 Alice Koenig1,2,3
Alice Koenig1,2,3 Olivier Thaunat1,2,3*
Olivier Thaunat1,2,3*
- 1Service de Transplantation, Néphrologie et Immunologie Clinique, Hôpital Edouard Herriot, Hospices Civils de Lyon, Lyon, France
- 2INSERM UMR1111, Lyon, France
- 3Université de Lyon, Lyon, France
The progressive organization of immune effectors into functional ectopic lymphoid structures, named tertiary lymphoid organs (TLO), has been observed in many conditions in which target antigens fail to be eliminated by the immune system. Not surprisingly, TLO have been recurrently identified in chronically rejected allografts. Although significant progress has been made over the last decades in understanding the molecular mechanisms involved in TLO development (a process named lymphoid neogenesis), the role of intragraft TLO (if any) in chronic rejection remains elusive. The prevailing dogma is that TLO contribute to graft rejection by generating and propagating local humoral and cellular alloimmune responses. However, TLO have been recently observed in long-term accepting allografts, suggesting that they might also be able to regulate alloimmune responses. In this review, we discuss our current understanding of how TLO are induced and propose a unified model in which TLO can play deleterious or regulatory roles and therefore actively modulate the kinetics of chronic rejection.
Introduction: The Challenge of Chronic Rejection in Transplantation
Vital organ failure is a life-threatening condition where a vital organ (i.e., kidney, heart, liver, or lung…) does not perform its expected function. Recent lifestyle changes in developed countries, and the increased incidence of chronic diseases such as hypertension, obesity, and diabetes, have set the stage for accelerated risk for, and the occurrence of, vital organ failure. As a result, vital organ failure is currently recognized as the leading cause of debility and premature death worldwide (www.who.int). In France alone, the personal, societal, and economic consequences of vital organ failure have a cost of more than €70 billion a year (25% of total health expenditures).
Transplantation consists in the restoration of vital physiologic functions through the surgical substitution of a defective organ by a functioning graft retrieved from a donor. Patients with end-stage vital organ failure depend on solid organ transplantation, which is their best (often their only) therapeutic option.
In clinical transplantation, the donor is from the same species but genetically different. Consequently, the immune system of the recipient inevitably recognizes the antigenic determinants (alloantigens) that differ between the recipient and the donor, particularly the highly polymorphic molecules from the major histocompatibility complex [i.e., human leukocyte antigen (HLA)] in humans. The alloimmune response that develops against the donor-specific HLA molecules is responsible for tissue damage, which leads to the failure of the transplanted organ, a process named “rejection.”
In the absence of a clinically applicable protocol able to induce the specific tolerance of the allogenic transplant by the recipient’s immune system (1, 2), the prevention of rejection is currently dependent upon immunosuppressive drugs (3). These drugs produce generalized immunosuppression, which means that any reduction in immune responsiveness to the allograft is accompanied by reduced immunity to infections and malignant diseases. Chronic immune injuries that result from the incomplete blockade of the recipient’s alloimmune response (i.e., chronic rejection) are currently the main factor limiting graft function duration (4). No significant progress has been made on this issue over the last decades as highlighted by the stagnation of graft half-life (5). A better understanding of the pathophysiology of chronic rejection is therefore a mandatory step in identifying innovative approaches that would prolong graft function duration.
Intragraft Tertiary Lymphoid Organs (TLO)
Rejected grafts are characterized by interstitial infiltration of cellular effectors, mainly T cells and macrophages, but also dendritic cells, NK cells, B cells, and plasma cells.
In contrast with acute rejection, where infiltrates exhibit no particular spatial organization, during chronic rejection immune cells tend to organize themselves in structures that morphologically resemble the secondary lymphoid organs.
Analyzing all sorts of human kidney grafts removed for terminal chronic rejection, we and others showed that in the majority of chronically rejected grafts the immune cells were grouped, conferring a nodular organization to the infiltrate (6, 7). These nodules exhibited a highly organized microarchitecture with clear cell subset segregation: the core, made of the B cells intermingled with a network of follicular dendritic cells, was surrounded by T cells and mature dendritic cells. CD138-expressing plasma cells were found within or in close vicinity to TLO, suggesting that part of these cells differentiated locally. As in canonical secondary lymphoid organs the compartmentalization of the different cell subsets appeared to be mediated by gradients of homeostatic chemokines CCL21 (in the T cell area) and CXCL13 (in the B cell area). Furthermore, neolymphatic vessels and PNAd-expressing high endothelial venules (HEVs) were observed in the periphery of the nodules (8).
The structural organization of immune effectors observed in chronically rejected renal grafts (Figure 1) does not seem specific of this type of transplant since similar lymphoid structures have been observed in chronically rejected pancreas, livers, hearts (7, 9–11), lungs (12), and even composite transplants (13–15). This phenomenon is not specific of the alloimmune setting either, since the very same lymphoid structures have previously been observed in various inflammatory conditions, including chronic infections, autoimmune diseases, and cancers (16, 17). Structural organization of immune effectors therefore appears as a generic response of the chronically stimulated immune system that cannot eradicate targeted antigens.
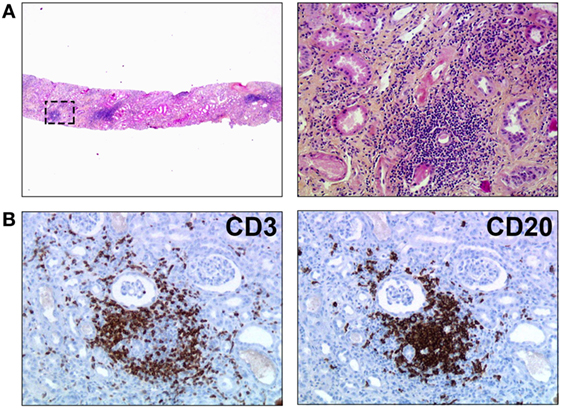
Figure 1. Tertiary lymphoid organs in a chronically rejected renal transplant. Biopsy of a renal transplant was performed for progressive deterioration of graft function, suggestive of chronic rejection. (A) HES staining revealed nodular infiltrates of mononuclear cells within graft parenchyma (original magnification: left panel, ×20; right panel, ×200). (B) Immunostainings unraveled the organized distribution of T cells (CD3+, left panel) and B cells (CD20, right panel). Original magnification: ×200.
Because the microarchitecture of organized immune infiltrates is highly reminiscent of that of secondary lymphoid organs, these lymphoid structures have been named TLO.
Molecular Mechanisms Involved in the Development of Secondary Lymphoid Organs
Primary immune responses are initiated in secondary lymphoid organs, which are located at strategic sites where antigens are most likely to be encountered.
The development of secondary lymphoid organs, a process named lymphoid organogenesis, is initiated during embryogenesis independently of antigen recognition at predetermined sites as a result of complex interactions between hematopoietic, mesenchymal, and endothelial cells (18, 19). Lymphoid organogenesis can be schematically divided into two consecutive steps: first the induction, then the organization phase.
The induction phase depends on lymphoid-tissue inducer cells, which arise in the fetal liver. Under the influence of TRANCE (at sites of peripheral lymph node development) or IL-7 (at mucosal sites) lymphoid-tissue inducer cells express membrane-bound lymphotoxin: a heterotrimer containing lymphotoxin α and lymphotoxin β that allow lymphoid-tissue inducer cells to interact with the lymphotoxin β receptor (LTβR) of stromal cells. Signaling through the LTβR initiates NFκB signaling in stromal cells, which promotes the production of homeostatic chemokines (18, 19).
Homeostatic chemokines are crucial for the organization phase. CXCL13 recruits circulating B cells to what becomes the B cell area of lymphoid tissues, and the T zone chemokines (CCL19 and CCL21) attract T and dendritic cells to shape the T cell area (18, 19). The lymphotoxin signaling pathway is also crucial in promoting the differentiation of HEVs, which are postcapillary venules expressing specific adhesion molecules (known as addressins) that have a crucial role in lymphocyte trafficking to secondary lymphoid organs (18, 19).
Molecular Mechanisms of Lymphoid Neogenesis in Transplantation
Chronic rejection provides optimal conditions for studying the molecular mechanisms involved in the development of TLO (Figure 2). Indeed, (i) TLO have systematically been detected in chronically rejected grafts; (ii) the antigens targeted by the immune system are known (recipient-mismatched HLA antigens of the transplanted tissues); and (iii) chronically rejected grafts are sometimes removed, providing a large amount of diseased tissue, which can be comprehensively analyzed.
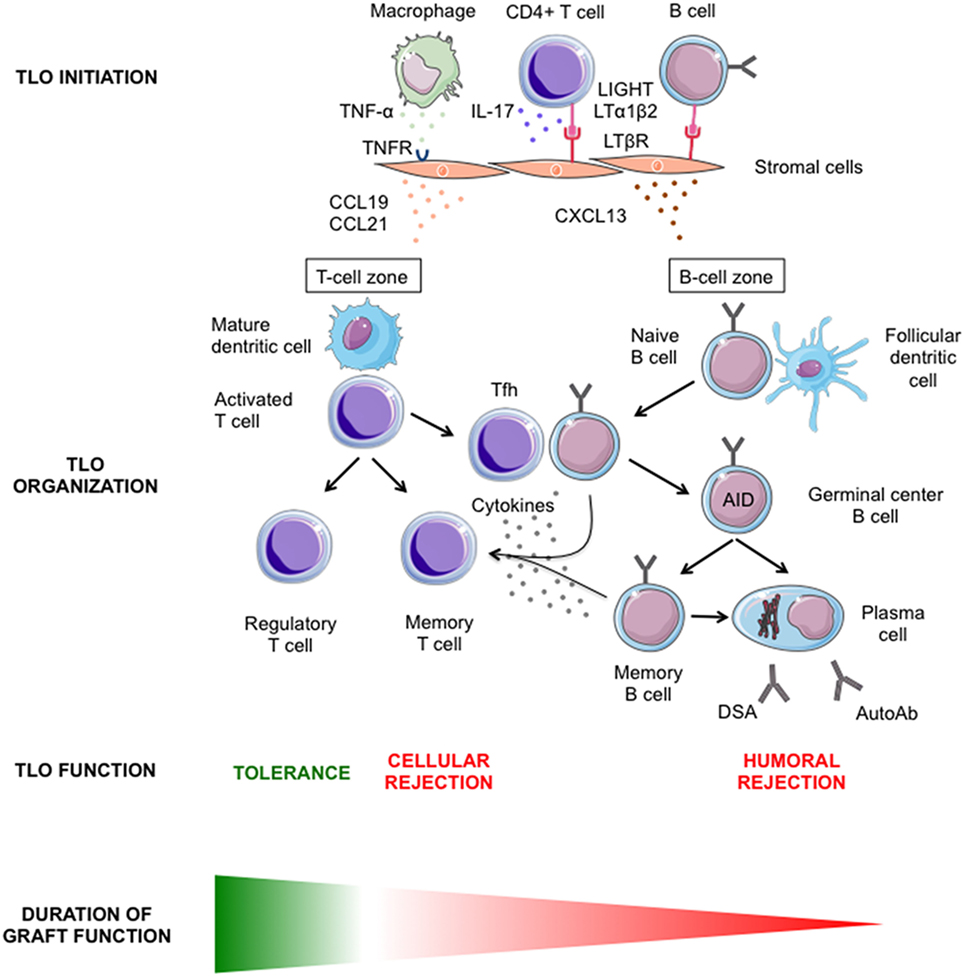
Figure 2. Graphical summary of our current understanding of intragraft lymphoid neogenesis. The main molecular mechanisms involved in the initiation (upper row) and the subsequent organization (middle row) of tertiary lymphoid organs (TLO) within transplanted organs are showed. The diverse functions of intragraft TLO and their respective impact on graft survival are presented (lower row).
In-depth analysis of a series of detransplanted human renal grafts revealed the heterogenous nature of the cellular composition of TLO (8). Two types of B cell nodules could be identified: nodules composed of a uniform CD20pos B cell population expressing IgD and Bcl-2 were similar to primary follicles, while nodules with a core of CD20posIgDnegBcl-2neg B cells, highly expressing Bcl-6 that had pushed aside the CD20posIgDposBcl-2pos B cells, resembled secondary follicles, i.e., germinal centers (8). The ratio between these two types of structures differed between samples, and the number of ectopic germinal centers did not increase with the quantity of primary nodules. The phenotypic heterogeneity of TLO correlated more with the expression profile of a set of genes (CCL19, CCL21, CXCL12, CXCL13, CCR7, CXCR4, and CXCR5) involved in the formation and the maintenance of canonical secondary lymphoid organs (i.e., the lymphoid organogenesis described in the previous section) (18, 19). The complete recapitulation of this genetic program in chronically rejected grafts resulted in the generation of fully functional ectopic germinal centers that allowed for the efficient maturation of B cells into memory B cells and plasma cells (Figure 2). In contrast, when this recapitulation was incomplete, local B cell maturation was impeded (8). These results highlighted the similarity between the molecular processes involved in the development of canonical secondary lymphoid organs and those involved in the organization of immune effectors during chronic inflammation, a process named lymphoid neogenesis.
If the molecular mechanisms responsible for the organization and maintenance of secondary lymphoid organs and TLO appear similar, the initiation of the cascade is likely to be different (Figure 2). The formation of secondary lymphoid organs in the embryo is developmentally programed and results from the interaction between lymphotoxin-α1β2-expressing lymphoid-tissue inducer cells and lymphotoxin-β receptor-expressing stromal organizer cells (18, 19). In contrast, TLO development seems independent of lymphoid-tissue inducer cells (20, 21). Yet, several studies have documented the importance of the lymphotoxin pathway in lymphoid neogenesis (21, 22), including in a transplantation setting (23), by demonstrating that the development of TLO was abolished by treatment with inhibitory LTβR–Ig fusion protein. We must then ask who provides lymphotoxin signaling in the chronic rejection setting. Beyond lymphoid-tissue inducer cells, lymphotoxin-α and lymphotoxin-β are also expressed by activated lymphocytes (24). It is therefore conceivable that activated T and/or B cells replace lymphoid-tissue inducer cells to initiate lymphoid neogenesis in rejected grafts (Figure 2) as already demonstrated for the induction of TLO in the gut (21). Another possibility is that lymphotoxin is dispensable for the formation of TLO. Lymphotoxin-α and lymphotoxin-β are two related members of the large TNF ligand family (25). Since homologous genes and gene products often have redundant physiological functions, it seems reasonable to propose that other ligands and/or receptors of the TNF superfamily could act as alternative pathways for TLO induction (Figure 2). In line with this hypothesis, the provision of the alternative LTβR ligand LIGHT (aka tumor necrosis factor superfamily member 14) by activated T cells infiltrating inflamed pancreas have been shown to be crucial for the formation of TLO (26). Furthermore, TNF-α, which is produced within rejected grafts (27), has been shown to be critical for the development of TLO in a murine model of atherosclerosis (20). TNF-α does not bind to LTβR but to distinct TNF receptors (25). Using apolipoprotein E-deficient mice, which spontaneously develop atherosclerotic lesions in their aorta, the Antonino Nicoletti’s group recently demonstrated that the blockade of LTβR signaling had no effect, whereas that of TNFR1/2 signaling reduced the expression of homeostatic chemokines and the subsequent development of TLO (20). Finally, it has recently been reported that IL-17 produced by CD4+ T cells (i.e., Th17 cells) was essential for the formation of both (i) TLO in the central nervous system of mice during experimental autoimmune encephalomyelitis (the animal model of multiple sclerosis) (28) and (ii) the development of inducible bronchus-associated lymphoid tissue, an ectopic lymphoid tissue that forms in the lungs after pulmonary inflammation (29, 30). In the latter setting, IL-17 acted by triggering the expression of homeostatic chemokines independently of lymphotoxin signaling (Figure 2). If this hypothesis was proven true in transplantation, initiation of lymphoid neogenesis in chronically rejected grafts could therefore be totally independent of both lymphoid-tissue inducer cells and the lymphotoxin/TNF pathway. Interestingly, we have recently reported that a Th17 polarization of CD4+ T cells infiltrating the graft was associated with increased TLO development during clinical chronic rejection (31).
It is conceivable that instead of conflicting with each other, these different works reveal the fact that several pathways can promote the initiation of TLO depending on the initiating events. This hypothesis was recently substantiated by the demonstration that the development of bronchus-associated lymphoid tissue was triggered by different pathways according to the pathogen responsible for lung inflammation (29).
While significant progress has been made in the identification of the molecular mechanisms that participate to the development of TLO, the endogenous signals capable of inhibiting the lymphoid neogenesis are far more elusive. Through evaluation of synovial tissues from rheumatoid arthritis patients it has been recently reported that low interleukin-27 (IL-27) expression corresponds with an increased incidence of TLO and gene signatures associated with their development and activity. The presence of synovial TLO was also noted in mice deficient in the IL-27 receptor after the onset of inflammatory arthritis (32). IL-27 might therefore represent a negative regulator of TLO development. Whether this is also true for chronic rejection remains to be demonstrated.
Do Intragraft TLO Promote Chronic Rejection?
Tertiary lymphoid organs differ from canonical secondary lymphoid organs inasmuch as they develop in an inflammatory milieu (31, 33), enriched in neoantigens released from injured tissue and trapped by defective lymphatic drainage (34). Comparing the cellular composition of TLO of chronically rejected grafts with one of the secondary lymphoid organs, we observed a drastic increase in the percentage of activated and memory CD4+ T cell in intragraft TLO and a symmetric decrease in T regulatory subsets (IL-10-producing Tr1 cells and Foxp3pos Tregs) in both a murine experimental model and human samples (33, 35).
These peculiarities suggest that the local immune response that develops in intragraft TLO might be less tightly regulated than in secondary lymphoid organs and are therefore more aggressive. In line with this hypothesis, we (33) and others (23) have shown that intragraft TLO are a major site where B cell tolerance breakdown occurs during chronic rejection (Figure 2). Interestingly, the generation of autoantibodies following solid organ transplantation has long been reported to correlate with chronic rejection, and the deleterious impact of some autoantibodies on graft survival has been demonstrated (36, 37). Furthermore, comparing the alloimmune responses elicited in intragraft TLO, spleen, and draining lymph nodes in a rat model of chronic rejection, our group observed increased production of anti-HLA antibodies in TLO as compared with canonical secondary lymphoid organs (35). Not only were the humoral alloimmune responses elicited in TLO quantitatively enhanced but they also displayed a more diverse repertoire, a finding that we confirmed in the clinical setting by the analysis of chronically rejected human kidney allografts (8).
Tertiary lymphoid organs could also contribute to chronic destruction of the graft through antibody-independent functions of B cells. B cells are indeed unique antigen-presenting cells because (i) they have an antigen-specific receptor (B cell receptor), which when engaged by surface-tethered antigens leads to the formation of an immunological synapse that coordinates cell signaling events and promotes antigen uptake for presentation on MHC class II molecules (38), even when the antigen is membrane-tethered or is present in limiting quantities and (ii) B cells have the capacity to clonally expand, thereby becoming the numerically dominant antigen-presenting cells. Interestingly, it has been reported that the presence of B cell clusters within the graft during rejection was associated with reduced graft survival and resistance to steroid therapy, independently of C4d (a breakdown product generated during classical complement pathway activation) deposition or alloantibody detection (39). Some authors have proposed that this could be due to the local presentation of antigen to effector T cells by intragraft B cells (40). This hypothesis is supported by experimental data from the group of Fadi Lakkis, who showed that in a murine skin graft model, TLO perpetuate the rejection process by supporting naïve T cell activation within the graft (41). Strikingly, the same authors also demonstrated that TLO generate T cell memory immune responses (41).
In addition to presenting antigen, B cells can also enhance T cell-mediated immune responses through the secretion of cytokines and chemokines. Studies from the group of Frances Lund (42) have shown that B cells can be functionally subdivided based on their cytokine profile. B cells activated in the presence of TH1-type cytokines (referred to as Be-1 cells) secrete IFNγ and IL-12 but not IL-4, IL-13, or IL-2. By contrast, B cells activated in the presence of TH2-type cytokines (Be-2 cells) secrete IL-2, lymphotoxin, IL-4, and IL-13 but make minimal amounts of IFNγ and IL-12. Both Be-1 and Be-2 cells seem able to produce IL-10, TNFα, and IL-6. The importance of B cell cytokines in promoting T cell responses has been illustrated in several models. For example, in vitro generated effector B cells that produced either TH1- or TH2-type cytokines were shown to promote the activation and differentiation of naïve T cells into effector TH1 and TH2 cells, respectively (43). The importance of B cell cytokines in promoting T cell responses has been confirmed in vivo. In a murine model of Toxoplasma gondii infection, TNF production by B cells was shown to be required for the generation of an optimal TH1 cell protective response (44). In another set of experiments, the generation of a protective TH2 memory response to H. polygyrus was shown to depend on IL-2-producing B cells (45). The exact role of cytokine-producing B cells in enhancing intra-TLO T cell responses remains to be evaluated.
Since grafts in which TLO were harboring germinal center reactions had a shorter life expectancy (Figure 2), we have proposed that lymphoid neogenesis could play a detrimental role during chronic rejection (8). However, the validity of this conclusion is limited by the fact that only explanted grafts have been analyzed, i.e., organs displaying extreme rejection damage that are sometimes (notably in the case of renal grafts) removed after immunosuppressive therapy withdrawal. The definitive demonstration that TLO are involved in the pathophysiology of chronic rejection would require selectively impairing the development of intragraft TLO while leaving the rest of the recipient’s immune system unaffected. Addressing this issue is not trivial because, as discussed above, TLO share many biological pathways with canonical lymphoid tissue, and hence an adequate experimental model is not currently available. Therefore, most of the attempts to validate the data obtained in murine experimental models and in human detransplanted grafts have relied on graft biopsies. The identification of TLO within the grafts before the development of the lesions indeed appears as a prerequisite for confirming the role of lymphoid neogenesis in chronic rejection. This implies a study of protocol biopsies, which has long been introduced as standard follow up in transplantation (46). Unfortunately, the numerous studies aiming at evaluating the correlation between the presence of TLO in protocol biopsies and the later development of chronic rejection have reached conflicting conclusions (Table 1).
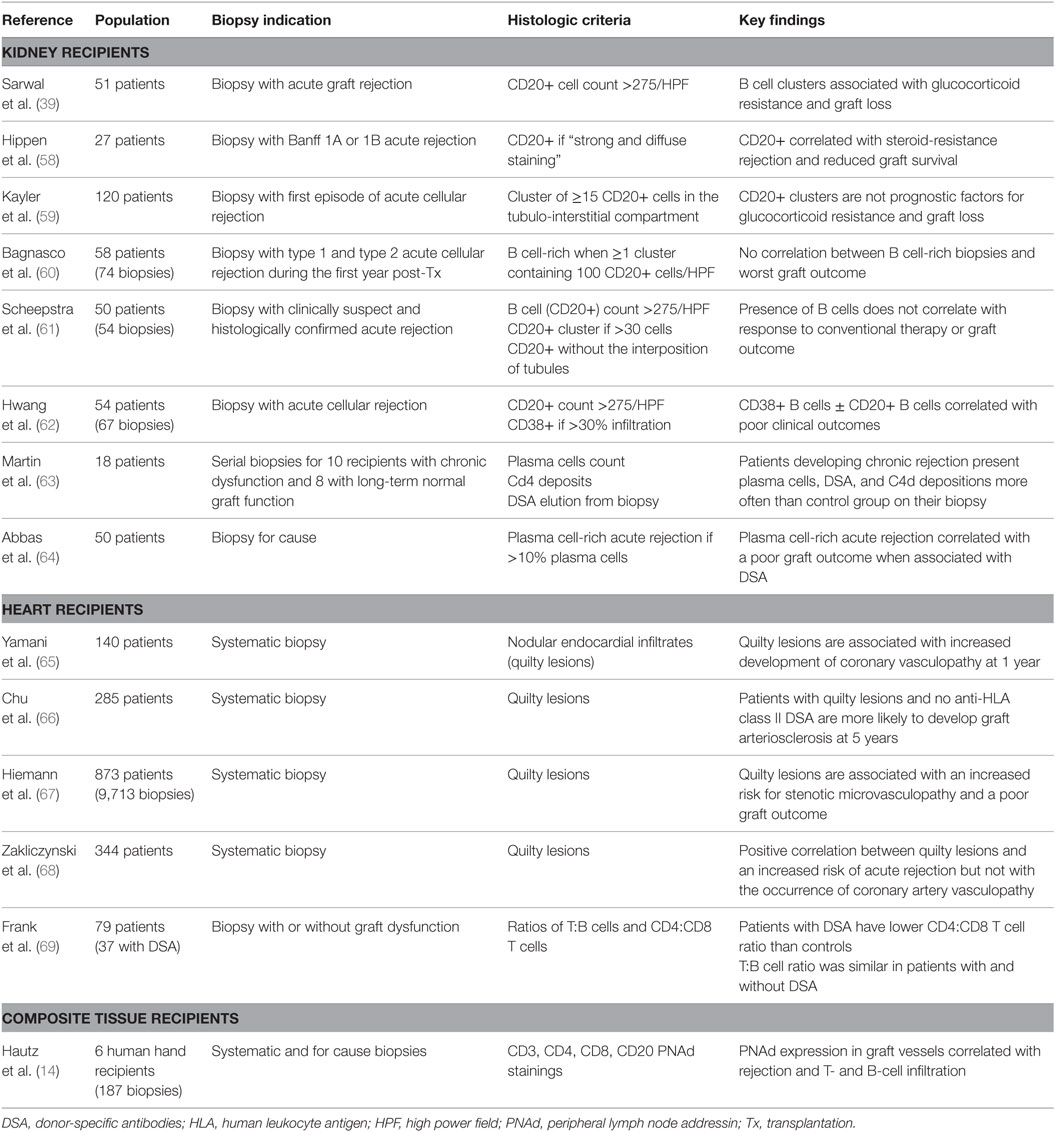
Table 1. Summary of biopsy-based studies evaluating the role of graft-infiltrating B cells.
The absence of an unequivocal deleterious role for B cell clusters has led to the conclusion that these structures could be like “fish in a sunken ship,” i.e., although fish are frequently seen in a sunken boat, they play no role in the process responsible for the shipwreck.
Intragraft TLO: Friends and Foes?
An alternative explanation could reconcile these apparently conflicting results. As discussed above, the proportion of B cells that infiltrate chronically rejected kidney grafts does not correlate with the functionality of intragraft TLO (8). The attraction of B cells within inflamed tissue appears therefore to be a generic phenomenon with no intrinsic deleterious consequences on the graft. However, when intragraft B cells meet the appropriate microenvironment, and upon the complete recapitulation of the lymphoid organogenesis program, B cell nodules organize themselves into functional ectopic germinal centers, which harbor the development of a local aggressive immune response. Because graft biopsies provide only a very limited amount of tissue (which is already an important limitation for evaluation in a patchy process such as lymphoid neogenesis), they do not allow for functional analysis of the ectopic lymphoid organs and are therefore inappropriate for analyzing the role of B cell clusters in rejected grafts.
Another layer of complexity has recently been brought into the picture by experimental evidence that certain B cell subsets are endowed with an immune regulatory role (47). For instance, IL-10-producing B cells have been shown to efficiently prevent the induction of autoimmune disease in several mouse models (48–50). Tolerance in transplantation is defined as the maintenance of graft function in the absence of therapeutic immunosuppression for at least 12 months. About 100 tolerant patients have been identified among renal transplant recipients over the last decade (51). These patients, defined as “operationally tolerant,” are healthy, do not exhibit more infections or malignancies than healthy volunteers, and do not display clinical evidence of immune incompetence (51). When compared with transplanted patients with stable graft function under pharmacologic immunosuppression, operationally tolerant patients exhibited an increase in both absolute number and frequency of total B cells (52). Furthermore, two independent microarray analyses of PBMC revealed a higher expression of B cell-related genes and their associated molecular pathways in tolerant recipients (53, 54). It is therefore conceivable that in certain conditions intragraft B cell infiltrate, instead of being neutral or deleterious, could actually promote graft survival (Figure 2). This theory has been nicely illustrated by murine experimental studies that recently reported the formation of TLO within tolerated allografts (55–57). If such a local protective response can prevent terminal failure of grafts, then not only would such samples having “tolerogenic” TLO be absent from the studies based on the analysis of detransplanted grafts but it could also explain the difficulty of biopsy-based studies to reach an unequivocal conclusion.
Conclusion
Transplanted organ expresses donor-specific alloantigens, which stimulate a recipient’s immune system. Prevention of acute rejection of the graft is achieved using a combination of non-specific immunosuppressive drugs that can only partially block the alloimmune effectors. The residual enduring alloimmune response promotes immune injuries known as chronic rejection, the main cause of late allograft loss. As in other chronic immune diseases, immune effectors within chronically rejected allografts progressively organize into functional TLO that display the same microarchitecture as secondary lymphoid organs, a process known as lymphoid neogenesis. Because biopsy-based studies have reached conflicting conclusions regarding the pathological significance of these TLO, it has been proposed that the presence of TLO in rejected grafts is a non-specific response to local inflammation-induced production of chemokines. While that can indeed sometimes be the case, it should not be excluded that under appropriate conditions, lymphoid neogenesis turns non-functional TLO into ectopic germinal centers, in which a local aggressive humoral immune response can be elicited. Alternatively, functional TLO can also regulate immune responses and slow down the destruction process.
Therefore, we propose that TLO be considered as active players, able to modulate the kinetics of the natural history of chronic rejection. Future works will determine if the versatility of TLO can be manipulated to design innovative therapeutic interventions that would improve graft life expectancy.
Author Contributions
All the authors listed have made substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to thank the Drs. Maud Rabeyrin and Brigitte McGregor, from the Pathology Department of Edouard Herriot Hospital (Lyon University Hospital), for their help in collecting the pathological samples. The authors wish to thank Mathilde Koenig and Servier Medical Art (creativecommons.org/licenses/by/3.0/fr/) for their contribution to the design of the figure.
Funding
OT is supported by the Agence Nationale pour la Recherche (ANR-12-PDOC-0019), the Agence de la Biomédecine (AOR-2013), the Société Françophone de Transplantation, and the Etablissement Français du Sang.
References
1. Leventhal J, Abecassis M, Miller J, Gallon L, Ravindra K, Tollerud DJ, et al. Chimerism and tolerance without GVHD or engraftment syndrome in HLA-mismatched combined kidney and hematopoietic stem cell transplantation. Sci Transl Med (2012) 4:124ra28. doi:10.1126/scitranslmed.3003509
2. Cetrulo CL, Drijkoningen T, Sachs DH. Tolerance induction via mixed chimerism in vascularized composite allotransplantation: is it time for clinical application? Curr Opin Organ Transplant (2015) 20:602–7. doi:10.1097/MOT.0000000000000248
3. Halloran PF. Immunosuppressive drugs for kidney transplantation. N Engl J Med (2004) 351:2715–29. doi:10.1056/NEJMra033540
4. Sellarés J, de Freitas DG, Mengel M, Reeve J, Einecke G, Sis B, et al. Understanding the causes of kidney transplant failure: the dominant role of antibody-mediated rejection and nonadherence. Am J Transplant (2012) 12:388–99. doi:10.1111/j.1600-6143.2011.03840.x
5. Lodhi SA, Lamb KE, Meier-Kriesche HU. Solid organ allograft survival improvement in the United States: the long-term does not mirror the dramatic short-term success. Am J Transplant (2011) 11:1226–35. doi:10.1111/j.1600-6143.2011.03539.x
6. Kerjaschki D, Regele HM, Moosberger I, Nagy-Bojarski K, Watschinger B, Soleiman A, et al. Lymphatic neoangiogenesis in human kidney transplants is associated with immunologically active lymphocytic infiltrates. J Am Soc Nephrol (2004) 15:603–12. doi:10.1097/01.ASN.0000113316.52371.2E
7. Thaunat O, Field A-C, Dai J, Louedec L, Patey N, Bloch M-F, et al. Lymphoid neogenesis in chronic rejection: evidence for a local humoral alloimmune response. Proc Natl Acad Sci U S A (2005) 102:14723–8. doi:10.1073/pnas.0507223102
8. Thaunat O, Patey N, Caligiuri G, Gautreau C, Mamani-Matsuda M, Mekki Y, et al. Chronic rejection triggers the development of an aggressive intragraft immune response through recapitulation of lymphoid organogenesis. J Immunol (2010) 185:717–28. doi:10.4049/jimmunol.0903589
9. Thaunat O, Patey N, Morelon E, Michel J-B, Nicoletti A. Lymphoid neogenesis in chronic rejection: the murderer is in the house. Curr Opin Immunol (2006) 18:576–9. doi:10.1016/j.coi.2006.07.006
10. Thaunat O, Nicoletti A. Lymphoid neogenesis in chronic rejection. Curr Opin Organ Transplant (2008) 13:16–9. doi:10.1097/MOT.0b013e3282f3df15
11. Wehner JR, Fox-Talbot K, Halushka MK, Ellis C, Zachary AA, Baldwin WM. B cells and plasma cells in coronaries of chronically rejected cardiac transplants. Transplantation (2010) 89:1141–8. doi:10.1097/TP.0b013e3181d3f271
12. Sato M, Hirayama S, Hwang DM, Lara-Guerra H, Wagnetz D, Waddell TK, et al. The role of intrapulmonary de novo lymphoid tissue in obliterative bronchiolitis after lung transplantation. J Immunol (2009) 182:7307–16. doi:10.4049/jimmunol.0803606
13. Kanitakis J, Karayannopoulou G, Lanzetta M, Petruzzo P. Graft vasculopathy in the skin of a human hand allograft: implications for diagnosis of rejection of vascularized composite allografts. Transpl Int (2014) 27:e118–23. doi:10.1111/tri.12399
14. Hautz T, Zelger BG, Nasr IW, Mundinger GS, Barth RN, Rodriguez ED, et al. Lymphoid neogenesis in skin of human hand, nonhuman primate, and rat vascularized composite allografts. Transpl Int (2014) 27:966–76. doi:10.1111/tri.12358
15. Thaunat O, Badet L, Dubois V, Kanitakis J, Petruzzo P, Morelon E. Immunopathology of rejection: do the rules of solid organ apply to vascularized composite allotransplantation? Curr Opin Organ Transplant (2015) 20:596–601. doi:10.1097/MOT.0000000000000242
16. Dieu-Nosjean M-C, Giraldo NA, Kaplon H, Germain C, Fridman WH, Sautès-Fridman C. Tertiary lymphoid structures, drivers of the anti-tumor responses in human cancers. Immunol Rev (2016) 271:260–75. doi:10.1111/imr.12405
17. Pitzalis C, Jones GW, Bombardieri M, Jones SA. Ectopic lymphoid-like structures in infection, cancer and autoimmunity. Nat Rev Immunol (2014) 14:447–62. doi:10.1038/nri3700
18. Randall TD, Carragher DM, Rangel-Moreno J. Development of secondary lymphoid organs. Annu Rev Immunol (2008) 26:627–50. doi:10.1146/annurev.immunol.26.021607.090257
19. Van de Pavert SA, Mebius RE. New insights into the development of lymphoid tissues. Nat Rev Immunol (2010) 10:664–74. doi:10.1038/nri2832
20. Guedj K, Khallou-Laschet J, Clement M, Morvan M, Gaston A-T, Fornasa G, et al. M1 macrophages act as LTβR-independent lymphoid tissue inducer cells during atherosclerosis-related lymphoid neogenesis. Cardiovasc Res (2014) 101:434–43. doi:10.1093/cvr/cvt263
21. Lochner M, Ohnmacht C, Presley L, Bruhns P, Si-Tahar M, Sawa S, et al. Microbiota-induced tertiary lymphoid tissues aggravate inflammatory disease in the absence of RORgamma t and LTi cells. J Exp Med (2011) 208:125–34. doi:10.1084/jem.20100052
22. Columba-Cabezas S, Griguoli M, Rosicarelli B, Magliozzi R, Ria F, Serafini B, et al. Suppression of established experimental autoimmune encephalomyelitis and formation of meningeal lymphoid follicles by lymphotoxin beta receptor-Ig fusion protein. J Neuroimmunol (2006) 179:76–86. doi:10.1016/j.jneuroim.2006.06.015
23. Motallebzadeh R, Rehakova S, Conlon TM, Win TS, Callaghan CJ, Goddard M, et al. Blocking lymphotoxin signaling abrogates the development of ectopic lymphoid tissue within cardiac allografts and inhibits effector antibody responses. FASEB J (2012) 26:51–62. doi:10.1096/fj.11-186973
24. Ware CF, Crowe PD, Grayson MH, Androlewicz MJ, Browning JL. Expression of surface lymphotoxin and tumor necrosis factor on activated T, B, and natural killer cells. J Immunol (1992) 149:3881–8.
25. Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell (2001) 104:487–501. doi:10.1016/S0092-8674(01)00237-9
26. Lee Y, Chin RK, Christiansen P, Sun Y, Tumanov AV, Wang J, et al. Recruitment and activation of naive T cells in the islets by lymphotoxin beta receptor-dependent tertiary lymphoid structure. Immunity (2006) 25:499–509. doi:10.1016/j.immuni.2006.06.016
27. Krams SM, Falco DA, Villanueva JC, Rabkin J, Tomlanovich SJ, Vincenti F, et al. Cytokine and T cell receptor gene expression at the site of allograft rejection. Transplantation (1992) 53:151–6. doi:10.1097/00007890-199201000-00031
28. Peters A, Pitcher LA, Sullivan JM, Mitsdoerffer M, Acton SE, Franz B, et al. Th17 cells induce ectopic lymphoid follicles in central nervous system tissue inflammation. Immunity (2011) 35:986–96. doi:10.1016/j.immuni.2011.10.015
29. Fleige H, Ravens S, Moschovakis GL, Bölter J, Willenzon S, Sutter G, et al. IL-17-induced CXCL12 recruits B cells and induces follicle formation in BALT in the absence of differentiated FDCs. J Exp Med (2014) 211:643–51. doi:10.1084/jem.20131737
30. Rangel-Moreno J, Carragher DM, de la Luz Garcia-Hernandez M, Hwang JY, Kusser K, Hartson L, et al. The development of inducible bronchus-associated lymphoid tissue depends on IL-17. Nat Immunol (2011) 12:639–46. doi:10.1038/ni.2053
31. Deteix C, Attuil-Audenis V, Duthey A, Patey N, McGregor B, Dubois V, et al. Intragraft Th17 infiltrate promotes lymphoid neogenesis and hastens clinical chronic rejection. J Immunol (2010) 184:5344–51. doi:10.4049/jimmunol.0902999
32. Jones GW, Bombardieri M, Greenhill CJ, McLeod L, Nerviani A, Rocher-Ros V, et al. Interleukin-27 inhibits ectopic lymphoid-like structure development in early inflammatory arthritis. J Exp Med (2015) 212:1793–802. doi:10.1084/jem.20132307
33. Thaunat O, Graff-Dubois S, Fabien N, Duthey A, Attuil-Audenis V, Nicoletti A, et al. A stepwise breakdown of B-cell tolerance occurs within renal allografts during chronic rejection. Kidney Int (2012) 81:207–19. doi:10.1038/ki.2011.317
34. Thaunat O, Kerjaschki D, Nicoletti A. Is defective lymphatic drainage a trigger for lymphoid neogenesis? Trends Immunol (2006) 27:441–5. doi:10.1016/j.it.2006.08.003
35. Thaunat O, Graff-Dubois S, Brouard S, Gautreau C, Varthaman A, Fabien N, et al. Immune responses elicited in tertiary lymphoid tissues display distinctive features. PLoS One (2010) 5:e11398. doi:10.1371/journal.pone.0011398
36. Dragun D, Müller DN, Bräsen JH, Fritsche L, Nieminen-Kelhä M, Dechend R, et al. Angiotensin II type 1-receptor activating antibodies in renal-allograft rejection. N Engl J Med (2005) 352:558–69. doi:10.1056/NEJMoa035717
37. Sicard A, Chen C-C, Morelon E, Thaunat O. Alloimmune-induced intragraft lymphoid neogenesis promotes B-cell tolerance breakdown that accelerates chronic rejection. Curr Opin Organ Transplant (2016) 21:368–74. doi:10.1097/MOT.0000000000000329
38. Carrasco YR, Batista FD. B cell recognition of membrane-bound antigen: an exquisite way of sensing ligands. Curr Opin Immunol (2006) 18:286–91. doi:10.1016/j.coi.2006.03.013
39. Sarwal M, Chua M-S, Kambham N, Hsieh S-C, Satterwhite T, Masek M, et al. Molecular heterogeneity in acute renal allograft rejection identified by DNA microarray profiling. N Engl J Med (2003) 349:125–38. doi:10.1056/NEJMoa035588
40. Zarkhin V, Kambham N, Li L, Kwok S, Hsieh S-C, Salvatierra O, et al. Characterization of intra-graft B cells during renal allograft rejection. Kidney Int (2008) 74:664–73. doi:10.1038/ki.2008.249
41. Nasr IW, Reel M, Oberbarnscheidt MH, Mounzer RH, Baddoura FK, Ruddle NH, et al. Tertiary lymphoid tissues generate effector and memory T cells that lead to allograft rejection. Am J Transplant (2007) 7:1071–9. doi:10.1111/j.1600-6143.2007.01756.x
42. Lund FE, Randall TD. Effector and regulatory B cells: modulators of CD4+ T cell immunity. Nat Rev Immunol (2010) 10:236–47. doi:10.1038/nri2729
43. Harris DP, Haynes L, Sayles PC, Duso DK, Eaton SM, Lepak NM, et al. Reciprocal regulation of polarized cytokine production by effector B and T cells. Nat Immunol (2000) 1:475–82. doi:10.1038/82717
44. Menard LC, Minns LA, Darche S, Mielcarz DW, Foureau DM, Roos D, et al. B cells amplify IFN-gamma production by T cells via a TNF-alpha-mediated mechanism. J Immunol (2007) 179:4857–66. doi:10.4049/jimmunol.179.7.4857
45. Wojciechowski W, Harris DP, Sprague F, Mousseau B, Makris M, Kusser K, et al. Cytokine-producing effector B cells regulate type 2 immunity to H. polygyrus. Immunity (2009) 30:421–33. doi:10.1016/j.immuni.2009.01.006
46. Thaunat O, Legendre C, Morelon E, Kreis H, Mamzer-Bruneel M-F. To biopsy or not to biopsy? Should we screen the histology of stable renal grafts? Transplantation (2007) 84:671–6. doi:10.1097/01.tp.0000282870.71282.ed
47. Rosser EC, Mauri C. Regulatory B cells: origin, phenotype, and function. Immunity (2015) 42:607–12. doi:10.1016/j.immuni.2015.04.005
48. Fillatreau S, Gray D, Anderton SM. Not always the bad guys: B cells as regulators of autoimmune pathology. Nat Rev Immunol (2008) 8:391–7. doi:10.1038/nri2315
49. Bouaziz J-D, Yanaba K, Tedder TF. Regulatory B cells as inhibitors of immune responses and inflammation. Immunol Rev (2008) 224:201–14. doi:10.1111/j.1600-065X.2008.00661.x
50. Thaunat O, Morelon E, Defrance T. Am“B”valent: anti-CD20 antibodies unravel the dual role of B cells in immunopathogenesis. Blood (2010) 116:515–21. doi:10.1182/blood-2010-01-266668
51. Brouard S, Pallier A, Renaudin K, Foucher Y, Danger R, Devys A, et al. The natural history of clinical operational tolerance after kidney transplantation through twenty-seven cases. Am J Transplant (2012) 12:3296–307. doi:10.1111/j.1600-6143.2012.04249.x
52. Pallier A, Hillion S, Danger R, Giral M, Racapé M, Degauque N, et al. Patients with drug-free long-term graft function display increased numbers of peripheral B cells with a memory and inhibitory phenotype. Kidney Int (2010) 78:503–13. doi:10.1038/ki.2010.162
53. Newell KA, Asare A, Kirk AD, Gisler TD, Bourcier K, Suthanthiran M, et al. Identification of a B cell signature associated with renal transplant tolerance in humans. J Clin Invest (2010) 120:1836–47. doi:10.1172/JCI39933
54. Sagoo P, Perucha E, Sawitzki B, Tomiuk S, Stephens DA, Miqueu P, et al. Development of a cross-platform biomarker signature to detect renal transplant tolerance in humans. J Clin Invest (2010) 120:1848–61. doi:10.1172/JCI39922
55. Brown K, Sacks SH, Wong W. Tertiary lymphoid organs in renal allografts can be associated with donor-specific tolerance rather than rejection. Eur J Immunol (2011) 41:89–96. doi:10.1002/eji.201040759
56. Le Texier L, Thebault P, Lavault A, Usal C, Merieau E, Quillard T, et al. Long-term allograft tolerance is characterized by the accumulation of B cells exhibiting an inhibited profile. Am J Transplant (2011) 11:429–38. doi:10.1111/j.1600-6143.2010.03336.x
57. Li W, Bribriesco AC, Nava RG, Brescia AA, Ibricevic A, Spahn JH, et al. Lung transplant acceptance is facilitated by early events in the graft and is associated with lymphoid neogenesis. Mucosal Immunol (2012) 5:544–54. doi:10.1038/mi.2012.30
58. Hippen BE, DeMattos A, Cook WJ, Kew CE, Gaston RS. Association of CD20+ infiltrates with poorer clinical outcomes in acute cellular rejection of renal allografts. Am J Transplant (2005) 5:2248–52. doi:10.1111/j.1600-6143.2005.01009.x
59. Kayler LK, Lakkis FG, Morgan C, Basu A, Blisard D, Tan HP, et al. Acute cellular rejection with CD20-positive lymphoid clusters in kidney transplant patients following lymphocyte depletion. Am J Transplant (2007) 7:949–54. doi:10.1111/j.1600-6143.2007.01737.x
60. Bagnasco SM, Tsai W, Rahman MH, Kraus ES, Barisoni L, Vega R, et al. CD20-positive infiltrates in renal allograft biopsies with acute cellular rejection are not associated with worse graft survival. Am J Transplant (2007) 7:1968–73. doi:10.1111/j.1600-6143.2007.01885.x
61. Scheepstra C, Bemelman FJ, van der Loos C, Rowshani AT, van Donselaar-Van der Pant KAMI, Idu MM, et al. B cells in cluster or in a scattered pattern do not correlate with clinical outcome of renal allograft rejection. Transplantation (2008) 86:772–8. doi:10.1097/TP.0b013e3181860a74
62. Hwang HS, Song JH, Hyoung BJ, Lee SY, Jeon YJ, Kang SH, et al. Clinical impacts of CD38+ B cells on acute cellular rejection with CD20+ B cells in renal allograft. Transplantation (2010) 89:1489–95. doi:10.1097/TP.0b013e3181dd35b8
63. Martin L, Charon-Barra C, Bocrie O, Guignier F, D’Athis P, Dautin G, et al. Detection of plasma cells, C4d deposits and donor-specific antibodies on sequential graft biopsies of renal transplant recipients with chronic dysfunction. Transpl Immunol (2010) 22:110–4. doi:10.1016/j.trim.2009.11.001
64. Abbas K, Mubarak M, Zafar MN, Aziz T, Abbas H, Muzaffar R, et al. Plasma cell-rich acute rejections in living-related kidney transplantation: a clinicopathological study of 50 cases. Clin Transplant (2015) 29:835–41. doi:10.1111/ctr.12589
65. Yamani MH, Ratliff NB, Starling RC, Tuzcu EM, Yu Y, Cook DJ, et al. Quilty lesions are associated with increased expression of vitronectin receptor (alphavbeta3) and subsequent development of coronary vasculopathy. J Heart Lung Transplant (2003) 22:687–90. doi:10.1016/S1053-2498(02)01181-6
66. Chu KE, Ho EK, de la Torre L, Vasilescu ER, Marboe CC. The relationship of nodular endocardial infiltrates (Quilty lesions) to survival, patient age, anti-HLA antibodies, and coronary artery disease following heart transplantation. Cardiovasc Pathol (2005) 14:219–24. doi:10.1016/j.carpath.2005.03.009
67. Hiemann NE, Knosalla C, Wellnhofer E, Lehmkuhl HB, Hetzer R, Meyer R. Quilty indicates increased risk for microvasculopathy and poor survival after heart transplantation. J Heart Lung Transplant (2008) 27:289–96. doi:10.1016/j.healun.2007.11.573
68. Zakliczynski M, Nozynski J, Konecka-Mrowka D, Pyka L, Trybunia D, Swierad M, et al. Quilty effect correlates with biopsy-proven acute cellular rejection but does not predict transplanted heart coronary artery vasculopathy. J Heart Lung Transplant (2009) 28:255–9. doi:10.1016/j.healun.2008.12.011
Keywords: transplantation, lymphoid neogenesis, tertiary lymphoid organs, chronic rejection, tolerance
Citation: Koenig A and Thaunat O (2016) Lymphoid Neogenesis and Tertiary Lymphoid Organs in Transplanted Organs. Front. Immunol. 7:646. doi: 10.3389/fimmu.2016.00646
Received: 03 October 2016; Accepted: 13 December 2016;
Published: 27 December 2016
Edited by:
Andreas Habenicht, Ludwig Maximilian University of Munich, GermanyReviewed by:
Matthias Lochner, Center for Experimental and Clinical Infection Research, GermanyMegan K. L. MacLeod, University of Glasgow, UK
Copyright: © 2016 Koenig and Thaunat. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Olivier Thaunat, b2xpdmllci50aGF1bmF0QGNodS1seW9uLmZy