 Vera Gallo1
Vera Gallo1 Laura Dotta2
Laura Dotta2 Giuliana Giardino1Emilia Cirillo1
Giuliana Giardino1Emilia Cirillo1 Vassilios Lougaris2Roberta D’Assante1Alberto Prandini2
Vassilios Lougaris2Roberta D’Assante1Alberto Prandini2 Rita Consolini3Emily G. Farrow4Isabelle Thiffault4
Rita Consolini3Emily G. Farrow4Isabelle Thiffault4 Carol J. Saunders4Antonio Leonardi5Alessandro Plebani2Raffaele Badolato2†
Carol J. Saunders4Antonio Leonardi5Alessandro Plebani2Raffaele Badolato2† Claudio Pignata1*†
Claudio Pignata1*†
- 1Department of Translational Medical Sciences, Federico II University, Naples, Italy
- 2Department of Clinical and Experimental Medicine, “Angelo Nocivelli” Institute for Molecular Medicine, University of Brescia, Brescia, Italy
- 3Department of Clinical and Experimental Medicine, University of Pisa, Pisa, Italy
- 4Center for Pediatric Genomic Medicine, Children’s Mercy Hospital, Kansas City, MO, USA
- 5Department of Molecular Medicine and Medical Biotechnology, Federico II University, Naples, Italy
Background: Recently, a growing number of novel genetic defects underlying primary immunodeficiencies (PIDs) have been identified, increasing the number of PID up to more than 250 well-defined forms. Next-generation sequencing (NGS) technologies and proper filtering strategies greatly contributed to this rapid evolution, providing the possibility to rapidly and simultaneously analyze large numbers of genes or the whole exome.
Objective: To evaluate the role of targeted NGS and whole exome sequencing (WES) in the diagnosis of a case series, characterized by complex or atypical clinical features suggesting a PID, difficult to diagnose using the current diagnostic procedures.
Methods: We retrospectively analyzed genetic variants identified through targeted NGS or WES in 45 patients with complex PID of unknown etiology.
Results: Forty-seven variants were identified using targeted NGS, while 5 were identified using WES. Newly identified genetic variants were classified into four groups: (I) variations associated with a well-defined PID, (II) variations associated with atypical features of a well-defined PID, (III) functionally relevant variations potentially involved in the immunological features, and (IV) non-diagnostic genotype, in whom the link with phenotype is missing. We reached a conclusive genetic diagnosis in 7/45 patients (~16%). Among them, four patients presented with a typical well-defined PID. In the remaining three cases, mutations were associated with unexpected clinical features, expanding the phenotypic spectrum of typical PIDs. In addition, we identified 31 variants in 10 patients with complex phenotype, individually not causative per se of the disorder.
Conclusion: NGS technologies represent a cost-effective and rapid first-line genetic approach for the evaluation of complex PIDs. WES, despite a moderate higher cost compared to targeted, is emerging as a valuable tool to reach in a timely manner, a PID diagnosis with a considerable potential to draw genotype–phenotype correlation. Nevertheless, a large fraction of patients still remains without a conclusive diagnosis. In these patients, the sum of non-diagnostic variants might be proven informative in future studies with larger cohorts of patients.
Introduction
Primary immunodeficiencies (PIDs) represent a heterogeneous group of monogenic disorders including more than 250 genetically defined diseases, mostly identified in recent years (1–3). A significant contribution to this rapid evolution is due to the integration of functional studies with the use of next-generation sequencing (NGS) technologies. The specific objective of this strategy is the identification of putative pathogenic alterations in known or novel genes implicated in well-defined biological pathways (4–7). However, despite the breadth of current knowledge, the diagnostic approach used for the identification of genetic causes underlying PID appears inefficient and time-consuming as the majority of PID cases remain without a diagnosis even after extensive clinical and genetic investigations (8). Moreover, even in a single PID gene mutation, a genotype–phenotype correlation is often missing, because various clinical phenotypes may be related to the same genetic defect and vice versa (9, 10). Nevertheless, early diagnosis in severe forms of PIDs is crucial to establish a proper treatment and improve the overall outcome (11, 12). In the traditional approach to PIDs, the molecular diagnosis has long been based on the sequencing of multiple genes by the Sanger method, a time-consuming strategy, which often results in delayed diagnosis (13).
Next-generation sequencing technology has become a valuable first-line diagnostic tool for the timely diagnosis of genetic disorders, in particular for complex clinical presentation (14). NGS allows rapid, cost-efficient, accurate, and high-throughput sequencing of millions of DNA fragments in a reasonably short time. Whole exome sequencing (WES) is limited to the coding regions and splice junctions of the genome, which despite accounting for only ~2% of the genome, contain about 85% of genetic alterations known as responsible for human diseases (15).
In PIDs, a targeted sequencing approach, restricted only to specific genes or to specific regions of interest is a reasonable possibility to identify putative pathogenic variants that explain a specific disorder, their related altered biological pathway, and the array of genes that are transcribed. This can be the first-line alternative as it involves a smaller dataset than WES or whole genome sequencing (WGS) and is easier for the management of the datasets (16–18). However, as the spectrum of distinct clinical entities and the presenting phenotypes are expanding because of the discovery of novel genes and of the identification of wider clinical phenotypes, the differential diagnosis and subsequent diagnostic targets must improve in parallel. This perspective makes the whole exome or genome sequencing strategy attractive options for the diagnosis of patients with PID (19).
Here, we report on a case series of 45 complex or atypical clinical phenotypes that were suggestive of PID, but difficult to diagnose in a timely manner using the current diagnostic procedures. Our aim was to use current NGS technology to help diagnose PID in this cohort of patients. We used T-NGS and WES in 27 and 18 patients, respectively.
Materials and Methods
Patients
We have studied 45 patients with a clinical history highly suggestive of a primary immunological defect, that were heterogeneous for ethnic origin, age, and sex. The patients were selected on the basis of clinical features and abnormal immune parameters (20). The clinical criteria included one or more of the following features: opportunistic infections, granuloma, chronic mucocutaneous candidiasis (CMC), intractable diarrhea, bronchiectasis, and severe autoimmunity. These symptoms were associated, in some patients, with non-immunological features. In six patients, a positive family history for a similar phenotype was observed. Clinical criteria were considered if associated with one or more of the following quantitative and/or qualitative immunological abnormalities: abnormal lymphocyte subsets (absolute count <2 SD of normal values according to ESID criteria); proliferative response to mitogens <10% of the levels measured in the control subject; absent/poor specific antibody response; hypogammaglobulinemia; elevated IgE levels (>2000 kU/l); severe impairment of cytolytic activity; and alteration of class switch recombination (CSR) with or without hyper-IgM. PID patients, selected on the basis of the above specified criteria, were subsequently grouped on the basis of NGS results, functional alterations, and consistency between genotype, phenotype, and immune assays.
The study was approved by the Institutional Ethical Committee “Carlo Romano” of Federico II University and by Ethical Committee of Spedali civili (Brescia) and conducted after informed consent was obtained.
DNA Extraction and Sequence Capture Array Design
Genomic DNA was isolated from peripheral blood lymphocytes with QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany). Quantity and quality were determined through the Epoch Microplate Spectrophotometer (BioTech Instruments, Winooski, VT, USA). A panel of 571 genes, including 68 genes known or predicted to be related to PIDs and/or immune regulation, was sequenced (Table S1 in Supplementary Material). Basically, broad searches in literature, PubMed queries and expert suggestions defined the gene panel. We used BioMart (Ontario Institute for Cancer Research/European Bioinformatics Institute) to retrieve the coordinates of all exons for the specified genes from Ensembl. Coordinates were based on the current human reference genome (hGRC37, hg19).
Next-Generation Sequencing and Bioinformatics Analysis
Samples were sequenced using either a targeted panel of 571 (TaGSCAN v2.0) or WES for enrichment. Briefly, TaGSCAN samples were prepared for sequencing using Illumina’s TruSight Inherited Disease panel according to manufacturer’s protocols (Illumina, San Diego, CA, USA). Samples were sequenced to at least 2.5 GB on an Illumina MiSeq with TruSeq MiSeq V3 reagents, yielding paired 250 nucleotide reads. Samples were prepared for exome sequencing using the TruSeq HT library preparation kit (Illumina; San Diego, CA, USA) followed by exome enrichment using the xGen Exome Research Panel V1.0 (Integrated DNA Technologies; Coralville, IA, USA) according to manufacturers’ protocols. Paired-end 2 bp × 125 bp sequencing was completed on an Illumina HiSeq 2500 instrument in high output mode using V4 Chemistry. Samples were sequenced to at least 7 GB of data resulting in a minimum target coverage of 50×. For all samples, sequence was assembled, aligned to reference gene sequences based on human genome build GRCh37/UCSC hg19, and analyzed using custom-developed software, RUNES and VIKING (21).
Alignment, variant calling, and analysis were performed, as described previously (21). In detail, variants were filtered by frequency and variant category (22). Variant analysis was confined to coding and splice variants with a minor allele frequency (MAF) of 1% or less in the CMH internal database, in the Exome Variant Server (EVS)1 and Exome Aggregation Consortium (ExAC).2 Other tools used to restrict NGS analysis to a set of gene-associated regions relevant to clinical presentations, was symptom- and sign-assisted genome analysis (SSAGA), which mapped the clinical features in ill neonates and children to disease genes, and Phenomizer databases (21, 23). Sorting Intolerant From Tolerant (SIFT) and Polymorphism Phenotyping v2 (PolyPhen2) have been used to provide information on the impact of the variants.
Functional Assays and Sanger Validation
Functional assays were performed to prove the impact of variants identified by NGS. Functional studies included lymphocyte proliferation assays performed through [3H]thymidine incorporation assay, natural killer cell-mediated lysis of target cells and lymphocyte apoptosis induced by FAS death receptor cross-linking, evaluated through flow cytometry, cytokines production after toll-like receptors (TLR) stimulation, evaluated through real-time PCR. Furthermore, protein expression was evaluated through western blot analysis and standard procedures. After identification of genetic variants that were predicted to be damaging, along with consistent genotype–phenotype correlation, mutations were validated by Sanger sequencing using standard protocols. In 9 patients, we detected 11 variants that were confirmed by Sanger sequencing.
Additional Phenotype-Based Variant Filtering Criteria
In order to identify potential causal mutations, annotated variants were further prioritized based on the following criteria:
– homozygous or heterozygous variants already reported that were related to any immunological clinical phenotype were selected to be further investigated by functional studies and validated by Sanger method;
– variants in genes implicated in a molecular pathway related to the phenotype were considered if associated with any functional alteration, which was even partially consistent, with the clinical phenotype;
– all the genetic variants of genes that were probably unrelated to the molecular pathway suspected to be involved in the pathogenesis of the disease were excluded from the functional studies, but reported in an ad hoc repository.
Results
NGS of 45 Patients
Genomic DNA from 27 patients was enriched for all exons from 571 genes, including genes involved in immunological pathways, while DNA from 18 patients was enriched for all nucleotides of the exome. DNA from six patients, analyzed with TaGSCAN, was subsequently sequenced for the entire exome and the bioinformatic analysis of data results is currently ongoing. For TaGSCAN, 98.9% of base pairs targeted had at least 10× coverage. Mean depth of coverage was 580×. WES covered 97.05% of exonic regions at 10× or greater on average. The overall analytic sensitivity for single nucleotide variants (SNVs) was 98.7%.
The results of NGS exon sequencing are shown in Tables 1–4. After bioinformatics analysis and filtering, the potential causative nucleotide changes needed to be further evaluated in each individual patient (Table 5). All of these variants were SNVs or small indel variants, with no other alteration such as large deletions or insertions identified.
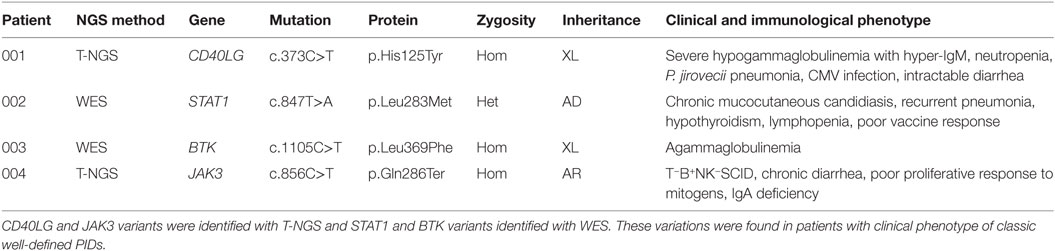
Table 1. Genetic variants associated with typical PID.
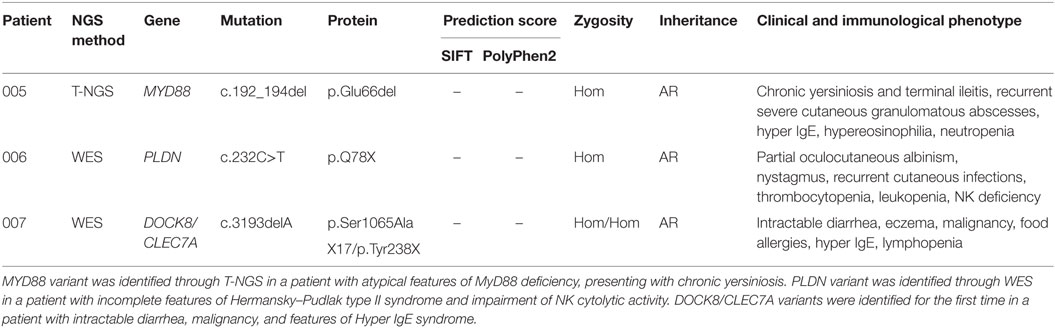
Table 2. Genetic variants associated with novel features of PID.
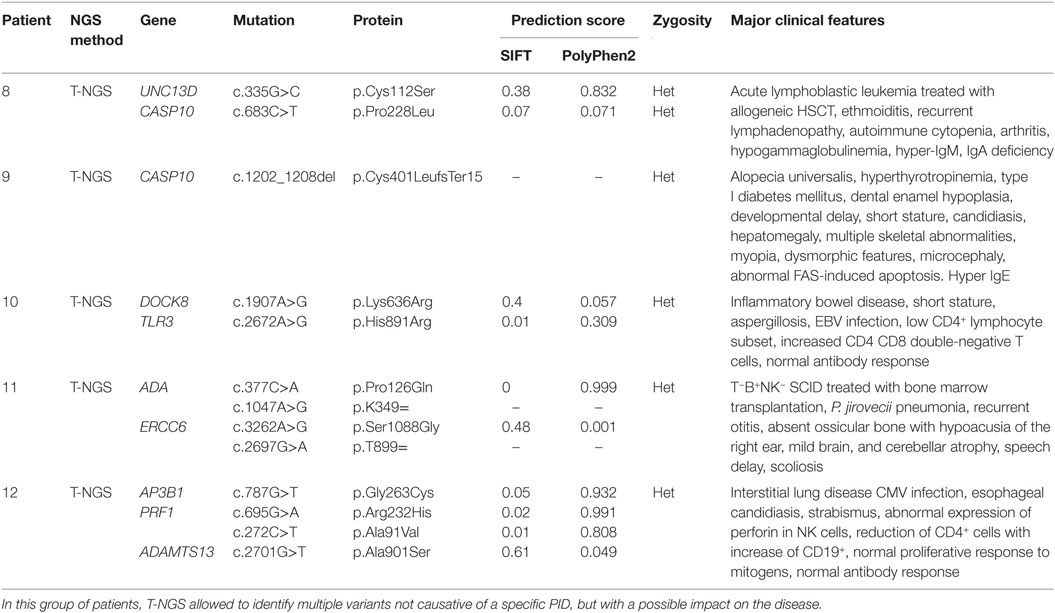
Table 3. Genetic variants potentially involved in the immunological features.
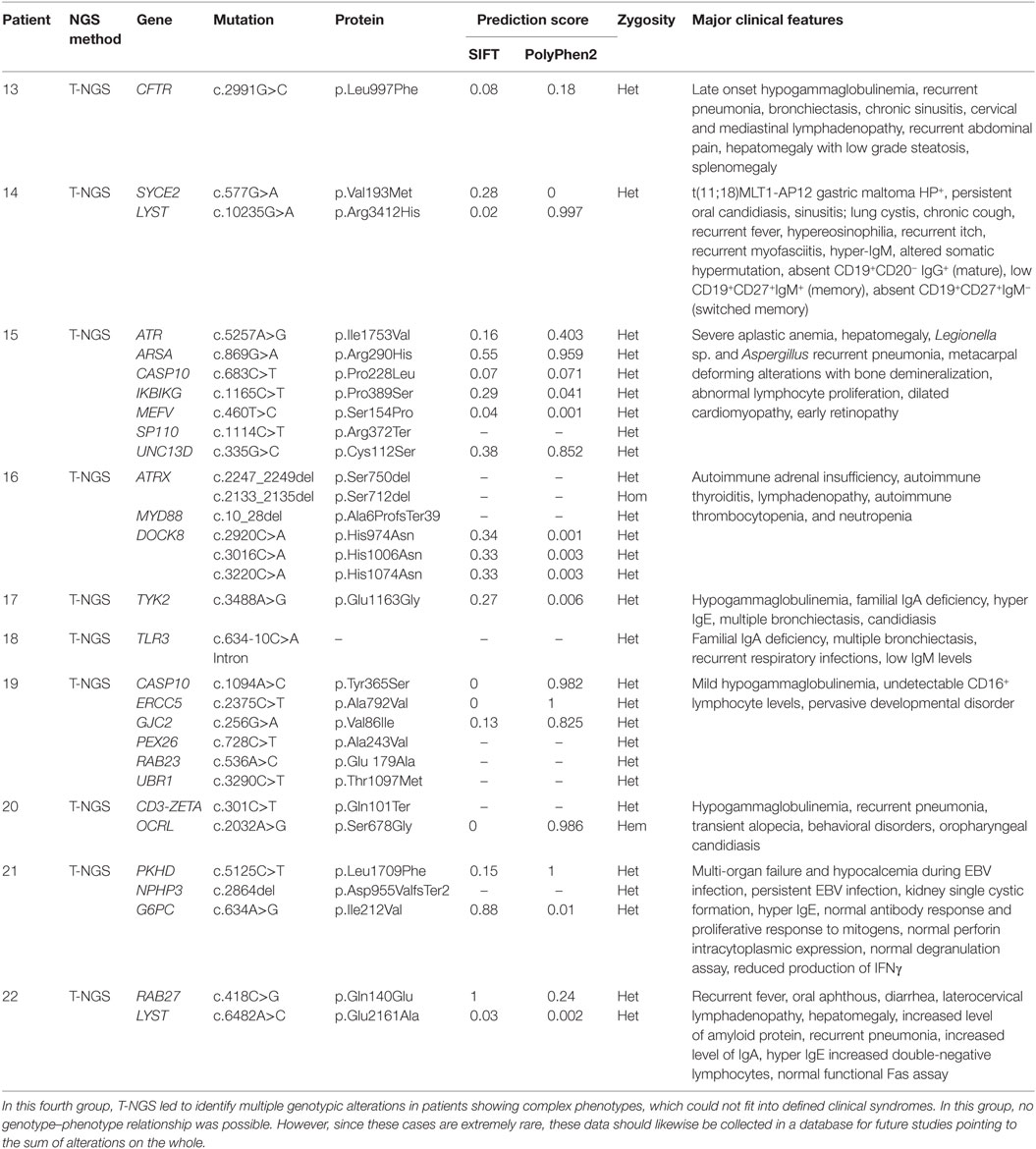
Table 4. Genetic variants not causative of PID with undetermined impact on the disease.
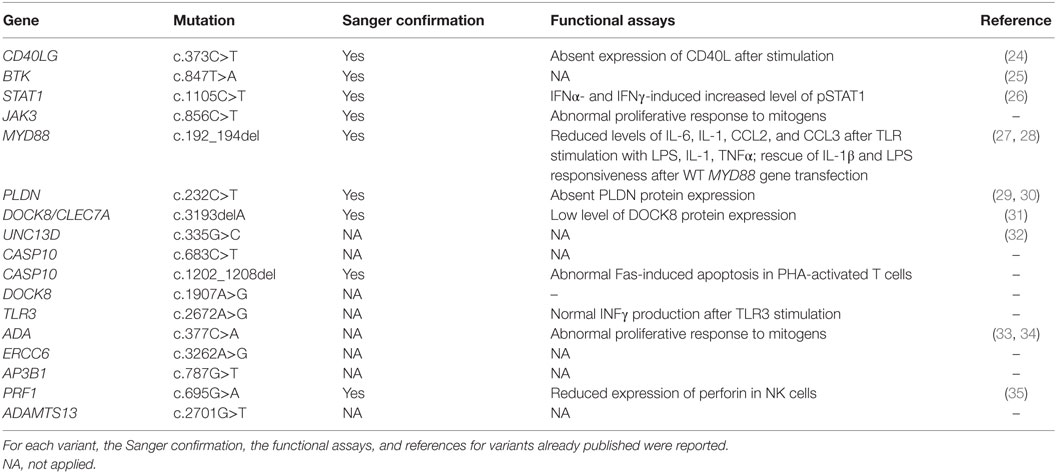
Table 5. Gene variants confirmed by Sanger sequencing, functional assays and/or previous report.
A satisfying molecular diagnosis of PID was achieved in 7 of the 45 patients (16%), including 3 patients with an atypical presentation. Three causative variations were identified through T-NGS, while five through WES. On the basis of the genetic findings and of the clinical phenotypes, patients were divided in four groups. Group 1 included subjects with a diagnostic genotype that was previously associated with an immunological and/or clinical phenotype already reported for that genetic disease. Group 2 included subjects with a diagnostic genotype associated with novel or atypical clinical features of a genetic disease. In the group 3, NGS revealed multiple genetic variants that were consistent only with some features of the clinical and immunological phenotype. Group 4 included patients with a complex unclassified disorder that was associated with multiple genetic variants, each of them, individually, was not per se causative of the disorder. However, regarding the variants of groups 3 and 4, it should be pointed out that, since parental cosegregation studies were not performed, the variants may not be truly disease-causing or disease-modifying alterations, especially if inherited together from an unaffected parent.
The first group included four subjects who have been identified for a genetic defect associated with a known well-established immunological and/or clinical phenotype of PID (Table 1). In detail, variants in CD40LG, BTK, STAT1 genes were already reported in literature as pathogenic (24–26). The JAK3 nonsense variant was predicted to result in nonsense-mediated decay resulting in no protein expression. This mutation, though not previously reported, was fully congruent with the classic JAK3–SCID phenotype observed in multiple affected family members who shared the same genotype (proband and two siblings).
The second group included other three subjects in whom genetic variants were found as associated with novel phenotypic features, in previously characterized phenotypes (Table 2). Specifically in this group, patient 005 was found to carry a homozygous mutation in MYD88 (c.192_194del; p.Glu66del), previously reported in several MYD88 deficiency affected families (27, 28). This patient was of a Rom ethnicity and had inherited the deletion from his unaffected consanguineous parents. But, early infant deaths due to severe infections were observed in the same family pedigree. Functionally, known mutations result in impairment of cytokine production after TLR stimulation (36, 37). Patients 006 and 007 were found to carry homozygous deleterious mutations in PLDN and DOCK8/CLEC7A gene, respectively, whose phenotypic peculiarity has been described in detail (29, 31).
The third group of subjects included five patients who carried multiple heterozygous variants affecting genes expressed in the hematopoietic system that were not consistent with a specific PID (Table 3). However, some of these patients might harbor a second mutation that was not identified by NGS or Sanger sequencing. In patient 008, we observed a heterozygous, probably, pathogenic variant in UNC13D (c.335G>C p.Cys112Ser), and a heterozygous, most likely benign variant in CASP10 gene (c.683C>T p.Pro228Leu; exon 10 not covered). This patient had a clinical phenotype consistent with an autosomal dominant type II ALPS (ALPS-II), associated with hypogammaglobulinemia and acute lymphoblastic leukemia. Even though the CASP10 variant alone in the in silico prediction programs, PolyPhen2 and SIFT, was predicted to be tolerated and was observed in 35 healthy individuals (ExAC), its significance in association with the second deleterious variant in UNC13D has never been described and could be potentially relevant. In fact, this UNC13D variant has been previously reported in association with heterozygous mutation of FAS and is considered a disease modifier for ALPS (32). In patient 009, we found by T-NGS a heterozygous frameshift variant in CASP10 causative of ALPS-II and categorized as likely pathogenic according to American College of Medical Genetics (ACMG) criteria (22). His immunological phenotype was characterized by very high IgE levels (>2000 IU). The functional analysis of Fas-induced apoptosis in PHA-activated T cells from the patient confirmed that the apoptotic pathway was impaired, since cell apoptosis upon triggering of Fas was impaired (92% survival; normal values: median 60%, 95th percentile 82%). His brother, who presented with similar manifestations, died early in life because of hemophagocytic lymphohistiocytosis. In addition, the patient 009 also had additional clinical features, such as developmental delay, microcephaly, peculiar facial dysmorphism, and skeletal abnormalities, which could not be at moment demonstrated or excluded to be directly explained by the variant. In patient 010, we found two heterozygous variants of unknown significance in DOCK8 and TLR3 with good coverage of each gene and no second variant. The TLR3 variant had multiple lines of computational evidence supporting a deleterious effect on gene/protein. Further studies are ongoing to find a possible correlation between the association of the two variants with the clinical phenotype, which was characterized by inflammatory bowel disease, susceptibility to viral infections, aspergillosis, T-cell lymphopenia, and increased CD4 and CD8 double-negative T cells. Alternatively, a second unidentified mutation of DOCK8 might account for the clinical manifestations of the patient. In patient 011 with a T−B+NK− SCID phenotype associated with deafness, microcephaly, brain and cerebellar atrophy, developmental delay, NGS revealed 2 heterozygous mutations of ADA gene, located in the same allele, the first one being likely pathogenic, and a further heterozygous variant of unknown significance of ERCC6 gene. The latter gene, which encodes for Cockayne syndrome B protein, has been recently described as essential for postnatal neuronal differentiation and neuritogenesis (38). Intriguingly, the patient showed also some neurological manifestations, such as macrocephaly and moderate grade cerebral atrophy. In patient 012 affected with hemophagocytic lymphohistiocytosis and marked reduction of perforin expression in NK cells, we identified two heterozygous mutations in PRF1 that were located in the same allele as shown by Sanger sequencing. In the same patient, we have also detected heterozygous variants in AP3B1 and ADAMTS13 genes that have been implicated in NK activity as well.
The last group of patients included 10 patients with a complex disorder that was not typical of any known syndrome and was associated with multiple genetic variants, each of them was not per se causative individually of that disorder (Table 4). In this group, it was not possible to draw any correlation between the genetic variations that were detected and the pathogenesis of the disorders. However, these alterations of unknown biologic significance were found in several genes implicated in immunological functions at different extent.
In the remaining 23 patients who were analyzed by NGS, including 9 studied by T-NGS and 15 by WES, we could not identify any candidate variants. These unsolved datasets will be re-analyzed when new bioinformatics tools become available and as new disease genes are described.
Discussion
The advent of NGS technologies has given the possibility to physicians to investigate multiple genes assay, to provide great opportunities for diagnosing patients affected with complex disorders of the immune system, and to increase our knowledge on the pathogenesis of these genetic disorders (39).
In this study, we used NGS technologies to identify potential disease-causing mutations in patients affected with clinical phenotypes highly suggestive of a PID, which were still not diagnosed after using traditional sequential Sanger sequencing procedures. Thanks to this novel diagnostic approach, we report that a definitive diagnosis of PID was achieved in a timely manner in 7 out of the 45 subjects. In three patients, the diagnosis was achieved through T-NGS, while, in the other four patients, the diagnosis was reached by WES. In all these subjects, the application of a clear-cut filtering strategy, consisting of targeted sequencing, bioinformatics analysis, phenotype-based filtering criteria, and confirmartory functional assays and Sanger sequencing, led to the identification of the underlying immune disorder (Figure 1). With this approach, eight of the overall group of variants resulted in potentially disease-causing mutations, distributed over seven patients.
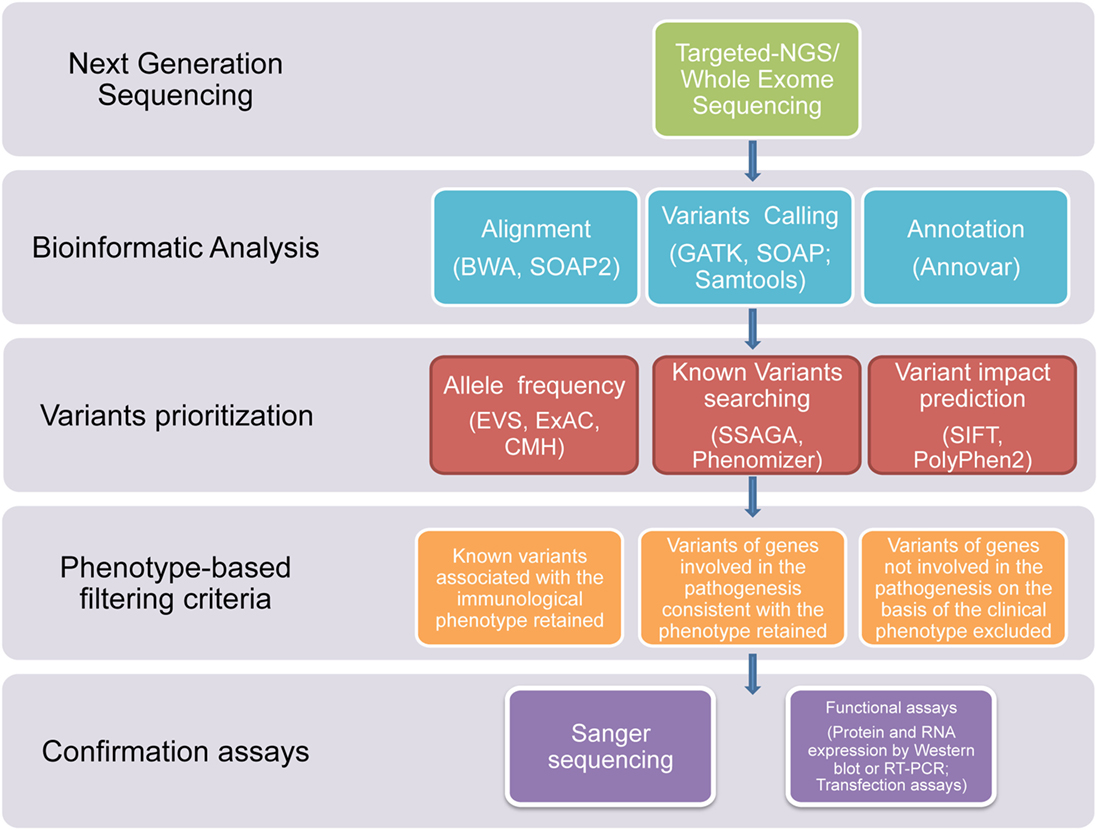
Figure 1. Flowchart of the filtering strategy. A schematic overview of the approach/clear-cut strategy used to filter variants identified through T-NGS or WES in order to identify potentially causative mutations.
We have divided NGS results into four categories: (I) genetic alterations associated with a canonical PID phenotype, (II) diagnostic genotype in atypical presentation, (III) genetic variants potentially involved in the immunological features, and (IV) multiple genetic variants, each of them was not per se causative individually of the disorder, even though the sum of variations of the different genes could be proven in the future of some pathogenic significance, as either causative or modifier factor.
The first and second groups included patients whose candidate genotypes were congruent with the immunologic features suggesting a causal relationship. Of note, exome sequencing allowed the identification of PLDN variants associated with a novel genetic cause of partial albinism and with PID (29). This study reported six patients who had undergone a diagnostic odyssey of 10–21 months driven by worldwide accepted protocols (40). Unfortunately, this candidate gene approach, although functionally driven, failed to yield a diagnosis. The diagnosis was finally made possible with NGS technology in a much more timely manner (2 months).
In the second category of patients, which included patients carrying genetic variants previously reported, but associated with novel features of previously established phenotypes, the molecular definition of the diagnosis contributed to expanding the overall knowledge of pathogenetic mechanisms underlying that specific disorder. In the patient with MYD88 deficiency, the atypical presentation was characterized by chronic yersiniosis resulting in terminal ileitis and recurrent neutropenia, in the absence of invasive pneumococcal disease, as is expected in this rare immunodeficiency. The atypical clinical presentation was responsible for the diagnostic delay. Moreover, Sanger sequencing of four candidate genes (STAT3, ELANE, RAG1, and RAG2) had given negative results (27). Homozygous mutations of DOCK8 and CLEC7A were already reported as genetic cause of two distinct PIDs, such as autosomal recessive Hyper IgE syndrome and of CMC, respectively, but never observed in a single patient. It is likely that these genetic variants affecting two distinct loci, and probably concurring to the clinical manifestations of the patient, could not be identified without the availability of NGS (31).
The cases highlighted herein contribute to the explanation of several phenotypes for very rare disorders. Such broadening of the phenotypic spectrum is a phenomenon shared with many other rare disorders, as atypical patients are identified through NGS. In the case of congenital immune disorders, at the beginning only the most severe forms are described (41). Only after years from the initial description of the syndrome milder features of the disease are recognized or extra-immunological clinical signs identified.
In the third group, NGS sequencing revealed multiple heterozygous genetic variants in each subject, some of them potentially involved in the immunological features. All the variants of this group are either single heterozygous or in cis, thus further studied are needed, including WES or microarray testing to try to find the second mutation or uncover mutation(s) in a separate gene that fully explain(s) the phenotype. In the patient 012, the PRF1 gene alteration was proven to have functional relevance, since perforin expression in NK cells was reduced. However, the patient was not placed in the group 1 since the two heterozygous PRF1 variations were in cis at the Sanger sequencing. Previously, our group has documented that this heterozygous variation may act as a susceptibility cofactor, which under certain circumstances may be associated with a functional alteration (42).
Finally, the fourth group included patients showing multiple genotypic alterations associated with complex phenotypes that could not fit into defined clinical phenotypes. In this group, no genotype–phenotype relationship was possible. However, since these cases are extremely rare, these data should likewise be collected in a database. The creation of such a database might improve the interpretation of NGS results in those cases currently interpreted as no causative of the disorder. The identification of different individuals with the same phenotype and mutations in the same array of genes would suggest that the sum of variations in different genes exerts a pathogenic role, either as causative or as modifier factor.
However, even though in our study in 7 out of the 45 patients, a diagnosis was achieved, it should be considered that in the majority of the patients, target NGS approach did not allow to identify the genetic basis of the disease. Several aspects should be considered to interpret this observation. The first limitation is the limited number of the genes included on our targeted NGS panel. Moreover, the sequencing techniques do not provide enough coverage for intronic, promoter, or regulator regions. To overcome these technical limitations, whole exome or genome sequencing might be better strategies to deeply investigate these cases as the second-line diagnostic tool (14). Moreover, all NGS techniques, due to the generation of short reads, show a low sensitivity to detect complex structural variations (deletions, insertions, and inversions), repeat sequences, or complex rearrangement (43).
Based on these considerations, the identification of genetic defects in patients with PIDs is still a major challenge, and the functional implication of the variation must be considered mandatory to definitely prove the relationship between the genetic alteration and the related phenotype.
Despite the above mentioned limitations, NGS technology represents a cost-effective and rapid first-line genetic approach for the evaluation of complex cases of PIDs. The advantage of this technique is the simultaneous sequencing of a panel of genes, perhaps leading to the rapid identification of a diagnosis that may not have been otherwise considered using the traditional phenotype-driven approach. Overall, in spite of a moderately higher cost, at moment WES could represent the first-line approach to initial PID management. The sequential investigation of several candidate genes is, by comparison, a very time- and cost-consuming process. Prompt diagnosis shows an unquestioned clinical advantage, allowing initiation of appropriate and often life-saving treatment.
Author Contributions
VG, GG, EC, VL, RC, and AP identified the pts and performed the immunological and phenotypic characterization; IT, EF, and CS performed the NGS and WES exps; RDA, AP, and VL performed the functional exps and the molecular confirmation exps; RB and CP interpreted the results; VG and CP wrote the manuscript; and RB and CP directed the project.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer JN and handling editor declared their shared affiliation, and the handling editor states that the process nevertheless met the standards of a fair and objective review.
Funding
This study was partially funded by “Associazione Immunodeficienze primitive.”
Supplementary Material
The Supplementary Material for this article can be found online at http://journal.frontiersin.org/article/10.3389/fimmu.2016.00466
Abbreviations
ACMG, American College of Medical Genetics; ALPS, autoimmune lymphoproliferative syndrome; CMC, chronic mucocutaneous candidiasis; MYD88, myeloid differentiation factor 88; NGS, next-generation sequencing; PIDs, primary immunodeficiencies; SNVs, single nucleotide variants; TLR, toll-like receptor; T-NGS, targeted next-generation sequencing; WES, whole exome sequencing; WGS, whole genome sequencing.
Footnotes
References
1. Al-Herz W, Bousfiha A, Casanova I, Chatila TA, Conley ME, Cunningham-Rundles C, et al. Primary immunodeficiency diseases: an update on the classification from the International Union of Immunological Societies Expert Committee for primary immunodeficiency. Front Immunol (2014) 5:162. doi:10.3389/fimmu.2014.00162
2. Cirillo E, Giardino G, Gallo V, D’Assante R, Grasso F, Romano R, et al. Severe combined immunodeficiency-an update. Ann N Y Acad Sci (2015) 1356:90–106. doi:10.1111/nyas.12849
3. Parvaneh N, Casanova JL, Notarangelo LD, Conley ME. Primary immunodeficiencies: a rapidly evolving story. J Allergy Clin Immunol (2013) 131:314–24. doi:10.1016/j.jaci.2012.11.051
4. de Greef JC, Wang J, Balog J, den Dunnen JT, Frants RR, Straasheijm KR, et al. Mutations in ZBTB24 are associated with immunodeficiency, centromeric instability, and facial anomalies syndrome type 2. Am J Hum Genet (2011) 88:796–804. doi:10.1016/j.ajhg.2011.04.018
5. Ghosh S, Krux F, Binder V, Gombert M, Niehues T, Feyen O, et al. Array-based sequence capture and generation sequencing for the identification of primary immunodeficiencies. Scand J Immunol (2012) 75:350–4. doi:10.1111/j.1365-3083.2011.02658.x
6. Maffucci P, Filion CA, Boisson B, Itan Y, Shang L, Casanova JL, et al. Genetic diagnosis using whole exome sequencing in common variable immunodeficiency. Front Immunol (2016) 7:220. doi:10.3389/fimmu.2016.00220
7. Moshous D, Martin E, Carpentier W, Lim A, Callebaut I, Canioni D, et al. Whole-exome sequencing identifies Coronin-1A deficiency in 3 siblings with immunodeficiency and EBV-associated B-cell lymphoproliferation. J Allergy Clin Immunol (2013) 131:1594–603. doi:10.1016/j.jaci.2013.01.042
8. Shearer WT, Dunn E, Notarangelo LD, Dvorak CC, Puck JM, Logan BR, et al. Establishing diagnostic criteria for severe combined immunodeficiency disease (SCID), leaky SCID, and Omenn syndrome: the primary immune deficiency treatment consortium experience. J Allergy Clin Immunol (2014) 133:1092–8. doi:10.1016/j.jaci.2013.09.044
9. Notarangelo LD. Primary immunodeficiencies. J Allergy Clin Immunol (2010) 125:S182–94. doi:10.1016/j.jaci.2009.07.053
10. Van der Burg M, Gennery AR. Educational paper. The expanding clinical and immunological spectrum of severe combined immunodeficiency. Eur J Pediatr (2011) 170:561–71. doi:10.1007/s00431-011-1452-3
11. Dvorak CC, Cowan MJ, Logan BR, Notarangelo LD, Griffith LM, Puck JM, et al. The natural history of children with severe combined immunodeficiency: baseline features of the first fifty patients of the Primary Immune Deficiency Treatment Consortium prospective study 6901. J Clin Immunol (2013) 33:1156–64. doi:10.1007/s10875-013-9917-y
12. Markert ML, Marques J, Neven B, Devlin B, McCarthy E, Chinn I, et al. First use of thymus transplantation therapy for Foxn1 deficiency (nude/SCID): a report of two cases. Blood (2011) 117:688–96. doi:10.1182/blood-2010-06-292490
13. Oliveira JB, Fleisher TA. Laboratory evaluation of primary immunodeficiencies. J Allergy Clin Immunol (2010) 125(2 Suppl 2):S297–305. doi:10.1016/j.jaci.2009.08.043
14. Chou J, Ohsumi TK, Geha RS. Use of whole exome and genome sequencing in the identification of genetic causes of primary immunodeficiency. Curr Opin Allergy Clin Immunol (2012) 12:623–8. doi:10.1097/ACI.0b013e3283588ca6
15. Bamshad MJ, Ng SB, Bigham AW, Tabor HK, Emond MJ, Nickerson DA, et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet (2011) 12:745–55. doi:10.1038/nrg3031
16. Moens L, Falk-Sorqvist E, Asplund AC, Bernatowska E, Smith CI, Nilsson M. Diagnostics of primary immunodeficiency disease: a sequencing capture approach. PLoS One (2014) 9:e114901. doi:10.1371/journal.pone.0114901
17. Nijman IJ, van Montfrans JM, Hoogstraat M, Boes ML, van de Corput L, Renner ED, et al. Target next-generation sequencing: a novel diagnostic tool for primary immunodeficiencies. J Allergy Clin Immunol (2014) 133:529–34. doi:10.1016/j.jaci.2013.08.032
18. Stoddard JL, Niemela JE, Fleisher TA, Rosenzweig SD. Target NGS: a cost-effective approach to molecular diagnosis of PIDs. Front Immunol (2014) 5:531. doi:10.3389/fimmu.2014.00531
19. Mousallem T, Urban TJ, McSweeney KM, Kleinstein SE, Zhu M, Adeli M, et al. Clinical application of whole-genome sequencing in patients with primary immunodeficiency. J Allergy Clin Immunol (2015) 136:476–9.e6. doi:10.1016/j.jaci.2015.02.040
20. Aloj G, Giardino G, Valentino L, Maio F, Gallo V, Esposito T, et al. Severe combined immunodeficiences: new and old scenarios. Int Rev Immunol (2012) 31:43–65. doi:10.3109/08830185.2011.644607
21. Saunders CJ, Miller NA, Soden SE, Dinwiddie DL, Noll A, Alnadi NA, et al. Rapid whole-genome sequencing for genetic disease diagnosis in neonatal intensive care units. Sci Transl Med (2012) 4:154ra135. doi:10.1126/scitranslmed.3004041
22. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med (2015) 17:405–24. doi:10.1038/gim.2015.30
23. Kingsmore SF, Dinwiddie DL, Miller NA, Soden SE, Saunders SE, Saunders CJ. Adopting orphans: comprehensive genetic testing of Mendelian disease of childhood by next-generation sequencing. Exp Rev Mol Diagn (2011) 11:855–68. doi:10.1586/erm.11.70
24. Hernandez A, Berron-Ruiz L, Staines-Boone T, Zarate-Hernandez M, Cordova-Calderon WO, Espinosa-Rosales FJ, et al. Clinical and genetic analysis of patients with X-linked hyper-IgM syndrome. Clin Genet (2013) 83:585–7. doi:10.1111/j.1399-0004.2012.01953.x
25. Holinski-Feder E, Weiss M, Brandau O, Jedele KB, Nore B, Bsckesjš CM, et al. Mutation screening of the BTK gene in 56 families with X-linked agammaglobulinemia (XLA): 47 unique mutations without correlation to clinical course. Pediatrics (1998) 101:276–84. doi:10.1542/peds.101.2.276
26. Dotta L, Scomodon O, Padoan R, Timpano S, Plebani A, Soresina A, et al. Clinical heterogeneity of dominant chronic mucocutaneous candidiasis disease: presenting as treatment-resistant candidiasis and chronic lung disease. Clin Immunol (2016) 164:1–9. doi:10.1016/j.clim.2015.12.010
27. Giardino G, Gallo V, Somma D, Farrow EG, Thiffault I, D’Assante R, et al. Target next-generation sequencing revealed MYD88 deficiency in a child with chronic yersiniosis and granulomatous lymphadenitis. J Allergy Clin Immunol (2016) 137:1591–5. doi:10.1016/j.jaci.2015.09.050
28. Yamamoto T, Tsutsumi N, Tochio H, Ohnishi H, Kubota K, Kato Z, et al. Functional assessment of the mutational effect of human IRAK4 and MyD88 genes. Mol Immunol (2014) 58:66–66. doi:10.1016/j.molimm.2013.11.008
29. Badolato R, Pradini A, Caracciolo S, Colombo F, Tabellini G, Giacomelli M, et al. Exome sequencing reveals a pallidin mutation in a Hermansky-Pudlak-like primary immunodeficiency syndrome. Blood (2012) 119:3185–7. doi:10.1182/blood-2012-01-404350
30. Cullinane AR, Curry JA, Carmona-Rivera C, Summers CG, Ciccone C, Cardillo ND, et al. A BLOC-1 mutation screen reveals that PLDN is mutated in Hermansky-Pudlak syndrome type. Am J Hum Genet (2011) 88:778–87. doi:10.1016/j.ajhg.2011.05.009
31. Dinwiddle DL, Kningsmore SF, Caracciolo S, Rossi G, Moratto D, Mazza C, et al. Combined DOCK8 and CLEC7A mutations causing immunodeficiency in 3 brothers with diarrhea, eczema, and infections. J Allergy Clin Immunol (2013) 131:594–7. doi:10.1016/j.jaci.2012.10.062
32. Arico’ M, Boggio E, Cetica V, Melensi M, Orilieri E, Clemente N, et al. Variations of the UNC13D gene in patients with autoimmune lymphoproliferative syndrome. PLoS One (2013) 8:e68045. doi:10.1371/journal.pone.0068045
33. Arredondo-Vega FX, Santisteban I, Daniels S, Toutain S, Hershfield MS. Adenosine deaminase deficiency: genotype-phenotype correlations based on expressed activity of 29 mutant alleles. Am J Hum Genet (1998) 63:1049–59. doi:10.1086/302054
34. Ozsahin H, Arredondo-Vega FX, Santisteban I, Fuhrer H, Tuchschmid P, Jochum W, et al. Adenosine deaminase deficiency in adults. Blood (1997) 89:2849–55.
35. Feldmann J, Le Deist F, Ouachze-Chardin M, Certain S, Alexander S, Quartier P, et al. Functional consequences of perforin gene mutations in 22 patients with familial heamophagocytic lymphohistiocytosis. Br J Haematol (2002) 117:965–72. doi:10.1046/j.1365-2141.2002.03534.x
36. Picard C, Casanova JL, Puel A. Infectious diseases in patients with IRAK-4, MyD88, NEMO, or IkBa deficiency. Clin Microbiol Rev (2011) 24:490–7. doi:10.1128/CMR.00001-11
37. von Bernuth H, Picard C, Jin Z, Pankla R, Xiao H, Ku CL, et al. Pyogenic bacterial infections in humans with MyD88 deficiency. Science (2008) 321:691–6. doi:10.1126/science.1158298
38. Ciaffardini F, Nicolai S, Caputo M, Canu G, Paccosi E, Costantino M, et al. The Cockayne syndrome B protein is essential for neuronal differentiation and neuritogenesis. Cell Death Dis (2014) 5:e1268. doi:10.1038/cddis.2014.228
39. O’Connell AE, Volpi S, Dobbs K, Fiorini C, Tsitsikov E, de Boer H, et al. Next generation sequencing reveals skewing of the T and B cell receptor repertoires in patients with Wiskott-Aldrich syndrome. Front Immunol (2014) 5:340. doi:10.3389/fimmu.2014.00340
40. De Vries E, Alvarez Cardona A, Abdul Latiff AH, Badolato R, Brodszki N, Cant AJ, et al. Patient-centred screening for primary immunodeficiency, a multi-stage diagnostic protocol designed for non-immunologists: 2011 update. Clin Exp Immunol (2012) 167:108–19. doi:10.1111/j.1365-2249.2011.04461.x
41. Maggina P, Gennery AR. Classification of primary immunodeficiencies: need for a revised approach? J Allergy Clin Immunol (2013) 131:292–4. doi:10.1016/j.jaci.2012.10.008
42. Busiello R, Adriani M, Locatelli F, Galgani M, Fimiani G, Clementi R, et al. Atypical features of familial hemophagocytic lymphohistiocytosis. Blood (2004) 103:4610–2. doi:10.1182/blood-2003-10-3551
Keywords: primary immunodeficiencies, genetic diagnosis, targeted next-generation sequencing, whole exome sequencing, genotype–phenotype correlation
Citation: Gallo V, Dotta L, Giardino G, Cirillo E, Lougaris V, D’Assante R, Prandini A, Consolini R, Farrow EG, Thiffault I, Saunders CJ, Leonardi A, Plebani A, Badolato R and Pignata C (2016) Diagnostics of Primary Immunodeficiencies through Next-Generation Sequencing. Front. Immunol. 7:466. doi: 10.3389/fimmu.2016.00466
Received: 11 July 2016; Accepted: 17 October 2016;
Published: 07 November 2016
Edited by:
Sergio Rosenzweig, National Institutes of Health, USAReviewed by:
Ivan K. Chinn, Duke University Medical Center, USAJulie E. Niemela, DHHS-NIH Clinical Center, USA
Copyright: © 2016 Gallo, Dotta, Giardino, Cirillo, Lougaris, D’Assante, Prandini, Consolini, Farrow, Thiffault, Saunders, Leonardi, Plebani, Badolato and Pignata. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Claudio Pignata, cGlnbmF0YUB1bmluYS5pdA==
†Raffaele Badolato and Claudio Pignata share senior authorship.