 Yarne Klaver
Yarne Klaver Sabine C. L. van Steenbergen1
Sabine C. L. van Steenbergen1 Reno Debets
Reno Debets Cor H. J. Lamers
Cor H. J. Lamers- 1Laboratory of Tumor Immunology, Department of Medical Oncology, Erasmus MC-Cancer Institute, Rotterdam, Netherlands
- 2Department of Medical Oncology, Erasmus MC-Cancer Institute, Rotterdam, Netherlands
Autologous T cells were genetically modified to express a chimeric antigen receptor (CAR) directed toward carboxy-anhydrase-IX (CAIX) and used to treat patients with CAIX-positive metastatic renal cell carcinoma. In this study, we questioned whether the T cell maturation stage in the pre-infusion product affected CAIX CAR expression and function in vitro as well as in vivo CAR T cell numbers and expansion. During the 14 days expansion of CAR T cells prior to administration, we observed shifts from a predominant CD4 to a CD8 T cell phenotype and from a significant fraction of naïve to central effector T cells. Surface expression of the CAR was equally distributed among different T cell subsets and T cell maturation stages. During T cell culture days 14–18 (which covered patient treatment days 1–5), T cells demonstrated a decline in CAR expression level per cell irrespective of T cell maturation stage, although the proportion of CAR-positive T cells and CAR-mediated T cell effector functions remained similar for both CD4 and CD8 T cell populations. Notably, patients with a higher fraction of naïve CD8 T cells at baseline (prior to genetic modification) or central effector CD8 T cells at 2 weeks of CAR T cell culture demonstrated a higher fold expansion and absolute numbers of circulating CAR T cells at 1 month after start of therapy. We conclude that the T cell maturation stage prior to and during CAR T cell expansion culture is related to in vivo CAR T cell expansion.
Introduction
Despite clinical successes in B-cell malignancies, adoptive transfer of T cells genetically modified with chimeric antigen receptors (CARs) or T cell receptors (TCRs) to treat solid tumors is challenged by limited patient responses (1). The efficacy of adoptive T cell therapy (in hematological malignancies) correlates with numbers and persistence of circulating modified T cells (2–5). Building on this notion, several approaches have been explored to improve persistence of genetically modified T cells in vivo. For example, the introduction in receptors of intracellular domains from the CD28 and/or CD137 co-stimulatory molecules has led to an increased CAR T cell persistence as well as expansion in vivo, and consequently in clinical responses (6, 7).
Furthermore, preclinical studies in mice and monkeys suggest that improved in vivo persistence and antitumor responses are obtained when T cells in early stages of differentiation (such as naïve or central memory cells) are used for genetic modification and treatment (8, 9). In fact, the differentiation state of CD8+ T cells appeared to be inversely related to their capacity to proliferate and persist (10, 11). We have previously generated CAR T cells directed against carboxy-anhydrase-IX (CAIX) and treated patients with CAIX-positive metastatic renal cell carcinoma (mRCC) (12). Here, we assessed T cell maturation stage prior to and during CAR T cell expansion cultures, analyzed whether the T cell maturation stage affects CAR expression and function in vitro as well as the in vivo properties of CAR T cells, in particular expansion potential and absolute numbers of circulating CAR T cells.
Materials and Methods
Patients and Treatment
Patients, diagnosed with CAIX-positive mRCC and for whom no standard treatment was available, were included in this phase-I trial. Patients were treated in three cohorts, and aimed to assess toxicity and to establish the maximum tolerated dose of the number of CAR T cells. Treatment schedule was previously presented (13) and was, in brief: in cohort 1, treatment consisted of intravenous administration of 2 × 107 T-cells at day 1, 2 × 108 T-cells at day 2, and 2 × 109 T-cells at days 3–5 (treatment cycle 1) and days 17–19 (treatment cycle 2). Simultaneously, patients received twice daily subcutaneous injections of interleukin-2 (IL-2) at 5 × 105 IU/m2 on days 1–10 and days 17–26. Because of liver toxicity (13), this schedule was changed in cohort 2, to a classic 3 × 3 dose-escalating phase I schedule starting at 1 × 108 CAR T-cells per infusion and extending to 2, 4, 8, 16, 20, 25, and 30 × 108 cells at subsequent dose levels, and applying a maximum of 10 T-cell infusions at days 1–5 and days 29–33 combined with sc IL-2 at 5 × 105 IU/m2 twice daily at days 1–10 and days 29–38. In cohort 3, patients were treated as in cohort 2, but received an extra i.v. infusion of 5 mg of the anti-CAIX mAb G250, 3 days before start of each series of CAR T-cell infusions, in order to block CAIX antigen in the liver and leaving accessible CAIX antigen at RCC tumor sites (14–16).
For the analyses of CAR T cell persistence, only patients treated in cohorts 2 and 3 were assessed until day 29, as from day 29 eight out of nine patients received a second treatment cycle of CAR T cells. Since patients in cohort 1 received varying numbers of CAR T cells, and one patient already received a second cycle of CAR T cells on days 17–19, cohort 1 was not assessed in the persistence analyses.
Patients did not receive lympho-depleting pretreatment. The clinical protocol and amendments were approved by governmental regulatory authorities (Central committee on research involving Human Subjects) as well as the Erasmus MC institutional medical ethical review board. The clinical protocol (DDHK97-29/P00.0040C) adheres to the Declaration of Helsinki protocols. Patient characteristics are detailed elsewhere (13).
T Cell Infusion Product and Post-Treatment Blood Sampling
Patient peripheral blood mononuclear cells (PBMC) from leukapheresis (n = 9) were activated at day 0 with soluble anti-CD3 mAb OKT3 (10 ng/mL) without IL-2. At days 2 and 3, T cells were retrovirally transduced with the CAIX CAR in the presence of 100 IU/ml IL-2. From days 4 to 18, T cells were expanded in medium supplemented with 360 IU/ml IL-2. Patients were treated with five daily infusions of “fresh” CAIX CAR T cells harvested from culture at days 14–18 (13, 17). We obtained blood samples at regular intervals before, during, and after treatment for direct flow cytometric (FCM) analysis and isolation and cryopreservation of PBMC in liquid nitrogen (18, 19).
Flow Cytometry
Carboxy-anhydrase-IX CAR-positive T cells in cultures and blood samples were assessed by FCM using the anti-CAIX CAR idiotype mAb NuH82, as described in Ref. (18) and Figure S1 in Supplementary Material. The starting T cell product (PBMC from leukapheresis) and T cell cultures were analyzed for various lymphocyte subsets, a.o. CD4+ and CD8+ T cells and T cell maturation subsets using following markers: CD27, CD28, CD45RA, CD45RO, CD62L, and CCR7. The starting T cell product (PBMC from leukapheresis) is referred to the “baseline” sample or measurement in the rest of this study. In addition, T cell maturation subsets were analyzed for CAR-expression (Figure S1 in Supplementary Material) and CAR-mediated effector function in response to CAIX+ RCC cell line (SKRC-17 MW1-clone4) by means of upregulated expression of CD107. Information on antibodies and staining combinations used in multi-color FCM is specified in Table S1 in Supplementary Material. Samples were measured on the FACS Canto, and analyzed with FCS express v. 4.07 software (De Novo software). Gating strategy to determine the T cell maturation stage is demonstrated in Figures S1A,B in Supplementary Material.
Statistical Analysis
Statistical analyses were performed using SPSS software (version 21) for Windows (IBM Corporation, IL, USA). Graphpad Prism v5.0 was used to prepare graphs.
Results
T Cell Phenotype
Patient PBMC at baseline (from leukapheresis) were activated, transduced with the CAIX CAR, and expanded. During the 14 days of expansion, the phenotype of the T cells shifted significantly. Although the extent varied per patient, T cell cultures reproducibly demonstrated a shift from a CD4+ to a CD8+ predominance compared to baseline (Figure 1A; Figure S2 in Supplementary Material). During the 18 days culture period, individual markers expressed on CD3+ T cells demonstrated a clear shift, especially during the first 14 days (Figure S3 in Supplementary Material). When assessing CD8 T cell maturation according to the markers CD45RA, CD45RO, CD27, and CD28, we observed that during culture the most prevalent subset drastically shifted from TES at baseline to TEM at day 14 [Figure 1B; see legend of Figure 1 for the definition of maturation stages: TN (naïve), TINT (intermediate between TN and TCM), TCM (central memory), TEM (effector memory), TCE (central effector), and TES (end stage) T cells]. Following T cell culture, there was also a decrease in the fraction of TN and an increase in the fraction of TCE, yet the overall change was in favor of younger T cells (TCM + TEM) at the expense of further maturated T cells (TCE + TES). CD4+ T cells also showed a culture-dependent decrease in the fraction of TN and an increase in the fraction of TEM, yet the most prevalent subset at baseline, i.e., TCM, remained unchanged (Figure 1C). Interestingly, in contrast to CD8+ T cells cultured CD4+ T cells harbored almost no TCE cells. Additional analysis with different T cell maturation markers [CD45RA, CCR7 (CD197), and CD62L] showed high concordance with the maturation stages and kinetics as described above (Figure S4 in Supplementary Material).
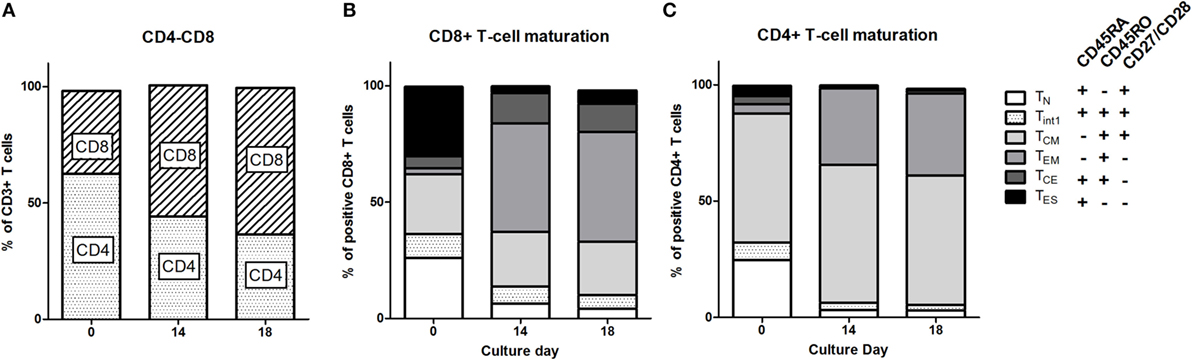
Figure 1. T cell phenotype and maturation during clinical T cell expansion cultures. (A) Proportions of CD4 and CD8 T cells at baseline (leukapheresis, day 0) and culture day 14. Data of individual patients are presented in Figure S1 in Supplementary Material. Maturation stages of CD8+ (B) and CD4+ (C) T cells at baseline and culture day 14 and at day 18 defined according to the expression of CD45RA, CD45RO, and CD27/CD28 as indicated in insert: Naïve, TN: CD45RA+, CD45RO−, CD27/CD28+; Central Memory, TCM: CD45RA−, CD45RO+, CD27/28+; Effector Memory, TEM: CD45RA−, CD45RO+, or CD27/CD28−; Central Effector, TCE: CD45RA+, CD45RO+, CD27/CD28−; and End Stage TES: CD45RA+, CD45RO−, CD27/CD28− T cells. A small fraction was CD45RA+, CD45RO+, and CD27/CD28+ and this population was defined as interphase (Int1) T cells. Data are presented as stacked bars of means of nine patients.
CAIX CAR Expression in T Cell Maturation Subsets
At culture day 14, the proportion of CAIX CAR-expressing cells was about equal for various T cell subpopulations, such as CD3 positive T cells in combination with CD4, CD8, CD56, CD57, TCRγδ, CD45RA, CD45RO, CD62L, or CCR7 (Figure S5 in Supplementary Material). The proportion of CAIX CAR-expressing cells within CD8+ and CD4+ T cells was stable during patient treatment (culture days 14–18), and appeared highest in the TINT+ TCM and lowest in the TES maturation stages (Figures 2A,B). When considering the CAR expression level per cell (mean fluorescence intensity, MFI), we observed a significant decrease of CAR expression between days 14 and 18 for almost all maturation stages with exception of CD8+ TN, CD8+ TES cells, and CD4+ TES cells (Figures 2C,D). This observation is in extension to a previous report on a general loss of CAR expression during the last 5 days of the T cell culture (20). Data at MFI level also reinforced the above observation that CAR expression was highest in the TINT + TCM and lowest in the TES maturation stages (Figures 2C,D). Further, we analyzed the CAIX CAR-mediated function (degranulation) and found no significant differences in CD107-upregulation between the different maturation stages or T cell culture times following co-culture of CAR T cells with a CAIX-positive RCC cell line or CAIX-negative K562 cells. Thus, the relatively small decrease in CAR expression did not result in a measurable decrease in CAR-mediated function (Figures 3A,B).
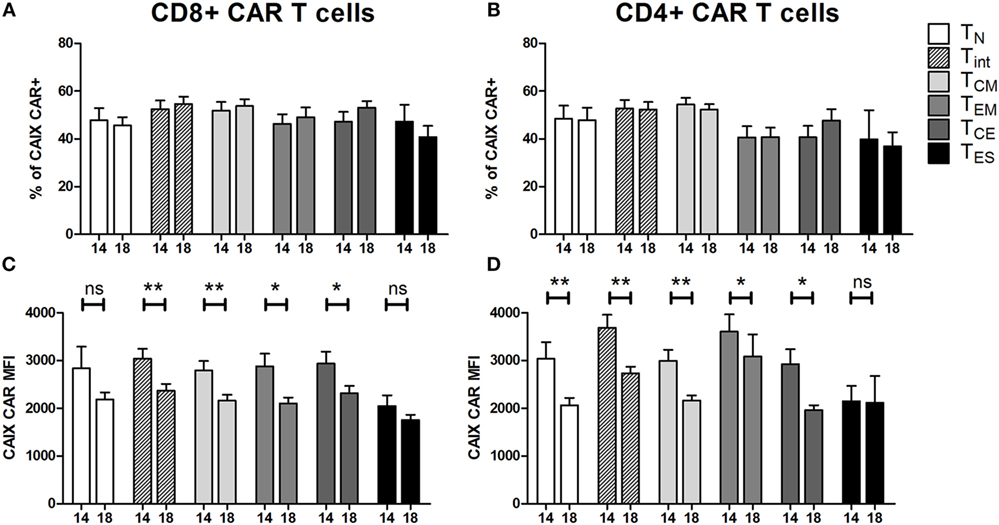
Figure 2. Carboxy-anhydrase-IX (CAIX) chimeric antigen receptor (CAR) expression of T cells in infusion products according to T cell maturation. Proportions of CAIX CAR-positive CD8+ (A) and CD4+ (B) T cells and CAIX CAR expression levels (expressed as mean fluorescence intensity) on CD8+ (C) and CD4+ (D) T cells at culture days 14 and 18 according to T cell maturation. Differences between culture days 14 and 18 with respect to paired continuous parameters were determined using an exact Wilcoxon rank-sum test. *p < 0.05; **p < 0.01; ns, not significant. Bars represent mean ± SEM from 16 independent clinical CAIX CAR T cell cultures for the treatment of nine patients, of which seven received infusions in two treatment cycles. See also legend to Figure 1.
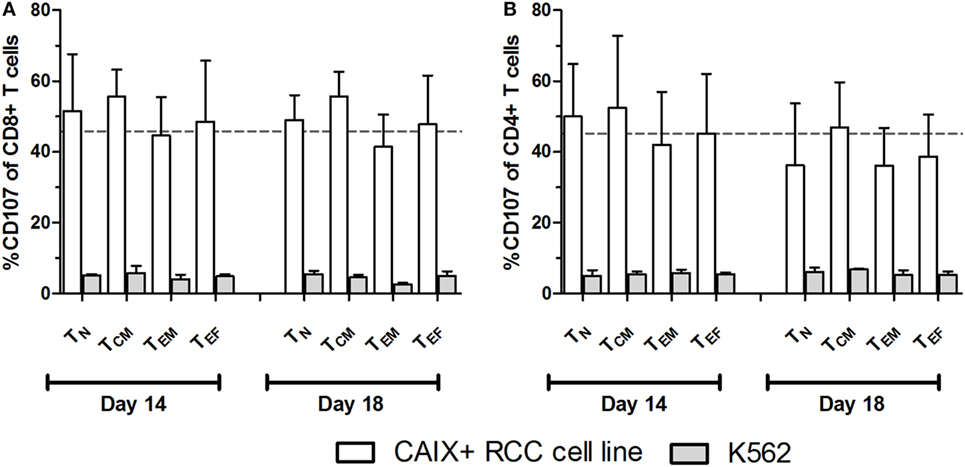
Figure 3. Carboxy-anhydrase-IX (CAIX) chimeric antigen receptor (CAR)-mediated function by T cell maturation subsets. CAIX CAR T cell cultures at days 14 and 18 were assayed for CAIX CAR-mediated cytolytic (degranulation) potential. CAIX CAR T cells were co-cultured (2 h) with the CAIX-positive RCC cell line (SKRC-17 MW1-clone4) or the CAIX-negative cell line K562 and subsequently analyzed by flow cytometry. Results are presented as proportions of CD107 positivity within differently matured CD8+ (A) and CD4+ (B) T cells. Dotted line represents the average proportion of CD107 positivity of CD8+ T cells after co-culture with the CAIX-positive RCC cell line. T cell maturations subsets were defined by the markers CD45RA and CCR7 as follows: TN: CD45RA+, CCR7+; TCM: CD45RA−, CCR7+; TEM: CD45RA−, CCR7−; TEF: CD45RA+, CCR7−. Bars represent mean ± SEM (n = 3).
T Cell Maturation Correlates with In Vivo CAR T Cell Expansion
We analyzed whether phenotype of T cells at baseline and after culture correlated with numbers of CAR T cells in patient blood and expansion (fold increase) of these CAR T cells during 5 days after the last infusion. Kinetics of circulating CAR T cell numbers irrespective of maturation stage in patients have been reported elsewhere (20). Here, we reveal a significant correlation between the fold increase in CAR T cell numbers in patients and the proportions of CD8+ TN cells at baseline (Figure 4A) and CD8+ TCE in the infusion product at culture day 14 (Figure 4B) and on day 18 [r = 0.683, p = 0.042 (data not shown)]. Patients with higher proportions of CD8+ TN cells in the baseline PBMC and CD8+ TCE in the infusion product, in general, had higher absolute numbers of circulating CAR T cells up to 29 days after the first infusion (Figures 4C,D). Our in vitro analyses considered both CD8+ and CD4+ T cells (Figures 1–3). Correlations between the relative occurrence of T cell maturation stages (prior to and during T cell processing) and numbers of T cells in post-treatment patient blood samples were assessed for both CD8+ and CD4+ T cells. Significant correlations, however, were found only for CD8+ T cells but not CD4+ T cells (data not shown). We found no correlations between proportion of CAR-positive T cells or CAR expression (MFI) in the infusion product and CAR T cell numbers or expansion in patients (data not shown).
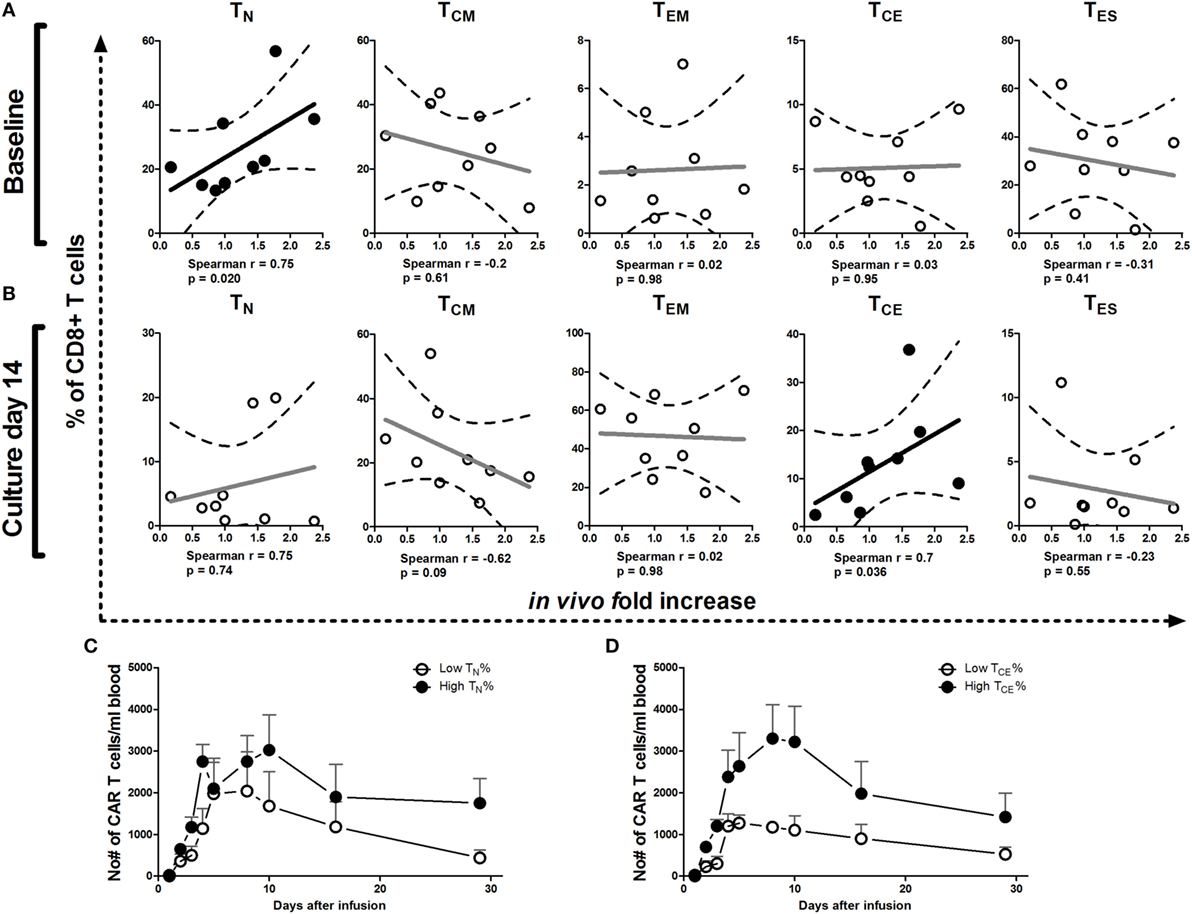
Figure 4. Correlations between T cell maturation (pre-infusion) and increase of chimeric antigen receptor (CAR) T cell numbers in patients. Correlation plots of proportions of TN, TCM, TEM, TCE, and TES cells at baseline (A) and culture day 14 (B) vs. increase of CAR T cell numbers in vivo during the first 5 days after the last T cell infusion of treatment cycle 1 (13, 20). Dotted lines represent the 95% confidence bands of the best fitted line. The Spearman correlation coefficient method was used to assess linear association. p-Values less than 0.05 were considered significant. Patients were divided into above or below median values (high and low, respectively) of TN at baseline (day 0) (C) or TCE cells at culture day 14 (D) and plotted for absolute numbers of circulating CAR T cells during treatment cycle 1 (days 1–29). See also legend to Figure 1 for further details.
Discussion
Here, we document T cell phenotypic changes during IL-2-supported CAIX CAR T cell cultures in preparation of a clinical trial to treat RCC patients. During the 2-week expansion period, T cells skewed from a CD4 to CD8 phenotype and the proportion of naïve CAR T cells (TN: CD45RA+, CD45RO−, CD27/CD28+) strongly declined, while the proportions of TEM (CD45RA−, CD45RO+, CD27/CD28−) and TCE (CD45RA+, CD45RO+, CD27/CD28−) cells significantly increased. We noted a shift from TN cells to more maturated stages as was described before for other transduced T cell products cultured with IL-2 (21). Interestingly, also the frequency of TES declined during the CAR T cell culture. We anticipate that the less maturated cells like TCM and TEM have proliferated faster, and thereby have overgrown the TES cells. We demonstrated a small decline in CAR expression level per cell during the 5-day CAR T cell culture covering the days of T cell administration (days 14–18) that was independent of T cell maturation stage and did not affect CAR-mediated function. We conclude that the five sequential and “freshly” prepared clinical preparations of CAIX CAR T cells had about equal phenotypic and functional properties.
In adoptive CAR T cell treatment, circulating numbers, persistence and in vivo expansion potential of infused CAR T cells are currently the only parameters revealed to be associated with improved clinical outcome (22). Most CAR T cell trials targeting CD19 in hematological malignancies show strong T cell expansion, mainly due to a high load and accessibility of target antigen, the nature of tumor cells (B cells being able to provide co-stimulation), and highly active (second generation) CAR T cells. Along these lines, we sought to define pre-infusion characteristics that may predict the in vivo behavior of infused CAR T cells. Here, we showed that patients with more CD8+ TN cells in baseline PBMC (from leukapheresis) or more CD8+ TCE in the infusion product at culture day 14, showed a higher fold increase in numbers of CAR T cells in vivo, resulting in higher blood levels of CAR T cells in these patients for at least 4 weeks. In fact, a correlation exists between characteristics of both baseline PBMC (proportion CD8+ TN) and infusion product (proportion CD8+ TCE) and numbers of circulation CAR T cells after treatment. Such findings bear clinical relevance, as younger T cells were shown to positively correlate with clinical effectiveness in adoptive T cell trials (1, 23, 24). To the best of our knowledge, our study is the first CAR T cell trial targeting a solid tumor, in which a correlation is demonstrated between pre-infusion and pre-expansion T cell characteristics and in vivo CAR T cell expansion potential. In only one other non-CD19 CAR trial, targeting neuroblastoma with GD2 CAR T cells, a correlation was described between in vivo CAR T cell persistence and the proportion of CD4+ or TCM cells in the infusion product (24). Our observation is in line with the report that effector T cells derived from naive rather than memory T cell subsets possess superior features for adoptive immunotherapy, though no correlations were made with in vivo parameters (25).
The observation that the proportion CD8+ TN in baseline PBMC determines the in vivo behavior of CAR T cells is intriguing and provides a means to manipulate leukapheresis products for a better clinical outcome, especially for patients with low proportions of TN cells in PBMC. This could include enrichment for TN cells before transduction or changing the culturing conditions by addition of cytokines such as IL-15 and IL-21 during expansion (21, 26). These data may provide clues to adapt the in vitro T cell processing toward optimal T cell fitness and may enable to develop an improved protocol for adoptive T cell therapy.
Ethics Statement
The manuscript contains clinical study data. The clinical protocol and amendments were approved by governmental regulatory authorities (Central committee on research involving Human Subjects) as well as the Erasmus MC institutional medical ethical review board. The clinical protocol (DDHK97-29/P00.0040C) adheres to the declaration of Helsinki protocols.
Author Contributions
YK contributed in conception and design of the study, the analysis, planned and directed the statistical analysis, was involved in interpretation of data, and drafted the manuscript. SCS was involved in acquisition of the data and assisted in drafting the manuscript. SS contributed to conception and design of the study, and assisted in revising of the manuscript. RD contributed in conception and design of the study, interpretation of data, and drafting and revising of the manuscript. CL contributed in conception and design of the study, analysis and interpretation of data, and assisted in drafting and revising the manuscript. All the authors read, critiqued, and approved the final manuscript. All the authors also agreed to be accountable for all aspects of the work.
Conflict of Interest Statement
The authors declare that research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Pascal van Elzakker, Brigitte van Krimpen, and Corrien Groot-van Ruyven for their technical assistance.
Funding
This work was funded in part by the Dutch Cancer Foundation (grant DDHK99-1865), the European Commission grants QLK3-1999-01262, FP7 Health F4-2012-305863 (ATTACK2), FP7 ATTRACT, and the Cancer Research Institute, New York, NY, USA (clinical investigation grant “Immuno-gene therapy of metastatic renal cell cancer patients”).
Supplementary Material
The Supplementary Material for this article can be found online at http://journal.frontiersin.org/article/10.3389/fimmu.2016.00648/full#supplementary-material.
Abbreviations
CAIX, carboxy-anhydrase-IX; CAR, chimeric antigen receptor; CD, cluster of differentiation; mRCC, metastatic renal cell carcinoma; PBMC, peripheral blood mononuclear cells; IL-2, interleukin-2; FCM, flow cytometry; TN, naïve T cells; TINT, intermediate T cells; TCM, central memory T cells; TEM, effector memory T cells; TCE, central effector T cells; TES, end stage T cells.
References
1. Klaver Y, Kunert A, Sleijfer S, Debets R, Lamers CH. Adoptive T-cell therapy: a need for standard immune monitoring. Immunotherapy (2015) 7(5):513–33. doi: 10.2217/imt.15.23
2. Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med (2013) 5(177):177ra38. doi:10.1126/scitranslmed.3005930
3. Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med (2013) 368(16):1509–18. doi:10.1056/NEJMoa1215134
4. Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med (2011) 3(95):95ra73. doi:10.1126/scitranslmed.3002842
5. Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med (2011) 365(8):725–33. doi:10.1056/NEJMoa1103849
6. Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci U S A (2009) 106(9):3360–5. doi:10.1073/pnas.0813101106
7. Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther (2009) 17(8):1453–64. doi:10.1038/mt.2009.83
8. Klebanoff CA, Gattinoni L, Torabi-Parizi P, Kerstann K, Cardones AR, Finkelstein SE, et al. Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc Natl Acad Sci U S A (2005) 102(27):9571–6. doi:10.1073/pnas.0503726102
9. Berger C, Jensen MC, Lansdorp PM, Gough M, Elliott C, Riddell SR. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J Clin Invest (2008) 118(1):294–305. doi:10.1172/JCI32103
10. Gattinoni L, Klebanoff CA, Palmer DC, Wrzesinski C, Kerstann K, Yu Z, et al. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J Clin Invest (2005) 115(6):1616–26. doi:10.1172/JCI24480
11. Cieri N, Camisa B, Cocchiarella F, Forcato M, Oliveira G, Provasi E, et al. IL-7 and IL-15 instruct the generation of human memory stem T cells from naive precursors. Blood (2013) 121(4):573–84. doi:10.1182/blood-2012-05-431718
12. Lamers CH, Klaver Y, Gratama JW, Sleijfer S, Debets R. Treatment of metastatic renal cell carcinoma (mRCC) with CAIX CAR-engineered T-cells – a completed study overview. Biochem Soc Trans (2016) 44(3):951–9. doi:10.1042/BST20160037
13. Lamers CH, Sleijfer S, van Steenbergen S, van Elzakker P, van Krimpen B, Groot C, et al. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity. Mol Ther (2013) 21(4):904–12. doi:10.1038/mt.2013.17
14. Steffens MG, Boerman OC, Oyen WJ, Kniest PH, Witjes JA, Oosterhof GO, et al. Intratumoral distribution of two consecutive injections of chimeric antibody G250 in primary renal cell carcinoma: implications for fractionated dose radioimmunotherapy. Cancer Res (1999) 59(7):1615–9.
15. Steffens MG, Boerman OC, Oosterwijk-Wakka JC, Oosterhof GO, Witjes JA, Koenders EB, et al. Targeting of renal cell carcinoma with iodine-131-labeled chimeric monoclonal antibody G250. J Clin Oncol (1997) 15(4):1529–37. doi:10.1200/jco.1997.15.4.1529
16. Steffens MG, Boerman OC, de Mulder PH, Oyen WJ, Buijs WC, Witjes JA, et al. Phase I radioimmunotherapy of metastatic renal cell carcinoma with 131I-labeled chimeric monoclonal antibody G250. Clin Cancer Res (1999) 5(10 Suppl):3268s–74s.
17. Lamers CH, van Elzakker P, Langeveld SC, Sleijfer S, Gratama JW. Process validation and clinical evaluation of a protocol to generate gene-modified T lymphocytes for imunogene therapy for metastatic renal cell carcinoma: GMP-controlled transduction and expansion of patient’s T lymphocytes using a carboxy anhydrase IX-specific scFv transgene. Cytotherapy (2006) 8(6):542–53. doi:10.1080/14653240601056396
18. Lamers CH, Gratama JW, Pouw NM, Langeveld SC, Krimpen BA, Kraan J, et al. Parallel detection of transduced T lymphocytes after immunogene therapy of renal cell cancer by flow cytometry and real-time polymerase chain reaction: implications for loss of transgene expression. Hum Gene Ther (2005) 16(12):1452–62. doi:10.1089/hum.2005.16.1452
19. Lamers CH, Langeveld SC, Groot-van Ruijven CM, Debets R, Sleijfer S, Gratama JW. Gene-modified T cells for adoptive immunotherapy of renal cell cancer maintain transgene-specific immune functions in vivo. Cancer Immunol Immunother (2007) 56(12):1875–83. doi:10.1007/s00262-007-0330-3
20. Klaver Y, van Steenbergen SC, Sleijfer S, Debets R, Lamers CH. Plasma IFN-gamma and IL-6 levels correlate with peripheral T-cell numbers but not toxicity in RCC patients treated with CAR T-cells. Clin Immunol (2016) 169:107–13. doi:10.1016/j.clim.2016.06.014
21. Lamers CH, van Steenbergen-Langeveld S, van Brakel M, Groot-van Ruijven CM, van Elzakker PM, van Krimpen B, et al. T cell receptor-engineered T cells to treat solid tumors: T cell processing toward optimal T cell fitness. Hum Gene Ther Methods (2014) 25(6):345–57. doi:10.1089/hgtb.2014.051
22. Porter DL, Hwang WT, Frey NV, Lacey SF, Shaw PA, Loren AW, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med (2015) 7(303):303ra139. doi:10.1126/scitranslmed.aac5415
23. Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, et al. Durable complete responses in heavily pretreted patients with metastatic melanoma using T cell transfer immunotherapy. Clin Cancer Res (2011) 17(13):4550–7. doi:10.1158/1078-0432.CCR-11-0116
24. Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood (2011) 118(23):6050–6. doi:10.1182/blood-2011-05-354449
25. Wu F, Zhang W, Shao H, Bo H, Shen H, Li J, et al. Human effector T cells derived from central memory cells rather than CD8(+)T cells modified by tumor-specific TCR gene transfer possess superior traits for adoptive immunotherapy. Cancer Lett (2013) 339(2):195–207. doi:10.1016/j.canlet.2013.06.009
Keywords: renal cell cancer, chimeric antigen receptor, carboxy-anhydrase-IX, T cell, immune monitoring, T cell maturation, T cell expansion, T cell persistence
Citation: Klaver Y, van Steenbergen SCL, Sleijfer S, Debets R and Lamers CHJ (2016) T Cell Maturation Stage Prior to and During GMP Processing Informs on CAR T Cell Expansion in Patients. Front. Immunol. 7:648. doi: 10.3389/fimmu.2016.00648
Received: 11 October 2016; Accepted: 13 December 2016;
Published: 26 December 2016
Edited by:
Giuseppe Sconocchia, National Research Council (Cnr), ItalyReviewed by:
Giulio Cesare Spagnoli, University of Basel, SwitzerlandJan Joseph Melenhorst, University of Pennsylvania, USA
Copyright: © 2016 Klaver, van Steenbergen, Sleijfer, Debets and Lamers. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cor H. J. Lamers, Yy5sYW1lcnNAZXJhc211c21jLm5s