may not be sufficient to delay the death program. Contradictory results have been reported for p38 MAPK; its action on neutrophil survival may be stimulus and/or context-specific (reviewed in Filep and El Kebir, 2010). For example, pro-survival function of p38 MAPK may include phosphorylation, and therefore inactivation of caspase-3 and caspase-8 (Alvarado-Kristensson et al., 2003). In other studies, constitutive or TNF-induced neutrophil apoptosis was found to be associated with phosphorylation of p38 MAPK (Khreiss et al., 2002; El Kebir et al., 2007). Activation of p38 MAPK by sodium salicylate is associated with reduced Mcl-1 expression and acceleration of apoptotic cell death (Derouet et al., 2006).
Neutrophil β2 Integrins Modulate Life and Death decisions
β2 Integrin Activation and Function
The β2 integrin (αβ) family consists of LFA-1 (leukocyte function-associated antigen 1, CD11a/CD18), Mac-1 (CD11b/CD18, αMβ2 integrin, ITAM antigen), p150,95 (CD11c/CD18, αXβ2 integrin, ITAX antigen), and αdβ2 (CD11d/CD18, ITAD antigen). The β2 integrins are in an inactive (low affinity) conformation on circulating leukocytes. Leukocyte agonists trigger inside-out signaling that through activation of Rap1 (reviewed in Evans et al., 2009) induces conformational changes that reflect the intermediate and high affinity states of Mac-1 (Xiong et al., 2001; Luo et al., 2007). Ligand occupancy, but not integrin clustering promotes switchblade-like extension of the Mac-1 extracellular domain and separation of the αM and β2 subunit cytoplasmic tails, structural hallmarks of integrin activation (Lefort et al., 2009). These lead to enhanced affinity for binding their ligand and/or regulation of avidity. Integrin activation is a complex and tightly regulated process which involves displacement of inhibitory proteins from the integrin cytoplasmic tail followed by targeting integrin activators or activator complexes, such as talin, kindlins, integrin-linked kinase, and migfilin (Kim et al., 2011). β2 integrins contribute to diverse neutrophil functions critical for innate immunity. Activated β2 integrins mediate leukocyte adhesion and transmigration across the endothelium through interactions with ICAM-1 on the activated endothelial cells (Ley et al., 2007; Abram and Lowell, 2009). Mac-1 also mediates other neutrophil adhesion-dependent functions, including binding to fibrinogen (Pluskota et al., 2004), immune complexes, and platelets (through Gp1b) (Mayadas and Cullere, 2005) and suppression of T cell proliferation (Pillay et al., 2012). Mac-1 and CD11c/CD18 are specific receptors for complement iC3b and mediate efficient phagocytosis of complement-opsonized targets, though they can also recognize many pathogens directly (Ross, 2000). Mac-1 functionally cooperates with other surface receptors, including TNF receptor, FcγRs, Toll-like receptor 2 (TLR2), and CD14 (Ehlers, 2000; Ross, 2000; Kobayashi et al., 2002; Salamone et al., 2004).
Mac-1-Mediated Pro-Survival Signaling
Outside-in signaling through Mac-1 could generate contrasting cues for neutrophils in a context-dependent fashion (Figure 1). Transendothelial migration of neutrophils (Watson et al., 1997; Yan et al., 2004) or neutrophil adherence to Mac-1 ligands, ICAM-, fibrinogen, and plasminogen, prolongs their lifespan by delaying apoptosis (Table 1). Binding of ICAM-1 to Mac-1 induces activation of the PI3k/Akt survival pathway (Whitlock et al., 2000). Fibrinogen-mediated suppression of neutrophil apoptosis also depends on Akt in addition to activation of NF-κB and the MAPK/ERK pathway (Rubel et al., 2003). Crosslinking activated Mac-1 with anti-Mac-1 antibody (Fab fragments) or clustering inactive Mac-1 in neutrophils in suspension signals survival cues through activation of Akt and ERK (Whitlock et al., 2000). Soluble fibrinogen activates neutrophils, as assessed by upregulation of Mac-1 expression and elevation of intracellular calcium concentration (Rubel et al., 2003; Pluskota et al., 2008), indicating that Mac-1-mediated adhesion per se is not a prerequisite for generation of survival signals. Engagement of both Mac-1 subunits with these ligands is a prerequisite for induction of pro-survival signals (Pluskota et al., 2008). Consistently, angiostatin, derived from plasminogen and neutrophil inhibitory factor (NIF), which interact primarily with the αM subunit do not trigger phosphorylation of ERK 1/2 and Akt and do not rescue neutrophils from constitutive apoptosis (Pluskota et al., 2008).
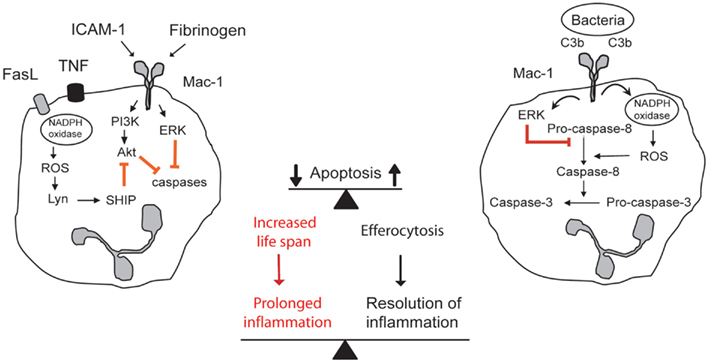
Figure 1. Contrasting outside-in signals through Mac-1 modulates survival and death decision in neutrophils. Aging neutrophils undergo constitutive apoptosis triggered by collapse of mitochondrial function. Engagement of Mac-1 with its ligands ICAM-1, fibrinogen, or MPO delays apoptosis by generating survival cues through activation of the PI3K/Akt and MEK/ERK pathways. Additional stimulation of death receptors with TNF or FasL evokes release of ROS, which through activation of lyn and SHIP leads to inhibition of Akt. Mac-1-mediated phagocytosis of opsonized bacteria evokes ROS production by NADPH oxidase, leading to ROS-mediated activation of caspase-8 and inhibition of ERK, and acceleration of the cell death program. Apoptotic neutrophils are recognized and phagocytosed by macrophages (efferocytosis), thus contributing to inflammatory resolution. Extended neutrophil longevity contributes to aggravation and prolongation of the inflammatory response.
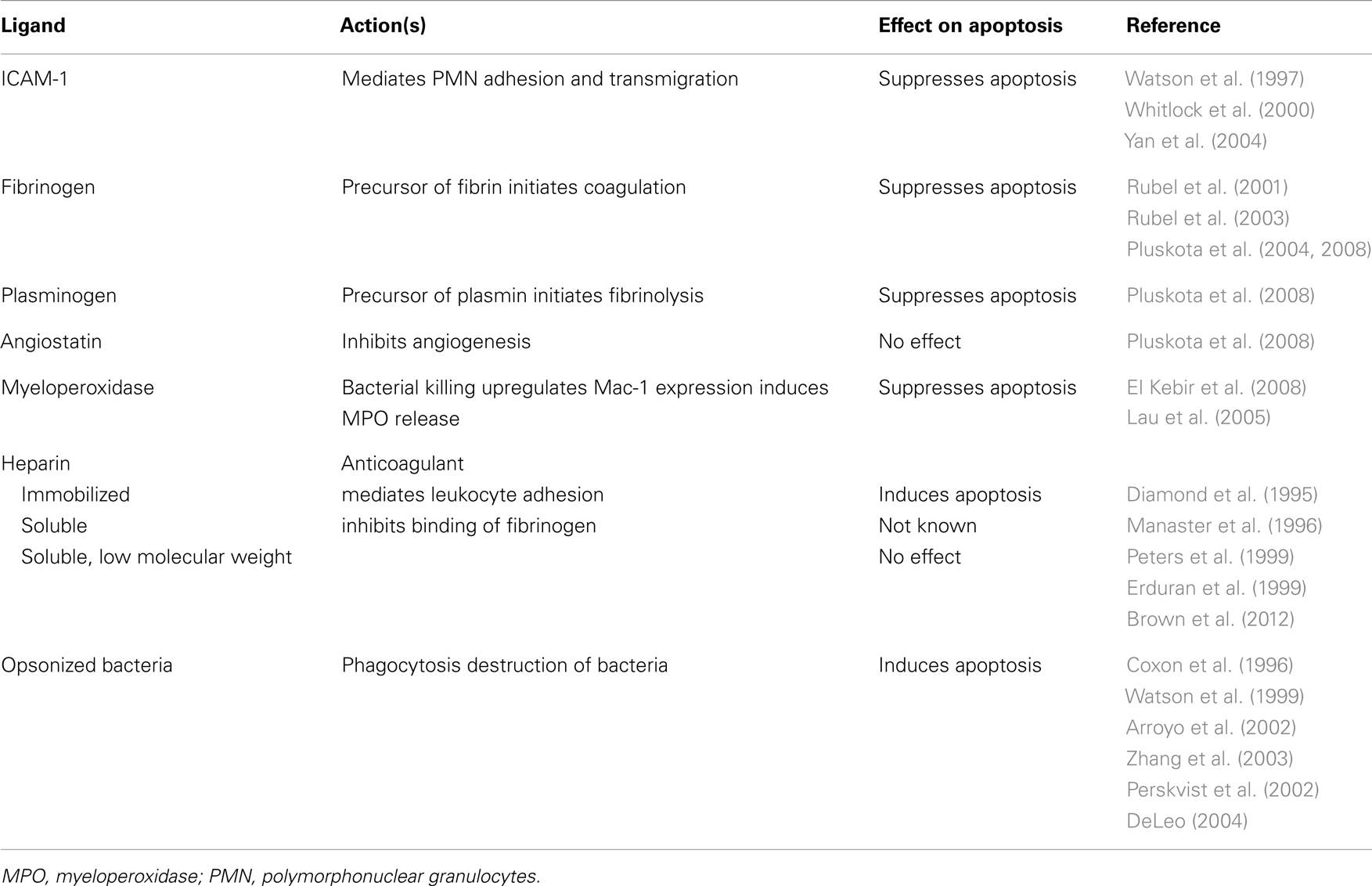
Table 1. Selective regulation of neutrophil apoptosis by different ligands of Mac-1.
Heparin also binds to Mac-1. Immobilized heparin can mediate leukocyte adhesion (Diamond et al., 1995), whereas unfractionated soluble heparin was reported to inhibit binding of fibrinogen and complement iC3b to Mac-1 (Peters et al., 1999). Inconsistent reports have been published on the effect of heparin on neutrophil lifespan. Unfractionated heparin was reported to induce apoptosis (Manaster et al., 1996), whereas low molecular weight heparin did not affect neutrophil apoptosis (Erduran et al., 1999; Brown et al., 2012).
In the presence of TNF or anti-Fas activating antibody, crosslinking Mac-1 with activating antibodies promotes neutrophil apoptosis (Whitlock et al., 2000). Activation of TNF receptor or Fas results in NADPH oxidase-mediated ROS generation and extracellular ROS release, leading to activation of SHIP [Src-homology 2(SH2)-containing inositol 5-phosphatase], which hydrolyzes PI3K products through the Src kinase Lyn (Gardai et al., 2002). This would lead to decreased Akt phosphorylation. Although Lyn was reported to exert anti-apoptotic actions in granulocytes (Daigle et al., 2002), exogenous H2O2 can activate Lyn independently of adhesion (Yan and Berton, 1996). ROS, most likely H2O2 that was released extracellularly might diffuse back into cells to suppress the PI3K/Akt survival signal (Zhu et al., 2006; Xu et al., 2010). Whether ROS affect signaling pathways leading to activation of PI3K or through activation of PTEN (phosphatase and tensin homolog) is still uncertain. PTEN converts phosphatidylinositol 3,4,5-triphosphate to phosphatidylinositol-4,5-diphosphate, thus preventing activation of Akt. PTEN-null neutrophils live longer than wildtype neutrophils (Zhu et al., 2006). In contrast, crosslinking Mac-1 with clustering non-activating antibodies or recombinant ICAM-1 results in partial attenuation of Fas-triggered apoptosis through sustained ERK activation and elevation in reduced glutathione (GSH) levels (Watson et al., 1997; Whitlock et al., 2000). The biological significance of differences in neutrophil responses to Mac-1 activating versus clustering antibodies remains to be investigated. TNF promotion of neutrophil apoptosis evoked by immune complexes or zymosan partially depends on Mac-1 (Salamone et al., 2004), suggesting functional cooperation of Mac-1 with Fcγ or zymosan receptors (Ehlers, 2000; Ross, 2000).
While most studies investigated Mac-1 signaling, ligation of LFA-1 may also generates contrasting cues for neutrophils. LFA-1-deficient mice exhibit neutrophilia and enhanced resistance to L. monocytogenes without changes in spontaneous apoptosis (Miyamoto et al., 2003). Similar to Mac-1, ligation of LFA-1 during transendothelial migration suppresses caspase-3 activation and thus delays apoptosis in neutrophils (Yan et al., 2004). In contrast, activation of ICAM-3 with a monoclonal antibody that recognizes an ICAM-3 epitope that binds its ligand LFA-1 was found to induce apoptosis (Kessel et al., 2006).
Mac-1-Mediated Acceleration of Apoptosis
Phagocytosis of opsonized bacteria or other targets accelerates apoptosis in neutrophils, also referred to as phagocytosis-induced cell death or PICD (Coxon et al., 1996; Watson et al., 1999; Perskvist et al., 2002; DeLeo, 2004). Mac-1-mediated phagocytosis evokes NADPH-dependent ROS generation within the phagolysosomes (Karlsson and Dahlgren, 2002) and is thought to contribute to killing bacteria (Nauseef, 2007) as well as to triggering PICD (Watson et al., 1996; Zhang et al., 2003). Consistently, patients with chronic granulomatous disease, who have low level of NADPH oxidase due to genetic mutations in gp91phox or other phox genes, suffer from recurrent infections (Heyworth et al., 2003) and their neutrophils do not undergo PICD (Coxon et al., 1996).
Mac-1-mediated phagocytosis evokes generation of NADPH oxidase-derived ROS, which, in turn, leads to activation of caspase-8 and subsequently caspase-3 (Arroyo et al., 2002; Zhang et al., 2003). ROS, most likely hydroxyl radicals and H2O2 (Watson et al., 1996; Perskvist et al., 2002) released within phagosomes might leak and trigger PICD. Superoxide released extracellularly is rapidly dismutated to H2O2, which might diffuse back to the cell to affect the redox status and signaling pathways. As caspase-8 activation is a signature of death receptor-mediated apoptosis, Mac-1 has been suggested to function as a death receptor, even though it does not contain a recognized death effector domain (Mayadas and Cullere, 2005). It should be noted that many stimuli that generate varying amounts of NADPH oxidase-derived ROS do not trigger neutrophil death. For example, GM-CSF enhances ROS production upon yeast phagocytosis, but inhibits PICD (Zhang et al., 2003). Thus, the amount, nature, and intra- or extracellular release of NADPH oxidase-derived ROS would likely determine their pro-apoptosis potential. PICD occurs despite phagocytosis-induced activation of the MAPK/ERK pathway (Zhang et al., 2003), indicating that ROS triggered pro-apoptosis signals effectively override survival cues. In contrast, GM-CSF evokes a more robust ERK phosphorylation in phagocytosing neutrophils, leading to generation of strong competing survival signals that shift the life-death balance toward survival (Zhang et al., 2003).
Myeloperoxidase: A Ligand for Mac-1 and Survival Signal for Neutrophils
An unexpected ligand for Mac-1 is myeloperoxidase (MPO), the most abundant granule enzyme in neutrophils (Schultz and Kaminker, 1962; Borregaard and Cowland, 1997). MPO, MPO-generated reactive oxidants, hypochlorous acid (HOCl) in particular and diffusible radical species have been implicated in the elimination of microbes (Klebanoff, 2005; Nauseef, 2007; Davies et al., 2008) as well as in inflicting tissue damage (Klebanoff, 2005; Winterbourn, 2008; Arnhold and Flemming, 2010). Non-activated neutrophils bind to MPO-coated surfaces (Johansson et al., 1997) or “free” circulating MPO through Mac-1 (Lau et al., 2005). Increased MPO association with neutrophil membrane was detected in blood from patients with inflammatory diseases, including sepsis, ischemia-reperfusion, or acute coronary syndromes compared with healthy controls (Lau et al., 2005). Membrane-bound MPO correlates with plasma MPO levels, indicating up-regulation of MPO export toward the plasma membrane as well as a potential for binding of free MPO to the neutrophil surface.
MPO binding to Mac-1 on human neutrophils leads to increased tyrosine phosphorylation (Lau et al., 2005), phosphorylation of p38 MAPK (Lau et al., 2005; El Kebir et al., 2008), ERK 1/2 and PI3K (El Kebir et al., 2008), and activation of NF-κB (Lau et al., 2005). Activation of p38 MAPK induces phosphorylation of p47phox, the cytoplasmic regulatory subunit of NADPH oxidase (Babior, 2004), leading to superoxide formation (Lau et al., 2005), and regulates NF-κB-mediated transcription of genes involved in the acute inflammatory response (Park et al., 2003). Intriguingly, MPO also upregulates surface expression of Mac-1 on neutrophils (Lau et al., 2005; El Kebir et al., 2009) through yet unidentified molecular mechanisms. MPO binding to Mac-1 induces release of elastase and MPO from the azurophilic granules (Lau et al., 2005). These findings are consistent with the central role of Mac-1-mediated outside-in signaling in degranulation (Harris et al., 2000), and imply an autocrine/paracrine mechanism for amplifying neutrophil responses to MPO (Figure 2) (El Kebir et al., 2008, 2009).
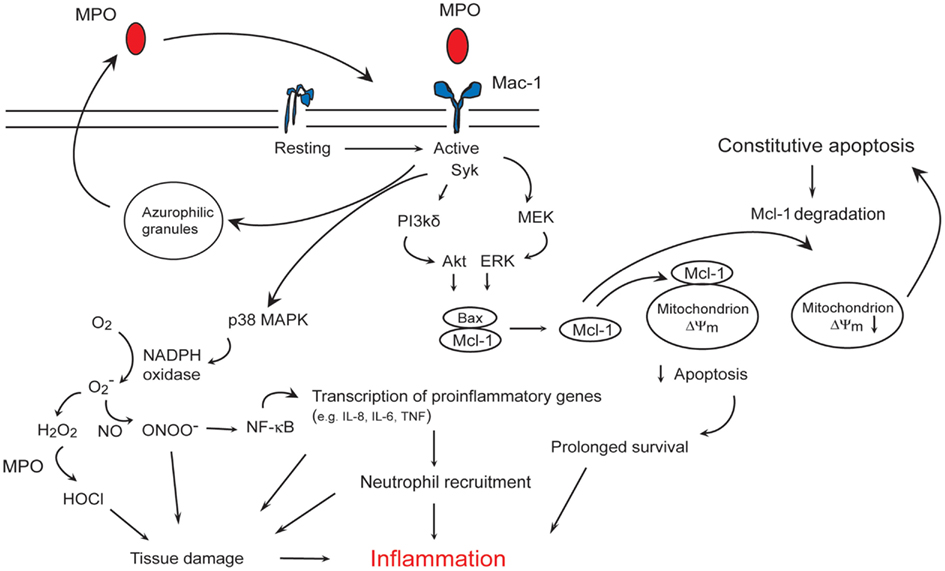
Figure 2. Myeloperoxidase (MPO)-Mac-1 self-amplifying circuit promotes neutrophils survival and inflammation. MPO binding to Mac-1 results in p38 MAPK-mediated NADPH oxidase, and PI3K/Akt and MEK/ERK-mediated preservation of the anti-apoptotic protein Mcl-1, leading to suppression of apoptosis. MPO also triggers MPO release from the azurophilic granules, and upregulates Mac-1 expression, thereby forming an autocrine/paracrine self-amplifying circuit. MPO catalyzed formation of HOCl, activation of NF-κB-regulated transcription of pro-inflammatory genes, recruitment of neutrophils, and prolongation of neutrophil longevity contribute to tissue damage and inflammation.
MPO, independent of its catalytic activity, rescues neutrophils from constitutive apoptosis through simultaneous activation of ERK 1/2 and Akt, Mcl-1 accumulation and suppression of the mitochondrial pathway of apoptosis (El Kebir et al., 2008). Consistently, MPO prevents cytochrome c release from the mitochondria and subsequent activation of caspase-3 (El Kebir et al., 2008). MPO-induced phosphorylation of p38 MAPK does not generate a survival signal, for pharmacological blockade of p38 MAPK retards neutrophil apoptosis both in the absence and presence of MPO (El Kebir et al., 2008). In contrast to these findings, MPO was found to mediate apoptosis in HL60 leukemia cells (Wagner et al., 2000; Kanayama and Miyamoto, 2007). While the involvement of Mac-1 in the pro-apoptosis action of MPO has not been elucidated, Mac-1 may play opposing roles in determining the fate of primary and leukemia cells likely by shifting the balance of pro-survival and pro-apoptosis cues. It is intriguing that MPO can prolong the lifespan of neutrophils, the predominant source of this enzyme and that function-blocking monoclonal anti-Mac-1 antibodies can almost completely prevent MPO-induced activation (Lau et al., 2005; El Kebir et al., 2008) and suppression of constitutive apoptosis of human neutrophils in vitro (El Kebir et al., 2008).
Myeloperoxidase Prolongs Neutrophil Lifespan and Delays Resolution of Inflammation
Acute elevation of plasma MPO levels to levels similar to those detected in patients with inflammatory vascular diseases (Brennan et al., 2001; Baldus et al., 2003) results in prolongation of the lifespan of rat neutrophils through suppression of apoptosis as assayed ex vivo (El Kebir et al., 2008). MPO also suppresses neutrophil apoptosis in a mouse model of carrageenan-induced lung injury and delays spontaneous self-resolution of pulmonary inflammation (El Kebir et al., 2008). Thus, combined administration of carrageenan and MPO evokes persisting lung injury/inflammation with few airway neutrophil exhibiting signs of apoptosis even 5 days post-injection, when pulmonary inflammation is almost completely resolved in the lungs of carrageenan-injected mice. The effects of MPO closely resemble those of zVAD-fmk, a pan-caspase inhibitor, which aggravates and prolongs carrageenan-elicited acute pleurisy (Rossi et al., 2006) and lung inflammation (El Kebir et al., 2008). MPO-deficient mice exhibit lower pulmonary bacterial colonization, reduced lung injury, and greater survival following intraperitoneal injection of Escherichia coli compared with wild type mice (Brovkovych et al., 2008). MPO deficiency also reduces ischemia/reperfusion-induced renal dysfunction and neutrophil accumulation in mice, but fails to abrogate apoptosis during early phases of reperfusion (Matthijsen et al., 2007). Intriguingly, MPO-deficient mice exhibit elevated baseline pulmonary inducible NO synthase expression and NO production that may partially compensate for the lack of HOCl-mediated bacterial killing (Brovkovych et al., 2008). The mechanism(s) responsible for upregulation of inducible NO synthase as well as the impact of enhanced NO production on the resolution of lung inflammation remains to be explored. Absence of MPO-derived oxidant production during E. coli septicemia in MPO-null mice is consistent with reduced lung injury and mortality. Further studies are required to investigate whether MPO deficiency could affect the lifespan of emigrated or circulating neutrophils, and whether changes in neutrophil longevity could contribute to protection against lung injury in this model of sepsis.
Targeting Mac-1 Signaling for Therapeutic Induction of Neutrophil Apoptosis
Accumulating experimental and clinical data suggest a causal relationship between neutrophil apoptosis and outcome of inflammation. Apoptosis of emigrated neutrophils has multiple pro-resolution actions. It renders neutrophils unresponsive to agonists and apoptotic neutrophils stop producing and releasing pro-inflammatory mediators. Apoptotic leukocytes sequester cytokines (Ariel et al., 2006; Ren et al., 2008) and phagocytosis of apoptotic cells induces macrophage polarization from a pro-inflammatory to a pro-resolution phenotype (Fadok et al., 1998; Ariel and Serhan, 2012; Sica and Mantovani, 2012). Injection of large number of apoptotic neutrophils protects mice against LPS-induced shock (Ren et al., 2008). Recent studies identified several classes of molecules for therapeutic induction of apoptosis in neutrophils for enhancing the resolution of inflammation.
Lipoxins Inhibit MPO Signaling through Mac-1 and Redirects Neutrophils to Apoptosis
The pivotal role of MPO in host defense and tissue injury makes it an attractive target for drug development. Indeed, while a number of promising compounds have been developed to inhibit the enzymatic activities of MPO, only limited information is available on their mechanisms of MPO inhibition and biological activities (reviewed in Malle et al., 2007). Targeting MPO signaling through Mac-1 has emerged as an alternative approach to counter the non-enzymatic activities of MPO.
Lipoxin A4 (LXA4) and aspirin-triggered 15-epi-LXA4 are typically generated by transcellular biosynthesis at sites of inflammation (Serhan et al., 2008; Serhan, 2011). In the aspirin-triggered pathway, acetylation of cyclooxygenase at Ser530 by aspirin (Clària and Serhan, 1995) or S-nitrosylation at Cys526 by atorvastatin (Birnbaum et al., 2006) catalyzes the conversion of arachidonate to 15R-HETE that can be converted by neutrophils and other cells to 15-epi-LXA4 and 15-epi-LXB4. LXA4 and 15-epi-LXA4 possess potent anti-inflammatory and pro-resolution actions predominantly through affecting the function of leukocytes. Lipoxins stimulate recruitment of monocytes and inhibit neutrophil trafficking and accumulation in inflamed tissues (reviewed in Serhan et al., 2007; Serhan et al., 2008). These actions are, in part, mediated through down-regulation of leukocyte Mac-1 expression (Fiore and Serhan, 1995; Filep et al., 1999). Accumulation of PGE2 at inflammatory sites induces a lipid mediator class switching from a predominantly 5-lipoxygenase activity to a 15-lipoxygenase activity generating LXA4 parallel with the resolution of inflammation (Levy et al., 2001) Thus, initiation of an inflammatory response would also activate subsequent pro-resolution mechanisms (Serhan and Savill, 2005).
Lipoxins exert multipronged actions to counter neutrophil responses to MPO. Down-regulation of Mac-1 expression on neutrophils and inhibition of neutrophil adhesion and transendothelial migration are important components of the anti-inflammatory activities of LXA4 and 15-epi-LXA4 (Serhan et al., 2008). 15-epi-LXA4 also prevents MPO-induced up-regulation of Mac-1 expression and MPO release, thereby interrupting MPO-mediated autocrine/paracrine loop for perpetuation of the inflammatory response (Figure 3) (El Kebir et al., 2009). 15-epi-LXA4 inhibition of NADPH oxidase-derived superoxide generation (Levy et al., 1999) and subsequent formation of ONOO− (József et al., 2002) result in reduced NF-κB activation and transcription of pro-inflammatory cytokines, such as IL-8 (József et al., 2002).
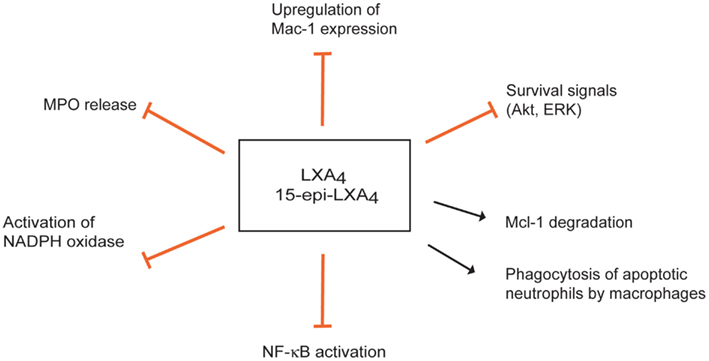
Figure 3. Multipronged actions of lipoxins to inhibit the myeloperoxidase (MPO)-Mac-1 circuit. Lipoxin A4 (LXA4) and aspirin-triggered 15-epi-LXA4 predominantly through FPR2/ALX attenuate MPO-stimulated degranulation, upregulation of surface expression of Mac-1, and superoxide formation, thus effectively interrupting this loop. LXA4 and15-epi-LXA4 redirect neutrophils to apoptosis by overriding the powerful survival signals from MPO through inducing loss of expression of Mcl-1 and aggravating mitochondrial dysfunction. Lipoxins also enhance phagocytosis of apoptotic neutrophils by macrophages.
Lipoxins themselves do not appear to interfere with the apoptotic machinery in neutrophils, whereas they can override the potent outside-in Mac-1-mediated survival signal and redirect neutrophils to apoptosis in vitro (El Kebir et al., 2009). 15-epi-LXA4 attenuates MPO-evoked ERK and Akt-mediated phosphorylation of the pro-apoptotic protein Bad and decreases Mcl-1 expression, critical events in enhancing neutrophil apoptosis. Non-phosphorylated Bad associates with Mcl-1 and prevents its anti-apoptotic actions (Reed, 2006). These would aggravate mitochondrial dysfunction, ultimately leading to caspase-3-mediated cell death (El Kebir et al., 2009; Wardle et al., 2011).
Treatment of mice with 15-epi-LXA4 at the peak of inflammation enhances resolution of carrageenan plus MPO-induced and E. coli septicemia-associated acute lung injury and improves the survival rate (El Kebir et al., 2009). 15-epi-LXA4 reduces pulmonary neutrophil accumulation with concomitant increases in the percentage of apoptotic neutrophils in the airways, facilitates recruitment of monocytes/macrophages and phagocytosis of apoptotic neutrophils and other cells (El Kebir et al., 2009), consistent with tissue repair (Godson et al., 2000; Mitchell et al., 2002). Furthermore, LXA4 released at sites of inflammation protects macrophages from apoptosis (Prieto et al., 2010). The beneficial actions of 15-epi-LXA4 can be prevented in the presence of a pan-caspase inhibitor, indicating the importance of neutrophil apoptosis in inflammatory resolution. Recent results indicate that aspirin or lovastatin reduction of acid aspiration-induced lung inflammation is, in part, mediated through stimulation of synthesis of 15-epi-LXA4 (Fukunaga et al., 2005; Planaguma et al., 2010). The direct effect of lovastatin on neutrophil apoptosis remains, however, to be investigated. Both aspirin and sodium salicylate promote neutrophil apoptosis and enhance their phagocytosis by macrophages in thioglycollate-induced peritonitis (Negrotto et al., 2006). A recent study reported that serum amyloid A acting through the formyl-peptide receptor 2/lipoxin receptor (FPR2/ALX) can overwhelm anti-inflammatory signaling by LXA4 to mediate exacerbation of glucocorticoid refractory lung inflammation in patients with chronic obstructive pulmonary disease (Bozinovski et al., 2012).
Resolvin E1 Promotes Phagocytosis-Induced Neutrophil Apoptosis
Resolvin E1 is synthesized from the ω-3 polyunsaturated fatty acid eicosapentaenoic acid during the resolution phase of acute inflammation with leukocyte 5-lipoxygenase playing a pivotal temporal role in the biosynthesis pathway (Serhan et al., 2000; Oh et al., 2011). RvE1 binds to the ChemR23 and (as a partial agonists/antagonist) the leukotriene B4 (LTB4) receptor BLT1 (Arita et al., 2005; Oh et al., 2011) and exhibits potent anti-inflammatory and pro-resolution activities. Thus, RvE1 inhibits neutrophil recruitment, facilitates efferocytosis (Serhan et al., 2002, 2008; Ohira et al., 2010; Oh et al., 2011; Serhan and Petasis, 2011), promotes mucosal surface clearance (Campbell et al., 2007), and induces generation of LXA4 (Haworth et al., 2008).These potent pro-resolution actions were also demonstrated in various experimental models, including peritonitis (Oh et al., 2011), polymicrobial sepsis (Schwab et al., 2007), and bacterial pneumonia (Seki et al., 2010) Moreover, studies on ChemR23-null mice demonstrated an endogenous anti-inflammatory role for ChemR23 (Cash et al., 2008).
Recent results indicate that RvE1 can modulate neutrophil apoptosis (El Kebir et al., 2012). While at low nanomolar concentrations, RvE1 per se does not affect the constitutive death program in neutrophils, it enhances Mac-1-mediated phagocytosis of complement-opsonized bacteria and yeast, leading to increased ROS generation by NADPH oxidase and subsequent activation of caspase-8 and caspase-3 (El Kebir et al., 2012). RvE1 also attenuates ERK and Akt-mediated survival cues generated by MPO and decreases Mcl-1 expression, thereby reinforcing the shift toward apoptosis (El Kebir et al., 2012). These actions of RvE1 are predominantly mediated via BLT1 in vitro, indicating that resolution mechanisms may also be activated via this type of LTB4 receptor. In contrast, RvE1 stimulation of phagocytosis of live E. coli and apoptotic neutrophils by macrophages leads to a macrophage phenotype switch without evoking apoptosis (Arita et al., 2005; Schwab et al., 2007; Oh et al., 2011). Since in macrophages RvE1 signals via ChemR23 (Ohira et al., 2010), RvE1 may exert different pro-resolution actions via distinct receptors, and concurrent activation of these circuits may be critical for optimal resolution. The neutrophil apoptosis-promoting action of RvE1 was also evident in experimental models of acute respiratory distress (ARDS) and pneumonia (El Kebir et al., 2012), in which MPO has been implicated in mediating lung injury. RvE1 administered at the peak of inflammation, promoted apoptosis in neutrophils in situ, enhanced recruitment of monocytes to the airways, and facilitated clearance of apoptotic neutrophils and other cells and tissue repair (El Kebir et al., 2012), consistent with the original properties defining RvE1 actions (Serhan et al., 2008). Efficient resolution of acute lung inflammation is intimately linked to apoptosis of neutrophils within the airways, as the pan-caspase inhibitor zVAD-fmk prevented RvE1-induced dramatic reduction in the number of infiltrating neutrophils (El Kebir et al., 2012) and aggravated lung injury likely due to persisting presence of neutrophils. Eicosapentaenoic acid is also a substrate for acetylated COX-2, which generates aspirin-triggered resolvins that shares anti-inflammatory actions of native resolvins (Spite and Serhan, 2010). These would raise the possibility that resolvin-triggered phagocytosis-induced neutrophil apoptosis could contribute to the beneficial actions of aspirin.
Other Approaches to Induce Neutrophil Apoptosis In vivo
Given the complexity of pathways involved in the regulation of neutrophil apoptosis, a number of agents have been identified that could shift the balance of survival and pro-apoptosis cues toward apoptosis. More importantly, some of these agents have been found to possess pro-resolution properties in diverse models of inflammation (see also Figure 4).
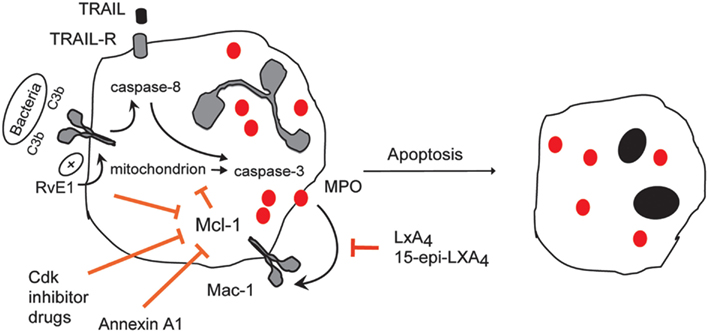
Figure 4. Therapeutic induction of neutrophil apoptosis with demonstrated pro-resolution actions in vivo. Treatment of experimental animals with CDK inhibitor drugs (e.g., R-roscovitine), 15-epi-LXA4, RvE1, rTRAIL, or annexin A1 enhance the resolution of inflammation by promoting apoptosis of neutrophils that have emigrated into tissues in various models of inflammation. Roscovitine through yet unidentified mechanisms, 15-epi-LXA4 through attenuating MPO-triggered Mac-1-mediated survival signaling and annexin A1 through dampening intracellular survival signaling down-regulate expression of the key survival protein Mcl-1. RvE1 enhances phagocytosis-induced apoptosis, leading to activation of caspase-8 and suppression of Mcl-1.rTRAIL activates caspase-8.
At the light of its pre-eminence as a key survival protein, Mcl-1 is an attractive target for therapeutic induction of apoptosis. The cyclin-dependent kinase inhibitor R-roscovitine (Seliciclib or CYC202) accelerates degradation of Mcl-1 and inhibits transcriptional activity in neutrophils by preventing cyclin-dependent kinase (CDK) 7 and 9-mediated phosphorylation of RNA polymerase II (Leitch et al., 2012), thereby inducing apoptosis in inflammatory cells in vitro (Rossi et al., 2006; Leitch et al., 2012). Consistently, treatment of with R-roscovitine enhances resolution of pleuritis and bleomycin-induced lung injury (Rossi et al., 2006) and pneumococcal meningitis in mice (Koedel et al., 2009), coinciding with increased numbers of apoptotic neutrophils in the airways and cerebrospinal fluid, respectively. R-roscovitine induces apoptosis in neutrophils from patients with cystic fibrosis, whereas the selective CFTR inhibitor CFTRInh172 does not affect constitutive apoptosis in neutrophils from healthy volunteers (Moriceau et al., 2010). Suppressed neutrophil apoptosis in cystic fibrosis is likely not simply a consequence of chronic infection. Since cystic fibrosis is associated with enhanced formation of MPO-generated oxidants, it would be interesting to know whether MPO is one of the yet unidentified modulatory factors intrinsic to CF.
The pro-resolution mediator annexin A1 binds to FPR2/ALX, which is also a receptor for LXA4 and 15-epi-LXA4, and accelerates neutrophil apoptosis in various murine models of inflammation by decreasing survival signals and Mcl-1 expression (Solito et al., 2003; Perretti and D’Acquisto, 2009; Perretti, 2012). A recent study reported that annexin A1 in inflammatory exudates promotes active resolution and augments neutrophil apoptosis in LPS-induced pleurisy in mice (Vago et al., 2012). Intriguingly, annexin A1-deficient mice exhibit an exaggerated inflammatory response and reduced Mac-1 expression on neutrophils (Hannon et al., 2003). Whether the pro-resolution actions of annexin A1 also involve modulation of signaling through Mac-1 remain to be explored. The phosphodiesterase 4 inhibitor rolipram has also recently been found to promote neutrophil apoptosis and resolution of LPS-induced pleurisy in mice by decreasing PI3K/Akt survival signaling and Mcl-1 expression (Sousa et al., 2010).
Another possibility is induction of caspase activity, the major effectors of apoptosis. Although selective caspase activators are currently not available, recent studies demonstrated the pro-resolving action of the death receptor ligand TRAIL, which may function as a physiological brake to limit the inflammatory response (Leitch et al., 2011). Thus, TRAIL-deficiency in mice is associated with delayed neutrophil apoptosis and exaggerated inflammatory response (Falschlehner et al., 2009; McGrath et al., 2011). Conversely, rTRAIL was found to facilitate neutrophil apoptosis through inducing activation of caspase-8 both in vitro and in vivo (Renshaw et al., 2003; McGrath et al., 2011). Consistent with enhanced neutrophil apoptosis, treatment with rTRAIL accelerated resolution of LPS-induced lung injury and zymosan-induced peritonitis in TRAIL-deficient mice (McGrath et al., 2011). Up-regulation of TRAIL has also been implicated in mediating TLR4 signaling through IFN-β to promote neutrophil apoptosis and limiting lung inflammation in a mouse model of ARDS (Leu et al., 2011).
Targeting pro-survival pathways to promote resolution has also been investigated at the level of MAPK and NF-κB signaling. As discussed above, pharmacological blockade of ERK 1/2 and/or PI3K prevents GM-CSF or MPO-induced neutrophil survival in vitro. The efficacy of the MEK/ERK inhibitor PD98059 in the resolution of inflammation was also demonstrated in the carrageenan-induced pleurisy model in rats (Sawatzky et al., 2006). NF-κB blockers have potential anti-inflammatory actions (Gosh and Hayden, 2008) and may also affect resolution. An oligonucleotide decoy to NF-κB was found to enhance neutrophil apoptosis and phagocytosis by macrophages in a rat model of chronic inflammation (Maiuri et al., 2004). Increased apoptosis correlated with increases in p53 or Bax expression and decreases in Bcl-2 protein expression. Systemic injection of a cell-permeable form of IκBα (Tat-srIκBα chimera) reduced leukocyte trafficking into the pleural cavity and increased caspase-3 activity and apoptosis in emigrated cells (Blackwell et al., 2004). In contrast, the Tat-srIκBα chimera produced only marginal reductions in neutrophil migration and apoptosis if administered locally (Blackwell et al., 2004), indicating that the route and timing of administration may be critical for exerting beneficial actions. In another study, selective NF-κB inhibitors failed to affect neutrophil accumulation in the pleural cavity (Sousa et al., 2010).
Conclusion
Intensive research during the past decade has revealed that inflammation does not terminate spontaneously; rather resolution is a tightly controlled active process. During the past decade, a number of novel mediators, including lipids, peptides, and proteins, and signaling circuits have been identified. Timely and efficient removal of migrated neutrophils requires these cells to undergo apoptosis. A growing body of evidence supports an important role for neutrophil apoptosis as a critical control point for the outcome of inflammation. Neutrophil surface receptors, including the adhesion molecule Mac-1 integrates opposing cues that modulate life and death decisions and therefore the outcome of inflammation. Outside-in signaling through Mac-1 is also a target for endogenous molecules, such as lipoxins, resolvin E1, and synthetic compounds to counter pro-survival and/or induce pro-apoptosis signals. Interfering with Mac-1 function may have two important benefits: inhibition of neutrophil trafficking into the inflamed site and acceleration of neutrophil clearance from inflamed tissues through the process of efferocytosis. Indeed, results from experimental models demonstrate that redirecting neutrophils to apoptosis and facilitating their clearance by macrophages are essential for enhancing resolution of acute inflammation. While clinical trials with these compounds remain distant, therapeutic induction of neutrophil apoptosis at the inflammatory site hold promise as a powerful pro-resolving intervention and may fulfill urgent, yet unmet clinical needs to prevent the deleterious consequences of inflammation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants MOP-67054 and MOP-97742 from the Canadian Institutes of Health Research (János G. Filep).
References
Abram, C. L., and Lowell, C. A. (2009). The ins and outs of leukocyte integrin signaling. Annu. Rev. Immunol. 27, 339–362.
Akgul, C., Moulding, D. A., and Edwards, S. W. (2001). Molecular control of neutrophil apoptosis. FEBS Lett. 487, 318–322.
Allen, L., Dockrell, D. H., Pattery, T., Lee, D. G., Cornelis, P., Hellewell, P. G., et al. (2005). Pyocyanin production by Pseudomonas aeruginosa induces neutrophil apoptosis and impairs neutrophil-mediated host defenses in vivo. J. Immunol. 174, 3643–3649.
Altznauer, F., Conus, S., Cavalli, A., Folkers, G., and Simon, H. U. (2004). Calpain-1 regulates Bax and subsequent Smac-dependent caspase-3 activation in neutrophil apoptosis. J. Biol. Chem. 279, 5947–5957.
Alvarado-Kristensson, M., Melander, F., Leandersson, K., Rónnstrand, L., Wernstedt, C., and Anderson, T. (2003). p38-MAPK signals survival by phosphorylation of caspase-8 and caspase-3 in human neutrophils. J. Exp. Med. 199, 449–458.
Ariel, A., Fredman, G., Sun, Y. P., Kantarci, A., Van Dyke, T. E., Luster, A. D., et al. (2006). Apoptotic neutrophils and T cells sequester chemokines during immune response resolution through modulation of CCR5 expression. Nat. Immunol. 7, 1209–1216.
Ariel, A., and Serhan, C. N. (2012). New lives given cell death: macrophage differentiation following their encounter with apoptotic leukocytes during the resolution of inflammation. Front. Immunol. 3:4. doi:10.3389/fimmu.2012.00004
Arita, M., Bianchini, F., Aliberti, J., Sher, A., Chiang, N., Hong, S., et al. (2005). Stereochemical assignment, antiinflammatory properties, and receptor for the ω3 lipid mediator resolvin E1. J. Exp. Med. 201, 713–722.
Arnhold, J., and Flemming, J. (2010). Human myeloperoxidase in innate and acquired immunity. Arch. Biochem. Biophys. 500, 92–106.
Arroyo, A., Modriansky, M., Serinkan, F. B., Bello, R. I., Matsura, T., Jiang, J., et al. (2002). NADPH oxidase-dependent oxidation and externalization of phosphatidylserine during apoptosis in Me2SO-differentiated HL-60 cells. Role in phagocytic clearance. J. Biol. Chem. 277, 49965–49975.
Baldus, S., Heeschen, C., Meinertz, T., Zeiher, A. M., Eiserich, J. P., Munzel, T., et al. (2003). Myeloperoxidase serum levels predict risk in patients with acute coronary syndromes. Circulation 108, 1440–1445.
Birnbaum, Y., Ye, Y., Lin, Y., Freeberg, S. Y., Nishi, S. P., Martinez, J. D., et al. (2006). Augmentation of myocardial production of 15-epi-lipoxin A4 by pioglitazone and atorvastatin in the rat. Circulation 114, 929–935.
Blackwell, N. M., Sembi, P., Newson, J. S., Lawrence, T., Gilroy, D. W., and Kabouridis, P. S. (2004). Reduced infiltration and increased apoptosis of leukocytes at sites of inflammation by systemic administration of a membrane-permeable IκBα repressor. Arthritis Rheum. 50, 2675–2684.
Borregaard, N., and Cowland, J. B. (1997). Granules of the human neutrophilic polymorphonuclear leukocyte. Blood 89, 3503–3521.
Bozinovski, S., Uddin, M., Vlahos, R., Thompson, M., McQualter, J. L., Merritt, A.-S., et al. (2012). Serum amyloid A opposes lipoxin A4 to mediate glucocorticoid refractory lung inflammation in chronic obstructive pulmonary disease. Proc. Natl. Acad. Sci. U.S.A. 109, 935–940.
Brennan, M. L., Anderson, M. M., Shih, D. M., Qu, X. D., Wang, X., Mehta, A. C., et al. (2001). Increased atherosclerosis in myeloperoxidase-deficient mice. J. Clin. Invest. 107, 419–430.
Brenner, D., and Mak, T. W. (2009). Mitochondrial cell death effectors. Curr. Opin. Cell Biol. 21, 871–877.
Brinkmann, V., Reichard, U., Goosmann, C., Fauler, B., Uhlemann, Y., Weiss, D. S., et al. (2004). Neutrophil extracellular traps kill bacteria. Science 303, 1532–1535.
Brovkovych, V., Gao, X. P., Ong, E., Brovkovych, S., Brennan, M. L., Su, X., et al. (2008). Augmented iNOS expression and increased NO production reduce sepsis-induced lung injury and mortality in myeloperoxidase-null mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 295, L96–L103.
Brown, R. A., Leung, E., Kankaanranta, H., Moilanen, E., and Page, C. P. (2012). Effects of heparin and related drugs on neutrophil function. Pulm. Pharmacol. Ther. 25, 185–192.
Brown, V., Elborn, J. S., Bradley, J., and Ennis, M. (2009). Dysregulated apoptosis and NF-κB expression in COPD subjects. Respir. Res. 10, 24.
Campbell, E. L., Louis, N. A., Tomassetti, S. E., Canny, G. O., Arita, M., Serhan, C. N., et al. (2007). Resolvin E1 promotes mucosal surface clearance of neutrophils: a new paradigm for inflammatory resolution. FASEB J. 21, 3162–3170.
Cartwright, G. E., Athens, J. W., and Wintrobe, M. M. (1964). The kinetics of granulopoesis in normal man. Blood 24, 780–803.
Cash, J. L., Hart, R., Russ, A., Dixon, J. P., Colledge, W. H., Doran, J., et al. (2008). Synthetic chemerin-derived peptides suppress inflammation through ChemR23. J. Exp. Med. 205, 767–775.
Catarzi, S., Marcucci, T., Papucci, L., Favilli, F., Donnini, M., Tonelli, F., et al. (2008). Apoptosis and Bax, Bcl-2, Mcl-1 expression in neutrophils of Crohn’s disease patients. Inflamm. Bowel Dis. 14, 819–825.
Chitnis, D., Dickerson, C., Munster, A. M., and Winchurch, R. A. (1996). Inhibition of apoptosis in polymorphonuclear neutrophils from burn patients. J. Leukoc. Biol. 59, 835–839.
Christenson, K., Björkman, L., Tängemo, C., and Bylund, J. (2008). Serum amyloid A inhibits apoptosis of human neutrophils via a P2X7-sensitive pathway independent of formyl peptide receptor-like 1. J. Leukoc. Biol. 83, 139–148.
Clària, J., and Serhan, C. N. (1995). Aspirin triggers previously undescribed bioactive eicosanoids by human endothelial cell-leukocyte interactions. Proc. Natl. Acad. Sci. U.S.A. 92, 9475–9479.
Colamussi, M. L., White, M. R., Crouch, E., and Hartshorn, K. L. (1999). Influenza A virus accelerates neutrophil apoptosis and markedly potentiates apoptotic effects of bacteria. Blood 93, 2395–2403.
Colotta, F., Re, F., Polentarutti, N., Sozzani, S., and Mantovani, A. (1992). Modulation of granulocyte survival and programmed cell death by cytokines and bacterial products. Blood 80, 2012–2020.
Conus, S., Perozzo, R., Reinheckel, T., Peters, C., Scapozza, L., Yousefi, S., et al. (2008). Caspase-8 is activated by cathepsin D initiating neutrophil apoptosis during the resolution of inflammation. J. Exp. Med. 205, 685–689.
Courtney, P. A., Crockard, A. D., Williamson, K., Irvine, A. E., Kennedy, R. J., and Bell, A. L. (1999). Increased apoptotic peripheral blood neutrophils in systemic lupus erythematosus: relations with disease activity, antibodies to double stranded DNA, and neutropenia. Ann. Rheum. Dis. 58, 309–314.
Coxon, A., Rieu, P., Barkalow, F. J., Askari, S., Sharpe, A. H., von Andrian, U. H., et al. (1996). A novel role for the beta 2 integrin, CD11b/CD18, in neutrophil apoptosis: a homeostatic mechanism in inflammation. Immunity 5, 653–666.
Daigle, I., Yousefi, S., Colonna, M., Green, D. R., and Simon, H. U. (2002). Death receptors bind SHP-1 and block cytokine-induced anti-apoptotic signaling in neutrophils. Nat. Med. 8, 61–67.
Davies, M. J., Hawkins, C. L., Pattison, D. I., and Rees, M. D. (2008). Mammalian heme oxidases: from molecular mechanisms to health implications. Antioxid. Redox Signal. 10, 1199–1234.
DeLeo, F. R. (2004). Modulation of phagocyte apoptosis by bacterial pathogens. Apoptosis 9, 399–413.
Derouet, M., Thomas, L., Moulding, D. A., Akgul, C., Cross, A., Moots, R. J., et al. (2006). Sodium salicylate promotes neutrophil apoptosis by stimulating caspase-dependant turn over of Mcl-1. J. Immunol. 176, 957–965.
Diamond, M. S., Alon, R., Parkos, C. A., Quinn, M. T., and Springer, T. A. (1995). Heparin is an adhesive ligand for the leukocyte integrin Mac-1. J. Cell Biol. 130, 1473–1481.
Dzhagalov, I., St John, A., and He, Y. W. (2007). The antiapoptotic protein Mcl-1 is essential for the survival of neutrophils but not macrophages. Blood 109, 1620–1626.
Edwards, S. W., Derouet, M., Howse, M., and Moots, R. J. (2004). Regulation of neutrophil apoptosis by Mcl-1. Biochem. Soc. Trans. 32, 489–492.
Ehlers, M. R. W. (2000). CR3: a general purpose adhesion-recognition receptor essential for innate immunity. Microbes Infect. 2, 289–294.
El Kebir, D., Gjorstrup, P., and Filep, J. G. (2012). Resolvin E1 promotes phagocytosis-induced neutrophil apoptosis and accelerates resolution of pulmonary inflammation. Proc. Natl. Acad. Sci. U.S.A. 109, 14983–14988.
El Kebir, D., József, L., Pan, W., and Filep, J. G. (2008). Myeloperoxidase delays neutrophil apoptosis through CD11b/CD18 integrins and prolongs inflammation. Circ. Res. 103, 352–359.
El Kebir, D., József, L., Pan, W., Petasis, N. A., Serhan, C. N., and Filep, J. G. (2007). Aspirin-triggered lipoxins override the apoptosis-delaying action of serum amyloid A in human neutrophils: a novel mechanism for resolution of inflammation. J. Immunol. 179, 616–622.
El Kebir, D., József, L., Pan, W., Wang, L., Petasis, N. A., Serhan, C. N., et al. (2009). 15-Epi-lipoxin A4 inhibits myeloperoxidase signaling and enhances resolution of acute lung injury. Am. J. Respir. Crit. Care Med. 180, 311–319.
Elbim, C., Katsikis, P. D., and Estaquier, J. (2009). Neutrophil apoptosis during viral infections. Open Virol. J. 3, 52–59.
Epling-Burnette, P. K., Zhong, B., Bai, F., Jiang, K., Bailey, R. D., Garcia, R., et al. (2001). Cooperative regulation of Mcl-1 by Janus kinase/stat and phosphatidylinositol 3-kinase contribute to granulocyte-macrophage colony-stimulating factor-delayed apoptosis in human neutrophils. J. Immunol. 166, 7486–7495.
Erduran, E., Tekelioglu, Y., Gedik, Y., and Yildiran, A. (1999). Apoptotic effects of heparin on lymphoblasts, neutrophils and mononuclear cells: results of a preliminary in vitro study. Am. J. Hematol. 61, 90–93.
Ertel, W., Keel, M., Infanger, M., Ungethum, U., Steckholzer, U., and Trentz, O. (1998). Circulating mediators in serum of injured patients with septic complications inhibit neutrophil apoptosis through up-regulation of protein-tyrosine phosphorylation. J. Trauma 44, 767–775.
Evans, R., Patzak, I., Svensson, L., De Filippo, K., Jones, K., McDowall, A., et al. (2009). Integrins in immunity. J. Cell. Sci. 122, 215–225.
Fadeel, B., Ahlin, A., Henter, J. I., Orrenius, S., and Hampton, M. B. (1998). Involvement of caspases in neutrophil apoptosis: regulation by reactive oxygen species. Blood 92, 4808–4818.
Fadok, V. A., Bratton, D. L., Konowal, A., Freed, P. W., Westcott, J. Y., and Henson, P. M. (1998). Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-β, PGE2, and PAF. J. Clin. Invest. 101, 890–898.
Falschlehner, C., Schaefer, U., and Walczak, H. (2009). Following TRAIL’s path in the immune system. Immunology 127, 145–154.
Filep, J. G., and El Kebir, D. (2009). Neutrophil apoptosis: a target for enhancing the resolution of inflammation. J. Cell. Biochem. 108, 1039–1046.
Filep, J. G., and El Kebir, D. (2010). Role of neutrophil apoptosis in the resolution of inflammation. ScientificWorldJournal 10, 1731–1748.
Filep, J. G., Zouki, C., Petasis, N. A., Hachicha, M., and Serhan, C. N. (1999). Anti-inflammatory actions of lipoxin A4 stable analogs are demonstrable in human whole blood: modulation of leukocyte adhesion molecules and inhibition of neutrophil-endothelial interactions. Blood 94, 4132–4142.
Fiore, S., and Serhan, C. N. (1995). Lipoxin A4 receptor activation is distinct from that of formyl peptide receptors in myeloid cells: inhibition of CD11/18 expression by lipoxin A4-lipoxin A4 receptor interaction. Biochemistry 34, 16678–16686.
Fossati, G., Moulding, D. A., Spiller, D. G., Moots, R. J., White, M. R., and Edwards, S. W. (2003). The mitochondrial network of human neutrophils: role in chemotaxis, phagocytosis, respiratory burst activation, and commitment to apoptosis. J. Immunol. 170, 1964–1972.
Fotouhi-Ardakani, N., El Kebir, D., Pierre-Charles, N., Wang, L., Ahern, S. P., Filep, J. G., et al. (2010). Role of myeloid nuclear differentiation antigen in the regulation of neutrophil apoptosis during sepsis. Am. J. Respir. Crit. Care Med. 182, 341–350.
Fox, S., Leitch, A. E., Duffin, R., Haslett, C., and Rossi, A. G. (2010). Neutrophil apoptosis: relevance to the innate immune response and inflammatory disease. J. Innate Immun. 2, 216–227.
Fuchs, T. A., Abed, U., Goosmann, C., Hurwitz, R., Schulze, I., Wahn, V., et al. (2007). Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 176, 231–241.
Fukunaga, K., Kohli, P., Bonnans, C., Fredenburgh, L. E., and Levy, B. D. (2005). Cyclooxygenase 2 plays a pivotal role in the resolution of acute lung injury. J. Immunol. 174, 5033–5039.
Furze, R. C., and Rankin, S. M. (2008). The role of bone marrow in neutrophil clearance under homeostatic conditions in the mouse. FASEB J. 22, 3111–3119.
Gardai, S., Whitlock, B. B., Helgason, C., Ambruso, D., Fadok, V., Bratton, D., et al. (2002). Activation of SHIP by NADPH oxidase-stimulated Lyn leads to enhanced apoptosis in neutrophils. J. Biol. Chem. 277, 5236–5246.
Garlichs, C. D., Eskafi, S., Cicha, I., Schmeisser, A., Walzog, B., Raaz, D., et al. (2004). Delay of neutrophil apoptosis in acute coronary syndromes. J. Leukoc. Biol. 75, 828–835.
Geering, B., and Simon, H.-U. (2011). Peculiarities of cell death mechanisms in neutrophils. Cell Death Differ. 18, 14657–11469.
Gilroy, D. W., Lawrence, T., Perretti, M., and Rossi, A. G. (2004). Inflammatory resolution: new opportunities for drug discovery. Nat. Rev. Drug Discov. 3, 401–416.
Godson, C., Mitchell, S., Harvey, K., Petasis, N. A., Hogg, N., and Brady, H. R. (2000). Cutting edge: lipoxins rapidly stimulate nonphlogistic phagocytosis of apoptotic neutrophils by monocyte-derived macrophages. J. Immunol. 164, 1663–1667.
Gosh, S., and Hayden, M. S. (2008). New regulators of NF-κB in inflammation. Nat. Rev. Immunol. 8, 837–848.
Hamasaki, A., Sendo, F., Nakayama, K., Ishida, N., Negishi, I., Nakayama, K., et al. (1998). Accelerated neutrophil apoptosis in mice lacking A1-a, a subtype of the bcl-2-related A1 gene. J. Exp. Med. 188, 1985–1992.
Hannon, R., Croxtall, J. D., Getting, S. J., Roviezzo, F., Yona, S., Paul-Clark, M. J., et al. (2003). Aberrant inflammation and resistance to glucocorticoids in Annexin 1−/− mouse. FASEB J. 17, 253–255.
Harris, E. S., McIntyre, T. M., Prescott, S. M., and Zimmerman, G. A. (2000). The leukocyte integrins. J. Biol. Chem. 275, 23409–23412.
Haslett, C. (1999). Granulocyte apoptosis and its role in the resolution and control of lung inflammation. Am. J. Respir. Crit. Care Med. 160, S5–S11.
Haworth, O., Cernadas, M., Yang, R., Serhan, C. N., and Levy, B. D. (2008). Resolvin E1 regulates interleukin 23, interferon gamma and lipoxin A4 to promote the resolution of allergic airway inflammation. Nat. Immunol. 8, 873–879.
Heyworth, P., Cross, A., and Curnutte, J. (2003). Chronic granulomatous disease. Curr. Opin. Immunol. 15, 578–584.
Johansson, M. W., Patarroyo, M., Oberg, F., Siegbahn, A., and Nilson, K. (1997). Myeloperoxidase mediates cell adhesion via the αMβ2 integrin (Mac-1, CD11b/CD18). J. Cell. Sci. 110, 1133–1139.
József, L., Khreiss, T., and Filep, J. G. (2004). CpG motifs in bacterial DNA delay apoptosis of neutrophil granulocytes. FASEB J. 18, 1776–1778.
József, L., Zouki, C., Petasis, N. A., Serhan, C. N., and Filep, J. G. (2002). Lipoxin A4 and aspirin-triggered 15-epi-lipoxin A4 inhibit peroxynitrite formation, NF-κB and AP-1 activation, and IL-8 gene expression in human leukocytes. Proc. Natl. Acad. Sci. U.S.A. 99, 13266–13271.
Kanayama, A., and Miyamoto, Y. (2007). Apoptosis triggered by phagocytosis-related oxidative stress through FLIPS down-regulation and JNK activation. J. Leukoc. Biol. 82, 1344–1352.
Karlsson, A., and Dahlgren, C. (2002). Assembly and activation of the neutrophil NADPH oxidase in granule membranes. Antioxid. Redox Signal. 4, 49–60.
Kasahara, Y., Iwai, K., Yachie, A., Ohta, K., Konno, A., Seki, H., et al. (1997). Involvement of reactive oxygen intermediates in spontaneous and CD95 (Fas/APO-1)-mediated apoptosis of neutrophils. Blood 89, 1748–1753.
Kato, T., Kutsuna, H., Oshitani, N., and Kitagawa, S. (2006). Cyclic AMP delays neutrophils apoptosis via stabilization of Mcl-1. FEBS Lett. 580, 4582–4586.
Kessel, J. M., Sedgwick, J. B., and Busse, W. W. (2006). Ligation of intercellular adhesion molecule 3 induces apoptosis of human blood eosinophils and neutrophils. J. Allergy Clin. Immunol. 118, 831–836.
Khreiss, T., József, L., Hossain, S., Chan, J. S. D., Potempa, L. A., and Filep, J. G. (2002). Loss of pentameric symmetry of C-reactive protein is associated with delayed apoptosis of human neutrophils. J. Biol. Chem. 277, 40775–40781.
Kim, C., Ye, F., and Grinsberg, M. H. (2011). Regulation of integrin activation. Annu. Rev. Cell Dev. Biol. 27, 321–345.
Klein, J. B., Rane, M. J., Scherzer, J. A., Coxon, P. Y., Kettritz, R., Mathiesen, J. M., et al. (2000). Granulocyte-macrophage colony-stimulating factor delays neutrophil constitutive apoptosis through phosphoinositide 3-kinase and extracellular signal-regulated kinase pathways. J. Immunol. 164, 4286–4291.
Kobayashi, S. D., Voyich, J. M., Buhl, C. L., Stahl, R. M., and DeLeo, F. R. (2002). Global changes in gene expression by human polymorphonuclear leukocytes during receptor-mediated phagocytosis: cell fate is regulated at the level of gene expression. Proc. Natl. Acad. Sci. U.S.A. 99, 6901–6906.
Koedel, U., Frankenburg, T., Kirschnek, S., Obermaier, B., Häcker, H., Paul, R., et al. (2009). Apoptosis is essential for neutrophil functional shutdown and determines tissue damage in experimental Pneumococcal meningitis. PLoS Pathog. 5:e1000461. doi:10.1371/journal.ppat.1000461
Laskay, T., van Zandbergen, G., and Solbach, W. (2008). Neutrophil granulocytes as host cells and transport vehicles for intracellular pathogens: apoptosis as infection-promoting factor. Immunobiology 213, 183–191.
Lau, D., Mollnau, H., Eiserich, J. P., Freeman, B. A., Daiber, A., Gehling, U. M., et al. (2005). Myeloperoxidase mediates neutrophil activation by association with CD11b/CD18 integrins. Proc. Natl. Acad. Sci. U.S.A. 102, 431–436.
Lee, A., Whyte, M. K., and Haslett, C. (1993). Inhibition of apoptosis and prolongation of neutrophil functional longevity by inflammatory mediators. J. Leukoc. Biol. 54, 283–288.
Lee, E., Lindo, T., Jackson, N., Meng-Choong, L., Reynolds, P., Hill, A., et al. (1999). Reversal of human neutrophil survival by leukotriene B4 receptor blockade and 5-lipoxygenase and 5-lipoxygenase activating protein inhibitors. Am. J. Respir. Crit. Care Med. 160, 2079–2085.
Lefort, C. T., Hyun, Y.-M., Schultz, J. B., Law, F.-Y., Waugh, R. E., Knauf, P. A., et al. (2009). Outside-in signal transmission by conformational changes in integrin Mac-1. J. Immunol. 183, 6460–6468.
Leitch, A. E., Lucas, C. D., Marwick, J. A., Duffin, R., Haslett, C., and Rossi, A. G. (2012). Cyclin-dependent kinases 7 and 9 specifically regulate neutrophils transcription and their inhibition drives apoptosis to promote resolution of inflammation. Cell Death Differ. 19, 1950–1961.
Leitch, A. E., Lucas, C. D., and Rossi, A. G. (2011). Neutrophil apoptosis: hot on the TRAIL of inflammatory resolution. J. Leukoc. Biol. 90, 841–843.
Leitch, A. E., Riley, N. A., Sheldrake, T. A., Festa, M., Fox, S., Duffin, R., et al. (2010). The cyclin-dependent kinase inhibitor R-roscovitine down-regulates Mcl-1 to override pro-inflammatory signaling and drive neutrophils apoptosis. Eur. J. Immunol. 40, 1127–1138.
Leu, S. W., Shi, L., Xu, C., Zhao, Y., Liu, B., Li, Y., et al. (2011). TLR4 through IFN-β promotes low molecular mass hyaluronan-induced neutrophil apoptosis. J. Immunol. 186, 556–562.
Leuenroth, S. J., Grutkoski, P. S., Ayala, A., and Simms, H. H. (2000). Suppression of PMN apoptosis by hypoxia is dependent on Mcl-1 and MAPK activity. Surgery 128, 171–177.
Levy, B. D., Clish, C. B., Schmidt, B., Gronert, C., and Serhan, C. N. (2001). Lipid mediator class switching during acute inflammation: signals in resolution. Nat. Immunol. 2, 612–619.
Levy, B. D., Fokin, V. V., Clark, J. M., Wakelam, M. J., Petasis, N. A., and Serhan, C. N. (1999). Polyisoprenyl phosphate (PIPP) signaling regulates phospholipase D activity: a “stop” signaling switch for aspirin-triggered lipoxin A4. FASEB J. 13, 903–911.
Ley, K., Laudanna, C., Cybulsky, M. I., and Nourshargh, S. (2007). Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat. Rev. Immunol. 7, 678–689.
Lindemans, C., Coffer, P. J., Schellens, I. M. M., de Graff, P. M. A., Kimpen, J. L. L., and Koenderman, L. (2006). Respiratory syncytial virus inhibits granulocyte apoptosis through a phosphatidylinositol 3-kinase and NF-κB-dependent mechanism. J. Immunol. 176, 5529–5537.
Lindsten, T., Ross, A. J., King, A., Zong, W. X., Rathmell, J. C., Shiels, H. A., et al. (2000). The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol. Cell 6, 1389–1399.
Luo, B. H., Carman, C. V., and Springer, T. A. (2007). Structural basis of integrin regulation and signaling. Annu. Rev. Immunol. 25, 619–647.
Maianski, N. A., Geissler, J., Srinivasula, S. M., Alnemri, E. S., Roos, D., and Kuijpers, T. W. (2004). Functional characterization of mitochondria in neutrophils: a role restricted to apoptosis. Cell Death Differ. 11, 143–153.
Maiuri, M. C., Tajana, G., Iuvone, T., De Stefano, D., Mele, G., Ribecco, M. T., et al. (2004). Nuclear factor-κB regulates inflammatory cell apoptosis and phagocytosis in rat carrageenan-sponge implant model. Am. J. Pathol. 165, 115–126.
Malle, E., Furtmüller, P. G., Sattler, W., and Obinger, C. (2007). Myeloperoxidase: a target for new drug development? Br. J. Pharmacol. 152, 838–854.
Manaster, J., Chezar, J., Schurtz-Swirski, R., Shapiro, G., Tendler, Y., Kristal, B., et al. (1996). Heparin induces apoptosis in human peripheral blood neutrophils. Br. J. Haematol. 94, 48–52.
Matthijsen, R. A., Huugen, D., Hoebers, N. T., de Vries, B., Peutz-Kootstra, C. J., Aratani, Y., et al. (2007). Myeloperoxidase is critically involved in the induction of organ damage after renal ischemia reperfusion. Am. J. Pathol. 171, 1743–1752.
Matute-Bello, G., Liles, W. C., Radella, F. II, Steinberg, K. P., Ruzinski, J. T., Jonas, M., et al. (1997). Neutrophil apoptosis in the acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 156, 1969–1977.
Mayadas, T. N., and Cullere, X. (2005). Neutrophil β2 integrins: moderators of life and death decisions. Trends Immunol. 26, 388–395.
McGrath, E. E., Marriott, H. M., Lawrie, A., Francis, S. E., Sabroe, I., Renshaw, S. A., et al. (2011). TNF-related apoptosis-inducing ligand (TRAIL) regulates inflammatory neutrophil apoptosis and enhances resolution of inflammation. J. Leukoc. Biol. 90, 855–865.
McKeon, D. J., Condliffe, A. M., Cowburn, A. S., Cadwallader, K. C., Farahi, N., Bilton, D., et al. (2008). Prolonged survival of neutrophils from patients with delta F508 CFTR mutations. Thorax 63, 660–661.
Melley, D. D., Evans, T. W., and Quinlan, G. J. (2005). Redox regulation of neutrophil apoptosis and the systemic inflammatory response syndrome. Clin. Sci. 108, 413–424.
Mitchell, S., Thomas, G., Harvey, K., Cottel, D., Reville, K., Berlasconi, G., et al. (2002). Lipoxins, aspirin-triggered epi-lipoxins, lipoxin stable analogues, and the resolution of inflammation: stimulation of macrophage phagocytosis of apoptotic neutrophils in vivo. J. Am. Soc. Nephrol. 13, 2497–2507.
Miyamoto, M., Emoto, M., Emoto, Y., Brinkmann, V., Yoshizawa, I., Seiler, P., et al. (2003). Neutrophilia in LFA-1-deficient mice confers resistance to listerosis: possible contribution of granulocyte-colony-stimulating factor and IL-17. J. Immunol. 170, 5228–5234.
Moriceau, S., Lenoir, G., and Witko-Sarsat, V. (2010). In cystic fibrosis homozygotes and heterozygotes, neutrophil apoptosis is delayed and modulated by diamide or roscovitine: evidence for an innate neutrophil disturbance. J. Innate Immun. 2, 260–266.
Moulding, D. A., Akgul, C., Derouet, M., White, M. R., and Edwards, S. W. (2001). BCL-2 family expression in human neutrophils during delayed and accelerated apoptosis. J. Leukoc. Biol. 70, 783–792.
Moulding, D. A., Giles, R. V., Spiller, D. G., White, M. R., Tidd, D. M., and Edwards, S. W. (2000). Apoptosis is rapidly triggered by antisense depletion of MCL-1 in differentiating U937 cells. Blood 96, 1756–1763.
Moulding, D. A., Qayle, J. A., Hart, C. A., and Edwards, S. W. (1989). Mcl-1 expression in human neutrophils: regulation by cytokines and correlation with cell survival. Blood 92, 2495–2502.
Nathan, C. (2006). Neutrophils and immunity: challenges and opportunities. Nat. Rev. Immunol. 6, 173–182.
Nauseef, W. M. (2007). How human neutrophils kill and degrade microbes: an integrated view. Immunol. Rev. 219, 88–102.
Negrotto, S., Malaver, E., Alvarez, M. E., Pacienza, N., D’Atri, L. P., Pozner, R. G., et al. (2006). Aspirin and salicylate suppress polymorphonuclear apoptosis delay mediated by proinflammatory stimuli. J. Pharmcol. Exp. Ther. 319, 972–979.
Oh, S. F., Pillai, P. S., Recchiuti, A., Yang, R., and Serhan, C. N. (2011). Pro-resolving actions and stereoselective biosynthesis of 18S E-series resolvins in human leukocytes and murine inflammation. J. Clin. Invest. 121, 569–581.
Ohira, T., Arita, M., Omori, K., Recchiuti, A., van Dyke, T. E., and Serhan, C. N. (2010). Resolvin E1 receptor activation signals phosphorylation and phagocytosis. J. Biol. Chem. 285, 3451–3461.
Park, J. S., Arcaroli, J., Yum, H. K., Yang, H., Wang, H., Yang, K. Y., et al. (2003). Activation of gene expression in human neutrophils by high mobility group box 1 protein. Am. J. Physiol. Cell Physiol. 284, C870–C879.
Perretti, M. (2012). To resolve or not to resolve: annexin A1 pushes resolution on track. J. Leukoc. Biol. 92, 245–247.
Perretti, M., and D’Acquisto, F. (2009). Annexin A1 and glucocorticoids as effectors of the resolution of inflammation. Nat. Rev. Immunol. 9, 62–70.
Perskvist, N., Long, M., Stendahl, O., and Zheng, L. (2002). Mycobacterium tuberculosis promotes apoptosis in human neutrophils by activating caspase-3 and altering expression of Bax/Bcl-xL via an oxygen-dependent pathway. J. Immunol. 168, 6358–6365.
Peters, K., Schwarz, M., Conradt, C., Nordt, T., Moser, M., Kübler, W., et al. (1999). Heparin inhibits ligand binding to the leukocyte integrin Mac-1 (CD11b/CD18). Circulation 100, 1533–1539.
Pillay, J., den Braber, I., Vrisekoop, N., Kwast, L. M., de Boer, R. J., Borghans, J. A., et al. (2010). In vivo labeling with 2H2O reveals a human neutrophil lifespan of 5.4 days. Blood 116, 625–627.
Pillay, J., Kamp, V. M., van Hoffen, E., Visser, T., Tak, T., Lammers, J. W., et al. (2012). A subset of neutrophils in human systemic inflammation inhibits T cell responses through Mac-1. J. Clin. Invest. 122, 327–336.
Planaguma, A., Pfeffer, M. A., Rubin, G., Croze, R., Uddin, M., Serhan, C. N., et al. (2010). Lovastatin decreases acute mucosal inflammation via 15-epi-lipoxin A4. Mucosal Immunol. 3, 270–279.
Pluskota, E., Soloviev, D. A., Bdeir, K., Cines, D. B., and Plow, E. F. (2004). Integrin αMβ2 orchestrates and accelerates plasminogen activation and fibrinolysis by neutrophils. J. Biol. Chem. 279, 18063–18072.
Pluskota, E., Soloviev, D. A., Szpak, D., Weber, C., and Plow, E. F. (2008). Neutrophil apoptosis: selective regulation by different ligands of integrin αMβ2. J. Immunol. 181, 3609–3619.
Prieto, P., Cuenca, J., Través, P. G., Fernández-Velasco, M., Martin-Saenz, P., and Boscá, L. (2010). Lipoxin A4 impairment of apoptotic signaling in macrophages: implication of the PI3K/Akt and the ERK/Nrf-2 defense pathways. Cell Death Differ. 17, 1179–1188.
Reed, J. C. (2006). Proapoptotic multidomain Bcl-1/Bax family proteins: mechanisms, physiological roles and therapeutic opportunities. Cell Death Differ. 13, 1378–1386.
Remijsen, Q., Van den Berghe, T., Wirawan, E., Asselbergh, B., Parthoens, E., De Rycke, R., et al. (2011). Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res. 21, 290–304.
Ren, Y., Xie, Y., Jiang, G., Fan, J., Yeung, J., Li, W., et al. (2008). Apoptotic cells protect mice against lipopolysaccharide-induced shock. J. Immunol. 180, 4978–4985.
Renshaw, S. A., Parmar, J. S., Singleton, V., Rowe, S. J., Dockrell, D. H., Dower, S. K., et al. (2003). Acceleration of human neutrophil apoptosis by TRAIL. J. Immunol. 170, 1027–1033.
Rosales, J. L., Ernst, J. D., Hallows, J., and Lee, K. Y. (2004). GTP-dependent secretion from neutrophils is regulated by Cdk5. J. Biol. Chem. 279, 53932–53936.
Ross, G. D. (2000). Regulation of the adhesion versus cytotoxic functions of Mac-1/CR-3/α-m β-2 integrin glycoprotein. Crit. Rev. Immunol. 20, 197–222.
Rossi, A. G., Sawatzky, D. A., Walker, A., Ward, C., Sheldrake, T. A., Riley, N. A., et al. (2006). Cyclin-dependent kinase inhibitors enhance the resolution of inflammation by promoting inflammatory cell apoptosis. Nat. Med. 12, 1056–1064.
Rubel, C., Fernandez, G. C., Dran, G., Bompadre, M. B., Isturiz, M. A., and Palermo, M. S. (2001). Fibrinogen promotes neutrophil activation and delays apoptosis. J. Immunol. 166, 2002–2010.
Rubel, C., Gomez, S., Fernandez, G. C., Isturiz, M. A., Caamano, J., and Palermo, M. S. (2003). Fibrinogen-CD11b/CD18 interaction activates the NF-κB pathway and delays apoptosis in human neutrophils. Eur. J. Immunol. 33, 1429–1438.
Rupp, J., Pfleiderer, L., Jugert, C., Moeller, S., Klinger, M., Dalhoff, K., et al. (2009). Chlamydia pneumoniae hides inside apoptotic neutrophils to silently infect and propagate in macrophages. PLoS ONE 4:e6020. doi:10.1371/journal.pone.0006020
Saelens, X., Festjens, N., Van de Walle, L., van Gurp, M., van Loo, G., and Vandenabeele, P. (2004). Toxic proteins released from mitochondria in cell death. Oncogene 23, 2861–2874.
Salamone, G., Trevani, A., Martinez, D., Vermeulen, M., Gamberale, R., Fernandez-Calotti, P., et al. (2004). Analysis of mechanisms involved in the stimulation of neutrophil apoptosis by tumor necrosis factor-α. Immunology 113, 355–362.
Savill, J., Dransfield, I., Gregory, C., and Haslett, C. (2002). A blast from the past: clearance of apoptotic cells regulates immune responses. Nat. Rev. Immunol. 2, 965–975.
Savill, J. S., Wyllie, A. H., Hanson, J. E., Walport, M. J., Hanson, P. M., and Haslett, J. C. (1989). Macrophage phagocytosis of aging neutrophils in inflammation. Programmed cell death in the neutrophils leads to its recognition by macrophages. J. Clin. Invest. 83, 865–875.
Sawatzky, D. A., Willoughby, D. A., Colville-Nash, P. R., and Rossi, A. G. (2006). The involvement of the apoptosis-modulating proteins ERK 1/2, Bcl-xL and Bax in the resolution of acute inflammation in vivo. Am. J. Pathol. 168, 33–41.
Schultz, J., and Kaminker, K. (1962). Myeloperoxidase of the leukocyte of normal human blood. I. Content and localization. Arch. Biochem. Biophys. 96, 465–467.
Schwab, J. M., Chiang, N., Arita, M., and Serhan, C. N. (2007). Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature 447, 869–874.
Seki, H., Fukunaga, K., Arita, M., Arai, H., Nakanishi, H., Taguchi, R., et al. (2010). The anti-inflammatory and proresolving mediator resolving E1 protects mice from bacterial pneumonia and acute lung injury. J. Immunol. 184, 836–843.
Serhan, C. N. (2011). The resolution of inflammation: the devil in the flask and in the details. FASEB J. 25, 1441–1448.
Serhan, C. N., Brain, S. D., Buckley, C. D., Gilroy, D. W., Haslett, C., O’Neill, L. A. J., et al. (2007). Resolution of inflammation: state of the art, definitions and terms. FASEB J. 21, 325–332.
Serhan, C. N., Chiang, N., and Van Dyke, T. (2008). Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat. Rev. Immunol. 8, 349–361.
Serhan, C. N., Clish, C. B., Brannon, J., Colgan, S. P., Chiang, N., and Gronert, C. (2000). Novel functional sets of lipid-derived mediators with antiinflammatory actions generated from ω-3 fatty acids via cyclooxygenase2-nonsteroidal antiinflammatory drugs and transcellular processing. J. Exp. Med. 192, 1197–1204.
Serhan, C. N., Hong, S., Gronert, K., Colgan, S. P., Devchand, P. R., Mirick, G., et al. (2002). Resolvins: a family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J. Exp. Med. 196, 1025–1037.
Serhan, C. N., and Petasis, N. A. (2011). Resolvins and protectins in inflammation resolution. Chem. Rev. 111, 5922–5943.
Serhan, C. N., and Savill, J. (2005). Resolution of inflammation: the beginning programs the end. Nat. Immunol. 6, 1191–1197.
Sica, A., and Mantovani, A. (2012). Macrophage plasticity and polarization: in vivo veritas. J. Clin. Invest. 122, 787–795.
Solito, E., Kamal, A., Russo-Marie, F., Buckingham, J. C., Marullo, S., and Perretti, M. (2003). A novel calcium-dependent proapoptotic effect of annexin 1 on human neutrophils. FASEB J. 17, 1544–1546.
Sousa, L. P., Lopes, F., Silva, D. M., Tavares, L. P., Vieira, A. T., Rezende, B. M., et al. (2010). PDE4 inhibition drives resolution of neutrophilic inflammation by inducing apoptosis in a PKA-PI3K/Akt-dependent and NF-κB-independent manner. J. Leukoc. Biol. 87, 895–904.
Spite, M., and Serhan, C. N. (2010). Novel lipid mediators promote resolution of acute inflammation. Impact of aspirin and statins. Circ. Res. 107, 1170–1184.
Steimer, D. A., Boyd, K., Takeuchi, O., Fisher, J. K., Zambetti, G. P., and Opferman, J. T. (2009). Selective roles for antiapoptotic MCL-1 during granulocyte development and macrophage effector function. Blood 113, 2805–2815.
Vago, J. P., Nogueira, C. R. C., Tavares, L. P., Soriani, F. M., Lopes, F., Russo, R. C., et al. (2012). Annexin A1 modulates natural and glucocorticoid-induce resolution of inflammation by enhancing neutrophil apoptosis. J. Leukoc. Biol. 92, 249–258.
Wagner, B. A., Buettner, G. R., Oberley, L. W., Darby, C. J., and Burns, P. C. (2000). Myeloperoxidase is involved in H2O2-induced apoptosis of HL-60 human leukemia cells. J. Biol. Chem. 275, 22461–22469.
Ward, C., Walker, A., Dransfield, I., Haslett, C., and Rossi, A. G. (2004). Regulation of granulocyte apoptosis by NF-κB. Biochem. Soc. Trans. 32, 465–467.
Wardle, D. J., Burgon, J., Sabroe, I., Bingle, C. D., Whyte, M. K. B., and Renshaw, S. A. (2011). Effective caspase inhibition blocks neutrophils apoptosis and reveals Mcl-1 as both a regulator and a target of neutrophils caspase activation. PLoS ONE 6:e15768. doi:10.1371/journal.pone.0015768
Watson, R. W., O’Neill, A., Brannigan, A. E., Coffey, R., Marshall, J. C., Brady, H. R., et al. (1999). Regulation of Fas antibody induced neutrophil apoptosis is both caspase and mitochondrial dependent. FEBS Lett. 453, 67–71.
Watson, R. W. G. (2002). Redox regulation of neutrophil apoptosis. Antioxid. Redox Signal. 4, 97–104.
Watson, R. W. G., Redmond, H. P., Wang, J. H., Condron, C., and Bouchier-Hayes, D. (1996). Neutrophils undergo apoptosis following ingestion of Escherichia coli. J. Immunol. 156, 3986–3992.
Watson, R. W. G., Rotstein, O. D., Nathens, A. B., Parodo, J., and Marshall, J. C. (1997). Neutrophil apoptosis is modulated by endothelial transmigration and adhesion molecule engagement. J. Immunol. 158, 945–953.
Whitlock, B. B., Gardai, S., Fadok, V., Bratton, D., and Henson, P. M. (2000). Differential roles for α(M)β(2) integrin clustering or activation in the control of apoptosis via regulation of Akt and ERK survival mechanisms. J. Cell Biol. 151, 1305–1320.
Winterbourn, C. C. (2008). Reconciling the chemistry and biology of reactive oxygen species. Nat. Chem. Biol. 4, 278–286.
Witko-Sarsat, V., Mocek, J., Bouayad, D., Tamassia, N., Ribeil, J. A., Candalh, C., et al. (2010). Proliferating cell nuclear antigen acts as a cytoplasmic platform controlling human neutrophil survival. J. Exp. Med. 207, 2631–2645.
Wong, S. H., Francis, N., Chahal, H., Raza, K., Salmon, M., Scheel-Toellner, D., et al. (2009). Lactoferrin is a survival factor for neutrophils in rheumatoid synovial fluid. Rheumatology 48, 39–44.
Xiong, J. P., Stehle, T., Diefenbach, B., Zhang, R., Dunker, R., Scott, D. L., et al. (2001). Crystal structure of the extracellular segment of integrin αVβ3. Science 294, 339–345.
Xu, Y., Loison, F., and Luo, H. R. (2010). Neutrophil spontaneous death is mediated by down-regulation of autocrine signaling through GPCR, PI3kγ, ROS, and actin. Proc. Natl. Acad. Sci. U.S.A. 107, 2950–2955.
Yan, S. R., and Berton, G. (1996). Regulation of src family tyrosine kinase activities in adherent human neutrophils: evidence that reactive oxygen intermediates produced by adherent neutrophils increase the activity of the p58-c-fgr and p53/56lyn tyrosine kinases. J. Biol. Chem. 271, 23464–23471.
Yan, S. R., Sapru, K., and Issekutz, A. C. (2004). The CD11/CD18 (beta2) integrins modulate neutrophil caspase activation and survival following TNF-alpha or endotoxin induced transendothelial migration. Immunol. Cell Biol. 82, 435–446.
Zhang, B., Hirahashi, J., Cullere, X., and Mayadas, T. N. (2003). Elucidation of molecular events leading to neutrophils apoptosis following phagocytosis. J. Biol. Chem. 278, 28443–28454.
Keywords: neutrophils, Mac-1, apoptosis, lipoxins, resolvins, myeloperoxidase, phagocytosis, resolution of inflammation
Citation: El Kebir D and Filep JG (2013) Modulation of neutrophil apoptosis and the resolution of inflammation through β2 integrins. Front. Immunol. 4:60. doi: 10.3389/fimmu.2013.00060
Received: 28 August 2012; Accepted: 21 February 2013;
Published online: 06 March 2013.
Edited by:
Lyle Leonard Moldawer, University of Florida College of Medicine, USAReviewed by:
Carolyn Louise Geczy, University of New South Wales, AustraliaDennis D. Taub, National Institutes of Health, USA
Copyright: © 2013 El Kebir and Filep. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: János G. Filep, Research Center, Maisonneuve-Rosemont Hospital, 5415 Boulevard de l’Assomption, Montreal, QC H1T 2M4, Canada. e-mail:amFub3MuZy5maWxlcEB1bW9udHJlYWwuY2E=