 Marie Wehenkel1
Marie Wehenkel1 Maripat Corr2
Maripat Corr2 Clifford S. Guy1
Clifford S. Guy1 Benjamin A. Edwards1Ashley H. Castellaw1Christopher Calabrese3
Benjamin A. Edwards1Ashley H. Castellaw1Christopher Calabrese3 Gilles Pagès4
Gilles Pagès4 Jacques Pouysségur4,5Peter Vogel3
Jacques Pouysségur4,5Peter Vogel3 Maureen A. McGargill1*
Maureen A. McGargill1*
- 1Department of Immunology, St. Jude Children’s Research Hospital, Memphis, TN, USA
- 2Division of Rheumatology, Allergy, and Immunology, University of California San Diego, La Jolla, CA, USA
- 3Department of Veterinary Pathology, St. Jude Children’s Research Hospital, Memphis, TN, USA
- 4Institute for Research of Cancer and Aging (IRCAN), University of Nice Sophia-Antipolis, Nice, France
- 5Centre Scientifique de Monaco (CSM), Monaco, France
Defects in cartilage homeostasis can give rise to various skeletal disorders including osteochondromas. Osteochondromas are benign bone tumors caused by excess accumulation of chondrocytes, the main cell type of cartilage. The extracellular signal-regulated kinase (ERK) pathway is a major signaling node that functions within chondrocytes to regulate their growth and differentiation. However, it is not known whether the ERK pathway in other cell types regulates cartilage homeostasis. We show here that mice with a germline deficiency of Erk1 and a conditional deletion of Erk2 in cells that express CD4, or expressed CD4 at one point in development, unexpectedly developed bone deformities. The bone lesions were due to neoplastic outgrowths of chondrocytes and disordered growth plates, similar to tumors observed in the human disease, osteochondromatosis. Chondrocyte accumulation was not due to deletion of Erk2 in the T cells. Rather, CD4cre was expressed in cell types other than T cells, including a small fraction of chondrocytes. Surprisingly, the removal of T cells accelerated osteochondroma formation and enhanced disease severity. These data show for the first time that T cells impact the growth of osteochondromas and describe a novel model to study cartilage homeostasis and osteochondroma formation.
Introduction
The differentiation and proliferation of cartilage cells is a highly ordered and tightly regulated process that is essential for bone growth. Defects in this process can cause a variety of disorders including osteochondromas, which are the most common type of benign bone tumor. These tumors are caused by excess accumulation of chondrocytes that erupt from the growth plate and are associated with significant pain, restriction of movement, impingement of blood vessels and nerves, and severe deformities. The most severe complication is malignant transformation into a secondary chondrosarcoma. Osteochondromas typically develop during childhood and adolescence as single tumors, although multiple tumors can develop in the heritable disease, multiple hereditary exostoses, also known as osteochondromatosis (1). The molecular and cellular mechanisms that maintain cartilage homeostasis and prevent osteochondroma formation are incompletely understood.
The extracellular signal-regulated kinase (ERK) pathway is transiently activated in response to extracellular cues in almost every type of cell. ERK is downstream of several growth factor receptors and is critical for cell-cycle progression, differentiation, lineage specification, and cell survival (2). Contrary to its role in most cell types, ERK activation decreases chondrocyte proliferation (3–5). In fact, sustained ERK signaling was associated with mutations that led to reduced chondrocyte proliferation and achondroplasia, a form of dwarfism (3, 6). On the other hand, inactivation of ERK signaling in chondrocytes led to an expansion of hypertrophic chondrocytes and enhanced growth of cartilaginous skeletal elements (5). These data suggest that ERK signaling within chondrocytes suppresses proliferation. In addition, deletion of Erk1 and Erk2 in osteo-chondroprogenitor cells blocked osteoblast differentiation and caused an accumulation of hypertrophic chondrocytes, indicating that ERK signaling is also important in lineage commitment and differentiation of chondrocytes (7).
While there is an intrinsic role for ERK signaling in the proliferation and differentiation of chondrocytes, it is not known whether this pathway is also important in other cells types to regulate cartilage homeostasis. We generated mice with a germline deletion of Erk1 and a conditional deletion of Erk2 mediated by CD4cre [Erk1−/−.Erk2fl/fl.CD4cre+, double knockout (DKOCD4)] (8, 9). CD4cre is expressed in all T cell subsets during development in the thymus, and therefore deletes genes in CD4+ T cells, CD8+ T cells, and NKT cells. Unexpectedly, we found that the DKOCD4 mice developed bone deformities due to hyperplastic outgrowths of epiphyseal chondrocytes. The bone lesions lacked mononuclear infiltrate and resembled the human disease, osteochondromatosis. Interestingly, removing the T cells by breeding the DKOCD4 mice to Rag1−/− mice did not prevent osteochondroma formation. In fact, tumor incidence and severity was enhanced in DKOCD4 mice lacking T cells, indicating that T cells play a role in regulating chondrocyte accumulation. While T cells are known to contribute to the destruction of cartilage in rheumatoid arthritis and other inflammatory bone disorders, thus far, a role for immune cells in the growth of cartilage or osteochondromas has not been described.
Materials and Methods
Mice
Erk1−/−.Erk2fl/fl.CD4cre+, double knockout (DKOCD4) mice were provided by Stephen Hedrick. 129.DKOCD4 mice were backcrossed to the 129 background for 6 generations and B6.DKOCD4 mice were backcrossed to the C57BL/6 strain for 12 generations. The ROSA.LSL.TdTomato (tdTom) reporter mice on the C57BL/6 background were provided by Dario Vignali and crossed to the N6 129.DKOCD4 mice. The offsprings were then intercrossed. The ROSA.LSL.YFP reporter mice on the C57BL/6 background were obtained from Jackson Laboratories and crossed to the N6 129.DKOCD4 mice. N6 129.DKOCD4 mice were crossed to C57BL/6 Rag1−/− mice (Jackson Laboratories), and the offsprings were intercrossed. Littermate controls (LMC) were used for all experiments. All mice were housed under specific pathogen-free conditions at St. Jude Children’s Research Hospital. Animal studies met the approval of the Animal Ethics Committee.
Incidence and Disease Severity Scoring
Mice of the indicated genotypes were monitored for disease incidence on a weekly basis. The symptoms tracked included swollen or deformed bones, abnormal gait, hunched posture, and dyspnea. Two consecutive weeks of a symptom being displayed, continuing through the remainder of the experimental observation period, was required to indicate disease incidence. Incidence was then denoted as the age at the first symptomatic observation. Disease severity was determined at the humane or experimental endpoint by addition of the number of symptomatic features, which could be detected visually, along with an additional value given for severe symptoms.
In Vivo CT
Mice were imaged using a 2,048 mm × 3,072 mm field of view (FOV) with whole body coverage achieved using automatic stitching of two bed positions (20% overlap). Projections were acquired at 80 kVp and 500 µA (165 ms exposure) over a rotation of 195° (390 steps) providing an isotropic resolution of 107 µm. Data were processed using Inveon Research Workplace (IRW) software for visualization. Isosurface (ISO) rendering is a raycast method where surfaces of similar density objects are rendered and the remaining materials are hidden. Maximum intensity projection (MIP) is a method where the color (in this case, white balance of grayscale) of the densest object along each raycast path is used to determine the final color of each pixel.
Ex Vivo CT
μCT micrographs of selected bones were generated using a specimen μCT scanner (LocusSP Specimen CT, GE Healthcare) at 14 µm isotropic voxel size, with 500 projections, 4 averages, an integration time of 3,000 ms, photon energy of 80 keV, and a current of 80 µA. Data were processed using MicroView software for visualization and bone morphometry quantification. Bone lengths were determined using manual electronic calipers built into the MicroView software system. For bone morphometry analysis, volumes of interest (VOIs) were generated covering 2.1 mm (proximal and distal femur and distal humerus) or 1.4 mm (proximal humerus). In-plane cortical bone thickness was manually determined at two independent points within the slice and three slices representative of proximal, medial, and distal components of the VOI. For cortical bone processing, images were automatically thresholded to select for cortical bone, segmented to this threshold, and cortical bone thickness across the VOI calculated. For bone volume and trabecular analyses, cortical bone was blanked from the VOI and whole volumes evaluated.
Histology
Bones were fixed in 10% neutral buffered formalin overnight and then decalcified in TBD-2 (Thermo Fisher Scientific, Waltham, MA, USA) for three days. The tissues were processed routinely, embedded in paraffin, sectioned at 4 µm, and stained with hematoxylin and eosin, Safranin O, or Alcian blue. Bone sections were examined by an experienced veterinary pathologist.
Pathology
Full necropsies were performed on selected animals [B6.LMC (n = 11), B6.DKOCD4 (n = 3); 129.LMC (n = 8), 129.DKOCD4 (n = 6)]. In addition to bones, the following tissues were examined: heart, skeletal muscle, tongue, trachea, lung, thyroid, liver, kidney, adrenal gland, salivary gland, lymph node, white adipose, brown adipose, aorta, thymus, spleen, pancreas, stomach, duodenum, jejunum, ileum, cecum, colon, urinary bladder, skin, brain, eye, nose, teeth, ear, bone, bone marrow, and either testis, epididymis, prostate gland, seminal vesicle, and vas deferens, or uterus and ovary.
In Vivo Radiography
Animals were anesthetized using avertin, and data were acquired at 50 µm2 in-plane resolution with 26 kV energy and ~12 s exposure time. Prone position data were acquired at shelf position 2 [beam coverage diameter (BC) 27.7 cm, source to object distance (SOD) (28.6 cm)], and lateral position data were acquired at shelf position 4 (BC = 10.4 cm, SOD = 14.3).
Chondrocyte Isolation and Analysis
Five- to seven-day-old mice were harvested individually for isolation and culture of chondrocytes as previously described (10). In addition, osteochondromas were identified in mice greater than 20 weeks of age by planar X-ray and microdissected. PCR was performed as previously described (8). Cells were lysed for western blotting using NP-40 lysis buffer with protease and phosphatase inhibitor cocktails (Calbiochem), and protein concentration was determined by BCA assay (Thermo Scientific). Equal protein concentrations (40 µg) were then denatured and separated by 10% polyacrylamide SDS gel electrophoresis (BioRad). After transfer to PVDF membrane, antibodies to ERK1/2 (Zymed 61-7400), COL2A1 (Santa Cruz M2139), and β-actin (Sigma AC-74) were used for analysis.
Immunofluorescence
Mice were euthanized via CO2 asphyxiation and perfused with 4% paraformaldehyde (Electron Microscopy Sciences) and the hindlimbs were bulk demuscled. Hindlimbs (femurs, tibiae, and fibulae) were fixed in 4% paraformaldehyde overnight at 4°C prior to decalcification with 0.5 M EDTA for an additional 24 h at 4°C. Samples were cryoprotected with 20% sucrose in PBS prior to embedding in tissue freezing medium (Electron Microscopy Sciences). Twenty micrometer cryosections were air dried for 1 h at room temperature prior to incubation with AF488-labeled wheat germ agglutinin (WGA) (Life Technologies; 2 µg/ml) for 1 h at room temperature. Slides were washed in TBS prior to mounting with hardset media containing DAPI (Vector Laboratories).
Widefield fluorescent images were acquired using a Nikon TiE microscope equipped with a Lambda LS lightsource (Sutter Instruments), 10 × 0.45 NA or 20 × 0.75 NA Plan Apo objectives, DU-897 EMCCD camera (Andor Technology), and standard DAPI/FITC/TRITC/Cy5 filter sets. Image analysis was performed using NiS Elements AR software (Nikon).
Confocal microscopy was performed using an AxioObserverZ.1 inverted microscope (Zeiss) equipped with a CSU-22 spinning disk (Yokagawa), 405, 473, 561, and 660 nm laser lines, and Evolve EMCCD camera (Photometrics). Images were acquired and analyzed using Slidebook software (Intelligent Imaging Innovations).
Flow Cytometry
Tissues were harvested and processed as described previously (11). After incubation with anti-FcReceptor, cells were stained with CD44-FITC, CD8α-PerCP (eBioscience), CD69-APC-Cy7, CD19-BV510, and CD4-BV711 (BioLegend) for 20 min on ice. For intracellular staining of FoxP3-PE (eBioscience), cells were fixed and permeabilized with Mouse Foxp3 Buffer set (BD Biosciences) after surface staining, according to the manufacturer’s instructions. Samples were run on a BD LSRFortessa analyzer and the results evaluated in FlowJo. Data were plotted in GraphPad Prism for statistical analysis.
Results
DKOCD4 Mice Develop Deformed Bones in Multiple Locations
Mice with a germline deletion of Erk1 were bred to mice with a conditional deletion of Erk2 mediated by CD4cre [Erk1−/−.Erk2fl/fl.CD4cre+, double knockout (DKOCD4)] (8, 9). CD4cre is expressed in developing T cells, and therefore deletes genes in all T cell subsets. Due to the requirement of ERK signaling in T cell development, DKOCD4 mice have a significant reduction in the number of T cells in the spleen and lymph nodes (Figures S1A,B in Supplementary Material) (8, 9). The residual T cells in the DKOCD4 mice exhibited an activated phenotype, likely due to the lymphopenic environment and homeostatic proliferation. While there was a reduction in the total number of regulatory T cells, a higher proportion of the CD4+ T cells expressed Foxp3 in the DKOCD4 mice compared to wild-type mice (Figure S1C in Supplementary Material), which is consistent with the fact that ERK2 signaling suppresses Foxp3 expression (12). Surprisingly, the DKOCD4 mice developed noticeable bone deformities between 10 and 20 weeks of age (Figure 1A). Approximately 40% of the DKOCD4 mice displayed grossly visible signs of disease by 28 weeks of age (Figure 1B). Importantly, neither Erk1−/− nor Erk2CD4 single knockout mice developed any signs of disease. Furthermore, Erk1−/− mice bred to mice with a conditional deletion of Erk2 mediated by CD11c-cre (DKOCD11c) did not develop disease (Figure 1B). As there was no bone phenotype in single knockout or wild-type mice, we used these mice interchangeably as littermate controls (LMC) in the remaining experiments.
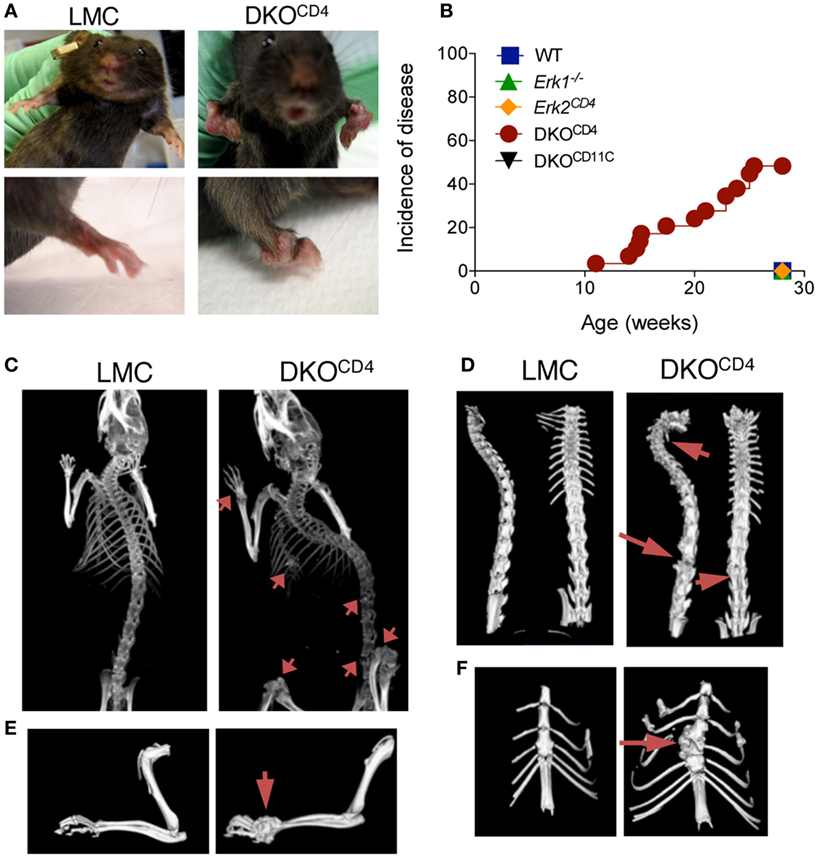
Figure 1. Joint deformities develop in DKOCD4 mice. (A) Anatomical deformities in the joints of a DKOCD4 mouse and littermate control (LMC) are pictured. (B) Mice were monitored weekly for swollen or deformed bones, abnormal gait, hunched posture, and dyspnea. The incidence of one of these symptoms is plotted for 29 DKOCD4 mice and at least 11 mice in the other groups. Data were analyzed with a Log-rank (Mantel–Cox) test. *P < 0.05 for DKOCD4 mice versus all other groups. (C) X-ray computed tomography (CT) in vivo images of LMC and DKOCD4 mice. Arrows indicate lesions in the radius, ulna, tibia, femur, vertebra, and sternum. (D) Isosurface rendering of ex vivo CT images of the vertebra, (E) forelimb, and (F) sternum. These images are representative of at least six mice per group.
X-ray computed tomography (CT) revealed pronounced areas of abnormal bone growth in various locations (Figure 1C), including the spinal column (Figure 1D), the radius and ulna (Figure 1E), and the sternum (Figure 1F). Virtual slicing through these deformities demonstrated that the lesions were contiguous with the bone and thus, likely arose from within it (Figure S2 in Supplementary Material).
Bone Lesions Are Composed of Chondrocytes
Histological examination of the DKOCD4 mice revealed nodular, disorganized accumulation of cartilage protruding from the bones (Figures 2A–E). The cartilage protrusions were continuous with the epiphyseal growth plate, and the intact perichondrium was visible in multiple lesions, suggesting that the cells that make up these protrusions are epiphyseal chondrocytes. Safranin O and Alcian blue staining confirmed that these lesions were chondrocytic, and showed the absence of non-cartilaginous cells within the lesion (Figure S3 in Supplementary Material). The presence of lesions in the radius, ulna, and sphenoid bone indicates these protrusions did not originate from the annulus fibrosis, which is absent from these locations. Notably, these nodules lacked mononuclear infiltrates, which was surprising as Erk2 deletion is mediated by CD4cre, which is primarily expressed in T cells. Overall, this pathology resembles the human disease, osteochondromatosis.
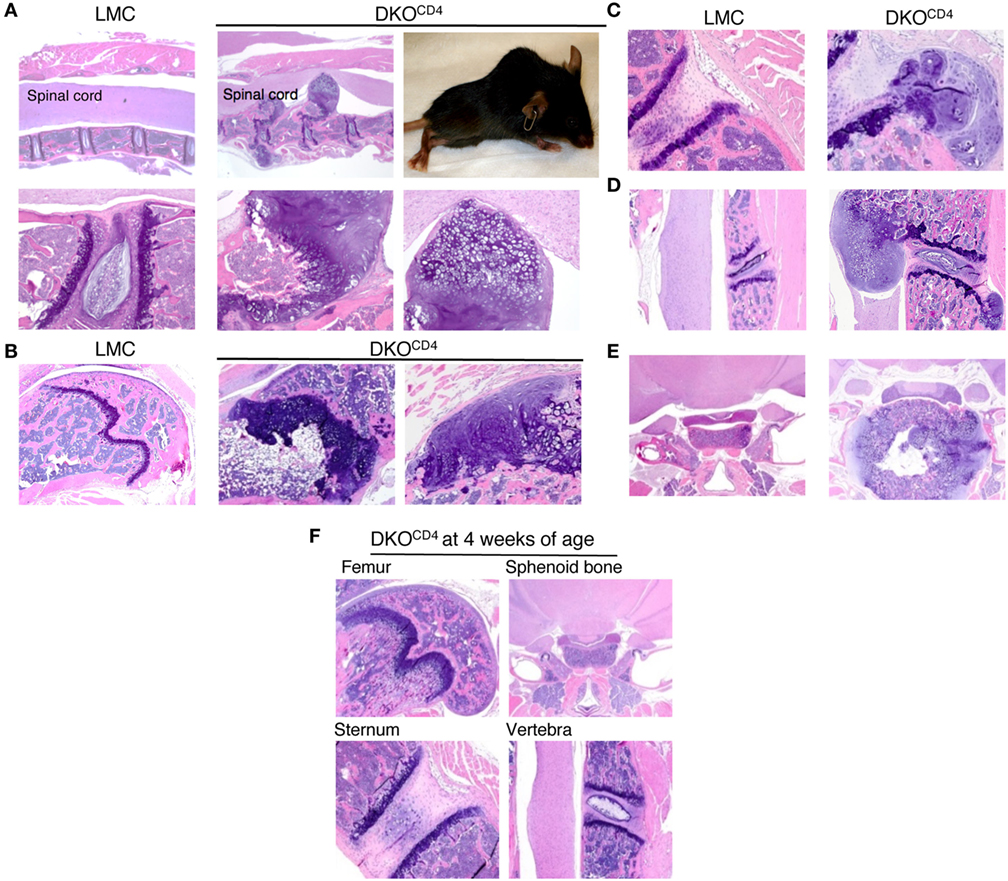
Figure 2. DKOCD4 mice exhibit ectopic accumulation of chondrocytes. Bones were removed from LMC and 129.DKOCD4 mice, fixed, decalcified, and stained with hematoxylin and eosin (H&E). (A) Sections of spinal column [2× (top) and 10× (bottom)], including a photograph of the paralyzed DKOCD4 mouse from which they were obtained, are shown. Other sections displayed include the (B) femur [4× and 10× (far right)], (C) sternum (10×), (D) vertebra (4×), and (E) sphenoid bone (2×). (F) H&E analysis of bones is depicted from 4-week-old 129.DKOCD4 mice, showing femur (4×), sphenoid bone (2×), sternum (10×), and vertebra (4×). Images are representative of at least five mice.
Histological analysis of a cohort of DKOCD4 mice showed that 70% of the DKOCD4 mice had at least one osteochondroma, which was significantly higher than the 40% incidence determined by physical examination (Figure 1B; Table 1). As in humans, the bones affected by the tumors varied between DKOCD4 mice and had the potential to cause severe morbidity. For example, lesions in the vertebrae caused paralysis by compressing the spinal cord (Figure 2A).
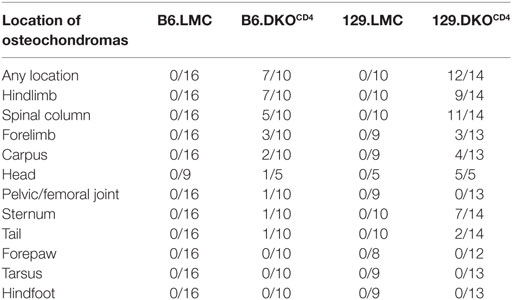
Table 1. Histopathological incidence of bone lesions at 23–44 weeks of age.
There were no obvious abnormalities in the growth plates or chondrocytes of 4-week-old DKOCD4 mice (Figure 2F), indicating that excessive chondrocyte accumulation does not occur early in DKOCD4 mice, unlike other models of osteochondromatosis where ERK signaling is disrupted within the chondrocytes (5, 7, 13). Together, these data indicate that germline deletion of Erk1 along with CD4cre-mediated deletion of Erk2 causes excessive accumulation of chondrocytes, resulting in osteochondromas and disorganized growth plates.
DKOCD4 Mice Do Not Display Overt Signs of Inflammation
Crossing the DKOCD4 mice onto the 129 background for two generations significantly exacerbated disease, resulting in 73% incidence by 28 weeks of age as determined by manual examination (Figure 3A), and 86% when analyzed by histology (Table 1). Since 129 mice typically exhibit more inflammation in various disease models compared to B6 mice (14–16), we examined all tissues from the B6.DKOCD4 and 129.DKOCD4 mice for evidence of mononuclear infiltrate or inflammation. Despite a thorough analysis, there was no indication of overt inflammation on either background. In addition, there was not an increase in anti-nuclear (ANA) antibodies, double-stranded DNA (dsDNA) autoantibodies, or inflammatory cytokines in the serum from 129.DKOCD4 mice compared to LMC (data not shown). These data indicate that there was no apparent inflammation in the 129.DKOCD4 mice that is typically associated with T cell-mediated autoimmunity.
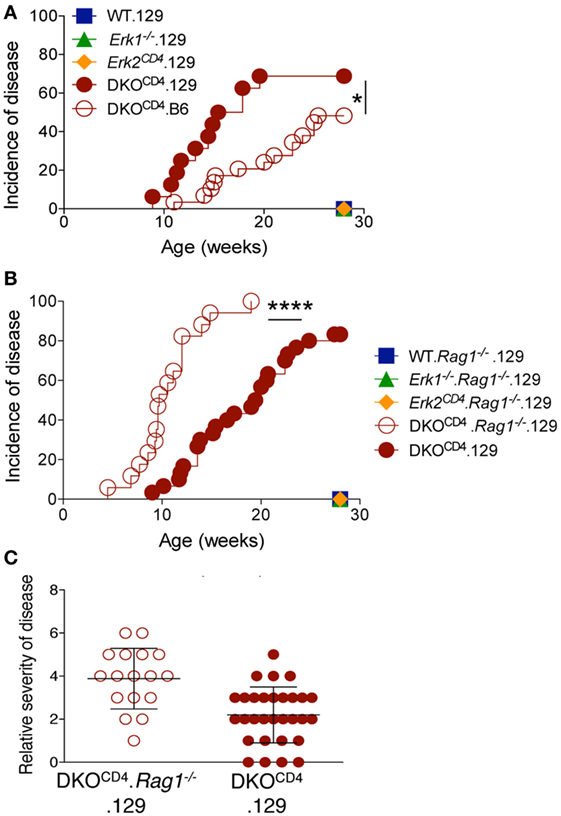
Figure 3. Osteochondroma formation is not due to deletion of Erk2 in T cells. (A) DKOCD4 mice were crossed to the 129 background and monitored as in Figure 1B. The incidence of symptoms is plotted for 16 129.DKOCD4 mice, 29 B6.DKOCD4 mice, and at least seven mice in the other groups. Data were analyzed with a Log-rank (Mantel–Cox) test (*P < 0.05). (B) 129.DKOCD4 mice were bred to Rag1−/− mice to create DKOCD4. Rag1−/− mice, which were monitored for disease. The incidence of symptoms is plotted for 30 129.DKOCD4 and 17 DKOCD4. Rag1−/− mice. Data were analyzed with a Log-rank (Mantel–Cox) test (*P < 0.0001). (C) Relative disease severity scores of the mice depicted in Figure 3B.
Osteochondroma Formation Is Not Due to Deletion of Erk2 in T Cells
Since the majority of cells that express CD4 are T cells, we tested whether deletion of Erk2 in the T cells induced osteochondromas by removing the T cells in DKOCD4 mice. DKOCD4 mice were bred to Rag1−/− mice to create DKOCD4. Rag1−/− mice, which lack T cells. Surprisingly, the DKOCD4. Rag1−/− mice not only developed osteochondromas, but these mice developed tumors faster, and with increased severity compared to DKOCD4 mice (Figures 3B,C). These data indicate that the osteochondromas are not due to deletion of Erk2 in T cells, and additionally that T cells may suppress the formation of osteochondromas. Furthermore, deletion of Erk2 in a cell type other than T cells induces osteochondroma formation. Subsets of myeloid cells and innate lymphoid cells also express CD4, and it is possible that deletion of Erk2 in these cells may result in dysregulation of growth factors, which causes excessive chondrocyte accumulation.
Majority of Chondrocytes Express ERK2 in DKOCD4 Mice
Contrary to other cell types, ERK activation in chondrocytes is associated with decreased proliferation and differentiation (3, 17). Although chondrocytes in DKOCD4 mice are Erk1-deficient, ERK2 expression in these cells is sufficient to regulate chondrocyte development, as indicated by the absence of osteochondromas in Erk1−/− mice. Likewise, other groups found that mice with single deletions of Erk1 or Erk2 specifically in chondrocytes did not display skeletal defects (5, 7). While chondrocytes are not known to express the CD4 protein, it was possible that CD4cre deleted Erk2 in chondrocytes, rendering these cells deficient in both Erk1 and Erk2. Therefore, we investigated Erk2 expression in chondrocytes isolated from wild-type, Erk1−/−.Erk2fl/fl.CD4cre− (E1), and DKOCD4 mice. Analysis of chondrocyte DNA revealed that the Erk2 gene was floxed in E1 and DKOCD4 chondrocytes; however, it was not excised by Cre recombinase as it was in CD8+ and CD4+ thymocytes from DKOCD4 mice (Figure 4A). Additionally, western blot analysis demonstrated that the ERK2 protein was expressed at similar levels in all chondrocytes examined (Figure 4B).
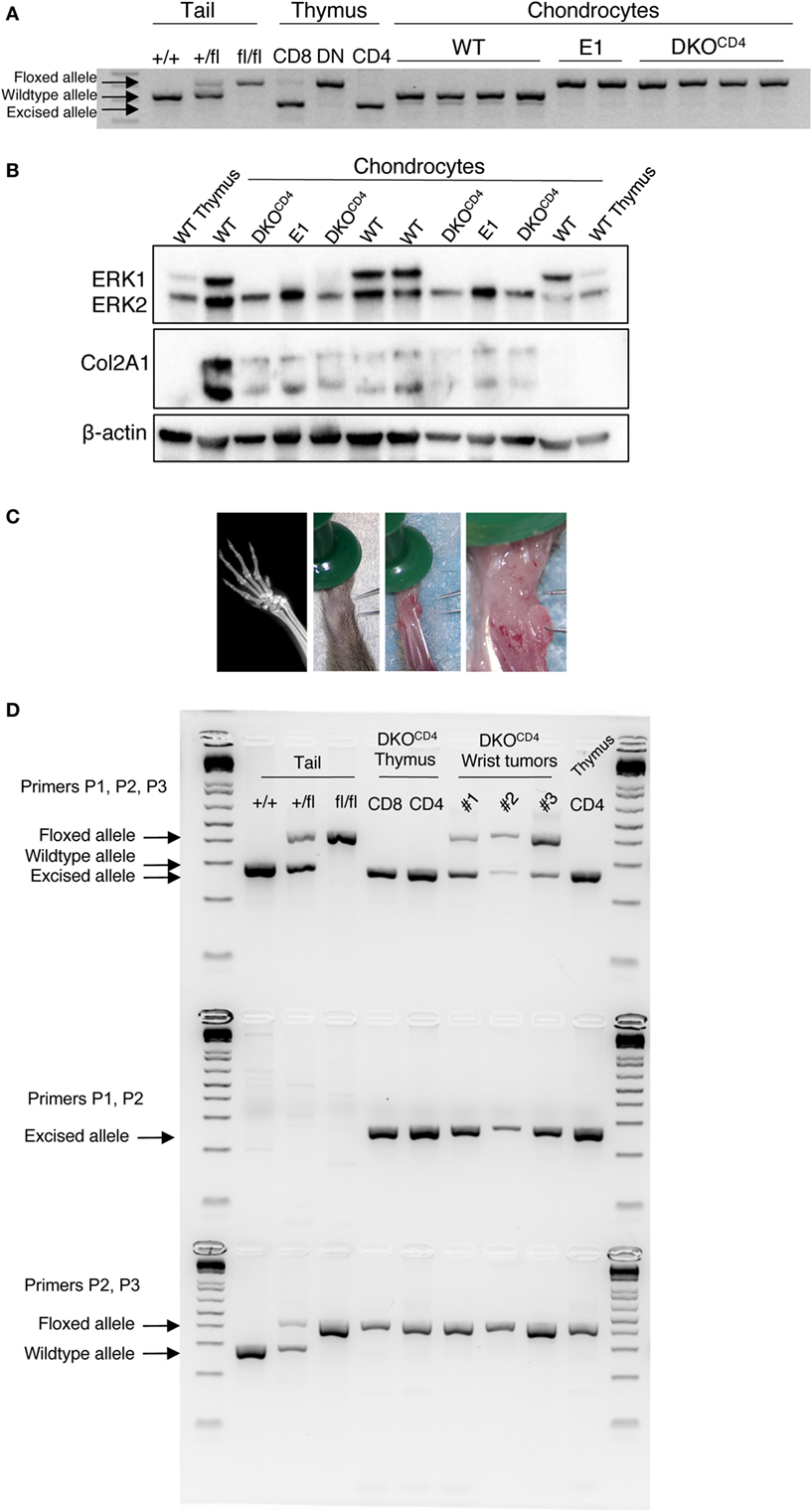
Figure 4. ERK2 is expressed in the majority of chondrocytes in DKOCD4 mice. (A) Chondrocytes were isolated from individual WT, Erk1−/−.Erk2fl/fl.CD4cre− (E1), or DKOCD4 mice and cultured in vitro. DNA was isolated and analyzed by PCR for the presence of the wild-type, floxed, or excised Erk2 allele. Tail DNA is shown as controls for the wild-type and floxed alleles, while DNA from thymocytes serves as the positive control for the excised allele. (B) Cultured chondrocytes were lysed and analyzed by western blot for ERK1, ERK2, type II collagen (Col2A1), and beta-actin. (C) Planar X-ray (left) and surgical microscopy images of a representative lesion from a DKOCD4 mouse. (D) Three microdissected tumors from individual mice greater than 20 weeks of age were analyzed by PCR to determine whether the Erk2 gene was wild-type, floxed, or excised by Cre recombinase. DNA from DKOCD4 thymocytes is shown as a positive control for Erk2 excision, while tail DNA is a negative control for excision. The first row contains primers that amplify the wild-type, floxed, or excised alleles, the second row is primers that only amplify the excised allele. The third row contains primers that amplify the wild-type or floxed alleles.
Further evidence that Erk2 is not deleted in the majority of chondrocytes in DKOCD4 mice is provided by the lack of characteristic bone deformities or alterations in bone length typical of mice that have reduced ERK signaling specifically in the chondrocytes (3, 5, 7). Others demonstrated that mice deficient in Erk1 and Erk2 specifically in the chondrocytes died at birth with severe bone deformities (7). In addition, deletion of Erk1 and Erk2 in mesenchymal cells (PrxCre) of the head and limb caused ectopic cartilage development and severe limb deformity at birth (7). Thus, if CD4cre deleted Erk2 in chondrocytes or osteo-chondroprogenitors, we would expect to see elongated bones, severely deformed bones, or disorganized growth plates in DKOCD4 mice beginning at a very young age. However, DKOCD4 mice did not have any differences in bone length, trabecular density or thickness, average cortical bone thickness, or the cortical area fraction compared to LMC (Figure S4 in Supplementary Material). Moreover, the growth plates of DKOCD4 mice at 4 weeks of age were indistinguishable from LMC (Figure 2F).
CD4cre Deletes Genes in Cells Other Than T Cells
To test whether osteochondromas were due to clonal expansion of rare chondrocytes that deleted the Erk2 allele, we analyzed chondrocytes from microdissected tumors by PCR that distinguishes the Erk2 wild-type, floxed, and excised alleles (Figure 4C). These tumors were dissected from mice greater than 20 weeks of age. We found that both the floxed and excised Erk2 alleles were detected in DNA from the chondrocytes in the tumors, suggesting that some of the chondrocytes deleted Erk2 (Figure 4D). Deletion of Ext1 or ptpn11 (SHP2) in only a proportion of chondrocytes can cause osteochondromas, enchondromas, or exostoses to form (18, 13, 19), indicating that deletion of genes in only a fraction of chondrocytes can cause abnormal accumulation. Thus, it is possible that the presence of a few chondrocytes that delete Erk2 is sufficient to cause abnormal chondrocyte accumulation and osteochondromas.
While we detected the excised Erk2 allele in DNA from osteochondromas, the PCR does not indicate what proportion of chondrocytes deleted the Erk2 allele. Therefore, we used Cre reporter mice to investigate what proportion of chondrocytes deleted Erk2. DKOCD4 mice were bred to ROSA.LSL.tdTomato (tdTom) or ROSA.LSL.YFP reporter mice that express a loxP-flanked stop cassette followed by the fluorescent protein. Cells that express CD4cre at any point in development excise the stop cassette and express tdTomato or YFP for the lifespan of that cell. Western blot analysis demonstrated that the tdTomato+ or YFP+ reporter splenocytes lacked ERK2 protein, while the tdTomato-negative or YFP-negative cells from these mice retained expression of ERK2 protein, indicating that the fluorescent reporters are effective indicators of Erk2 deletion (Figure S5 in Supplementary Material). Sections of hindlimbs of 5-week-old Erk1+/+Erk2+/+.tdTomfl/fl.CD4cre+ (WTCD4.tdTom) and Erk1−/−Erk2fl/fl.tdTomfl/fl.CD4cre+ (DKOCD4.tdTom) mice were analyzed via microscopy by staining with WGA to visualize chondrocytes, and DAPI to identify nuclei. Composite widefield sections showed tdTomato+ cells in the bones of both WTCD4.tdTom and DKOCD4.tdTom mice (Figures 5A,B). While the majority of the chondrocytes did not express the tdTomato reporter, there were tdTomato+ cells in the bone marrow and in the area of the chondrocytes in the growth plate. Confocal microscopy also showed that the majority of chondrocytes in the growth plates were not tdTomato+; however, a small proportion did express the reporter (Figure 5C). These data suggest that a minor fraction of chondrocytes may be Erk2-deficient, which may contribute to osteochondroma formation.
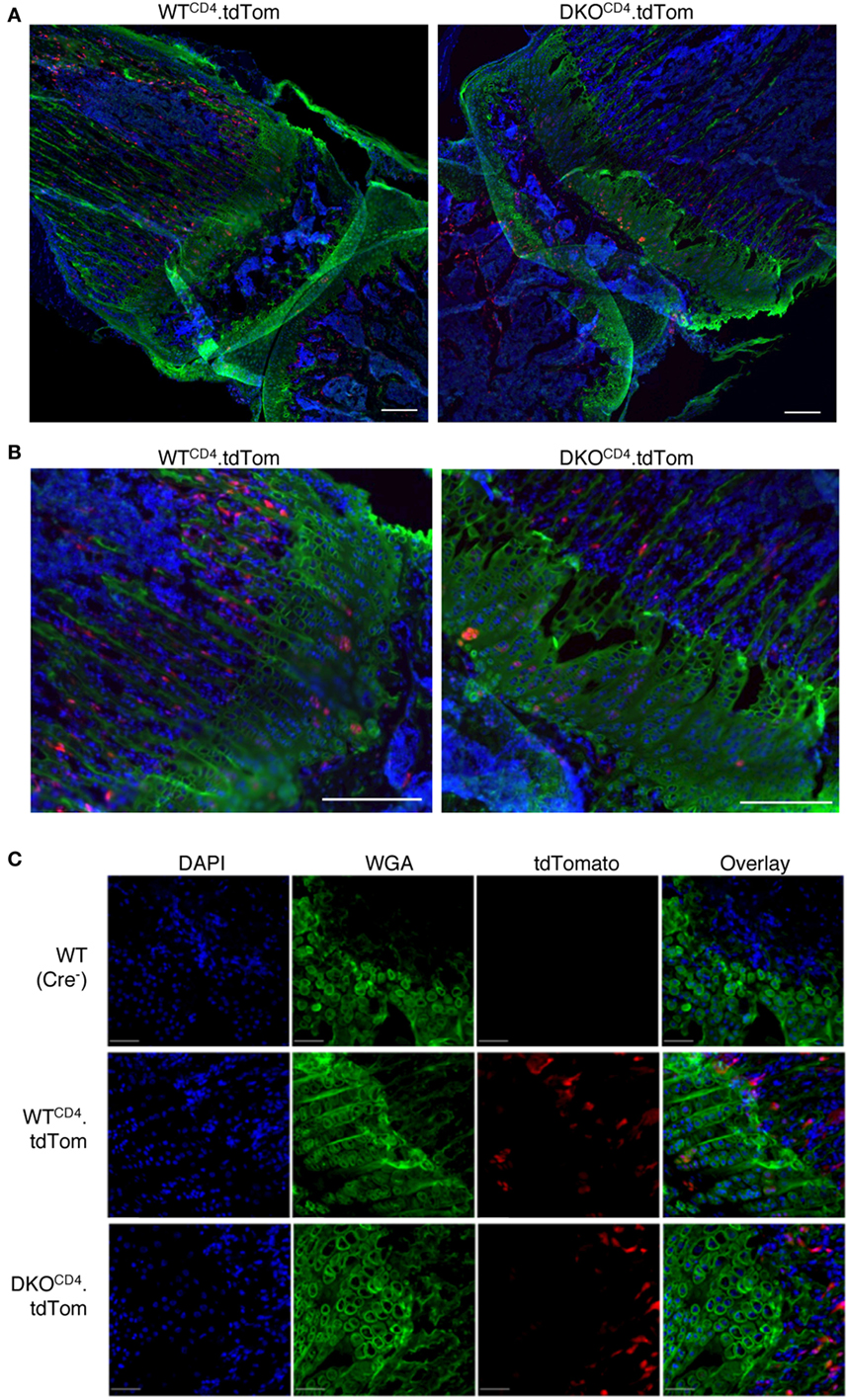
Figure 5. CD4-lineage positive cells are in close proximity to the epiphyseal growth plate. Hindlimbs from 5-week-old WTCD4.tdTom and DKOCD4.tdTom mice were cryosectioned then stained with DAPI and wheat germ agglutinin. (A) Composite widefield sections of tibia obtained at 10× were digitally stitched together. (B) Sections from (A) were electronically zoomed. (C) Confocal images of femur sections taken at 20×. Representative images of at least three mice per group are presented.
To further assess what cells other than T cells express CD4cre, we bred DKOCD4. Rag1−/− mice to the ROSA.LSL.YFP reporter mouse to create Erk1−/−Erk2fl/fl.Rag1−/−. YFPfl/fl.CD4cre+ (DKOCD4. Rag1−/−.YFP) mice and analyzed cells from the bone marrow, spleen, and small intestine intraepithelial (IEL) and lamina propria (LP) for YFP expression. Although the proportion of YFP+ cells was low in all of the organs, an YFP+ population was consistently present in all of the mice we analyzed (Figure 6A). The degree of CD4cre expression varied between mice, but was not influenced by the absence of Erk2 (Figure 6B). The majority of YFP+ cells in the bone marrow were CD45+ and either CD11b+ or CD11c+ (Figure 6C). The YFP+ cells were not Thy1+, suggesting that they were not ILCs. Additionally, DKOCD4.YFP mice that were Rag1-sufficient also had a significant proportion of cells other than T cells that expressed the Cre reporter (data not shown), suggesting that CD4cre is deleting genes in a small fraction of cells other than T cells, which may impact the phenotype if the gene plays an essential role in other cell types.
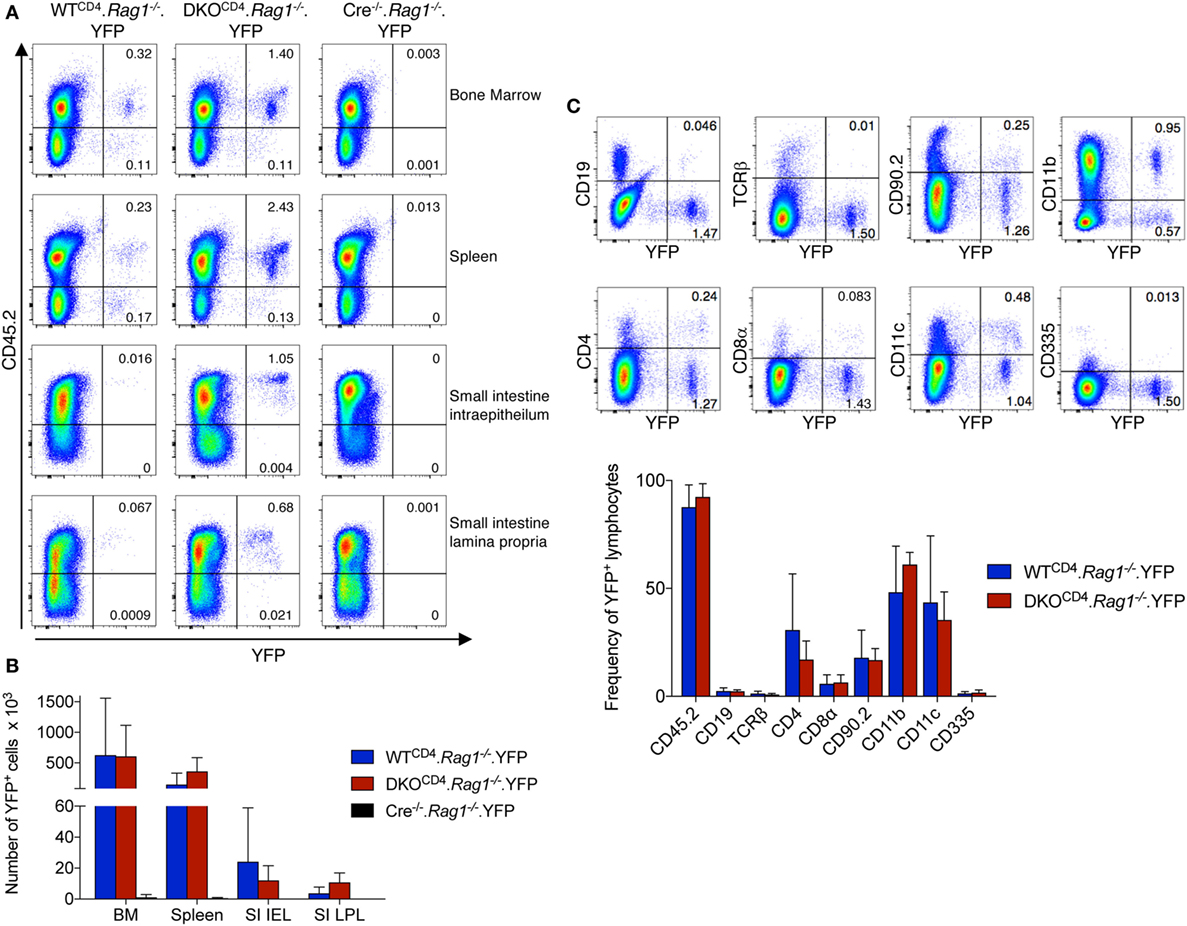
Figure 6. CD4cre is expressed in multiple immune cell types. (A) Lymphocytes from the bone marrow, spleen, small intestine intraepithelium, and small intestine lamina propria of WTCD4. Rag1−/− and DKOCD4. Rag1−/− mice were examined for expression of YFP by flow cytometry. Representative dot plots are shown with frequencies of YFP+ populations of total lymphocytes based on forward and side scatter. Cre−/−. Rag1−/− mice are shown as a negative control for YFP expression. (B) Quantification of the total numbers of YFP+ cells per tissue from plots shown in (A). n = 4 mice per group for WTCD4. Rag1−/− and DKOCD4. Rag1−/− mice and n = 2 for the Cre−/−. Rag1−/− group. (C) Representative dot plots from the bone marrow of a DKOCD4. Rag1−/− mouse are shown. Cells are electronically gated on lymphocytes based on forward and side scatter, and single cells. Numbers represent frequencies of YFP+ populations of the lymphocyte population. The average proportion of YFP+ cells that is positive for each subset in the bone marrow is plotted in the graph (n = 4 per group).
Discussion
The data presented here reveal two novel levels of chondrocyte regulation. First, ERK signaling in a cell that expresses CD4 is critical for regulating excess cartilage accumulation in the growth plate. Second, the presence of T cells delays the development of osteochondromas, demonstrating a critical impact of the immune system on cartilage homeostasis.
While there is a clear role for ERK signaling within chondrocytes to regulate proliferation and differentiation, it is likely that there are multiple levels of chondrocyte regulation. We propose that the growth of chondrocytes is regulated by signaling pathways within the chondrocytes, as well as external cues from cells within the environment. The fact that osteochondromas only developed in DKOCD4 mice, but not mice that only lack Erk2 in CD4-expressing cells, suggests that the chondrocytes must also lack Erk1 in order for osteochondromas to form. The absence of Erk1 in the chondrocytes may render these cells more susceptible to ectopic accumulation; however, disease does not occur unless Erk2 is also deleted in a cell type that expressed CD4 at one point during development.
Although chondrocytes and their progenitors are not known to express CD4, we observed CD4cre expression in a small fraction of chondrocytes in both WTCD4 and DKOCD4 mice, suggesting that a proportion of chondrocytes may be deficient in both Erk1 and Erk2. However, our data show that the majority of the chondrocytes in DKOCD4 mice maintain Erk2 expression. In fact, chondrocytes isolated from young mice showed no deletion of Erk2 (Figure 4A). Furthermore, if both Erk1 and Erk2 were deleted in the majority of chondrocytes, we would expect to see elongated bones, deformed bones, or disorganized growth plates in DKOCD4 mice beginning at a very young age as others observed when ERK signaling was impaired specifically within the chondrocytes (7).
While it is clear that Erk2 is not deleted in the majority of the chondrocytes in the DKOCD4 mice, osteochondromas can form when only a proportion of chondrocytes lack critical regulators such as ptpn11 (SHP2) or Ext1. Mice with deletion of these genes in 10–50% of chondrocytes developed cartilaginous tumors composed of a mix of wild-type and mutated chondrocytes (18, 19). Although it is not confirmed in these models that deletion of these genes in a small fraction of chondrocytes caused osteochondromas, it is possible that deletion of Erk2 in only a proportion of the chondrocytes in DKOCD4 mice induced osteochondromas. It is also conceivable that deletion of Erk2 in cells other than chondrocytes regulates cartilage homeostasis. Sections of growth plates from the Cre reporter mice showed a significant population of cells that deleted the gene, but were not chondrocytes. These cells are in close proximity to the chondrocytes and could contribute to chondrocyte accumulation.
Whether the osteochondromas form due to deletion of Erk2 in the chondrocytes or another subset of immune cells, our data show that the presence of T cells delays the onset and development of osteochondromas. This is surprising as it has not been shown that T cells, or any cell of the immune system, alter development of osteochondromas. T cells may impact osteochondroma function directly by suppressing tumor growth through cytolytic mechanisms. However, there is little evidence of infiltrating T cells in osteochondromas, and therefore, it is likely that the T cells affect the formation of osteochondromas indirectly by expressing a regulatory cytokine or influencing the development or function of a cell type that promotes osteochondroma formation.
Interestingly, deletion of Sos1/2 via CD4cre also caused development of osteochondromas similar to DKOCD4 mice (20). Sos-1/2 are Ras-guanine exchange factors that contribute to activation of ERK signaling (21). Deletion of Sos1/2 reduces ERK activation, and deletion of either of these genes via CD4cre results in spontaneous osteochondroma formation (20). These data highlight that this pathway is important in cells that expressed CD4 in order to regulate chondrocyte accumulation.
Together, these data suggest that although Erk1 deletion may render chondrocytes more sensitive to ectopic accumulation, disease does not occur unless Erk2 is also deleted in a cell type that expressed CD4 at one point during development. Importantly, the fact that T cells delay the onset and decrease the severity of osteochondromas reveals a previously unknown role for the immune system in regulating cartilage homeostasis.
Ethics Statement
This study was carried out in accordance with the recommendations of IACUC. The protocol was approved by the St. Jude IACUC committee.
Author Contributions
MW designed and performed research, analyzed data, and wrote the paper. AC and MC designed research, performed research, and analyzed data. CG, BE, and CC performed research and analyzed data. GP and JP contributed new reagents. PV analyzed data. MM designed and performed research, analyzed data, secured funding, and wrote the paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Stephen Hedrick for DKOCD4 mice; Jieun Kim, Melissa Johnson, Malireddi Subbarao, Sean Savage, and the Veterinary Pathology core for technical assistance. This work was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under award number R01AR069614-01 to MM, and by ALSAC. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Supplementary Material
The Supplementary Material for this article can be found online at http://journal.frontiersin.org/article/10.3389/fimmu.2017.00482/full#supplementary-material.
References
1. Jones KB. Glycobiology and the growth plate: current concepts in multiple hereditary exostoses. J Pediatr Orthop (2011) 31(5):577–86. doi: 10.1097/BPO.0b013e31821c7738
2. Yoon S, Seger R. The extracellular signal-regulated kinase: multiple substrates regulate diverse cellular functions. Growth Factors Chur Switz (2006) 24(1):21–44. doi:10.1080/02699050500284218
3. Krejci P. The paradox of FGFR3 signaling in skeletal dysplasia: why chondrocytes growth arrest while other cells over proliferate. Mutat Res Rev Mutat Res (2014) 759:40–8. doi:10.1016/j.mrrev.2013.11.001
4. Krejci P, Masri B, Salazar L, Farrington-Rock C, Prats H, Thompson LM, et al. Bisindolylmaleimide I suppresses fibroblast growth factor-mediated activation of Erk MAP kinase in chondrocytes by preventing Shp2 association with the Frs2 and Gab1 adaptor proteins. J Biol Chem (2007) 282(5):2929–36. doi:10.1074/jbc.M606144200
5. Sebastian A, Matsushita T, Kawanami A, Mackem S, Landreth GE, Murakami S. Genetic inactivation of ERK1 and ERK2 in chondrocytes promotes bone growth and enlarges the spinal canal. J Orthop Res (2011) 29(3):375–9. doi:10.1002/jor.21262
6. Murakami S, Balmes G, McKinney S, Zhang Z, Givol D, de Crombrugghe B. Constitutive activation of MEK1 in chondrocytes causes Stat1-independent achondroplasia-like dwarfism and rescues the Fgfr3-deficient mouse phenotype. Genes Dev (2004) 18(3):290–305. doi:10.1101/gad.1179104
7. Matsushita T, Chan YY, Kawanami A, Balmes G, Landreth GE, Murakami S. Extracellular signal-regulated kinase 1 (ERK1) and ERK2 play essential roles in osteoblast differentiation and in supporting osteoclastogenesis. Mol Cell Biol (2009) 29(21):5843–57. doi:10.1128/MCB.01549-08
8. Fischer AM, Katayama CD, Pagès G, Pouysségur J, Hedrick SM. The role of erk1 and erk2 in multiple stages of T cell development. Immunity (2005) 23(4):431–43. doi:10.1016/j.immuni.2005.08.013
9. McGargill MA, Ch’en IL, Katayama CD, Pagès G, Pouysségur J, Hedrick SM. Cutting edge: extracellular signal-related kinase is not required for negative selection of developing T cells. J Immunol (2009) 183(8):4838–42. doi:10.4049/jimmunol.0902208
10. Gosset M, Berenbaum F, Thirion S, Jacques C. Primary culture and phenotyping of murine chondrocytes. Nat Protoc (2008) 3(8):1253–60. doi:10.1038/nprot.2008.95
11. Mucida D, Park Y, Kim G, Turovskaya O, Scott I, Kronenberg M, et al. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science (2007) 317(5835):256–60. doi:10.1126/science.1145697
12. Chang C-F, D’Souza WN, Ch’en IL, Pages G, Pouyssegur J, Hedrick SM. Polar opposites: Erk direction of CD4 T cell subsets. J Immunol (2012) 189(2):721–31. doi:10.4049/jimmunol.1103015
13. Yang W, Wang J, Moore DC, Liang H, Dooner M, Wu Q, et al. Ptpn11 deletion in a novel progenitor causes metachondromatosis by inducing hedgehog signalling. Nature (2013) 499(7459):491–5. doi:10.1038/nature12396
14. Esworthy RS, Smith DD, Chu F-F. A strong impact of genetic background on gut microflora in mice. Int J Inflamm (2010) 2010(2010):986046. doi:10.4061/2010/986046
15. Carlucci F, Cortes-Hernandez J, Fossati-Jimack L, Bygrave AE, Walport MJ, Vyse TJ, et al. Genetic dissection of spontaneous autoimmunity driven by 129-derived chromosome 1 loci when expressed on C57BL/6 Mice. J Immunol (2007) 178(4):2352–60. doi:10.4049/jimmunol.178.4.2352
16. Davidson S, Crotta S, McCabe TM, Wack A. Pathogenic potential of interferon αβ in acute influenza infection. Nat Commun (2014) 5. Available from: http://www.nature.com/ncomms/2014/140521/ncomms4864/full/ncomms4864.html
17. Foldynova-Trantirkova S, Wilcox WR, Krejci P. Sixteen years and counting: the current understanding of fibroblast growth factor receptor 3 (FGFR3) signaling in skeletal dysplasias. Hum Mutat (2012) 33(1):29–41. doi:10.1002/humu.21636
18. Matsumoto K, Irie F, Mackem S, Yamaguchi Y. A mouse model of chondrocyte-specific somatic mutation reveals a role for Ext1 loss of heterozygosity in multiple hereditary exostoses. Proc Natl Acad Sci U S A (2010) 107(24):10932–7. doi:10.1073/pnas.0914642107
19. Bowen ME, Ayturk UM, Kurek KC, Yang W, Warman ML. SHP2 regulates chondrocyte terminal differentiation, growth plate architecture and skeletal cell fates. PLoS Genet (2014) 10(5):e1004364. doi:10.1371/journal.pgen.1004364
20. Guittard G, Gallardo DL, Li W, Melis N, Lui JC, Kortum RL, et al. Unexpected Cartilage Phenotype in CD4-Cre-Conditional SOS-Deficient Mice. Front Immunol (2017) 8:343. doi:10.3389/fimmu.2017.00343
Keywords: cartilage, extracellular signal-regulated kinase, osteochondroma, chondrocyte, animal model, bone
Citation: Wehenkel M, Corr M, Guy CS, Edwards BA, Castellaw AH, Calabrese C, Pagès G, Pouysségur J, Vogel P and McGargill MA (2017) Extracellular Signal-Regulated Kinase Signaling in CD4-Expressing Cells Inhibits Osteochondromas. Front. Immunol. 8:482. doi: 10.3389/fimmu.2017.00482
Received: 22 February 2017; Accepted: 07 April 2017;
Published: 01 May 2017
Edited by:
Bernard Malissen, Centre d’Immunologie de Marseille Luminy, FranceReviewed by:
Sho Yamasaki, Kyushu University, JapanJeroen Roose, University of California San Francisco, USA
Copyright: © 2017 Wehenkel, Corr, Guy, Edwards, Castellaw, Calabrese, Pagès, Pouysségur, Vogel and McGargill. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maureen A. McGargill, bWF1cmVlbi5tY2dhcmdpbGxAc3RqdWRlLm9yZw==