 Ralf J. Ludwig1*
Ralf J. Ludwig1* Karen Vanhoorelbeke2
Karen Vanhoorelbeke2 Frank Leypoldt3,4,5
Frank Leypoldt3,4,5 Ziya Kaya6
Ziya Kaya6 Katja Bieber1
Katja Bieber1 Sandra M. McLachlan7
Sandra M. McLachlan7 Lars Komorowski8
Lars Komorowski8 Jie Luo9
Jie Luo9 Otavio Cabral-Marques10
Otavio Cabral-Marques10 Christoph M. Hammers11
Christoph M. Hammers11 Jon M. Lindstrom9
Jon M. Lindstrom9 Peter Lamprecht10
Peter Lamprecht10 Andrea Fischer6Gabriela Riemekasten10
Andrea Fischer6Gabriela Riemekasten10 Claudia Tersteeg2
Claudia Tersteeg2 Peter Sondermann12Basil Rapoport7Klaus-Peter Wandinger13Christian Probst8
Peter Sondermann12Basil Rapoport7Klaus-Peter Wandinger13Christian Probst8 Asmaa El Beidaq14
Asmaa El Beidaq14 Enno Schmidt1Alan Verkman15,16
Enno Schmidt1Alan Verkman15,16 Rudolf A. Manz14Falk Nimmerjahn17
Rudolf A. Manz14Falk Nimmerjahn17
- 1Lübeck Institute of Experimental Dermatology, University of Lübeck, Lübeck, Germany
- 2Laboratory for Thrombosis Research, IRF Life Sciences, KU Leuven Campus Kulak Kortrijk, Kortrijk, Belgium
- 3Neuroimmunology, Institute of Clinical Chemistry, University Hospital Schleswig-Holstein, Kiel, Germany
- 4Neuroimmunology, Institute of Clinical Chemistry, University Hospital Schleswig-Holstein, Lübeck, Germany
- 5Department of Neurology, University of Kiel, Kiel, Germany
- 6Department of Internal Medicine III, University of Heidelberg, Heidelberg, Germany
- 7Thyroid Autoimmune Disease Unit, Cedars-Sinai Medical Center, UCLA School of Medicine, Los Angeles, CA, United States
- 8Institute for Experimental Immunology, Affiliated to Euroimmun AG, Lübeck, Germany
- 9Department of Neuroscience, University of Pennsylvania Medical School, Philadelphia, PA, United States
- 10Department of Rheumatology, University of Lübeck, Lübeck, Germany
- 11Department of Dermatology, University of Lübeck, Lübeck, Germany
- 12SuppreMol GmbH, Martinsried, Germany
- 13Department of Neurology, Institute of Clinical Chemistry, University Medical-Centre Schleswig-Holstein, Lübeck, Germany
- 14Institute for Systemic Inflammation Research, University of Lübeck, Lübeck, Germany
- 15Department of Medicine, University of California, San Francisco, CA, United States
- 16Department of Physiology, University of California, San Francisco, CA, United States
- 17Department of Biology, Institute of Genetics, University of Erlangen-Nuremberg, Erlangen, Germany
Autoantibodies are frequently observed in healthy individuals. In a minority of these individuals, they lead to manifestation of autoimmune diseases, such as rheumatoid arthritis or Graves’ disease. Overall, more than 2.5% of the population is affected by autoantibody-driven autoimmune disease. Pathways leading to autoantibody-induced pathology greatly differ among different diseases, and autoantibodies directed against the same antigen, depending on the targeted epitope, can have diverse effects. To foster knowledge in autoantibody-induced pathology and to encourage development of urgently needed novel therapeutic strategies, we here categorized autoantibodies according to their effects. According to our algorithm, autoantibodies can be classified into the following categories: (1) mimic receptor stimulation, (2) blocking of neural transmission, (3) induction of altered signaling, triggering uncontrolled (4) microthrombosis, (5) cell lysis, (6) neutrophil activation, and (7) induction of inflammation. These mechanisms in relation to disease, as well as principles of autoantibody generation and detection, are reviewed herein.
Autoantibody-Mediated Diseases: One Major Medical Burden, a Congregation of Different Pathways to Disease Manifestation
Over the past decades, a sharp increase in autoimmune diseases has been noted worldwide (1, 2). The cumulative prevalence of autoimmune diseases caused by autoantibodies is well over 2.5% (3). Despite developing insights into the pathogenesis of autoantibody-mediated autoimmune diseases (reviewed herein), systemic immunosuppression, i.e., with high doses of corticosteroids, is still the backbone of the treatment. Consequently, patients suffer from a high, and partially treatment-associated, morbidity and face an increased mortality (4). Thus, there is a high, and thus far, unmet medical need for development of novel treatments for patients suffering from autoantibody-mediated autoimmune diseases.
However, autoantibodies induce disease through a multitude of pathophysiological pathways. These differ among autoimmune diseases, yet within diseases multiple mechanisms may contribute to clinical manifestation. To disentangle these different autoantibody-mediated disease mechanisms, we aimed to categorize autoantibodies according to their main pathologic features. In brief, albeit surely not complete (Table 1), autoantibodies specific for a range of autoantigens induce pathology by a variety of mechanisms (Figure 1).
1. Mimic hormone stimulation of receptor: thyroid-stimulating autoantibodies (TSAb) in Graves’ disease.
2. Blockade of neural transmission by receptor blockade or alteration of the synaptic structures: antibodies against muscle nicotinic acetylcholine receptors (AChRs) in myasthenia gravis or against N-methyl-d-aspartate-(NMDA)-receptor in anti-NMDA encephalitis.
3. Induction of altered signaling: antibodies against desmoglein-3 in pemphigus.
4. Triggering uncontrolled microthrombosis: autoantibodies against ADAMTS13 in acquired thrombotic thrombopenic purpura.
5. Cell lysis: anti-platelet autoantibodies in autoimmune idiopathic thrombocytopenia.
6. Uncontrolled neutrophil activation: antineutrophil cytoplasmatic autoantibodies in granulomatosis with polyangitis.
7. Induction of inflammation at the site of autoantibody binding: autoantibodies against structural proteins of the skin in PD, or autoantibodies targeting myosin in myocarditis or autoantibodies recognizing citrullinated proteins in rheumatoid arthritis (RA) or autoantibodies targeting aquaporin-4 (AQP4) in neuromyelitis optica (NMO).
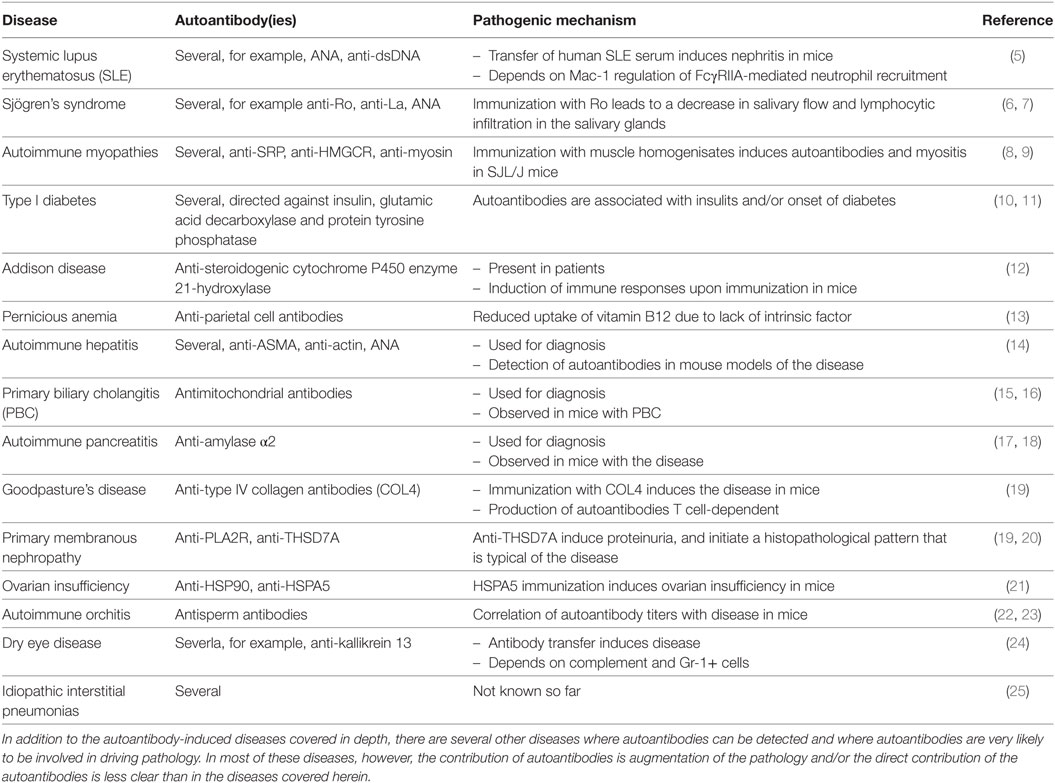
Table 1. Autoantibody-mediated diseases not discussed in this review.
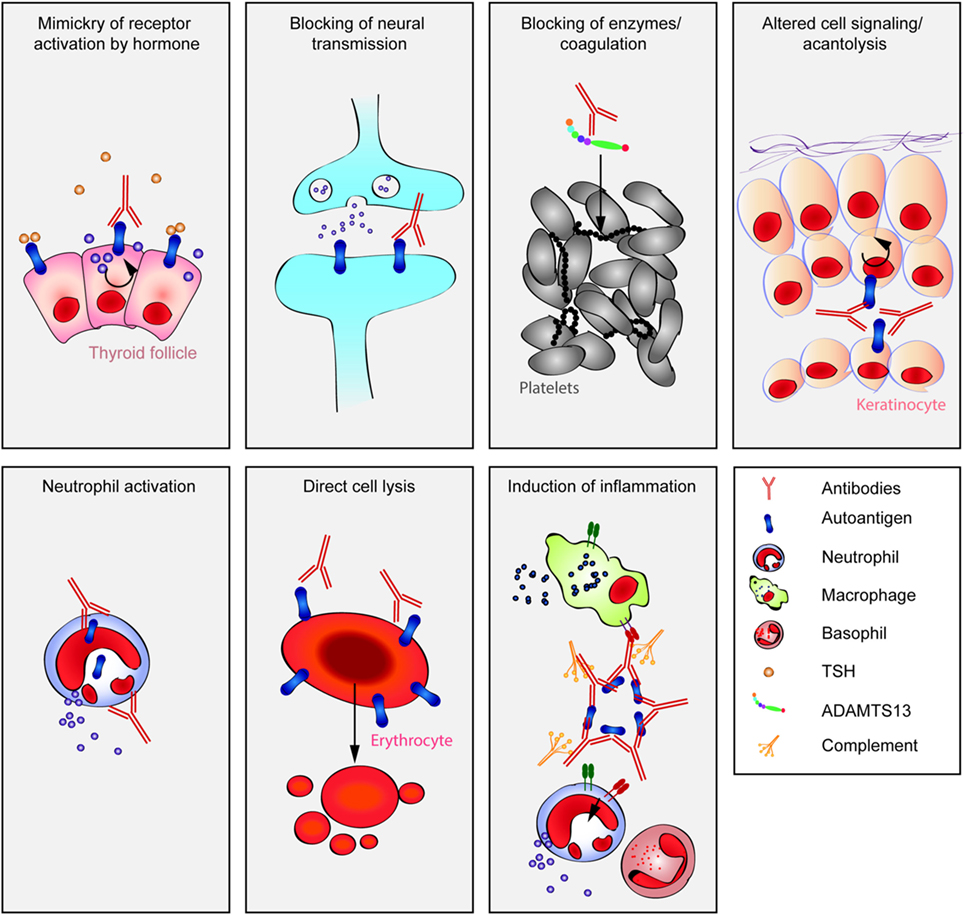
Figure 1. Multiple pathways lead to autoantibody-induced pathology. Depending on the targeted autoantigen, and sometimes even depending on the targeted epitope within a single autoantigen, autoantibodies induce pathology through specific and distinct mechanisms. Some are highlighted in this cartoon (from upper left to lower right): antibodies against the thyrotropin receptor (TSHR) mimic hormone stimulation of the TSHR receptor leading to hyperthyroidism, blockade of neural transmission by autoantibody binding to the corresponding receptors may lead to severe neurological diseases such as anti-N-methyl-d-aspartate encephalitis, autoantibody-mediated blockade of enzymes of the primary hemostasis may trigger uncontrolled microthrombosis, in pemphigus, autoantibodies induce an altered signaling in keratinocytes, which either reflects or leads to, a loss of cell–cell adhesion, resulting in severe skin blistering, autoantibodies to antigens expressed by neutrophils can lead to their uncontrolled activation, resulting in severe tissue injury, in autoimmune idiopathic thrombocytopenia autoantibodies trigger thrombocytopenia and severe bleeding, Fcγ-mediated functions may trigger tissue inflammation in many autoimmune diseases, e.g., rheumatoid arthritis and pemphigoid disease.
With this review, we intend to foster knowledge in an increasingly important medical field. Mostly, we encourage clinicians and scientists alike to build on the current knowledge on autoantibody-mediated pathology summarized here to develop urgently needed novel treatment modalities for patients suffering from autoantibody-mediated autoimmune diseases. We begin by briefly reviewing mechanisms leading to the generation and maintenance of autoantibodies, then discuss pathophysiological pathways in relation the corresponding diseases, and close by reviewing the diagnostic tools for autoantibody detection.
Generation and Maintenance of Autoantibodies
The presence of autoantibodies in serum reflects leakiness of central and/or peripheral tolerance mechanisms, allowing the maturation of autoantibody-producing B cells and their subsequent differentiation into antibody-secreting plasma cells. The mechanisms that normally mediate B cell tolerance include multiple selection steps that deplete or functionally silence autoreactive B cells. These well-orchestrated processes occur at the B cell immature, transitional, and mature stages (26, 27), and possibly also after activation of B cells in the periphery before they enter the plasma cell compartment in bone marrow (28, 29). The complex mechanisms contributing to B cell tolerance are multifaceted and may involve receptor editing, controlled migration, and limited availability of BAFF, CD22, Siglec-G, miRNA, and follicular regulatory T cells (30–36). These mechanisms have been extensively discussed elsewhere and are beyond the scope of this review. Nevertheless, we stress here that autoreactive B cells are selected against according to their receptor binding affinities (i.e., membrane-bound antibodies) to self-antigens. B cells producing antibodies that bind with high affinity to self-antigens are efficiently eliminated or undergo anergy, while those B cells that produce autoantibodies with medium or low binding affinity may escape, even in non-autoimmune individuals (37, 38). Many of these low-affinity autoantibodies are polyreactive and recognize self-antigens as well as pathogen-derived antigens (38, 39).
Autoantibodies are not necessarily pathogenic. Natural polyreactive/autoreactive IgM antibodies can sometimes protect from autoimmune diseases (40). There is also increasing evidence that immunoglobulin (Ig)G autoantibodies can exhibit anti-inflammatory capacities, depending on their IgG subclass (isotype) and the extent of glycosylation/sialylation of the Fc glycan linked to Asn297 (41–43). These properties modulate antibody binding to a variety of different Fc-receptors on innate effector cells (43). This receptor family includes FcγRI (CD64), FcγRIIIA (CD16a), and FcγRIIIB (CD16b) that mediate activating signals, but also includes the inhibiting receptors FcγRIIA and FcγRIIB (CD32). Antibodies exhibiting a distinct glycosylation/sialylation status and/or belonging to different IgG subclasses bind to activating and inhibiting Fc-receptors with different affinities (41, 44). Consequently, IgG subclass and glycosylation/sialylation patterns determine whether an autoantibody exhibits FcγR-mediated pro-or anti-inflammatory functions (45). Hence, differential autoantibody glycosylation might be an important regulator of autoimmune disorders (46–48).
So far, little is known about the mechanisms that control antibody glycosylation/sialylation patterns. IgGs derived in the context of T-independent immune reactions in murine models were shown to exhibit a high degree of sialylation that mediates anti-inflammatory properties, while IgGs generated in the context of T-dependent reactions were poorly sialylated and pro-inflammatory. In mice deficient for both the IFN-γ and the IL-17 receptors, T-dependent IgGs exhibit a high degree of sialylation, suggesting that T cell-derived IFN-γ and IL-17 are involved in the regulation of antibody sialylation (49). These experiments could not formally rule out the possibility that these cytokines were derived from cells other than T cells. Nevertheless, these observations suggest that similar to what is long known for antibody class switch (50), antibody sialylation is also modulated by the interaction of activated B cells with T follicular helper cells (49). Interestingly, both T cell differentiation into follicular helper T cells and cytokine profiles are modulated by activated B cells and plasma cells themselves, via presentation of antigen, co-stimulatory molecules, and B cell-derived cytokines (51–56). Following TLR stimulation, B cells produce different cytokines than dendritic cells (57). Dendritic cells might be the most important antigen-presenting cells during T cell priming. However, there is evidence that later, antigen presentation by B cells is important to promote the expansion of activated T cell clones, the development of robust T effector responses, and normal T cell memory compartments (58–60). Moreover, it was shown that TLR-signals in murine B cells promote IFN-γ production from T cells and in consequence control antibody isotype switching to IgG2 in vivo (57). Hence, the cross talk between activated B and T lymphocytes seems to be crucial for the outcome of antibody responses and their pathogenic potential, i.e., the antibody class and glycosylation pattern (Figure 2).
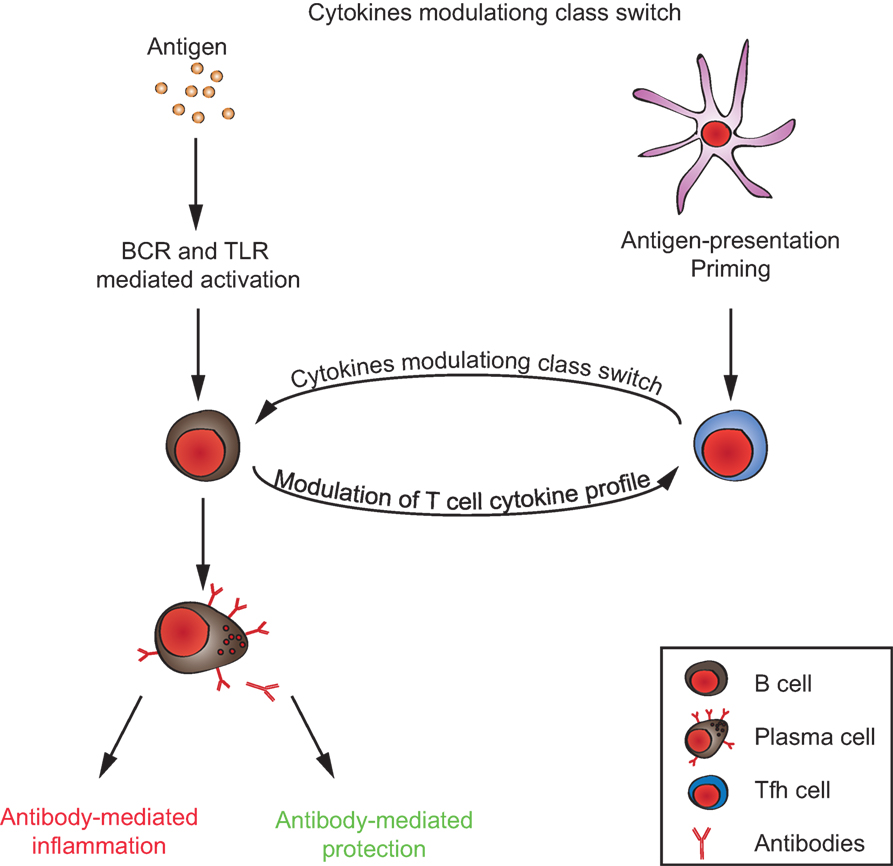
Figure 2. T/B cell cross talk in generating the autoimmune response. Autoantibodies can either promote or inhibit inflammation, depending on their immunoglobulin-isotype and glycosylation/sialylation patterns of their Fc-regions. Signals from both antigen-presenting B cells to T cells and from T cells to B cells, together, determine the inflammatory/anti-inflammatory property of the antibody response.
Measurement of autoantibodies is a major diagnostic tool in many diseases. However, autoantibodies are often found in otherwise healthy individuals (61–64). Considering the importance of the autoantibody subclass and glycosylation pattern for the pathogenic potential of a particular antibody, it might be helpful to include these parameters into the diagnostic analysis.
Once the production of pathogenic autoantibodies has started, it could be maintained either by ongoing activation of autoreactive B cells resulting in the continuous formation of short-lived plasma cells or through the formation of long-lived plasma cells, or both (65, 66). While B cell activation and short-lived plasma cell responses are suppressed by current therapeutic treatment options, long-lived plasma cells remain a therapeutic challenge (67, 68). A novel approach to deplete long-lived plasma cells and otherwise refractory autoantibodies is treatment with the proteasome inhibitor bortezomib. This drug was shown to deplete short-lived and long-lived plasma cells in murine models of systemic lupus erythematosus (SLE) and experimental autoimmune myasthenia gravis. In these experiments, the capacity of bortezomib to suppress lupus nephritis and myasthenic symptoms has been demonstrated (69–71).
Results of the first clinical investigations using bortezomib for the treatment of refractory SLE and thrombotic thrombocytopenic purpura (TTP) are promising (72–74). However, additional controlled studies are required to elucidate the potential of bortezomib to eliminate otherwise refractory autoantibodies. Bortezomib affects neither naïve nor memory B cells. Once long-lived plasma cells are depleted, these cell types could differentiate into new plasma cells. Accordingly, bortezomib treatment is able to result in long-lasting depletion of autoantibodies only if applied in combination with B cell depletion (75, 76). The development of proteasome inhibiting drugs exhibiting fewer side effects than bortezomib might be necessary to allow application of therapeutic proteasome inhibition to a broader set of patients.
Autoantibody-Induced Stimulation of Receptors
Grave’s Disease
Thyroid autoimmunity involves a breakdown in self-tolerance to three thyroid proteins: thyroglobulin, thyroid peroxidase (TPO), and the thyroid-stimulating hormone receptor (TSHR) (77). Autoantibodies to TPO and/or thyroglobulin are invariably associated with Hashimoto’s thyroiditis, but with a lower prevalence in Graves’ disease. Autoantibodies that arise spontaneously to the TSHR, the direct cause of Graves’ disease, have a number of unusual properties.
First, in Graves’ disease, TSAb mimic TSH and activate the TSH receptor in an unregulated manner, thereby causing hyperthyroidism (78) (Figure 3A). Transplacental transfer of TSAb in a mother with Graves’ disease may lead to transient neonatal hyperthyroidism (79). This confirms the role of TSAb in thyroid stimulation in vivo. Second, the TSHR itself plays a role in the breakdown in self-tolerance to the receptor in genetically susceptible individuals. The highly glycosylated extracellular TSHR A-subunit, formed by intramolecular cleavage of the holoreceptor, is shed and is the autoantigen primarily responsible for TSAb induction in Graves’ disease (80–82) (Figure 3B). Third, in rare patients, non-activating TSHR autoantibodies can compete for TSH binding and block TSH stimulation [TSH blocking antibodies (TBAb)], thereby causing hypothyroidism with an atrophic goiter (83, 84) (Figure 3A). Hashimoto’s thyroiditis, which is a common autoimmune condition, may also lead to hypothyroidism by TPO-specific T cells and possibly TPO autoantibody-mediated damage by NK cells and activation of the complement cascade (85). Fourth, the assays used to measure TSHR autoantibodies for clinical purposes are unusual. Unlike most autoantibodies that can be detected by enzyme-linked immunosorbent assay (ELISA) or western blotting (see below), pathogenic TSHR antibodies are measured by the inhibition of TSH binding to its receptor (“TBI”) or in bioassays involving the generation of cAMP from monolayers of TSHR-expressing cells (“TSAb” activity) (86). Fifth, in experimental models, TSHR autoantibodies induced by conventional immunization using TSHR protein and a variety of adjuvants do not stimulate the thyroid gland and do not induce experimental Graves’ disease (87). Instead, TSAb and Graves’-like hyperthyroidism can be induced in mice or hamsters by injecting intact eukaryotic cells expressing the TSHR or by injecting plasmid/adenoviral vectors encoding the TSHR or, more efficiently, its A-subunit (77, 87). Valuable monoclonal antibodies (MAbs) have been isolated from mice immunized using these novel approaches (88, 89).
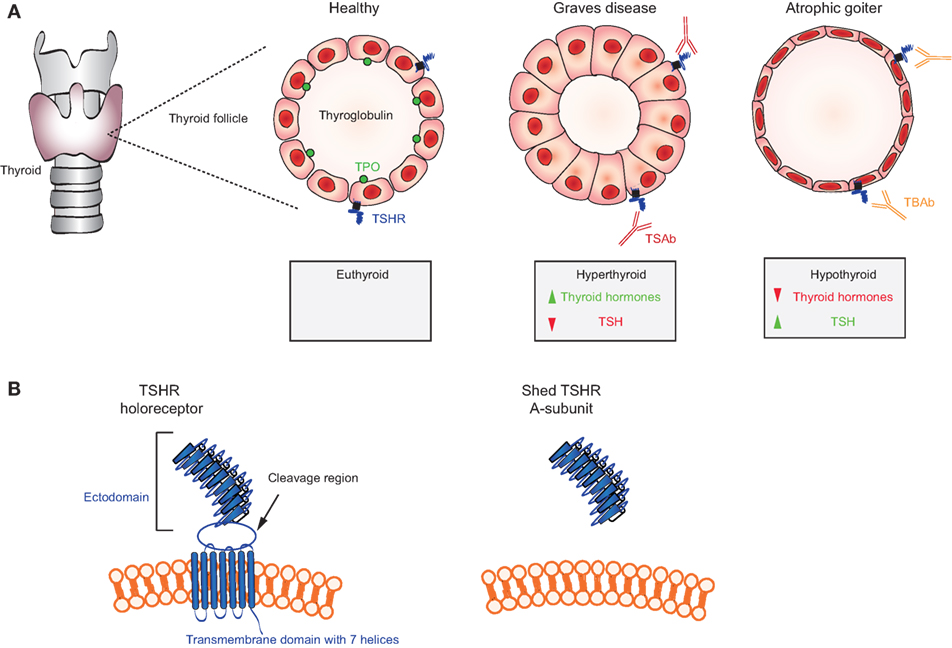
Figure 3. Mechanisms in thyroid autoimmunity. (A) Location of the thyroid gland in relation to the larynx and trachea and schematic representations of a thyroid follicle in a healthy individual (cuboidal cells) with the location of the thyroid-stimulating hormone receptor (TSHR), thyroglobulin, and thyroid peroxidase (TPO); a stimulated follicle in a patient with thyroid-stimulating autoantibodies (TSAb) (Graves disease; columnar epithelial cells); and a follicle in a patient with TSH blocking antibodies (TBAb) (atrophic goiter; very thin epithelial cells). Thyroid hormones (T4, T3) are elevated and TSH levels are very low in Graves’ disease; conversely, T4 and T3 are low and TSH is elevated TSH levels in atrophic goiter. (B) Representation of the TSH holoreceptor including its transmembrane domain (left) and the TSHR A-subunit (right) shed after cleavage.
It should be noted that the human TSHR or its A-subunit are used for immunization in almost all studies. Mice immunized with adenovirus encoding mouse TSHR A-subunit fail to develop TSHR antibodies because of central tolerance to the endogenous TSHR. TSHR-deficient mice injected with adenovirus encoding mouse TSHR A-subunit develop TSHR antibodies (90). Adoptive transfer of splenic T cells from immunized TSHR-deficient donors to wild-type recipients on the same genetic background reveals the stimulatory ability of these induced TSHR autoantibodies (91). Finally, although TSHR autoantibodies are unusual, Graves’ hyperthyroidism is a common condition, with a prevalence of ~1% (92). Moreover, TSHR autoantibodies play a role in Graves’ exophthalmos and Graves’ dermopathy by interacting with the TSHR expressed on orbital fibroblasts (93) and dermal fibroblasts (94). It is notable that patients with Graves’ exophthalmos and dermopathy usually have very high circulating levels of TSHR autoantibodies (94).
Properties of TSHR Autoantibodies
The immunological properties of TSAb and TBAb, based on patients’ sera and human MAbs derived from Graves’ patients, are described in detail elsewhere (95, 96). In brief, TSAb are present at very low (ng/ml) concentrations in serum (97–99). In contrast, TBAb are present at much higher concentrations, μg/ml (80, 99). TSHR autoantibodies are predominantly IgG although IgA- and IgE-class TSHR autoantibodies have been observed by flow cytometry (100). Both serum TSHR autoantibodies and human mAb have extremely high affinities, as would be expected for antibodies that compete with TSH for binding to its receptor. “Switching” from TBAb to TSAb (or vice versa) is a rare phenomenon observed in some patients. These changes involve differences in the concentrations of TSAb versus TBAb, as well as their affinities and/or potencies in individual patients (86). Anti-thyroid drugs or immunosuppression/hemodilution in pregnancy reduce initially low TSAb levels even further while higher concentration TBAb may persist. In contrast, if TSAb emerge during levothyroxine administration for TBAb-induced hypothyroid, these stimulating antibodies may be sufficient to swing the pendulum to hyperthyroidism.
Thyroid-stimulating autoantibodies and TBAb interact with conformational epitopes and do not bind to synthetic TSHR peptides. Crystallization of both types of mAb with recombinant TSHR-A-subunit protein reveal closely overlapping epitopes with subtle differences expected for a TSAb (M22) and a TBAb (K1-70) (101, 102). Unlike spontaneously arising TSHR autoantibodies, some TSHR mAb derived from immunized hamsters bind to a linear peptide in the region deleted from the single chain TSH holoreceptor after intramolecular cleavage into A- and B-subunits. These “neutral” or “cleavage region” TSHR antibodies neither stimulate the thyroid nor block TSH stimulation but may play a role in signaling cascades leading to apoptosis (103). Because the epitope of these neutral antibodies (unlike TSAb and TBAb) is lost after TSHR cleavage into subunits, they can only interact with the single chain, uncleaved TSHR. Whether the TSHR in vivo exists in the uncleaved form has been the subject of debate (82). In human disease, confirmation of the existence of neutral TSHR autoantibodies will require information on their concentrations or affinities. To date, no human “neutral/cleavage” TSHR autoantibodies have been cloned.
Current and Future Approaches to Treat Graves’ Disease
Several approaches are available to treat the hyperthyroidism in Graves disease including thionamide drugs, thyroid ablation by Radioiodine (131-I), or subtotal thyroidectomy as well as β-blockers to inhibit the actions of excess thyroid hormone (104). In general, these therapies are effective, thionamide drugs are inexpensive, and all approaches have been used for many years. Radioiodine therapy frequently leads to permanent hypothyroidism, subsequently leading to life-long thyroid hormone replacement. Rarely, development of serious side effects with thionamide drugs requires one of the alternative forms of treatment. Atrophic goiter is treated by replacement of thyroid hormone (levothyroxine, LT4), which is safe and inexpensive.
Three novel forms of therapy for Graves’ hyperthyroidism are currently under investigation: (i) small molecule inhibitors of TSHR function (105–107), (ii) monoclonal TBAb, such as K1-70 (96) that block the binding and function of TSAb, and (iii) inhibitors of components of the adaptive immune system, such as rituximab, which targets B-lymphocytes prior to their maturation into plasma cells (Table 2).
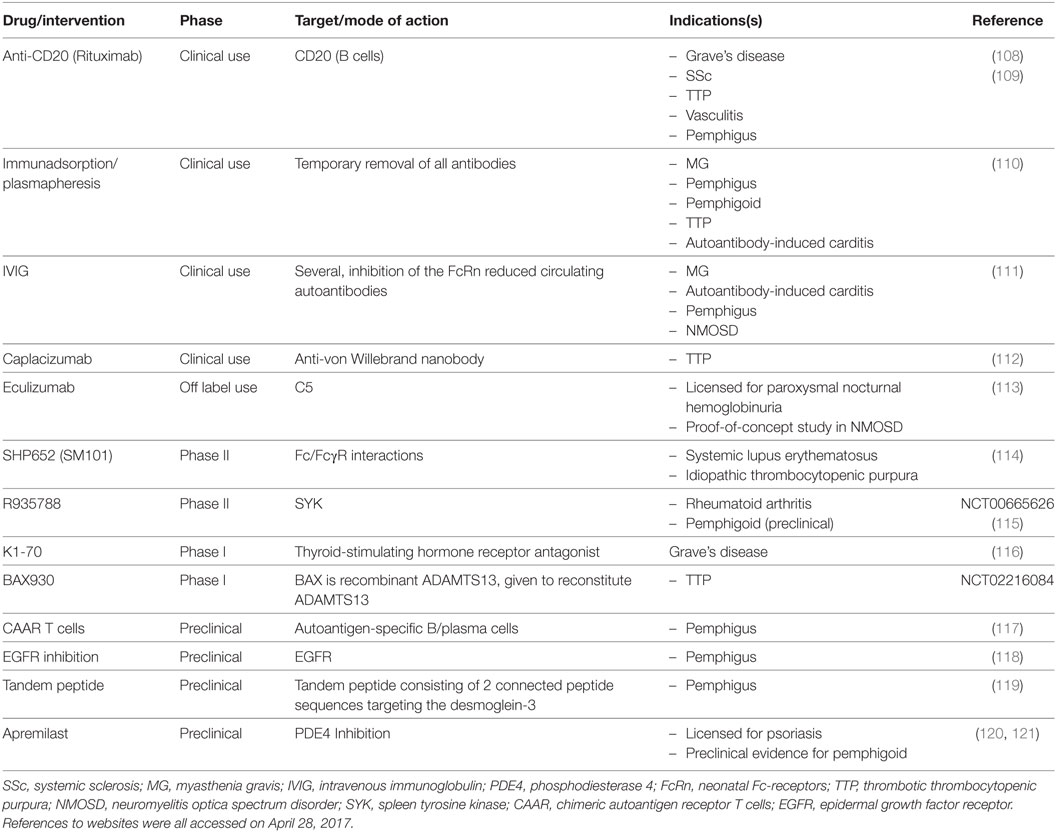
Table 2. Selected drugs targeting autoantibody-induced pathology.
Small molecule inhibitors and monoclonal TBAb target the proximate cause of hyperthyroidism, the TSHR. Both of these new forms of therapy, if successful, are likely to be more expensive than the presently available and effective therapies. In addition, because small molecule inhibitors interact with the heptahelical transmembrane domain of the TSHR, this treatment option carries the risk of side effects consequent to potential cross-reactivity with others in the very large G-protein-coupled receptor family. Graves’ ophthalmopathy occurs with varying degrees of severity and can be very distressing symptomatically and cosmetically, as well as (fortunately rarely) leading to loss of vision. Options for the treatment of Graves’ ophthalmopathy include, in addition to the restoration of the euthyroid state, corticosteroids, orbital irradiation, and orbital surgical decompression. Because none of these therapeutic modalities are optimal, there is presently much interest in the use of antibodies to CD20 (rituximab) that cause B cell depletion. Two recent double-blind clinical trials with rituximab have been reported: in patients with long-standing Graves’ eye disease (10 months), rituximab was no more effective than control saline injections (122). In contrast, in patients in whom eye disease duration was short (4.5 months), rituximab was more effective at reducing clinical symptoms than methylprednisone (123). The different outcomes of these two studies emphasize the importance of early intervention, which, in turn, requires early disease diagnosis.
The challenge in Graves’ disease is to specifically inhibit the development, or ongoing production, of TSHR autoantibodies. Such an approach has been attempted in mouse models of induced Graves’ disease. First, injecting neonatal BALB/c mice with TSHR A-subunit protein induced tolerance that could not be broken by subsequent immunization with adenovirus encoding A-subunit (124). Second, hyperthyroidism was attenuated in adult mice by injecting TSHR A-subunit protein before A-subunit adenovirus immunization. The latter involved deviating pathogenic antibodies to non-functional antibodies and was only effective if applied before immunization, not after hyperthyroidism had been established (125). However, in a mouse model that spontaneously develops pathogenic TSHR antibodies (126), injecting TSHR A-subunit protein failed to divert the autoantibody response to a non-pathogenic form, highlighting critical differences between induced and spontaneous Graves’ disease models, with implications for potential immunotherapy in humans (127).
Systemic Sclerosis (SSc)
Systemic sclerosis or scleroderma is one of the most lethal rheumatic diseases characterized by microvascular dysfunction, dysregulation of innate and adaptive immunity, and interstitial and perivascular fibrosis in the skin and internal organs (128). Mechanistically, a hallmark SSc feature is the presence of high serum concentrations of multiple autoantibodies (129). Among these autoantibodies, increased titers of stimulating autoantibodies targeting both angiotensin II type 1 receptor (AT1R) and endothelin-1 type A receptor (ETAR) have been reported to contribute to SSc pathogenesis and suggested as biomarkers for risk assessment of disease progression (130). These antibodies can be detected by ELISA (see below). Anti-AT1R and anti-ETAR are strong predictors of digital ulcers development (131), they can be considered as predictive and prognostic biomarkers of (SSc)-associated pulmonary arterial hypertension (132) and are linked to the development of lung fibrosis and vasculopathies in patients with SSc (133). Data from mice models show that anti-AT1R and anti-ETAR antibody-positive SSc-IgG cause structural alterations of the lungs including induction of interstitial lung disease and obliterative vasculopathy, increased cellular density, and enhanced interstitial cellular infiltrations following passive transfer of IgG from SSc patients to wild-type mice (134, 135).
Autoantibodies targeting ETAR and AT1R increase the sensitivity of both AT1R and ETAR to their natural ligands (angiotensin II and endothelin-1) and have synergistic effects in the presence of these ligands (132, 136). Anti-AT1R and anti-ETAR autoantibodies activate signaling molecules and transcription factors such as protein kinase C-α (PKC-α), extracellular signal-regulated kinase 1/2, nuclear factor-κB (NF-κB), and activator protein 1, which are involved in several pathophysiologic processes (130, 136–138). Increased number of reports have recently linked a variety of anti-AT1R and anti-ETAR cellular and systemic effects to SSc pathogenesis (139). Among them, anti-AT1R and anti-ETAR from SSc patients (SSc-IgG) contribute to vasculopathies by regulating cell migration through interleukin-8 (IL-8) production and expression of vascular cell adhesion molecule-1 by human microvascular endothelial cells (HMEC-1). Consistent with these findings, IL-8 release by PBMCs, regulation of wound repair, and T-cell chemotaxis have been also observed following IgG-induced ETAR and AT1R activation (133, 134). Anti-AT1R and anti-ETAR autoantibodies directly induce collagen production by skin fibroblasts (134) and regulate the synthesis of profibrotic factors such as transforming growth factor β (TGF-β) by human dermal microvascular endothelial cells (130), which are pathological mechanisms in SSc patients who develop lung fibrosis. Another important mechanism of autoantibody-induced pathology triggered by anti-AT1R and anti-ETAR is the production of chemokine (C-C motif) ligand 18 (CCL18) (133). CCL18 levels in bronchoalveolar fluid and sera directly reflect pulmonary fibrotic activity and are predictive for lung disease progression and mortality in patients with SSc (140–142). Additional pathological mechanisms triggered by anti-AT1R and anti-ETAR have been reported such as the increase of neutrophil migration and reactive oxygen species (ROS) production by neutrophils (134). An overview of the pathological mechanisms triggered by anti-AT1R and anti-ETAR autoantibodies is shown in Figure 4.
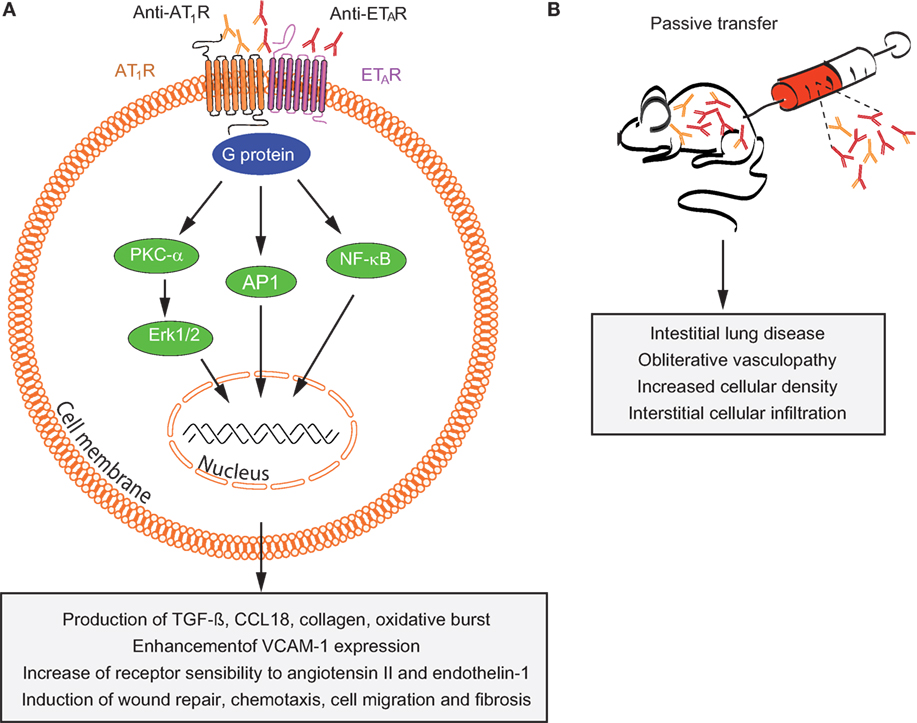
Figure 4. Overview of mechanisms by which anti-AT1R and anti-ETAR induce systemic sclerosis (SSc) pathogenesis. (A) In vitro and (B) in vivo effects of autoantibodies targeting ETAR and AT1R in SSc are shown. Abbreviations: AT1R, angiotensin II type 1 receptor; ETAR, endothelin-1 type A receptor; PKC-α, protein kinase C-α; ERK1/2, extracellular signal-regulated kinase 1/2; NF-κB, nuclear factor-κB; AP-1, activator protein 1; TGF-β, transforming growth factor β; CCL18, chemokine (C-C motif) ligand 18; VCAM-1, vascular cell adhesion molecule-1.
In addition to the levels of anti-AT1R and anti-ETAR autoantibodies, AT1R and ETAR expression needs to be considered in SSc pathogenesis. The cellular effects triggered by anti-AT1R and anti-ETAR autoantibodies depend on the AT1R and ETAR expression levels. For instance, imbalanced AT1R/AT2R and ETAR/ETBR expression ratios in SSc influence autoantibody-mediated effects such as secretion of profibrotic CCL18 (143). So far, a therapy that specifically inhibits anti-AT1R and anti-ETAR autoantibodies or/and regulates AT1R/AT2R and ETAR/ETBR expression is not available. Expansion of the currently state of knowledge about the cell signaling pathways and mechanism of autoantibody-induced pathology triggered by anti-AT1R and anti-ETAR will be essential to develop new therapies specifically targeting autoantibody effects simultaneously maintaining the renin–angiotensin and endothelin systems physiological response. Achieving this goal will be essential for the management of SSc patients in order to improve their quality of life and reduce SSc mortality rates. This achievement will also provide new therapeutic options for other diseases in which high circulating levels of anti-AT1R and anti-ETAR autoantibodies have pathological effects (138, 144–148).
Disruption of Neural Transmission by Autoantibodies
Myasthenia Gravis (MG)
Myasthenia gravis is an uncommon autoimmune disorder characterized by muscle weakness and abnormal fatigability that worsens with use of affected muscles and improves with rest. It is caused by the presence of autoantibodies to components of the postsynaptic muscle endplate localized at the neuromuscular junction (149, 150). The annual incidence of MG is approximately 1.7–34 per million, and the prevalence ranges from 15 to 320 per million population (151–153). However, MG remains underdiagnosed and the prevalence shows a steady increase over time, which is likely reflective of better recognition of the condition, aging of the population, and the longer life span of patients, possibly due to improved disease treatment.
Myasthenia gravis is a chronic affliction that affects people of all ages and both sexes (154). Studies of large groups of patients show that the age of onset is characterized by a bimodal distribution with female predominance in the second to third decade of life and slight male predominance in the sixth to eighth decades. MG, especially early-onset MG, is often associated with other autoimmune diseases, most commonly autoimmune thyroid disease, SLE, RA, Addison’s disease, Guillain–Barré syndrome, type 1 diabetes mellitus, and NMO (155).
Several subgroups of MG have been identified based on clinical presentation, age of onset, autoantibody profile, and thymic pathology. Over two-thirds of all patients with MG begin with symptoms relating to their eye muscles (156). The symptoms usually progress to other muscles during the first 2 years, resulting in generalized MG. In about 15% of MG patients, symptoms are restricted to the eye muscles, and this condition is termed ocular MG. In most cases, a specific cause of MG cannot be identified (157). There is strong evidence that the pathogenesis of MG involves a combination of multiple genotypes of low penetrance and largely unidentified environmental factors (158). Patients with early-onset MG are usually associated with HLA-B8DR3 and thymic hyperplasia (155, 159). Approximately 15% of patients with MG have a thymoma, and 50% of thymoma patients develop MG (160). Improving the clinical symptoms by thymectomy in some MG patients, although questioned by many, also suggests a specific role of the thymus in MG (161, 162).
About 90% of generalized MG is caused by pathogenic autoantibodies to muscle nicotinic AChRs. MG patients who do not have detectable autoantibodies to AChRs are referred to as seronegative. For many years, the cause of MG remained a mystery. In 1960, Simpson suggested that MG was caused by autoantibodies to AChRs acting as competitive antagonists (163). In 1973, Patrick and Lindstrom demonstrated that MG is an autoimmune disease by showing that rabbits immunized with muscle-like AChR purified from fish electric organ developed MG-like symptoms (164). Experimental autoimmune MG (EAMG) in rabbits, was reproduced in other species, especially inbred Lewis rats where the most detailed studies have been completed (165). EAMG shares many clinical and immunopathological features with those of MG. Clinical, electrophysiological, histological, pharmacological, and immunological analysis of EAMG led to extensive understanding of the pathological mechanisms in MG (150, 166). These findings promoted the use of immunosuppressive treatments in MG.
The pathogenic role of autoantibodies to AChRs is clearly established. Most patients with MG have circulating antibodies to AChR (167). Injection of patients’ IgG or isolated autoantibodies to AChR from patients into laboratory animals passively transfers several features of MG from human to the recipients (168, 169). Immune complexes (IgG and complement) co-localize with AChRs on the postsynaptic membrane (170, 171). Plasmapheresis that removes circulating antibodies leads to a substantial, but temporary, improvement in muscle function (172). EAMG can be induced by immunization with purified AChR (164) or by injection of mAbs to AChR (173). However, among MG patients, there is not a close correlation between the absolute concentration of autoantibodies to AChR and severity (167, 174). Variability in specificities, affinity, and isotypes of the autoantibodies to AChR may contribute to this lack of correlation (175).
Autoantibodies to AChR can differ in their isotype, affinity, and specificity to various AChR epitopes. Pathological autoantibodies are directed at conformation-dependent extracellular epitopes of AChRs, especially the main immunogenic region (MIR) on AChR α1 subunits (176). Half or more of the autoantibodies to AChR in MG target the MIR. These autoantibodies impair neuromuscular transmission primarily by two mechanisms: (1) focal complement-mediated lysis of the postsynaptic membrane that destroys AChRs and disrupts synaptic morphology (166, 171), (2) cross-linking AChRs by the autoantibodies on the surface of postsynaptic membrane accelerating endocytosis and lysosomal destruction of AChRs, termed antigenic modulation (177) (Figure 5). Some autoantibodies inhibit AChR function by direct blockage of acetylcholine (ACh) binding sites, but these appear to play a minor role (178). No cellular infiltration at the target muscle tissue of MG patients indicates that tissue damage is not caused by direct harmful effects of cytotoxic cells (170). However, autoreactive T cells provide help to B cells that produce autoantibodies to AChR (179). Autoantibodies directed at cytoplasmic epitopes of human AChR are also present in MG sera (180). However, these autoantibodies are not pathogenic because the cytoplasmic domain of AChR is not accessible to these autoantibodies in intact muscle. Additionally, rats repeatedly immunized with the cytoplasmic domains of AChR in adjuvant do not develop EAMG and serum antibodies from these rats do not bind the MIR, passively transfer EAMG, or cause antigenic modulation of AChR, although these antibodies bind to solubilized native AChRs (175, 181).
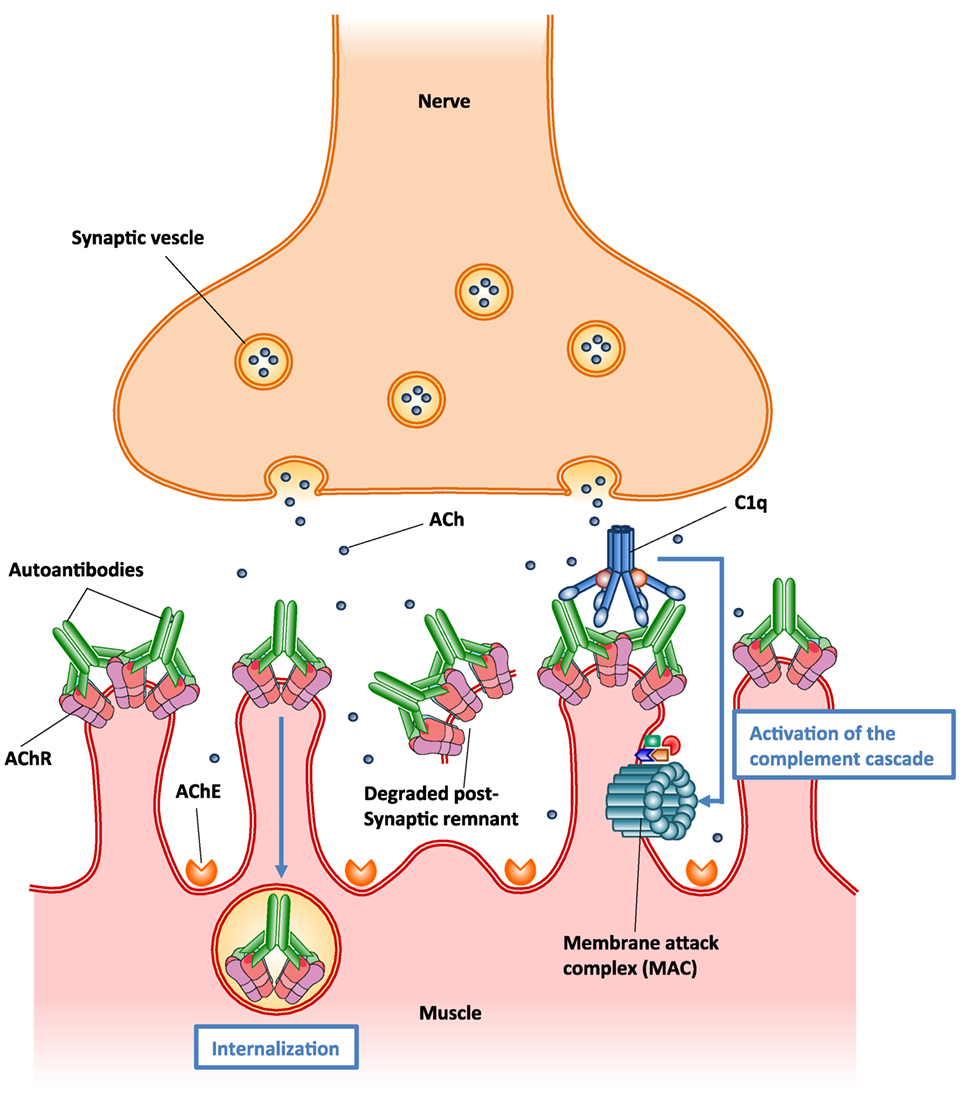
Figure 5. Schematic illustration of the two major pathogenic mechanisms of autoantibodies to acetylcholine receptor (AChR). (1) Autoantibody binding to the AChR on the surface of postsynaptic muscle membrane activates the complement cascade, resulting in the formation of membrane attack complex (MAC) and localized destruction of the postsynaptic membrane. The immune assault releases shedding of membrane fragments containing AChRs into the synaptic cleft, and leads to a simplified, altered morphology of the postsynaptic membrane. (2) Autoantibodies cross-link AChR molecules on the postsynaptic muscle membrane, causing endocytosis of the cross-linked AChR molecules and their degradation (antigenic modulation). This leads to a reduced number of AChR molecules on the postsynaptic membrane.
In MG patients, the predominant isotypes of autoantibodies to AChR are IgG1 and IgG3, which fix complement (182). A series of studies have suggested that accumulation of autoantibodies to AChR and subsequent activation of the complement cascade on postsynaptic membrane result in the assembly of membrane attack complex (MAC), thereby destroying AChRs and disrupting synaptic morphology (166, 171, 183) (Figure 5). It seems likely that complement-mediated destruction of the postsynaptic muscle membrane is more pathologically significant than antigenic modulation in MG pathogenesis (175, 184). These findings have promoted the development of novel therapies based on inhibition of complement pathways in MG treatment (185).
Half of AChR seronegative patients have antibodies to muscle-specific kinase (MuSK), which is a component of the agrin receptor complex that mediates clustering and stabilization of AChR in developing muscle (186). Autoantibodies to MuSK and those to AChR rarely coexist in the same patient. MuSK-related MG differs from AChR-related MG primarily by demonstrating more focal involvement and wasting of the involved muscles (187). This type of MG is predominantly in females and usually reported in adults. There is a HLA association with HLA-DQ5 (188). No thymus pathological changes have been reported and patients usually have no response to thymectomy (189). Autoantibodies to MuSK are mainly of the IgG4 isotype, which does not activate complement, and the IgG4 fraction alone is sufficient for passive transfer of the disease (188). Autoantibodies to MuSK directly suppress the postsynaptic tyrosine kinase pathway thus indirectly reduce the metabolic stability of endplate AChRs (190). Complement is not necessary for pathogenesis in mice (191). Patients with MuSK-related MG respond better to plasmapheresis than to intravenous immunoglobulin (IVIG) (192). Rituximab appears to be more effective in patients with MuSK-related MG than in patients with AChR-related MG (193).
Low-density lipoprotein receptor-related protein 4 (LRP4), which belongs to a family of the low-density lipoprotein receptor family, is a receptor for nerve-derived agrin and an activator of MUSK, and is necessary to maintain AChR function (194). Autoantibodies to LRP4 have been detected in some of double-seronegative MG patients who do not have antibodies to AChR or MuSK (195, 196). LRP4-related MG has a female preponderance. Most of these patients present with ocular or generalized mild MG. This type of MG appears to be clinically similar to AChR-related MG, but there is no clear role for thymectomy. Autoantibodies to LRP4 are predominantly of the IgG1 isotype, which fixes complement, and thus may damage the postsynaptic muscle membrane by a similar mechanism as those to the AChR. Inhibition of argin-induced AChR clustering may also be a potential pathophysiological mechanism (197).
Some seronegative patients have low-affinity antibodies to AChR that are not detectable by the classical immunoprecipitation assay. These autoantibodies can be identified by cell-based assay (CBA) only (198). These patients are similar to patients with AChR-related MG with respect to their clinical presentation, their response to treatment, and their thymic abnormalities. Some seronegative patients probably have pathogenic antibodies to as-yet-unidentified antigens in the postsynaptic membrane. Antibodies to agrin and cortactin are often identified in combination with other autoantibodies (199). Their contribution to MG pathogenesis is still unclear. Some patients with AChR-related MG have antibodies to titin and ryanodine receptor (200). These antibodies cannot enter the muscle cell in vivo and thus might not cause any muscle weakness, but rather could be markers of the autoimmune disease directed at muscle surface resulting in exposure of cytoplasmic proteins.
Treatment options available today rely primarily on a combination of symptomatic therapies and general, non-specific immunosuppression. Many MG patients respond favorably to treatment (149). Symptomatic treatment of MG with acetylcholinesterase inhibitor temporarily enhances neuromuscular transmission by increasing the availability of ACh to compensate for loss of AChRs, but does not induce complete or sustained relief of MG symptoms in most patients and does not alter disease progression (149, 154). Patients with MuSK-related MG may not respond to cholinesterase inhibitor. At high doses, cholinesterase inhibitor may actually cause more muscle weakness through desensitization of AChRs (201). Most patients require additional immunosuppressive treatment. High doses of corticosteroids and chronic treatment with non-specific immunosuppressive drugs are usually required to maintain disease control. Non-specific immunosuppressive drugs primarily suppress lymphocyte activation and proliferation and have little effect on long-lived plasma cells that are terminally differentiated cells and continue producing pathogenic antibodies (202). Therefore, the use of these drugs is often hampered by delayed clinical response. Plasmapheresis and IVIG are used for acute severe exacerbations in generalized MG. For a chronic disease like MG, the current treatment has a high cost (203), as most patients during the long-term treatment suffer several undesirable side effects (204). A distinct subset of patients often referred to as having refractory MG, do not respond well to current treatments (205).
Advances in understanding of the pathogenesis of the various forms of MG and breakthrough use of mAbs specific for different aspects of the immune system involved in antibody production and antibody-mediated tissue damage provide an opportunity to develop more specific treatment with long-lasting benefits and fewer adverse effects. Rituximab, a chimeric murine-human mAb that depletes B cells by binding to their CD20 surface marker, has been reported to be effective in patients with severe or refractory MG, especially in those with MuSK-related MG (193, 205). Yale University is sponsoring a multicenter clinical trial (NCT02110706) to determine whether rituximab is a safe and beneficial therapeutic for MG. BAFF is a potent survival factor for B cells and belimumab, a human mAb targeting BAFF, reduces B cell activation and differentiation into antibody-producing plasma cells. A phase II study of belimumab in AChR- and MuSK-related MG is in progress (NCT01480596). A placebo-controlled phase II study in patients with refractory generalized MG has shown that eculizubmab, a humanized mAb against C5 complement, is effective, promoting an ongoing phase III study (NCT01997229). Additionally, a trial of hematopoietic stem cell therapy in MG patients is in progress (NCT00424489).
Like many autoimmune diseases described in this review, MG is an ideal disease for antigen-specific immunotherapy because of its clearly defined autoantigen. Theoretically, an antigen-specific immunotherapy would eliminate the pathogenic autoimmune response to autoantigen specifically without affecting the other functions of the immune system, and thus avoiding severe adverse effects. Such antigen-specific immunosuppressive therapy is not yet available for MG (206). The idea of specific immunosuppression of autoimmune response to AChR by administration of AChR peptides or fragments has been investigated in EAMG for many years (181). However, translation into the clinic has been hampered by concerns about the potential for exacerbation of pathogenic autoimmune response, because the rationale behind these studies is based on a hypothesis that specific immunosuppression of autoimmune response requires the use of disease-inducing sequences (207). Recently, a vaccine using AChR cytoplasmic domains has been shown to be effective at specifically suppressing EAMG (175, 181, 208). Immunization with the vaccine in adjuvant prevents development of chronic EAMG, rapidly inhibits established EAMG, and prevents reinduction of EAMG for at least 6 months. Therapeutic effects might result from inhibition of production of pathological antibodies by a combination of antibody-mediated feedback suppression (Figure 6) and regulatory T cell-mediated active suppression. These studies indicate that not all autoantibodies are harmful. Autoantibodies to AChR cytoplasmic domain may play a therapeutic role in the antigen-specific immunotherapy. This approach may be applicable to other antibody-mediated autoimmune responses to other transmembrane proteins.
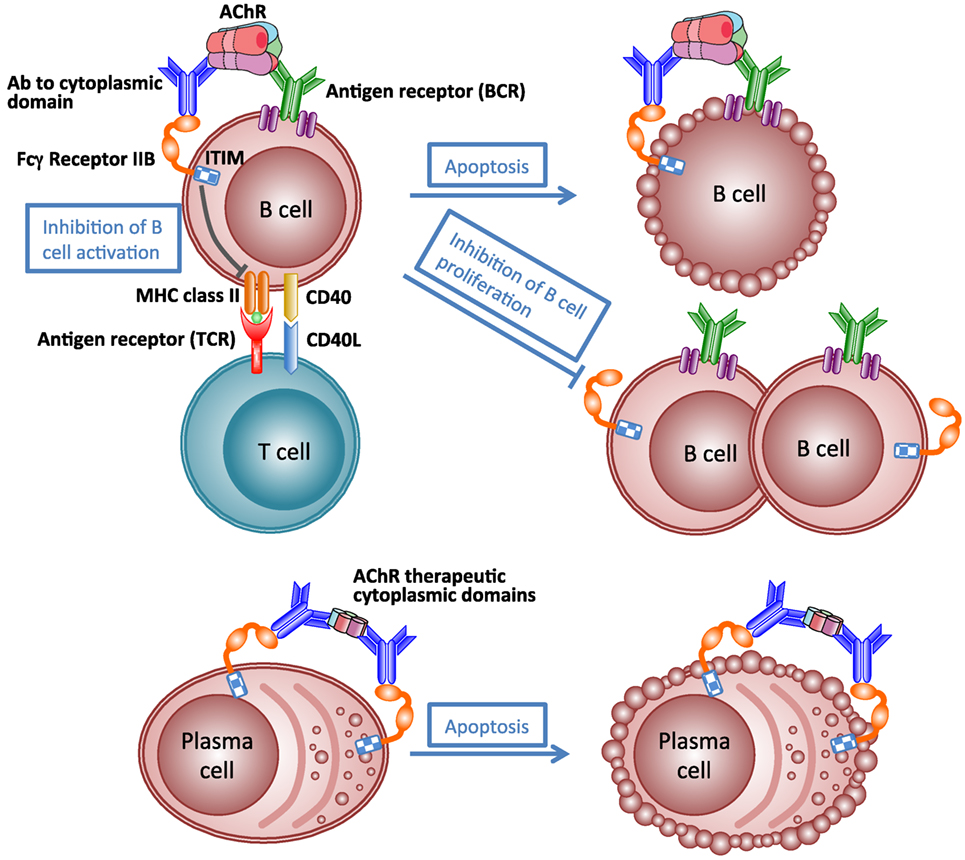
Figure 6. Schematic illustration of mechanism of antibody-mediated feedback suppression. Cross-linking of the B-cell receptor and the inhibitory IgG receptor (FcγRIIB) on the B cell surface by antigen–antibody complex may result in apoptosis of antigen-specific B cells, inhibition of B cell activation by helper T cells, and inhibition of B cell proliferation. Cross-linking FcγRIIB on the surface of plasma cells by immune complexes induces apoptosis of plasma cells.
Anti-NMDA Encephalitis
The recently discovered anti-N-methyl-d-aspartate-receptor (NMDAR) encephalitis—is the most common antibody-associated acute autoimmune encephalitis (209). Incidence has been estimated at 3–5/1.000.000/year (210). Although a rare disease, its description has greatly influenced neuroimmunology and neuroscience. It represents a model disease for a group of syndromes characterized by detection of autoantibodies targeting synaptic structures, hence called synaptic encephalitis (211).
Anti-NMDAR encephalitis preferentially occurs in young adults and children, predominantly women (80%). Approximately 70% of the patients develop prodromal symptoms, e.g., headache or fever, followed by rapid change of behavior including anxiety, hallucinations, and psychosis. Abnormal movements (orofacial dyskinesias, chorea, stereotyped movements) and eventually decrease of consciousness, coma, and severe global autonomic dysregulation (sometimes leading to hypoventilation and asystolia) ensue. Seizures and status epilepticus may occur at any stage of the disease. Brain MRI is normal in most but cerebrospinal fluid (CSF) shows non-specific inflammatory changes in almost all cases. Around 40% of the patients have an underlying neoplasm. The majority of tumors are ovarian teratomas. Approximately, 50% of patients respond well to IVIGs, steroids, or plasma exchange and the other 50% require rituximab alone or combined with cyclophosphamide. During recovery and after the acute symptoms have resolved, patients continue with deficits of memory and attention, impulsivity, behavioral disinhibition, and executive dysfunction that usually improve over many months. However, in some patients, recovery is incomplete, may take years, and mortality due to intensive care complications can be as high as 7% (212–214).
Diagnosis of anti-NMDAR encephalitis relies on the detection of IgG autoantibodies targeting a highly conserved and very restricted epitope on the aminoterminal-domain of the GluN1 subunit of the heterotetrameric ionotropic glutamate receptor NMDAR in patients’ CSF and serum by indirect fluorescence or ELISA (212, 215). A strong line of experimental and clinical evidence has been gathered, showing that NMDAR autoantibodies directly—without any other humoral or cellular components of the immune system involved—and reversibly interfere with NMDAR mediated synaptic function and disrupt central nervous system network function.
The pathophysiology of anti-NMDAR encephalitis can be divided into (1) initiation of systemic immune response, (2) propagation of antibody-producing cells into the central nervous system, and (3) effects of the antibodies on synaptic function (Figure 7).
(1) In paraneoplastic anti-NMDAR encephalitis, ovarian teratomas show ectopic expression of NMDAR and a dense infiltrate of T-, B-lymphocytes, and macrophages together with complement deposition in the tumors (212). The tumor-induced inflammation likely provides co-stimulation to autoreactive NMDAR specific T- and B-lymphocytes causing a breach of endogenous tolerance, systemic immune response, generation of plasma cells, and autoantibodies (Figure 7, steps 1–3). However, not all ovarian teratomas induce NMDAR antibodies presumably due to tumor-intrinsic and host-intrinsic factors (e.g., MHC class II haplotypes) (Figure 7A) (217). The initial trigger in idiopathic cases is currently unknown (Figure 7B). Interestingly, HSV-1 encephalitis can induce secondary anti-NMDAR encephalitis (218–222), and further post-infectious encephalitis variants are emerging (e.g., Japanese B-encephalitis). Furthermore, idiopathic anti-NMDAR encephalitis appears to occur seasonally in children (223). Thus, it is intriguing to speculate that in some “idiopathic” cases, yet unidentified viral infections might prove to be a trigger of anti-NMDAR encephalitis.
(2) A systemic NMDAR-directed immune response by itself does not appear to be sufficient to cause symptoms (Figure 7, step 4). Serum NMDAR antibodies can persist long after symptoms have resolved and serum titers do not correlate well with symptoms (224). The intact blood–brain barrier (BBB) prevents systemic antibodies and complement from reaching the synapse [CNS IgG levels are 400-fold lower than in serum (225)]. However, CSF NMDAR titers are correlated with disease activity (224). They are produced by dense infiltrates of plasma cells found perivascular in the CNS (Figure 7, steps 6–8) (226). The driving force behind establishing these CNS-local plasmablasts and -cells is unknown. A “second hit” hypothesis postulates a secondary infectious or inflammatory condition attracting preexisting NMDAR-antibody-producing cells to the CNS (Figure 7, step 5). Some support to this hypothesis is provided by detection of high levels of the B-cell-attracting chemokine CXCL13 in the CSF of anti-NMDAR encephalitis patients (227). Other chemokines and cytokines are also likely to be involved. Elucidation of the responsible “second hit,” the chemokines involved and the possible contribution of T-lymphocytes and long-lived plasma cells hidden behind the BBB would provide a new angle and rationale for treatment of patients, e.g., IL6-directed treatment or proteasome inhibitor treatment. It might furthermore help to explain the beneficial effects of the CD20-antibody rituximab in this condition in spite of its inability to cross the intact BBB.
(3) For anti-NMDAR encephalitis, a direct pathogenic role of GluN1 IgG antibodies has been established in cultures of neurons and after cerebroventricular infusion of patients’ antibodies to rodents (Figure 7, step 9). The antibodies are mainly composed of complement-fixing IgG1 and IgG3 isotypes, yet complement deposition is not a pathological feature of the disease (228–230). This is likely due to the low concentration of complement components in the CSF. Using cultured neurons, patients’ antibodies cause cross-linking and selective internalization of NMDARs that correlate with the antibody titers. These effects were reversible after removing the antibodies and dependent on the cross-linking ability but not the Fc-terminus of the antibodies (230). In contrast to the intense effects on NMDAR, patients’ antibodies did not alter the localization or expression of other synaptic proteins, number of synapses, dendritic spines, dendritic complexity, or cell survival (216, 231, 232). A transfer murine model using continuous ventricular infusion (14 days via osmotic pumps) of CSF from patients with anti-NMDAR encephalitis showed profound effects on memory and behavior in parallel to progressive hippocampal antibody binding and a decrease of total and synaptic NMDARs, without affecting PSD95 or AMPAR. These effects gradually improved after stopping the antibody infusion, with reversibility of symptoms accompanied by restoration of NMDAR levels, establishing the pathogenicity of the antibodies (233). A need to develop new therapies targeting this direct pathogenic effect of autoantibodies derives from the fact that some patients suffer from severe symptoms and prolonged intensive care treatment in spite of aggressive immunosuppressive therapy. In these, long-lasting and therapy-refractive local autoantibody production in the CNS by plasma cells hidden within the BBB might be the cause (224). Future therapies could (1) interfere with binding of NMDAR-antibodies to its target antigen as has been successfully shown in vivo with the co-administration of Ephrin-B2 in an animal model (234); (2) apply BBB-permeable decoy approaches reducing effective NMDAR-antibody burden in the CNS; and (3) counteract symptoms via modulating antagonistic neurotransmitters to rebalance disturbed network functions.
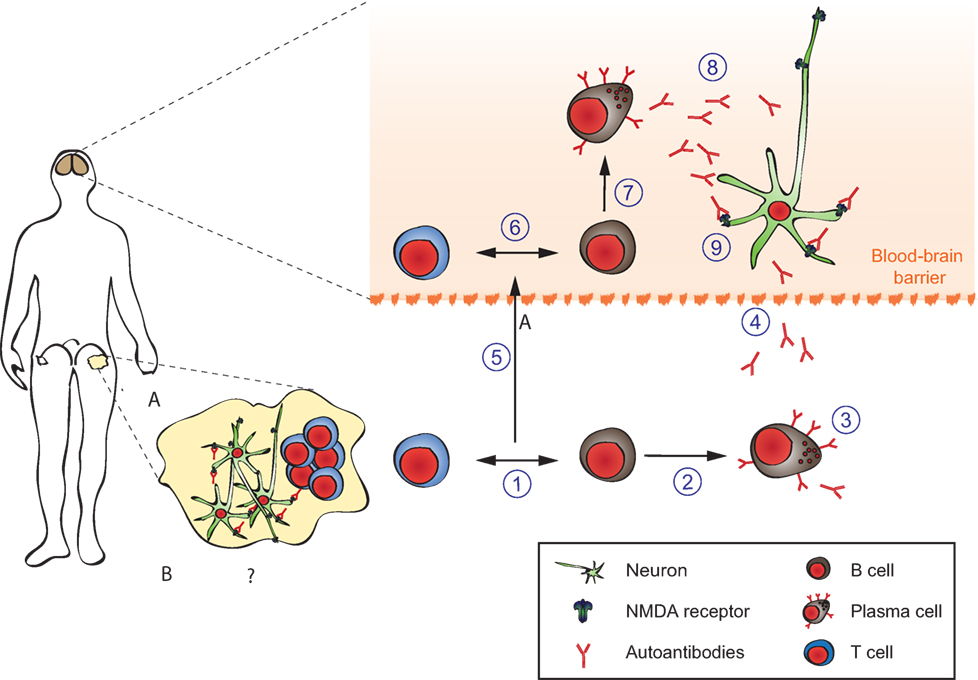
Figure 7. Pathophysiology of anti-N-methyl-d-aspartate-receptor (NMDAR) encephalitis. Details are explained in the text, where the numbering corresponds to the labeling used here. In brief, ectopic expression of NMDA receptors in ovarian teratomas together with co-stimulatory signals (A) or unknown triggers (B) lead to systemic immune response and formation of antigen-specific circulating B-, T-, and plasma cells producing NMDAR antibodies (1–4). The latter do not reach neurons in sufficient concentratiosn to exert effects due to the blood–brain barrier (dashed line). An unknown secondary trigger (e.g., systemic infections, 5) eventually mediates transition of B-, T-, and plasma cells into the brain. The resulting antibody production effectively increases intrathecal NMDAR antibody concentration above a threshold overwhelming neuronal compensation mechanisms and leading to net loss of surface NMDAR resulting in neurological symptoms. Adapted from Moscato et al. (216).
Induction of Structural Changes and Alterations in Signaling by Autoantibodies in Pemphigus Diseases
Epidemiology, Clinical Presentation, Histology, and Immunohistology of Pemphigus
Pemphigus diseases are a group of rare, but prototypical autoimmune blistering, skin conditions with autoantibodies against defined structural antigens of epidermal keratinocytes (235). There are two major types of pemphigus diseases, pemphigus vulgaris (PV) and pemphigus foliaceus (PF). The incidence depends on the population and is 1.6 per 100,000 adults in the Jerusalem area (236), but 0.7 for the total UK population (237) in PV. For PF, there is an endemic form (i.e., fogo selvagem) in Brazil, accounting for an incidence of 1–4 cases per 1,200 person years (238). Clinically, in PV, oral mucous membranes are usually affected, with facultative involvement of the skin (235). Affections in PF are only seen on the skin and blisters are even more fragile. Here, patients usually present with scaly crusted lesions, because blisters break very early after formation (235).
Histologically, pemphigus lesions show loss of intercellular adhesion of epidermal keratinocytes (239). All patients show intercellular staining of IgG within a biopsy of perilesional epidermis, whereas more than 80% of patients feature circulating IgG directed against keratinocyte surfaces in their serum (240). Multiple lines of evidence exist for the finding, that the main antigens targeted in pemphigus are desmogleins (Dsgs), desmosomal transmembrane glycoproteins that mediate epidermal cell–cell adhesion, and different expression (and compensation) of Dsg3 and Dsg1 within epidermis and mucous membranes explains the localization of blisters in PV and PF patients (235, 240, 241). Other antigens targeted in a minority of pemphigus patients may include other desmosomal cadherins (e.g., desmocollins, Dscs), classical cadherins or even other autoantigens (242–248). Knowledge of these antigens targeted in pemphigus patients allowed for development of serological tests as immunofluorescence and ELISA, and titers measured usually correlate with disease activity (see below) (249–251).
Brief Overview on Standard Treatment Options in Pemphigus
Standard present treatment is reviewed extensively elsewhere (235, 252), but critically depends on immunosuppression and modulation. Corticosteroids are effective within days when given systemically, likely because of increased synthesis of targeted Dsgs by keratinocytes, counteracting the desmosome-depleting effects of anti-Dsg antibodies in patients (253). Anti-CD20 therapy with rituximab represents potentially very effective therapy, leading to complete remission off all therapy in about 90% of pemphigus patients treated with one or more cycles of intravenous CD20+ B-cell-depleting rituximab (109). All of these options have their own limitations and challenges, and there are patients resistant to multiple regimens of therapy illustrating the need for a better understanding of the pathophysiology of pemphigus on a molecular level -including signaling pathways implicated into autoantibody-induced blistering- hopefully resulting in more specific therapy.
Autoantibodies Alone Cause Skin Pathology in Pemphigus
Early experiments using passive transfer of human IgG from pemphigus patients into mice demonstrated dose-dependent recapitulation of human pathology (254). Disease induction is possible with bivalent F(ab′)2 and monovalent Fab′ fragments purified from PF patients’ sera as well with monovalent scFv fragments cloned by antibody phage display from pemphigus patients (255–258). Consequently, Fc-dependent mechanisms are dispensable in pemphigus pathophysiology, and autoantibodies directly mediate disease by interfering with the interaction of desmoglein molecules on the outside of keratinocytes, either by interference with homophilic trans- or cis-interaction of Dsg molecules (Figure 8) (259–261). Further evidence for direct interference with adhesion comes from epitope-mapping studies of pemphigus autoantibodies on domain-swapped and point-mutated Dsg1/Dsg3 molecules, mapping most of the dominant epitopes bound by patients’ sera to the aminoterminal ectodomains of Dsg1/3, which are critical for adhesion (262). Finally, pemphigus antibodies bind to calcium-dependent conformational epitopes, something seen in adhesion mediated by cadherins as well, again suggesting that patients’ autoantibodies do bind to domains important for adhesion. Interestingly, Dsg3-specific T cells have also been demonstrated to cause cutaneous pathology by inducing interface dermatitis when injected into lymphocyte-deficient mice (263).
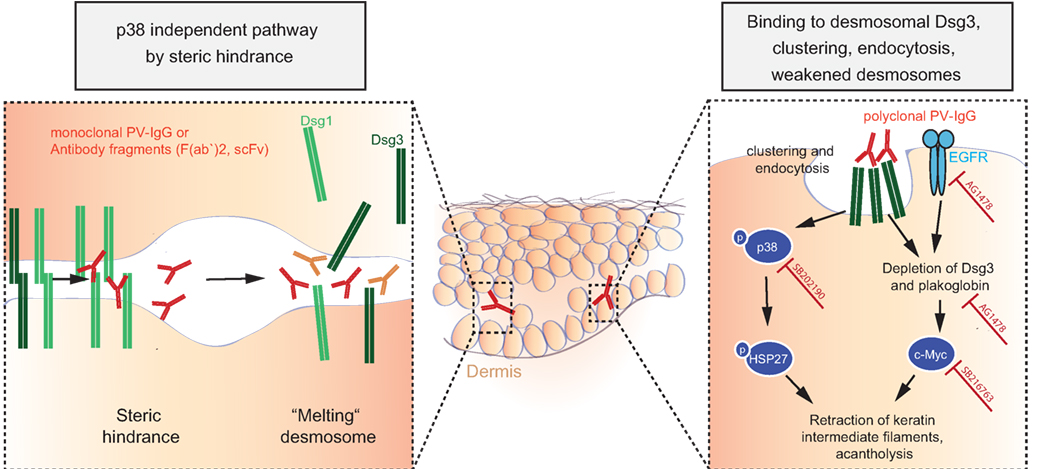
Figure 8. Autoantibody-induced loss of cell–cell adhesion in pemphigus. For explanation, please refer to the text.
Clustering, Internalization, and Depletion of Desmosomal Dsgs by Autoantibodies
Various researchers have been proposing other models, which are most likely not opposing the model of steric hindrance but rather complementing observed pathologies in pemphigus: the Dsg non-assembly depletion hypothesis describes how divalent anti-Dsg antibodies cross-link and cluster Dsgs, leading not only to internalization of non-junctional membrane-bound Dsgs but also resulting in prevention of incorporation of newly synthesized Dsgs into forming desmosomes (264, 265). Consequently, desmosomes become depleted of Dsgs and fail to provide adhesion. This model is supported by data from patients and from cell culture experiments, showing clustering of Dsg3/1 by Dsg3/1-specific autoantibodies (266–268), and by the observation that forced expression of Dsg3 by adenoviral delivery can prevent autoantibody-mediated Dsg3 depletion of desmosomes and acantholysis (267).
Autoantibody-Mediated Changes in Cell Signaling Contribute to Loss of Cell Adhesion in Pemphigus
The observations that polyclonal IgG from PV patients causes retraction of keratin intermediate filaments (thus contributing to intercellular loss of adhesion) in cultured murine wild-type keratinocytes and that plakoglobin (PG)-deficient mice do not show this reaction pointed to intracellular signaling mechanisms contributing to acantholysis (269). Further research then demonstrated that PG is a suppressor of c-Myc expression and that PV autoantibodies trigger c-Myc upregulation by depletion of PG together with Dsg3; increased c-Myc then leads to cell proliferation and weakened intercellular adhesion. These findings were corroborated by pharmacological inhibition of c-Myc, resulting in an inhibition of the ability of PV autoantibodies to cause acantholysis in mice (270, 271). As another signaling cascade extensively studied in pemphigus pathophysiology is the p38MAPK-signaling pathway (272–274). HSP27 and p38MAPK have been shown to be phosphorylated upon incubation of human keratinocyte cell cultures with PV-IgG (272, 273) and to be linked to internalization of Dsg3 (275). Blister formation was blocked in vivo by pharmacological inhibition of p38MAPK and its downstream targets when studied in the passive transfer mouse model for PV and PF (274, 276, 277), and this signaling cascade was also shown to be functional in patients’ skin (278). Downstream of p38, epidermal growth factor receptor (EGFR) signaling was shown to be activated in human keratinocytes after addition of PV autoantibodies, and again, inhibition of EGFR signaling prevented blistering induced by PV-IgG in mice (118). What has not been fully resolved is the issue of whether p38 activation is a primary event causing acantholysis or is secondary to initial loss of cell adhesion (235).
Attempting to integrate published, but sometimes seemingly contradictory data, recent research has pointed out that (i) steric hindrance and (ii) Dsg3 clustering, depletion, and signaling are distinct events. Monoclonal pathogenic antibodies can cause loss of intercellular adhesion through steric hindrance without relying on p38MAPK signaling, whereas polyclonal PV-IgG autoantibodies cause Dsg3 clustering and endocytosis via a p38MAPK-dependent pathway (235, 260). Recent data support these findings and suggested that, after induction of acantholysis and activation of p38MAPK signaling by polyclonal PV-IgG, effective modulation of the signaling pathways involved in acantholysis may even outbalance direct inhibition of Dsg3 binding by PV-IgG and rescue cell adhesion (279), potentially by upregulating other cell adhesion molecules not targeted by anti-Dsg3 antibodies (e.g., Dsg1, Dscs, cadherins).
Emerging Treatments and Novel Therapeutic Targets Based on Modulation of Signaling and Cell–Cell Adhesion
All these cell biological studies of signaling and desmosomal adhesion in pemphigus have led to innovative and new approaches to treatment. For example, cross-linking the adhesive interfaces of Dsgs by use of a specific tandem peptide successfully stabilized adhesion and inhibited PV-IgG-mediated activation of the central p38MAPK pathway and skin blistering (119). Overexpression of plakophilin-1, an intracellular armadillo protein that links desmosomal cadherins to keratin intermediate filaments of keratinocytes, resulted in hyperadhesive desmosomes that were significantly less prone to PV-IgG-mediated pathology (280). Introducing a point mutation into DP (DP-S2849G) led to inhibition of both Dsg3 depletion from the cell surface and keratin filament retraction caused by PV-IgG (281), by preventing protein kinase C-dependent phosphorylation of DP at that specific site. Since the protein kinase C inhibitor Bim-X has the same inhibiting effects, this compound may serve as an important new pharmacological tool in pemphigus.
Interestingly, studying the precise mechanisms of seemingly “boring” but very effective corticosteroid therapy in pemphigus disease can yield new therapeutic insights as well. As previously assumed, corticosteroids do upregulate Dsg3 transcription in primary human keratinocytes. In addition, recent data point to inhibition of Stat3 as a key mechanism. Similarly, inhibition of mTOR by rapamycin (i.e., sirolimus) is Stat3-dependent and upregulating Dsg3 transcription, explaining how PV-IgG-mediated effects are antagonized by rapamycin and corticosteroids (282).
Very recently, researchers in the pemphigus field have made an important contribution to the treatment of all autoimmune disease: engineered human T cells that express a chimeric autoantibody receptor (CAAR), consisting of the PV autoantigen, desmoglein (Dsg) 3, fused to CD137–CD3z signaling domains were generated. These Dsg3 CAAR-T cells showed specific cytotoxicity against cells expressing anti-Dsg3 BCRs in vitro and expand, persist, and specifically eliminate Dsg3-specific B cells in vivo. Thus, CAAR-T cells may provide an effective and also common strategy for specific targeting of autoreactive B cells in antibody-mediated autoimmune disease (117).
Taken together, these data on autoantibody-mediated changes of cell signaling in a group of prototypic autoimmune diseases illustrate that studying the potentially multifaceted roles of the target antigens is of the utmost importance: as shown for the desmosomal autoantigens in pemphigus diseases, Dsgs are not just structural components critical for adhesion but also critical regulators of signal transduction, affecting differentiation, cell homeostasis, and carcinogenesis (283–285). Integrating all these and dissecting the precise mechanisms of standard therapy may suggest promising new targets for therapy and modulation of autoimmunity.
Autoantibody-Induced TTP: Anti-ADAMTS13 Antibodies
Clinical Presentation, Epidemiology, Treatment, and Treatment Challenges
Thrombotic thrombocytopenic purpura is a thrombotic microangiopathic disorder caused by a deficiency in the multidomain metalloprotease ADAMTS13 (A Disintegrin And Metalloprotease with ThromboSpondin type 1 repeats, number 13) (286, 287) (Figure 9A). In the absence of ADAMTS13, ultra-large hyperactive von Willebrand factor (VWF) multimers accumulate in the circulation and spontaneously bind platelets. The resulting VWF-rich microthrombi block capillaries and arterioles in different organs, thereby preventing oxygen supply (Figure 9B). Patients suffer from recurrent episodes of severe organ failure, which without urgent treatment can be fatal. Organ involvement is variable and can comprise multiple organs, most predominantly brain, heart, and kidney (287). TTP is clinically diagnosed by the occurrence of severe thrombocytopenia and hemolytic anemia (288, 289). It is assumed that platelets are consumed in the microthrombi and that red blood cells rupture as they are pushed through the blocked microcapillaries and arterioles resulting in the appearance of schistocytes, as well as increased levels of hemoglobin and haptoglobin. Tissue damage resulting from occlusive VWF-rich microthrombi furthermore results in increased levels of lactate dehydrogenase. TTP diagnosis is ascertained by determination of ADAMTS13 activity (<10%) allowing differentiating TTP from other microangiopathic disorders like hemolytic uremic syndrome (286). Congenital TTP is caused by the presence of mutations in the ADAMTS13 gene and occurs in ~5% of the TTP patients. About 95% of TTP patients suffer from acquired TTP. These patients develop anti-ADAMTS13 autoantibodies and hence suffer from the autoimmune form of the disease (290). Acquired TTP with severe ADAMTS13 deficiency is an orphan disease with an incidence rate of around 2–6/1,000,000 patients per year (286, 291). Since in a subset of TTP patients in remission, ADAMTS13 activity is still below 10%, secondary triggers like pregnancy, infection, and surgery have been suggested to induce TTP.
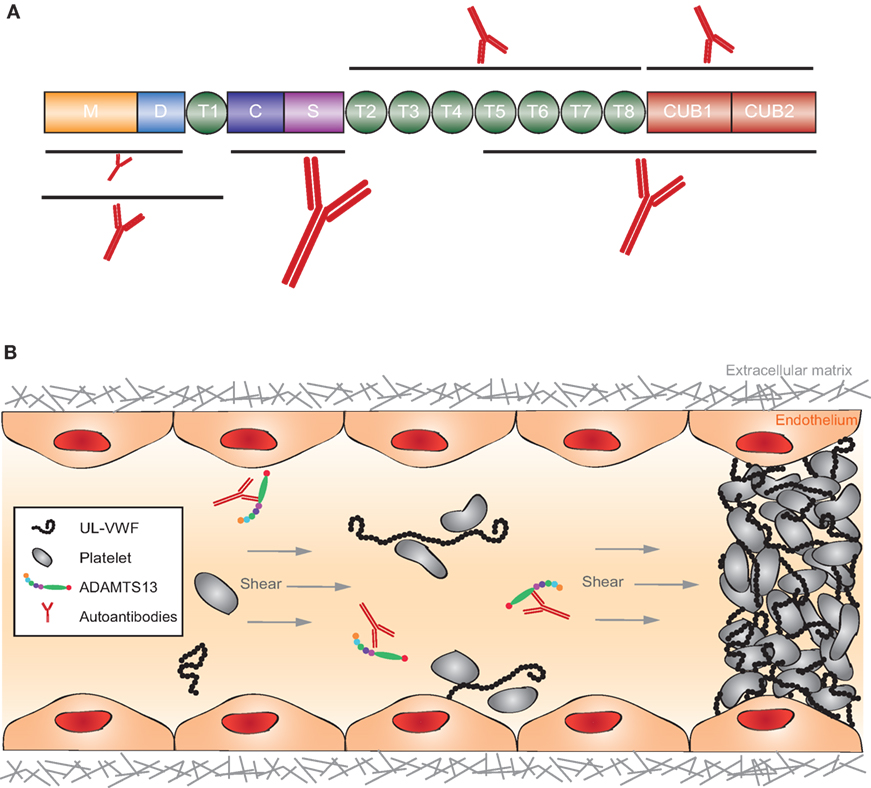
Figure 9. Structure of ADAMTS13 and the frequency of autoantibodies targeting specific domains and graphical representation of the pathophysiology of acquired thrombotic thrombocytopenic purpura (TTP). (A) ADAMTS13 consists of a metalloprotease (M; orange) and disintegrin-like (D; blue) domain, a thrombospondin type-1 repeat (T1; green), a cysteine-rich (C; purple) and spacer (S; pink) domain, seven additional thrombospondin type-1 repeats (T2–T8; green), and two CUB domains (CUB1-2; red). Epitope mapping of anti-ADAMTS13 autoantibodies revealed that most patients (90–100%) have autoantibodies against the cysteine-spacer domain (indicated with the largest antibody). The size of the other antibodies in this figure demonstrates the relative frequency of these autoantibodies in the plasma of acquired TTP patients. (B) In normal conditions, UL-VWF released from activated endothelial cells is directly proteolysed by ADAMTS13, preventing spontaneous platelet binding. In the pathophysiology of acquired TTP, autoantibodies inhibit ADAMTS13 activity. UL-VWF multimers accumulate in the circulation and spontaneously bind platelets. This process results in the formation of VWF-rich microthrombi blocking the circulation in microcapillaries and arterioles.
Infusion with fresh frozen plasma is the treatment of choice for congenital TTP patients to provide sufficient levels of functional ADAMTS13. Plasma exchange, where patient plasma is exchanged with fresh frozen plasma, is used to treat acquired TTP patients in order to remove anti-ADAMTS13 autoantibodies and to supply active ADAMTS13. To treat the underlying autoimmune disorder in acquired TTP, an immunosuppressive corticosteroid therapy is started together with the plasma exchange therapy (288, 289, 292). Rituximab, the anti-CD20 mAb targeting mature and memory B-cells, is now additionally administered to those acquired TTP patients experiencing a suboptimal response to plasma exchange and corticosteroid therapy and to patients suffering from TTP relapses (293–295). Although standard therapy (plasma exchange and immunosuppressive therapy) reduces the mortality rate in 80–90% of TTP patients, mortality remains at 10–20% (292). In addition, plasma exchange is a challenging therapy for TTP patients as the patients have a poor clinical condition and a high number of plasma exchanges is required with large volumes of plasma. Major complications linked with plasma exchange are observed and are related to systemic infection, venous catheter obstruction, hypotension, hypoxia, venous thrombosis, non-fatal cardiac arrest, and anaphylaxis (288).
Pathogenesis of Acquired TTP
Autoantibodies play a predominant role in the pathophysiology of acquired TTP. Anti-ADAMTS13 autoantibodies either inhibit ADAMTS13 function and/or clear ADAMTS13 from the circulation (296). Both processes result in the absence of active ADAMTS13 in patient plasma leading to the accumulation of prothrombotic ultra-large VWF multimers and spontaneous VWF-rich microthrombi formation, the hallmark of acute TTP episodes. The presence of both, free anti-ADAMTS13 autoantibodies and circulating autoantibody–antigen immune complexes, has been described in plasma of acquired TTP patients (297). Anti-ADAMTS13 autoantibodies are mainly of the IgG class, with IgG4 being the most prevalent, but IgM and IgA autoantibodies have also been reported (298). The IgG4 subclass is also most predominantly observed in circulating immune complexes (297). Accordingly, moderately elevated complement activation is observed during acute TTP episodes (299–301). It is however unclear whether the circulating autoantibody–ADAMTS13 immune complexes activate complement and play a role in the pathophysiology of TTP.
Antibody titers change during acute phases and remission. The autoimmune response in acquired TTP is polyclonal but with immunodominant epitopes in the ADAMTS13 spacer domain (Figure 9A) (290, 302). Plasma screening of acquired TTP patients against the different ADAMTS13 domains revealed that 90–100% of acquired TTP patients have autoantibodies with an epitope in the spacer domain (Figure 9A) while 30–50% of the patients additionally have antibodies directed against other ADAMTS13 domains (M, D, T, C, T2-8, and CUB1-2, Figure 9A) (290, 296, 302). Many anti-spacer autoantibodies have been cloned from acquired TTP patients and were shown to have a strong, weak, or absent in vitro inhibitory effect on ADAMTS13 function (296, 302–305). The ADAMTS13 spacer domain contains a major exosite for binding to its substrate VWF explaining why anti-spacer domain autoantibodies can inhibit ADAMTS13 function. Non-inhibitory cloned anti-spacer antibodies are expected to clear ADAMTS13 from the circulation as it was demonstrated by Thomas et al. that the IgG fraction isolated from acquired TTP patients contains non-inhibitory anti-ADAMTS13 autoantibodies and that ADAMTS13 antigen levels were low in these patients (296). Only a few anti-ADAMTS13 autoantibodies with an epitope outside the spacer domain have been cloned. These comprise autoantibodies with an epitope in the ADAMTS13 metalloprotease, disintegrin, and CUB domains, but their effect on ADAMTS13 function has not yet been studied (305, 306). Anti-metalloprotease autoantibodies could inhibit ADAMTS13 function, as the metalloprotease domain harbors the active site of the enzyme. Anti-disintegrin or anti-CUB autoantibodies could have similar effects, as these domains are involved in binding and docking ADAMTS13 to VWF, respectively. However, in addition, autoantibodies could merely clear ADAMTS13 from the circulation. Whether deposition of circulating immune complexes in tissues also play a role in the pathophysiology of acquired TTP remains to be determined.
We showed that injection of a murine anti-metalloprotease mAb that potently inhibited ADAMTS13 function in vitro could induce acquired TTP in baboons (307). This experiment shows that anti-ADAMTS13 antibodies play a central role in the pathophysiology of acquired TTP. Recently, it was shown that injection of a cloned inhibitory anti-spacer single chain (sc) Fv autoantibody into mice also resulted in acquired TTP when TTP symptoms were triggered with Shiga toxin or recombinant VWF (305, 308). TTP symptoms presented due to full inhibition of ADAMTS13 activity in both animal models, as animals injected with non-inhibitory control antibodies did not develop TTP symptoms. However, not all acquired TTP patients have strong inhibitory anti-spacer autoantibodies and 30–50% of the patients also have autoantibodies against other ADAMTS13 domains.
Hence, animal models where individual autoantibodies or mixtures of these autoantibodies are injected need to be performed to further unravel the contribution of all these autoantibodies in the pathophysiology of acquired TTP. Indeed, since the immune response in acquired TTP is polyclonal, it is reasonable to speculate that multiple autoantibodies will contribute to inhibition and/or clearance of ADAMTS13 in 30–50% of the patients. It is currently less obvious to study the clearance of anti-ADAMTS13 autoantibodies as most available cloned autoantibodies are either mounted on an IgG1 or IgG4 scaffold, or are scFv fragments. Mouse models cannot be used to perform these studies as clearance mechanisms in mice are too different from those in humans.
The mechanism of autoantibody development in acquired TTP patients is currently not known but both environmental and genetic factors will probably play a role as shown in other immune diseases. Indeed, HLA-DRB1*11 has been shown to be overrepresented in patients with acquired TTP compared to controls (309). The role of specific CD4+ T-cells in acquired TTP has only recently been investigated. It was shown that ADAMTS13 is endocytosed by dendritic cells via the macrophage mannose receptor (310). Peptides derived from the carboxy-terminal CUB2 domain of ADAMTS13 were preferentially presented, showing that the CUB2 domain contains potential immunodominant T cell epitopes (310). Investigating ADAMTS13 reactive CD4+ T cells from acquired TTP patients showed that CUB2 domain reactive CD4+ T cells might be involved in the etiology of acquired TTP (311).
Emerging Treatments and Novel Therapeutic Targets
Recombinant (r)ADAMTS13 is currently under development as an alternative treatment for TTP. It is effective to treat congenital TTP symptoms in mice (312) and acquired TTP symptoms in rats, where rADAMTS13 was able to overcome circulating inhibitors and reconstitute ADAMTS13 activity (313). A Phase I clinical study is currently going on to assess rADAMTS13 in the treatment and prophylaxis of congenital TTP (NCT02216084). Apart from providing active ADAMTS13 to patients, a treatment has been developed that prevents the formation of VWF-rich microthrombi, which play a detrimental role in TTP pathophysiology. Others and we showed that administration of an antibody blocking the binding between VWF and platelets results in the prevention and treatment of acquired TTP in preclinical animal models (314, 315). The inhibitory anti-VWF nanobody caplacizumab in combination with standard therapy was tested in clinical trials and induced a faster resolution of acute TTP episodes compared to placebo in acquired TTP patients (112). A second therapy that aims at preventing or reducing VWF–platelet interactions is the FDA approved drug N-acetylcysteine (NAC) currently used to treat acetaminophen overdose or to decrease the viscosity of mucous secretions in respiratory disorders. NAC reduces disulfide bonds in mucins and was shown to also reduce disulfide bonds in VWF multimers, thereby reducing the formation of VWF-platelet thrombi in mesenteric venules of mice (316). NAC has been administered to patients with refractory TTP but the outcome has been variable (317–320). To determine the clinical value of NAC, patients are currently being recruited (ClinicalTrials.gov: NCT1808521). Another drug that is evaluated to treat refractory TTP is the proteasome inhibitor bortezomib. Although case reports have suggested a benefit for the treatment of refractory TTP (73, 320), possibly by eliminating autoreactive plasma cells producing anti-ADAMTS13 antibodies (321), prospective clinical trials are needed to investigate the effectiveness of this drug in the treatment of refractory TTP.
Autoantibody-Induced Cell Lysis: Idiopathic Thrombocytopenic Purpura (ITP) and Autoimmune Hemolytic Anemia
Immune thrombocytopenic purpura (formerlyITP) and autoimmune hemolytic anemia (AIHA) are autoimmune diseases that are characterized by reduced levels of erythrocytes or platelets resulting in anemia or platelet deficiency, respectively. AIHA and ITP may both appear as secondary disease in consequence of a different medical condition or can be induced by medical treatments. In contrast to these secondary forms, the pathogenesis of the primary diseases is not fully understood but in the majority of patients, autoantibodies directed against erythrocytes or platelets, respectively, can be detected.
Primary AIHA seems to be exclusively caused by such autoantibodies while in primary ITP these can only be detected in 60% of the patients (322). In the residual cases, a contribution of cytotoxic T cells is considered (323) but it is likely that autoantibody levels are simply undetectable due to the quantitative binding to platelets and their subsequent fast elimination. The removal and destruction of erythrocytes or platelets in AIHA or ITP, respectively, is mainly mediated by Fcγ-receptor (FcγR)-based mechanisms in cases where the autoantibodies are of the IgG class. While the role of complement in the removal of platelets does not play a prominent role in ITP (324), the situation in AIHA is somewhat different. In a considerable fraction of patients, IgM autoantibodies are involved that efficiently can activate complement, which is not the case when erythrocytes are opsonized by IgG (325). As the different pathways of red blood cell destruction require also different treatments, the determination of the involved autoantibody Ig class is essential.
The contribution of FcγR-mediated effector functions to the pathogenesis of ITP triggered by opsonized platelets has identified an association with reduced expression levels or impaired signaling of inhibitory FcγRIIB (326, 327) as well as the presence of a functional copy of the activating FcγRIIC gene (328). Similar genetic association studies have been conducted for the role of the activating FcγRs. For the FcγRIIA H/R131 dimorphism, an elevated prevalence of the FcγRIIA-R131 allele, which has a lower affinity for IgG2 and a higher one for IgG3, was observed (329, 330), while for that of FcγRIIIA (F/V158) diverging results have been reported in ITP patients (331–333).
While the impact of activating FcγRs on both human heterogeneous diseases remains unclear, studies in mouse models of experimental AIHA and ITP could demonstrate a contribution to disease severity mediated by activating FcγRs, namely, FcγRIIIA (329, 334–337). In a therapeutic approach, the hypothesis was tested if blockade of FcγRIII can improve ITP in chronic refractory patients. Using an anti-FcγRIIIA antibody, around 50% of the included patients responded with significantly improved platelet levels but treatment was accompanied by severe adverse events. Using several engineered versions of the anti-FcγRIII antibody, the authors demonstrated that the observed adverse events were independent of Fc-mediated effector functions or immunogenicity (338) but rather depended on the dimerization of FcγRIII by both Fab fragments of the used antibody. This finding was supported by a study using a passive ITP mouse model in which a monovalent anti-FcγRIII antibody was used. This construct which blocks immune complex-mediated engagement of FcγRIII mediated an efficient inhibition of antibody-dependent platelet removal without triggering adverse events (339).
In a related approach, a soluble FcγR was used with the intention to compete with cellular FcγRs for the binding of opsonized platelets. As a consequence, the platelet binding to FcγRs is blocked and destruction and cellular activation of the immune cells is inhibited (340). In chronic ITP patients, the drug was well tolerated and induced a sustained platelet response during the 3-month follow-up period (341). Both approaches underline the relevance of the FcγR system in autoimmune diseases and may provide new treatment options for the management of ITP, AIHA, and eventually other antibody-mediated autoimmune diseases.
Autoantibody-Induced Vascular Inflammation
Antineutrophil cytoplasmic autoantibody (ANCA)-associated vasculitides (AAV) comprise a group of three different rare and potentially life-threatening chronic inflammatory vessel diseases of unknown etiology: granulomatosis with polyangiitis (GPA), microscopic polyangiitis (MPA), and eosinophilic granulomatosis with polyangiitis (EGPA). GPA is associated with proteinase 3 (PR3)-specific ANCA (PR3-ANCA), whereas MPA and—less commonly—EGPA are associated with myeloperoxidase (MPO)-specific ANCA (MPO-ANCA). Usually P33 and PR3-ANCA are detected by indirect fluorescence (see below). AAV predominantly affect small vessels, i.e., intraparenchymal small arteries, arterioles, capillaries, and venules. The vasculitis is histopathologically characterized by fibrinoid necrosis and paucity of immune complexes (“pauci-immune vasculitis”). In addition to systemic vasculitis, GPA displays a peculiar propensity for a predominantly extravascular necrotizing granulomatous inflammation mainly affecting the upper and/or lower respiratory tract. In EGPA, extravascular eosinophilic infiltration is found in various tissues. In contrast, MPA displays no extravascular granulomatous inflammation (342–344).
In central Europe, GPA is the most common form of AAV (incidence 10 per million, prevalence 98 per million), followed by MPA (incidence 2 per million, prevalence 28 per million) and EGPA (incidence 1 per million, prevalence 23 per million). However, these numbers may underestimate the true incidence and prevalence of AAV due to referral bias. The mean age at disease manifestation lies between 50 and 70 years. Women and men are equally affected (345). AAV as a group are characterized by diversity and at the same time considerable overlap of clinical and pathological features. Malaise, flu-like symptoms, arthralgia, and myalgia often herald the onset of AAV. Respiratory tract manifestations are the most frequent presenting features in GPA. Involvement of upper and/or lower airways with rhinitis, sinusitis, and pulmonary infiltrates is a distinctive feature of GPA. Asthma, pulmonary infiltrates, and hypereosinophilia with eosinophilic vasculitis and inflammation affecting various organs are characteristic features of EGPA. Pulmonary-renal syndrome with alveolar hemorrhage as a result of capillaritis and rapidly declining renal function due to focal segmental necrotizing glomerulonephritis with crescent formation affects the majority of patients with full-blown GPA and MPA. Renal involvement is less common in EGPA. Owing to its systemic nature, the vasculitis may affect any organ in AAV. Localized forms of GPA restricted to the upper and/or lower respiratory tract and a renal-limited form of MPA are less commonly encountered (343).
Prior to the introduction of immunosuppressive therapy, AAV used to be an inevitably fatal condition. With the advent of corticosteroids and establishment of immunosuppressive cytotoxic treatment protocols in randomized trials, the outcome and survival rates have considerably improved (343, 346). However, the mortality ratio continues to be increased (2.6, 95% CI 2.2–3.1) compared with an age- and sex-matched general population (347). Despite further advances in treatment such as the use of rituximab for the induction and maintenance of remission, the course of AAV remains characterized by chronicity, risk of relapse, and complicating comorbidities. Thus, major goals of research are the prevention of disease through a personalized medical approach allowing the evaluation of predisposing genetic risk factors for disease manifestation and relapse, the determination of triggering environmental factors, and the development of combined targeted therapies intervening early in the break of tolerance and with mechanisms fostering chronic non-resolving inflammation in AAV (346).
Antineutrophil cytoplasmic autoantibodies are highly sensitive and specific for AAV. Combining immunofluorescence technique and ELISA (see below) for the detection of ANCA yields a sensitivity of 70% and specificity of 99% for AAV (348). ANCA levels do not strongly reflect disease activity. Therefore, ANCAs cannot be used for the guidance of immunosuppressive therapy in clinical practice (349). However, persistence of ANCA after the induction of remission is strongly associated with an increased risk for relapse in AAV (350). ANCAs are rarely detected in healthy controls. Interestingly, there is no evident co-occurrence with other autoantibodies suggesting differences in the break of tolerance between different autoimmune diseases (63). ANCA with specificity for a subset of immunodominant epitopes are associated with disease activity in AAV (351). ANCAs induce respiratory burst and degranulation of cytokine- and complement factor C5a-primed neutrophils in vitro (352, 353). Prestimulation of neutrophils with cytokines promotes translocation of the target antigens of ANCA on the cellular surface. Interaction of ANCA with both its target antigen on the cellular membrane and Fcγ-receptors IIa or IIIb is required for the full activation and subsequent degranulation of neutrophils (354, 355). Binding of ANCA to both, the membrane-bound ANCA targets MPO and PR3 and Fcγ-receptors IIa or IIIb, generates activation signals distinct form conventional Ig-induced activation via Fcγ-receptors (356). Genes of pro-inflammatory mediators are upregulated following stimulation with ANCA (357). ANCAs facilitate adhesion and transmigration of cytokine-primed neutrophils across the endothelial cell barrier in vitro and in vivo as observed in flow-chamber experiments and by intravital microscopy (358, 359). Transfer of MPO-ANCA generated from MPO-knockout mice immunized with mouse MPO induces glomerulonephritis and vasculitis in susceptible mice. The genetic background of the mouse determines the severity of glomerulonephritis in these models (360). The alternative complement pathway is activated in AAV. Complement factor C5a receptor deficiency ameliorates MPO-ANCA-induced experimental glomerulonephritis in mice (361). Owing to structural and biologic differences between human PR3 and its rodent homolog, the development of models for PR3-ANCA-induced vasculitis has remained a challenge. Pathogenicity of PR3-ANCA resulting in acute vascular damage has been demonstrated in immunodeficient chimeric mice following injection of human hematopoietic stem cells and PR3-ANCA (362). However, extravascular necrotizing granulomatous inflammation was not found in the respiratory tract of these mice, suggesting that the pathogenesis of granulomatous inflammation may be separate from acute systemic vasculitis and in particular T-cell-dependent in GPA (362, 363).
Clinical observations, the efficacy of B-cell-depleting therapy, and evidence from in vitro and in vivo experiments taken together all lend support to the concept that ANCAs are pathogenic and induce a necrotizing small-vessel vasculitis. However, unresolved questions concern the cascade of events leading to the induction of pathogenic ANCA and the pathogenesis of extravascular granulomatosis in GPA and EGPA. Thus, key elements of the pathogenesis of AAV and its characteristic immune pathology remains to be elucidated (346). While the uptake of apoptotic cells and debris, a process known as efferocytosis, is usually immunologically silent in healthy individuals, upregulation of PR3 on the cell surface of apoptotic neutrophils interferes with normal efferocytosis in GPA. Increased PR3 expression on apoptotic neutrophils induces the secretion of inflammatory cytokines including granulocyte-colony stimulating factor (G-CSF) by macrophages in vitro and in vivo. In turn, G-CSF upregulates PR3 expression on maturing neutrophils from GPA patients in the bone marrow. Thus, a G-CSF-driven autoamplificatory loop sustains non-resolving inflammation in GPA. Moreover, PR3-expressiong apoptotic cells skew the cytokine response of plasamocytoid dendritic cells toward the generation of Th2 and Th9 cytokines. In the presence of PR3-ANCA, the cytokine response was further skewed toward a Th17 response in a murine model. Accordingly, skewing of the cytokines response with an increase in circulating Th2, Th9, and Th17 cells was also detected in GPA patients (364). Notably, ectopic lymphoid structures and plasma cells displaying signs of autoreactivity are found in granulomatous inflammation rich in neutrophils in GPA (365). Once tolerance is broken and pathological autoreactivity toward PR3 and MPO established in AAV, the autoreactive immune response is further self-amplified by neutrophil extracellular traps (NETs). NETs are released by ANCA-stimulated neutrophils and contain the targeted autoantigens PR3 and MPO (366).
Taken together, the abovementioned studies suggest an immunological paradox in which cell death, i.e., necrosis, is not only the endpoint of ANCA-induced neutrophil degranulation and inflammation in AAV but also at the same time represents the starting point of a cascade of pathophysiological events leading to chronic non-resolving extravascular and vascular inflammation and the breakdown in self-tolerance. This paradox, however, could explain why neutrophil-derived antigens become the target of pathological autoreactivity and inflammation is sustained and does not resolve in AAV. The cascade may initially be triggered by environmental factors, e.g., infections, in genetically susceptible persons. G-CSF and/or IL-17 could become new therapeutic targets for the treatment of AAV. It is inferred that targeting G-CSF and IL-17 interferes with fundamental pathophysiologic mechanisms driving non-resolving chronic inflammation and the break of tolerance in AAV.
Autoantibody-Induced Inflammation
Autoantibody-Induced Cutaneous Inflammation: Pemphigoid Diseases (PD)
Pemphigoid diseases are characterized and caused by autoantibodies against distinct structural components of the dermal–epidermal junction. Junction proteins link the cytoskeleton of the basal keratinocytes to the extracellular matrix of the dermis. Binding of PD autoantibodies leads to the separation of the epidermis and dermis by a complex, yet relatively well-understood process. PDs comprise eight distinct disorders for which the molecular target antigens have been identified (367). Of these, due to the availability of well-defined animal models, bullous pemphigoid [BP, autoimmunity against type XVII collagen (COL17, BP180)] and epidermolysis bullosa acquisita [EBA, autoimmunity against type VII collagen (COL7)] are particularity well studied (368–371). We here will focus on EBA because the expression of COL7 beyond the skin, i.e., in the gastrointestinal tract (372, 373), leads to a severe and difficult-to-treat clinical presentation. The cutaneous manifestations in EBA are heterogeneous: two major clinical subtypes have been described. The mechanobullous (non-inflammatory, classical) variant of EBA is characterized by skin fragility, tense blisters, scaring, and milia formation preferably localized to trauma-prone sites. The inflammatory variant mimics other PDs, and widespread vesiculobullous eruptions are present. Independent of the clinical variation, extracutaneous manifestations are frequently observed. These include ocular, oral mucosa, esophagus, anal, vaginal, tracheal, and laryngeal lesions, which can lead to blindness, esophageal strictures, hoarseness, impaired phonation, and may led to irreversible respiratory distress (374). Overall, EBA is notoriously difficult to treat, and often long-term use of high doses of corticosteroids in combination with other immunosuppressants is required to achieve clinical remission (375, 376). Thus, there is a high, and so far unmet medical need, for the development of novel treatment strategies for patients with EBA.
One of the breakthrough discoveries in EBA was the identification of COL7 as the autoantigens in the late 1980 (377). Based on this insight, the pathogenic relevance of COL7 autoantibodies has been demonstrated (i) in vitro by demonstrating digestion of the dermal–epidermal junction in skin specimen incubated with anti-COL7 IgG or IgA and neutrophils (378, 379), (ii) in vivo by induction of inflammation and blistering in mice by transfer of anti-COL7 IgG (380, 381) or by immunization (382, 383), and (iii) clinically by the observation of a correlation of circulating autoantibody titers with clinical disease severity (384, 385). Subsequently, use of these animal models has greatly contributed to our current understanding of EBA pathogenesis. The pathogenesis of blistering and inflammation in EBA can be divided in the following steps (Figure 10).
(1) The pathology-triggering event in EBA is without doubt the binding of the autoantibodies directed against COL7.
(2) This binding triggers the generation and/or the release of pro- and anti-inflammatory mediators, such as cytokines and complement. Of these, C5a, GM-CSF, CXCL1/2, TNF, and leukotriene B4 promote inflammation (44, 380, 386–389), while IL-1ra and IL-6 have profound anti-inflammatory activities (390, 391). These insights have almost exclusively been obtained from EBA mouse models, where inhibition of C5a, GM-CSF, CXCL1/2, or TNF impaired disease induction and/or ameliorated or even improved already manifest EBA. Conversely, blockade of IL-6 led to a deteriorating clinical EBA phenotype, while treatment with recombinant IL-6 or anakinra had beneficial effects (371). So far, no data on the source of these mediators have been published. Recent data, however, suggest that the diversity of the cutaneous microbiome has a significant impact on the expression of inflammatory mediators in the skin (392): in immunization-induced experimental EBA, the clinical outcome, i.e., development of clinically manifest disease, was associated with the diversity of the cutaneous microbiome. Mice with a relatively low diversity of cutaneous microbiota developed clinical EBA, while mice with a relatively high diversity were protected from disease induction. Of note, low diversity of cutaneous microbiota was also associated with higher cutaneous expression of TNF and CXCL1, as well as CD11c. This indicates that skin-resident cells or keratinocytes may be a source of these cytokines known to modulate blistering and inflammation in EBA. In addition, excess production of IL-10, i.e., induced by polyclonal B cell activation, has been shown do directly dampen C5a-driven inflammatory responses in EBA (56).
(3) Overall, this mediator release prompts a vascular response, characterized by an increased expression of endothelial adhesion molecules in cutaneous vessels (393), which allows the CD18-dependent extravasation of Gr-1+ myeloid cells into the skin (394).
(4) Within the skin, myeloid cells attach to the skin-bound immune complexes via specific Fc gamma receptors (395, 396). Regarding the activating Fc gamma receptors, the Fc gamma receptors IIA and IIIB mediate blistering in man, and the Fc gamma receptor IV in the mouse. At least in the mouse, the inhibitory Fc gamma receptor IIB has protective effects. These findings identified activating Fc gamma receptors as a potential drug target in EBA and other PDs. Indeed, if the binding of activating Fc gamma receptors to the skin-bound immune complexes is blocked, i.e., by removal of the sugar residues as Asn297 (397, 398), or by treatment with recombinant CD32-Fc (399), the induction of blistering can be prevented or even improved in mice with experimental EBA. Furthermore, the inhibitory Fc gamma receptor IIB is required to mediate the protective effect of high doses IgG in this model (400, 401).
(5) The Fc gamma receptor-mediated engagement of the myeloid effector cells to the tissue-bound immune complexes triggers a signaling process that initially triggers Syk and Src family kinases (115, 402). Downstream signaling involves PI3K beta (403), PDE4 (120), as well as RORα (391). These insights into the pathogenesis of EBA have led to the identification of several novel therapeutic targets for the treatment of the disease. For example, pharmacological inhibition of all of the above pathways hinders disease induction, and in some cases even improves already established disease when therapeutically applied.
(6) Ultimately, this process leads to the release of ROS via activation of the NADPH oxidase (394) and proteases (404), which cause blistering. For example, mice deficient for NCF1, a subunit of the neutrophil NADPH oxidase, are completely protected from induction of antibody transfer-induced EBA (394). Regarding proteases, blockade of neutrophil elastase or gelaninase B almost completely abolished dermal–epidermal separation of cryosections of skin incubated with EBA patient sera and leukocytes (404). In line, neutrophil elastase deficient mice are completely protected from blister induction by transfer of anti-type XVII collagen antibodies (405). Recent data also suggest that T cells can modulate this myeloid-driven inflammation. More specifically, γδ and NK-T cells promote inflammation and blistering by enhancing the migratory capabilities of myeloid cells (406).
(7) Quite recently, the actin remodeling protein Flightless I (Flii), known to be involved in would healing (407), has been identified as a potential therapeutic target for the treatment of EBA. In accordance to observations in would healing, overexpression of Flii led to a more severe clinical phenotype in antibody transfer-induced EBA (408), while pharmacological inhibition had therapeutic effects in mice with EBA (409). These findings indicate that processes controlling would healing are important to resolve inflammation and blistering, opening new avenues in our understanding of EBA pathogenesis, as well as pointing to so far neglected potential therapeutic targets.
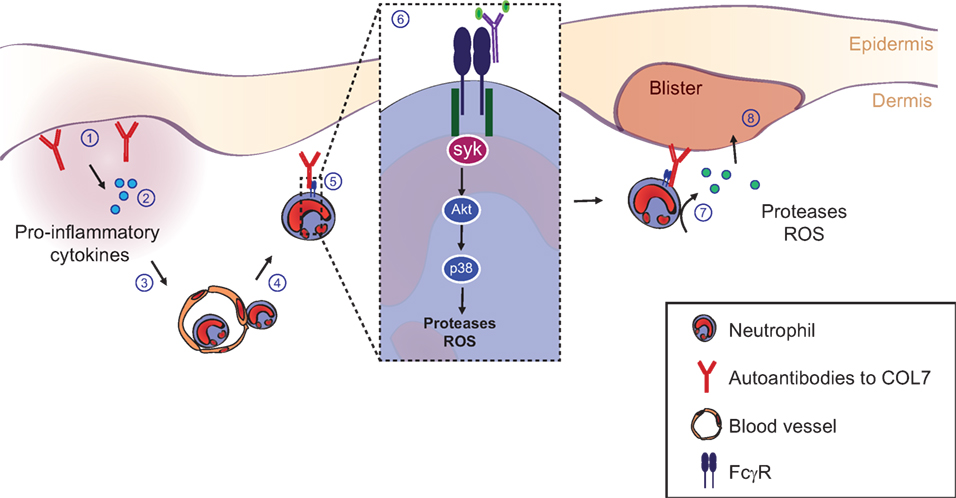
Figure 10. Pathogenesis of inflammation and blistering in the pemphigoid disease epidermolysis bullosa acquisita. Details are explained in the text, where the numbering corresponds to the labeling used here.
Based on this detailed understanding of autoantibody-induced pathology in PD, several therapeutic targets have emerged. In my personal opinion, topical treatment using small molecules inhibiting signal transduction will have a significant impact on the management on patient care.
Autoantibody-Induced Carditis
Myocarditis, defined as an inflammation of the myocardium, is a major cause of heart failure in young adults. Furthermore, myocarditis can lead to dilated cardiomyopathy (DCM), being the most frequent cause for heart transplantation (410). DCM is characterized by progressive depression of myocardial contractile function and ventricular dilation. Clinical manifestation of myocarditis ranges from patients without any relevant symptoms to acute cardiogenic shock. Most patients display flu-like symptoms, palpitations, or arrhythmias. Myocarditis is diagnosed by histological examination of endomyocardial biopsies (EMB). In echocardiography, CT and MRI an impaired wall movement and reduced ejection fraction might be observed. Changes in electrocardiogram can include ST elevation, heart block, and low voltage. Elevated levels of troponin T (TnT) and troponin I (TnI) suggest unspecific myocyte damage (410–412). Apart from infectious agents, like virus or bacteria, myocarditis can be caused by autoimmune reactions. Such an autoimmune myocarditis is characterized as histological confirmed myocarditis with no detectable viral genome in EMB (413) and detection of autoantibodies.
The induction of an autoimmune response against the heart can be a consequence of cardiac injury induced by endogenous or exogenous factors (Figure 11). Triggering agents could be acute infections and toxic or ischemic events resulting in presentation of potentially antigenic determinants to the immune systems. Furthermore, molecular mimicry and cross-reactivity may play an important role. The resulting autoimmune reaction could lead to perpetuation of immune-mediated cardiac damage involving either cellular (e.g., T-cell), and/or humoral (e.g., B-cell) immune responses. Co-activation of both the innate and the adaptive immune system is possible (414). Thus, autoantibodies could be observed in all forms of inflammatory cardiomyopathies (415). These autoantibodies are targeted against various self-antigens, some of which are specific for heart cells. Already in 1987, Neu and colleagues demonstrated the presence of heart-specific autoantibodies following murine Coxsackievirus B3 myocarditis (416). Furthermore, it has been reported that heart-reactive antibodies were present in 59% of patients with myocarditis when rat tissue was used as substrate, whereas 20% of patients with DCM and less than 5% of healthy controls displayed antibodies against rat heart tissue (417). A study by Caforio et al. showed a higher frequency of circulating autoantibodies in relatives of DCM patients, which might be a possible predisposition to develop DCM (418). Another possible genetic predisposition for developing an autoimmune myocarditis could be observed in animal models. Here, mice of different genetic background showed variation in susceptibility to develop an inflammation of the myocardium after viral infection or immunization with heart-specific autoantigenes (419, 420).
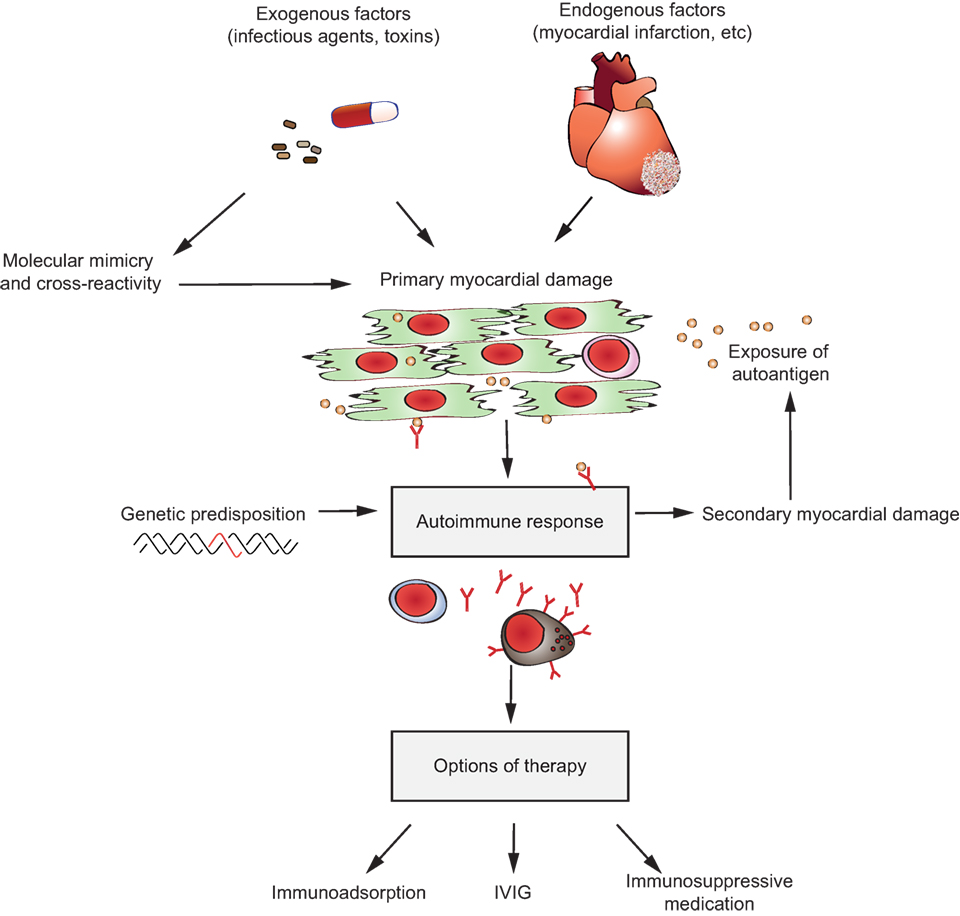
Figure 11. Potential pathomechanisms in autoantibody-induced carditis and therapeutic options. Release of self-antigens through cardiomyocte damage caused by exogenous or endogenous factors can induce an autoimmune response against myocardial tissue in genetic predisposed patients leading to secondary myocardial damage. Therapeutic options could be immunoadsorption, treatment with intravenous immunoglobulin (IVIG) and/or immunosuppressive medication.
Negative inotropic effects can cause impairment of cardiac function through autoantibodies. Moreover, apoptosis of cardiomyocytes and activation of the complement system may play a role (414). In fact, autoantibodies against adenine-nucleotide translocase induce functional consequences, which could be shown in vitro (421).
The “Etiology, Titre-Course, and Survival Study” investigates the prevalence and kinetics of autoantibodies in different forms of heart failure in a large multicenter trial (422). Moreover, various studies have investigated the treatment of autoimmune myocarditis beyond regular heart failure medication. The use of anti-inflammatory drugs like Igs, corticosteroids, azathioprine, and cyclosporine is one therapeutic approach (423, 424). However, for an immunosuppressive therapy, patients have to be virus-negative, because patients with biopsy-based exclusion of virus respond much better to the therapy (425, 426). Another approach is the treatment with IVIG. Here, an improved recovery of left ventricular function with a better survival tendency during the first year could be observed (427). The mode of action of IVIG preparation may be based on the binding and neutralization of circulating autoantibodies (428). In addition to IVIG therapy, immunoadsorption (IA) can be also used to eliminate cardiotoxic antibodies in the patient’s plasma. Here, the antibodies bind to a column adsorbing IgG with higher affinity than IgA and IgM. At least 18 h after the last IA treatment, patients receive polyclonal IgG infusion to restore their IgG levels. Due to IA, functionally active cardiac autoantibodies are removed resulting in improved hemodynamic parameters and biopsy-proven decrease in lymphocytic infiltration and expression of cellular adhesion molecules (423). According to this, a study by Wallukat et al. showed that IA removed circulating IgG3 antibodies targeting β1-AR. The clearance of these antibodies led to an improvement of heart function and a shift to a lower NYHA state (429).
Despite various research approaches, the pathogenesis of autoimmune myocarditis is yet not fully understood. A better understanding of the mechanisms leading to an autoimmune response against cardiac antigens may contribute to a more specific therapy. To investigate the role of autoantibodies and their influence on disease progression, different mouse models, e.g., immunization with cardiac peptides derived from TnI or myosin, have been established (430).
Taken together, different studies demonstrate an association between autoimmune damage related to cardiovascular disorders and the generation of functional antibodies against various proteins. Some therapeutic approaches reached clinical relevance but further studies are needed to confirm their benefit.
Autoantibody-Induced Joint Inflammation: RA
With an estimated incidence of 0.6–1%, RA represents a major challenge for an aging Western population (431). Despite the availability of highly efficient therapies, such as the blockade of pro-inflammatory cytokines, B cell depletion, or a more generalized immunosuppression, many patients do not respond to therapy or become refractory to treatment. Thus, an in-depth understanding of the underlying mechanisms causing joint inflammation and bone destruction may be critical to identify novel therapeutic avenues. Although the final proof of an essential role of autoantibodies in the disease pathology of RA in humans is lacking, the strong association of autoantibodies recognizing cyclic citrullinated proteins (CCP-specific antibodies), which were shown to directly impact osteoclast activity, or the presence of rheumatoid factor antibodies, makes this scenario highly likely (431). Moreover, certain activating FcγR alleles were shown to be associated with the incidence or severity of disease, providing further indirect evidence for a contribution of autoantibodies to disease pathology (432). The strongest evidence for a critical role of the humoral immune system in RA stems from preclinical model systems, which have permitted studying the complex disease mechanisms involved in the molecular and cellular processes ultimately resulting in joint inflammation and bone destruction (433). In these studies, the passive transfer of serum from mice with inflammatory arthritis into healthy animals was sufficient for induction of joint inflammation and bone destruction.
Using a range of knockout mouse strains deficient in Fcγ-receptors or components of the complement pathway has helped to elucidate critical mechanisms responsible for triggering inflammation. Of note, both the complement pathway and cellular Fc-receptors were shown to participate in autoantibody-dependent inflammation and bone destruction (434). With respect to the involvement of complement, not the classical antibody-dependent pathway but rather non-classical pathways were shown to be essential for tissue inflammation, quite similar to what was observed for autoantibody-mediated skin blistering diseases (434). With respect to Fc-receptors, a hierarchy of the involvement of different receptors and cell types has emerged (Figure 12). Immediately after autoantibody injection, a mast cell-dependent opening of the vasculature was observed, which was abrogated in FcγR but not in complement-deficient animals (435, 436). Similar to the human disease, IL-1β, IL-6, and TNFα were suggested to be crucial for initiation and maintenance of inflammation (437). Interestingly, IL-1 secreted by mast cells was suggested as a key for initiation of inflammation (438). Apart from mast cells, tissue-resident macrophages were also suggested to be a key component during the very early phase of inflammation (439). Establishment and maintenance of inflammation is driven by neutrophils via activating Fc-receptors through activated complement components C3a and C5a (Figure 9). More recently, several studies have documented a critical role for Fc-receptors in the process of bone destruction (440–443). Consistent with the essential role of neutrophils and activating FcγRs in disease pathology, Syk or PLCg-deficient mice or animals with a neutrophil-specific deletion of Syk were protected from arthritis development (444, 445).
Figure 12. Autoantibody-dependent pathology in inflammatory arthritis. Shown are the autoantibody-driven processes that contribute to inflammation and bone destruction in inflammatory arthritis. Upon autoantibody deposition in the joints (1) tissue resident macrophages or mast cells become activated via Fcγ-receptors and release pro-inflammatory cytokines (including activated complement components) and chemokines (2). This leads to the recruitment of neutrophils and classical monocytes from the blood, leading to full-blown joint inflammation (3). The cytokine milieu favors the fusion of classical monocytes and their differentiation into immature osteoclasts (4). Immune complexes present in the joint bind to immature osteoclasts via FcgRs, which enhances their maturation into mature bone resorbing osteoclasts (5). Refer to the text, for a more detailed description.
During chronic joint inflammation, osteoclasts differentiation from monocyte precursor cells recruited to the joint is favored by the pro-inflammatory milieu, which is the key event ultimately resulting in excessive bone resorption. It was demonstrated that especially inflammatory monocytes, which are recruited via the chemokine CCR2, differentiate into osteoclasts and are responsible for bone destruction (441). Moreover, autoantibodies modulated this process by binding to activating FcγRs expressed on immature and mature osteoclasts. Demonstrating the therapeutic potential of this approach, depletion of inflammatory monocytes or blocking autoantibody binding to osteoclast FcγRs diminished bone destruction (441). Moreover, characteristic changes in (auto)antibody glycosylation have been noted in mice and humans with arthritis (446, 447). Especially pro-inflammatory IgG glycoforms with low levels of terminal sialic acid and galactose residues, which interact efficiently with activating FcγRs were shown to dominate during active disease. More recent studies suggest that not only the enhanced triggering of pro-inflammatory effector functions but also the lack of anti-inflammatory sialic acid rich IgG glycovariants may contribute to the induction of excessive inflammation (448). Thus, infusion of IVIGs pooled from thousands of donors (IVIG therapy) was shown to induce resolution of inflammatory arthritis and of other autoantibody-dependent autoimmune diseases by restoring highly sialylated IgG glycovariants (400). As at least one part of the mechanism, an upregulation of the inhibitory FcγRIIb on joint infiltrating innate immune effector cells was shown which would increase the threshold for cell activation via autoantibody immune complexes (449). Ultimately this would reduce the downstream recruitment of neutrophils and osteoclast precursor cells from the blood and result in resolution of inflammation (448).
Taken together, activating FcγRs and activated complement components have been demonstrated to be of critical importance during all phases of autoantibody-dependent joint inflammation and may therefore represent promising therapeutic targets. Potential strategies may include blocking autoantibody interaction with activating FcγRs or inhibiting their downstream activating signals via small molecules. Alternatively, lowering autoantibody half-life by blocking antibody access to the neonatal Fc-receptor (FcRn) may also help to reduce inflammation and tissue destruction (450). In addition, a more in depth understanding of pathways essential for initiating resolution of inflammation (as induced by IVIG for example) may identify further treatment options to stop autoantibody pathology (448). As a most straightforward approach, IVIG variants with enhanced levels of anti-inflammatory IgG glycovariants may help to obtain enhanced therapeutic activity (451).
Autoantibody-Induced Inflammation of the Optic Nerve and Spinal Cord: Neuromyelitis Optica Spectrum Disorder (NMOSD)
NMOSD: Epidemiology, Clinical Presentation, and Treatment Challenges
There is a rare neurological disorder caused by autoantibodies directed against a membrane protein expressed on astrocytes. The disease, NMO, recently renamed NMOSD, is an inflammatory demyelinating disease of the central nervous system that primarily affects optic nerves and spinal cord, and to a lesser extent brain (452, 453). The major clinical manifestations of NMOSD include recurrent bouts of eye and back pain with visual and motor impairment that can lead to blindness, paralysis, and death. Some patients with brain involvement manifest intractable vomiting, hiccups, and other symptoms. The prevalence of NMOSD is ~1–8 per 100,000 individuals, with a female:male ratio ~8:1 and a median age of presentation of 30–40 years, though the disease can present in the children and the elderly (454, 455). NMOSD was originally thought to be a subtype of multiple sclerosis, another inflammatory demyelinating disease of the central nervous system, but it is now clear that the diseases have distinct clinical signs and symptoms, pathogenesis, and responses to therapeutics. The mainstay of NMOSD therapy includes immunosuppression, plasma exchange, and B-cell depletion therapy, though new therapeutics are in the pipeline as discussed below. The current therapies are reasonably good, particularly B-cell depletion with rituximab, though many patients continue to have exacerbations and neurological deficit even with multiple drugs, and immunosuppressants can have significant side effects with long-term use.
NMOSD Pathogenesis
The major breakthrough in NMOSD was the discovery that the majority of patients have a circulating IgG1 autoantibody directed against AQP4, a water channel expressed at the plasma membrane of astrocytes throughout the central nervous system, including spinal cord, optic nerve, and brain (456). AQP4 is also expressed in various peripheral tissues including stomach, kidney, airways, and skeletal muscle. AQP4 functions a bidirectional water channel that facilitates water movement across cell plasma membranes in response to osmotic gradients produced by solute transport. Phenotype studies in AQP4 knockout mice have demonstrated its involvement in brain water movement, neuroexcitatory phenomena, and astrocyte migration (457). For example, mice lacking AQP4 are partially protected from cytotoxic (cell swelling) brain edema in stroke and other brain injuries (458). While most NMOSD patients are seropositive for anti-AQP4 autoantibodies (called AQP4-IgG), a small subset of patients are seropositive for anti-MOG (myelin oligodendrocyte glycoprotein) antibodies, and other patients show no detectable antibodies against AQP4 or MOG, though it is recognized that assay sensitivity remains imperfect and antibody titers vary over time.
The major pathological features of NMOSD lesions include astrocyte damage with loss of AQP4 and glial fibrillary acidic protein, inflammation with prominent granulocyte and macrophage infiltration, vasculocentric deposition of activated complement, and demyelination; in later stages, there is neuronal loss and scarring. The NMOSD pathogenesis mechanism in AQP4-IgG seropositive patients does not involve antibody effects on AQP4 function, but rather primary damage to astrocytes by complement- and cell-mediated mechanisms, as shown in Figure 13. Evidence from human pathological specimens and rodent models of passive AQP4-IgG transfer suggests that binding of AQP4-IgG to AQP4 on astrocytes initiates complement-dependent cytotoxicity (CDC) and antibody-dependent cellular cytotoxicity (ADCC), astrocyte damage, inflammation, and BBB disruption, which leads to oligodendrocyte injury and demyelination (452, 453). Complement activation plays a central role in NMOSD pathology, both by formation of a terminal attack complex on astrocytes, as well as by elaboration of anaphylatoxins that attract and activate inflammatory leukocytes. There may also be involvement of AQP4-sensitized T-cells (459), though it is not clear whether T-cells are involved in permeabilization of the BBB and/or in astrocyte cytotoxicity (460). Major, largely unanswered, questions in NMOSD pathogenesis include why disease is localized mainly to spinal cord and optic nerve without involvement of peripheral, tissues expressing AQP4, how early oligodendrocyte damage is caused by AQP4-IgG binding on astrocytes, and how peripherally generated AQP4-IgG enters the central nervous system to initiate disease.
Figure 13. Neuromyelitis optica spectrum disorder pathogenesis mechanisms. Scheme shows astrocyte aquaporin-4 (AQP4) as the target of AQP4-IgG autoantibodies. Antibody binding initiates complement and cell-mediated cytotoxicity and an inflammatory response, resulting in oligodendrocyte injury and demyelination. Abbreviations: CDC, complement-dependent cytotoxicity; ADCC, antibody-dependent cellular cytotoxicity.
Emerging Treatments and Novel Therapeutic Targets
Several NMOSD therapeutics in the development pipeline, many of which emerged from the improved understanding of pathogenesis (461, 462). Given the central role of complement, the C5 inhibitor Eculizumab is in clinical trials following encouraging data in a small open-label study in AQP4-IgG seropositive patients having frequent disease exacerbations (113). Anti-IL6 receptor antagonists that target antibody-producing plasma cells are in clinical studies. The repurposing of other approved drugs for NMOSD is under evaluation, including antibodies targeting VEGF and CD-19/20, small molecule drugs targeting neutrophils and eosinophils, and IVIG. Therapeutics in preclinical development include antibodies (aquaporumab) targeting AQP4-IgG binding to AQP4 (463), and AQP4-IgG inactivation by enzymatic deglycosylation (464). Other potential targets for NMO therapeutics include AQP4 cell surface expression and its supramolecular aggregation, complement inhibitor protein CD59, and various components of the CDC, ADCC, and inflammation pathways. Recently, experimental animal data support the possibility of a remyelination approach in NMOSD to limit neuron loss (465). Lastly, the possibility of antigen-specific tolerization against AQP4 is a theoretical possibility, as is autologous hematopoietic stem cell transplantation.
Diagnostic Tools for Detection of Autoantibodies
Indirect Immunofluorescence
One of the “gold standards” for autoantibody determination maintains to be the indirect immunofluorescence assay (IFA) using cryosections of mammalian tissue or cultured cell lines, e.g., HEp-2, which is the standard substrate for the screening of antinuclear antibodies (Figure 14). The immunoassay principle essentially consists of two steps: in the initial incubation a human sample like blood or CSF is brought into contact with the substrate. Subsequently, unbound sample constituents are washed off and the bound antibodies are visualized with fluorochrome-labeled secondary antibodies. The evaluation is done by fluorescence microscopy leading to the interpretation of the staining patterns. The approach has three main advantages: (i) all autoantigens in a given substrate are present and displayed in their native environment, (ii) it is possible to screen for antibodies against unidentified autoantigens based on characteristic staining patterns such that even de novo screenings of patient cohorts are possible, and (iii) a negative outcome rules out the presence of multiple autoantibody specificities with a single analysis. The concentration of the respective autoantibody can be estimated by titering-out the sample. However, in some cases of autoantibodies against soluble antigens or against those that are prone to denaturation by necessary fixation steps, the use of IFA is limited. Lately, the introduction of automated digitalization together with the computer-assisted evaluation of the results (466–468) and the development of genetically modified cells expressing the necessary autoantigens—often designated as CBA—have rejuvenated the use of IFA (456, 469–475). Another support for the use of IFA as a routine diagnostic platform is the availability of incubation automates that decrease hands-on time and lower the total costs per analysis.
Figure 14. Detection of autoantibodies. Details are explained in the text. Abbreviations: ELISA, enzyme-linked immunosorbent assay; FIPA, fluorescence immunoprecipitation assay; IFA, indirect immunfluorescence assay; RIA, radioimmunoassay.
Immunoprecipitation
Due to the highly sensitive detection of radioactive substances, radioimmunoassays (RIAs) based on specifically labeled antigens were developed decades ago and are still a good option for the detection of autoantibodies against membrane proteins. Commonly, a radiolabel like 125-iodine is chemically or enzymatically attached to a purified ligand, e.g., 125-I-α-bungarotoxin for the labeling of AChR (476). The ligand is then incubated with tissue or cell extracts containing the desired membrane protein in a conformation that is able to specifically bind the ligand. Alternatively, in vitro-translated 35-S-methionine labeled antigens can be used with the general limitation to simple (intracellular) antigens (477). The radiolabeled antigen is then brought into contact with a human sample and the immune complexes formed in this step as well as free Igs are precipitated by a secondary antibody or an affinity matrix like agarose-bound staphylococcal protein G. The amount of precipitated radioactivity is a quantitative measure of the antibody against the labeled target antigen in the sample. Classically, RIAs have been used for the sensitive detection of autoantibodies against both intracellular and surface-exposed target antigens, e.g., against double-stranded DNA in SLE or the nicotinic AChR in MG. As an alternative to the radiolabel, fluorophores can be introduced to the target antigen, either by chemical labeling of the isolated target antigen or by its recombinant expression in frame with a fluorescent fusion protein, e.g., a variant of the green fluorescent protein (478).
ELISA, Fluorescence Immunoassay (FIA), Chemilumenescence Immunoassay (CLIA), and Bead-Based Assays
Another classical method for the semiquantitative determination of autoantibodies is the ELISA. Basically, a target antigen isolated from a native biosource or a recombinant host by a combination of biochemical procedures is initially immobilized on a solid phase, e.g., in cavities of plastic microplates or on the surface of membranes. The immobilization allows for sophisticated blocking, washing, and stabilization steps. The sample to be investigated is eventually brought into contact with the target antigen, unbound sample constituents are removed by washing and bound antibodies are detected with the help of a secondary antibody coupled to a reporter. Microplate ELISA typically makes use of reporting enzymes that convert a colorless reagent to a chromophore that is subsequently quantified with a spectrophotometer. Membrane-based ELISA (commonly designated “lineblot” or “dot blot”) instead commonly employ a precipitating chromophore that can be quantified by densitometric analysis of electronic images produced with the help of a flatbed scanner or a camera system (479). Alternative protocols are based on reagents that produce a chemilumenscent reaction upon conversion by the reporter enzyme in a CLIA or make use of a fluorophore-reporter in FIA, in some cases combined with fluorescent beads in the form of addressable laser-bead immunoassays (ALBIA) (480, 481). Alternatively, paramagnetic microparticles were developed as an additional solid phase that allows for magnetic separation of the target antigen from other reagents in subsequent steps. This bead-based assay platform is in most cases combined with chemilumenescence detection (482). All of the different methods and platforms can be partnered with sophisticated incubation, handling and detection devices or are integrated into commercially available test systems that allow for semi- or fully automated incubation and evaluation of the samples resulting in comprehensive output.
Western Blot
A further classical method for qualitative autoantibody determination is the Western blot (482). For this method, tissue or cell extracts containing one or several target antigens are separated by gel electrophoresis and subsequently transferred to membranes. The sample that is to be investigated for antibodies is then incubated and bound antibodies are visualized with the help of a secondary antibody coupled to a reporter, e.g., an enzyme converting a colorless reagent to a precipitating chromophore. Generally, the denaturing effect of the electrophoresis limits the determination of antibodies to those binding denaturation-insensitive structures (“linear epitopes”).
Multiplexing
In most cases, screening and confirmation assays are combined for autoantibody determination to achieve a meaningful result with regard to the clinical problem. Modern test systems combine both approaches in one. Slides that carry more than one substrate per incubation field enable multiplexed testing in the IFA format. For example, the combination of different tissues like rat kidney, liver, and stomach—as commonly employed for the determination of antibodies associated with autoimmune liver diseases—allows for a more differentiated, highly specific evaluation with just one analysis. The biochip format made of mosaics (471, 483–485) of millimeter-sized glass chips coated with the diagnostically relevant antigenic substrates even allows the combined use of tissue cryosections, cultured (genetically modified and unmodified control) cells, and directly immobilized purified antigens for a comprehensive analysis covering the whole spectrum of autoantibodies relevant for differential diagnostics of autoimmune syndromes. Moreover, microplate and membrane-based ELISA as well as ALBIA can be used to form antigen profiles for the detection of multiple antibodies in parallel. Multiparametric lineblots have been developed for this purpose carrying more than 20 individually optimized membrane chips on a plastic layer and harboring antigen mixtures, individual antigens and even lipids (486–488).
It should be emphasized that none of the above approaches were used to discover TSHR autoantibodies nor can they be used clinically to measure TSHR antibodies in Graves’ patients. In 1956, using guinea pigs as a read-out, Graves sera were found to contain a thyroid-stimulating factor with a duration of action more prolonged than TSH [“long-acting thyroid stimulator” (LATS)] (489). LATS was later found to be an IgG molecule. After the discovery of the TSHR, it was shown that LATS (like TSH) activated thyrocyte adenylyl cyclase and competed for TSH binding to the TSHR. This background to TSHR antibodies suggests that, like Graves’ patients, some patients described as seronegative for a particular autoantibody may have functional antibodies not detectable by the conventional approaches listed above.
Conclusion
As highlighted herein, autoantibodies may cause pathology through a wide range of mechanisms. Insights into these pathways identified several novel therapeutic targets, such as FcγR-induced neutrophil activation, which go beyond unspecific immunosuppression and are already in clinical trials (340). Preclinical models even point toward the possibility to specifically target autoantibody-specific B cells and/or plasma cells, which could be considered as curative treatment (117). Based on the vast amount of insights into pathomechanism of autoantibody-induced pathology, some of which have been reviewed herein, we expect that these concepts will be evaluated in clinical trials, which will significantly improve the still not satisfying therapeutic options currently used to treat autoimmunity.
Author Contributions
All authors listed, have made substantial, direct, and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
RL received speakers honoraria from and/or has consultancy agreements with AbbVie, Allmiral, ArGEN-X, Biogen Idec, Biotest, Novartis, UCB Phrama, Topadur, and TxCell and holds the patent “Soluble Fc gamma receptor for treatment of autoimmune bullous diseases” (US 15/029,994). FL has received speakers’ honoraria from Grifols, Biogen, Teva, and Roche. His institution offers commercial antibody testing, and he does not receive personal benefits from this. All other authors declare no conflicts of interest. CP and LK are employees of the Euroimmun AG, a company that develops, manufactures, and markets human diagnostics. JL and JML have a patent application for an antigen-specific immunosuppressive therapy for MG.
Funding
The work was supported by the following grants: The Excellence Cluster Inflammation at Interfaces (EXC 306/2), the Research Training Programs Modulation of Autoimmunity (GRK 1727/2) and Genes, Environment and Inflammation (GRK 1743/1), the Clinical Research Unit Pemphigoid Diseases (KFO 303/1), DFG LU 877/(9-12)-1 to RL—all from the Deutsche Forschungsgemeinschaft. DK 54684 to SM and DK19289 to BR—from the National Institutes of Health; Research Foundation (FWO) Flanders, Belgium (G.0D13.15N), and the Horizon 2020 Marie Skłodowska-Curie Innovative Training Network grant PROFILE (675746) to KV; CT is a postdoctoral fellow supported by the Research Foundation—Flanders (FWO), Belgium (12N0715N); DFG KA 1797/(2-9)-1 to ZK, DFG-CRC1181-A07 to FN.
References
1. Bach JF. The effect of infections on susceptibility to autoimmune and allergic diseases. N Engl J Med (2002) 347:911–20. doi:10.1056/NEJMra020100
2. Vento S, Cainelli F. Autommune diseases in low and middle income countries: a neglected issue in global health. Isr Med Assoc J (2016) 18(1):54–5.
3. Eaton WW, Rose NR, Kalaydjian A, Pedersen MG, Mortensen PB. Epidemiology of autoimmune diseases in Denmark. J Autoimmun (2007) 29:1–9. doi:10.1016/j.jaut.2007.05.002
4. Joly P, Roujeau JC, Benichou J, Picard C, Dreno B, Delaporte E, et al. A comparison of oral and topical corticosteroids in patients with bullous pemphigoid. N Engl J Med (2002) 346:321–7. doi:10.1056/NEJMoa011592
5. Rosetti F, Tsuboi N, Chen K, Nishi H, Ernandez T, Sethi S, et al. Human lupus serum induces neutrophil-mediated organ damage in mice that is enabled by mac-1 deficiency. J Immunol (2012) 189:3714–23. doi:10.4049/jimmunol.1201594
6. Mavragani CP, Moutsopoulos HM. Sjögren’s syndrome. Annu Rev Pathol (2014) 9:273–85. doi:10.1146/annurev-pathol-012513-104728
7. Scofield RH, Asfa S, Obeso D, Jonsson R, Kurien BT. Immunization with short peptides from the 60-kDa Ro antigen recapitulates the serological and pathological findings as well as the salivary gland dysfunction of Sjogren’s syndrome. J Immunol (2005) 175:8409–14. doi:10.4049/jimmunol.175.12.8409
8. Simon JP, Marie I, Jouen F, Boyer O, Martinet J. Autoimmune myopathies: where do we stand. Front Immunol (2016) 7:234. doi:10.3389/fimmu.2016.00234
9. Rosenberg NL, Ringel SP, Kotzin BL. Experimental autoimmune myositis in SJL/J mice. Clin Exp Immunol (1987) 68:117–29.
10. Taplin CE, Barker JM. Autoantibodies in type 1 diabetes. Autoimmunity (2008) 41:11–8. doi:10.1080/08916930701619169
11. Robles DT, Eisenbarth GS, Dailey NJ, Peterson LB, Wicker LS. Insulin autoantibodies are associated with islet inflammation but not always related to diabetes progression in NOD congenic mice. Diabetes (2003) 52:882–6. doi:10.2337/diabetes.52.3.882
12. Bratland E, Bredholt G, Mellgren G, Knappskog PM, Mozes E, Husebye ES. The purification and application of biologically active recombinant steroid cytochrome P450 21-hydroxylase: the major autoantigen in autoimmune Addison’s disease. J Autoimmun (2009) 33:58–67. doi:10.1016/j.jaut.2009.02.018
13. Bunn HF. Vitamin B12 and pernicious anemia – the dawn of molecular medicine. N Engl J Med (2014) 370:773–6. doi:10.1056/NEJMcibr1315544
14. Cancado EL, Abrantes-Lemos CP, Terrabuio DR. The importance of autoantibody detection in autoimmune hepatitis. Front Immunol (2015) 6:222. doi:10.3389/fimmu.2015.00222
15. Cancado EL, Harriz M. The importance of autoantibody detection in primary biliary cirrhosis. Front Immunol (2015) 6:309. doi:10.3389/fimmu.2015.00309
16. Oertelt S, Lian ZX, Cheng CM, Chuang YH, Padgett KA, He XS, et al. Anti-mitochondrial antibodies and primary biliary cirrhosis in TGF-beta receptor II dominant-negative mice. J Immunol (2006) 177:1655–60. doi:10.4049/jimmunol.177.3.1655
17. Sánchez Castañón M, Zuliani V, Amodio A, Campagnola P, Granato A, Gabbrielli A, et al. Role of amylase-α2A autoantibodies in the diagnosis of autoimmune pancreatitis. Pancreas (2015) 44:1078–82. doi:10.1097/MPA.0000000000000417
18. Kurisaki H, Nagao Y, Nagafuchi S, Mitsuyama M. Autoimmune gastro-pancreatitis with anti-protein disulfide isomerase-associated 2 autoantibody in aire-deficient BALB/cAnN mice. PLoS One (2013) 8:e73862. doi:10.1371/journal.pone.0073862
19. Mastroianni-Kirsztajn G, Hornig N, Schlumberger W. Autoantibodies in renal diseases – clinical significance and recent developments in serological detection. Front Immunol (2015) 6:221. doi:10.3389/fimmu.2015.00221
20. Tomas NM, Hoxha E, Reinicke AT, Fester L, Helmchen U, Gerth J, et al. Autoantibodies against thrombospondin type 1 domain-containing 7A induce membranous nephropathy. J Clin Invest (2016) 126:2519–32. doi:10.1172/JCI85265
21. Silva CA, Yamakami LY, Aikawa NE, Araujo DB, Carvalho JF, Bonfá E. Autoimmune primary ovarian insufficiency. Autoimmun Rev (2014) 13:427–30. doi:10.1016/j.autrev.2014.01.003
22. Silva CA, Cocuzza M, Carvalho JF, Bonfá E. Diagnosis and classification of autoimmune orchitis. Autoimmun Rev (2014) 13:431–4. doi:10.1016/j.autrev.2014.01.024
23. Hirai S, Naito M, Terayama H, Hatayama N, Qu N, Musha M, et al. Serum autoantibodies in mice immunized with syngeneic testicular germ cells alone. Am J Reprod Immunol (2013) 70:509–17. doi:10.1111/aji.12145
24. Stern ME, Schaumburg CS, Siemasko KF, Gao J, Wheeler LA, Grupe DA, et al. Autoantibodies contribute to the immunopathogenesis of experimental dry eye disease. Invest Ophthalmol Vis Sci (2012) 53:2062–75. doi:10.1167/iovs.11-9299
25. Fischer A, Antoniou KM, Brown KK, Cadranel J, Corte TJ, du Bois RM, et al. An official European Respiratory Society/American Thoracic Society research statement: interstitial pneumonia with autoimmune features. Eur Respir J (2015) 46:976–87. doi:10.1183/13993003.00150-2015
26. Pelanda R, Torres RM. Central B-cell tolerance: where selection begins. Cold Spring Harb Perspect Biol (2012) 4:a007146. doi:10.1101/cshperspect.a007146
27. Yurasov S, Nussenzweig MC. Regulation of autoreactive antibodies. Curr Opin Rheumatol (2007) 19:421–6. doi:10.1097/BOR.0b013e328277ef3b
28. Scheid JF, Mouquet H, Kofer J, Yurasov S, Nussenzweig MC, Wardemann H. Differential regulation of self-reactivity discriminates between IgG+ human circulating memory B cells and bone marrow plasma cells. Proc Natl Acad Sci U S A (2011) 108:18044–8. doi:10.1073/pnas.1113395108
29. Tiller T, Kofer J, Kreschel C, Busse CE, Riebel S, Wickert S, et al. Development of self-reactive germinal center B cells and plasma cells in autoimmune Fc gammaRIIB-deficient mice. J Exp Med (2010) 207:2767–78. doi:10.1084/jem.20100171
30. Basten A, Silveira PA. B-cell tolerance: mechanisms and implications. Curr Opin Immunol (2010) 22:566–74. doi:10.1016/j.coi.2010.08.001
31. Gonzalez-Martin A, Adams BD, Lai M, Shepherd J, Salvador-Bernaldez M, Salvador JM, et al. The microRNA miR-148a functions as a critical regulator of B cell tolerance and autoimmunity. Nat Immunol (2016) 17:433–40. doi:10.1038/ni.3385
32. Müller J, Nitschke L. The role of CD22 and Siglec-G in B-cell tolerance and autoimmune disease. Nat Rev Rheumatol (2014) 10:422–8. doi:10.1038/nrrheum.2014.54
33. Sage PT, Paterson AM, Lovitch SB, Sharpe AH. The coinhibitory receptor CTLA-4 controls B cell responses by modulating T follicular helper, T follicular regulatory, and T regulatory cells. Immunity (2014) 41:1026–39. doi:10.1016/j.immuni.2014.12.005
34. Vaeth M, Müller G, Stauss D, Dietz L, Klein-Hessling S, Serfling E, et al. Follicular regulatory T cells control humoral autoimmunity via NFAT2-regulated CXCR5 expression. J Exp Med (2014) 211:545–61. doi:10.1084/jem.20130604
35. Mackay F, Schneider P. Cracking the BAFF code. Nat Rev Immunol (2009) 9:491–502. doi:10.1038/nri2572
36. Stadanlick JE, Cancro MP. BAFF and the plasticity of peripheral B cell tolerance. Curr Opin Immunol (2008) 20:158–61. doi:10.1016/j.coi.2008.03.015
37. Tiller T, Tsuiji M, Yurasov S, Velinzon K, Nussenzweig MC, Wardemann H. Autoreactivity in human IgG+ memory B cells. Immunity (2007) 26:205–13. doi:10.1016/j.immuni.2007.01.009
38. Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, Nussenzweig MC. Predominant autoantibody production by early human B cell precursors. Science (2003) 301:1374–7. doi:10.1126/science.1086907
39. Mietzner B, Tsuiji M, Scheid J, Velinzon K, Tiller T, Abraham K, et al. Autoreactive IgG memory antibodies in patients with systemic lupus erythematosus arise from nonreactive and polyreactive precursors. Proc Natl Acad Sci U S A (2008) 105:9727–32. doi:10.1073/pnas.0803644105
40. Mannoor K, Xu Y, Chen C. Natural autoantibodies and associated B cells in immunity and autoimmunity. Autoimmunity (2013) 46:138–47. doi:10.3109/08916934.2012.748753
41. Strait RT, Posgai MT, Mahler A, Barasa N, Jacob CO, Köhl J, et al. IgG1 protects against renal disease in a mouse model of cryoglobulinaemia. Nature (2015) 517:501–4. doi:10.1038/nature13868
42. Rademacher TW, Williams P, Dwek RA. Agalactosyl glycoforms of IgG autoantibodies are pathogenic. Proc Natl Acad Sci U S A (1994) 91:6123–7. doi:10.1073/pnas.91.13.6123
43. Nimmerjahn F, Ravetch JV. Anti-inflammatory actions of intravenous immunoglobulin. Annu Rev Immunol (2008) 26:513–33. doi:10.1146/annurev.immunol.26.021607.090232
44. Karsten CM, Pandey MK, Figge J, Kilchenstein R, Taylor PR, Rosas M, et al. Anti-inflammatory activity of IgG1 mediated by Fc galactosylation and association of FcgammaRIIB and dectin-1. Nat Med (2012) 18:1401–6. doi:10.1038/nm.2862
45. Anthony RM, Ravetch JV. A novel role for the IgG Fc glycan: the anti-inflammatory activity of sialylated IgG Fcs. J Clin Immunol (2010) 30(Suppl 1):S9–14. doi:10.1007/s10875-010-9405-6
46. Goulabchand R, Vincent T, Batteux F, Eliaou JF, Guilpain P. Impact of autoantibody glycosylation in autoimmune diseases. Autoimmun Rev (2014) 13:742–50. doi:10.1016/j.autrev.2014.02.005
47. Böhm S, Kao D, Nimmerjahn F. Sweet and sour: the role of glycosylation for the anti-inflammatory activity of immunoglobulin G. Curr Top Microbiol Immunol (2014) 382:393–417. doi:10.1007/978-3-319-07911-0_18
48. Collin M, Ehlers M. The carbohydrate switch between pathogenic and immunosuppressive antigen-specific antibodies. Exp Dermatol (2013) 22:511–4. doi:10.1111/exd.12171
49. Hess C, Winkler A, Lorenz AK, Holecska V, Blanchard V, Eiglmeier S, et al. T cell-independent B cell activation induces immunosuppressive sialylated IgG antibodies. J Clin Invest (2013) 123:3788–96. doi:10.1172/JCI65938
50. McHeyzer-Williams LJ, Pelletier N, Mark L, Fazilleau N, McHeyzer-Williams MG. Follicular helper T cells as cognate regulators of B cell immunity. Curr Opin Immunol (2009) 21:266–73. doi:10.1016/j.coi.2009.05.010
51. Fillatreau S, Gray D, Anderton SM. Not always the bad guys: B cells as regulators of autoimmune pathology. Nat Rev Immunol (2008) 8:391–7. doi:10.1038/nri2315
52. Matsumoto M, Baba A, Yokota T, Nishikawa H, Ohkawa Y, Kayama H, et al. Interleukin-10-producing plasmablasts exert regulatory function in autoimmune inflammation. Immunity (2014) 41:1040–51. doi:10.1016/j.immuni.2014.10.016
53. Pelletier N, McHeyzer-Williams LJ, Wong KA, Urich E, Fazilleau N, McHeyzer-Williams MG. Plasma cells negatively regulate the follicular helper T cell program. Nat Immunol (2010) 11:1110–8. doi:10.1038/ni.1954
54. Winer DA, Winer S, Shen L, Wadia PP, Yantha J, Paltser G, et al. B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat Med (2011) 17:610–7. doi:10.1038/nm.2353
55. Yanaba K, Bouaziz JD, Haas KM, Poe JC, Fujimoto M, Tedder TF. A regulatory B cell subset with a unique CD1dhiCD5+ phenotype controls T cell-dependent inflammatory responses. Immunity (2008) 28:639–50. doi:10.1016/j.immuni.2008.03.017
56. Kulkarni U, Karsten CM, Kohler T, Hammerschmidt S, Bommert K, Tiburzy B, et al. IL-10 mediates plasmacytosis-associated immunodeficiency by inhibiting complement-mediated neutrophil migration. J Allergy Clin Immunol (2016) 137:1487–97.e6. doi:10.1016/j.jaci.2015.10.018
57. Barr TA, Brown S, Ryan G, Zhao J, Gray D. TLR-mediated stimulation of APC: distinct cytokine responses of B cells and dendritic cells. Eur J Immunol (2007) 37:3040–53. doi:10.1002/eji.200636483
58. Crawford A, Macleod M, Schumacher T, Corlett L, Gray D. Primary T cell expansion and differentiation in vivo requires antigen presentation by B cells. J Immunol (2006) 176:3498–506. doi:10.4049/jimmunol.176.6.3498
59. Barr TA, Brown S, Mastroeni P, Gray D. TLR and B cell receptor signals to B cells differentially program primary and memory Th1 responses to Salmonella enterica. J Immunol (2010) 185:2783–9. doi:10.4049/jimmunol.1001431
60. Barr TA, Gray M, Gray D. B cells: programmers of CD4 T cell responses. Infect Disord Drug Targets (2012) 12:222–31. doi:10.2174/187152612800564446
61. Nagele EP, Han M, Acharya NK, DeMarshall C, Kosciuk MC, Nagele RG. Natural IgG autoantibodies are abundant and ubiquitous in human sera, and their number is influenced by age, gender, and disease. PLoS One (2013) 8:e60726. doi:10.1371/journal.pone.0060726
62. Sakata S, Matsuda M, Ogawa T, Takuno H, Matsui I, Sarui H, Rentzsch K, Juhl D, Henschler R, et al. Prevalence of thyroid hormone autoantibodies in healthy subjects. Clin Endocrinol (Oxf) (1994) 41:365–70. doi:10.1111/j.1365-2265.1994.tb02558.x
63. Prussmann J, Prussmann W, Recke A, Rentzsch K, Juhl D, Henschler R, et al. Co-occurrence of autoantibodies in healthy blood donors. Exp Dermatol (2014) 23:519–21. doi:10.1111/exd.12445
64. Prüßmann W, Prüßmann J, Koga H, Recke A, Iwata H, Juhl D, et al. Prevalence of pemphigus and pemphigoid autoantibodies in the general population. Orphan J Rare Dis (2015) 10:63. doi:10.1186/s13023-015-0278-x
65. Hoyer BF, Moser K, Hauser AE, Peddinghaus A, Voigt C, Eilat D, et al. Short-lived plasmablasts and long-lived plasma cells contribute to chronic humoral autoimmunity in NZB/W mice. J Exp Med (2004) 199:1577–84. doi:10.1084/jem.20040168
66. Manz RA, Hauser AE, Hiepe F, Radbruch A. Maintenance of serum antibody levels. Annu Rev Immunol (2005) 23:367–86. doi:10.1146/annurev.immunol.23.021704.115723
67. Mumtaz IM, Hoyer BF, Panne D, Moser K, Winter O, Cheng QY, et al. Bone marrow of NZB/W mice is the major site for plasma cells resistant to dexamethasone and cyclophosphamide: implications for the treatment of autoimmunity. J Autoimmun (2012) 39:180–8. doi:10.1016/j.jaut.2012.05.010
68. Hiepe F, Dörner T, Hauser AE, Hoyer BF, Mei H, Radbruch A. Long-lived autoreactive plasma cells drive persistent autoimmune inflammation. Nat Rev Rheumatol (2011) 7:170–8. doi:10.1038/nrrheum.2011.1
69. Neubert K, Meister S, Moser K, Weisel F, Maseda D, Amann K, et al. The proteasome inhibitor bortezomib depletes plasma cells and protects mice with lupus-like disease from nephritis. Nat Med (2008) 14:748–55. doi:10.1038/nm1763
70. Bontscho J, Schreiber A, Manz RA, Schneider W, Luft FC, Kettritz R. Myeloperoxidase-specific plasma cell depletion by bortezomib protects from anti-neutrophil cytoplasmic autoantibodies-induced glomerulonephritis. J Am Soc Nephrol (2011) 22:336–48. doi:10.1681/ASN.2010010034
71. Gomez AM, Vrolix K, Martínez-Martínez P, Molenaar PC, Phernambucq M, van der Esch E, et al. Proteasome inhibition with bortezomib depletes plasma cells and autoantibodies in experimental autoimmune myasthenia gravis. J Immunol (2011) 186:2503–13. doi:10.4049/jimmunol.1002539
72. Alexander T, Sarfert R, Klotsche J, Kühl AA, Rubbert-Roth A, Lorenz HM, et al. The proteasome inhibitior bortezomib depletes plasma cells and ameliorates clinical manifestations of refractory systemic lupus erythematosus. Ann Rheum Dis (2015) 74:1474–8. doi:10.1136/annrheumdis-2014-206016
73. Patriquin CJ, Thomas MR, Dutt T, McGuckin S, Blombery PA, Cranfield T, et al. Bortezomib in the treatment of refractory thrombotic thrombocytopenic purpura. Br J Haematol (2016) 173:779–85. doi:10.1111/bjh.13993
74. Patel PP, Becker J, Freyer C, Griffiths E, Thompson JE, Wang ES. Rituximab-refractory thrombotic thrombocytopenic purpura responsive to intravenous but not subcutaneous bortezomib. Transfusion (2016) 56:970–4. doi:10.1111/trf.13465
75. Khodadadi L, Cheng Q, Alexander T, Sercan-Alp Ö, Klotsche J, Radbruch A, et al. Bortezomib plus continuous B cell depletion results in sustained plasma cell depletion and amelioration of lupus nephritis in NZB/W F1 mice. PLoS One (2015) 10:e0135081. doi:10.1371/journal.pone.0135081
76. Taddeo A, Khodadadi L, Voigt C, Mumtaz IM, Cheng Q, Moser K, et al. Long-lived plasma cells are early and constantly generated in New Zealand Black/New Zealand White F1 mice and their therapeutic depletion requires a combined targeting of autoreactive plasma cells and their precursors. Arthritis Res Ther (2015) 17:39. doi:10.1186/s13075-015-0551-3
77. McLachlan SM, Rapoport B. Breaking tolerance to thyroid antigens: changing concepts in thyroid autoimmunity. Endocr Rev (2014) 35:59–105. doi:10.1210/er.2013-1055
78. Rapoport B, Chazenbalk GD, Jaume JC, McLachlan SM. The thyrotropin (TSH) receptor: interaction with TSH and autoantibodies. Endocr Rev (1998) 19:673–716. doi:10.1210/edrv.19.6.0352
79. Zakarija M, McKenzie JM, Eidson MS. Transient neonatal hypothyroidism: characterization of maternal antibodies to the thyrotropin receptor. J Clin Endocrinol Metab (1990) 70:1239–46. doi:10.1210/jcem-70-5-1239
80. Chazenbalk GD, Pichurin P, Chen CR, Latrofa F, Johnstone AP, McLachlan SM, et al. Thyroid-stimulating autoantibodies in Graves disease preferentially recognize the free A subunit, not the thyrotropin holoreceptor. J Clin Invest (2002) 110:209–17. doi:10.1172/JCI0215745
81. Chen CR, Pichurin P, Nagayama Y, Latrofa F, Rapoport B, McLachlan SM. The thyrotropin receptor autoantigen in Graves disease is the culprit as well as the victim. J Clin Invest (2003) 111:1897–904. doi:10.1172/JCI200317069
82. Rapoport B, McLachlan SM. TSH receptor cleavage into subunits and shedding of the A-subunit; A molecular and clinical perspective. Endocr Rev (2016) 37:114–34. doi:10.1210/er.2015-1098
83. Orgiazzi J, Williams DE, Chopra IJ, Solomon DH. Human thyroid adenyl cyclase-stimulating activity in immunoglobulin G of patients with Graves’ disease. J Clin Endocrinol Metab (1976) 42:341–54. doi:10.1210/jcem-42-2-341
84. Endo K, Kasagi K, Konishi J, Ikekubo K, Okuno T, Takeda Y, et al. Detection and properties of TSH-binding inhibitor immunoglobulins in patients with Graves’ disease and Hashimoto’s thyroiditis. J Clin Endocrinol Metab (1978) 46:734–9. doi:10.1210/jcem-46-5-734
85. McLachlan SM, Rapoport B. The molecular biology of thyroid peroxidase: cloning, expression and role as autoantigen in autoimmune thyroid disease. Endocr Rev (1992) 13:192–206. doi:10.1210/er.13.2.192
86. McLachlan SM, Rapoport B. Thyrotropin-blocking autoantibodies and thyroid-stimulating autoantibodies: potential mechanisms involved in the pendulum swinging from hypothyroidism to hyperthyroidism or vice versa. Thyroid (2013) 23:14–24. doi:10.1089/thy.2012.0374
87. Nagayama Y. Graves’ animal models of Graves’ hyperthyroidism. Thyroid (2007) 17:981–8. doi:10.1089/thy.2007.0161
88. McLachlan SM, Rapoport B. Thyroid stimulating monoclonal antibodies: overcoming the road blocks and the way forward. Clin Endocrinol (Oxf) (2004) 61:10–8. doi:10.1111/j.1365-2265.2004.02028.x
89. Gilbert JA, Gianoukakis AG, Salehi S, Moorhead J, Rao PV, Khan MZ, et al. Monoclonal pathogenic antibodies to the thyroid-stimulating hormone receptor in Graves’ disease with potent thyroid-stimulating activity but differential blocking activity activate multiple signaling pathways. J Immunol (2006) 176:5084–92. doi:10.4049/jimmunol.176.8.5084
90. Nakahara M, Mitsutake N, Sakamoto H, Chen CR, Rapoport B, McLachlan SM, et al. Enhanced response to mouse thyroid-stimulating hormone (TSH) receptor immunization in TSH receptor-knockout mice. Endocrinology (2010) 151:4047–54. doi:10.1210/en.2010-0315
91. Nakahara M, Johnson K, Eckstein A, Taguchi R, Yamada M, Abiru N, et al. Adoptive transfer of antithyrotropin receptor (TSHR) autoimmunity from TSHR knockout mice to athymic nude mice. Endocrinology (2012) 153:2034–42. doi:10.1210/en.2011-1846
92. Hollowell JG, Staehling NW, Flanders WD, Hannon WH, Gunter EW, Spencer CA, et al. Serum TSH, T(4), and thyroid antibodies in the United States population (1988 to 1994): National Health and Nutrition Examination Survey (NHANES III). J Clin Endocrinol Metab (2002) 87:489–99. doi:10.1210/jcem.87.2.8182
94. Rapoport B, Alsabeh R, Aftergood D, McLachlan SM. Elephantiasic pretibial myxedema: insight into and a hypothesis regarding the pathogenesis of the extrathyroidal manifestations of Graves’ disease. Thyroid (2000) 10:685–92. doi:10.1089/10507250050137761
95. Sanders J, Evans M, Betterle C, Sanders P, Bhardwaja A, Young S, et al. A human monoclonal autoantibody to the thyrotropin receptor with thyroid-stimulating blocking activity. Thyroid (2008) 18:735–46. doi:10.1089/thy.2007.0327
96. Evans M, Sanders J, Tagami T, Sanders P, Young S, Roberts E, et al. Monoclonal autoantibodies to the TSH receptor, one with stimulating activity and one with blocking activity, obtained from the same blood sample. Clin Endocrinol (Oxf) (2010) 73:404–12. doi:10.1111/j.1365-2265.2010.03831.x
97. de Forteza R, Smith CU, Amin J, McKenzie JM, Zakarija M. Visualization of the thyrotropin receptor on the cell surface by potent autoantibodies. J Clin Endocrinol Metab (1994) 78:1271–3. doi:10.1210/jcem.78.5.7909819
98. Jaume JC, Kakinuma A, Chazenbalk GD, Rapoport B, McLachlan SM. Thyrotropin receptor autoantibodies in serum are present at much lower levels than thyroid peroxidase autoantibodies: analysis by flow cytometry. J Clin Endocrinol Metab (1997) 82:500–7. doi:10.1210/jcem.82.2.3740
99. Nakatake N, Sanders J, Richards T, Burne P, Barrett C, Pra CD, et al. Estimation of serum TSH receptor autoantibody concentration and affinity. Thyroid (2006) 16:1077–84. doi:10.1089/thy.2006.16.1077
100. Metcalfe R, Jordan N, Watson P, Gullu S, Wiltshire M, Crisp M, et al. Demonstration of immunoglobulin G, A, and E autoantibodies to the human thyrotropin receptor using flow cytometry. J Clin Endocrinol Metab (2002) 87:1754–61. doi:10.1210/jcem.87.4.8411
101. Sanders J, Chirgadze DY, Sanders P, Baker S, Sullivan A, Bhardwaja A, et al. Crystal structure of the TSH receptor in complex with a thyroid-stimulating autoantibody. Thyroid (2007) 17:395–410. doi:10.1089/thy.2007.0041
102. Sanders P, Young S, Sanders J, Kabelis K, Baker S, Sullivan A, et al. Crystal structure of the TSH receptor (TSHR) bound to a blocking-type TSHR autoantibody. J Mol Endocrinol (2011) 46:81–99. doi:10.1530/JME-10-0127
103. Morshed SA, Davies TF. Graves’ disease mechanisms: the role of stimulating, blocking, and cleavage region TSH receptor antibodies. Horm Metab Res (2015) 47:727–34. doi:10.1055/s-0035-1559633
104. Burch HB, Cooper DS. Management of graves disease: a review. JAMA (2015) 314:2544–54. doi:10.1001/jama.2015.16535
105. van Zeijl CJ, van Koppen CJ, Surovtseva OV, de Gooyer ME, Plate R, Conti P, et al. Complete inhibition of rhTSH-, Graves’ disease IgG-, and M22-induced cAMP production in differentiated orbital fibroblasts by a low-molecular-weight TSHR antagonist. J Clin Endocrinol Metab (2012) 97:E781–5. doi:10.1210/jc.2011-2931
106. van Koppen CJ, de Gooyer ME, Karstens WJ, Plate R, Conti PG, van Achterberg TA, et al. Mechanism of action of a nanomolar potent, allosteric antagonist of the thyroid-stimulating hormone receptor. Br J Pharmacol (2012) 165:2314–24. doi:10.1111/j.1476-5381.2011.01709.x
107. Turcu AF, Kumar S, Neumann S, Coenen M, Iyer S, Chiriboga P, et al. A small molecule antagonist inhibits thyrotropin receptor antibody-induced orbital fibroblast functions involved in the pathogenesis of Graves ophthalmopathy. J Clin Endocrinol Metab (2013) 98:2153–9. doi:10.1210/jc.2013-1149
108. Reichert JM. Marketed therapeutic antibodies compendium. MAbs (2012) 4:413–5. doi:10.4161/mabs.19931
109. Joly P, Maho-Vaillant M, Prost-Squarcioni C, Hebert V, Houivet E, Calbo S, et al. First-line rituximab combined with short-term prednisone versus prednisone alone for the treatment of pemphigus (Ritux 3): a prospective, multicentre, parallel-group, open-label randomised trial. Lancet (2017). doi:10.1016/S0140-6736
110. Meyersburg D, Schmidt E, Kasperkiewicz M, Zillikens D. Immunoadsorption in dermatology. Ther Apher Dial (2012) 16:311–20. doi:10.1111/j.1744-9987.2012.01075.x
111. Zuercher AW, Spirig R, Baz Morelli A, Käsermann F. IVIG in autoimmune disease – potential next generation biologics. Autoimmun Rev (2016) 15:781–5. doi:10.1016/j.autrev.2016.03.018
112. Peyvandi F, Scully M, Kremer Hovinga JA, Cataland S, Knöbl P, Wu H, et al. Caplacizumab for acquired thrombotic thrombocytopenic purpura. N Engl J Med (2016) 374:511–22. doi:10.1056/NEJMoa1505533
113. Pittock SJ, Lennon VA, McKeon A, Mandrekar J, Weinshenker BG, Lucchinetti CF, et al. Eculizumab in AQP4-IgG-positive relapsing neuromyelitis optica spectrum disorders: an open-label pilot study. Lancet Neurol (2013) 12:554–62. doi:10.1016/S1474-4422(13)70076-0
114. Shire, Research and Development Pipeline (2017). Available from: https://www.shire.com/research-and-development/pipeline.
115. Ludwig RJ. Signaling and targeted-therapy of inflammatory cells in epidermolysis bullosa acquisita. Exp Dermatol (2017). doi:10.1111/exd.13335
116. ClinicalTrials.gov. K1-70 – A Study in Subjects with Graves’ Disease. (2017). Available from: https://clinicaltrials.gov/ct2/show/NCT02904330?term=k1-70&rank=1
117. Ellebrecht CT, Bhoj VG, Nace A, Choi EJ, Mao X, Cho MJ, et al. Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science (2016) 353:179–84. doi:10.1126/science.aaf6756
118. Bektas M, Jolly PS, Berkowitz P, Amagai M, Rubenstein DS. A pathophysiologic role for epidermal growth factor receptor in pemphigus acantholysis. J Biol Chem (2013) 288:9447–56. doi:10.1074/jbc.M112.438010
119. Spindler V, Rötzer V, Dehner C, Kempf B, Gliem M, Radeva M, et al. Peptide-mediated desmoglein 3 crosslinking prevents pemphigus vulgaris autoantibody-induced skin blistering. J Clin Invest (2013) 123:800–11. doi:10.1172/JCI60139
120. Koga H, Recke A, Vidarsson G, Pas HH, Jonkman MF, Hashimoto T, et al. PDE4 inhibition as potential treatment of epidermolysis bullosa acquisita. J Invest Dermatol (2016) 136:2211–20. doi:10.1016/j.jid.2016.06.619
121. Papp K, Cather JC, Rosoph L, Sofen H, Langley RG, Matheson RT, et al. Efficacy of apremilast in the treatment of moderate to severe psoriasis: a randomised controlled trial. Lancet (2012) 380:738–46. doi:10.1016/S0140-6736(12)60642-4
122. Stan MN, Garrity JA, Carranza Leon BG, Prabin T, Bradley EA, Bahn RS. Randomized controlled trial of rituximab in patients with Graves’ orbitopathy. J Clin Endocrinol Metab (2015) 100:432–41. doi:10.1210/jc.2014-2572
123. Salvi M, Vannucchi G, Currò N, Campi I, Covelli D, Dazzi D, et al. Efficacy of B-cell targeted therapy with rituximab in patients with active moderate to severe Graves’ orbitopathy: a randomized controlled study. J Clin Endocrinol Metab (2015) 100:422–31. doi:10.1210/jc.2014-3014
124. Wu L, Xun L, Yang J, Xu L, Tian Z, Gao S, et al. Induction of murine neonatal tolerance against Graves’ disease using recombinant adenovirus expressing the TSH receptor A-subunit. Endocrinology (2011) 152:1165–71. doi:10.1210/en.2010-0737
125. Misharin AV, Nagayama Y, Aliesky HA, Mizutori Y, Rapoport B, McLachlan SM. Attenuation of induced hyperthyroidism in mice by pretreatment with thyrotropin receptor protein: deviation of thyroid-stimulating to nonfunctional antibodies. Endocrinology (2009) 150:3944–52. doi:10.1210/en.2009-0181
126. Rapoport B, Aliesky HA, Banuelos B, Chen CR, McLachlan SM. A unique mouse strain that develops spontaneous, iodine-accelerated, pathogenic antibodies to the human thyrotrophin receptor. J Immunol (2015) 194:4154–61. doi:10.4049/jimmunol.1500126
127. Rapoport B, Banuelos B, Aliesky HA, Hartwig Trier N, McLachlan SM. Critical differences between induced and spontaneous mouse models of Graves’ disease with implications for antigen-specific immunotherapy in humans. J Immunol (2016) 197:4560–8. doi:10.4049/jimmunol.1601393
128. Ho YY, Lagares D, Tager AM, Kapoor M. Fibrosis – a lethal component of systemic sclerosis. Nat Rev Rheumatol (2014) 10:390–402. doi:10.1038/nrrheum.2014.53
129. Günther J, Rademacher J, van Laar JM, Siegert E, Riemekasten G. Functional autoantibodies in systemic sclerosis. Semin Immunopathol (2015) 37:529–42. doi:10.1007/s00281-015-0513-5
130. Riemekasten G, Philippe A, Näther M, Slowinski T, Müller DN, Heidecke H, et al. Involvement of functional autoantibodies against vascular receptors in systemic sclerosis. Ann Rheum Dis (2011) 70:530–6. doi:10.1136/ard.2010.135772
131. Avouac J, Riemekasten G, Meune C, Ruiz B, Kahan A, Allanore Y. Autoantibodies against endothelin 1 type A receptor are strong predictors of digital ulcers in systemic sclerosis. J Rheumatol (2015) 42:1801–7. doi:10.3899/jrheum.150061
132. Becker MO, Kill A, Kutsche M, Guenther J, Rose A, Tabeling C, et al. Vascular receptor autoantibodies in pulmonary arterial hypertension associated with systemic sclerosis. Am J Respir Crit Care Med (2014) 190:808–17. doi:10.1164/rccm.201403-0442OC
133. Günther J, Kill A, Becker MO, Heidecke H, Rademacher J, Siegert E, et al. Angiotensin receptor type 1 and endothelin receptor type A on immune cells mediate migration and the expression of IL-8 and CCL18 when stimulated by autoantibodies from systemic sclerosis patients. Arthritis Res Ther (2014) 16:R65. doi:10.1186/ar4503
134. Kill A, Tabeling C, Undeutsch R, Kühl AA, Günther J, Radic M, et al. Autoantibodies to angiotensin and endothelin receptors in systemic sclerosis induce cellular and systemic events associated with disease pathogenesis. Arthritis Res Ther (2014) 16:R29. doi:10.1186/ar4457
135. Kill A, Riemekasten G. Functional autoantibodies in systemic sclerosis pathogenesis. Curr Rheumatol Rep (2015) 17:34. doi:10.1007/s11926-015-0505-4
136. Wenzel K, Rajakumar A, Haase H, Geusens N, Hubner N, Schulz H, et al. Angiotensin II type 1 receptor antibodies and increased angiotensin II sensitivity in pregnant rats. Hypertension (2011) 58:77–84. doi:10.1161/HYPERTENSIONAHA.111.171348
137. Dechend R, Viedt C, Müller DN, Ugele B, Brandes RP, Wallukat G, et al. AT1 receptor agonistic antibodies from preeclamptic patients stimulate NADPH oxidase. Circulation (2003) 107:1632–9. doi:10.1161/01.CIR.0000058200.90059.B1
138. Dragun D, Müller DN, Bräsen JH, Fritsche L, Nieminen-Kelhä M, Dechend R, et al. Angiotensin II type 1-receptor activating antibodies in renal-allograft rejection. N Engl J Med (2005) 352:558–69. doi:10.1056/NEJMoa035717
139. Cabral-Marques O, Riemekasten G. Vascular hypothesis revisited: role of stimulating antibodies against angiotensin and endothelin receptors in the pathogenesis of systemic sclerosis. Autoimmun Rev (2016) 15:690–4. doi:10.1016/j.autrev.2016.03.005
140. Prasse A, Pechkovsky DV, Toews GB, Schäfer M, Eggeling S, Ludwig C, et al. CCL18 as an indicator of pulmonary fibrotic activity in idiopathic interstitial pneumonias and systemic sclerosis. Arthritis Rheum (2007) 56:1685–93. doi:10.1002/art.22559
141. Luzina IG, Atamas SP, Wise R, Wigley FM, Xiao HQ, White B. Gene expression in bronchoalveolar lavage cells from scleroderma patients. Am J Respir Cell Mol Biol (2002) 26:549–57. doi:10.1165/ajrcmb.26.5.4683
142. Schupp J, Becker M, Günther J, Müller-Quernheim J, Riemekasten G, Prasse A. Serum CCL18 is predictive for lung disease progression and mortality in systemic sclerosis. Eur Respir J (2014) 43:1530–2. doi:10.1183/09031936.00131713
143. Rademacher J, Kill A, Mattat K, Dragun D, Siegert E, Günther J, et al. Monocytic angiotensin and endothelin receptor imbalance modulate secretion of the profibrotic chemokine ligand 18. J Rheumatol (2016) 43:587–91. doi:10.3899/jrheum.150474
144. Wallukat G, Homuth V, Fischer T, Lindschau C, Horstkamp B, Jüpner A, et al. Patients with preeclampsia develop agonistic autoantibodies against the angiotensin AT1 receptor. J Clin Invest (1999) 103:945–52. doi:10.1172/JCI4106
145. Xiong J, Liang Y, Yang H, Zhu F, Wang Y. The role of angiotensin II type 1 receptor-activating antibodies in patients with lupus nephritis. [letter]. Int J Clin Pract (2013) 67(10):1066–7. doi:10.1111/ijcp.12242
146. Guo L, Li M, Chen Y, Wang Q, Tian Z, Pan S, et al. Anti-endothelin receptor type A autoantibodies in systemic lupus erythematosus-associated pulmonary arterial hypertension. Arthritis Rheumatol (2015) 67:2394–402. doi:10.1002/art.39212
147. Zhao L, Xu C, Xu J. Autoantibodies against β1 receptor and AT1 receptor in type 2 diabetes patients with left ventricular dilatation. Cardiology (2014) 129:191–6. doi:10.1159/000365782
148. Budding K, van de Graaf EA, Hoefnagel T, Kwakkel-van Erp JM, van Kessel DA, Dragun D, et al. Anti-ETAR and anti-AT1R autoantibodies are elevated in patients with endstage cystic fibrosis. J Cyst Fibros (2015) 14:42–5. doi:10.1016/j.jcf.2014.07.007
149. Conti-Fine BM, Milani M, Kaminski HJ. Myasthenia gravis: past, present, and future. J Clin Invest (2006) 116:2843–54. doi:10.1172/JCI29894
150. Lindstrom JM. Acetylcholine receptors and myasthenia. Muscle Nerve (2000) 23:453–77. doi:10.1002/(SICI)1097-4598(200004)23:4<453::AID-MUS3>3.0.CO;2-O
151. Breiner A, Widdifield J, Katzberg HD, Barnett C, Bril V, Tu K. Epidemiology of myasthenia gravis in Ontario, Canada. Neuromuscul Disord (2016) 26:41–6. doi:10.1016/j.nmd.2015.10.009
152. Carr AS, Cardwell CR, McCarron PO, McConville J. A systematic review of population based epidemiological studies in myasthenia gravis. BMC Neurol (2010) 10:46. doi:10.1186/1471-2377-10-46
153. Heldal AT, Eide GE, Gilhus NE, Romi F. Geographical distribution of a seropositive myasthenia gravis population. Muscle Nerve (2012) 45:815–9. doi:10.1002/mus.23271
154. Keesey JC. Clinical evaluation and management of myasthenia gravis. Muscle Nerve (2004) 29:484–505. doi:10.1002/mus.20030
155. Nacu A, Andersen JB, Lisnic V, Owe JF, Gilhus NE. Complicating autoimmune diseases in myasthenia gravis: a review. Autoimmunity (2015) 48:362–8. doi:10.3109/08916934.2015.1030614
156. Berrih-Aknin S, Le Panse R. Myasthenia gravis: a comprehensive review of immune dysregulation and etiological mechanisms. J Autoimmun (2014) 52:90–100. doi:10.1016/j.jaut.2013.12.011
157. Cufi P, Dragin N, Weiss JM, Martinez-Martinez P, De Baets MH, Roussin R, et al. Implication of double-stranded RNA signaling in the etiology of autoimmune myasthenia gravis. Ann Neurol (2013) 73:281–93. doi:10.1002/ana.23791
158. Zagoriti Z, Kambouris ME, Patrinos GP, Tzartos SJ, Poulas K. Recent advances in genetic predisposition of myasthenia gravis. Biomed Res Int (2013) 2013:404053. doi:10.1155/2013/404053
159. Verschuuren JJ, Huijbers MG, Plomp JJ, Niks EH, Molenaar PC, Martinez-Martinez P, et al. Pathophysiology of myasthenia gravis with antibodies to the acetylcholine receptor, muscle-specific kinase and low-density lipoprotein receptor-related protein 4. Autoimmun Rev (2013) 12:918–23. doi:10.1016/j.autrev.2013.03.001
160. Romi F. Thymoma in myasthenia gravis: from diagnosis to treatment. Autoimmune Dis (2011) 2011:474512. doi:10.4061/2011/474512
161. Diaz A, Black E, Dunning J. Is thymectomy in non-thymomatous myasthenia gravis of any benefit. Interact Cardiovasc Thorac Surg (2014) 18:381–9. doi:10.1093/icvts/ivt510
162. Kuks JB, Oosterhuis HJ, Limburg PC, The TH. Anti-acetylcholine receptor antibodies decrease after thymectomy in patients with myasthenia gravis. Clinical correlations. J Autoimmun (1991) 4:197–211. doi:10.1016/0896-8411(91)90018-8
163. Simpson JA. Myasthenia gravis: a new hypothesis. Scott Med J (1960) 5:419–36. doi:10.1177/003693306000501001
164. Patrick J, Lindstrom J. Autoimmune response to acetylcholine receptor. Science (1973) 180:871–2. doi:10.1126/science.180.4088.871
165. Lennon VA, Lindstrom JM, Seybold ME. Experimental autoimmune myasthenia: a model of myasthenia gravis in rats and guinea pigs. J Exp Med (1975) 141:1365–75. doi:10.1084/jem.141.6.1365
166. Engel AG. Myasthenia gravis and myasthenic syndromes. Ann Neurol (1984) 16:519–34. doi:10.1002/ana.410160502
167. Lindstrom JM, Seybold ME, Lennon VA, Whittingham S, Duane DD. Antibody to acetylcholine receptor in myasthenia gravis. Prevalence, clinical correlates, and diagnostic value. Neurology (1976) 26:1054–9. doi:10.1212/WNL.26.11.1054
168. Toyka KV, Drachman DB, Griffin DE, Pestronk A, Winkelstein JA, Fishbeck KH, et al. Myasthenia gravis. Study of humoral immune mechanisms by passive transfer to mice. N Engl J Med (1977) 296:125–31. doi:10.1056/NEJM197701202960301
169. Kordas G, Lagoumintzis G, Sideris S, Poulas K, Tzartos SJ. Direct proof of the in vivo pathogenic role of the AChR autoantibodies from myasthenia gravis patients. PLoS One (2014) 9:e108327. doi:10.1371/journal.pone.0108327
170. Engel AG, Sahashi K, Fumagalli G. The immunopathology of acquired myasthenia gravis. Ann N Y Acad Sci (1981) 377:158–74. doi:10.1111/j.1749-6632.1981.tb33730.x
171. Engel AG, Arahata K. The membrane attack complex of complement at the endplate in myasthenia gravis. Ann N Y Acad Sci (1987) 505:326–32. doi:10.1111/j.1749-6632.1987.tb51301.x
172. Newsom-Davis J, Pinching AJ, Vincent A, Wilson SG. Function of circulating antibody to acetylcholine receptor in myasthenia gravis: investigation by plasma exchange. Neurology (1978) 28:266–72. doi:10.1212/WNL.28.3.266
173. Tzartos S, Hochschwender S, Vasquez P, Lindstrom J. Passive transfer of experimental autoimmune myasthenia gravis by monoclonal antibodies to the main immunogenic region of the acetylcholine receptor. J Neuroimmunol (1987) 15:185–94. doi:10.1016/0165-5728(87)90092-0
174. Sanders DB, Burns TM, Cutter GR, Massey JM, Juel VC, Hobson-Webb L, et al. Does change in acetylcholine receptor antibody level correlate with clinical change in myasthenia gravis. Muscle Nerve (2014) 49:483–6. doi:10.1002/mus.23944
175. Luo J, Lindstrom J. Antigen-specific immunotherapeutic vaccine for experimental autoimmune myasthenia gravis. J Immunol (2014) 193:5044–55. doi:10.4049/jimmunol.1401392
176. Luo J, Taylor P, Losen M, de Baets MH, Shelton GD, Lindstrom J. Main immunogenic region structure promotes binding of conformation-dependent myasthenia gravis autoantibodies, nicotinic acetylcholine receptor conformation maturation, and agonist sensitivity. J Neurosci (2009) 29:13898–908. doi:10.1523/JNEUROSCI.2833-09.2009
177. Drachman DB, Angus CW, Adams RN, Michelson JD, Hoffman GJ. Myasthenic antibodies cross-link acetylcholine receptors to accelerate degradation. N Engl J Med (1978) 298:1116–22. doi:10.1056/NEJM197805182982004
178. Drachman DB, Adams RN, Josifek LF, Self SG. Functional activities of autoantibodies to acetylcholine receptors and the clinical severity of myasthenia gravis. N Engl J Med (1982) 307:769–75. doi:10.1056/NEJM198209233071301
179. Conti-Fine BM, Milani M, Wang W. CD4+ T cells and cytokines in the pathogenesis of acquired myasthenia gravis. Ann N Y Acad Sci (2008) 1132:193–209. doi:10.1196/annals.1405.042
180. Tzartos SJ, Remoundos M. Detection of antibodies directed against the cytoplasmic region of the human acetylcholine receptor in sera from myasthenia gravis patients. Clin Exp Immunol (1999) 116:146–52. doi:10.1046/j.1365-2249.1999.00846.x
181. Luo J, Lindstrom J. AChR-specific immunosuppressive therapy of myasthenia gravis. Biochem Pharmacol (2015) 97:609–19. doi:10.1016/j.bcp.2015.07.011
182. Rødgaard A, Nielsen FC, Djurup R, Somnier F, Gammeltoft S. Acetylcholine receptor antibody in myasthenia gravis: predominance of IgG subclasses 1 and 3. Clin Exp Immunol (1987) 67:82–8.
183. Tüzün E, Scott BG, Goluszko E, Higgs S, Christadoss P. Genetic evidence for involvement of classical complement pathway in induction of experimental autoimmune myasthenia gravis. J Immunol (2003) 171:3847–54. doi:10.4049/jimmunol.171.7.3847
184. Tüzün E, Christadoss P. Complement associated pathogenic mechanisms in myasthenia gravis. Autoimmun Rev (2013) 12:904–11. doi:10.1016/j.autrev.2013.03.003
185. Soltys J, Kusner LL, Young A, Richmonds C, Hatala D, Gong B, et al. Novel complement inhibitor limits severity of experimentally myasthenia gravis. Ann Neurol (2009) 65:67–75. doi:10.1002/ana.21536
186. Strochlic L, Cartaud A, Cartaud J. The synaptic muscle-specific kinase (MuSK) complex: new partners, new functions. Bioessays (2005) 27:1129–35. doi:10.1002/bies.20305
187. Vincent A, Bowen J, Newsom-Davis J, McConville J. Seronegative generalised myasthenia gravis: clinical features, antibodies, and their targets. Lancet Neurol (2003) 2:99–106. doi:10.1016/S1474-4422(03)00306-5
188. Klooster R, Plomp JJ, Huijbers MG, Niks EH, Straasheijm KR, Detmers FJ, et al. Muscle-specific kinase myasthenia gravis IgG4 autoantibodies cause severe neuromuscular junction dysfunction in mice. Brain (2012) 135:1081–101. doi:10.1093/brain/aws025
189. Reddel SW, Morsch M, Phillips WD. Clinical and scientific aspects of muscle-specific tyrosine kinase-related myasthenia gravis. Curr Opin Neurol (2014) 27:558–65. doi:10.1097/WCO.0000000000000136
190. Ghazanfari N, Morsch M, Reddel SW, Liang SX, Phillips WD. Muscle-specific kinase (MuSK) autoantibodies suppress the MuSK pathway and ACh receptor retention at the mouse neuromuscular junction. J Physiol (2014) 592:2881–97. doi:10.1113/jphysiol.2013.270207
191. Mori S, Kubo S, Akiyoshi T, Yamada S, Miyazaki T, Hotta H, et al. Antibodies against muscle-specific kinase impair both presynaptic and postsynaptic functions in a murine model of myasthenia gravis. Am J Pathol (2012) 180:798–810. doi:10.1016/j.ajpath.2011.10.031
192. Evoli A, Tonali PA, Padua L, Monaco ML, Scuderi F, Batocchi AP, et al. Clinical correlates with anti-MuSK antibodies in generalized seronegative myasthenia gravis. Brain (2003) 126:2304–11. doi:10.1093/brain/awg223
193. Iorio R, Damato V, Alboini PE, Evoli A. Efficacy and safety of rituximab for myasthenia gravis: a systematic review and meta-analysis. J Neurol (2015) 262:1115–9. doi:10.1007/s00415-014-7532-3
194. Shen C, Xiong WC, Mei L. LRP4 in neuromuscular junction and bone development and diseases. Bone (2015) 80:101–8. doi:10.1016/j.bone.2015.05.012
195. Higuchi O, Hamuro J, Motomura M, Yamanashi Y. Autoantibodies to low-density lipoprotein receptor-related protein 4 in myasthenia gravis. Ann Neurol (2011) 69:418–22. doi:10.1002/ana.22312
196. Pevzner A, Schoser B, Peters K, Cosma NC, Karakatsani A, Schalke B, et al. Anti-LRP4 autoantibodies in AChR- and MuSK-antibody-negative myasthenia gravis. J Neurol (2012) 259:427–35. doi:10.1007/s00415-011-6194-7
197. Shen C, Lu Y, Zhang B, Figueiredo D, Bean J, Jung J, et al. Antibodies against low-density lipoprotein receptor-related protein 4 induce myasthenia gravis. J Clin Invest (2013) 123:5190–202. doi:10.1172/JCI66039
198. Leite MI, Jacob S, Viegas S, Cossins J, Clover L, Morgan BP, et al. IgG1 antibodies to acetylcholine receptors in ‘seronegative’ myasthenia gravis. Brain (2008) 131:1940–52. doi:10.1093/brain/awn092
199. Cossins J, Belaya K, Zoltowska K, Koneczny I, Maxwell S, Jacobson L, et al. The search for new antigenic targets in myasthenia gravis. Ann N Y Acad Sci (2012) 1275:123–8. doi:10.1111/j.1749-6632.2012.06833.x
200. Gilhus NE, Verschuuren JJ. Myasthenia gravis: subgroup classification and therapeutic strategies. Lancet Neurol (2015) 14:1023–36. doi:10.1016/S1474-4422(15)00145-3
201. Guptill JT, Soni M, Meriggioli MN. Current treatment, emerging translational therapies, and new therapeutic targets for autoimmune myasthenia gravis. Neurotherapeutics (2016) 13:118–31. doi:10.1007/s13311-015-0398-y
202. Gomez AM, Willcox N, Molenaar PC, Buurman W, Martinez-Martinez P, De Baets MH, et al. Targeting plasma cells with proteasome inhibitors: possible roles in treating myasthenia gravis. Ann N Y Acad Sci (2012) 1274:48–59. doi:10.1111/j.1749-6632.2012.06824.x
203. Guptill JT, Sharma BK, Marano A, Soucy A, Krueger A, Sanders DB. Estimated cost of treating myasthenia gravis in an insured U.S. population. Muscle Nerve (2012) 45:363–6. doi:10.1002/mus.22327
204. Gold R, Hohlfeld R, Toyka KV. Progress in the treatment of myasthenia gravis. Ther Adv Neurol Disord (2008) 1:36–51. doi:10.1177/1756285608093888
205. Silvestri NJ, Wolfe GI. Treatment-refractory myasthenia gravis. J Clin Neuromuscul Dis (2014) 15:167–78. doi:10.1097/CND.0000000000000034
206. Steinman L, Merrill JT, McInnes IB, Peakman M. Optimization of current and future therapy for autoimmune diseases. Nat Med (2012) 18:59–65. doi:10.1038/nm.2625
207. Dalakas MC. Future perspectives in target-specific immunotherapies of myasthenia gravis. Ther Adv Neurol Disord (2015) 8:316–27. doi:10.1177/1756285615605700
208. Luo J, Kuryatov A, Lindstrom JM. Specific immunotherapy of experimental myasthenia gravis by a novel mechanism. Ann Neurol (2010) 67:441–51. doi:10.1002/ana.21901
209. Granerod J, Ambrose HE, Davies NW, Clewley JP, Walsh AL, Morgan D, et al. Causes of encephalitis and differences in their clinical presentations in England: a multicentre, population-based prospective study. Lancet Infect Dis (2010) 10:835–44. doi:10.1016/S1473-3099(10)70222-X
210. Granerod J, Cousens S, Davies NW, Crowcroft NS, Thomas SL. New estimates of incidence of encephalitis in England. Emerg Infect Dis (2013) 19. doi:10.3201/eid1909.130064
211. Leypoldt F, Armangue T, Dalmau J. Autoimmune encephalopathies. Ann N Y Acad Sci (2015) 1338:94–114. doi:10.1111/nyas.12553
212. Dalmau J, Lancaster E, Martinez-Hernandez E, Rosenfeld MR, Balice-Gordon R. Clinical experience and laboratory investigations in patients with anti-NMDAR encephalitis. Lancet Neurol (2011) 10:63–74. doi:10.1016/S1474-4422(10)70253-2
213. Titulaer MJ, McCracken L, Gabilondo I, Armangué T, Glaser C, Iizuka T, et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol (2013) 12:157–65. doi:10.1016/S1474-4422(12)70310-1
214. Armangue T, Titulaer MJ, Málaga I, Bataller L, Gabilondo I, Graus F, et al. Pediatric anti-N-methyl-d-aspartate receptor encephalitis-clinical analysis and novel findings in a series of 20 patients. J Pediatr (2013) 162:850–6.e2. doi:10.1016/j.jpeds.2012.10.011
215. Gleichman AJ, Spruce LA, Dalmau J, Seeholzer SH, Lynch DR. Anti-NMDA receptor encephalitis antibody binding is dependent on amino acid identity of a small region within the GluN1 amino terminal domain. J Neurosci (2012) 32:11082–94. doi:10.1523/JNEUROSCI.0064-12.2012
216. Moscato EH, Jain A, Peng X, Hughes EG, Dalmau J, Balice-Gordon RJ. Mechanisms underlying autoimmune synaptic encephalitis leading to disorders of memory, behavior and cognition: insights from molecular, cellular and synaptic studies. Eur J Neurosci (2010) 32:298–309. doi:10.1111/j.1460-9568.2010.07349.x
217. Mangler M, Trebesch de Perez I, Teegen B, Stöcker W, Prüss H, Meisel A, et al. Seroprevalence of anti-N-methyl-d-aspartate receptor antibodies in women with ovarian teratoma. J Neurol (2013) 260:2831–5. doi:10.1007/s00415-013-7074-0
218. Prüss H, Finke C, Höltje M, Hofmann J, Klingbeil C, Probst C, et al. N-methyl-d-aspartate receptor antibodies in herpes simplex encephalitis. Ann Neurol (2012) 72:902–11. doi:10.1002/ana.23689
219. Leypoldt F, Titulaer MJ, Aguilar E, et al. Herpes simplex virus-1 encephalitis can trigger anti-NMDA receptor encephalitis: case report. Neurology (2013) 81:1637–9. doi:10.1212/WNL.0b013e3182a9f531
220. Armangue T, Leypoldt F, Málaga I, Raspall-Chaure M, Marti I, Nichter C, et al. Herpes simplex virus encephalitis is a trigger of brain autoimmunity. Ann Neurol (2014) 75:317–23. doi:10.1002/ana.24083
221. Armangue T, Moris G, Cantarín-Extremera V, Conde CE, Rostasy K, Erro ME, et al. Autoimmune post-herpes simplex encephalitis of adults and teenagers. Neurology (2015) 85:1736–43. doi:10.1212/WNL.0000000000002125
222. Hacohen Y, Deiva K, Pettingill P, et al. N-methyl-d-aspartate receptor antibodies in post-herpes simplex virus encephalitis neurological relapse. Mov Disord (2014) 29:90–6. doi:10.1002/mds.25626
223. Adang LA, Lynch DR, Panzer JA. Pediatric anti-NMDA receptor encephalitis is seasonal. Ann Clin Transl Neurol (2014) 1:921–5. doi:10.1002/acn3.100
224. Gresa-Arribas N, Titulaer MJ, Torrents A, Aguilar E, McCracken L, Leypoldt F, et al. Antibody titres at diagnosis and during follow-up of anti-NMDA receptor encephalitis: a retrospective study. Lancet Neurol (2014) 13:167–77. doi:10.1016/S1474-4422(13)70282-5
225. Reiber H, Peter JB. Cerebrospinal fluid analysis: disease-related data patterns and evaluation programs. J Neurol Sci (2001) 184:101–22. doi:10.1016/S0022-510X(00)00501-3
226. Martinez-Hernandez E, Horvath J, Shiloh-Malawsky Y, Sangha N, Martinez-Lage M, Dalmau J. Analysis of complement and plasma cells in the brain of patients with anti-NMDAR encephalitis. Neurology (2011) 77:589–93. doi:10.1212/WNL.0b013e318228c136
227. Leypoldt F, Höftberger R, Titulaer MJ, Armangue T, Gresa-Arribas N, Jahn H, et al. Investigations on CXCL13 in anti-N-methyl-d-aspartate receptor encephalitis: a potential biomarker of treatment response. JAMA Neurol (2015) 72:180–6. doi:10.1001/jamaneurol.2014.2956
228. Dalmau J, Gleichman AJ, Hughes EG, Rossi JE, Peng X, Lai M, et al. Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol (2008) 7:1091–8. doi:10.1016/S1474-4422(08)70224-2
229. Manto M, Dalmau J, Didelot A, Rogemond V, Honnorat J. In vivo effects of antibodies from patients with anti-NMDA receptor encephalitis: further evidence of synaptic glutamatergic dysfunction. Orphanet J Rare Dis (2010) 5:31. doi:10.1186/1750-1172-5-31
230. Hughes EG, Peng X, Gleichman AJ, Lai M, Zhou L, Tsou R, et al. Cellular and synaptic mechanisms of anti-NMDA receptor encephalitis. J Neurosci (2010) 30:5866–75. doi:10.1523/JNEUROSCI.0167-10.2010
231. Mikasova L, De Rossi P, Bouchet D, Georges F, Rogemond V, Didelot A, et al. Disrupted surface cross-talk between NMDA and Ephrin-B2 receptors in anti-NMDA encephalitis. Brain (2012) 135:1606–21. doi:10.1093/brain/aws092
232. Moscato EH, Peng X, Jain A, Parsons TD, Dalmau J, Balice-Gordon RJ. Acute mechanisms underlying antibody effects in anti-N-methyl-d-aspartate receptor encephalitis. Ann Neurol (2014) 76:108–19. doi:10.1002/ana.24195
233. Planagumà J, Leypoldt F, Mannara F, Gutiérrez-Cuesta J, Martín-García E, Aguilar E, et al. Human N-methyl d-aspartate receptor antibodies alter memory and behaviour in mice. Brain (2015) 138:94–109. doi:10.1093/brain/awu310
234. Planagumà J, Haselmann H, Mannara F, Petit-Pedrol M, Grünewald B, Aguilar E, et al. Ephrin-B2 prevents N-methyl-d-aspartate receptor antibody effects on memory and neuroplasticity. Ann Neurol (2016) 80:388–400. doi:10.1002/ana.24721
235. Hammers CM, Stanley JR. Mechanisms of disease: pemphigus and bullous pemphigoid. Annu Rev Pathol (2016) 11:175–97. doi:10.1146/annurev-pathol-012615-044313
236. Pisanti S, Sharav Y, Kaufman E, Posner LN. Pemphigus vulgaris: incidence in Jews of different ethnic groups, according to age, sex, and initial lesion. Oral Surg Oral Med Oral Pathol (1974) 38:382–7. doi:10.1016/0030-4220(74)90365-X
237. Langan SM, Smeeth L, Hubbard R, Fleming KM, Smith CJ, West J. Bullous pemphigoid and pemphigus vulgaris – incidence and mortality in the UK: population based cohort study. BMJ (2008) 337:a180. doi:10.1136/bmj.a180
238. Aoki V, Millikan RC, Rivitti EA, Hans-Filho G, Eaton DP, Warren SJ, et al. Environmental risk factors in endemic pemphigus foliaceus (fogo selvagem). J Investig Dermatol Symp Proc (2004) 9:34–40. doi:10.1111/j.1087-0024.2004.00833.x
239. Lever WF. Pemphigus. Medicine (Baltimore) (1953) 32:1–123. doi:10.1097/00005792-195302000-00001
240. Stanley JR, Amagai M. Pemphigus, bullous impetigo, and the staphylococcal scalded-skin syndrome. N Engl J Med (2006) 355:1800–10. doi:10.1056/NEJMra061111
241. Stanley JR, Nishikawa T, Diaz LA, Amagai M. Pemphigus: is there another half of the story. J Invest Dermatol (2001) 116(4):489. doi:10.1046/j.1523-1747.2001.01307.x
242. Mao X, Nagler AR, Farber SA, Choi EJ, Jackson LH, Leiferman KM, et al. Autoimmunity to desmocollin 3 in pemphigus vulgaris. Am J Pathol (2010) 177:2724–30. doi:10.2353/ajpath.2010.100483
243. Kozlowska A, Hashimoto T, Jarzabek-Chorzelska M, Amagai A, Nagata Y, Strasz Z, et al. Pemphigus herpetiformis with IgA and IgG antibodies to desmoglein 1 and IgG antibodies to desmocollin 3. J Am Acad Dermatol (2003) 48:117–22. doi:10.1067/mjd.2003.23
244. Rafei D, Müller R, Ishii N, Llamazares M, Hashimoto T, Hertl M, et al. IgG autoantibodies against desmocollin 3 in pemphigus sera induce loss of keratinocyte adhesion. Am J Pathol (2011) 178:718–23. doi:10.1016/j.ajpath.2010.10.016
245. Hisamatsu Y, Amagai M, Garrod DR, Kanzaki T, Hashimoto T. The detection of IgG and IgA autoantibodies to desmocollins 1-3 by enzyme-linked immunosorbent assays using baculovirus-expressed proteins, in atypical pemphigus but not in typical pemphigus. Br J Dermatol (2004) 151:73–83. doi:10.1111/j.1365-2133.2004.05995.x
246. Evangelista F, Dasher DA, Diaz LA, Prisayanh PS, Li N. E-cadherin is an additional immunological target for pemphigus autoantibodies. J Invest Dermatol (2008) 128:1710–8. doi:10.1038/sj.jid.5701260
247. Kalantari-Dehaghi M, Anhalt GJ, Camilleri MJ, Chernyavsky AI, Chun S, Felgner PL, et al. Pemphigus vulgaris autoantibody profiling by proteomic technique. PLoS One (2013) 8:e57587. doi:10.1371/journal.pone.0057587
248. Chen Y, Chernyavsky A, Webber RJ, Grando SA, Wang PH. Critical role of FcRn in the pathogenic action of anti-mitochondrial autoantibodies synergizing with anti-desmoglein autoantibodies in pemphigus vulgaris. J Biol Chem (2015) 290:23826–37. doi:10.1074/jbc.M115.668061
249. Ishii K, Amagai M, Hall RP, Hashimoto T, Takayanagi A, Gamou S, et al. Characterization of autoantibodies in pemphigus using antigen-specific enzyme-linked immunosorbent assays with baculovirus-expressed recombinant desmogleins. J Immunol (1997) 159:2010–7.
250. Cheng SW, Kobayashi M, Kinoshita-Kuroda K, Tanikawa A, Amagai M, Nishikawa T. Monitoring disease activity in pemphigus with enzyme-linked immunosorbent assay using recombinant desmogleins 1 and 3. Br J Dermatol (2002) 147:261–5. doi:10.1046/j.1365-2133.2002.04838.x
251. Abasq C, Mouquet H, Gilbert D, Tron F, Grassi V, Musette P, et al. ELISA testing of anti-desmoglein 1 and 3 antibodies in the management of pemphigus. Arch Dermatol (2009) 145:529–35. doi:10.1001/archdermatol.2009.9
252. Almugairen N, Hospital V, Bedane C, Duvert-Lehembre S, Picard D, Tronquoy AF, et al. Assessment of the rate of long-term complete remission off therapy in patients with pemphigus treated with different regimens including medium- and high-dose corticosteroids. J Am Acad Dermatol (2013) 69:583–8. doi:10.1016/j.jaad.2013.05.016
253. Nguyen VT, Arredondo J, Chernyavsky AI, Kitajima Y, Pittelkow M, Grando SA. Pemphigus vulgaris IgG and methylprednisolone exhibit reciprocal effects on keratinocytes. J Biol Chem (2004) 279:2135–46. doi:10.1074/jbc.M309000200
254. Anhalt GJ, Labib RS, Voorhees JJ, Beals TF, Diaz LA. Induction of pemphigus in neonatal mice by passive transfer of IgG from patients with the disease. N Engl J Med (1982) 306:1189–96. doi:10.1056/NEJM198205203062001
255. Rock B, Labib RS, Diaz LA. Monovalent Fab′ immunoglobulin fragments from endemic pemphigus foliaceus autoantibodies reproduce the human disease in neonatal Balb/c mice. J Clin Invest (1990) 85:296–9. doi:10.1172/JCI114426
256. Ishii K, Lin C, Siegel DL, Stanley JR. Isolation of pathogenic monoclonal anti-desmoglein 1 human antibodies by phage display of pemphigus foliaceus autoantibodies. J Invest Dermatol (2008) 128:939–48. doi:10.1038/sj.jid.5701132
257. Payne AS, Ishii K, Kacir S, Lin C, Li H, Hanakawa Y, et al. Genetic and functional characterization of human pemphigus vulgaris monoclonal autoantibodies isolated by phage display. J Clin Invest (2005) 115:888–99. doi:10.1172/JCI24185
258. Yamagami J, Payne AS, Kacir S, Ishii K, Siegel DL, Stanley JR. Homologous regions of autoantibody heavy chain complementarity-determining region 3 (H-CDR3) in patients with pemphigus cause pathogenicity. J Clin Invest (2010) 120:4111–7. doi:10.1172/JCI44425
259. Heupel WM, Zillikens D, Drenckhahn D, Waschke J. Pemphigus vulgaris IgG directly inhibit desmoglein 3-mediated transinteraction. J Immunol (2008) 181:1825–34. doi:10.4049/jimmunol.181.3.1825
260. Saito M, Stahley SN, Caughman CY, Mao X, Tucker DK, Payne AS, et al. Signaling dependent and independent mechanisms in pemphigus vulgaris blister formation. PLoS One (2012) 7:e50696. doi:10.1371/journal.pone.0050696
261. Di Zenzo G, Di Lullo G, Corti D, Calabresi V, Sinistro A, Vanzetta F, et al. Pemphigus autoantibodies generated through somatic mutations target the desmoglein-3 cis-interface. J Clin Invest (2012) 122:3781–90. doi:10.1172/JCI64413
262. Sekiguchi M, Futei Y, Fujii Y, Iwasaki T, Nishikawa T, Amagai M. Dominant autoimmune epitopes recognized by pemphigus antibodies map to the N-terminal adhesive region of desmogleins. J Immunol (2001) 167:5439–48. doi:10.4049/jimmunol.167.9.5439
263. Takahashi H, Kouno M, Nagao K, Wada N, Hata T, Nishimoto S, et al. Desmoglein 3-specific CD4+ T cells induce pemphigus vulgaris and interface dermatitis in mice. J Clin Invest (2011) 121:3677–88. doi:10.1172/JCI57379
264. Oktarina DA, van der Wier G, Diercks GF, Jonkman MF, Pas HH. IgG-induced clustering of desmogleins 1 and 3 in skin of patients with pemphigus fits with the desmoglein nonassembly depletion hypothesis. Br J Dermatol (2011) 165:552–62. doi:10.1111/j.1365-2133.2011.10463.x
265. van der Wier G, Pas HH, Kramer D, Diercks GF, Jonkman MF. Smaller desmosomes are seen in the skin of pemphigus patients with anti-desmoglein 1 antibodies but not in patients with anti-desmoglein 3 antibodies [letter]. J Invest Dermatol (2014) 134(8):2287–90. doi:10.1038/jid.2014.140
266. Aoyama Y, Kitajima Y. Pemphigus vulgaris-IgG causes a rapid depletion of desmoglein 3 (Dsg3) from the Triton X-100 soluble pools, leading to the formation of Dsg3-depleted desmosomes in a human squamous carcinoma cell line, DJM-1 cells. J Invest Dermatol (1999) 112:67–71. doi:10.1046/j.1523-1747.1999.00463.x
267. Jennings JM, Tucker DK, Kottke MD, Saito M, Delva E, Hanakawa Y, et al. Desmosome disassembly in response to pemphigus vulgaris IgG occurs in distinct phases and can be reversed by expression of exogenous Dsg3. J Invest Dermatol (2011) 131:706–18. doi:10.1038/jid.2010.389
268. Stahley SN, Saito M, Faundez V, Koval M, Mattheyses AL, Kowalczyk AP. Desmosome assembly and disassembly are membrane raft-dependent. PLoS One (2014) 9:e87809. doi:10.1371/journal.pone.0087809
269. Caldelari R, de Bruin A, Baumann D, Suter MM, Bierkamp C, Balmer V, et al. A central role for the armadillo protein plakoglobin in the autoimmune disease pemphigus vulgaris. J Cell Biol (2001) 153:823–34. doi:10.1083/jcb.153.4.823
270. Williamson L, Raess NA, Caldelari R, Zakher A, de Bruin A, Posthaus H, et al. Pemphigus vulgaris identifies plakoglobin as key suppressor of c-Myc in the skin. EMBO J (2006) 25:3298–309. doi:10.1038/sj.emboj.7601224
271. Williamson L, Hunziker T, Suter MM, Müller EJ. Nuclear c-Myc: a molecular marker for early stage pemphigus vulgaris [letter]. J Invest Dermatol (2007) 127(6):1549–55. doi:10.1038/sj.jid.5700735
272. Berkowitz P, Hu P, Liu Z, Diaz LA, Enghild JJ, Chua MP, et al. Desmosome signaling. Inhibition of p38MAPK prevents pemphigus vulgaris IgG-induced cytoskeleton reorganization. J Biol Chem (2005) 280:23778–84. doi:10.1074/jbc.M501365200
273. Rubenstein DS, Diaz LA. Pemphigus antibody induced phosphorylation of keratinocyte proteins. Autoimmunity (2006) 39:577–86. doi:10.1080/08916930600971885
274. Mao X, Li H, Sano Y, Gaestel M, Mo Park J, Payne AS. MAPKAP kinase 2 (MK2)-dependent and -independent models of blister formation in pemphigus vulgaris. J Invest Dermatol (2014) 134:68–76. doi:10.1038/jid.2013.224
275. Jolly PS, Berkowitz P, Bektas M, Lee HE, Chua M, Diaz LA, et al. p38MAPK signaling and desmoglein-3 internalization are linked events in pemphigus acantholysis. J Biol Chem (2010) 285:8936–41. doi:10.1074/jbc.M109.087999
276. Berkowitz P, Hu P, Warren S, Liu Z, Diaz LA, Rubenstein DS. p38MAPK inhibition prevents disease in pemphigus vulgaris mice. Proc Natl Acad Sci U S A (2006) 103:12855–60. doi:10.1073/pnas.0602973103
277. Berkowitz P, Chua M, Liu Z, Diaz LA, Rubenstein DS. Autoantibodies in the autoimmune disease pemphigus foliaceus induce blistering via p38 mitogen-activated protein kinase-dependent signaling in the skin. Am J Pathol (2008) 173:1628–36. doi:10.2353/ajpath.2008.080391
278. Berkowitz P, Diaz LA, Hall RP, Rubenstein DS. Induction of p38MAPK and HSP27 phosphorylation in pemphigus patient skin [letter]. J Invest Dermatol (2008) 128(3):738–40. doi:10.1038/sj.jid.5701080
279. Vielmuth F, Waschke J, Spindler V. Loss of desmoglein binding is not sufficient for keratinocyte dissociation in pemphigus. J Invest Dermatol (2015) 135:3068–77. doi:10.1038/jid.2015.324
280. Tucker DK, Stahley SN, Kowalczyk AP. Plakophilin-1 protects keratinocytes from pemphigus vulgaris IgG by forming calcium-independent desmosomes. J Invest Dermatol (2014) 134:1033–43. doi:10.1038/jid.2013.401
281. Dehner C, Rötzer V, Waschke J, Spindler V. A desmoplakin point mutation with enhanced keratin association ameliorates pemphigus vulgaris autoantibody-mediated loss of cell cohesion. Am J Pathol (2014) 184:2528–36. doi:10.1016/j.ajpath.2014.05.016
282. Mao X, Cho MJT, Ellebrecht CT, Mukherjee EM, Payne AS. Stat3 regulates desmoglein 3 transcription in epithelial keratinocytes. JCI Insight (2017) 2(9):92253. doi:10.1172/jci.insight.92253
283. Harmon RM, Simpson CL, Johnson JL, Koetsier JL, Dubash AD, Najor NA, et al. Desmoglein-1/Erbin interaction suppresses ERK activation to support epidermal differentiation. J Clin Invest (2013) 123:1556–70. doi:10.1172/JCI65220
284. Hammers CM, Stanley JR. Desmoglein-1, differentiation, and disease. J Clin Invest (2013) 123:1419–22. doi:10.1172/JCI69071
285. Dusek RL, Attardi LD. Desmosomes: new perpetrators in tumour suppression. Nat Rev Cancer (2011) 11:317–23. doi:10.1038/nrc3051
286. Mariotte E, Veyradier A. Thrombotic thrombocytopenic purpura: from diagnosis to therapy. Curr Opin Crit Care (2015) 21:593–601. doi:10.1097/MCC.0000000000000255
287. Crawley JT, Scully MA. Thrombotic thrombocytopenic purpura: basic pathophysiology and therapeutic strategies. Hematology Am Soc Hematol Educ Program (2013) 2013:292–9. doi:10.1182/asheducation-2013.1.292
288. George JN. How I treat patients with thrombotic thrombocytopenic purpura: 2010. Blood (2010) 116:4060–9. doi:10.1182/blood-2010-07-271445
289. Sayani FA, Abrams CS. How I treat refractory thrombotic thrombocytopenic purpura. Blood (2015) 125:3860–7. doi:10.1182/blood-2014-11-551580
290. Tersteeg C, Verhenne S, Roose E, Schelpe AS, Deckmyn H, De Meyer SF, et al. ADAMTS13 and anti-ADAMTS13 autoantibodies in thrombotic thrombocytopenic purpura – current perspectives and new treatment strategies. Expert Rev Hematol (2016) 9:209–21. doi:10.1586/17474086.2016.1122515
291. Reese JA, Muthurajah DS, Kremer Hovinga JA, Vesely SK, Terrell DR, George JN. Children and adults with thrombotic thrombocytopenic purpura associated with severe, acquired Adamts13 deficiency: comparison of incidence, demographic and clinical features. Pediatr Blood Cancer (2013) 60:1676–82. doi:10.1002/pbc.24612
292. Scully M, Goodship T. How I treat thrombotic thrombocytopenic purpura and atypical haemolytic uraemic syndrome. Br J Haematol (2014) 164:759–66. doi:10.1111/bjh.12718
293. Hie M, Gay J, Galicier L, Provôt F, Presne C, Poullin P, et al. Preemptive rituximab infusions after remission efficiently prevent relapses in acquired thrombotic thrombocytopenic purpura. Blood (2014) 124:204–10. doi:10.1182/blood-2014-01-550244
294. Froissart A, Veyradier A, Hié M, Benhamou Y, Coppo P; French Reference Center for Thrombotic Microangiopathies. Rituximab in autoimmune thrombotic thrombocytopenic purpura: a success story. Eur J Intern Med (2015) 26:659–65. doi:10.1016/j.ejim.2015.07.021
295. Page EE, Kremer Hovinga JA, Terrell DR, Vesely SK, George JN. Rituximab reduces risk for relapse in patients with thrombotic thrombocytopenic purpura [letter]. Blood (2016) 127(24):3092–4. doi:10.1182/blood-2016-03-703827
296. Thomas MR, de Groot R, Scully MA, Crawley JT. Pathogenicity of anti-ADAMTS13 autoantibodies in acquired thrombotic thrombocytopenic purpura. EBioMedicine (2015) 2:942–52. doi:10.1016/j.ebiom.2015.06.007
297. Ferrari S, Palavra K, Gruber B, Kremer Hovinga JA, Knöbl P, Caron C, et al. Persistence of circulating ADAMTS13-specific immune complexes in patients with acquired thrombotic thrombocytopenic purpura. Haematologica (2014) 99:779–87. doi:10.3324/haematol.2013.094151
298. Ferrari S, Mudde GC, Rieger M, Veyradier A, Kremer Hovinga JA, Scheiflinger F. IgG subclass distribution of anti-ADAMTS13 antibodies in patients with acquired thrombotic thrombocytopenic purpura. J Thromb Haemost (2009) 7:1703–10. doi:10.1111/j.1538-7836.2009.03568.x
299. Réti M, Farkas P, Csuka D, Rázsó K, Schlammadinger Á, Udvardy ML, et al. Complement activation in thrombotic thrombocytopenic purpura. J Thromb Haemost (2012) 10:791–8. doi:10.1111/j.1538-7836.2012.04674.x
300. Cataland SR, Holers VM, Geyer S, Yang S, Wu HM. Biomarkers of terminal complement activation confirm the diagnosis of aHUS and differentiate aHUS from TTP. Blood (2014) 123:3733–8. doi:10.1182/blood-2013-12-547067
301. Westwood JP, Langley K, Heelas E, Machin SJ, Scully M. Complement and cytokine response in acute thrombotic thrombocytopenic purpura. Br J Haematol (2014) 164:858–66. doi:10.1111/bjh.12707
302. Pos W, Crawley JT, Fijnheer R, Voorberg J, Lane DA, Luken BM. An autoantibody epitope comprising residues R660, Y661, and Y665 in the ADAMTS13 spacer domain identifies a binding site for the A2 domain of VWF. Blood (2010) 115:1640–9. doi:10.1182/blood-2009-06-229203
303. Schaller M, Vogel M, Kentouche K, Lämmle B, Kremer Hovinga JA. The splenic autoimmune response to ADAMTS13 in thrombotic thrombocytopenic purpura contains recurrent antigen-binding CDR3 motifs. Blood (2014) 124:3469–79. doi:10.1182/blood-2014-04-561142
304. Casina VC, Hu W, Mao JH, Lu RN, Hanby HA, Pickens B, et al. High-resolution epitope mapping by HX MS reveals the pathogenic mechanism and a possible therapy for autoimmune TTP syndrome. Proc Natl Acad Sci U S A (2015) 112:9620–5. doi:10.1073/pnas.1512561112
305. Ostertag EM, Kacir S, Thiboutot M, Gulendran G, Zheng XL, Cines DB, et al. ADAMTS13 autoantibodies cloned from patients with acquired thrombotic thrombocytopenic purpura: 1. Structural and functional characterization in vitro. Transfusion (2016) 56:1763–74. doi:10.1111/trf.13584
306. Luken BM, Kaijen PH, Turenhout EA, Kremer Hovinga JA, van Mourik JA, Fijnheer R, et al. Multiple B-cell clones producing antibodies directed to the spacer and disintegrin/thrombospondin type-1 repeat 1 (TSP1) of ADAMTS13 in a patient with acquired thrombotic thrombocytopenic purpura. J Thromb Haemost (2006) 4:2355–64. doi:10.1111/j.1538-7836.2006.02164.x
307. Feys HB, Roodt J, Vandeputte N, Pareyn I, Lamprecht S, van Rensburg WJ, et al. Thrombotic thrombocytopenic purpura directly linked with ADAMTS13 inhibition in the baboon (Papio ursinus). Blood (2010) 116:2005–10. doi:10.1182/blood-2010-04-280479
308. Pickens B, Mao Y, Li D, Siegel DL, Poncz M, Cines DB, et al. Platelet-delivered ADAMTS13 inhibits arterial thrombosis and prevents thrombotic thrombocytopenic purpura in murine models. Blood (2015) 125:3326–34. doi:10.1182/blood-2014-07-587139
309. Coppo P, Busson M, Veyradier A, Wynckel A, Poullin P, Azoulay E, et al. HLA-DRB1*11: a strong risk factor for acquired severe ADAMTS13 deficiency-related idiopathic thrombotic thrombocytopenic purpura in Caucasians. J Thromb Haemost (2010) 8(4):856–9. doi:10.1111/j.1538-7836.2010.03772.x
310. Sorvillo N, van Haren SD, Kaijen PH, ten Brinke A, Fijnheer R, Meijer AB, et al. Preferential HLA-DRB1*11-dependent presentation of CUB2-derived peptides by ADAMTS13-pulsed dendritic cells. Blood (2013) 121:3502–10. doi:10.1182/blood-2012-09-456780
311. Verbij FC, Turksma AW, de Heij F, Kaijen P, Lardy N, Fijnheer R, et al. CD4+ T cells from patients with acquired thrombotic thrombocytopenic purpura recognize CUB2 domain-derived peptides. Blood (2016) 127:1606–9. doi:10.1182/blood-2015-10-668053
312. Schiviz A, Wuersch K, Piskernik C, Dietrich B, Hoellriegl W, Rottensteiner H, et al. A new mouse model mimicking thrombotic thrombocytopenic purpura: correction of symptoms by recombinant human ADAMTS13. Blood (2012) 119:6128–35. doi:10.1182/blood-2011-09-380535
313. Tersteeg C, Schiviz A, De Meyer SF, Plaimauer B, Scheiflinger F, Rottensteiner H, et al. Potential for recombinant ADAMTS13 as an effective therapy for acquired thrombotic thrombocytopenic purpura. Arterioscler Thromb Vasc Biol (2015) 35:2336–42. doi:10.1161/ATVBAHA.115.306014
314. Feys HB, Roodt J, Vandeputte N, Pareyn I, Mottl H, Hou S, et al. Inhibition of von Willebrand factor-platelet glycoprotein Ib interaction prevents and reverses symptoms of acute acquired thrombotic thrombocytopenic purpura in baboons. Blood (2012) 120:3611–4. doi:10.1182/blood-2012-04-421248
315. Callewaert F, Roodt J, Ulrichts H, Stohr T, van Rensburg WJ, Lamprecht S, et al. Evaluation of efficacy and safety of the anti-VWF Nanobody ALX-0681 in a preclinical baboon model of acquired thrombotic thrombocytopenic purpura. Blood (2012) 120:3603–10. doi:10.1182/blood-2012-04-420943
316. Chen J, Reheman A, Gushiken FC, Nolasco L, Fu X, Moake JL, et al. N-acetylcysteine reduces the size and activity of von Willebrand factor in human plasma and mice. J Clin Invest (2011) 121:593–603. doi:10.1172/JCI41062
317. Li GW, Rambally S, Kamboj J, Reilly S, Moake JL, Udden MM, et al. Treatment of refractory thrombotic thrombocytopenic purpura with N-acetylcysteine: a case report. Transfusion (2014) 54:1221–4. doi:10.1111/trf.12440
318. Rottenstreich A, Hochberg-Klein S, Rund D, Kalish Y. The role of N-acetylcysteine in the treatment of thrombotic thrombocytopenic purpura. J Thromb Thrombolysis (2016) 41:678–83. doi:10.1007/s11239-015-1259-6
319. Chapin J, Weksler B, Magro C, Laurence J. Eculizumab in the treatment of refractory idiopathic thrombotic thrombocytopenic purpura. Br J Haematol (2012) 157:772–4. doi:10.1111/j.1365-2141.2012.09084.x
320. Shortt J, Oh DH, Opat SS. ADAMTS13 antibody depletion by bortezomib in thrombotic thrombocytopenic purpura. N Engl J Med (2013) 368:90–2. doi:10.1056/NEJMc1213206
321. Rosenberg AS, Pariser AR, Diamond B, Yao L, Turka LA, Lacana E, et al. A role for plasma cell targeting agents in immune tolerance induction in autoimmune disease and antibody responses to therapeutic proteins. Clin Immunol (2016) 165:55–9. doi:10.1016/j.clim.2016.02.009
322. Coopamah MD, Garvey MB, Freedman J, Semple JW. Cellular immune mechanisms in autoimmune thrombocytopenic purpura: an update. Transfus Med Rev (2003) 17:69–80. doi:10.1053/tmrv.2003.50004
323. Ji X, Zhang L, Peng J, Hou M. T cell immune abnormalities in immune thrombocytopenia. J Hematol Oncol (2014) 7:72. doi:10.1186/s13045-014-0072-6
324. Cooper N, Bussel J. The pathogenesis of immune thrombocytopaenic purpura. Br J Haematol (2006) 133:364–74. doi:10.1111/j.1365-2141.2006.06024.x
325. Berentsen S, Sundic T. Red blood cell destruction in autoimmune hemolytic anemia: role of complement and potential new targets for therapy. Biomed Res Int (2015) 2015:363278. doi:10.1155/2015/363278
326. Bruin M, Bierings M, Uiterwaal C, Révész T, Bode L, Wiesman ME, et al. Platelet count, previous infection and FCGR2B genotype predict development of chronic disease in newly diagnosed idiopathic thrombocytopenia in childhood: results of a prospective study. Br J Haematol (2004) 127:561–7. doi:10.1111/j.1365-2141.2004.05235.x
327. Wu Z, Zhou J, Prsoon P, Wei X, Liu X, Peng B. Low expression of FCGRIIB in macrophages of immune thrombocytopenia-affected individuals. Int J Hematol (2012) 96:588–93. doi:10.1007/s12185-012-1187-6
328. Breunis WB, van Mirre E, Bruin M, Geissler J, de Boer M, Peters M, et al. Copy number variation of the activating FCGR2C gene predisposes to idiopathic thrombocytopenic purpura. Blood (2008) 111:1029–38. doi:10.1182/blood-2007-03-079913
329. Williams Y, Lynch S, McCann S, Smith O, Feighery C, Whelan A. Correlation of platelet Fc gammaRIIA polymorphism in refractory idiopathic (immune) thrombocytopenic purpura. Br J Haematol (1998) 101:779–82. doi:10.1046/j.1365-2141.1998.00802.x
330. Carlsson LE, Santoso S, Baurichter G, Kroll H, Papenberg S, Eichler P, et al. Heparin-induced thrombocytopenia: new insights into the impact of the FcgammaRIIa-R-H131 polymorphism. Blood (1998) 92:1526–31.
331. Fujimoto TT, Inoue M, Shimomura T, Fujimura K. Involvement of Fc gamma receptor polymorphism in the therapeutic response of idiopathic thrombocytopenic purpura. Br J Haematol (2001) 115:125–30. doi:10.1046/j.1365-2141.2001.03109.x
332. Foster CB, Zhu S, Erichsen HC, Lehrnbecher T, Hart ES, Choi E, et al. Polymorphisms in inflammatory cytokines and Fcgamma receptors in childhood chronic immune thrombocytopenic purpura: a pilot study. Br J Haematol (2001) 113:596–9. doi:10.1046/j.1365-2141.2001.02807.x
333. Gruel Y, Pouplard C, Lasne D, Magdelaine-Beuzelin C, Charroing C, Watier H. The homozygous FcgammaRIIIa-158V genotype is a risk factor for heparin-induced thrombocytopenia in patients with antibodies to heparin-platelet factor 4 complexes. Blood (2004) 104:2791–3. doi:10.1182/blood-2004-01-0058
334. Hazenbos WL, Heijnen IA, Meyer D, Hofhuis FM, Renardel de Lavalette CR, Schmidt RE, et al. Murine IgG1 complexes trigger immune effector functions predominantly via Fc gamma RIII (CD16). J Immunol (1998) 161:3026–32.
335. Clynes R, Ravetch JV. Cytotoxic antibodies trigger inflammation through Fc receptors. Immunity (1995) 3:21–6. doi:10.1016/1074-7613(95)90155-8
336. Fossati-Jimack L, Ioan-Facsinay A, Reininger L, Chicheportiche Y, Watanabe N, Saito T, et al. Markedly different pathogenicity of four immunoglobulin G isotype-switch variants of an antierythrocyte autoantibody is based on their capacity to interact in vivo with the low-affinity Fcgamma receptor III. J Exp Med (2000) 191:1293–302. doi:10.1084/jem.191.8.1293
337. Meyer D, Schiller C, Westermann J, Izui S, Hazenbos WL, Verbeek JS, et al. FcgammaRIII (CD16)-deficient mice show IgG isotype-dependent protection to experimental autoimmune hemolytic anemia. Blood (1998) 92:3997–4002.
338. Nakar CT, Bussel JB. 3G8 and GMA161, anti FcγRIII inhibitory monoclonal antibodies in the treatment of chronic refractory ITP. (Summary of 2 pilot studies). Blood (2009) 114:Abstract 2404.
339. Yu X, Menard M, Prechl J, Bhakta V, Sheffield WP, Lazarus AH. Monovalent Fc receptor blockade by an anti-Fcγ receptor/albumin fusion protein ameliorates murine ITP with abrogated toxicity. Blood (2016) 127:132–8. doi:10.1182/blood-2015-08-664656
340. Sondermann P. The FcγR/IgG interaction as target for the treatment of autoimmune diseases. J Clin Immunol (2016) 36(Suppl 1):95–9. doi:10.1007/s10875-016-0272-7
341. Konstaninova TS, Leonidovna IV, Hellmann A, Kyrcz-Krzemien S, Tillmanns S, Sonderman P. Interim results from a phase Ib/IIa clinical trial with the soluble Fc-Gamma IIb receptor SM101 for the treatment of primary immune thrombocytopenia. Blood (2012) 120:3388.
342. Jennette JC, Falk RJ. Pathogenesis of antineutrophil cytoplasmic autoantibody-mediated disease. Nat Rev Rheumatol (2014) 10:463–73. doi:10.1038/nrrheum.2014.103
343. Kallenberg CG. Key advances in the clinical approach to ANCA-associated vasculitis. Nat Rev Rheumatol (2014) 10:484–93. doi:10.1038/nrrheum.2014.104
344. Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, et al. 2012 revised international chapel hill consensus conference nomenclature of vasculitides. Arthritis Rheum (2013) 65:1–11. doi:10.1002/art.37715
345. Herlyn K, Buckert F, Gross WL, Reinhold-Keller E. Doubled prevalence rates of ANCA-associated vasculitides and giant cell arteritis between 1994 and 2006 in northern Germany. Rheumatology (Oxford) (2014) 53:882–9. doi:10.1093/rheumatology/ket440
346. Lamprecht P. [Systemic vasculitides: revised nomenclature, new therapeutic strategies]. Dtsch Med Wochenschr (2013) 138:651–4. doi:10.1055/s-0032-1332966
347. Flossmann O, Berden A, de Groot K, Hagen C, Harper L, Heijl C, et al. Long-term patient survival in ANCA-associated vasculitis. Ann Rheum Dis (2011) 70:488–94. doi:10.1136/ard.2010.137778
348. Csernok E, Lamprecht P, Gross WL. Diagnostic significance of ANCA in vasculitis. Nat Clin Pract Rheumatol (2006) 2:174–5. doi:10.1038/ncprheum0159
349. Finkielman JD, Merkel PA, Schroeder D, Hoffman GS, Spiera R, St Clair EW, et al. Antiproteinase 3 antineutrophil cytoplasmic antibodies and disease activity in Wegener granulomatosis. Ann Intern Med (2007) 147:611–9. doi:10.7326/0003-4819-147-9-200711060-00005
350. Sanders JS, Huitma MG, Kallenberg CG, Stegeman CA. Prediction of relapses in PR3-ANCA-associated vasculitis by assessing responses of ANCA titres to treatment. Rheumatology (Oxford) (2006) 45:724–9. doi:10.1093/rheumatology/kei272
351. Roth AJ, Ooi JD, Hess JJ, van Timmeren MM, Berg EA, Poulton CE, et al. Epitope specificity determines pathogenicity and detectability in ANCA-associated vasculitis. J Clin Invest (2013) 123:1773–83. doi:10.1172/JCI65292
352. Falk RJ, Terrell RS, Charles LA, Jennette JC. Anti-neutrophil cytoplasmic autoantibodies induce neutrophils to degranulate and produce oxygen radicals in vitro. Proc Natl Acad Sci U S A (1990) 87:4115–9. doi:10.1073/pnas.87.11.4115
353. Hao J, Meng LQ, Xu PC, Chen M, Zhao MH. p38MAPK, ERK and PI3K signaling pathways are involved in C5a-primed neutrophils for ANCA-mediated activation. PLoS One (2012) 7:e38317. doi:10.1371/journal.pone.0038317
354. Csernok E, Ernst M, Schmitt W, Bainton DF, Gross WL. Activated neutrophils express proteinase 3 on their plasma membrane in vitro and in vivo. Clin Exp Immunol (1994) 95:244–50. doi:10.1111/j.1365-2249.1994.tb06518.x
355. Porges AJ, Redecha PB, Kimberly WT, Csernok E, Gross WL, Kimberly RP. Anti-neutrophil cytoplasmic antibodies engage and activate human neutrophils via Fc gamma RIIa. J Immunol (1994) 153:1271–80.
356. Ben-Smith A, Dove SK, Martin A, Wakelam MJ, Savage CO. Antineutrophil cytoplasm autoantibodies from patients with systemic vasculitis activate neutrophils through distinct signaling cascades: comparison with conventional Fcgamma receptor ligation. Blood (2001) 98:1448–55. doi:10.1182/blood.V98.5.1448
357. Surmiak M, Kaczor M, Sanak M. Proinflammatory genes expression in granulocytes activated by native proteinase-binding fragments of anti-proteinase 3 IgG. J Physiol Pharmacol (2015) 66:609–15.
358. Radford DJ, Luu NT, Hewins P, Nash GB, Savage CO. Antineutrophil cytoplasmic antibodies stabilize adhesion and promote migration of flowing neutrophils on endothelial cells. Arthritis Rheum (2001) 44:2851–61. doi:10.1002/1529-0131(200112)44:12<2851::AID-ART473>3.0.CO;2-2
359. Little MA, Smyth CL, Yadav R, Ambrose L, Cook HT, Nourshargh S, et al. Antineutrophil cytoplasm antibodies directed against myeloperoxidase augment leukocyte-microvascular interactions in vivo. Blood (2005) 106:2050–8. doi:10.1182/blood-2005-03-0921
360. Xiao H, Ciavatta D, Aylor DL, Hu P, de Villena FP, Falk RJ, et al. Genetically determined severity of anti-myeloperoxidase glomerulonephritis. Am J Pathol (2013) 182:1219–26. doi:10.1016/j.ajpath.2012.12.006
361. Schreiber A, Xiao H, Jennette JC, Schneider W, Luft FC, Kettritz R. C5a receptor mediates neutrophil activation and ANCA-induced glomerulonephritis. J Am Soc Nephrol (2009) 20:289–98. doi:10.1681/ASN.2008050497
362. Little MA, Al-Ani B, Ren S, Al-Nuaimi H, Leite M Jr, Alpers CE, et al. Anti-proteinase 3 anti-neutrophil cytoplasm autoantibodies recapitulate systemic vasculitis in mice with a humanized immune system. PLoS One (2012) 7:e28626. doi:10.1371/journal.pone.0028626
363. Salama AD, Little MA. Animal models of antineutrophil cytoplasm antibody-associated vasculitis. Curr Opin Rheumatol (2012) 24:1–7. doi:10.1097/BOR.0b013e32834d2d52
364. Millet A, Martin KR, Bonnefoy F, Saas P, Mocek J, Alkan M, et al. Proteinase 3 on apoptotic cells disrupts immune silencing in autoimmune vasculitis. J Clin Invest (2015) 125:4107–21. doi:10.1172/JCI78182
365. Mueller A, Brieske C, Schinke S, Csernok E, Gross WL, Hasselbacher K, et al. Plasma cells within granulomatous inflammation display signs pointing to autoreactivity and destruction in granulomatosis with polyangiitis. Arthritis Res Ther (2014) 16:R55. doi:10.1186/ar4490
366. Kessenbrock K, Krumbholz M, Schönermarck U, Back W, Gross WL, Werb Z, et al. Netting neutrophils in autoimmune small-vessel vasculitis. Nat Med (2009) 15:623–5. doi:10.1038/nm.1959
367. Schmidt E, Zillikens D. Pemphigoid diseases. Lancet (2013) 381:320–32. doi:10.1016/S0140-6736(12)61140-4
368. Heimbach L, Li N, Diaz A, Liu Z. Experimental animal models of bullous pemphigoid. G Ital Dermatol Venereol (2009) 144:423–31.
369. Ujiie H, Nishie W, Shimizu H. Pathogenesis of bullous pemphigoid. Dermatol Clin (2011) 29:439–46, ix. doi:10.1016/j.det.2011.03.008
370. Ludwig RJ, Kalies K, Köhl J, Zillikens D, Schmidt E. Emerging treatments for pemphigoid diseases. Trends Mol Med (2013) 19:501–12. doi:10.1016/j.molmed.2013.06.003
371. Kasperkiewicz M, Sadik CD, Bieber K, Ibrahim SM, Manz RA, Schmidt E, et al. Epidermolysis bullosa acquisita: from pathophysiology to novel therapeutic options. J Invest Dermatol (2016) 136:24–33. doi:10.1038/JID.2015.356
372. Chen M, O’Toole EA, Sanghavi J, Mahmud N, Kelleher D, Weir D, et al. The epidermolysis bullosa acquisita antigen (type VII collagen) is present in human colon and patients with Crohn’s disease have autoantibodies to type VII collagen. J Invest Dermatol (2002) 118:1059–64. doi:10.1046/j.1523-1747.2002.01772.x
373. Ishii N, Recke A, Mihai S, Hirose M, Hashimoto T, Zillikens D, et al. Autoantibody-induced intestinal inflammation and weight loss in experimental epidermolysis bullosa acquisita. J Pathol (2011) 224:234–44. doi:10.1002/path.2857
374. Ludwig RJ. Clinical presentation, pathogenesis, diagnosis, and treatment of epidermolysis bullosa acquisita. ISRN Dermatology (2013) 2013:812029. doi:10.1155/2013/812029
375. Kim JH, Kim YH, Kim SC. Epidermolysis bullosa acquisita: a retrospective clinical analysis of 30 cases. Acta Derm Venereol (2011) 91:307–12. doi:10.2340/00015555-1065
376. Iranzo P, Herrero-González JE, Mascaró-Galy JM, Suárez-Fernández R, España A. Epidermolysis bullosa acquisita: a retrospective analysis of 12 patients evaluated in four tertiary hospitals in Spain. Br J Dermatol (2014) 171:1022–30. doi:10.1111/bjd.13144
377. Woodley DT, Burgeson RE, Lunstrum G, Bruckner-Tuderman L, Reese MJ, Briggaman RA. Epidermolysis bullosa acquisita antigen is the globular carboxyl terminus of type VII procollagen. J Clin Invest (1988) 81:683–7. doi:10.1172/JCI113373
378. Sitaru C, Kromminga A, Hashimoto T, Brocker EB, Zillikens D. Autoantibodies to type VII collagen mediate Fcgamma-dependent neutrophil activation and induce dermal-epidermal separation in cryosections of human skin. Am J Pathol (2002) 161:301–11. doi:10.1016/S0002-9440(10)64182-X
379. Recke A, Trog LM, Pas HH, Vorobyev A, Abadpour A, Jonkman MF, et al. Recombinant human IgA1 and IgA2 autoantibodies to type VII collagen induce subepidermal blistering ex vivo. J Immunol (2014) 193:1600–8. doi:10.4049/jimmunol.1400160
380. Sitaru C, Mihai S, Otto C, Chiriac MT, Hausser I, Dotterweich B, et al. Induction of dermal-epidermal separation in mice by passive transfer of antibodies specific to type VII collagen. J Clin Invest (2005) 115:870–8. doi:10.1172/JCI200521386
381. Woodley DT, Chang C, Saadat P, Ram R, Liu Z, Chen M. Evidence that anti-type VII collagen antibodies are pathogenic and responsible for the clinical, histological, and immunological features of epidermolysis bullosa acquisita. J Invest Dermatol (2005) 124:958–64. doi:10.1111/j.0022-202X.2005.23702.x
382. Sitaru C, Chiriac MT, Mihai S, Büning J, Gebert A, Ishiko A, et al. Induction of complement-fixing autoantibodies against type VII collagen results in subepidermal blistering in mice. J Immunol (2006) 177:3461–8. doi:10.4049/jimmunol.177.5.3461
383. Iwata H, Bieber K, Tiburzy B, Chrobok N, Kalies K, Shimizu A, et al. B cells, dendritic cells, and macrophages are required to induce an autoreactive CD4 helper T cell response in experimental epidermolysis bullosa acquisita. J Immunol (2013) 191:2978–88. doi:10.4049/jimmunol.1300310
384. Marzano AV, Cozzani E, Fanoni D, De Pità O, Vassallo C, Berti E, et al. Diagnosis and disease severity assessment of epidermolysis bullosa acquisita by ELISA for anti-type VII collagen autoantibodies: an Italian multicentre study. Br J Dermatol (2013) 168:80–4. doi:10.1111/bjd.12011
385. Kim JH, Kim YH, Kim S, Noh EB, Kim SE, Vorobyev A, et al. Serum levels of anti-type VII collagen antibodies detected by enzyme-linked immunosorbent assay in patients with epidermolysis bullosa acquisita are correlated with the severity of skin lesions. J Eur Acad Dermatol Venereol (2013) 27:e224–30. doi:10.1111/j.1468-3083.2012.04617.x
386. Hirose M, Brandolini L, Zimmer D, Götz J, Westermann J, Allegretti M, et al. The allosteric CXCR1/2 inhibitor DF2156A improves experimental epidermolysis bullosa acquisita. J Genet Syndr Gene Ther (2013). doi:10.4172/2157-7412.S3-005
387. Samavedam UK, Iwata H, Müller S, Schulze FS, Recke A, Schmidt E, et al. GM-CSF modulates autoantibody production and skin blistering in experimental epidermolysis bullosa acquisita. J Immunol (2014) 192:559–71. doi:10.4049/jimmunol.1301556
388. Hirose M, Kasprick K, Beltisou F, Dieckhoff Schulze K, Schulze FS, Samavedam UK, et al. Reduced skin blistering in experimental epidermolysis bullosa acquisita after anti-TNF treatment. Mol Med (2016). doi:10.2119/molmed.2015.00206
389. Sezin T, Krajewski M, Wutkowski A, Mousavi S, Chakievska L, Bieber K, et al. The leukotriene B4 and its receptor BLT1 act as critical drivers of neutrophil recruitment in murine bullous pemphigoid-like epidermolysis bullosa acquisita. J Invest Dermatol (2017) 137:1104–13. doi:10.1016/j.jid.2016.12.021
390. Samavedam UK, Kalies K, Scheller J, Sadeghi H, Gupta Y, Jonkman MF, et al. Recombinant IL-6 treatment protects mice from organ specific autoimmune disease by IL-6 classical signalling-dependent IL-1ra induction. J Autoimmun (2013) 40:74–85. doi:10.1016/j.jaut.2012.08.002
391. Sadeghi H, Gupta Y, Möller M, Samavedam UK, Behnen M, Kasprick A, et al. The retinoid-related orphan receptor alpha is essential for the end-stage effector phase of experimental epidermolysis bullosa acquisita. J Pathol (2015) 237:111–22. doi:10.1002/path.4556
392. Ellebrecht CT, Srinivas G, Bieber K, Banczyk D, Kalies K, Künzel S, et al. Skin microbiota-associated inflammation precedes autoantibody induced tissue damage in experimental epidermolysis bullosa acquisita. J Autoimmun (2016) 68:14–22. doi:10.1016/j.jaut.2015.08.007
393. Sadeghi H, Lockmann A, Hund AC, Samavedam UK, Pipi E, Vafia K, et al. Caspase-1-independent IL-1 release mediates blister formation in autoantibody-induced tissue injury through modulation of endothelial adhesion molecules. J Immunol (2015) 194:3656–63. doi:10.4049/jimmunol.1402688
394. Chiriac MT, Roesler J, Sindrilaru A, Scharffetter-Kochanek K, Zillikens D, Sitaru C. NADPH oxidase is required for neutrophil-dependent autoantibody-induced tissue damage. J Pathol (2007) 212:56–65. doi:10.1002/path.2157
395. Yu X, Holdorf K, Kasper B, Zillikens D, Ludwig RJ, Petersen F. FcgammaRIIA and FcgammaRIIIB are required for autoantibody-induced tissue damage in experimental human models of bullous pemphigoid. J Invest Dermatol (2010) 130:2841–4. doi:10.1038/jid.2010.230
396. Kasperkiewicz M, Nimmerjahn F, Wende S, Hirose M, Iwata H, Jonkman MF, et al. Genetic identification and functional validation of FcγRIV as key molecule in autoantibody-induced tissue injury. J Pathol (2012) 228:8–19. doi:10.1002/path.4023
397. Hirose M, Vafia K, Kalies K, Groth S, Westermann J, Zillikens D, et al. Enzymatic autoantibody glycan hydrolysis alleviates autoimmunity against type VII collagen. J Autoimmun (2012) 39:304–14. doi:10.1016/j.jaut.2012.04.002
398. Schulze FS, Beckmann T, Nimmerjahn F, Ishiko A, Collin M, Köhl J, et al. Fc gamma receptors III and IV mediate tissue destruction in a novel adult mouse model of bullous pemphigoid. Am J Pathol (2014) 184:2185–96. doi:10.1016/j.ajpath.2014.05.007
399. Iwata H, Pipi E, Mockel N, Sondermann P, Vorobyev A, van Beek N, et al. Recombinant soluble CD32 suppresses disease progression in experimental epidermolysis bullosa acquisita. J Invest Dermatol (2015) 135:916–9. doi:10.1038/jid.2014.451
400. Schwab I, Mihai S, Seeling M, Kasperkiewicz M, Ludwig RJ, Nimmerjahn F. Broad requirement for terminal sialic acid residues and FcgammaRIIB for the preventive and therapeutic activity of intravenous immunoglobulins in vivo. Eur J Immunol (2014) 44:1444–53. doi:10.1002/eji.201344230
401. Hirose M, Tiburzy B, Ishii N, Pipi E, Wende S, Rentz E, et al. Effects of intravenous immunoglobulins on mice with experimental epidermolysis bullosa acquisita. J Invest Dermatol (2015) 135:768–75. doi:10.1038/jid.2014.453
402. Kovács M, Németh T, Jakus Z, Sitaru C, Simon E, Futosi K, et al. The Src family kinases Hck, Fgr, and Lyn are critical for the generation of the in vivo inflammatory environment without a direct role in leukocyte recruitment. J Exp Med (2014) 211:1993–2011. doi:10.1084/jem.20132496
403. Kulkarni S, Sitaru C, Jakus Z, Andersson KE, Damoulakis G, Davidson K, et al. Essential role for PI3Kβ in neutrophil activation by immune complexes. Sci Signal (2011) 4:ra23. doi:10.1126/scisignal.2001617
404. Shimanovich I, Mihai S, Oostingh GJ, Ilenchuk TT, Bröcker EB, Opdenakker G, et al. Granulocyte-derived elastase and gelatinase B are required for dermal-epidermal separation induced by autoantibodies from patients with epidermolysis bullosa acquisita and bullous pemphigoid. J Pathol (2004) 204:519–27. doi:10.1002/path.1674
405. Liu Z, Shapiro SD, Zhou X, Twining SS, Senior RM, Giudice GJ, et al. A critical role for neutrophil elastase in experimental bullous pemphigoid. J Clin Invest (2000) 105:113–23. doi:10.1172/JCI3693
406. Bieber K, Witte M, Sun S, Hundt JE, Kalies K, Dräger S, et al. T cells mediate autoantibody-induced cutaneous inflammation and blistering in epidermolysis bullosa acquisita. Sci Rep (2016) 6:38357. doi:10.1038/srep38357
407. Kopecki Z, Arkell R, Powell BC, Cowin AJ. Flightless I regulates hemidesmosome formation and integrin-mediated cellular adhesion and migration during wound repair. J Invest Dermatol (2009) 129:2031–45. doi:10.1038/jid.2008.461
408. Kopecki Z, Arkell RM, Strudwick XL, Hirose M, Ludwig RJ, Kern JS, et al. Overexpression of the Flii gene increases dermal-epidermal blistering in an autoimmune ColVII mouse model of epidermolysis bullosa acquisita. J Pathol (2011) 225:401–13. doi:10.1002/path.2973
409. Kopecki Z, Ruzehaji N, Turner C, Iwata H, Ludwig RJ, Zillikens D, et al. Topically applied flightless I neutralizing antibodies improve healing of blistered skin in a murine model of epidermolysis bullosa acquisita. J Invest Dermatol (2013) 133:1008–16. doi:10.1038/jid.2012.457
410. Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, et al. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association scientific statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation (2006) 113:1807–16. doi:10.1161/CIRCULATIONAHA.106.174287
411. Hufnagel G, Pankuweit S, Richter A, Schönian U, Maisch B. The European study of epidemiology and treatment of cardiac inflammatory diseases (ESETCID). First epidemiological results. Herz (2000) 25:279–85. doi:10.1007/s000590050021
412. Magnani JW, Dec GW. Myocarditis: current trends in diagnosis and treatment. Circulation (2006) 113:876–90. doi:10.1161/CIRCULATIONAHA.105.584532
413. Caforio AL, Pankuweit S, Arbustini E, Basso C, Gimeno-Blanes J, Felix SB, et al. Current state of knowledge on aetiology, diagnosis, management, and therapy of myocarditis: a position statement of the European Society of Cardiology Working Group on myocardial and pericardial diseases. Eur Heart J (2648) 2013(34):2636–48. doi:10.1093/eurheartj/eht210
414. Jahns R, Boivin V, Schwarzbach V, Ertl G, Lohse MJ. Pathological autoantibodies in cardiomyopathy. Autoimmunity (2008) 41:454–61. doi:10.1080/08916930802031603
415. Neumann DA, Burek CL, Baughman KL, Rose NR, Herskowitz A. Circulating heart-reactive antibodies in patients with myocarditis or cardiomyopathy. J Am Coll Cardiol (1990) 16:839–46. doi:10.1016/S0735-1097(10)80331-6
416. Neu N, Beisel KW, Traystman MD, Rose NR, Craig SW. Autoantibodies specific for the cardiac myosin isoform are found in mice susceptible to Coxsackievirus B3-induced myocarditis. J Immunol (1987) 138:2488–92.
417. Rose NR. Myocarditis: infection versus autoimmunity. J Clin Immunol (2009) 29:730–7. doi:10.1007/s10875-009-9339-z
418. Caforio AL, Mahon NG, Baig MK, Tona F, Murphy RT, Elliott PM, et al. Prospective familial assessment in dilated cardiomyopathy: cardiac autoantibodies predict disease development in asymptomatic relatives. Circulation (2007) 115:76–83. doi:10.1161/CIRCULATIONAHA.106.641472
419. Neu N, Rose NR, Beisel KW, Herskowitz A, Gurri-Glass G, Craig SW. Cardiac myosin induces myocarditis in genetically predisposed mice. J Immunol (1987) 139:3630–6.
420. Fairweather D, Frisancho-Kiss S, Rose NR. Viruses as adjuvants for autoimmunity: evidence from coxsackievirus-induced myocarditis. Rev Med Virol (2005) 15:17–27. doi:10.1002/rmv.445
421. Schulze K, Becker BF, Schauer R, Schultheiss HP. Antibodies to ADP-ATP carrier – an autoantigen in myocarditis and dilated cardiomyopathy – impair cardiac function. Circulation (1990) 81:959–69. doi:10.1161/01.CIR.81.3.959
422. Deubner N, Berliner D, Schlipp A, Gelbrich G, Caforio AL, Felix SB, et al. Cardiac beta1-adrenoceptor autoantibodies in human heart disease: rationale and design of the Etiology, Titre-Course, and Survival (ETiCS) Study. Eur J Heart Fail (2010) 12:753–62. doi:10.1093/eurjhf/hfq072
423. Schultheiss HP, Kühl U, Cooper LT. The management of myocarditis. Eur Heart J (2011) 32:2616–25. doi:10.1093/eurheartj/ehr165
424. Wojnicz R, Nowalany-Kozielska E, Wojciechowska C, Glanowska G, Wilczewski P, Niklewski T, et al. Randomized, placebo-controlled study for immunosuppressive treatment of inflammatory dilated cardiomyopathy: two-year follow-up results. Circulation (2001) 104:39–45. doi:10.1161/01.CIR.104.1.39
425. Frustaci A, Chimenti C, Calabrese F, Pieroni M, Thiene G, Maseri A. Immunosuppressive therapy for active lymphocytic myocarditis: virological and immunologic profile of responders versus nonresponders. Circulation (2003) 107:857–63. doi:10.1161/01.CIR.0000048147.15962.31
426. Stanton C, Mookadam F, Cha S, McNamara D, Aukrust P, Wojnicz R, et al. Greater symptom duration predicts response to immunomodulatory therapy in dilated cardiomyopathy. Int J Cardiol (2008) 128:38–41. doi:10.1016/j.ijcard.2007.05.016
427. Drucker NA, Colan SD, Lewis AB, Beiser AS, Wessel DL, Takahashi M, et al. Gamma-globulin treatment of acute myocarditis in the pediatric population. Circulation (1994) 89:252–7. doi:10.1161/01.CIR.89.1.252
428. Kaya Z, Leib C, Katus HA. Autoantibodies in heart failure and cardiac dysfunction. Circ Res (2012) 110:145–58. doi:10.1161/CIRCRESAHA.111.243360
429. Wallukat G, Reinke P, Dörffel WV, Luther HP, Bestvater K, Felix SB, et al. Removal of autoantibodies in dilated cardiomyopathy by immunoadsorption. Int J Cardiol (1996) 54:191–5. doi:10.1016/0167-5273(96)02598-3
430. Muller AM, Fischer A, Katus HA, Kaya Z. Mouse models of autoimmune diseases – autoimmune myocarditis. Curr Pharm Des (2015) 21:2498–512. doi:10.2174/1381612821666150316123711
431. McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med (2011) 365:2205–19. doi:10.1056/NEJMra1004965
432. Nimmerjahn F. Activating and inhibitory FcgammaRs in autoimmune disorders. Springer Semin Immunopathol (2006) 28:305–19. doi:10.1007/s00281-006-0052-1
433. Monach PA, Benoist C, Mathis D. The role of antibodies in mouse models of rheumatoid arthritis, and relevance to human disease. Adv Immunol (2004) 82:217–48. doi:10.1016/S0065-2776(04)82005-4
434. Ji H, Ohmura K, Mahmood U, Lee DM, Hofhuis FM, Boackle SA, et al. Arthritis critically dependent on innate immune system players. Immunity (2002) 16:157–68. doi:10.1016/S1074-7613(02)00275-3
435. Lee DM, Friend DS, Gurish MF, Benoist C, Mathis D, Brenner MB. Mast cells: a cellular link between autoantibodies and inflammatory arthritis. Science (2002) 297:1689–92. doi:10.1126/science.1073176
436. Binstadt BA, Patel PR, Alencar H, Nigrovic PA, Lee DM, Mahmood U, et al. Particularities of the vasculature can promote the organ specificity of autoimmune attack. Nat Immunol (2006) 7:284–92. doi:10.1038/ni1306
437. Ji H, Pettit A, Ohmura K, Ortiz-Lopez A, Duchatelle V, Degott C, et al. Critical roles for interleukin 1 and tumor necrosis factor alpha in antibody-induced arthritis. J Exp Med (2002) 196:77–85. doi:10.1084/jem.20020439
438. Nigrovic PA, Binstadt BA, Monach PA, Johnsen A, Gurish M, Iwakura Y, et al. Mast cells contribute to initiation of autoantibody-mediated arthritis via IL-1. Proc Natl Acad Sci U S A (2007) 104:2325–30. doi:10.1073/pnas.0610852103
439. Feyerabend TB, Weiser A, Tietz A, Stassen M, Harris N, Kopf M, et al. Cre-mediated cell ablation contests mast cell contribution in models of antibody- and T cell-mediated autoimmunity. Immunity (2011) 35:832–44. doi:10.1016/j.immuni.2011.09.015
440. Harre U, Georgess D, Bang H, Bozec A, Axmann R, Ossipova E, et al. Induction of osteoclastogenesis and bone loss by human autoantibodies against citrullinated vimentin. J Clin Invest (2012) 122:1791–802. doi:10.1172/JCI60975
441. Seeling M, Hillenhoff U, David JP, Schett G, Tuckermann J, Lux A, et al. Inflammatory monocytes and Fcγ receptor IV on osteoclasts are critical for bone destruction during inflammatory arthritis in mice. Proc Natl Acad Sci U S A (2013) 110:10729–34. doi:10.1073/pnas.1301001110
442. Harre U, Lang SC, Pfeifle R, Rombouts Y, Frühbeißer S, Amara K, et al. Glycosylation of immunoglobulin G determines osteoclast differentiation and bone loss. Nat Commun (2015) 6:6651. doi:10.1038/ncomms7651
443. Negishi-Koga T, Gober HJ, Sumiya E, Komatsu N, Okamoto K, Sawa S, et al. Immune complexes regulate bone metabolism through FcRγ signalling. Nat Commun (2015) 6:6637. doi:10.1038/ncomms7637
444. Elliott ER, Van Ziffle JA, Scapini P, Sullivan BM, Locksley RM, Lowell CA. Deletion of Syk in neutrophils prevents immune complex arthritis. J Immunol (2011) 187:4319–30. doi:10.4049/jimmunol.1100341
445. Jakus Z, Simon E, Balázs B, Mócsai A. Genetic deficiency of Syk protects mice from autoantibody-induced arthritis. Arthritis Rheum (2010) 62:1899–910. doi:10.1002/art.27438
446. Parekh RB, Dwek RA, Sutton BJ, Fernandes DL, Leung A, Stanworth D, et al. Association of rheumatoid arthritis and primary osteoarthritis with changes in the glycosylation pattern of total serum IgG. Nature (1985) 316:452–7. doi:10.1038/316452a0
447. Kaneko Y, Nimmerjahn F, Ravetch JV. Anti-inflammatory activity of immunoglobulin G resulting from Fc sialylation. Science (2006) 313:670–3. doi:10.1126/science.1129594
448. Schwab I, Nimmerjahn F. Intravenous immunoglobulin therapy: how does IgG modulate the immune system. Nat Rev Immunol (2013) 13:176–89. doi:10.1038/nri3401
449. Anthony RM, Kobayashi T, Wermeling F, Ravetch JV. Intravenous gammaglobulin suppresses inflammation through a novel T(H)2 pathway. Nature (2011) 475:110–3. doi:10.1038/nature10134
450. Vaccaro C, Zhou J, Ober RJ, Ward ES. Engineering the Fc region of immunoglobulin G to modulate in vivo antibody levels. Nat Biotechnol (2005) 23:1283–8. doi:10.1038/nbt1143
451. Washburn N, Schwab I, Ortiz D, Bhatnagar N, Lansing JC, Medeiros A, et al. Controlled tetra-Fc sialylation of IVIg results in a drug candidate with consistent enhanced anti-inflammatory activity. Proc Natl Acad Sci U S A (2015) 112:E1297–306. doi:10.1073/pnas.1422481112
452. Papadopoulos MC, Verkman AS. Aquaporin 4 and neuromyelitis optica. Lancet Neurol (2012) 11:535–44. doi:10.1016/S1474-4422(12)70133-3
453. Zekeridou A, Lennon VA. Aquaporin-4 autoimmunity. Neurol Neuroimmunol Neuroinflamm (2015) 2:e110. doi:10.1212/NXI.0000000000000110
454. Flanagan EP, Cabre P, Weinshenker BG, St Sauver J, Jacobson DJ, Majed M, et al. Epidemiology of aquaporin-4 autoimmunity and neuromyelitis optica spectrum. Ann Neurol (2016) 79:775–83. doi:10.1002/ana.24617
455. Wingerchuk DM, Banwell B, Bennett JL, Cabre P, Carroll W, Chitnis T, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology (2015) 85:177–89. doi:10.1212/WNL.0000000000001729
456. Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med (2005) 202:473–7. doi:10.1084/jem.20050304
457. Papadopoulos MC, Verkman AS. Aquaporin water channels in the nervous system. Nat Rev Neurosci (2013) 14:265–77. doi:10.1038/nrn3468
458. Manley GT, Fujimura M, Ma T, Noshita N, Filiz F, Bollen AW, et al. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat Med (2000) 6:159–63. doi:10.1038/72256
459. Zeka B, Hastermann M, Hochmeister S, Kögl N, Kaufmann N, Schanda K, et al. Highly encephalitogenic aquaporin 4-specific T cells and NMO-IgG jointly orchestrate lesion location and tissue damage in the CNS. Acta Neuropathol (2015) 130:783–98. doi:10.1007/s00401-015-1501-5
460. Sagan SA, Winger RC, Cruz-Herranz A, Nelson PA, Hagberg S, Miller CN, et al. Tolerance checkpoint bypass permits emergence of pathogenic T cells to neuromyelitis optica autoantigen aquaporin-4. Proc Natl Acad Sci U S A (2016) 113:14781–6. doi:10.1073/pnas.1617859114
461. Papadopoulos MC, Bennett JL, Verkman AS. Treatment of neuromyelitis optica: state-of-the-art and emerging therapies. Nat Rev Neurol (2014) 10:493–506. doi:10.1038/nrneurol.2014.141
462. Kessler RA, Mealy MA, Levy M. Treatment of neuromyelitis optica spectrum disorder: acute, preventive, and symptomatic. Curr Treat Options Neurol (2016) 18:2. doi:10.1007/s11940-015-0387-9
463. Tradtrantip L, Zhang H, Saadoun S, Phuan PW, Lam C, Papadopoulos MC, et al. Anti-aquaporin-4 monoclonal antibody blocker therapy for neuromyelitis optica. Ann Neurol (2012) 71:314–22. doi:10.1002/ana.22657
464. Tradtrantip L, Ratelade J, Zhang H, Verkman AS. Enzymatic deglycosylation converts pathogenic neuromyelitis optica anti-aquaporin-4 immunoglobulin G into therapeutic antibody. Ann Neurol (2013) 73:77–85. doi:10.1002/ana.23741
465. Yao X, Su T, Verkman AS. Clobetasol promotes remyelination in a mouse model of neuromyelitis optica. Acta Neuropathol Commun (2016) 4:42. doi:10.1186/s40478-016-0309-4
466. Voigt J, Krause C, Rohwäder E, Saschenbrecker S, Hahn M, Danckwardt M, et al. Automated indirect immunofluorescence evaluation of antinuclear autoantibodies on HEp-2 cells. Clin Dev Immunol (2012) 2012:651058. doi:10.1155/2012/651058
467. Krause C, Ens K, Fechner K, Voigt J, Fraune J, Rohwäder E, et al. EUROPattern suite technology for computer-aided immunofluorescence microscopy in autoantibody diagnostics. Lupus (2015) 24:516–29. doi:10.1177/0961203314559635
468. Op De Beéck K, Vermeersch P, Verschueren P, Westhovens R, Mariën G, Blockmans D, et al. Antinuclear antibody detection by automated multiplex immunoassay in untreated patients at the time of diagnosis. Autoimmun Rev (2012) 12:137–43. doi:10.1016/j.autrev.2012.02.013
469. Radzimski C, Probst C, Teegen B, Rentzsch K, Blöcker IM, Dähnrich C, et al. Development of a recombinant cell-based indirect immunofluorescence assay for the determination of autoantibodies against soluble liver antigen in autoimmune hepatitis. Clin Dev Immunol (2013) 2013:572815. doi:10.1155/2013/572815
470. Dalmau J, Tüzün E, Wu HY, Masjuan J, Rossi JE, Voloschin A, et al. Paraneoplastic anti-N-methyl-d-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol (2007) 61:25–36. doi:10.1002/ana.21050
471. van Beek N, Rentzsch K, Probst C, Komorowski L, Kasperkiewicz M, Fechner K, et al. Serological diagnosis of autoimmune bullous skin diseases: prospective comparison of the BIOCHIP mosaic-based indirect immunofluorescence technique with the conventional multi-step single test strategy. Orphanet J Rare Dis (2012) 7:49. doi:10.1186/1750-1172-7-49
472. Waters PJ, McKeon A, Leite MI, Rajasekharan S, Lennon VA, Villalobos A, et al. Serologic diagnosis of NMO: a multicenter comparison of aquaporin-4-IgG assays. Neurology (2012) 78:665–71; discussion 669. doi:10.1212/WNL.0b013e318248dec1
473. Dähnrich C, Komorowski L, Probst C, Seitz-Polski B, Esnault V, Wetzels JF, et al. Development of a standardized ELISA for the determination of autoantibodies against human M-type phospholipase A2 receptor in primary membranous nephropathy. Clin Chim Acta (2013) 421:213–8. doi:10.1016/j.cca.2013.03.015
474. Probst C, Komorowski L, de Graaff E, van Coevorden-Hameete M, Rogemond V, Honnorat J, et al. Standardized test for anti-Tr/DNER in patients with paraneoplastic cerebellar degeneration. Neurol Neuroimmunol Neuroinflamm (2015) 2:e68. doi:10.1212/NXI.0000000000000068
475. Komorowski L, Teegen B, Probst C, Aulinger-Stöcker K, Sina C, Fellermann K, et al. Autoantibodies against exocrine pancreas in Crohn’s disease are directed against two antigens: the glycoproteins CUZD1 and GP2. J Crohns Colitis (2013) 7:780–90. doi:10.1016/j.crohns.2012.10.011
476. Beeson D, Jacobson L, Newsom-Davis J, Vincent A. A transfected human muscle cell line expressing the adult subtype of the human muscle acetylcholine receptor for diagnostic assays in myasthenia gravis. Neurology (1996) 47:1552–5. doi:10.1212/WNL.47.6.1552
477. Betteridge ZE, Gunawardena H, McHugh NJ. Novel autoantibodies and clinical phenotypes in adult and juvenile myositis. Arthritis Res Ther (2011) 13:209. doi:10.1186/ar3275
478. Waters P, Jarius S, Littleton E, Leite MI, Jacob S, Gray B, et al. Aquaporin-4 antibodies in neuromyelitis optica and longitudinally extensive transverse myelitis. Arch Neurol (2008) 65:913–9. doi:10.1001/archneur.65.7.913
479. Hanke K, Brückner CS, Dähnrich C, Huscher D, Komorowski L, Meyer W, et al. Antibodies against PM/Scl-75 and PM/Scl-100 are independent markers for different subsets of systemic sclerosis patients. Arthritis Res Ther (2009) 11:R22. doi:10.1186/ar2614
480. Stinton LM, Barr SG, Tibbles LA, Yilmaz S, Sar A, Benedikttson H, et al. Autoantibodies in lupus nephritis patients requiring renal transplantation. Lupus (2007) 16:394–400. doi:10.1177/0961203307078391
481. Behnert A, Schiffer M, Müller-Deile J, Beck LH, Mahler M, Fritzler MJ. Antiphospholipase A2 receptor autoantibodies: a comparison of three different immunoassays for the diagnosis of idiopathic membranous nephropathy. J Immunol Res (2014) 2014:143274. doi:10.1155/2014/143274
482. Schmidt E, Zillikens D. Modern diagnosis of autoimmune blistering skin diseases. Autoimmun Rev (2010) 10:84–9. doi:10.1016/j.autrev.2010.08.007
483. Damoiseaux J, Steller U, Buschtez M, Vaessen M, Rosemann A, van Paassen P, et al. EUROPLUS ANCA BIOCHIP mosaic: PR3 and MPO antigen microdots improve the laboratory diagnostics of ANCA-associated vasculitis. J Immunol Methods (2009) 348:67–73. doi:10.1016/j.jim.2009.07.001
484. Granieri L, Marnetto F, Valentino P, Frau J, Patanella AK, Nytrova P, et al. Evaluation of a multiparametric immunofluorescence assay for standardization of neuromyelitis optica serology. PLoS One (2012) 7:e38896. doi:10.1371/journal.pone.0038896
485. Pavlidis P, Komorowski L, Teegen B, Liaskos C, Koutsoumpas AL, Smyk DS, et al. Diagnostic and clinical significance of Crohn’s disease-specific pancreatic anti-GP2 and anti-CUZD1 antibodies. Clin Chem Lab Med (2016) 54:249–56. doi:10.1515/cclm-2015-0376
486. Ghirardello A, Bendo R, Rampudda ME, Bassi N, Zampieri S, Doria A. Commercial blot assays in the diagnosis of systemic rheumatic diseases. Autoimmun Rev (2009) 8:645–9. doi:10.1016/j.autrev.2009.02.012
487. Villalta D, Sorrentino MC, Girolami E, Tampoia M, Alessio MG, Brusca I, et al. Autoantibody profiling of patients with primary biliary cirrhosis using a multiplexed line-blot assay. Clin Chim Acta (2015) 438:135–8. doi:10.1016/j.cca.2014.08.024
488. Villalta D, Imbastaro T, Di Giovanni S, Lauriti C, Gabini M, Turi MC, et al. Diagnostic accuracy and predictive value of extended autoantibody profile in systemic sclerosis. Autoimmun Rev (2012) 12:114–20. doi:10.1016/j.autrev.2012.07.005
Keywords: autoimmunity, autoantibodies, treatment, pathogenesis, mouse models, B cells, diagnosis
Citation: Ludwig RJ, Vanhoorelbeke K, Leypoldt F, Kaya Z, Bieber K, McLachlan SM, Komorowski L, Luo J, Cabral-Marques O, Hammers CM, Lindstrom JM, Lamprecht P, Fischer A, Riemekasten G, Tersteeg C, Sondermann P, Rapoport B, Wandinger K-P, Probst C, El Beidaq A, Schmidt E, Verkman A, Manz RA and Nimmerjahn F (2017) Mechanisms of Autoantibody-Induced Pathology. Front. Immunol. 8:603. doi: 10.3389/fimmu.2017.00603
Received: 13 February 2017; Accepted: 08 May 2017;
Published: 31 May 2017
Edited by:
Massimo Gadina, National Institute of Arthritis and Musculoskeletal and Skin Diseases, United StatesReviewed by:
Simone Anna Mader, Feinstein Institute for Medical Research, United StatesAmir Sharabi, Harvard Medical School, United States
Copyright: © 2017 Ludwig, Vanhoorelbeke, Leypoldt, Kaya, Bieber, McLachlan, Komorowski, Luo, Cabral-Marques, Hammers, Lindstrom, Lamprecht, Fischer, Riemekasten, Tersteeg, Sondermann, Rapoport, Wandinger, Probst, El Beidaq, Schmidt, Verkman, Manz and Nimmerjahn. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ralf J. Ludwig, cmFsZi5sdWR3aWdAdWtzaC5kZQ==