 Gisela von Hoven1†
Gisela von Hoven1† Claudia Neukirch1†Martina Meyenburg1Sabine Füser1Maria Bidna Petrivna1Amable J. Rivas1Alexey Ryazanov2
Claudia Neukirch1†Martina Meyenburg1Sabine Füser1Maria Bidna Petrivna1Amable J. Rivas1Alexey Ryazanov2 Randal J. Kaufman3Raffi V. Aroian4*
Randal J. Kaufman3Raffi V. Aroian4* Matthias Husmann1*
Matthias Husmann1*
- 1University Medical Center, Institute of Medical Microbiology and Hygiene, Johannes Gutenberg-University, Mainz, Germany
- 2Department of Pharmacology, Rutgers Robert Wood Johnson Medical School, Piscataway, NJ, USA
- 3Degenerative Diseases Program, Sanford-Burnham Medical Research Institute, La Jolla, CA, USA
- 4University of Massachusetts Medical School, Worcester, MA, USA
We report on the role of conserved stress–response pathways for cellular tolerance to a pore forming toxin. First, we observed that small molecular weight inhibitors including of eIF2α-phosphatase, jun-N-terminal kinase (JNK), and PI3-kinase sensitized normal mouse embryonal fibroblasts (MEFs) to the small pore forming S. aureus α-toxin. Sensitization depended on expression of mADAM10, the murine ortholog of a proposed high-affinity receptor for α-toxin in human cells. Similarly, eIF2αS51A/S51A MEFs, which harbor an Ala knock-in mutation at the regulated Ser51 phosphorylation site of eukaryotic translation initiation factor 2α, were hyper-sensitive to α-toxin. Inhibition of translation with cycloheximide did not mimic the tolerogenic effect of eIF2α-phosphorylation. Notably, eIF2α-dependent tolerance of MEFs was toxin-selective, as wild-type MEFs and eIF2αS51A/S51A MEFs exhibited virtually equal sensitivity to Vibrio cholerae cytolysin. Binding of S. aureus α-toxin to eIF2αS51A/S51A MEFs and toxicity in these cells were enhanced as compared to wild-type cells. This led to the unexpected finding that the mutant cells carried more ADAM10. Because basal phosphorylation of eIF2α in MEFs required amino acid deprivation-activated eIF2α-kinase 4/GCN2, the data reveal that basal activity of this kinase mediates tolerance of MEFs to α-toxin. Further, they suggest that modulation of ADAM10 is involved. During infection, bacterial growth may cause nutrient shortage in tissues, which might activate this response. Tolerance to α-toxin was robust in macrophages and did not depend on GCN2. However, JNKs appeared to play a role, suggesting differential cell type and toxin selectivity of tolerogenic stress responses. Understanding their function or failure will be important to comprehend anti-bacterial immune responses.
Introduction
Membrane perforation by pore forming toxins (PFT) is an ancient mode of attack employed by many bacteria, which helps them to establish or sustain infection (1–3). PFT represent a large group of bacterial toxins, which can be divided into various structural families (2). Many PFT have been discovered based on their ability not only to lyse red blood cells but they also affect nucleated cells, with effects ranging from induction of cell death to proliferation, time scales of occurrence from seconds to days after attack (4). Of particular relevance in the present context, cell autonomous defenses are in place to limit or reverse damage of nucleated target cells of PFT (4–13); they have been discussed as an integral part of the innate immune system, defending against bacteria (14). Work in C. elegans identified MAPK as master regulators of defense against PFT (7, 15). Whereas the importance of p38 MAPK is well established (3, 4, 9, 16–18), data on the role of jun-N-terminal kinases (JNKs) are somewhat conflicting (19–21). Large scale analyses of perforated cells have identified multiple additional changes taking place in response to PFT (15, 20, 22, 23), many of which appear to be triggered by the drop of cytosolic potassium (11, 20, 24, 25). Although the contribution of the various pathways to cell autonomous defense against PFT remains to be established in most cases, a basic concept emerges according to which removal of membrane pores (3, 10, 13, 26–32) and metabolic homeostasis (10, 12, 13, 20, 33, 34) are cornerstones of early cell autonomous defense against PFT. Importantly, mechanisms involved in pore removal depend on PFT and cell type (4, 8, 29). It appears that MAPK p38 and autophagy are required if the recovery process is prolonged as with S. aureus α-toxin and aerolysin (8, 20).
Phosphorylation of eukaryotic translation initiation factor 2 α (eIF2α) is a conserved stress–response activated by various PFT (12, 13, 20, 33–35). How this pathway impacts survival of target cells remains incompletely understood. Eukaryotic translation initiation requires assembly of a 43S ternary pre-initiation complex, consisting of met-t-RNAi(Met), eIF2, and GTP. In mammalian cells, this step is controlled through phosphorylation of eIF2α at serine 51 by 1 of 4 eIF2α-kinases (GCN2, PERK, PKR, and HRI), which respond to different types of stress (36). GCN2 serves as nutritional sensor, which is activated by uncharged t-RNAs (37, 38). Several lines of evidence indicate that membrane stress triggers this pathway: first, mutations that affect vesicular transport in yeast trigger phosphorylation of eIF2α (39). Second, in mammalian cells, plasma membrane perforation by bacterial PFT leads to activation of GCN2 (12, 33), phosphorylation of eIF2α, transient attenuation of translation, and activation of autophagy (12, 13, 20). Also, membrane damage by chlorpromazine or detergent triggers GCN2 (40). S. aureus α-toxin inhibited uptake of leucine by cells, providing an explanation for activation of GCN2 in target cells of PFT (12). In human epithelial cells, eIF2α, eIF2α-kinases and the regulatory eIF2α-phosphatase subunit CReP/Ppp1r15B are all required for efficient recovery from α-toxin-dependent loss of ATP (13). Surprisingly, these proteins served to remove membrane pores, thus, linking control of translation initiation and membrane traffic (13).
Conspicuously, many rodent cell types are not affected even by comparably high concentrations (micromolar range) of α-toxin; but the cause is not known. Receptor density on murine cells might be low, or murine ADAM10 might be an inefficient receptor as compared to its human counterpart. Alternatively, murine cells might be particularly tolerant to the consequences of successful attack. At any rate, to better understand results of in vivo experiments with S. aureus or α-toxin in mice, it is important to comprehend the mechanisms underlying tolerance of murine cells.
In ecoimmunology, “tolerance” denotes the ability of an organism to cope with high-pathogen load and resulting damage (41–43). To elucidate mechanisms of tolerance, it will be important to investigate the phenomenon at the cellular level using defined noxious agents. Here, we focus on cellular tolerance to PFT. A PFT may fail to cause overt damage to a cell if it cannot bind to, or attack, membranes in the first place. Alternatively, target cells may be able to cope with membrane perforation. In both cases, we consider the target cell “tolerant to the PFT.” The term “susceptibility” shall denote responsiveness of a cell to a PFT as measured by loss of ATP or loss of potassium ions from the cytosol.
In the present work, we have investigated tolerance of mouse cells to S. aureus α-toxin. The results support a broader protective function of MAPK and document a cell type- and toxin-selective role of eIF2α.
Results
Normal Mouse Embryonal Fibroblasts are Tolerant to S. aureus α-Toxin
Exposure of human keratinocytes (HaCaT) to nanomolar concentrations of α-toxin for 2 h leads to significant loss of ATP. In contrast, murine keratinocytes (PDV) or mouse embryonal fibroblasts (MEFs) appeared to be not susceptible to α-toxin (Figure 1A). In contrast, MEFs were exquisitely susceptible to pVCC (Figure 1B), another PFT of the small β-barrel family (44).
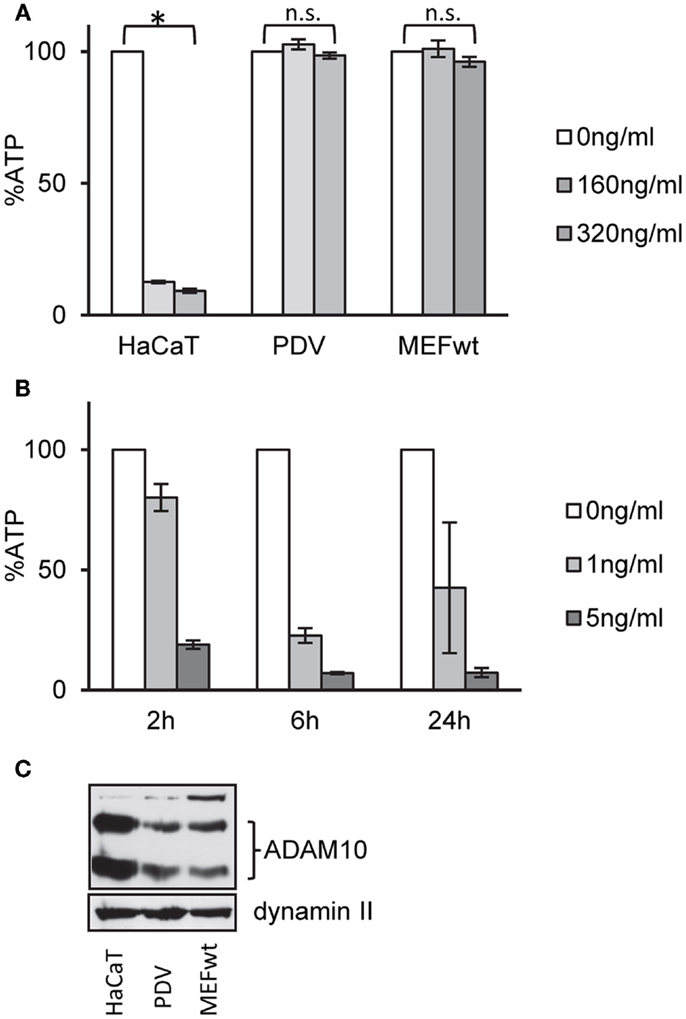
Figure 1. Wild-type MEFs are tolerant to α-toxin. (A) HaCaT cells (human keratinocytes), PDV cells (murine keratinocytes), or MEFs wt cells (mouse embryonal fibroblasts) were treated or not with α-toxin. Cellular ATP levels were determined after 2 h; data are percent of untreated controls; mean values ±SE; n ≥ 4; asterisk denotes p ≤ 0.05. (B) MEFs wt cells were treated with indicated doses of pVCC and cellular ATP levels were determined after 2, 6, or 24 h; shown are percent of untreated controls; mean values ±SE; n = 3. (C) Cell lysates of wt, PDV, and HaCaT were analyzed by Western blot for ADAM10 and dynamin II (loading control).
Inhibitors of Various Signaling Pathways Sensitize MEFs for α-Toxin
One potential explanation for selective tolerance of MEF to α-toxin could be the lack of an appropriate receptor, or lower expression levels of receptor. Amino acid sequences of human and murine ADAM10, the proposed high-affinity receptor of α-toxin, are not identical. Western blots with an antibody against ADAM10 yielded weaker bands with the murine cells (Figure 1C). Although it cannot be excluded that the antibody binds human and murine cells with different efficiency, it is therefore possible that qualitative or quantitative differences in receptor expression could explain lower susceptibility of murine vs. human cells to α-toxin. Alternatively, however, efficient ongoing repair of membrane damage and fitness to cope with metabolic stress could also play a role. Therefore, we tested a panel of small molecular weight compounds that we knew to inhibit recovery from α-toxin-dependent ATP-depletion in HaCaT cells. Several (combinations of) inhibitors sensitized MEFs for α-toxin (Figure 2A). The effect depended on the concentration of inhibitor, as exemplified for JNK3XIISR3576 (Figure 2B). Although JNK3XIISR3576 is selective for JNK3 if applied at nanomolar concentrations, MAPK other than JNK3 seem to be involved here because MEFs do not express JNK3 (45). As shown for Sal/Dyn, inhibitors sensitized wild-type MEF for α-toxin, provided they expressed ADAM10 (Figure 2C). Thus, inhibitors did not sensitize cells by increasing unspecific toxicity of α-toxin.
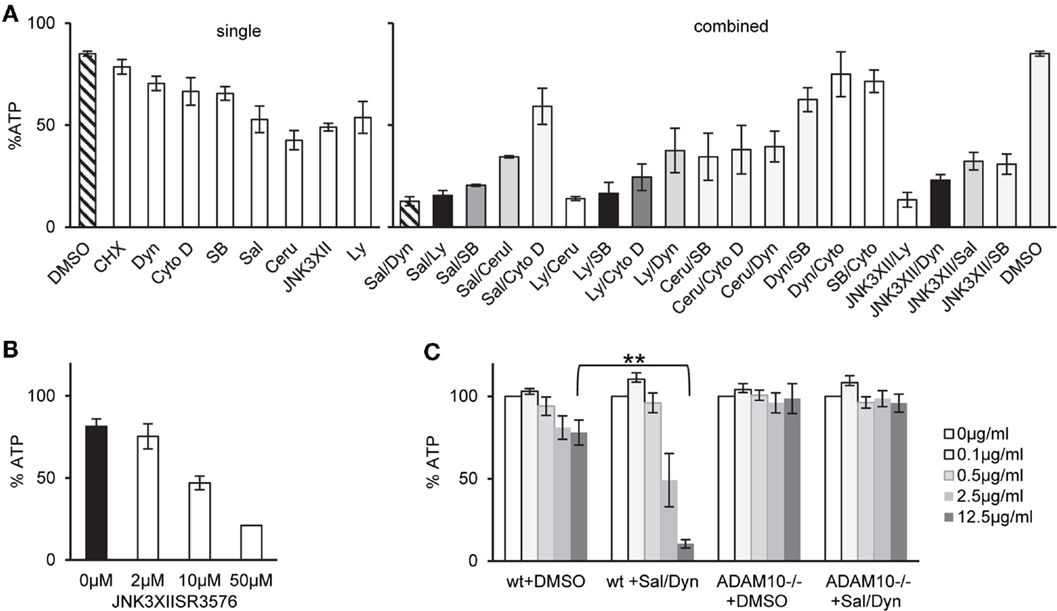
Figure 2. Inhibitors of various pathways sensitize MEFs to α-toxin. (A) wt cells were treated as indicated either with single inhibitors or combinations of two inhibitors listed below and treated with 10 μg/ml α-toxin. Cellular ATP levels were determined after 2 h; data are percent of untreated controls; shown are mean values for single inhibitors ±SE with n ≥ 6, or mean values of n ≥ 2 for combinations of inhibitors, SE was <30% throughout. Inhibitor concentrations: Salubrinal (40 μM), Dynasore (80 μM), Ly29400L (100 μM), SB203580 (20 μM), Cerulenin (20 μM), Cytochalasin D (20 μM), and JNK3XIISR3576 (10 μM). (B) MEFs wt were treated with indicated doses of JNK3XIISR3576 and incubated with 10 μg/ml α-toxin. Cellular ATP levels were determined after 2 h; data are percent of controls treated with similar concentrations of JNK3XIISR3576 but not treated with α-toxin; mean values ±SE; n ≥ 3. (C) MEFs wt or MEFs ADAM10−/− (mouse embryonal fibroblasts) were pretreated with 40 μM Salubrinal and 80 μM Dynasore or solvent alone for 30 min and treated or not with indicated doses of α-toxin. ATP levels were determined after 2 h; shown are percent of untreated controls; mean values ±SE; n = 3; two asterisks denote p-values ≤0.001.
Lack of Phosphorylatable eIF2α, GCN2, or Ppp1r15B Sensitizes MEFs for α-Toxin
That Salubrinal, an inhibitor of eIF2α-phosphatases (46) sensitized MEFs to α-toxin, and similar observations in keratinocytes (13), prompted us to investigate the response of MEFs with defined genetic modifications affecting expression or function of proteins involved in regulation of translation: first, in eIF2αS51A/S51A cells, the eIF2α locus is replaced with a non-phosphorylatable version, thus precluding regulated attenuation of translation via P-eIF2α. Second, GCN2−/− MEFs lack nutrient sensitive eIF2α-kinase GCN2/EIF2AK4, thereby blunting phosphorylation of eIF2α in response to amino acid deprivation. Third, Ppp1r15B−/− MEFs are devoid of the sole constitutive regulatory subunits of eIF2α-phosphatase (47); this defect leads to constitutively higher eIF2α-phosphorylation levels. Fourth, EeF2K−/− MEFs lack eukaryotic elongation factor 2 kinase (EeF2K), which functions downstream of mTOR to control protein synthesis (48).
First, we compared ATP levels in these MEFs to assess metabolic perturbation after treatment with α-toxin. EeF2K−/− cells were not susceptible to 10 μg/ml of α-toxin and thus behaved like wild-type MEFs. In contrast, lack of GCN2 or Ppp1r15B both sensitized cells to α-toxin. The strongest effect was observed with eIF2αS51A/S51A MEFs, which lost up to ~80% of ATP (Figure 3A). Although α-toxin led to dose-dependent loss of ATP in eIF2αS51A/S51A cells, there was barely any effect on wild-type cells (Figure 3B). Notably, pVCC-dependent loss of ATP was similar in wild-type or mutant MEFs (Figure 3C). Next, we determined the frequency of sub-G1 events, a measure of DNA fragmentation, in cells treated with α-toxin for 48 h. No significant difference was observed between wild-type and eIF2αS51A/S51A cells in the presence of β-ME and non-essential amino acids (NEAA). However, without these supplements, the number of sub-G1 events was doubled in eIF2αS51A/S51A cells (Figure 3D), although toxin-dependent loss of ATP and phosphorylation of eIF2α were equal in media with or without additives (data not shown). Additives thus appear to protect cells from secondary damage.
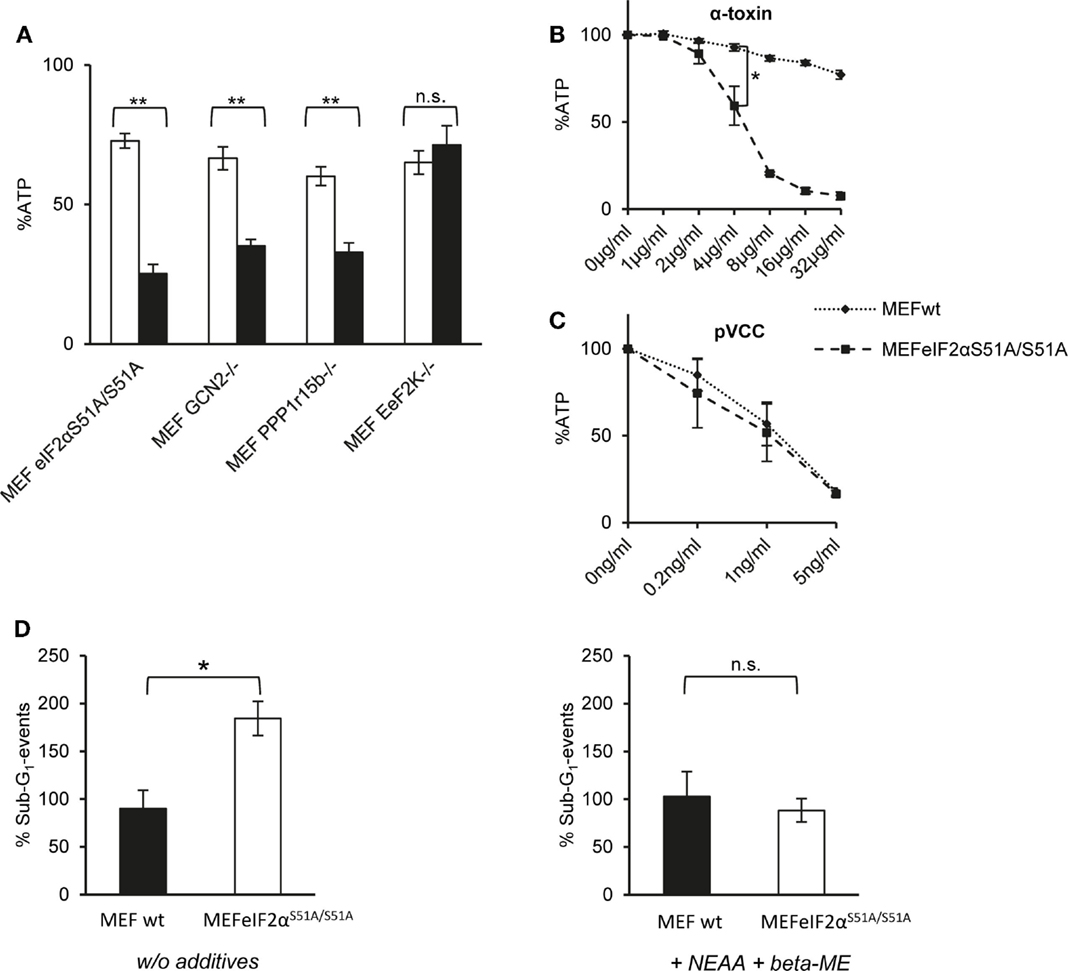
Figure 3. Dysregulation of eIF2α-phosphorylation increases sensitivity for α-toxin. (A) MEFs eIF2αS51A/S51A, MEFs GCN2−/−, MEFs Ppp1r15b−/−, or MEFs EeF2K−/− were treated or not with 10 μg/ml α-toxin. ATP levels were determined after 2 h; shown are percent of untreated controls; mean values ±SE; n ≥ 4. Black bars show data with MEFs cell variants as indicated; white bars corresponding control cells; two asterisks denote p-values ≤0.001. (B) MEFs wt or eIF2αS51A/S51A were treated, or not with indicated doses of α-toxin. Cellular ATP levels were determined after 2 h; data are percent of untreated control; mean values ±SE; n = 5; asterisk: p = 0.026. (C) MEFs wt or MEFs eIF2αS51A/S51A treated or not with indicated doses of pVCC. Cellular ATP levels were determined after 2 h; data are percent of untreated control; mean values ±SE; n = 3. (D) MEFs wt or MEFs eIF2αS51A/S51A were cultured in standard media or in presence of β-ME and additional non-essential amino acids and treated with 10 μg/ml α-toxin for 48 h, stained with Propidium iodide and subsequently frequency of Sub-G1-DNA was determined; data show percent of untreated controls; mean values ±SE; n = 3; asterisk denotes p ≤ 0,05.
GCN2/EIF2AK4 Contains α-Toxin-Dependent Stress
Next, we assessed basal or toxin-dependent eIF2α-phosphorylation in the various MEFs lines. In wild-type cells, some basal phosphorylation of eIF2α was noted, which was moderately increased by α-toxin; after normalization for eIF2α, the effect was, however, statistically insignificant. As expected, P-eIF2α was not detected in eIF2αS51A/S51A MEFs (Figure 4A). No P-eIF2α was also discerned with samples of untreated GCN2−/− cells; this indicated that basal phosphorylation of eIF2α in cultured MEFs depends on GCN2. Paradoxically, however, α-toxin-dependent phosphorylation of eIF2α was enhanced in cells lacking this eIF2α-kinase. This showed that increased susceptibility of GCN2−/− MEFs to α-toxin (Figure 3A) cannot be accounted for by diminished toxin-dependent eIF2α-phosphorylation. Membranes were re-probed with antibodies against p38, which becomes phosphorylated in response to PFT. Strikingly, α-toxin caused robust phosphorylation of stress activated protein kinase p38 in GCN2−/− MEFs, Ppp1r15B MEFs, and eIF2αS51A/S51A MEFs (Figure 4A), but not in wild-type MEFs, supporting the notion that cells with imbalanced eIF2α-phosphorylation experienced more severe toxin-dependent stress. p70S6K, substrate of mTORC1 became de-phosphorylated (Figure 4A), indicating that α-toxin inhibits mTORC1, master regulator of translation and autophagy.
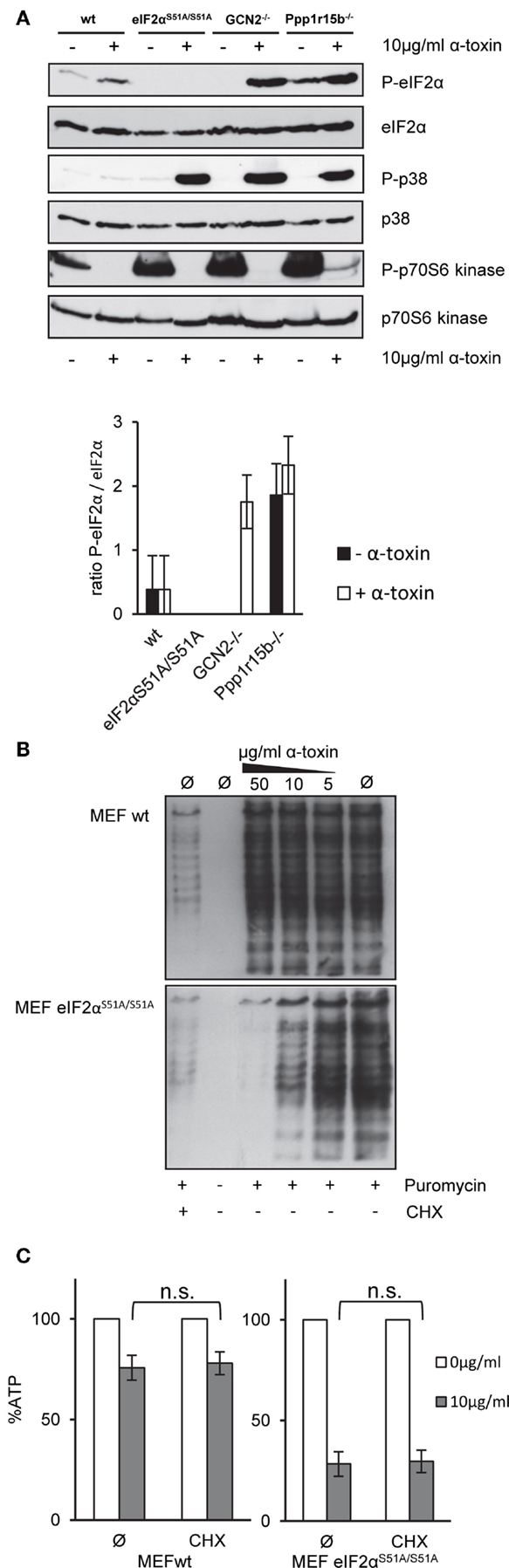
Figure 4. α-toxin causes translational arrest in MEFs eIF2αS51A/S51A. Cycloheximide does not sensitize wild-type MEFs (A) MEFs wt, MEFs eIF2αS51A/S51A, MEFs GCN2−/−, or MEFs Ppp1r15b−/− were treated or not with 10 μg/ml α-toxin for 2 h. Cell lysates were analyzed by Western blot for P-eIF2α, eIF2α, P-p70S6 kinase, p70S6 kinase P-p38, and p-38. One of three similar blots is shown; lower panel summarizes densitometric data for (P)-eIF2α, mean ± SE; n = 3 (B) MEFs wt or MEFs eIF2αS51A/S51A cells were treated or not with indicated concentrations of α-toxin and incubated for 1 h at 37°C. Treatment with CHX served as positive control for translational arrest. Subsequently, the cells were incubated for 1 h at 37°C with 10 μg/ml puromycin, which incorporates during ongoing synthesis into nascent proteins. Eventually, cells were analyzed by Western blot for puromycin. (C) MEFs wt or MEFs eIF2αS51A/S51A cells were treated or not with 10 μg/ml α-toxin and incubated with or without CHX at 37°C. Cellular ATP levels were determined after 2 h; data are percent of untreated controls; mean values ±SE; n = 5.
CHX Does Not Tolerize eIF2αS51A/S51A MEFs to α-Toxin
Results from Western blots (Figure 4A) raised the question whether α-toxin impacts translation differentially in eIF2αS51A/S51A vs. wild-type MEFs. Paradoxically, treatment with α-toxin caused dose-dependent attenuation of translation in eIF2αS51A/S51A MEFs, but had no such effect on wild-type MEFs (Figure 4B). This suggested that translation was inhibited in response to α-toxin through a mechanism that was independent of eIF2α-phosphorylation. Together with results from the foregoing ATP-assays, this also revealed that attenuation of translation per se is insufficient to maintain metabolic homeostasis upon attack by α-toxin. Conversely, toxin-dependent ATP-loss in eIF2αS51A/S51A MEFs is not a consequence of translational arrest, because treatment of wild-type cells with CHX stops translation (Figure 4B), but does not hyper-sensitize MEFs for α-toxin (Figure 4C).
Wild-Type eIF2α Modulates Binding and Action of α-Toxin
That inhibition of translation did not protect eIF2αS51A/S51A MEFs provoked the question how phosphorylation of eIF2α tolerizes MEF. So, we investigated a primary event underlying many of the rapid molecular changes induced by PFT, i.e., disturbance of natural ion gradients (4). Loss of potassium appears to be one major trigger (11, 20, 24, 49). We measured loss of intracellular potassium by flame photometry. In line with results from ATP-assays, net loss of potassium was enhanced in GCN2−/− or Ppp1r15B−/− cells and even more so in eIF2αS51A/S51A MEFs (Figure 5A), suggesting that membrane damage was more severe in cells with deficiencies in regulation of eIF2α-phosphorylation.
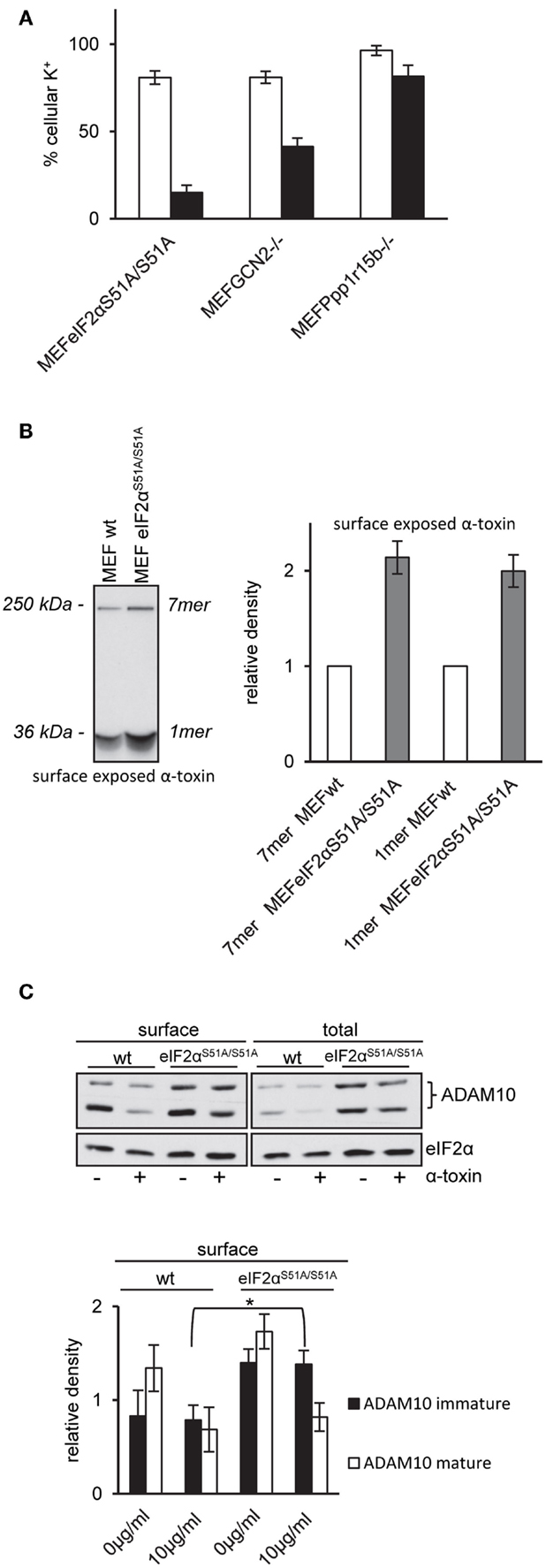
Figure 5. MEFs eIF2αS51A/S51A over-express ADAM10 and bind higher amounts of α-toxin. (A) MEFs eIF2αS51A/S51A, MEFs GCN2−/−, MEFs Ppp1r15b−/−, or corresponding control cells were treated or not with 10 μg/ml α-toxin. Potassium levels were determined after 2 h, shown are percent of untreated controls; mean values ±SE; n ≥ 4. Black bars show data of MEFs cell variants as indicated; white bars corresponding control cells. (B) Right: MEFs wt or MEFs eIF2αS51A/S51A cells were incubated with radio-labeled α-toxin 2 and 8 μg/ml α-toxin at 37°C for 15 min. Subsequently, cells were surface biotinylated, lysates were obtained and subjected to sequential neutravidin-pulldown (NP) and immunoprecipitation (IP) followed by PAGE/fluography, as described in Kloft et al. (13). Left: band intensities were measured by densitometry using ImageJ software. Shown are mean values ±SE; n = 4. Variations of loading with toxin and plating of cells were <1 and <10%, respectively. (C) MEFs wt or MEFs eIF2αS51A/S51A were treated or not with 10 μg/ml α-toxin for 2 h. Subsequently, cells were surface biotinylated or not and lysed. Lysates were subjected to NP; both lysates and precipitation were analyzed by Western blot for ADAM10 and eIF2α (loading control); upper panel: one of four similar blots; lower panel: bar chart summarizing data (mean ± SE; n = 4).
Pore formation by α-toxin depends on oligomerization and insertion into the plasma membrane. Because oligomers resist SDS, they can be detected by SDS-PAGE. Using cell surface labeling after incubation with internally radio-labeled α-toxin, we compared the amount of α-toxin on the surface of wild-type MEFs and eIF2αS51A/S51A MEFs. The fluorographic analysis shown in Figure 5B reveals that more α-toxin is present at the surface of eIF2αS51A/S51A MEFs as compared to wild-type cells, providing a straightforward explanation for enhanced loss of potassium and ATP from these cells.
Wild-Type eIF2α Modulates Expression of ADAM10
Increased amounts of toxin associated with eIF2αS51A/S51A MEFs could be due to higher abundance of α-toxin receptors. Therefore, we compared ADAM10 expression in eIF2αS51A/S51A MEFs and wild-type MEFs. Western-blot analysis revealed that ADAM10 is over-expressed in eIF2αS51A/S51A MEFs; and more ADAM10 is exposed on the cell surface of eIF2αS51A/S51A MEFs as compared to wild-type cells (Figure 5C). No significant differences between wild-type cells and eIF2αS51A/S51A MEFs were found in a lipidomics analysis (data not shown). Treatment with α-toxin led to down-regulation of ADAM10 at the cell surface of both wild-type and mutant cells. Because basal eIF2α-phosphorylation in MEFs depends on GCN2 (Figure 4A), the collective data indicate that nutrient stress or basal activity of GCN2 modulates levels of ADAM10 in MEFs.
Role of GCN2 for Tolerance to α-Toxin is Not Conserved in BMDM
Like MEFs, BMDM proved to be highly tolerant to α-toxin although they were susceptible to Vibrio cholera cytolysin (VCC) (Figure 6A). However, lack of GCN2-expression in BMDM did not significantly alter susceptibility to α-toxin (Figures 6A,D); α-toxin-dependent phosphorylation of eIF2α appeared slightly reduced (Figure 6B). Further, infection of BMDM by S. aureus was equally efficient with both strains whether or not bacteria produced toxin or not (Figure 6C). Therefore, GCN2 seemed to play no major role for resistance or tolerance to α-toxin of BMDMs.
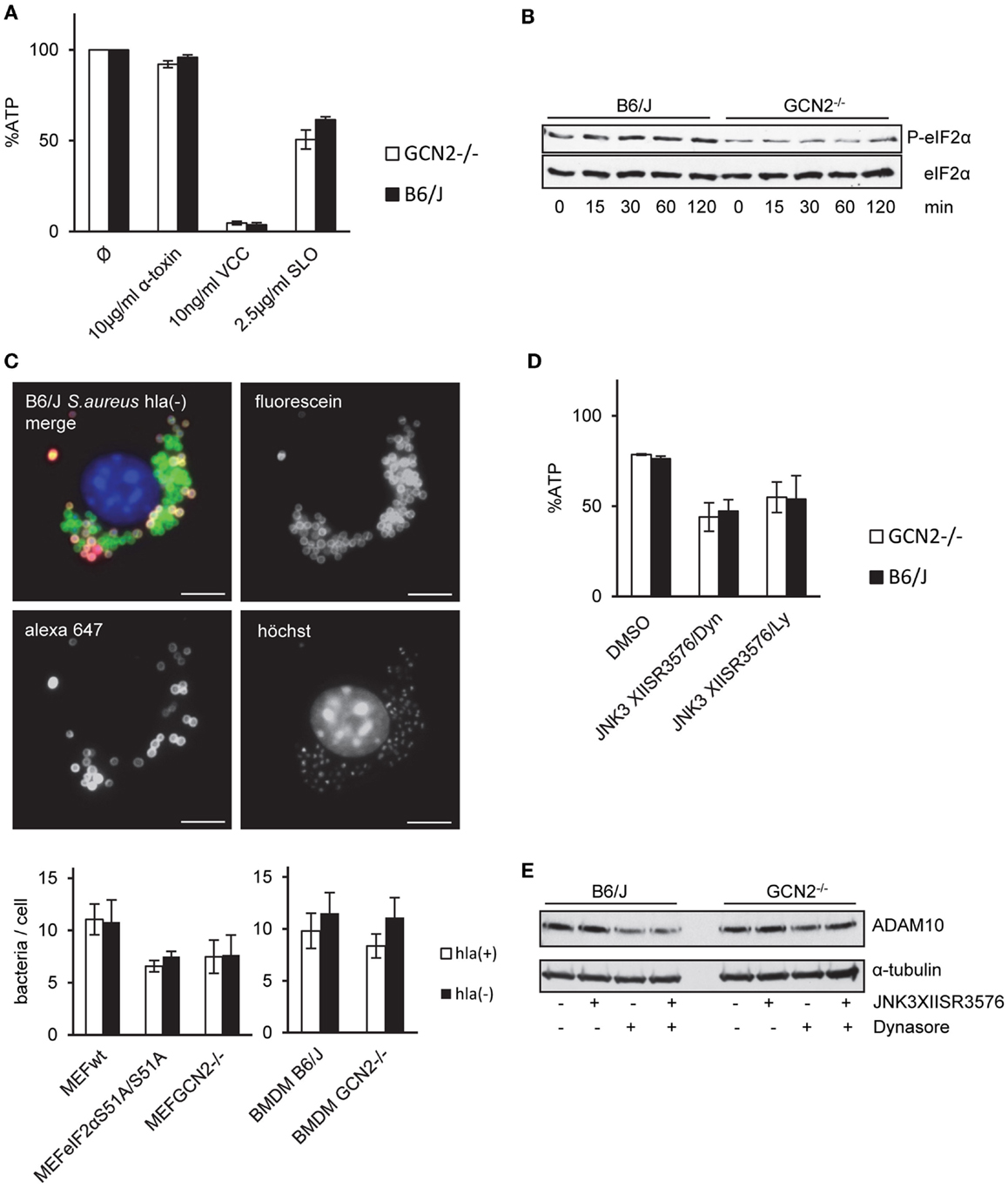
Figure 6. GCN2 does not render BMDMs tolerant to α-toxin. (A) BMDMs isolated from GCN2−/− mice or control mice (B6/J) were treated or not with indicated doses of α-toxin, VCC, or SLO. Cellular ATP levels were determined after 2 h. White bars show BMDMs from GCN2−/− mice and black bars show BMDMs from control mice; mean values ±SE, n ≥ 3 (B) BMDMs of GCN2−/− mice or control mice were incubated with 10 μg/ml α-toxin for indicated times. Cell lysates were analyzed by Western blot for P-eIF2α and eIF2α. (C) MEFs variants (right graph) or BMDMs of GCN2−/− and control mice (left graph) were incubated with fluorescein/biotin labeled S. aureus hla(−) or hla(+) strains (MOI 1:30) for 1 h, washed and incubated for an additional hour at 37°C. After fixation, extracellular bacteria were stained with streptavidin-coupled Alexa 647. Upper: exemplary picture of solely fluorescein-stained S. aureus, representing intracellular bacteria, and extracellular S. aureus that were accessible for Alexa 647-Streptavidin. Lower: graphs show counts of solely fluorescein-stained (intracellular) bacteria per cell; mean values ±SE; BMDMs n = 2; MEFs n = 3. (D) BMDMs of GCN2−/− mice or control mice were incubated with combinations of JNK3XIISR3576 (10 μM), Dynasore (80 μM), JNK3 XIISR3576 (10 μM), and Ly29400L (100 μM) or solvent alone and treated with 10 μg/ml α-toxin. Cellular ATP levels were determined after 2 h (percentage of controls). (Mean values ±SE; n = 3). (E) BMDMs of GCN2−/− and control mice were treated with the combination of Dynasore (80 μM) and JNK3XIISR3576 (10 μM), Dynasore (80 μM) alone, JNK3XIISR3576 (10 μM) alone, or solvent alone (DMSO) for 2.5 h. Cell lysates were analyzed by Western blot for ADAM10 and α-tubulin (loading control).
Small MW inhibitors of JNK and dynamin, a cocktail, which efficiently breaks tolerance to α-toxin in MEFs, moderately sensitized BMDM to purified α-toxin (Figure 6D). Inhibitors did not enhance ADAM10 expression (Figure 6E). Thus, MAPK enhance natural tolerance of BMDM to α-toxin, but modulation of ADAM10 expression appears not to be involved.
Discussion
One conclusion of this work is that regulators of translation initiation (eIF2α, GCN2, and Ppp1r15B) render murine embryonic fibroblasts tolerant to S. aureus α-toxin. This is consistent with our previous finding in human epithelial cells that these proteins promote recovery from successful attack (13). Although it remains to be investigated whether a priori tolerance of MEFs to α-toxin is likewise based on efficient endocytic removal of α-toxin pores from the cell surface, the present results document that the tolerogenic effect of eIF2α, GCN2, and Ppp1r15B is conserved in mice and man, and that it is observable in fibroblasts. That both lack of an eIF2α-kinase and of constitutive eIF2α-phosphatase reduce tolerance to α-toxin could be interpreted to show that balanced phosphorylation of eIF2α, or cycling of eIF2α between phosphorylated and unphosphorylated state is required for the tolerogenic effect.
A role of translational regulation for various aspects of innate immunity, and a protective function against PFT has been discussed in the recent literature (34, 50, 51). One established function of eIF2α-phosphorylation and the integrated stress–response is to reprogram expression of genes, including of genes that regulate immunity. Translational attenuation in host cells might also help them to conserve energy, as proposed by some authors (20). However, (P)-eIF2α-dependent tolerance of MEF could not be explained by toxin-dependent translational attenuation per se: actually, α-toxin caused inhibition of translation in eIF2αS51A/S51A MEFs, but not in wild-type MEFs, and CHX did neither protect nor hyper-sensitize wild-type cells from/for α-toxin. Yet, eIF2αS51A/S51A MEFs proved to be significantly less tolerant to α-toxin. Probably as a consequence, protein synthesis was halted through alternative pathways. This could occur, for instance, by deactivation of mTOR, as indeed suggested by α-toxin-dependent dephosphorylation of p70S6K, a target of mTOR. The data may reflect a hierarchy of stress responses: Activation of eIF2α via nutritional sensor GCN2 contains α-toxin-dependent damage and stress, which would otherwise lead to exaggerated hyper-phosphorylation of eIF2α by (an)other eIF2α-kinase(s), possibly PERK and PKR. If eIF2α-phosphorylation fails, translation would be halted via robust deactivation of mTOR. Sustained inhibition of translation is obviously not tolerated by cells (35), but marked α-toxin-dependent inhibition of translation, observed in eIF2αS51A/S51A MEFs, does not explain the increased loss of ATP and potassium from these cells, because CHX did not affect these parameters in wild-type cells.
The fact that lack of phosphorylatable eIF2α was associated with increased toxin-dependent loss of potassium and ATP prompted us to measure the binding of α-toxin and expression of ADAM10, the proposed high-affinity receptor for α-toxin (52). This led to the unexpected finding that ADAM10 levels were significantly higher in eIF2αS51A/S51A cells. Therefore, tolerance to α-toxin in MEFs may be well due to GCN2/P-eIF2α-dependent modulation of its receptor, ADAM10. Whether or not the apparent link between basal nutrient stress and expression of ADAM10 was shaped by co-evolution of S. aureus and humans, it may also bear on functions of ADAM10 that are not related to infection.
How basal phosphorylation of eIF2α-levels modulates ADAM10-expression remains to be elucidated. Because P-eIF2α is required for starvation-dependent autophagy (53), a possible role of eIF2α in this context could be to maintain basal autophagic flux, which in turn could impact ADAM10 levels. Recently, Atg16L1, a protein essential for autophagy, has been shown to confer tolerance to α-toxin; the authors proposed that autophagy constitutively dampens ADAM10 levels in a cell-type selective manner (54). Similarly, eIF2α-dependent modulation of ADAM10 shown in the present work is constitutive and cell-type selective. Together this seems to suggest that basal eIF2α-phosphorylation functions upstream of autophagy to mediate tolerance. Alternatively, P-eIF2α, eIF2α-kinases, and -phosphatases could function through mechanisms acting in parallel to autophagy, for instance, by regulating endocytosis of membrane pores, as has been shown in epithelial cells (13). Whatever the effector mechanism(s) downstream of eIF2α are, our data reveal that GCN2-dependent basal phophorylation of eIF2α in MEF modulates ADAM10 levels as well as binding and action of α-toxin. Because GCN2 is activated by low levels of amino acids in cells, basal nutrient stress might be the driving force; the potential links discussed here are summarized in a model (Figure 7).
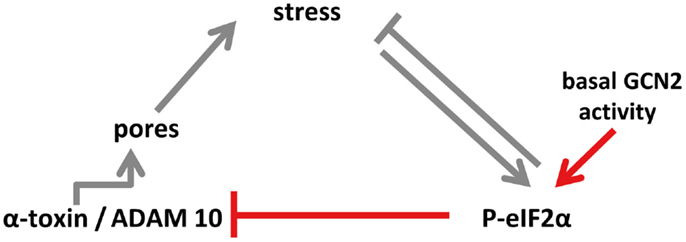
Figure 7. Model of eIF2α-dependent cellular tolerance to α-toxin. Disruption of membrane integrity by insertion of membrane pores causes various forms of stresses in target cells (e.g., loss of potassium, starvation), which trigger an array of conserved responses, including phosphorylation of MAPK and of eIF2α. Not only may these pathways feed back to alleviate stress but also eIF2α may modulate formation of S. aureus α-toxin membrane pores (this work; highlighted in red in the scheme), or persistence of lesions (13), root causes of α-toxin-dependent stress. Notably, basal activity of GCN2 maintains low levels of ADAM10, resulting in bated binding of α-toxin. Thus, basal nutrient stress in cells could serve as a preemptive stimulus of cellular tolerance to S. aureus α-toxin. This link, which might have evolved from mutual adaptation of S. aureus and humans, is selective, as suggested by the fact that Vibrio cholerae cytolysin breaks cell autonomous defense.
Inhibitors of various pathways hyper-sensitized MEFs to S. aureus α-toxin, which supports the notion that multiple signaling pathways are required to confer cellular tolerance to α-toxin. This raises the possibility that drugs used in pharmaco-therapy may have potential tolerance-modulating effects, an issue that warrants further investigation. The present data support a tolerogenic role of JNKs in both MEFs and BMDM. More work is required to understand the underlying mechanisms, but they seem to be distinct from eIF2α-dependent cellular tolerance to α-toxin. Conserved stress responses may fail to protect against some PFT, as exemplified here by V. cholerae cytolysin. Whether cells of the immune system tolerate attack of a given PFT will co-determine the ability of an organism to mount an effective “classic” immune response to infection with corresponding bacteria.
Materials and Methods
Antibodies
Antibodies against P-eIF2α (phosphorylated at Ser51), eIF2α, P-p38 (phosphorylated at Thr180/Tyr182), p-38, P-p70S6K (phosphorylated at Thr389), p70S6K, ADAM10, and α-tubulin were from Cell Signaling Technology. Antibodies against dynamin II were purchased from Santa-Cruz Biotechnology. Anti-Puromycin-antibody was from Merck Millipore. Antibodies against LC3 were bought from Sigma. HRP-conjugated secondary antibodies were from Santa-Cruz Biotechnology (mouse) and Cell Signaling Technology (rabbit).
Inhibitors
Salubrinal (SAL), Cycloheximid (CHX), JNK3XIISR3576 (JNK3XII), and SB203580 (SB) were obtained from Calbiochem. Ly29400L (Ly) was from Cell Signaling. Bafilomycin, Cytochalasin D (Cyto D), and Cerulenin (Ceru) were from Sigma and Dynasore (Dyn) was from Tocris bioscience.
Chemicals
RNase, propidium iodide, and puromycin were purchased from Sigma. NHS-fluorescein and EZ-Link Sulfo-NHS-LC-biotin were from Thermo Fisher Scientific and Streptavidin-Alexa 647 was obtained from Molecular Probes. Rapamycin was from Calbiochem.
Toxins
α-toxin, internally radio-labeled α-toxin, streptolysin (SLO), and VCC were made as published elsewhere (10, 12).
S. aureus
In this study, S. aureus strain DU1090 (55) and α-toxin producing S. aureus strain, plasmid transformed derivative of DU1090 (55), were employed and referred to as hla(−) and hla(+), respectively.
Cells and Culture
Mouse embryonal fibroblasts GCN2−/− (56) were purchased from ATCC and PDV from CLS Cell Lines Service GmbH. MEFs eIF2αS51A/S51A and corresponding control cells MEFs eIF2αS51/S51(57), MEFs Ppp1r15b−/− (58), MEFs EeF2K−/− (59), and MEFs ADAM10−/− (60) were kindly provided by Heather Harding and David Ron, Randal Kaufman, Alexey Ryazanov, and Paul Saftig, respectively. All MEFs cell lines were cultured in DMEM GlutaMAX™-I medium with 10% fetal calf serum, 1% HEPES buffer, 1% penicillin/streptomycin, 1% MEM NEAA, and 55 mM 2-mercaptoethanol. Under these conditions, we did not note significant differences in morphology, viability, or growth rate. PDV (murine keratinocyte cell line) were grown in DMEM GlutaMAX™-I medium with 10% fetal calf serum, 1% HEPES buffer, 1% penicillin/streptomycin without MEM NEAA, and 2-mercaptoethanol. HaCaT (non-virally transformed HaCaT) (61) were cultured in DMEM/F-12 GlutaMAX™-I medium with 10% fetal calf serum, 1% HEPES buffer, and 1% penicillin/streptomycin. All media and medium additives were obtained from Gibco by life technologies™. BMDMs were isolated from C57BL/6J (B6/J) or B6.129S6-EIF2αk4tm1.2Dron/j (GCN2−/−) mice (Charles River laboratories) and cultured in DMEM GlutaMAX™-I supplemented with 20% fetal calf serum and M-CSF by adding 10% supernatant of L929 cells.
Western Blot
For Western blots, cell lysates were mixed with 2× SDS-loading buffer [65 mM Tris, 10% (v/v) glycerol, 5% (v/v) 2-mercaptoethanol, 2% (w/v) SDS, and bromphenol blue] and boiled for 5 min at 95°C. Proteins were separated by SDS-PAGE (10%) and electroblotted onto nitrocellulose membrane. After blocking for 1 h at room temperature in BSA or skim milk in TBST [Tris 50 μM, NaCl 0.15M, Tween 0.1% (v/v)], membrane was incubated with a primal antibody in BSA or skim milk in TBST, washed three times in TBST and incubated with HRP-conjugated second antibody for 1 h at room temperature. After three washing steps, bound antibody was detected by ECL (Roche Applied Science).
Puromycin Incorporation
The assay has been described elsewhere (62). MEFs eIF2αS51A/S51A cells were seeded at a density of 2 × 105 cells/well into six-well plates. Cells were incubated as indicated with several concentrations of pVCC or α-toxin for 1 h at 37°C. Thereafter, 10 μg/ml puromycin was added and cells were incubated for an additional hour at 37°C. Subsequently, medium was removed; cells were washed with PBS and analyzed by Western blot using anti-Puromycin-antibody in 2.5% (w/v) skim milk in TBST.
ATP-Measurements
Measurements of intracellular ATP were performed, as described elsewhere (22).
Potassium Efflux Measurements
For measurements of intracellular potassium, cells were seeded at a density of 3 × 105 cells/well into six-well plates. Cells were incubated or not with 8 μM Rapamycin for 3 h before adding 10 μg/ml α-toxin. After 2 h incubation at 37°C, cells were washed three times on ice with cold Choline-chlorid-buffer and solubilized in 2 ml Choline-chlorid-buffer containing 0.5% Triton X-100 (63) for 30 min at room temperature under constant shaking. Potassium content of supernatants of centrifuged cell lysates was determined using Sherwood single channel flame photometer M401.
Quantitation of Surface-Exposed or Internalized α-Toxin/ADAM10
Surface labeling, neutravidin pull-down, and fluorometric detection of α-toxin have been described previously (13). Variation of input (labeled S35-Met-α-toxin) was <1%; equal amounts of total protein were loaded.
Quantification of Internalized Bacteria
The assay has been performed, as described elsewhere (64).
Sub-G1 Analysis
Cells were seeded at a density of 1 × 105 cells/well into six-well plates and treated or not with 10 μg/ml α-toxin permanently for 48 h at 37°C. Cells were harvested including the detached cells in the supernatant, washed in PBS and centrifuged. Cells were resuspended and fixed in cold 70% ethanol in PBS/EDTA for 2 h at 4°C. Subsequently, the cells were washed and treated with RNase for 10 min at 37°C. Then, propidium iodide was added and cells were incubated for 5 min at room temperature. Eventually, cells were analyzed using a FACScan flow cytometer (BD Biosciences) and CellQuest software.
Statistical Analysis
Two-sided, unpaired Student’s t-test was employed to assess statistical significance of differences between mean values. Significance was assumed at p < 0.05.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Research reported in this publication was supported by the National Institutes of Health NIH/NIFMS under award number 7-R0I-GM071603-09 to RA and MH (subrecipient); DK042394, DK088227, and HL052173 to RK; MH and GH received support by University Medical Center Mainz. We like to thank Dr. H. Harding and Dr. D. Ron for generously providing Ppp1r15b−/− MEFs, Dr. P. Saftig for ADAM10−/− MEFs, and Dr. B. Bruegger for a large scale lipid analysis. Further, we gratefully acknowledge support by Dr. A. Diefenbach.
References
1. Bhakdi S, Tranum-Jensen J. Membrane damage by pore-forming bacterial cytolysins. Microb Pathog (1986) 1:5–14. doi:10.1016/0882-4010(86)90027-6
2. Iacovache I, Van Der Goot FG, Pernot L. Pore formation: an ancient yet complex form of attack. Biochim Biophys Acta (2008) 1778:1611–23. doi:10.1016/j.bbamem.2008.01.026
3. Los FC, Randis TM, Aroian RV, Ratner AJ. Role of pore-forming toxins in bacterial infectious diseases. Microbiol Mol Biol Rev (2013) 77:173–207. doi:10.1128/MMBR.00052-12
4. Bischofberger M, Iacovache I, Van Der Goot FG. Pathogenic pore-forming proteins: function and host response. Cell Host Microbe (2012) 12:266–75. doi:10.1016/j.chom.2012.08.005
5. Thelestam M, Mollby R. Survival of cultured-cells after functional and structural disorganization of plasma-membrane by bacterial hemolysins and phospholipases. Toxicon (1983) 21:805–15. doi:10.1016/0041-0101(83)90069-7
6. Walev I, Palmer M, Martin E, Jonas D, Weller U, Hohn-Bentz H, et al. Recovery of human fibroblasts from attack by the pore-forming alpha-toxin of Staphylococcus aureus. Microb Pathog (1994) 17:187–201. doi:10.1006/mpat.1994.1065
7. Huffman DL, Abrami L, Sasik R, Corbeil J, Van Der Goot FG, Aroian RV. Mitogen-activated protein kinase pathways defend against bacterial pore-forming toxins. Proc Natl Acad Sci U S A (2004) 101:10995–1000. doi:10.1073/pnas.0404073101
8. Husmann M, Dersch K, Bobkiewicz W, Beckmann E, Veerachato G, Bhakdi S. Differential role of p38 mitogen activated protein kinase for cellular recovery from attack by pore-forming S. aureus alpha-toxin or streptolysin O. Biochem Biophys Res Commun (2006) 344:1128–34. doi:10.1016/j.bbrc.2006.03.241
9. Aroian R, Van Der Goot FG. Pore-forming toxins and cellular non-immune defenses (CNIDs). Curr Opin Microbiol (2007) 10:57–61. doi:10.1016/j.mib.2006.12.008
10. Husmann M, Beckmann E, Boller K, Kloft N, Tenzer S, Bobkiewicz W, et al. Elimination of a bacterial pore-forming toxin by sequential endocytosis and exocytosis. FEBS Lett (2009) 583:337–44. doi:10.1016/j.febslet.2008.12.028
11. Kloft N, Busch T, Neukirch C, Weis S, Boukhallouk F, Bobkiewicz W, et al. Pore-forming toxins activate MAPK p38 by causing loss of cellular potassium. Biochem Biophys Res Commun (2009) 385:503–6. doi:10.1016/j.bbrc.2009.05.121
12. Kloft N, Neukirch C, Bobkiewicz W, Veerachato G, Busch T, Von Hoven G, et al. Pro-autophagic signal induction by bacterial pore-forming toxins. Med Microbiol Immunol (2010) 199:299–309. doi:10.1007/s00430-010-0163-0
13. Kloft N, Neukirch C, Von Hoven G, Bobkiewicz W, Weis S, Boller K, et al. A subunit of eukaryotic translation initiation factor 2alpha-phosphatase (CreP/PPP1R15B) regulates membrane traffic. J Biol Chem (2012) 287:35299–317. doi:10.1074/jbc.M112.379883
14. Stuart LM, Paquette N, Boyer L. Effector-triggered versus pattern-triggered immunity: how animals sense pathogens. Nat Rev Immunol (2013) 13:199–206. doi:10.1038/nri3398
15. Kao CY, Los FC, Huffman DL, Wachi S, Kloft N, Husmann M, et al. Global functional analyses of cellular responses to pore-forming toxins. PLoS Pathog (2011) 7:e1001314. doi:10.1371/journal.ppat.1001314
16. Gonzalez MR, Bischofberger M, Pernot L, Van Der Goot FG, Freche B. Bacterial pore-forming toxins: the (w)hole story? Cell Mol Life Sci (2008) 65:493–507. doi:10.1007/s00018-007-7434-y
17. Porta H, Cancino-Rodezno A, Soberon M, Bravo A. Role of MAPK p38 in the cellular responses to pore-forming toxins. Peptides (2011) 32:601–6. doi:10.1016/j.peptides.2010.06.012
18. Von Hoven G, Kloft N, Neukirch C, Ebinger S, Bobkiewicz W, Weis S, et al. Modulation of translation and induction of autophagy by bacterial exoproducts. Med Microbiol Immunol (2012) 201:409–18. doi:10.1007/s00430-012-0271-0
19. Stassen M, Muller C, Richter C, Neudorfl C, Hultner L, Bhakdi S, et al. The streptococcal exotoxin streptolysin O activates mast cells to produce tumor necrosis factor alpha by p38 mitogen-activated protein kinase- and protein kinase C-dependent pathways. Infect Immun (2003) 71:6171–7. doi:10.1128/IAI.71.11.6171-6177.2003
20. Gonzalez MR, Bischofberger M, Freche B, Ho S, Parton RG, Van Der Goot FG. Pore-forming toxins induce multiple cellular responses promoting survival. Cell Microbiol (2011) 13(7):1026–43. doi:10.1111/j.1462-5822.2011.01600.x
21. Nagahama M, Shibutani M, Seike S, Yonezaki M, Takagishi T, Oda M, et al. The p38 MAPK and JNK pathways protect host cells against Clostridium perfringens beta-toxin. Infect Immun (2013) 81:3703–8. doi:10.1128/IAI.00579-13
22. Haugwitz U, Bobkiewicz W, Han SR, Beckmann E, Veerachato G, Shaid S, et al. Pore-forming Staphylococcus aureus alpha-toxin triggers epidermal growth factor receptor-dependent proliferation. Cell Microbiol (2006) 8:1591–600. doi:10.1111/j.1462-5822.2006.00733.x
23. Richter E, Harms M, Ventz K, Gierok P, Chilukoti RK, Hildebrandt JP, et al. A multi-omics approach identifies key hubs associated with cell type-specific responses of airway epithelial cells to staphylococcal alpha-toxin. PLoS One (2015) 10:e0122089. doi:10.1371/journal.pone.0122089
24. Hamon MA, Cossart P. K+ efflux, is required for histone-H3 dephosphorylation by Listeria LLO and other pore forming toxins. Infect Immun (2011) 79(7):2839–46. doi:10.1128/IAI.01243-10
25. Imre G, Heering J, Takeda AN, Husmann M, Thiede B, Zu Heringdorf DM, et al. Caspase-2 is an initiator caspase responsible for pore-forming toxin-mediated apoptosis. EMBO J (2012) 31(11):2615–28. doi:10.1038/emboj.2012.93
26. Idone V, Tam C, Goss JW, Toomre D, Pypaert M, Andrews NW. Repair of injured plasma membrane by rapid Ca2+-dependent endocytosis. J Cell Biol (2008) 180:905–14. doi:10.1083/jcb.200708010
27. Tam C, Idone V, Devlin C, Fernandes MC, Flannery A, He X, et al. Exocytosis of acid sphingomyelinase by wounded cells promotes endocytosis and plasma membrane repair. J Cell Biol (2010) 189:1027–38. doi:10.1083/jcb.201003053
28. Babiychuk EB, Monastyrskaya K, Potez S, Draeger A. Blebbing confers resistance against cell lysis. Cell Death Differ (2011) 18:80–9. doi:10.1038/cdd.2010.81
29. Draeger A, Monastyrskaya K, Babiychuk EB. Plasma membrane repair and cellular damage control: the annexin survival kit. Biochem Pharmacol (2011) 81:703–12. doi:10.1016/j.bcp.2010.12.027
30. Corrotte M, Fernandes MC, Tam C, Andrews NW. Toxin pores endocytosed during plasma membrane repair traffic into the lumen of MVBs for degradation. Traffic (2012) 13:483–94. doi:10.1111/j.1600-0854.2011.01323.x
31. Corrotte M, Almeida PE, Tam C, Castro-Gomes T, Fernandes MC, Millis BA, et al. Caveolae internalization repairs wounded cells and muscle fibers. Elife (2013) 2:e00926. doi:10.7554/eLife.00926
32. Castro-Gomes T, Koushik AB, Andrews NW. ESCRT: nipping the wound in the bud? Trends Biochem Sci (2014) 39:307–9. doi:10.1016/j.tibs.2014.06.001
33. Tattoli I, Sorbara MT, Vuckovic D, Ling A, Soares F, Carneiro LA, et al. Amino acid starvation induced by invasive bacterial pathogens triggers an innate host defense program. Cell Host Microbe (2012) 11:563–75. doi:10.1016/j.chom.2012.04.012
34. Lemaitre B, Girardin SE. Translation inhibition and metabolic stress pathways in the host response to bacterial pathogens. Nat Rev Microbiol (2013) 11:365–9. doi:10.1038/nrmicro3029
35. Chakrabarti S, Liehl P, Buchon N, Lemaitre B. Infection-induced host translational blockage inhibits immune responses and epithelial renewal in the Drosophila gut. Cell Host Microbe (2012) 12:60–70. doi:10.1016/j.chom.2012.06.001
36. Wek RC, Jiang HY, Anthony TG. Coping with stress: eIF2 kinases and translational control. Biochem Soc Trans (2006) 34:7–11. doi:10.1042/BST0340007
37. Dever TE, Feng L, Wek RC, Cigan AM, Donahue TF, Hinnebusch AG. Phosphorylation of initiation factor 2 alpha by protein kinase GCN2 mediates gene-specific translational control of GCN4 in yeast. Cell (1992) 68:585–96. doi:10.1016/0092-8674(92)90193-G
38. Dong J, Qiu H, Garcia-Barrio M, Anderson J, Hinnebusch AG. Uncharged tRNA activates GCN2 by displacing the protein kinase moiety from a bipartite tRNA-binding domain. Mol Cell (2000) 6:269–79. doi:10.1016/S1097-2765(00)00028-9
39. Deloche O, De La Cruz J, Kressler D, Doere M, Linder P. A membrane transport defect leads to a rapid attenuation of translation initiation in Saccharomyces cerevisiae. Mol Cell (2004) 13:357–66. doi:10.1016/S1097-2765(04)00008-5
40. De Filippi L, Fournier M, Cameroni E, Linder P, De Virgilio C, Foti M, et al. Membrane stress is coupled to a rapid translational control of gene expression in chlorpromazine-treated cells. Curr Genet (2007) 52:171–85. doi:10.1007/s00294-007-0151-0
41. Schneider DS, Ayres JS. Two ways to survive infection: what resistance and tolerance can teach us about treating infectious diseases. Nat Rev Immunol (2008) 8:889–95. doi:10.1038/nri2432
42. Raberg L, Graham AL, Read AF. Decomposing health: tolerance and resistance to parasites in animals. Philos Trans R Soc Lond B Biol Sci (2009) 364:37–49. doi:10.1098/rstb.2008.0184
43. Medzhitov R, Schneider DS, Soares MP. Disease tolerance as a defense strategy. Science (2012) 335:936–41. doi:10.1126/science.1214935
44. De S, Olson R. Crystal structure of the Vibrio cholerae cytolysin heptamer reveals common features among disparate pore-forming toxins. Proc Natl Acad Sci U S A (2011) 108:7385–90. doi:10.1073/pnas.1017442108
45. Tournier C, Hess P, Yang DD, Xu J, Turner TK, Nimnual A, et al. Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science (2000) 288:870–4. doi:10.1126/science.288.5467.870
46. Boyce M, Bryant KF, Jousse C, Long K, Harding HP, Scheuner D, et al. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science (2005) 307:935–9. doi:10.1126/science.1101902
47. Jousse C, Oyadomari S, Novoa I, Lu P, Zhang Y, Harding HP, et al. Inhibition of a constitutive translation initiation factor 2alpha phosphatase, CReP, promotes survival of stressed cells. J Cell Biol (2003) 163:767–75. doi:10.1083/jcb.200308075
48. Ryazanov AG, Shestakova EA, Natapov PG. Phosphorylation of elongation factor 2 by EF-2 kinase affects rate of translation. Nature (1988) 334:170–3. doi:10.1038/334170a0
49. Vadia S, Seveau S. Fluxes of Ca2+ and K+ are required for the listeriolysin O-dependent internalization pathway of Listeria monocytogenes. Infect Immun (2014) 82:1084–91. doi:10.1128/IAI.01067-13
50. Dunbar TL, Yan Z, Balla KM, Smelkinson MG, Troemel ER. C. elegans detects pathogen-induced translational inhibition to activate immune signaling. Cell Host Microbe (2012) 11:375–86. doi:10.1016/j.chom.2012.02.008
51. Arguello RJ, Rodriguez Rodrigues C, Gatti E, Pierre P. Protein synthesis regulation, a pillar of strength for innate immunity? Curr Opin Immunol (2015) 32:28–35. doi:10.1016/j.coi.2014.12.001
52. Wilke GA, Bubeck Wardenburg J. Role of a disintegrin and metalloprotease 10 in Staphylococcus aureus alpha-hemolysin-mediated cellular injury. Proc Natl Acad Sci U S A (2010) 107:13473–8. doi:10.1073/pnas.1001815107
53. Talloczy Z, Jiang W, Virgin HWT, Leib DA, Scheuner D, Kaufman RJ, et al. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc Natl Acad Sci U S A (2002) 99:190–5. doi:10.1073/pnas.012485299
54. Maurer K, Reyes-Robles T, Alonzo F III, Durbin J, Torres VJ, Cadwell K. Autophagy mediates tolerance to Staphylococcus aureus alpha-toxin. Cell Host Microbe (2015) 17:429–40. doi:10.1016/j.chom.2015.03.001
55. Jursch R, Hildebrand A, Hobom G, Tranum-Jensen J, Ward R, Kehoe M, et al. Histidine residues near the N terminus of staphylococcal alpha-toxin as reporters of regions that are critical for oligomerization and pore formation. Infect Immun (1994) 62:2249–56.
56. Jiang HY, Wek SA, McGrath BC, Scheuner D, Kaufman RJ, Cavener DR, et al. Phosphorylation of the alpha subunit of eukaryotic initiation factor 2 is required for activation of NF-kappaB in response to diverse cellular stresses. Mol Cell Biol (2003) 23:5651–63. doi:10.1128/MCB.23.16.5651-5663.2003
57. Scheuner D, Song B, McEwen E, Liu C, Laybutt R, Gillespie P, et al. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol Cell (2001) 7:1165–76. doi:10.1016/S1097-2765(01)00265-9
58. Harding HP, Zhang Y, Scheuner D, Chen JJ, Kaufman RJ, Ron D. Ppp1r15 gene knockout reveals an essential role for translation initiation factor 2 alpha (eIF2alpha) dephosphorylation in mammalian development. Proc Natl Acad Sci U S A (2009) 106:1832–7. doi:10.1073/pnas.0809632106
59. Chu HP, Liao Y, Novak JS, Hu Z, Merkin JJ, Shymkiv Y, et al. Germline quality control: eEF2K stands guard to eliminate defective oocytes. Dev Cell (2014) 28:561–72. doi:10.1016/j.devcel.2014.01.027
60. Hartmann D, De Strooper B, Serneels L, Craessaerts K, Herreman A, Annaert W, et al. The disintegrin/metalloprotease ADAM 10 is essential for Notch signalling but not for alpha-secretase activity in fibroblasts. Hum Mol Genet (2002) 11:2615–24. doi:10.1093/hmg/11.21.2615
61. Boukamp P, Petrussevska RT, Breitkreutz D, Hornung J, Markham A, Fusenig NE. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J Cell Biol (1988) 106:761–71. doi:10.1083/jcb.106.3.761
62. Schmidt EK, Clavarino G, Ceppi M, Pierre P. SUnSET, a nonradioactive method to monitor protein synthesis. Nat Methods (2009) 6:275–7. doi:10.1038/nmeth.1314
63. Abrami L, Fivaz M, Glauser PE, Parton RG, Van Der Goot FG. A pore-forming toxin interacts with a GPI-anchored protein and causes vacuolation of the endoplasmic reticulum. J Cell Biol (1998) 140:525–40. doi:10.1083/jcb.140.3.525
Keywords: pore forming toxins, S. aureus α-toxin, cellular tolerance, EIF2AK4, MAPK
Citation: von Hoven G, Neukirch C, Meyenburg M, Füser S, Petrivna MB, Rivas AJ, Ryazanov A, Kaufman RJ, Aroian RV and Husmann M (2015) eIF2α confers cellular tolerance to S. aureus α-toxin. Front. Immunol. 6:383. doi: 10.3389/fimmu.2015.00383
Received: 28 May 2015; Accepted: 13 July 2015;
Published: 27 July 2015
Edited by:
Katharina F. Kubatzky, University Hospital Heidelberg, GermanyReviewed by:
Falko Hochgräfe, University Greifswald, GermanyKen Cadwell, NYU School of Medicine, USA
Copyright: © 2015 von Hoven, Neukirch, Meyenburg, Füser, Petrivna, Rivas, Ryazanov, Kaufman, Aroian and Husmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matthias Husmann, Mainz, Germany,aHVzbWFubkB1bmktbWFpbnouZGU=;
Raffi V. Aroian, Worcester, MA, USA,cmFmZmkuYXJvaWFuQHVtYXNzbWVkLmVkdQ==
†Gisela von Hoven and Claudia Neukirch have contributed equally to this work.