 Fiona J. Whelan
Fiona J. Whelan Nicholas V. L. Yap
Nicholas V. L. Yap Michael G. Surette
Michael G. Surette G. Brian Golding2
G. Brian Golding2 Dawn M. E. Bowdish
Dawn M. E. Bowdish- 1Department of Biochemistry and Biomedical Sciences, McMaster University, Hamilton, ON, Canada
- 2Department of Biology, McMaster University, Hamilton, ON, Canada
- 3Department of Pathology and Molecular Medicine, McMaster University, Hamilton, ON, Canada
Bioinformatics includes a suite of methods, which are cheap, approachable, and many of which are easily accessible without any sort of specialized bioinformatic training. Yet, despite this, bioinformatic tools are under-utilized by immunologists. Herein, we review a representative set of publicly available, easy-to-use bioinformatic tools using our own research on an under-annotated human gene, SCARA3, as an example. SCARA3 shares an evolutionary relationship with the class A scavenger receptors, but preliminary research showed that it was divergent enough that its function remained unclear. In our quest for more information about this gene – did it share gene sequence similarities to other scavenger receptors? Did it contain conserved protein domains? Where was it expressed in the human body? – we discovered the power and informative potential of publicly available bioinformatic tools designed for the novice in mind, which allowed us to hypothesize on the regulation, structure, and function of this protein. We argue that these tools are largely applicable to many facets of immunology research.
Introduction
Although public perception indicates that bioinformatics is a relatively new discipline borne out of the “omics” age, bioinformatics is more than just “data crunching” and, in some form, has been around longer than our understanding of how DNA translates into protein. The term “bioinformatics” was coined in 1970 by Hogeweg and Hesper to mean “the study of informatic processes in biotic systems” (1). In this sense, the interdisciplinary approach characteristic of bioinformatic’s combination of information science, mathematics, and biology is not a new venture. Even before the term was ever used, Erwin Schrodinger, recognizable for his thought experiments and developments in quantum mechanics (2), gave a series of lectures in war-time Ireland entitled What is Life? (3), encouraging many classically trained physicists and chemists, including Francis Crick and Rosalind Franklin, to turn their interests toward biology. These new recruits became some of the first interdisciplinary scientists. Since then, it has been used for a broad range of applications, including the Human Genome Project (4), the discovery of new drugs (5), and further elucidation of Darwin’s Tree of Life (6).
Just as bioinformatics can be applied to the study of human genetics and evolution, it can also be used to inform immunology research. This combination of immunology and computational biology is sometimes referred to as “immunomics” or “computational immunology.” Bioinformatic techniques have been used to model how major histocompatibility complex (MHC) heterozygosity affects one’s interaction with bacteria (7) and the influenza virus (8), how host stress affects the pathogenicity of Pseudomonas aeruginosa in the human gut (9), and why the frequency of staphylococcal-induced toxic stress response is low even though infections by these bacteria are high (10). While some of these investigations require a user to have extensive knowledge of computational science, increasingly, bioinformatic tools are equipped with intuitive graphical user interfaces and so are more accessible to those without such a background. Many powerful and informative results can be generated with an Internet connection and a DNA sequence of interest. The plethora of publicly available, easy-to-use bioinformatic tools that investigate nucleotide or protein sequences, can provide information about potential post-translational modifications, predict protein structure and gene expression, and document genetic variation within a population, species, or kingdom. Within minutes, information can be generated to guide in vitro experiments, which can save the typical bench scientist both time and resources.
This review uses recent examples of our own quest to seek out information on a potential member of the class A scavenger receptor family, SCARA3, via publicly available bioinformatic tools. The scavenger receptors are a family of proteins required for host defense and phagocytosis of senescent cells and modified proteins (11). Although SCARA3 is a member of this family, there is very little information on its structure or function. Through an example of our bioinformatic analyses of the SCARA3 gene, this review aims to explain how approachable and accessible bioinformatic tools can be used to obtain sequence and structural information, gene expression patterns, genetic variation across human populations and, most importantly, to generate informed hypotheses that can be tested bench-side.
Sequence Analysis
Acquiring a FASTA Sequence from a Public Online Database
The FASTA file format was originally described by William R. Pearson as part of his 1990 bioinformatic software package of the same name (12). Since this time, it has become the de facto file format for most, if not all, bioinformatic sequence analyses. Simply put, this format is a description of a sequence preceded by a greater-than (“ >”) symbol, followed by the sequence in the standard IUPAC nucleotide or protein code.
An accurately annotated and appropriately formatted sequence of the gene(s) of interest is a prerequisite of many bioinformatic techniques. Since 2007, the National Center for Biotechnology Information (NCBI) has made the nucleotide sequences of more than 260,000 organisms accessible through its publicly available database, GenBank (13). GenBank’s global coverage of sequence data is ensured by daily exchanges of information with the European Molecular Biology Laboratory’s (EMBL) Nucleotide Sequence Database, and the DNA Data Bank of Japan (DDBJ) (13). The information stored in GenBank is made accessible through Entrez, NCBI’s comprehensive search engine (13). Users of Entrez have the option of searching within specific databases, such as nucleotide and protein sequences, Expressed Sequence Tags (ESTs), and macromolecular structures (14).
One such database is Entrez Gene, which provides gene-centered information (15). Entrez Gene includes only those gene records corresponding to genomes which have been fully sequenced or to genes that have active research groups associated with them (15); searches of this or other curated databases avoid poor search results. Additionally, because some annotations in complete genomes are quite suspect, the use of Entrez Gene prevents the use of inappropriately annotated or low quality sequences. Searching this database provides useful information such as the “Genomic regions, transcripts, and products” section, which is helpful in visualizing the exonic structure and chromosomal orientation of a gene. The “Bibliography” section summarizes peer-reviewed articles in which the gene is at the forefront. Additionally, a multiple sequence alignment of the gene of interest to known homologs can be generated by choosing the “Homology” section under “General gene information”; this may be of interest to those conducting cross-species or evolutionary studies.
When gathering sequence data, the user should refer to the section entitled “NCBI Reference Sequences (RefSeq)” (Figure 1). Using RefSeqs is important because these sequences meet a stringent standard set by NCBI, including the assurance that supporting evidence for the gene is available (16). Here, at least one set of mRNA and protein sequences will be displayed; isoforms of a given protein are displayed with multiple entries.
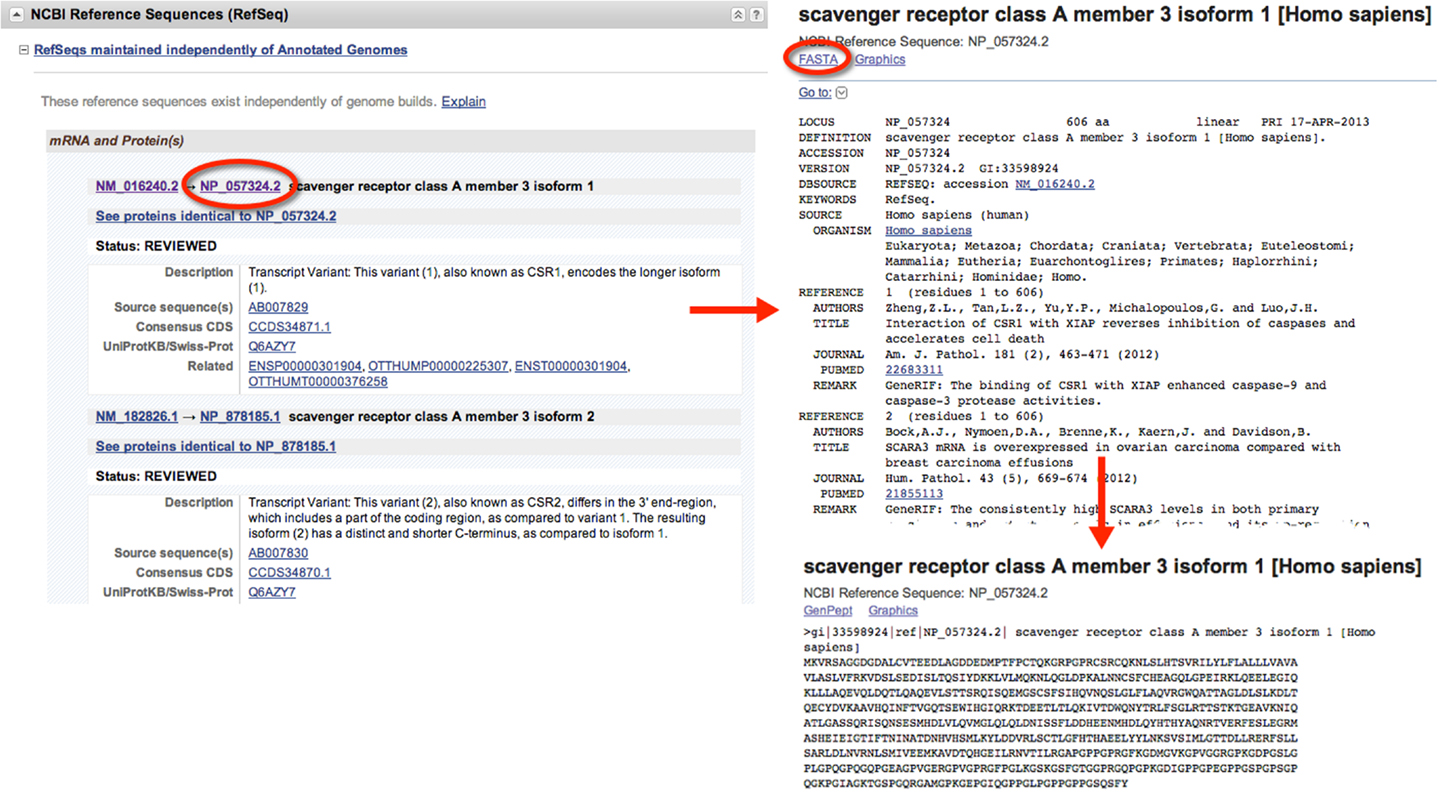
Figure 1. Retrieval of nucleic acid and protein FASTA formatted sequences from an Entrez Gene search. Upon searching for and selecting the Homo sapiens SCARA3 gene, a variety of information can be retrieved including identifiers for the Ensembl, Mendelian Inheritance of Man (MIM), and Human Protein Reference Database, in addition to information about the genomic context of the gene. From the “NCBI Reference Sequences (RefSeq)” section, the most up-to-date and thoroughly curated FASTA formatted sequences may be obtained. Sequences with Accession Identifiers beginning with NM or XM are mRNA and NP or XP are protein. Multiple RefSeq entries may be present in the case of gene isoforms. Selecting the NP_057324.2 Accession Identifier, information concerning the SCARA3 isoform 1, protein is displayed, including links to publications involving this protein. By selecting “FASTA” at the top of the page, the FASTA formatted sequence is provided, which includes the reference number, species, and name. This sequence is suitable for input into most online bioinformatic tools.
Although we have chosen to use the NCBI’s Entrez platform in this example it should be noted that there are other equally appropriate databases available. Although it is beyond the scope of this review to describe them in detail, Table 1 provides an overview.

Table 1. Public databases containing DNA, mRNA and protein sequences.
Predicting Post-Translational Modifications
Post-translational modifications of a protein can include phosphorylation, glycosylation, ubiquitination, methylation, and lipidation amongst many others. Post-translational modification may change the function, cellular localization, or abundance of a protein. Just as understanding protein domains and genomic context can inform the function of a protein, understanding how a protein is post-translationally modified may provide important clues regarding function. For example, signal transduction mediated by the immunoreceptor tyrosine-based activation motif (ITAM) of the T-cell receptor, requires the dual phosphorylation of two of its tyrosine residues [reviewed in Ref. (17)]. Predictions as to which of the many possible post-translational modifications are statistically likely in a given protein may explain cellular localization patterns, regulation of protein abundance, and indicate whether the protein contains specific signaling properties.
As an example, previous research has demonstrated that the prototypical member of the class A scavenger receptors, SRAI, has a serine in the cytoplasmic domain of this protein, which, when phosphorylated, is essential for its phagocytic function (18, 19). However, it is not known whether the other members of the class A scavenger receptor family, such as SCARA3, contain similar sites of post-translational modifications. Knowledge of such sites would suggest that SCARA3, like SRAI, is also a phagocytic receptor whose signaling pathways are conserved within this receptor family. The SCARA3 FASTA formatted protein sequence obtained from NCBI was analyzed using the NetPhos 2.0 Server (Figure 2). This tool was built on the knowledge that the 7- to 12-amino acids neighboring a phosphorylated residue tend to have a specified composition in order to be recognized by specific kinases and phosphatases (20). Using this information, NetPhos predicts sites of phosphorylation in a protein sequence. In the case of SCARA3, multiple sites were identified over the threshold probability value defined by the software to be serine (S)-, threonine (T)-, or tyrosine (Y)-phosphorylated (Figure 2), indicating that even though these residues differ from those identified in SRAI, SCARA3 may possess similar functionality.
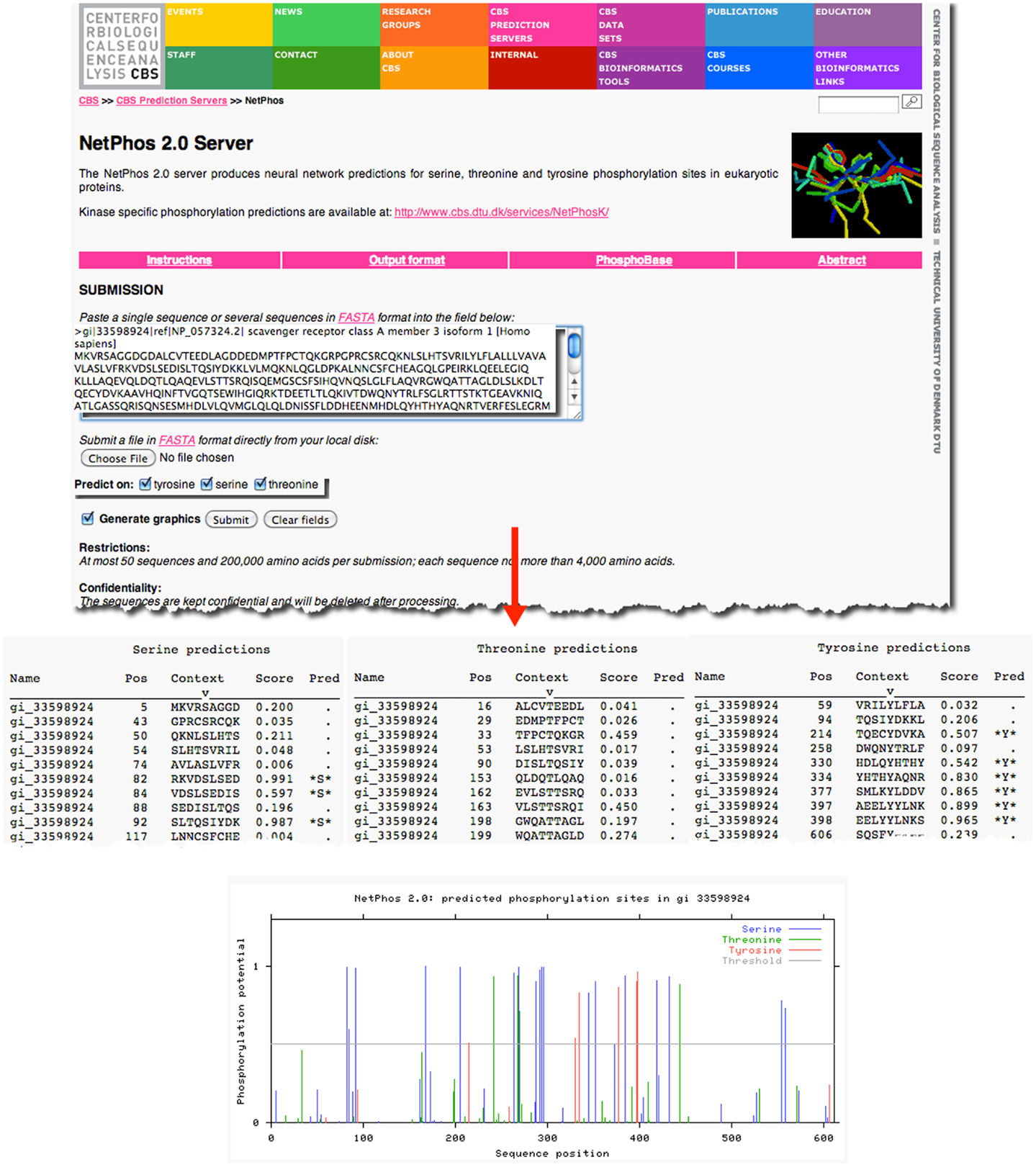
Figure 2. Prediction of post-translational modifications in SCARA3. The FASTA formatted sequence of SCARA3 from Homo sapiens was entered into the NetPhos 2.0 Server to predict serine (S), threonine (T), and tyrosine (Y) residues that may be phosphorylated. Each instance of these residues and surrounding sequences are displayed under the “Context” column. Scores above 0.5 are considered to be significant and those residues are highlighted in the “Pred” column with asterisks. The Server also displays the output graphically, including a horizontal line to indicate the 0.5 score threshold. Multiple residues in SCARA3 reach this threshold of significance, and may guide further in vitro analysis of this protein.
In addition to NetPhos, there are many post-translational modification prediction tools publically available which require the sole input of a protein sequence. A representative collection of these tools is summarized in Table 2.
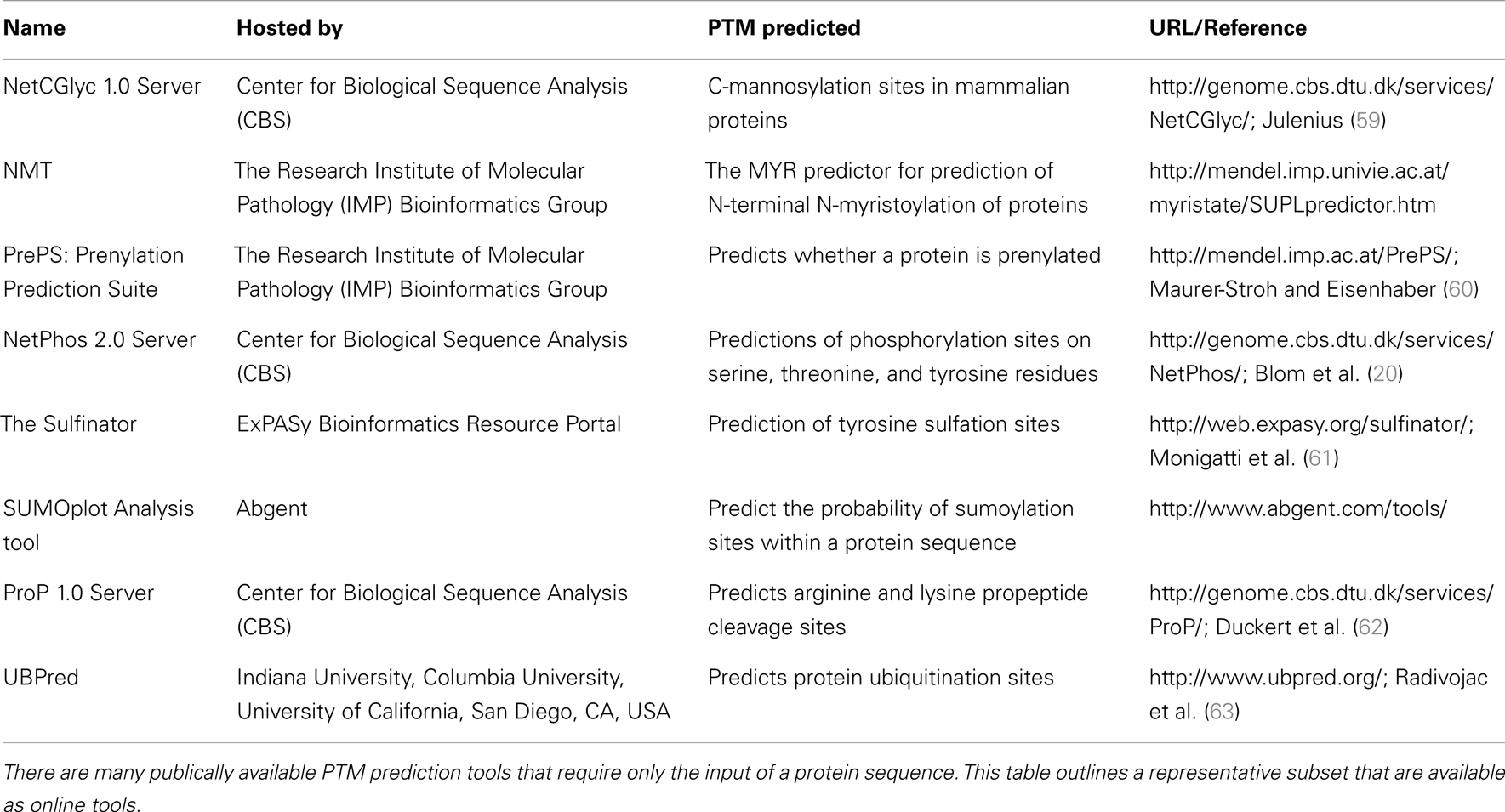
Table 2. A representative collection of bioinformatic tools for post-translational modification (PTM) prediction.
Identifying Conserved Motifs
Some regions of a gene are more susceptible to the accumulation of mutational change over evolutionary time than others and protection from change is largely due to the biological importance of such a region (21). Highly conserved regions have generally been demonstrated to encode for areas essential for a protein’s expression or function where even slight changes would threaten the organism’s survival. In contrast, in other areas of a protein, neutral mutations that do not affect protein function may accumulate over time (21). By examining areas of conservation in a protein of interest across its orthologs (i.e., genes separated by a speciation event; the same gene in different species) and paralogs (i.e., genes separated by a gene duplication event; similar genes in the same species) one can predict regions that are important for expression or function (22).
This is accomplished by performing sequence alignments. An alignment of sequences simply put, is the addition of gaps (represented as “-”s) at variable positions in a set of input sequences in order to maximize the number of similar residues per column in the alignment (22). These alignments come in a variety of forms: first, they can either be “pairwise,” involve only two sequences, or “multiple,” involve more than two sequences. Second, they can be “global,” which means the full length of all sequences are aligned, or “local,” indicating that the best alignment is displayed, even if that means only aligning a portion of the inputted sequences to each other (23). The use of pairwise versus multiple sequence alignments depends on how many closely related proteins the user has at their disposal; the more sequences, if they are closely related, will better inform the alignment. However, the choice of local versus global alignments is not as straightforward. The results of local alignments are often more meaningful because the method emphasizes regions of high similarity between sequences (23). These types of alignments are quite informative when comparing divergent protein sequences that are hypothesized to share a specific protein domain. However, often a researcher is interested in comparing full-length sequences of high similarity to each other, in which case a global alignment must be employed.
In our case, we were interested in the similarities of SCARA3 to the other members of the class A scavenger receptors (its paralogs) that, to date, have been better characterized in terms of biological function and expression. Any similarities between specific regions of SCARA3 and these well-characterized cousins would allow us to hypothesize that these regions perform similar functions in both proteins. As such, we computed a global alignment of the human SCARA3 protein with the other four members of this protein family (Figure 3). A global sequence alignment is used in this case because previous research has suggested that these proteins have evolved in parallel for many millions of years, resulting in some similar biological functions, suggesting that they share areas of similarity across the full lengths of these proteins (11, 24).
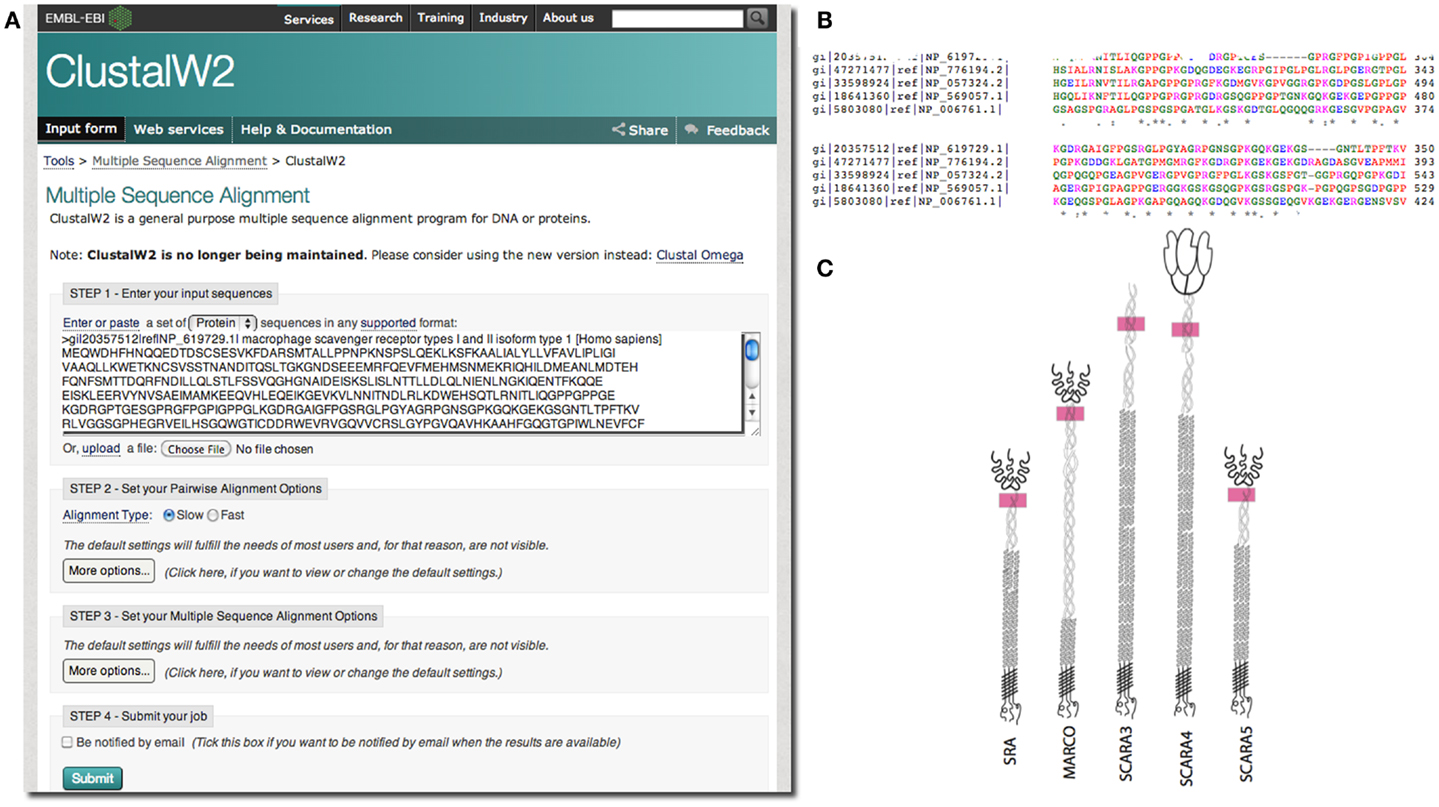
Figure 3. Use of multiple sequence alignments to discover regions of evolutionary conservation and presumed functionality. FASTA formatted protein sequences of the scavenger receptors were obtained as described previously for SRAI (NP_619729.1), MARCO (NP_006761.1), SCARA3 (NP_057324.2), SCARA4 (NP_569057.1), and SCARA5 (NP_776194.2) and inputted into the Multiple Sequence Alignment tool, ClustalW2 (A). The sequences were aligned; a portion of the alignment with the highest conservation across all five sequences is shown (B). The user may choose to view colored output, where red represents small, hydrophobic amino acids (AVFPMILW), blue represents acidic amino acids (DE), magenta represents basic amino acids (RK), and green represents STYHCNGQ (hydroxyl, sulfhydryl, amine, and glycine). Coloring allows the viewer to visualize the distribution of charge and hydrophobicity in the protein. In this example, we see that there is an orderly distribution of hydrophobic amino acids (red). The degree of consensus is represented with symbols. (*) Indicates positions which have a single, fully conserved residue; (:) indicates conservation between groups of strongly similar properties; (.) represents conservation between groups of amino acids with weakly similar properties. The fact that all five members of this family share this highly conserved region at locations in these proteins indicated with pink rectangles, (C), and that it is the highest area of conservation within the proteins is strongly suggestive of a conserved function.
European Molecular Biology Laboratory’s European Bioinformatics Institute (EBI) has a set of tools available for both pairwise1 and multiple sequence alignments2. In the example in Figure 3, we perform a global multiple sequence alignment of the class A scavenger receptor protein sequences from Homo sapiens using the ClustalW2 tool (Figure 3A). ClustalW2 was chosen because it is suitable for “medium-length” alignments, which is perfect for analysis of the scavenger receptors, which are approximately 500 base pairs in length. Additionally, ClustalW2 produces a colorful output, which makes it easy to visualize conserved residues and patterns of charge or residue repeats by visual inspection. A portion of the results of this alignment can be visualized in Figure 3B. Notably, this alignment identified an area of conservation at the C-terminal region of the collagenous domain across all five members of the class A scavenger receptors (Figure 3C). This area, consisting of predominantly charged amino acids, has been previously implicated in ligand binding in SRAI (25). Consequently we might predict that this region is a ligand-binding site not only in SRAI, but also in the other four members of this protein family.
Another approach to the identification of conserved motifs, especially useful when no known homologs exist, are specialized tools that examine an input sequence for known domains. An example of such a tool is NCBI’s Conserved Domain Search (CD-search) which compares a user-provided sequence against an NCBI-curated database of known domains (26). These tools do not find the intricacies of sequence alignments but can, however, be very informative.
Structural Analysis
Acquiring Publically Available Macromolecular Structures
Of course, while clues to a protein’s function can be hidden within its sequence, at the end of the day, it’s the protein’s structure that dictates its function. Because of the ease of DNA and protein sequencing given today’s technologies, there is more sequence data available compared to structural evidence; however, databases with structural information are available. The Protein Data Bank (PDB) is a worldwide collection of macromolecular structures governed by the Research Collaboratory for Structural Bioinformatics (RCSB). This online, searchable database3 has come a long way from its meager beginnings as a repository established in 1971 for seven structures, as it is now home to 92104 structures and counting (27). Each experimentally validated entry is assigned a PDB Identifier that can be used to search against the database. Alternatively, information such as the molecule name or author may be used.
A quick search of PDB with the search term “SCARA3” resulted in no hits. This is unsurprising given that little work has been done with this protein. However, since we know from our sequence analyses that there are regions of homology between SCARA3 and the other receptors, it is worth searching for these proteins as well. A search for “MARCO” revealed a structure (PDB ID: 2OY3) of the SRCR domain of the mouse MARCO protein (Figure 4). The PDB entry for this structure includes information such as the citation to the original publication, the functional classification of this region, its molecular weight, and an exportable macromolecular structure. Structures can be downloaded in a variety of formats, including as a form of coded text saved as a .pdb file or as a static.jpg image. The .pdb file gives the user a chance to interact with the structure by moving it along an axis, coloring based on amino acid type, or calculating potential protein-ligand interaction partners. These types of manipulations can be implemented in freely available software such as UCSF’s Chimera (28) or others summarized in Table 3.
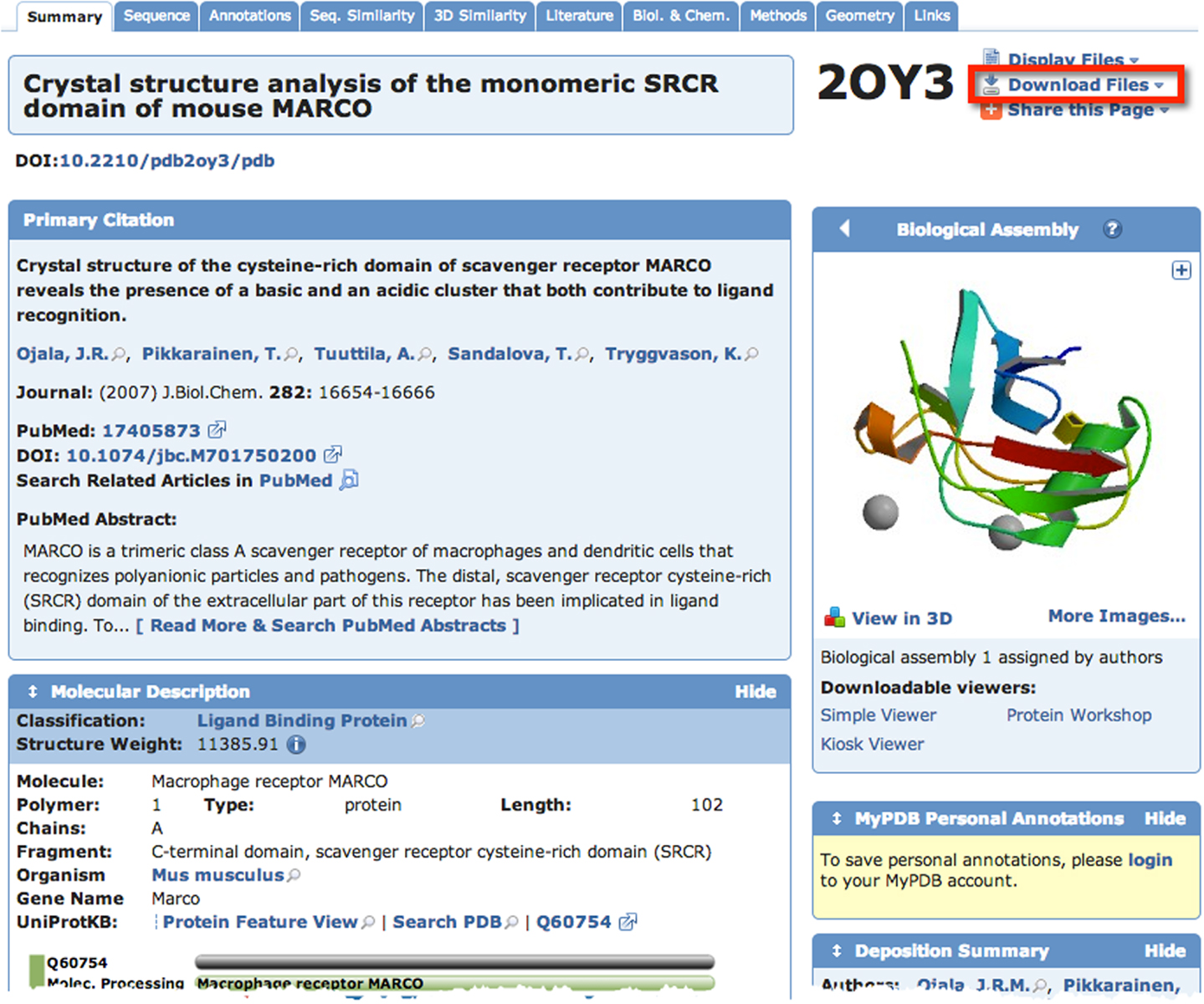
Figure 4. The Protein Data Bank (PDB) entry for a macromolecular structure of a scavenger receptor. Because crystal structures of proteins are more difficult to obtain than their protein sequences, the PDB database is less populated than sequence databases such as NCBI’s Entrez. However, PDB is still an excellent resource. Here, an example of the detailed entry for PDB ID 2OY3 is displayed after a search for “MARCO” was performed. Information is displayed such as the primary citation from which this structure was submitted, and a small visualization of the structure. Further, more detailed visualizations can be created easily by the user by downloading the .pdb formatted file from the top right of an entry, and displaying it in software such as UCSF Chimera.
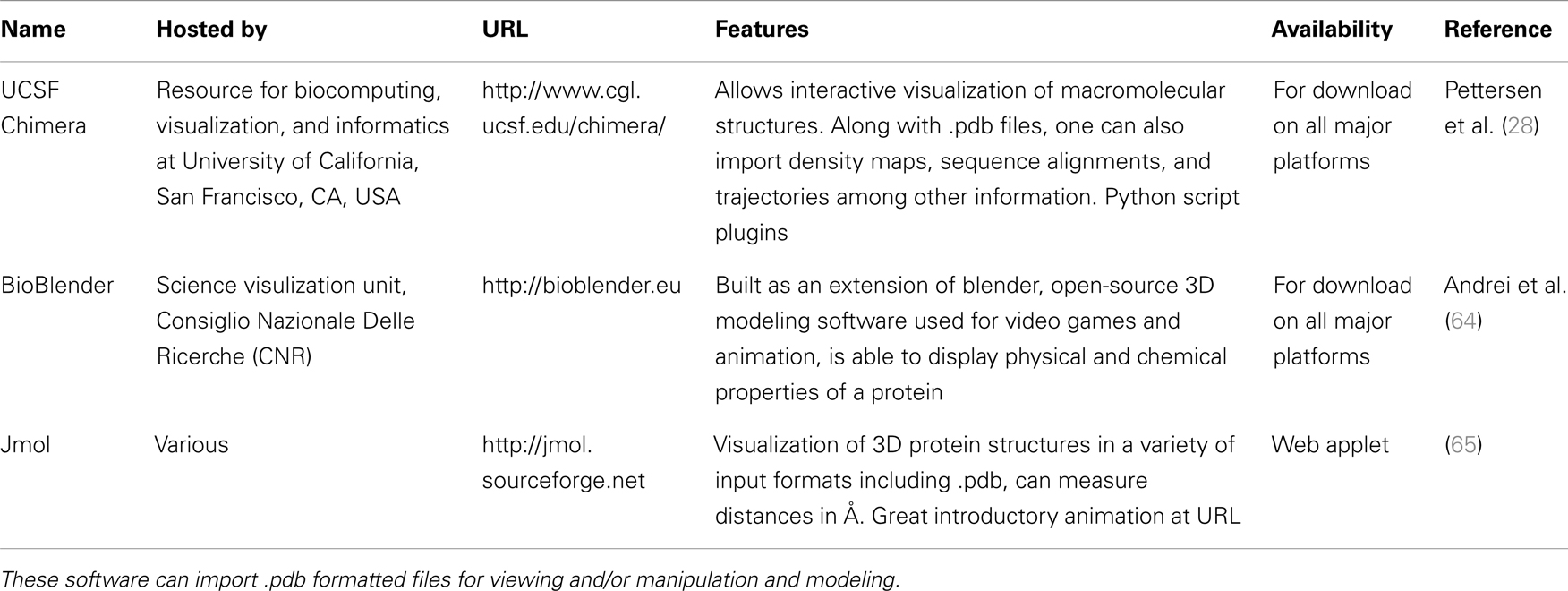
Table 3. Summary of publicly available software for the modeling of macromolecular structures.
Unfortunately for our explorations of SCARA3, our previous sequence analyses indicate that the SRCR domain of MARCO –the only current macromolecular structure of a scavenger receptor – is not a region that is shared between these two receptors and, thus, it does not indicate any new information about our protein of interest. As structural prediction technologies improve, and more experiments are conducted, the size of PDB will grow, but even in its current state it is an excellent resource for structural information.
Protein Structural Predictions
However, even if an experimentally verified protein structure such as those in PDB does not exist for a protein of interest, predictions as to the potential secondary structure of a protein can still be made based on the primary protein sequence. One common method is the reliance on identifying similar motifs in a protein sequence of interest when compared to a well-studied protein with known function (29). However, use of this method risks the transfer of incorrectly annotated information from protein to protein, thus potentially causing the corruption of genome databases if perpetuated (30). Other methods are based on highly complex algorithmic analyses, which make simplifying assumptions that exchange some accuracy for an algorithmic solution (31). These algorithms take into account certain patterns characteristic of a secondary structure, which tend to be represented in the primary sequence. For example, collagen, the main constituent of connective tissue, is generally encoded as a combination of glycine, proline, hydroxyproline, and hydroxylysine (32). These patterns allow bioinformatic tools to predict certain secondary structures such as collagenous regions from a primary sequence.
Psipred is an excellent example of such a predictive tool. Psipred is an online resource, which combines multiple secondary structure prediction methods into one, easy-to-use web-interface (33). First, psipred generates a sequence profile of the user’s sequence using BLAST, which determines areas of conservation and variation (33). Conserved areas denote areas of functionality, as well as areas that form the core of the protein; whereas, variable regions not responsible for specific folds, or the integrity of the protein structure generally exist on the surface (33). These sequence profiles give this tool its first hints as to the protein’s structure. Subsequently, an algorithmic approach is used to compare those patterns found in the sequence of interest to those identified in other proteins.
The results of inputting the human SCARA3 protein sequence into the online Psipred tool gave us an indication of which segments of the sequence formed α-helices and β-sheets (Figure 5). When we were analyzing the protein sequences of all the scavenger receptors as part of our determination of the evolution of the protein family (24), we were able to build off of this information to discover that some of the predicted α-helix segments were indeed coiled-coil motifs based on the form HxxHcccH where hydrophobic (H) residues were interspersed with other amino acids (x), some of which were more likely to be charged (c) (34, 35). There are a few other tools that work in a similar fashion to Psipred, which we have reviewed in Table 4.
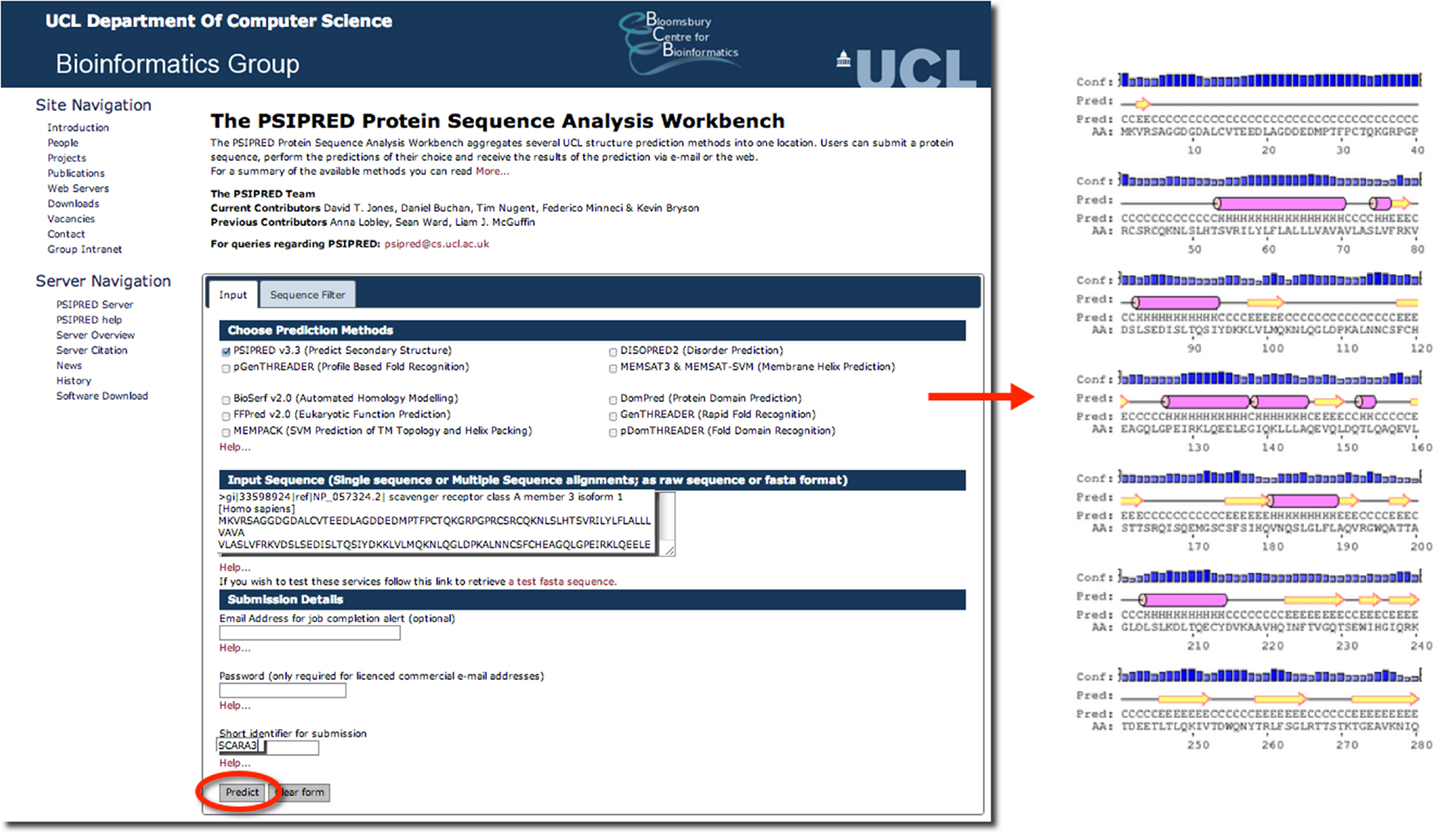
Figure 5. The use of Psipred for the prediction of the secondary protein structure of SCARA3. The Psipred tool combines various secondary protein prediction algorithms into one web-interface. Upon inputting the NCBI RefSeq protein sequence of SCARA3, Psipred outputted structural predictions, including the location of α-helices (pink cylinders) and β-sheets (yellow arrows).
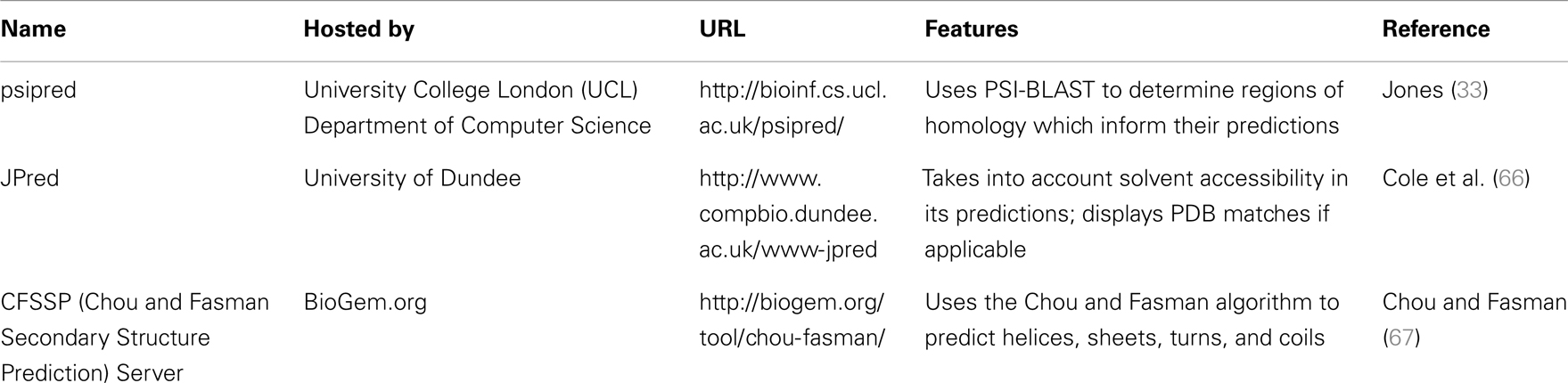
Table 4. Tools for the prediction of secondary structure characteristics.
In addition to these general tools, there are others that focus on predicting specific aspects of different types of proteins. The TMHMM Server, for example, focuses on the prediction of transmembrane domains using a statistical model (36). Output from this tool, indicates whether a protein has a transmembrane domain and its predicted location. Additionally, tools such as SignalP focus on the prediction of signal peptide cleavage sites within an amino acid sequence, which can add to the user’s knowledge of a protein’s structure (37).
Transcriptomics
Gene Expression Profiles to Answer Immunological Questions
Studies of global gene expression (“transcriptomics”) using microarrays, RNA sequencing (RNAseq), and other platforms have been a valuable tool for immunologists. Transcriptomics can be used to discover “gene signatures” of disease states or to provide mechanistic insight into disease etiology. Because variability within individuals dictate symptoms and disease progression, it is very rare that changes in expression of a single gene will be sufficiently robust for diagnosis; however, combinatorial changes that indicate a common mode of regulation are more robust and allow for the formation of “gene signatures.” For example, an “interferon signature” of gene expression was discovered in lupus when type I interferon inducible genes were found to be elevated in the peripheral blood mononuclear cells (PBMCs) of patients with lupus compared to healthy controls (38). Other notable discoveries in immunology made using transcriptomics include the discovery of the mechanisms of genetic regulation associated with lipopolysaccharide (LPS) tolerance (39), predicting long-term survival from breast and other cancers (40), and studying changes in microbial gene expression over the course of disease (41). As the immunology community’s use of transcriptomic data increases, public repositories such as the NCBI’s Gene Expression Omnibus4, EBI’s Gene Expression Atlas5, and other specialized sites such as http://www.macrophages.com/ contain a rich amount of data waiting to be mined. These resources include transcriptional profiles of different immunological cell types and activation states in a wide range of organisms. Although there are challenges with comparing microarray data from different platforms and sources (42) the cost savings of reproducing publicly available experiments have increased the appeal of utilizing public resources.
Transcriptomics has also fed the immunologist’s obsession with characterizing leukocyte subsets and lineage. In some cases, defining cells by their transcriptional profile has proven to be as effective as sorting by flow cytometry (42). These data have inspired researchers to search for the holy grail of transcriptional profiles that characterize subsets of immune cells and are more specific than surface markers. Although this approach has been somewhat successful [e.g., in identifying a novel subset of NK cells; (43), for cell types such as macrophages and dendritic cells that seem to have a more plastic phenotype and ontogeny, the usefulness of this approach has been a subject of debate (44, 45)]. Nonetheless this quest has inspired the creation of the Immunological Genome Project6 (46). This consortium of researchers is characterizing the transcriptional profile of immune cells based on rigid sorting and purification profiles, and although these data consist almost entirely of mouse genes in the steady state, it is a valuable resource to the immunology community. In our attempt to learn about SCARA3, we used the “Gene Skyline” and “Modules and Regulators” tools (Figure 6A) to find that transcripts of SCARA3 are expressed broadly across a wide range of cells at relatively low abundance (Figure 6B). There is no published data describing how SCARA3 is transcriptionally regulated; however, four transcription factor binding sites (NFIA, TAL1, KLF4, and LMO2) and two regulatory regions are predicted to occur in the promotor region of SCARA3 (Figure 6C). The Immgen database allows researchers to glean a considerable amount of data about their gene of interest with very little investment or specialized knowledge.
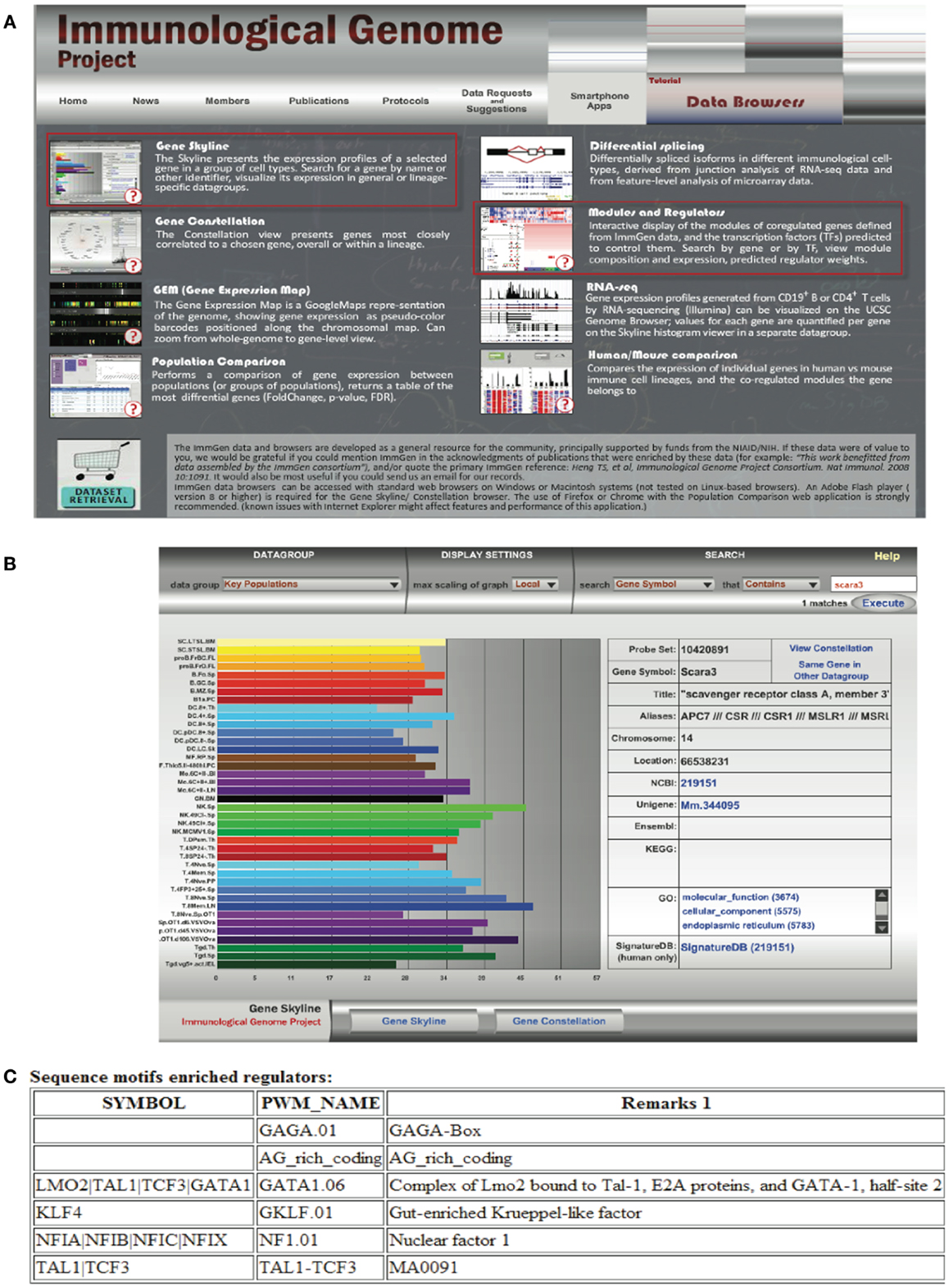
Figure 6. Querying the Immunological Genome Project (http://immgen.org) for data on expression and transcriptional regulation of SCARA3. (A) The Immunological Genome project has a number of ways to browse the data and visualize patterns of gene expression and transcriptional regulation. (B) Using the “Gene Skyline” browser we see that the transcript for SCARA3 is expressed at low levels in most cell types in the database. (C) Using the “Modules and Regulators” browser we see that there are four predicted transcription factor binding sites (NF1.01, GATA1.06, GKLF.01, and TAL1-TCF3) and two regulatory regions (GAGA.01, AG_rich_coding) in the promotor of SCARA3.
Although the Immgen database is probably the most user friendly, it is dominated by mouse immune cell subsets. Other resources such as IRIS (Immune response in silico) take a similar approach to characterizing the transcriptional profiles of human leukocyte subsets and include different activation states (47).
Genetic Variation
Analysis of Single-Nucleotide Polymorphism
The most common type of variation within the human genome are single-nucleotide polymorphisms (SNPs), which occur, on average, every 1200 base pairs (48). SNPs can be non-synonymous or synonymous; non-synonymous SNPs result in a change in the amino acid sequence of the translated protein, while synonymous SNPs do not alter the amino acid composition because of the redundancy of the genetic code.
Single-nucleotide polymorphism analysis of a protein can greatly aid in the understanding of its function as these small alterations can result in substantial changes in the functionality of the protein. For example, a SNP at a receptor’s binding site may alter the original protein such that it would be able to bind a pathogen that it previously was unable to, or, in contrast, may abolish its ability to bind its usual binding partner. In one study, researchers studied differences in SNP frequencies of Mal/TIRAP to explain differences in TLR2 and TLR4 signaling between European and African populations (49). After cloning the two variants, S180L and S180, results indicated that S180L heterozygous individuals had a higher cytokine production level than S180 homozygous individuals (49). Lower allele frequencies of S180L in African and Asian populations might indicate selection occurred after humans migrated from Africa since the variant may have granted added bacterial resistance in the changing habitat (49). This study demonstrates how SNP analyses can be used to identify functional domains of a protein as well as uncover a protein’s potential evolutionary history.
There are several publicly available online databases for the analysis of SNPs in a protein of interest (summarized in Table 5); here, we use The University of California, Santa Cruz (UCSC) Genome Browser7 to perform an analysis of SNPs present within SCARA3. Regions of interest can be searched for by entering the name of a gene or its corresponding chromosomal position. The Genome Browser contains multiple “tracks” that contain different types of annotation, including those based on NCBI RefSeqs, mRNA alignments, and UCSC Genes (50) (Figure 7). In addition, the browser can display reports regarding gene expression, regulation, and variation, among other information (50). The UCSC Genome Browser includes an annotated SNP track with over 23 million reference SNPs from NCBI’s SNP Database (dbSNP) (50) (Figure 7B). SNPs are annotated using a refSNP cluster ID number (rs#) which represents all SNPs, often from multiple population studies, that map to the same location in the gene. Additionally, each individual SNP within a cluster is associated with a SNP Accession number (ss#) (48). Selecting a refSNP cluster within the Genome Browser will display information such as the nucleotide change, chromosomal position, and type of variant as well as a link to the dbSNP database (Figure 8), which contains further detail on the population studies associated with the SNP, including observed allele frequencies and links to other resources such as GenBank and PubMed (48). The dbSNP database can also be accessed externally through NCBI, and individual SNPs can be searched for using their SNP Accession number, population study name, or via a BLAST search (51).
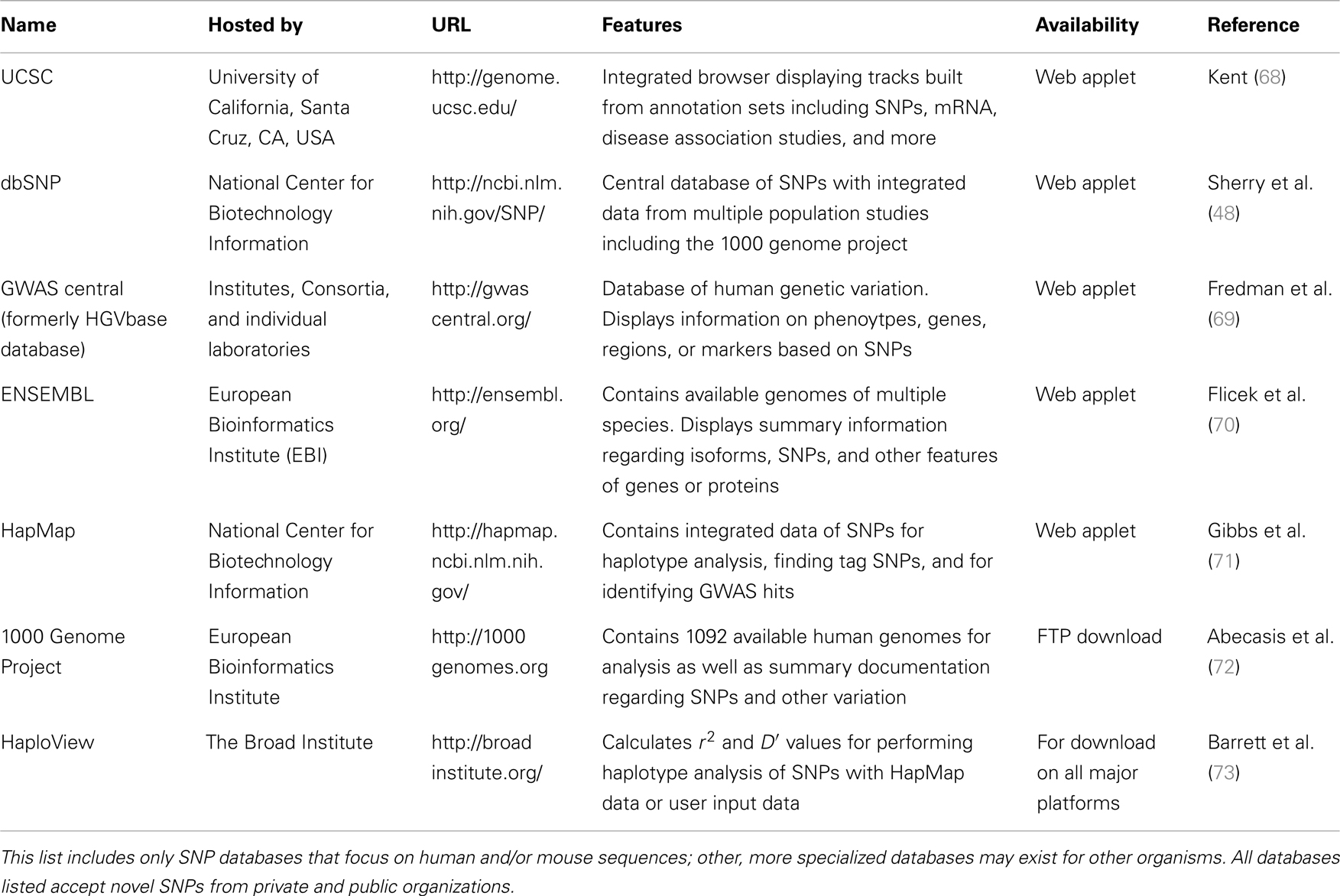
Table 5. Publicly available single-nucleotide polymorphism (SNP) databases.
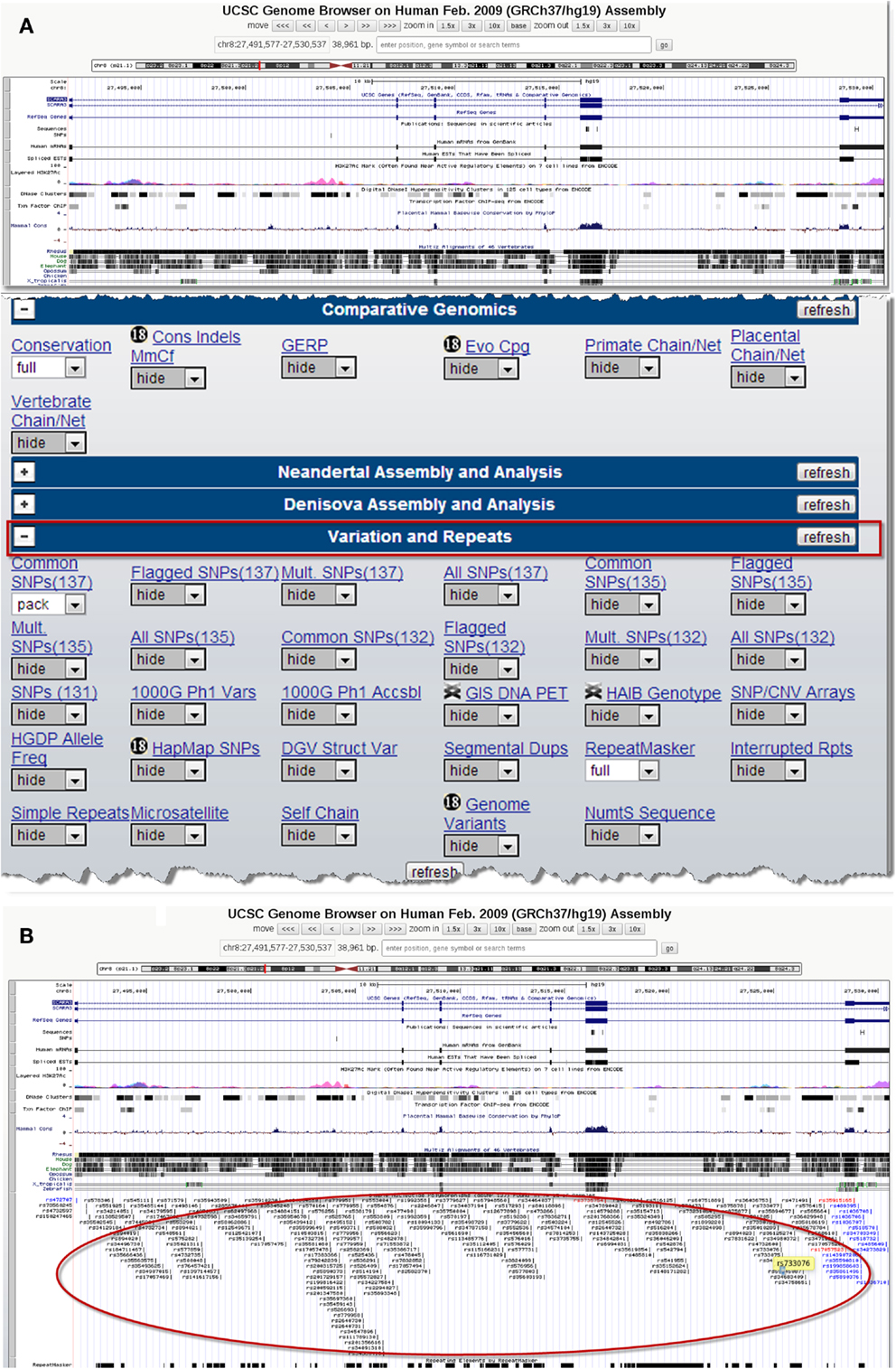
Figure 7. Using the UCSC Genome Browser to search for single-nucleotide polymorphisms (SNPs) in SCARA3. This browser contains multiple “tracks,” including the location of SNPs across the length of a protein. Here we show the output from inputting the NCBI RefSeq for SCARA3 isoform (A). Further options to hide or show more annotation tracks are available directly below the graphical output. Under the “Variation and Repeats” tab, selecting “pack” under the “Common SNPs” option updates the output to include a full display of SNPs represented by their refSNP cluster ID numbers [(B), circled]. Clicking on any of the refSNP cluster IDs leads to a link displaying further information regarding the SNP as well as a link to NCBI’s dbSNP database.
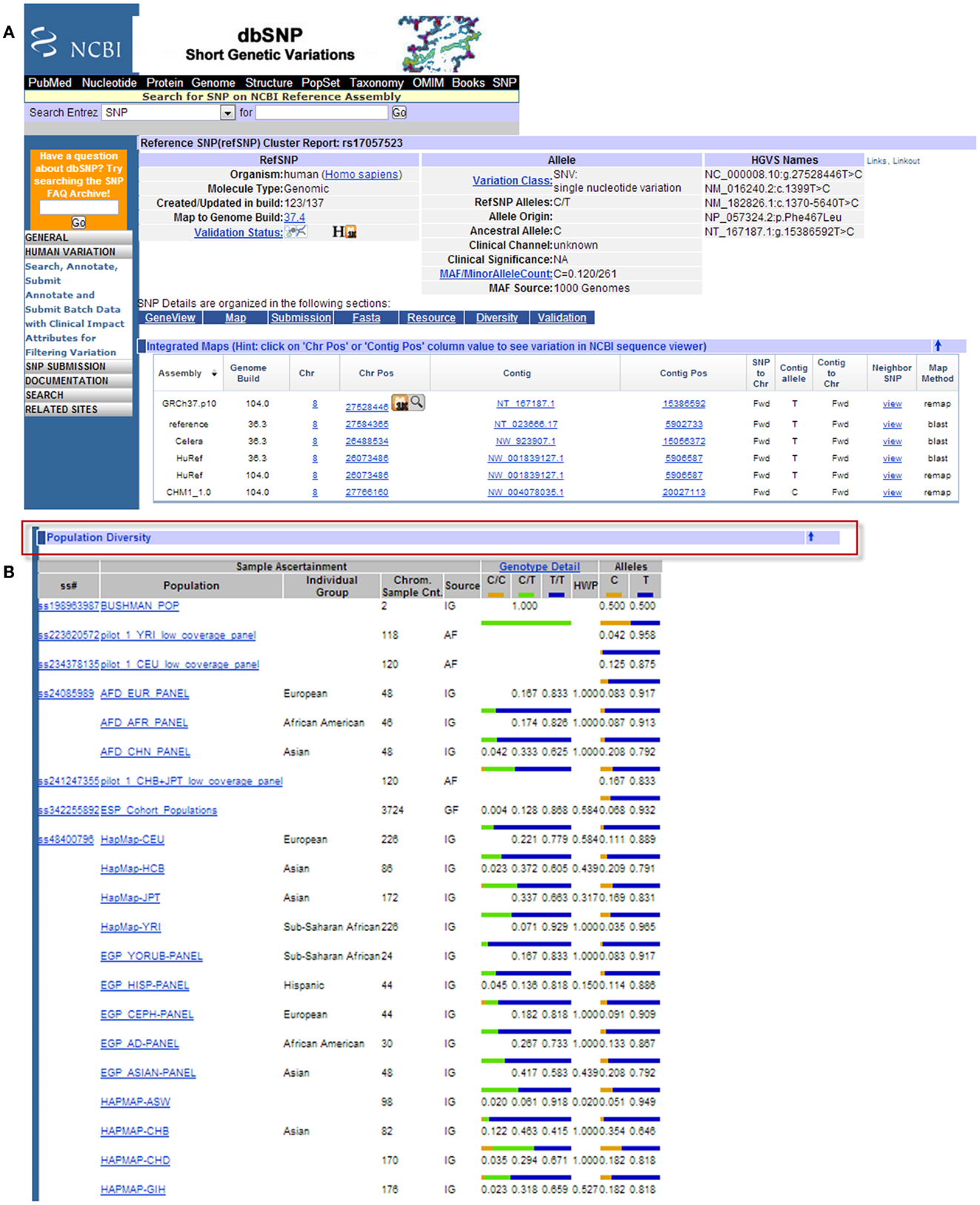
Figure 8. Example results page from the NCBI dbSNP database for SCARA3 SNP rs17057523. By following the link from the UCSC Genome Browser to dbSNP, more information is provided for SNP rs17057523 including allele frequencies, ancestral alleles, and chromosomal position (A). Following this information on the database website, are other tabs that show more information regarding the SNP that may be useful to investigators. The “Population Diversity” section displays information regarding allele frequencies from different sampled populations (B). Clicking any of the population links shows information on how the SNP was genotyped, the population sample size, and other experimental conditions used.
When the UCSC Genome Browser is used to search for SCARA3, the resulting SNP track shows all of the reported SNPs within the gene (Figure 7B). Most of the annotated SNPs within SCARA3 are intronic variants, which would not alter the resultant protein; however intronic regions have been shown to be involved in regulatory processes. Of the three SNPs found in the exons of SCARA3, rs17057523 has the highest global minor allele frequency of 0.120 based on The 1000 Genome Project phase 1 data. Following the external link to dbSNP’s “Population Diversity” section shows that the SNP is found at higher frequencies in Asian populations, with allele frequencies up to 0.222 while other populations remain close to 0.1 (Figure 8). Additionally, the “Multiz Alignment” track shows areas of conservation between multiple vertebrates and suggests that SNP rs17057523 is present within a conserved area of SCARA3. Further testing by cloning the variant can help determine the function of this domain by examining functional differences between the SNP and wildtype allele.
Further Analyses
What has been covered here represents the basic knowledge upon which most bioinformatic analyses will be conducted. As in any field, there are a plethora of examples of highly specialized bioinformatic tools and software that have been developed for the various sub-fields of immunology. For example, HLA peptide binding predictions can be made using various tools such as that available from the National Institute of Health8 (52). While an exhaustive list of such programs cannot be given, we suggest that the reader referred to other, more specialized reviews of such tools [(53–55) for example].
Concluding Remarks
In our opinion, bioinformatics is a methodology that is under-utilized in immunological studies. Far from being inaccessible and complicated, many bioinformatic tools are straightforward and available via online servers, meaning that a researcher can obtain results instantaneously without fear of the often-steep learning curve associated with installable software. Although a strong background in computer science is an asset for more complicated techniques, in order to perform the analyses that we have described here, a passing familiarity with the cut and paste function is all that is required. If the reader is interested in going beyond this, there are excellent, freely available resources such as Software Carpentry9, Rosalind10, and online courses such as those available at Coursera11 and edX12. Acquiring vocabulary is probably the most challenging aspect of venturing into bioinformatics; however, one might argue that this is considerably easier to master than the language of immunology with its interminable number of interleukins, CD numbers, and signaling pathways. The goal of this review is to demonstrate some basic principles and techniques that are easily incorporated into the average bench scientist’s research and to encourage immunologists and cell biologists to consider using in silico approaches to generate and test hypotheses and answer research questions. Of course, like all hypotheses, those generated with in silico approaches must be experimentally tested. Whether in silico approaches are more or less accurate that traditional methods of hypothesis generation are yet to be evaluated. Our inquiry into the properties of SCARA3 indicates that these tools are immensely useful in generating hypotheses that can then be tested bench-side. Although many researchers have decried the lack of trained bioinformaticians and bioinformaticists, perhaps the best way to overcome the current shortage may be for scientists to become conversant in some of the basic techniques of bioinformatics in much the same way that we must be knowledgeable of the statistical tools required to analyze and understand our research.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was funded by a Natural Sciences and Engineering Research Council grant to Dawn M. E. Bowdish. Fiona Whelan was funded by an Ontario Graduate Scholarship (OGS). Additionally, work in the Bowdish laboratory is supported in by the McMaster Immunology Research Centre (MIRC) and the Michael G. DeGroote Institute for Infectious Disease Research (IIDR). Nicholas Yap was funded by a Natural Sciences and Engineering Research Council Discovery grant to G. Brian Golding. Fiona J. Whelan and Dawn M. E. Bowdish conceived and designed this article. Fiona J. Whelan, Nicholas V. L. Yap, G. Brian Golding, and Dawn M. E. Bowdish drafted the manuscript. All authors read, edited, and approved the final manuscript.
Footnotes
- ^http://www.ebi.ac.uk/Tools/psa
- ^http://www.ebi.ac.uk/Tools/msa
- ^http://www.pdb.org
- ^http://www.ncbi.nlm.nih.gov/geo/
- ^https://www.ebi.ac.uk/gxa/
- ^www.immgen.org
- ^http://genome.ucsc.edu
- ^http://www-bimas.cit.nih.gov/index.shtml
- ^http://software-carpentry.org/
- ^http://rosalind.info/
- ^http://www.coursera.org
- ^http://www.edx.org
References
3. Schrodinger E. What is Life?: The Physical Aspects of Living Cell with Mind and Matter and Autobiographical Sketches. Cambridge: Cambridge University Press (1967).
4. Olson MV. The human genome project. Proc Natl Acad Sci U S A (1993) 90:4338–44. doi: 10.1073/pnas.90.10.4338
5. Wishart DS. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res (2006) 34:D668–72. doi:10.1093/nar/gkj067
6. Roach J, Glusman G, Rowen L, Kaur A, Purcell M, Smith K, et al. The evolution of vertebrate Toll-like receptors. Proc Natl Acad Sci USA (2005) 102:9577. doi:10.1073/pnas.0502272102
7. Levasseur A, Pontarotti P. Was the ancestral MHC involved in innate immunity? Eur J Immunol (2010) 40:2682–5. doi:10.1002/eji.201040856
8. Rapin N, Lund O, Bernaschi M, Castiglione F. Computational immunology meets bioinformatics: the use of prediction tools for molecular binding in the simulation of the immune system. PLoS One (2010) 5:e9862. doi:10.1371/journal.pone.0009862
9. Seal JB, Alverdy JC, Zaborina O, An G. Agent-based dynamic knowledge representation of Pseudomonas aeruginosa virulence activation in the stressed gut: towards characterizing host-pathogen interactions in gut-derived sepsis. Theor Biol Med Model (2011) 8:33. doi:10.1186/1742-4682-8-33
10. Chau TA, McCully ML, Brintnell W, An G, Kasper KJ, Vinés ED, et al. Toll-like receptor 2 ligands on the staphylococcal cell wall downregulate superantigen-induced T cell activation and prevent toxic shock syndrome. Nat Med (2009) 15:641–8. doi:10.1038/nm.1965
11. Bowdish D, Gordon S. Conserved domains of the class A scavenger receptors: evolution and function. Immunol Rev (2009) 227:19–31. doi:10.1111/j.1600-065X.2008.00728.x
12. Pearson WR. Searching protein sequence libraries: comparison of the sensitivity and selectivity of the Smith-Waterman and FASTA algorithms. Genomics (1991) 11:635–50. doi:10.1016/0888-7543(91)90071-L
13. Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Wheeler DL. GenBank. Nucleic Acids Res (2008) 36:D25–30. doi:10.1093/nar/gkm929
14. Schuler GD, Epstein JA, Ohkawa H, Kans JA. Entrez: molecular biology database and retrieval system. Methods Enzymol (1996) 266:141–62.
15. Maglott D, Ostell J, Pruitt KD, Tatusova T. Entrez Gene: gene-centered information at NCBI. Nucleic Acids Res (2007) 35:D26–31. doi:10.1093/nar/gkl993
16. Pruitt KD, Maglott DR. RefSeq and LocusLink: NCBI gene-centered resources. Nucleic Acids Res (2001) 29:137–40. doi:10.1093/nar/29.1.137
17. Cambier JC. Antigen and Fc receptor signaling. The awesome power of the immunoreceptor tyrosine-based activation motif (ITAM). J Immunol (1995) 155:3281–5.
18. Fong LG. Modulation of macrophage scavenger receptor transport by protein phosphorylation. J Lipid Res (1996) 37:574–87.
19. Fong LG, Le D. The processing of ligands by the class A scavenger receptor is dependent on signal information located in the cytoplasmic domain. J Biol Chem (1999) 274:36808–16. doi:10.1074/jbc.274.51.36808
20. Blom N, Gammeltoft S, Brunak S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J Mol Biol (1999) 294(5):1351–62. doi:10.1006/jmbi.1999.3310
21. Kimura M. The Neutral Theory of Molecular Evolution. Cambridge: Cambridge University Press (1984).
23. Mount DW. Bioinformatics: Sequence and Genome Analysis. New York: Cold Spring Harbor Laboratory Press (2004).
24. Whelan FJ, Meehan CJ, Golding GB, McConkey BJ, Bowdish DM. The evolution of the class A scavenger receptors. BMC Evol Biol (2012) 12:227. doi:10.1186/1471-2148-12-227
25. Acton S, Resnick D, Freeman M, Ekkel Y, Ashkenas J, Krieger M. The collagenous domains of macrophage scavenger receptors and complement component C1q mediate their similar, but not identical, binding specificities for polyanionic ligands. J Biol Chem (1993) 268:3530.
26. Marchler-Bauer A, Lu S, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, et al. CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res (2010) 39:D225–9. doi:10.1093/nar/gkq1189
27. Westbrook J. The protein data bank and structural genomics. Nucleic Acids Res (2003) 31:489–91. doi:10.1093/nar/gkg068
28. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF chimera: a visualization system for exploratory research and analysis. J Comput Chem (2004) 25:1605–12. doi:10.1002/jcc.20084
29. Whisstock J, Lesk A. Prediction of protein function from protein sequence and structure. Q Rev Biophys (2003) 36:307–40. doi:10.1017/S0033583503003901
30. Hegyi H, Gerstein M. The relationship between protein structure and function: a comprehensive survey with application to the yeast genome 1. J Mol Biol (1999) 288:147–64. doi:10.1006/jmbi.1999.2661
31. Burkowski FJ. Structural Bioinformatics: An Algorithmic Approach. Boca Raton, FL: Chapman & Hall/CRC (2008).
32. Gross J, Dumsha B, Glazer N. Comparative biochemistry of collagen some amino acids and carbohydrates. Biochim Biophys Acta (1958) 30:293–7. doi:10.1016/0006-3002(58)90053-2
33. Jones DT. Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol (1999) 292(2):195–202. doi:10.1006/jmbi.1999.3091
34. McAlinden A. Helical coiled-coil oligomerization domains are almost ubiquitous in the collagen superfamily. J Biol Chem (2003) 278:42200–7. doi:10.1074/jbc.M302429200
35. Parry DAD, Fraser RDB, Squire JM. Fifty years of coiled-coils and α-helical bundles: a close relationship between sequence and structure. J Struct Biol (2008) 163:258–69. doi:10.1016/j.jsb.2008.01.016
36. Krogh A, Larsson B, Heijne Von G, Sonnhammer E. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes1. J Mol Biol (2001) 305:567–80. doi:10.1006/jmbi.2000.4315
37. Petersen TN, Brunak S, Heijne von G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods (2011) 8:785–6. doi:10.1038/nmeth.1701
38. Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci U S A (2003) 100:2610–5. doi:10.1073/pnas.0337679100
39. Foster SL, Hargreaves DC, Medzhitov R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature (2007) 447(7147):972–8. doi:10.1038/nature05836
40. van’t Veer LJ, Dai H, Van De Vijver MJ, He YD, Hart AA, Mao M, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature (2002) 415:530–6. doi:10.1038/415530a
41. Orihuela CJ, Radin JN, Sublett JE, Gao G, Kaushal D, Tuomanen EI. Microarray analysis of pneumococcal gene expression during invasive disease. Infect Immun (2004) 72:5582–96. doi:10.1128/IAI.72.10.5582-5596.2004
42. Wang Y, Joshi T, Zhang XS, Xu D, Chen L. Inferring gene regulatory networks from multiple microarray datasets. Bioinformatics (2006) 22:2413–20. doi:10.1093/bioinformatics/btl396
43. Koopman LA, Kopcow HD, Rybalov B, Boyson JE, Orange JS, Schatz F, et al. Human decidual natural killer cells are a unique NK cell subset with immunomodulatory potential. J Exp Med (2003) 198:1201–12. doi:10.1084/jem.20030305
44. Hume DA, Mabbott N, Raza S, Freeman TC. Can DCs be distinguished from macrophages by molecular signatures? Nat Immunol (2013) 14:187–9. doi:10.1038/ni0813-876d
45. Randolph G, Merad M. Can DCs be distinguished from macrophages by molecular signatures? Nat Immunol (2013) 14:189–90. doi:10.1038/ni.2517
46. Heng TS, Painter MW, Elpek K, Lukacs-Kornek V, Mauermann N, Turley SJ, et al. The Immunological Genome Project: networks of gene expression in immune cells. Nat Immunol (2008) 9:1091–4. doi:10.1038/ni1008-1091
47. Abbas AR, Baldwin D, Ma Y, Ouyang W, Gurney A, Martin F, et al. Immune response in silico (IRIS): immune-specific genes identified from a compendium of microarray expression data. Genes Immun (2005) 6:319–31. doi:10.1038/sj.gene.6364173
48. Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res (2001) 29:308–11. doi:10.1093/nar/29.1.308
49. Ferwerda B, Alonso S, Banahan K, McCall MB, Giamarellos-Bourboulis EJ, Ramakers BP, et al. Functional and genetic evidence that the Mal/TIRAP allele variant 180L has been selected by providing protection against septic shock. Proc Natl Acad Sci U S A (2009) 106:10272–7. doi:10.1073/pnas.0811273106
50. Fujita PA, Rhead B, Zweig AS, Hinrichs AS, Karolchik D, Cline MS, et al. The UCSC Genome Browser database: update 2011. Nucleic Acids Res (2010) 39:D876–82. doi:10.1093/nar/gkq963
52. Parker KC, Bednarek MA, Coligan JE. Scheme for ranking potential HLA-A2 binding peptides based on independent binding of individual peptide side-chains. J Immunol (1994) 152:163–75.
53. Korber B, LaBute M, Yusim K. Immunoinformatics comes of age. PLoS Comput Biol (2006) 2:e71. doi:10.1371/journal.pcbi.0020071
54. Tong JC, Ren EC. Immunoinformatics: current trends and future directions. Drug Discov Today (2009) 14:684–9. doi:10.1016/j.drudis.2009.04.001
55. Tomar N, De RK. Immunoinformatics: an integrated scenario. Immunology (2010) 131:153–68. doi:10.1111/j.1365-2567.2010.03330.x
56. Kulikova T. The EMBL nucleotide sequence database. Nucleic Acids Res (2004) 32:27D–30D. doi:10.1093/nar/gkh120
57. Miyazaki S. DNA Data Bank of Japan (DDBJ) in XML. Nucleic Acids Res (2003) 31:13–6. doi:10.1093/nar/gkg088
58. Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, et al. The human genome browser at UCSC. Genome Res (2002) 12:996–1006. doi:10.1101/gr.229102
59. Julenius K. NetCGlyc 1.0: prediction of mammalian C-mannosylation sites. Glycobiology (2007) 17:868–76. doi:10.1093/glycob/cwm050
60. Maurer-Stroh S, Eisenhaber F. Refinement and prediction of protein prenylation motifs. Genome Biol (2005) 6:R55. doi:10.1186/gb-2005-6-6-r55
61. Monigatti F, Gasteiger E, Bairoch A, Jung E. The Sulfinator: predicting tyrosine sulfation sites in protein sequences. Bioinformatics (2002) 18:769–70. doi:10.1093/bioinformatics/18.5.769
62. Duckert P, Brunak S, Blom N. Prediction of proprotein convertase cleavage sites. Protein Eng Des Sel (2004) 17:107–12. doi:10.1093/protein/gzh013
63. Radivojac P, Vacic V, Haynes C, Cocklin RR, Mohan A, Heyen JW, et al. Identification, analysis, and prediction of protein ubiquitination sites. Proteins (2010) 78:365–80. doi:10.1002/prot.22555
64. Andrei RM, Loni T, Callieri M, Zini MF, Maraziti G, Pan MC. BioBlender: A Software for Intuitive Representation of Surface Properties of Biomolecules. (2010). p. 1–19. Available at: http://cds.cern.ch/record/1294402
65. Jmol: An Open-Source Java Viewer for Chemical Structures in 3D. Available at: http://www.jmol.org/
66. Cole C, Barber JD, Barton GJ. The Jpred 3 secondary structure prediction server. Nucleic Acids Res (2008) 36:W197–201. doi:10.1093/nar/gkn238
67. Chou P, Fasman G. Prediction of protein conformation. Biochemistry (1974) 13:222–45. doi:10.1021/bi00699a002
68. Kent WJ. BLAT – the BLAST-like alignment tool. Genome Res (2002) 12:656–64. doi:10.1101/gr.229202
69. Fredman D, Siegfried M, Yuan YP, Bork P, Lehväslaiho H, Brookes AJ. HGVbase: a human sequence variation database emphasizing data quality and a broad spectrum of data sources. Nucleic Acids Res (2002) 30:387–91. doi:10.1093/nar/30.1.387
70. Flicek P, Ahmed I, Amode MR, Barrell D, Beal K, Brent S, et al. Ensembl 2013. Nucleic Acids Res (2012) 41:D48–55. doi:10.1093/nar/gks1236
71. Gibbs RA, Belmont JW, Hardenbol P, Willis TD, Yu F, Yang H, et al. The international HapMap project. Nature (2003) 426:789–96. doi:10.1038/nature02168
72. Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, et al. An integrated map of genetic variation from 1,092 human genomes. Nature (2012) 490:56–65. doi:10.1038/nature11632
Keywords: bioinformatics, immunology, sequence alignments, single-nucleotide polymorphisms, transcriptional profiling, scavenger receptor
Citation: Whelan FJ, Yap NVL, Surette MG, Golding GB and Bowdish DME (2013) A guide to bioinformatics for immunologists. Front. Immunol. 4:416. doi: 10.3389/fimmu.2013.00416
Received: 10 September 2013; Accepted: 13 November 2013;
Published online: 04 December 2013.
Edited by:
Fabrizio Mattei, Istituto Superiore di Sanità, ItalyReviewed by:
Geanncarlo Lugo-Villarino, Centre National de la Recherche Scientifique, FranceCan Peng, Tongji University, China
Copyright: © 2013 Whelan, Yap, Surette, Golding and Bowdish. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dawn M. E. Bowdish, Department of Pathology and Molecular Medicine, McMaster Immunology Research Centre, M. G. DeGroote Institute for Infectious Disease Research, McMaster University, 1280 Main Street West, Hamilton, ON L8S 4K1, Canada e-mail:Ym93ZGlzaEBtY21hc3Rlci5jYQ==