 Matthias Klein
Matthias Klein Tobias Bopp
Tobias Bopp- University Medical Center, Institute for Immunology, Johannes Gutenberg-University, Mainz, Germany
T regulatory (Treg) cells are one of the key players in the immune tolerance network, and a plethora of manuscripts have described their development and function in the course of the last two decades. Nevertheless, it is still a matter of debate as to which mechanisms and agents are employed by Treg cells, providing the basis of their suppressive potency. One of the important candidates is cyclic AMP (cAMP), which is long known as a potent suppressor at least of T cell activation and function. While this suppressive function by itself is widely accepted, the source and the mechanism of action of cAMP are less clear, and a multitude of seemingly contradictory data allow for, in principle, two different scenarios of cAMP-mediated suppression. In one scenario, Treg cells contain high amounts of cAMP and convey this small molecule via gap junction intercellular communication directly to the effector T cells (Teff) leading to their suppression. Alternatively, it was shown that Treg cells represent the origin of considerable amounts of adenosine, which trigger the adenylate cyclases in Teff cells via A2A and A2B receptors, thus strongly increasing intracellular cAMP. This review will present and discuss initial findings and recent developments concerning the function of cAMP for Treg cells and its impact on immune regulation.
Introduction
The immune system commands an impressive armament to repel invading microorganisms. The overwhelming part of such an antimicrobial immune response is coordinated and initiated by CD4+ T helper (Th) cells, which recognize foreign antigens through their T cell receptor after presentation by antigen-presenting cells. A huge Th cell receptor repertoire ensures that almost all pathogens can be recognized and subsequently be eliminated. However, this virtually exhaustless T cell receptor repertoire obviously bears the risk of containing autoaggressive T cells (1). This was demonstrated by the transfer of naive Th cells, which represent approximately 90% of all peripheral CD4+ Th cells into T cell-deficient mice that led to a multitude of autoimmune diseases indicating that a considerable part of this Th cell population is potentially autoreactive (2–4). These potentially autoreactive naive Th cells are usually kept in check when CD25+ peripheral Th cells are cotransferred together with CD25− T cells, thus indicating that this residual T cell compartment that was separated from naive T cells before transfer is capable to prevent autoimmune symptoms (5, 6). Today, we know that this suppressive Th cell population mainly consists of T regulatory (Treg) cells which were found to control T effector (Teff) cells in vitro in a contact-dependent manner. The Treg/Teff cell interaction was shown to suppress preferentially IL-2 production and proliferation of the Teff cells – a hallmark of clonal T cell expansion (7). Concerning the suppressive mechanism(s), the usage of cytokine-deficient and cytokine receptor-deficient mice could exclude that IL-10 and TGF-β – at least in vitro – mediated the suppressive properties of Treg cells (7, 8). Subsequently, the characterization of the transcription factor forkhead box protein 3 (FOXP3), as a lineage-specific marker for Treg cells, and the generation of FOXP3 reporter mice strongly boosted Treg cell research. Continuative analyses revealed that FOXP3 is crucial for Treg cell development and function (9–11). These findings provided the opportunity to screen the FOXP3-regulated Treg cell transcriptome which revealed that the expression of a cyclic AMP (cAMP)-degrading phosphodiesterase (PDE3b) is strongly repressed in Treg cells, whereas the expression of ectonucleotidases (CD39 and CD73) as well as expression of adenylyl cyclase 9 (AC9), an enzyme promoting de novo generation of intracellular cAMP, was upregulated (12, 13). Therefore, reduced expression of phosphodiesterase (PDE3b) implied a decreased degradation of intracellular cAMP accompanied by a strong production of cAMP due to strong expression of AC9, while elevated expression of CD39/CD73 should lead to an increased generation of extracellular adenosine in the proximity of Treg cells. Hence, FOXP3-dependent transcriptional profiling suggested that the suppressive properties of Treg cells is based at least partially on comparatively high amounts of intracellular cAMP concomitantly with an enhanced ability to generate extracellular adenosine from adenosine triphosphate (ATP) [reviewed in Ref. (14, 15)].
Intracellular cAMP Enables Treg Cells to Maintain the Balance of the Immune Tolerance Network During Immune Homeostasis
Intracellular cAMP has long been recognized as a potent inhibitor of T cell activation. Especially, agents that elevated cAMP in T cells like cholera toxin, prostaglandin E2, and forskolin were found to strongly impair IL-2 production and T cell proliferation (16–19). Comparative analyses of intracellular cAMP revealed that Treg cells contained high intracellular amounts of cAMP, while it was hardly detectable in Teff cells (20). In addition, co-activation of cocultured Treg and Teff cells resulted in a considerable intracellular increase of cAMP in Teff cells, suggesting a cell contact-dependent transfer of cAMP. One possibility for cell contact-dependent transfer was gap junction intercellular communication (GJIC). GJIC was demonstrated by employing the fluorescent dye calcein which can only be transferred between T cells by gap junctions (21, 22). The functional consequence of such a GJIC-mediated transfer of cAMP between Treg and Teff cells was a strong reduction of IL-2 expression, and as a consequence, inhibition of proliferation, which was both reversed in the presence of the GJIC inhibitor GAP27. In addition, it was shown that the coculture of murine Treg cells and dendritic cells (DC) led to a strong elevation of cAMP in DC concomitantly with an immediate downregulation of CD80/CD86 costimulators (23). This Treg cell-mediated suppression of DC activation via transfer of cAMP was suggested to be decisively involved in the control of a Graft-versus-host disease (GvHD) by Treg cells. Accordingly, the potency of Treg cells to ameliorate a GvHD was found to be strongly increased in the presence of PDE-inhibitors like rolipram (24). In agreement with these findings, it was shown that neonatal human Treg cells suppress DC activation by CTLA-4 and cAMP (25).
The importance of GJIC for Treg-mediated suppression was recently emphasized by the finding that diabetic NOD mice showed an impaired expression of connexin 43 (Cx43), an important component of gap junctions, leading in consequence to a strongly reduced suppressive potency of their Treg cells (26). Overexpression of Cx43 in such Treg cells increased their suppressive properties roughly to the level of non-diabetic young NOD mice. The same outcome could be observed when Cx43-mediated GJIC was strengthened via the treatment with alpha-connexin carboxyl-terminal peptide 1 (αCT-1). αCT-1 is a unique synthetic peptide that mimics a cytoplasmic regulatory domain of Cx43 and that specifically disrupts the interaction between Cx43 und its binding partner zonula occludens (ZO)-1, ultimately leading to an enhanced gap junction aggregation (27, 28). However, treatment with αCT-1 failed when Cx43-deficient Treg cells were used, indicating the specificity of action of this agent (26).
The comparatively high content of cAMP in Treg cells was described to result from a strong FOXP3-mediated reduction of PDE3b expression concomitantly with a considerable suppressive activity of this T cell type (29). Further analyses of the underlying molecular mechanisms revealed that FOXP3 not only binds and regulates the Pde3b locus but also additionally downregulates expression of the miRNA miR-142-3p, a potent inhibitor of AC9, which catalyzes the conversion of ATP to 3′,5′-cAMP. As a consequence of the impaired targeting of AC9 mRNA due to reduced miR-142-3p expression in Treg cells, these cells contain high levels of AC9, driving elevated intracellular cAMP level. Accordingly, transfection of Treg cells with miR-142-3p not only reduced AC9 expression and intracellular cAMP levels but also considerably impaired the suppressive properties of these Treg cells. The same effect was observed when Treg cells were transfected with AC9 siRNA, again preventing de novo generation of intracellular cAMP by AC9. Thus, FOXP3 strongly promotes AC9-dependent intracellular cAMP production by suppressing expression of miR-142-3p and PDE3b as well. Concerning human Treg cells, it was shown that polyclonal activation as well as activation via CD4, known to induce tolerance, strongly increased the concentration of intracellular cAMP. The irreversible blockade of adenylate cyclases (AC) by MDL12 inhibited this upregulation of cAMP concomitantly with a strong impairment of their suppressive potency – in vitro as well as in vivo – and led to an increased proliferation of these AC-blocked Treg cells (30). Nevertheless, such Treg cells regained their suppressive properties after several days of expansion probably as a consequence of the dilution of the AC inhibitor MDL12. Similar to MDL12, the ectopic expression of cAMP-degrading phosphodiesterase, PDE4c, also prevented the increase of intracellular cAMP in human Treg cells and strongly impaired their suppressive capacity. By contrast, it could be shown that the inhibition of PDE4 by rolipram strongly enhanced the suppressive potency of Treg cells in vitro and in vivo. Transfer of rolipram-treated Treg cells strongly inhibited asthma symptoms in a prophylactic as well as a therapeutic preclinical animal model of experimental asthma (31). As a consequence of Treg–Th2 interaction in vivo, a strong increase of intracellular cAMP could be observed in lung-resident Th2 cells. Additional studies revealed that asthma-promoting Th1 and Th2 cells exhibited a similar increase of intracellular cAMP after contact with Treg cells in vitro and in vivo, but that Th1 cells were far more sensitive to Treg-mediated suppression (32). This was in agreement with the finding that comparatively high cAMP contents are needed to strongly inhibit Th2-derived IL-4 production, while comparatively low cAMP concentrations are sufficient in suppressing IL-2 production and subsequent proliferation of Th1 cells (18).
Extracellular Adenosine is Required to Limit a Potentially Self-Destructive Inflammatory Immune Response
Inflammatory immune reactions are accompanied by the release of high amounts of ATP in the extracellular space where it is converted into AMP by CD39 and dephosphorylated to adenosine by CD73 (33). Adenosine has strong anti-inflammatory influence on immune cells preferentially by triggering A2A and A2B receptors leading to the generation of intracellular cAMP. Interestingly, the anti-inflammatory action of adenosine is exploited by tumors where adenosine was shown to accumulate, thereby preventing immunological tumor regression (34, 35). The tumor-protective role of adenosine was underlined using mice deficient for the genes Adora2a, encoding for A2A or Adora2b, encoding for A2B receptors which show enhanced elimination of tumors compared to A2A and A2B receptor sufficient wild-type mice (35–37). Among mouse T cells, CD39 and CD73 were described to be preferentially co-expressed in Treg cells (38). This was in agreement with the finding that the expression of Entpd1 (CD39) and Nt5e (CD73) was shown to be upregulated by FOXP3 (12). In line with this, inhibitors of CD39 and CD73 decreased the suppressive capacity of Tregs (39–41). Conclusively, Entpd1-deficient (CD39-deficient) Treg cells exhibited impaired suppressive properties in vitro in a coculture assay as well as in vivo using an allogeneic skin graft model upon cotransfer of Treg/Teff cells indicating the importance of this ectonucleotidase for the suppressive function of Treg cells. Similarly, in a model of acute lung injury (ALI), adoptive transfer of Nt5e-deficient (CD73-deficient) Treg cells failed to resolve ALI adequately, whereas transfer of wild-type Treg cells led to normal resolution (42). Furthermore, it was shown in a model of acute kidney injury that DC treated with an A2A receptor agonist could protect the kidney by suppressing DC-dependent NKT cell activation, suggesting a strong immunosuppressive influence of adenosine on the accessory functions of such DC (43).
Interestingly, Teff cells that represent a major target of Treg cell suppressive activity were found to express the T cell-typical adenosine receptor A2A not before day 4 after activation, thus excluding immediate early suppressive influence by this mechanism (38). Accordingly, the authors stated that the adenosine-driven suppression was most probably functional during the late phases of Teff cell activation. Nonetheless, the importance of A2A receptors on Teff cells for Treg cell-mediated suppression in the late phases of an adaptive immune response in vivo was demonstrated in an adoptive T cell transfer model of chronic colitis. Herein, wild-type colitogenic CD4+ Teff cells were considerably suppressed after cotransfer of Treg cells in SCID mice, whereas their Adora2a (A2A receptor)-deficient counterparts could not be inhibited by the cotransferred Treg cells so that the transfer of such Teff cells led to the development of colitis (44). In addition, and in line with the aforementioned results, A2A receptor-deficient Treg cells could not prevent colitis caused by pathogenic wild-type Teff cells, suggesting that adenosine is also of central importance for the suppressive function of Treg cells in this model of inflammatory bowel disease. Hence, it can be reasonably assumed that in addition to reduced PDE3b expression and enhanced AC9 expression, high intracellular levels of cAMP in Treg cells also depend on adenosine-driven elevation of cAMP. This assumption was corroborated by a recent publication of Sitkovsky’s group demonstrating that adenosine stimulated the expansion of Treg cells and concomitantly raised their suppressive capacity (45). Thus, adenosine can directly inhibit Teff cell activation and simultaneously improve the suppressive function of the Treg cell compartment most possibly by affecting cAMP production.
In conclusion, high intracellular cAMP levels in Treg cells provide a major contribution to a balanced immune tolerance network (ITN) in the course of immune homeostasis when Treg cells are preferentially stimulated via the T cell receptor complex in combination with rather low costimulatory influences (Figure 1). However, in the course of a local inflammation which is additionally boosted by a myriad of potent microorganism-derived costimulators (bacterial, viral, fungal), an immune response can easily get out of control leading to collateral damage and with that to detrimental consequences. To prevent such a fatal scenario, the suppressive capacity of Treg cells can obviously be strongly improved by adenosine which originates from the CD39/CD73-mediated degradation of extracellular ATP that is characteristically released in huge amounts during such collateral damage of inflamed tissue. This ATP-derived adenosine profoundly increased the overall cAMP content of the involved cells as well as the suppressive arsenal of the local Treg cell compartment which is automatically reduced when inflammation is resolved enabling the ITN to return to immune homeostasis. Thus, the ITN follows an escalating suppressive strategy that allows for the initiation of an effective immune response by breaking a relatively weak suppression that is based on intracellular cAMP which subsequently exploits the locally originating ATP leading to the generation of immune suppressive adenosine to further strengthen intracellular cAMP levels, and by that, curbing a potentially life threatening immune reaction.
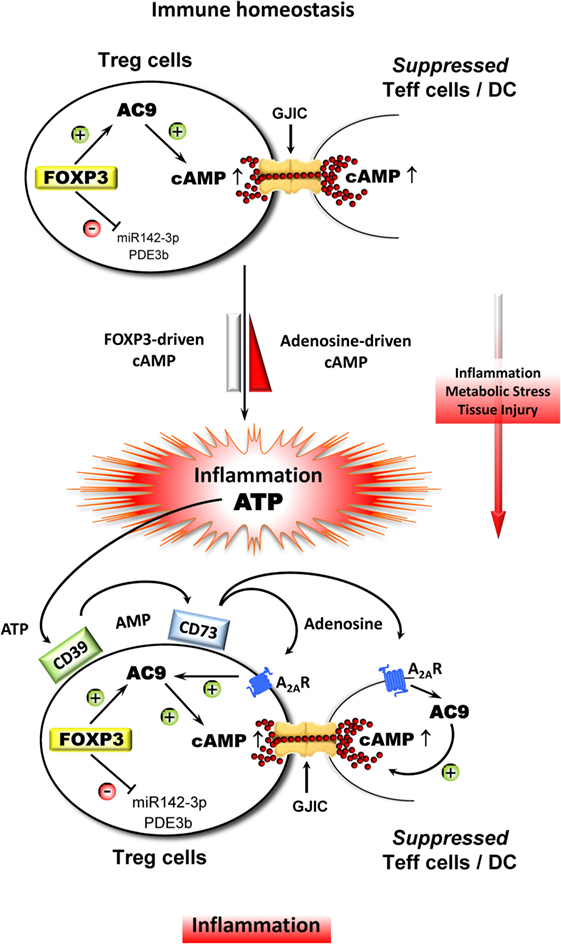
Figure 1. Adenosine strongly improves the suppressive influence of Treg cell-derived cAMP in the course of inflammation. During immune homeostasis, Treg cells stabilize the ITN with the aid of endogenous cAMP that is driven by FOXP3, which indirectly upregulates adenylate cylase 9 (AC9) through the inhibition of miR142-3p and which concomitantly downregulates cAMP-degrading phosphodiesterase 3b (PDE3b). As a result, Treg cells contain comparatively high amounts of cAMP leading to the suppression of Teff cells and DC via gap junctional intercellular communication (GJIC). Inflammation in combination with metabolic stress and tissue injury results in a massive release of ATP, which represents a powerful danger signal that serves as an additional local inflammatory booster that bears the risk of collateral damage by uncontrollable immune reactions. Therefore, metabolization of ATP by ectonucleotidases CD39 and CD73 that leads to increased local amounts of adenosine prevents such a fatal development especially through triggering the adenosine receptor A2A (A2AR) on Treg and Teff cells and DC as well. The A2AR-mediated elevation of intracellular cAMP inhibits the activation of Teff cells and impairs the accessory function of DC and simultaneously strongly improves the suppressive activity of Treg cells.
Author Contributions
All authors listed have made substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Karen Lingnau for critical reading of our manuscript.
Funding
This work was supported by the Deutsche Forschungsgemeinschaft (DFG BO 3306/1-1 and SCHM1014/7-1).
References
1. Mason D. A very high level of crossreactivity is an essential feature of the T-cell receptor. Immunol Today (1998) 19:395–404. doi:10.1016/S0167-5699(98)01299-7
2. Sakaguchi S, Fukuma K, Kuribayashi K, Masuda T. Organ-specific autoimmune diseases induced in mice by elimination of T cell subset. I. Evidence for the active participation of T cells in natural self-tolerance; deficit of a T cell subset as a possible cause of autoimmune disease. J Exp Med (1985) 161:72–87. doi:10.1084/jem.161.1.72
3. Sugihara S, Izumi Y, Yoshioka T, Yagi H, Tsujimura T, Tarutani O, et al. Autoimmune thyroiditis induced in mice depleted of particular T cell subsets. I. Requirement of Lyt-1 dull L3T4 bright normal T cells for the induction of thyroiditis. J Immunol (1988) 141:105–13.
4. Powrie F, Mason D. OX-22high CD4+ T cells induce wasting disease with multiple organ pathology: prevention by the OX-22low subset. J Exp Med (1990) 172:1701–8. doi:10.1084/jem.172.6.1701
5. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol (1995) 155:1151–64.
6. Asano M, Toda M, Sakaguchi N, Sakaguchi S. Autoimmune disease as a consequence of developmental abnormality of a T cell subpopulation. J Exp Med (1996) 184:387–96. doi:10.1084/jem.184.2.387
7. Thornton AM, Shevach EM. CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J Exp Med (1998) 188:287–96. doi:10.1084/jem.188.2.287
8. Piccirillo CA, Letterio JJ, Thornton AM, McHugh RS, Mamura M, Mizuhara H, et al. CD4(+)CD25(+) regulatory T cells can mediate suppressor function in the absence of transforming growth factor beta1 production and responsiveness. J Exp Med (2002) 196:237–46. doi:10.1084/jem.20020590
9. Khattri R, Cox T, Yasayko SA, Ramsdell F. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat Immunol (2003) 4:337–42. doi:10.1038/ni909
10. Ramsdell F. Foxp3 and natural regulatory T cells: key to a cell lineage? Immunity (2003) 19:165–8. doi:10.1016/S1074-7613(03)00207-3
11. Fontenot JD, Rasmussen JP, Williams LM, Dooley JL, Farr AG, Rudensky AY. Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity (2005) 22:329–41. doi:10.1016/j.immuni.2005.01.016
12. Gavin MA, Rasmussen JP, Fontenot JD, Vasta V, Manganiello VC, Beavo JA, et al. Foxp3-dependent programme of regulatory T-cell differentiation. Nature (2007) 445:771–5. doi:10.1038/nature05543
13. Zheng Y, Josefowicz SZ, Kas A, Chu T-T, Gavin MA, Rudensky AY. Genome-wide analysis of Foxp3 target genes in developing and mature regulatory T cells. Nature (2007) 445:936–40. doi:10.1038/nature05563
14. Wehbi VL, Taskén K. Molecular mechanisms for cAMP-mediated immunoregulation in T cells – role of anchored protein kinase A signaling units. Front Immunol (2016) 7:222. doi:10.3389/fimmu.2016.00222
15. Rueda CM, Jackson CM, Chougnet CA. Regulatory T-cell-mediated suppression of conventional T-cells and dendritic cells by different cAMP intracellular pathways. Front Immunol (2016) 7:216. doi:10.3389/fimmu.2016.00216
16. Johnson KW, Davis BH, Smith KA. cAMP antagonizes interleukin 2-promoted T-cell cycle progression at a discrete point in early G1. Proc Natl Acad Sci U S A (1988) 85:6072–6. doi:10.1073/pnas.85.16.6072
17. Mary D, Aussel C, Ferrua B, Fehlmann M. Regulation of interleukin 2 synthesis by cAMP in human T cells. J Immunol (1987) 139:1179–84.
18. Muñoz E, Zubiaga AM, Merrow M, Sauter NP, Huber BT. Cholera toxin discriminates between T helper 1 and 2 cells in T cell receptor-mediated activation: role of cAMP in T cell proliferation. J Exp Med (1990) 172:95–103. doi:10.1084/jem.172.1.95
19. Raker VK, Becker C, Steinbrink K. The cAMP pathway as therapeutic target in autoimmune and inflammatory diseases. Front Immunol (2016) 7:123. doi:10.3389/fimmu.2016.00123
20. Bopp T, Becker C, Klein M, Klein-Hessling S, Palmetshofer A, Serfling E, et al. Cyclic adenosine monophosphate is a key component of regulatory T cell mediated suppression. J Exp Med (2007) 204:1303–10. doi:10.1084/jem.20062129
21. Fonseca PC, Nihei OK, Savino W, Spray DC, Alves LA. Flow cytometry analysis of gap junction-mediated cell-cell communication: advantages and pitfalls. Cytometry A (2006) 69:487–93. doi:10.1002/cyto.a.20255
22. Bedner P, Niessen H, Odermatt B, Kretz M, Willecke K, Harz H. Selective permeability of different connexin channels to the second messenger cyclic AMP. J Biol Chem (2006) 281:6673–81. doi:10.1074/jbc.M511235200
23. Fassbender M, Gerlitzki B, Ullrich N, Lupp C, Klein M, Radsak MP, et al. Cyclic adenosine monophosphate and IL-10 coordinately contribute to nTreg cell-mediated suppression of dendritic cell activation. Cell Immunol (2010) 265:91–6. doi:10.1016/j.cellimm.2010.07.007
24. Weber M, Lupp C, Stein P, Kreft A, Bopp T, Wehler TC, et al. Mechanisms of cyclic nucleotide phosphodiesterases in modulating T cell responses in murine graft-versus-host disease. PLoS One (2013) 8:e58110. doi:10.1371/journal.pone.0058110
25. Rueda CM, Moreno-Fernandez ME, Jackson CM, Kallapur SG, Jobe AH, Chougnet CA. Neonatal regulatory T cells have reduced capacity to suppress dendritic cell function. Eur J Immunol (2015) 45:2582–92. doi:10.1002/eji.201445371
26. Kuczma M, Wang C-Y, Ignatowicz L, Gourdie R, Kraj P. Altered connexin 43 expression underlies age-dependent decrease of regulatory T cell suppressor function in nonobese diabetic mice. J Immunol (2015) 194:5261–71. doi:10.4049/jimmunol.1400887
27. Hunter AW, Barker RJ, Zhu C, Gourdie RG. Zonula occludens-1 alters connexin43 gap junction size and organization by influencing channel accretion. Mol Biol Cell (2005) 16:5686–98. doi:10.1091/mbc.E05-08-0737
28. Rhett JM, Jourdan J, Gourdie RG. Connexin 43 connexon to gap junction transition is regulated by zonula occludens-1. Mol Biol Cell (2011) 22:1516–28. doi:10.1091/mbc.E10-06-0548
29. Huang B, Zhao J, Lei Z, Shen S, Li D, Shen G-X, et al. miR-142-3p restricts cAMP production in CD4+CD25- T cells and CD4+CD25+ TREG cells by targeting AC9 mRNA. EMBO Rep (2009) 10:180–5. doi:10.1038/embor.2008.224
30. Klein M, Vaeth M, Scheel T, Grabbe S, Baumgrass R, Berberich-Siebelt F, et al. Repression of cyclic adenosine monophosphate upregulation disarms and expands human regulatory T cells. J Immunol (2012) 188:1091–7. doi:10.4049/jimmunol.1102045
31. Bopp T, Dehzad N, Reuter S, Klein M, Ullrich N, Stassen M, et al. Inhibition of cAMP degradation improves regulatory T cell-mediated suppression. J Immunol (2009) 182:4017–24. doi:10.4049/jimmunol.0803310
32. Dehzad N, Bopp T, Reuter S, Klein M, Martin H, Ulges A, et al. Regulatory T cells more effectively suppress Th1-induced airway inflammation compared with Th2. J Immunol (2011) 186:2238–44. doi:10.4049/jimmunol.1002027
33. Antonioli L, Pacher P, Vizi ES, Haskó G. CD39 and CD73 in immunity and inflammation. Trends Mol Med (2013) 19:355–67. doi:10.1016/j.molmed.2013.03.005
34. Blay J, White TD, Hoskin DW. The extracellular fluid of solid carcinomas contains immunosuppressive concentrations of adenosine. Cancer Res (1997) 57:2602–5.
35. Ohta A, Gorelik E, Prasad SJ, Ronchese F, Lukashev D, Wong MKK, et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc Natl Acad Sci U S A (2006) 103:13132–7. doi:10.1073/pnas.0605251103
36. Ryzhov S, Novitskiy SV, Zaynagetdinov R, Goldstein AE, Carbone DP, Biaggioni I, et al. Host A(2B) adenosine receptors promote carcinoma growth. Neoplasia (2008) 10:987–95. doi:10.1593/neo.08478
37. Cekic C, Sag D, Li Y, Theodorescu D, Strieter RM, Linden J. Adenosine A2B receptor blockade slows growth of bladder and breast tumors. J Immunol (2012) 188:198–205. doi:10.4049/jimmunol.1101845
38. Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med (2007) 204:1257–65. doi:10.1084/jem.20062512
39. Kobie JJ, Shah PR, Yang L, Rebhahn JA, Fowell DJ, Mosmann TR. T regulatory and primed uncommitted CD4 T cells express CD73, which suppresses effector CD4 T cells by converting 5′-adenosine monophosphate to adenosine. J Immunol (2006) 177:6780–6. doi:10.4049/jimmunol.177.10.6780
40. Mandapathil M, Hilldorfer B, Szczepanski MJ, Czystowska M, Szajnik M, Ren J, et al. Generation and accumulation of immunosuppressive adenosine by human CD4+CD25highFOXP3+ regulatory T cells. J Biol Chem (2010) 285:7176–86. doi:10.1074/jbc.M109.047423
41. Kinsey GR, Huang L, Jaworska K, Khutsishvili K, Becker DA, Ye H, et al. Autocrine adenosine signaling promotes regulatory T cell-mediated renal protection. J Am Soc Nephrol (2012) 23:1528–37. doi:10.1681/ASN.2012010070
42. Ehrentraut H, Clambey ET, McNamee EN, Brodsky KS, Ehrentraut SF, Poth JM, et al. CD73+ regulatory T cells contribute to adenosine-mediated resolution of acute lung injury. FASEB J (2013) 27:2207–19. doi:10.1096/fj.12-225201
43. Li L, Huang L, Ye H, Song SP, Bajwa A, Lee SJ, et al. Dendritic cells tolerized with adenosine A2AR agonist attenuate acute kidney injury. J Clin Invest (2012) 122:3931–42. doi:10.1172/JCI63170
44. Naganuma M, Wiznerowicz EB, Lappas CM, Linden J, Worthington MT, Ernst PB. Cutting edge: critical role for A2A adenosine receptors in the T cell-mediated regulation of colitis. J Immunol (2006) 177:2765–9. doi:10.4049/jimmunol.177.5.2765
Keywords: cyclic AMP, regulatory T cells, FOXP3, adenosine, immune regulation, immune tolerance network, suppression
Citation: Klein M and Bopp T (2016) Cyclic AMP Represents a Crucial Component of Treg Cell-Mediated Immune Regulation. Front. Immunol. 7:315. doi: 10.3389/fimmu.2016.00315
Received: 15 June 2016; Accepted: 02 August 2016;
Published: 29 August 2016
Edited by:
Josef Bodor, BIOCEV, Czech RepublicReviewed by:
Viktor Umansky, German Cancer Research Center (HZ), GermanyJacques A. Nunes, Centre de Recherche en Cancerologie de Marseille, France
Copyright: © 2016 Klein and Bopp. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tobias Bopp, Ym9wcHRAdW5pLW1haW56LmRl