 J. Luis Espinoza
J. Luis Espinoza Mika Minami
Mika Minami- 1Department of Hematology and Rheumatology, Faculty of Medicine, Kindai University, Osakasayama, Japan
- 2Faculty of Medicine, Kindai University, Higashi-osaka, Japan
The human genome is constantly exposed to exogenous and endogenous DNA damaging factors that frequently cause DNA damages. Unless repaired, damaged DNA can result in deleterious mutations capable of causing malignant transformation. Accordingly, cells have developed an advanced and effective surveillance system, the DNA damage response (DDR) pathway, which maintains genetic integrity. In addition to well-defined outcomes, such as cell cycle arrest, apoptosis, and senescence, another consequence of DDR activation is the induction of natural killer group 2 member D ligands (NKG2D-Ls) on the surface of stressed cells. Consequently, NKG2D-Ls-expressing cells are recognized and eliminated by NKG2D receptor-expressing immune cells, including NK cells, and various subsets of T-cells. Recent pieces of evidence indicate that commensal microbial imbalance (known as dysbiosis) can trigger DDR activation in host cells, which may result in sustained inflammatory responses. Therefore, dysbiosis can be seen as an important source of DNA damage agents that may be partially responsible for the overexpression of NKG2D-Ls on intestinal epithelial cells that is frequently observed in patients with inflammatory bowel disease and other disorders associated with altered human microbiota, including the development of colorectal cancer. In this article, we discuss recent evidence that appears to link an altered human microbiota with autoimmunity and carcinogenesis via the activation of DDR signals and the induction of NKG2D-Ls in stressed cells.
Introduction
The DNA damage response (DDR) is a highly efficient network of cellular pathways that play a crucial role in maintaining DNA integrity (1, 2). This surveillance system is responsible for monitoring, detecting and repairing DNA lesions, in order to prevent the generation of potentially deleterious mutations, which otherwise may result in the irreversible damage of DNA molecules, leading to cancer and other alterations in cell behavior (3, 4). The accumulation of un-repaired DNA damages in non-replicating cells, such as most of the cells in the brains or muscles of adults, is believed to contribute to the aging process in humans (5, 6). In highly replicating cells, such as hematopoietic stem cells and epithelial cells, DNA mutations that result from unrepaired DNA damages play a crucial role in malignant transformation and cancer progression (5, 7, 8). Endogenous agents capable of harming DNA, such as reactive oxygen species (ROS), lipid peroxidation products, and reactive nitrogen species (RNS) are naturally released during cell metabolic activities or hydrolytic processes (1, 9). In addition, DDR activation can be triggered by thousands of exogenous agents, including ionizing radiation, chemotherapy, virus infections, and chronic inflammation (10–13).
DNA damage response activation is controlled by three protein kinases: ataxia telangiectasia mutated (ATM), DNA-dependent protein kinase (DNA-PK), and ATM- and Rad3-related (ATR) (7, 14). Both ATM and DNA-PK are recruited by DNA double strand breaks (DSB), however, whereas DNA-PK coordinates DSB repair via non-homologous coupling, ATM promotes homologous recombination and cell cycle arrest at various checkpoints (14). ATR is activated in response to persistent single-stranded DNA and acts at the S-phase checkpoint (14). Upon DNA damage recognition, these kinases activate various downstream mediators including p53, CHK1, CHK2, BRCA, and H2AX, which (depending on the extent of DNA damage) may lead to cell-cycle arrest, DNA repair, senescence, or apoptosis (14–16). A key mediator of ATM signal is the checkpoint kinase Chk2, which induces G1/S checkpoint via Cdk2 inactivation or can block cell cycle at G2/M by preventing cyclinB1/Cdk1 complex formation (17). On the other hand, Chk1, triggered by ATR signal, activates Cdc25A phosphatase and Treslin, which induce G2 and S phase arrest (7).
Another consequence of DDR and ATM/ATR activation is the induction of cell stress molecules that are proteins expressed on the surface of damaged cells (18, 19). These stress ligands, which are usually absent in normal cells, are specifically recognized by either, the natural killer group 2 member D (NKG2D) and the DNAX accessory molecule immunoreceptors (18–20).
Natural killer group 2 member D, also known as Klrk1, is a C-type lectin-like type II transmembrane protein constitutively expressed by NK cells, activated macrophages and various T-cell subsets, such as NKT cells, CD8+ αβ, CD4+αβ, and γδ T lymphocytes (21–23). Upon engagement of specific NKG2D ligands (NKG2D-Ls), NKG2D receptor activates downstream signaling pathways resulting in effector immune responses like cytokine releases and cellular cytotoxicity (22, 24).
Recent evidence has linked various bacterial pathogens with DDR activation caused by either the direct effect of microbe produced genotoxins (25–28) or indirectly by ROS or RNS that result from the prolonged or excessive activation of host immune cells in response to certain microbes or their metabolic end-products (29, 30). This bacterial-induced DDR is not limited to highly pathogenic bacteria, since genotoxic damage induced by certain members of the commensal bacteria community (termed the “microbiota”) have been also documented (31, 32). Notably, increased expression of NKG2D-Ls on the surface of intestinal epithelial cells and its recognition by NKG2D receptor-expressing immune cells is believed to contribute to the pathogenesis of inflammatory bowel diseases (IBDs), such as ulcerative colitis and Crohn’s disease (33–35) and dysregulated gut microbiota has been etiologically linked to IBD and colorectal cancer (CRC) (27, 36, 37). In this article, we discuss recent pieces of evidence that appear to link alterations in gut microbiota with activation of the DDR. The potential effects that perturbations in this network have on the development of autoimmunity and cancer immunosurveillance are also discussed.
NKG2D-Ls Expression
In humans, multiple families of structurally unrelated NKG2D-Ls have been identified, including the MHC class I chain-related molecules (MICA and MICB), and the UL-16 binding proteins (ULBP1, -2, -3, -4, -5, and -6) (24, 38–40). NKG2D-Ls are absent or poorly expressed on the surfaces of normal cells but they are induced under certain pathological conditions like heat shock, virus infection, oxidative stress, and malignant transformation (39, 41). The elimination of NKG2D-Ls expressing cells by NKG2D receptor-expressing immune cells is one of the underlying grounds of the concept of cancer immunosurveillance (42–44). NKG2D-Ls upregulation has been described in various human cancers, including carcinomas of the breast (45), lung, colon (46) and prostate cancer (47), as well as in melanomas (48), gliomas (49), leukemias (18), and cervix cancer (50). The expression of these molecules is tightly regulated by mechanisms that control gene transcription, mRNA stability, protein translation, and stabilization (20, 39). Intriguingly, NKG2D-Ls expression has also been documented in certain normal cells. For example, in primary bronchial epithelial cells, MICA and ULBP1-4 are detectable mainly at intracellular level, but become detectable on the cell surface when the cells are exposed to oxidative stress (51). NKG2D-Ls (mainly ULBP1) have also been detected in peripheral blood cells (52) and these proteins are particularly upregulated in activated T cells and B cells (20, 53, 54). In addition, normal gut epithelium constitutively expresses MICA, although most cells appear to express these proteins intracellularly (55). On the other hand, the aberrant expression of NKG2D-Ls has been documented in certain autoimmune diseases, especially in the damaged tissues of patients with inflammatory bowel disease (IBD) that includes Crohn’s disease (35, 56) and ulcerative colitis (33). In these disorders, NKG2D-Ls expression correlates with increased number of infiltrating NKG2D+ lymphocytes in the damaged tissues (33, 35). Consistent with these observations, a randomized controlled clinical trial recently showed that a single dose of an anti-NKG2D blocking monoclonal antibody, significantly reduced disease activity in patients with active Crohn’s disease (57). Despite the relatively small size of this study (78 patients) and the fact that patients with UC were not included, these encouraging data support the involvement of NKG2D/NKG2D-Ls axis in the pathogenesis or clinical course of IBD.
In patients with active Celiac disease, MICA is strongly expressed on the surface of intestinal epithelial cells and it is further upregulated by wheat gliadin, which triggers the activation of intraepithelial NKG2D+ lymphocytes, leading to epithelial damage and villous atrophy (55). Notably, the probiotics Lactobacillus fermentum and Bifidobacterium lactis were found to directly inhibit the toxic effects of gliadin in intestinal cells (58) and a gluten-free diet strongly downregulated NKG2D-Ls in intestinal epithelial cells and concomitantly decreased NKG2D receptor expression on infiltrating NK cells (59).
Dysbiosis and NKG2D-Ls Expression
The community of commensal microorganisms living within the human intestines, known as gut microbiota, plays critical roles in maintaining immune tolerance and epithelial integrity (60–62).
Significant upregulation of NKG2D-Ls was observed in the intestinal mucosa of germ-free mice lacking commensal microbiota, as well as in commensal-depleted animals (ampicillin-treated mice), and low ligands expression level was restored when ampicillin treatment was stopped. Strikingly, the same study found low levels of NKG2D-Ls in animals treated with vancomycin, which was attributed to the selective propagation of the vancomycin-resistant bacterium Akkermansia muciniphila in mice intestines (31), indicating that NKG2D-Ls expression, at least in intestinal tissues, is largely influenced by the gut microbiota composition. Interestingly, A. muciniphila has been linked with anti-inflammatory protective properties against IBD (63).
The loss of microbial balance and the overgrowth of pathogenic bacteria (known as dysbiosis) is often associated with the development of autoimmune disorders and the development of CRC (62, 64, 65). Strikingly, direct microbe-induced NKG2D-Ls upregulation has been documented in human intestinal epithelial cells exposed to Escherichia coli strains, where the interaction between bacterial adhesin AfaE and its cellular receptor CD55 results in MICA expression (66).
Another study showed that Pseudomonas aeruginosa infection increased NKG2D-Ls (Rae1) in mouse airway epithelial cells in vivo and upregulated ULBP2 in human airway epithelial cells in vitro, although the mechanism of ligand induction by this pathogen is unknown (67).
Propionibacterium acnes was recently linked with Corpus-dominant lymphocytic gastritis (CDLG), a Helicobacter pylori negative entity and typically characterized by extensive infiltration of CD8+ T-cells in the stomach epithelium. Interestingly, P. acnes infection correlated with increased levels of IL-15 and the upregulation of NKG2D-Ls in the inflamed gastric epithelium. Although the mechanisms leading to NKG2D-L upregulation in this entity remains unclear, a microbe-derived stimuli, probably live P. acnes or microbial-derived short-chain fatty acids were proposed as triggering factors (68). Notably, CDLG frequently coexists with autoimmune disorders with altered microbiota including Celiac disease (69) and Crohn’s disease (70, 71). Moreover, propionic acid, derived from the fermentation of plant-derived dietary fiber mainly under the presence Propionibacterium, upregulated MICA/B in human cells including, activated T lymphocytes and different cancer lines (72). Mycobacterium tuberculosis (M. tuberculosis)-infected dendritic and airway epithelial cells also upregulate MICA expression in vitro and in vivo, and ligand recognition by Vγ2Vδ2 T cells expressing NKG2D receptor induces a potent inflammatory reaction (73).
Of note, albeit the above studies indicate that NKG2D-Ls upregulation is frequently observed in host cells exposed to various bacteria or their products, the molecular mechanisms of this phenomenon have not been elucidated. In addition, the exact mechanism that determines the fate of host cells exposed to dysbiosis (cell cycle arrest, apoptosis, malignant transformation or NKG2D-Ls upregulation) is currently unknown. Current data suggest that the extent of DNA damage and the resultant cellular responses determine cell fate under these stress conditions, hence in the context of dysbiosis, it is conceivable that cell fate may be dependent on the specific bacterium or group of bacteria dysregulated in the host.
As mentioned above, commensal bacteria play critical roles maintaining gut homeostasis, and this particular feature can be exploited for therapeutic purposes. The oral administration of commensal lactic acid bacteria effectively protected mice from dextran sulfate sodium-induced experimental colitis, which was attributed to the enhanced interferon-β production triggered by double-stranded RNA derived from commensal lactic acid bacteria (74). Although this study did not explore NKG2D-Ls expression on intestinal cells, it is worth mentioning that type I interferons have been shown to downregulate NKG2D-Ls expression impairing NK cells-dependent killing of target cells (38).
Bacterial Genotoxins and DDR Activation
Various intestinal bacteria are known to release genotoxins (bacterial products capable of targeting host DNA), which together with the induction of sustained inflammation, promotes genomic instability and ultimately autoimmunity or cancer (Table 1). The first characterized bacterial genotoxin was cytolethal distending toxins (CDT), which is produced by several Gram-negative bacteria, including E. coli, Campylobacter sp., helicobacter sp., Shigella dysenteriae, and Haemophilus ducreyi. CDT induces DNA DSB in exposed host cells that may lead to transient cell cycle arrest or malignant transformation (32, 36, 75). Mouse liver cells exposed to CDT producing helicobacter develop dysplasia (76) and fibroblasts or intestinal epithelial cells chronically exposed to large concentrations of CDT, in the absence of immune cell clearance, show genomic instability, fail to activate DDR, and eventually become prone to malignant transformation (32).
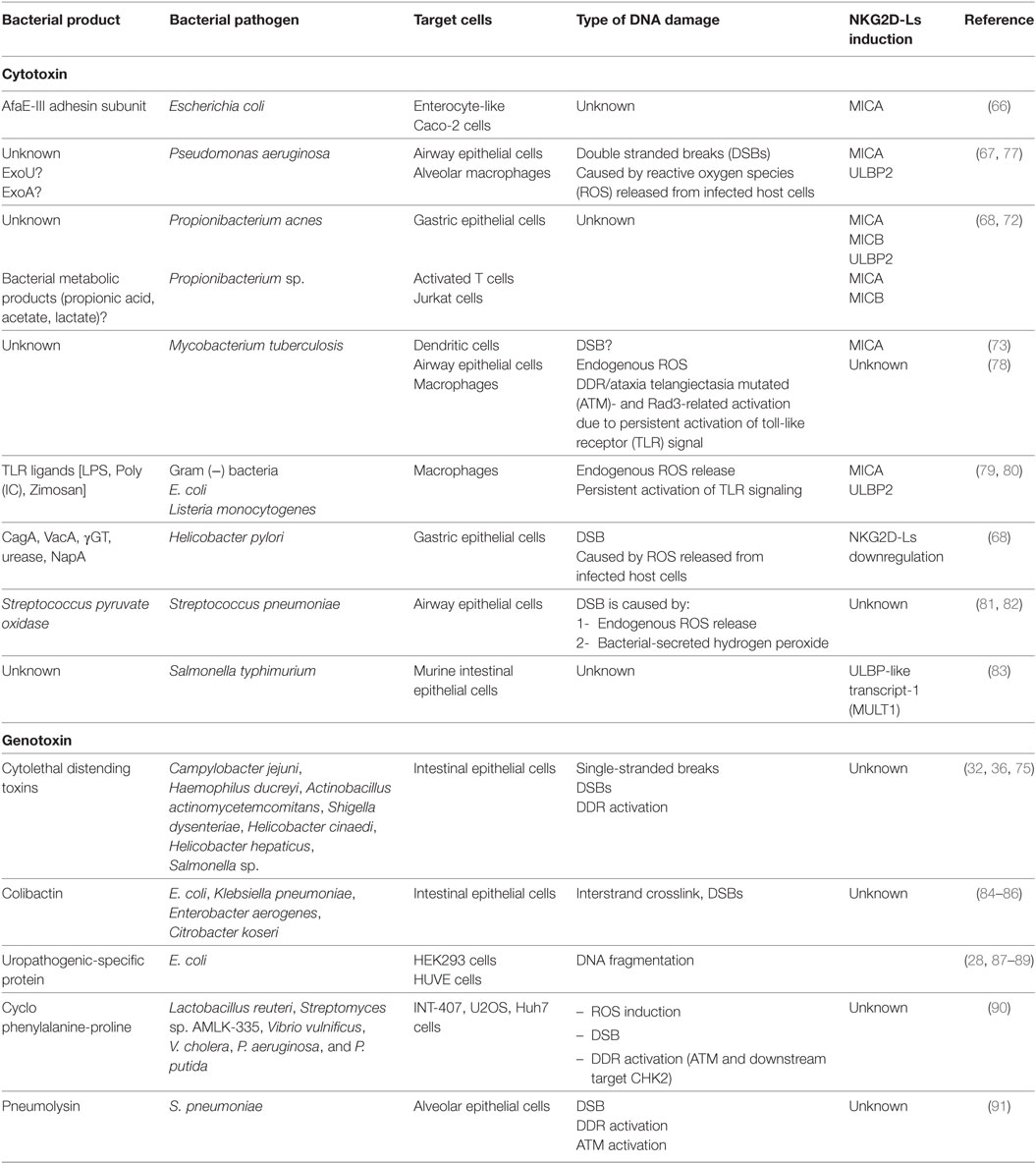
Table 1. Bacterial pathogens or their products that activate DNA damage response (DDR) and may induce NKG2D ligands (NKG2D-Ls) expression in host cells.
Another bacterial-derived genotoxin is colibactin produced by E. coli strains of the B2 phylogroup harboring the polyketide synthetase island (pks), which is also found in other Enterobacteriaceae members such as Proteus mirabilis and Klebsiella pneumoniae (84–86). Infection with E. coli harboring this genomic cluster generates DSB leading to DDR activation, cell cycle arrest and genomic instability (36, 85). Notably, E. coli harboring pks are frequently detectable in patients with IBD, as well as in patients with CRC, suggesting that pks is directly related to disease pathogenesis (26, 92).
Escherichia coli uropathogenic-specific protein (Usp) is another bacterial toxin that induces genotoxic stress and activates DDR in exposed cells. This genotoxin is produced by E. coli strains associated with pyelonephritis, prostatitis, and bacteremia (87, 88). Purified Usp cleaves linearized naked DNA in vitro and causes DNA fragmentation in mammalian cells (28).
Interestingly, compared with normal intestinal samples where toxin-producing bacteria constitute a minority of the commensal microbiota, human CRC tissues contain a high expression of these microorganisms (93).
Cyclo phenylalanine-proline (cFP) is other genotoxin produced by various bacteria such as Lactobacillus reuteri, Streptomyces sp. AMLK-335, Vibrio vulnificus, V. cholera, P. aeruginosa, and P. putida. Mammalian cells like INT-407, U2OS, and Huh7 cells exposed to cFP develop DSB and eventually activate ATM-CHK2 (90).
Pneumolysin, a toxin produced by Streptococcus pneumoniae and a key virulence factor against host cells, induces DSBs and ATM-mediated H2AX phosphorylation in epithelial alveolar cells. Consequently pneumolysin-exposed cells undergo cell cycle arrest and apoptosis, although the induction of NKG2D-Ls was not investigated in this study (91). Interestingly, pyruvate oxidase, another cytotoxin released by S. pneumoniae, induces DSBs and contributes to pneumolysin release (81).
Bacteria may also trigger DDR activation by inducing the expression of enzymes that enhance ROS in host cells, which lead to DNA damage or the induction of chronic inflammation (36, 94, 95). Bacterial-induced DNA damage can be further amplified by ROS released from immune cells at sites of chronic inflammation since inflammatory especially macrophages and neutrophils constitute a constant source of ROS, RNS, and cytokines that can develop in response to dysbiosis (94). Granuloma formation associated with M. tuberculosis infection was recently linked to the persistent inflammatory signals mediated by toll-like receptor (TLR) signals, which promotes macrophage polyploidy by regulating DDR signals via ATR activation (78). Although this study did not assess the expression of NKG2D-Ls in polyploidy macrophages, previous studies have shown NKG2D-Ls upregulation in macrophages exposed to bacterial-derived products via TLR signal activation (79, 80).
In addition, convincing evidence have established a link between infections with certain bacteria such as E. coli, Bacteroides fragilis, and Fusobacterium nucleatum with the development of CRC (26), indicating that dysbiosis may either cause carcinogenesis or autoimmunity. In some circumstances, bacterial can interrupt the DDR activation in host cells, thus allowing the cell cycle to progress in cells with unrepaired or in repaired with errors DNA resulting in mutations of critical genes associated with malignant transformation (26, 32). It is conceivable that the chronic exposure to genotoxin-secreting bacteria in host cells with fully functional DDR may result in NKG2D-Ls overexpression, via ATM activation, which ultimately increases the risk of autoimmunity. Conversely, in host cells with failed DDR, DSB may result in the survival of cells with unrepaired DNA and elevated risk of malignant transformation (Figure 1). This assumption is supported by the observations that epithelial cells exposed to Chlamydia trachomatis, a bacteria associated with cervical and ovarian cancer development, undergo DNA damaged but fail to activate DDR, due to bacterial-induced impaired DNA repair. Consequently, infected cells continue to proliferate in an environment favorable for malignant transformation (96).Thus, the NKG2D/NKG2D-Ls axis maintains a delicate immune equilibrium, which is crucial for cancer immunosurveillance but that, under certain conditions, it can eventually promote autoimmunity (97).
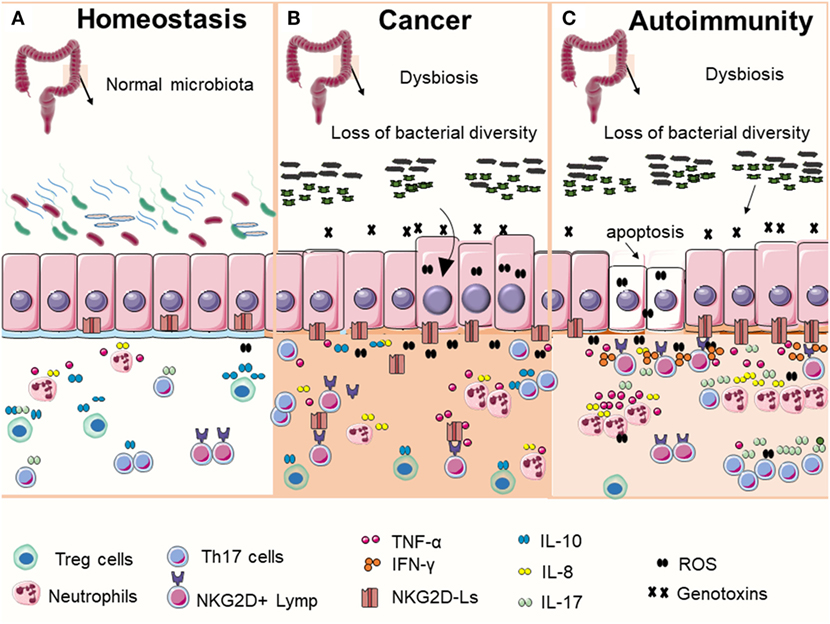
Figure 1. Commensal bacteria play an important role in maintaining gut homeostasis (A). In normal condition, intestinal epithelial cells express low levels of NKG2D ligands (NKG2D-Ls) (mostly intracellular) and beneficial bacterial contribute to immune education and help to maintain immune tolerance by promoting the induction and accumulation of regulatory T cells (Treg cells). In the context of microbial imbalance (dysbiosis), pathogenic bacteria may release genotoxins that generate DNA damage in host cells. DNA damage response (DDR) is then activated, which may lead to cell cycle arrest, apoptosis, or NKG2D-Ls induction in exposed cells. These events, together with the sustained immune activation in response to dysbiosis may eventually contribute to the development of autoimmune disorders or malignant transformation. Although the precise mechanisms that determine why cells take one of these two contrasting cell fates are unclear, current data appear to suggest that (B) bacteria-induced DNA damage in cells with failed DDR (caused by mutations or by inhibitory factors secreted by bacteria) may result in the survival and proliferation of cells with unrepaired DNA, increasing the risk of malignant transformation. Transformed cells may release or shade NKG2D-Ls that impairs NKG2D receptor-mediated functions leading to failed immune surveillance and tumor growth. (C) Alternatively, the chronic exposure to genotoxin-secreting bacterial in host cells with fully functional DDR may result in NKG2D-Ls overexpression via ataxia telangiectasia mutated activation leading to increased risk of autoimmunity.
Despite the above studies have consistently shown that bacterial genotoxin can activate DDR, unfortunately, none of them explored the potential NKG2D-Ls upregulation in host cells exposed to those bacterial products; therefore, further studied are needed to elucidate the immunological impact of genotoxins.
Concluding Remarks
Cells are continuously exposed to hostile environmental stressors, including extremes of temperature, toxins, and oxygen or nutrient deprivation. As a result, cells have evolved a wide range of molecular changes and stress responses to minimize damage which, depending on the severity and duration of stress confronted, can range from the activation of survival-promoting pathways to eliciting cell senescence or programmed cell death (98). During the last few years, remarkable progress has been made in our understanding of the molecular mechanisms of DDR activation and its role in various cellular processes like aging and cancer development, and it has become apparent that the immune system constitutes an important component of the cellular response to DNA damage stressors (99). In this regard, the recognition of NKG2D-Ls overexpressed on the surface of host cells as a consequence of DDR activation constitutes one of the mechanisms by which the immune cells expressing the NKG2D receptor can sense DNA damage in host cells (100). Importantly, dysbiosis has been causally liked with DDR activation, via either the release of genotoxin or by promoting chronic inflammation (92). Several questions about the causal link of dysbiosis with NKG2D-Ls induction via DDR remain unanswered. For example, the precise mechanisms by which DDR induced by bacterial genotoxins or dysbiosis may result in malignant transformation or autoimmunity have not been fully elucidated. In addition, the specific role of dysbiosis in the upregulation of NKG2D-Ls in the intestinal epithelial cells of patients with IBD needs to be clarified. The polymorphism rs1049174 in the NKG2D gene (generating HNK and LNK genotypes) influences NKG2D receptor expression on immune cells and is implicated in individual susceptibility to certain cancers. The HNK genotype is associated with greater NK cells cytotoxic activity and lower prevalence of epithelial cells-derived malignancies, in comparison with the low cytotoxic genotype LNK (23, 50, 101, 102). It is currently unknown if HNK and LNK genotypes can affect NKG2D receptor-mediated immune responses in the context of dysbiosis and if they are implicated in the development of autoimmune disorders like IBD. Future studies on this regard are warranted as genetic variants of human genes involved in immunity and gut architecture are associated with an altered composition of the gut microbiome (103).
Compelling evidence indicate that dysbiosis is implicated in the pathogenesis of several human diseases, ranging from metabolic disorders, autoimmunity, and cancer (64, 104, 105) and various studies have shown that manipulating human microbiota, for example by using probiotics or fecal transplantation, has promising therapeutic potential (65, 106, 107). Further research is needed to uncover the specific microbes within a dysbiotic microbiota that are directly implicated in disease etiology. Designing optimal interventions aimed to remove pathogenic microorganisms or for replacing them with beneficial ones will have enormous therapeutic potential.
Author Contributions
The authors contributed extensively to the work presented in this paper. JE: conceived and designed the study; created and drew figures and wrote the manuscript. MM: wrote the manuscript, searched and collected bibliography.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Roos WP, Kaina B. DNA damage-induced apoptosis: from specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett (2012) 332(2):237–48. doi:10.1016/j.canlet.2012.01.007
2. Pearl LH, Schierz AC, Ward SE, Al-Lazikani B, Pearl FM. Therapeutic opportunities within the DNA damage response. Nat Rev Cancer (2015) 15(3):166–80. doi:10.1038/nrc3891
3. Gasser S, Raulet D. The DNA damage response, immunity and cancer. Semin Cancer Biol (2006) 16(5):344–7. doi:10.1016/j.semcancer.2006.07.004
4. Cirombella R, Montrone G, Stoppacciaro A, Giglio S, Volinia S, Graziano P, et al. Fhit loss in lung preneoplasia: relation to DNA damage response checkpoint activation. Cancer Lett (2010) 291(2):230–6. doi:10.1016/j.canlet.2009.10.017
5. Iyama T, Wilson DM. DNA repair mechanisms in dividing and non-dividing cells. DNA Repair (Amst) (2013) 12(8):620–36. doi:10.1016/j.dnarep.2013.04.015
6. Vahidi Ferdousi L, Rocheteau P, Chayot R, Montagne B, Chaker Z, Flamant P, et al. More efficient repair of DNA double-strand breaks in skeletal muscle stem cells compared to their committed progeny. Stem Cell Res (2014) 13(3 Pt A):492–507. doi:10.1016/j.scr.2014.08.005
7. Delia D, Mizutani S. The DNA damage response pathway in normal hematopoiesis and malignancies. Int J Hematol (2017) 106(3):328–34. doi:10.1007/s12185-017-2300-7
8. Mo D, Zhao Y, Balajee AS. Human RecQL4 helicase plays multifaceted roles in the genomic stability of normal and cancer cells. Cancer Lett (2017) 413:1–10. doi:10.1016/j.canlet.2017.10.021
9. Lonkar P, Dedon PC. Reactive species and DNA damage in chronic inflammation: reconciling chemical mechanisms and biological fates. Int J Cancer (2011) 128(9):1999–2009. doi:10.1002/ijc.25815
10. Goldstein M, Kastan MB. The DNA damage response: implications for tumor responses to radiation and chemotherapy. Annu Rev Med (2015) 66:129–43. doi:10.1146/annurev-med-081313-121208
11. Sprung CN, Ivashkevich A, Forrester HB, Redon CE, Georgakilas A, Martin OA. Oxidative DNA damage caused by inflammation may link to stress-induced non-targeted effects. Cancer Lett (2015) 356(1):72–81. doi:10.1016/j.canlet.2013.09.008
12. Luftig MA. Viruses and the DNA damage response: activation and antagonism. Annu Rev Virol (2014) 1(1):605–25. doi:10.1146/annurev-virology-031413-085548
13. Williamson L, Saponaro M, Boeing S, East P, Mitter R, Kantidakis T, et al. UV irradiation induces a non-coding RNA that functionally opposes the protein encoded by the same gene. Cell (2017) 168(5):843–55.e13. doi:10.1016/j.cell.2017.01.019
14. Blackford AN, Jackson SP. ATM, ATR, and DNA-PK: the trinity at the heart of the DNA damage response. Mol Cell (2017) 66(6):801–17. doi:10.1016/j.molcel.2017.05.015
15. Shiloh Y, Ziv Y. The ATM protein: the importance of being active. J Cell Biol (2012) 198(3):273–5. doi:10.1083/jcb.201207063
16. Ditch S, Paull TT. The ATM protein kinase and cellular redox signaling: beyond the DNA damage response. Trends Biochem Sci (2012) 37(1):15–22. doi:10.1016/j.tibs.2011.10.002
17. Takagi M. DNA damage response and hematological malignancy. Int J Hematol (2017) 106(3):345–56. doi:10.1007/s12185-017-2226-0
18. Luis Espinoza J, Takami A, Trung LQ, Nakao S. Ataxia-telangiectasia mutated kinase-mediated upregulation of NKG2D ligands on leukemia cells by resveratrol results in enhanced natural killer cell susceptibility. Cancer Sci (2013) 104(6):657–62. doi:10.1111/cas.12141
19. Gasser S, Orsulic S, Brown EJ, Raulet DH. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature (2005) 436(7054):1186–90. doi:10.1038/nature03884
20. Cerboni C, Fionda C, Soriani A, Zingoni A, Doria M, Cippitelli M, et al. The DNA damage response: a common pathway in the regulation of NKG2D and DNAM-1 ligand expression in normal, infected, and cancer cells. Front Immunol (2014) 4:508. doi:10.3389/fimmu.2013.00508
21. Zingoni A, Ardolino M, Santoni A, Cerboni C. NKG2D and DNAM-1 activating receptors and their ligands in NK-T cell interactions: role in the NK cell-mediated negative regulation of T cell responses. Front Immunol (2012) 3:408. doi:10.3389/fimmu.2012.00408
22. Espinoza JL, Takami A, Yoshioka K, Nakata K, Sato T, Kasahara Y, et al. Human microRNA-1245 downregulates the NKG2D receptor in NK cells and impairs NKG2D-mediated functions. Haematologica (2012) 97(9):1295–303. doi:10.3324/haematol.2011.058529
23. Espinoza JL, Takami A, Onizuka M, Sao H, Akiyama H, Miyamura K, et al. NKG2D gene polymorphism has a significant impact on transplant outcomes after HLA-fully-matched unrelated bone marrow transplantation for standard risk hematologic malignancies. Haematologica (2009) 94(10):1427–34. doi:10.3324/haematol.2009.008318
24. Jelenčić V, Lenartić M, Wensveen FM, Polić B. NKG2D: a versatile player in the immune system. Immunol Lett (2017) 189:48–53. doi:10.1016/j.imlet.2017.04.006
25. Žgur-Bertok D. DNA damage repair and bacterial pathogens. PLoS Pathog (2013) 9(11):e1003711. doi:10.1371/journal.ppat.1003711
26. Gagnaire A, Nadel B, Raoult D, Neefjes J, Gorvel JP. Collateral damage: insights into bacterial mechanisms that predispose host cells to cancer. Nat Rev Microbiol (2017) 15(2):109–28. doi:10.1038/nrmicro.2016.171
27. Ray D, Kidane D. Gut microbiota imbalance and base excision repair dynamics in colon cancer. J Cancer (2016) 7(11):1421–30. doi:10.7150/jca.15480
28. Grasso F, Frisan T. Bacterial genotoxins: merging the DNA damage response into infection biology. Biomolecules (2015) 5(3):1762–82. doi:10.3390/biom5031762
29. Fahrer J, Huelsenbeck J, Jaurich H, Dörsam B, Frisan T, Eich M, et al. Cytolethal distending toxin (CDT) is a radiomimetic agent and induces persistent levels of DNA double-strand breaks in human fibroblasts. DNA Repair (Amst) (2014) 18:31–43. doi:10.1016/j.dnarep.2014.03.002
30. Kalisperati P, Spanou E, Pateras IS, Korkolopoulou P, Varvarigou A, Karavokyros I, et al. Inflammation, DNA damage, Helicobacter pylori and gastric tumorigenesis. Front Genet (2017) 8:20. doi:10.3389/fgene.2017.00020
31. Hansen CH, Holm TL, Krych Ł, Andresen L, Nielsen DS, Rune I, et al. Gut microbiota regulates NKG2D ligand expression on intestinal epithelial cells. Eur J Immunol (2013) 43(2):447–57. doi:10.1002/eji.201242462
32. Guidi R, Guerra L, Levi L, Stenerlöw B, Fox JG, Josenhans C, et al. Chronic exposure to the cytolethal distending toxins of Gram-negative bacteria promotes genomic instability and altered DNA damage response. Cell Microbiol (2013) 15(1):98–113. doi:10.1111/cmi.12034
33. La Scaleia R, Stoppacciaro A, Oliva S, Morrone S, Di Nardo G, Santoni A, et al. NKG2D/Ligand dysregulation and functional alteration of innate immunity cell populations in pediatric IBD. Inflamm Bowel Dis (2012) 18(10):1910–22. doi:10.1002/ibd.22899
34. Ito Y, Kanai T, Totsuka T, Okamoto R, Tsuchiya K, Nemoto Y, et al. Blockade of NKG2D signaling prevents the development of murine CD4+ T cell-mediated colitis. Am J Physiol Gastrointest Liver Physiol (2008) 294(1):G199–207. doi:10.1152/ajpgi.00286.2007
35. Allez M, Tieng V, Nakazawa A, Treton X, Pacault V, Dulphy N, et al. CD4+NKG2D+ T cells in Crohn’s disease mediate inflammatory and cytotoxic responses through MICA interactions. Gastroenterology (2007) 132(7):2346–58. doi:10.1053/j.gastro.2007.03.025
36. Gagnière J, Raisch J, Veziant J, Barnich N, Bonnet R, Buc E, et al. Gut microbiota imbalance and colorectal cancer. World J Gastroenterol (2016) 22(2):501–18. doi:10.3748/wjg.v22.i2.501
37. Carding S, Verbeke K, Vipond DT, Corfe BM, Owen LJ. Dysbiosis of the gut microbiota in disease. Microb Ecol Health Dis (2015) 26:26191. doi:10.3402/mehd.v26.26191
38. Zhang J, Basher F, Wu JD. NKG2D ligands in tumor immunity: two sides of a coin. Front Immunol (2015) 6:97. doi:10.3389/fimmu.2015.00097
39. Huergo-Zapico L, Acebes-Huerta A, López-Soto A, Villa-Álvarez M, Gonzalez-Rodriguez AP, Gonzalez S. Molecular bases for the regulation of NKG2D ligands in cancer. Front Immunol (2014) 5:106. doi:10.3389/fimmu.2014.00106
40. Lanier LL. NKG2D receptor and its ligands in host defense. Cancer Immunol Res (2015) 3(6):575–82. doi:10.1158/2326-6066.CIR-15-0098
41. Ardolino M, Zingoni A, Cerboni C, Cecere F, Soriani A, Iannitto ML, et al. DNAM-1 ligand expression on Ag-stimulated T lymphocytes is mediated by ROS-dependent activation of DNA-damage response: relevance for NK-T cell interaction. Blood (2011) 117(18):4778–86. doi:10.1182/blood-2010-08-300954
42. Sagiv A, Burton DG, Moshayev Z, Vadai E, Wensveen F, Ben-Dor S, et al. NKG2D ligands mediate immunosurveillance of senescent cells. Aging (Albany NY) (2016) 8(2):328–44. doi:10.18632/aging.100897
43. Weil S, Memmer S, Lechner A, Huppert V, Giannattasio A, Becker T, et al. Natural killer group 2D ligand depletion reconstitutes natural killer cell immunosurveillance of head and neck squamous cell carcinoma. Front Immunol (2017) 8:387. doi:10.3389/fimmu.2017.00387
44. Guerra N, Tan Y, Joncker N, Choy A, Gallardo F, Xiong N, et al. NKG2D-deficient mice are defective in tumor surveillance in models of spontaneous malignancy. Immunity (2008) 28(4):571–80. doi:10.1016/j.immuni.2008.02.016
45. Breunig C, Pahl J, Küblbeck M, Miller M, Antonelli D, Erdem N, et al. MicroRNA-519a-3p mediates apoptosis resistance in breast cancer cells and their escape from recognition by natural killer cells. Cell Death Dis (2017) 8(8):e2973. doi:10.1038/cddis.2017.364
46. Kim GR, Ha GH, Bae JH, Oh SO, Kim SH, Kang CD. Metastatic colon cancer cell populations contain more cancer stem-like cells with a higher susceptibility to natural killer cell-mediated lysis compared with primary colon cancer cells. Oncol Lett (2015) 9(4):1641–6. doi:10.3892/ol.2015.2918
47. Chávez-Blanco A, De la Cruz-Hernández E, Domínguez GI, Rodríguez-Cortez O, Alatorre B, Pérez-Cárdenas E, et al. Upregulation of NKG2D ligands and enhanced natural killer cell cytotoxicity by hydralazine and valproate. Int J Oncol (2011) 39(6):1491–9. doi:10.3892/ijo.2011.1144
48. Paschen A, Baingo J, Schadendorf D. Expression of stress ligands of the immunoreceptor NKG2D in melanoma: regulation and clinical significance. Eur J Cell Biol (2014) 93(1–2):49–54. doi:10.1016/j.ejcb.2014.01.009
49. Zhang X, Rao A, Sette P, Deibert C, Pomerantz A, Kim WJ, et al. IDH mutant gliomas escape natural killer cell immune surveillance by downregulation of NKG2D ligand expression. Neuro Oncol (2016) 18(10):1402–12. doi:10.1093/neuonc/now061
50. Espinoza JL, Nguyen VH, Ichimura H, Pham TT, Nguyen CH, Pham TV, et al. A functional polymorphism in the NKG2D gene modulates NK-cell cytotoxicity and is associated with susceptibility to human papilloma virus-related cancers. Sci Rep (2016) 6:39231. doi:10.1038/srep39231
51. Borchers MT, Harris NL, Wesselkamper SC, Vitucci M, Cosman D. NKG2D ligands are expressed on stressed human airway epithelial cells. Am J Physiol Lung Cell Mol Physiol (2006) 291(2):L222–31. doi:10.1152/ajplung.00327.2005
52. Nowbakht P, Ionescu MC, Rohner A, Kalberer CP, Rossy E, Mori L, et al. Ligands for natural killer cell-activating receptors are expressed upon the maturation of normal myelomonocytic cells but at low levels in acute myeloid leukemias. Blood (2005) 105(9):3615–22. doi:10.1182/blood-2004-07-2585
53. Raulet DH, Gasser S, Gowen BG, Deng W, Jung H. Regulation of ligands for the NKG2D activating receptor. Annu Rev Immunol (2013) 31:413–41. doi:10.1146/annurev-immunol-032712-095951
54. Cerboni C, Zingoni A, Cippitelli M, Piccoli M, Frati L, Santoni A. Antigen-activated human T lymphocytes express cell-surface NKG2D ligands via an ATM/ATR-dependent mechanism and become susceptible to autologous NK-cell lysis. Blood (2007) 110(2):606–15. doi:10.1182/blood-2006-10-052720
55. Hüe S, Mention JJ, Monteiro RC, Zhang S, Cellier C, Schmitz J, et al. A direct role for NKG2D/MICA interaction in villous atrophy during celiac disease. Immunity (2004) 21(3):367–77. doi:10.1016/j.immuni.2004.06.018
56. Vadstrup K, Galsgaard ED, Jensen H, Lanier LL, Ryan JC, Chen SY, et al. NKG2D ligand expression in Crohn’s disease and NKG2D-dependent stimulation of CD8(+) T cell migration. Exp Mol Pathol (2017) 103(1):56–70. doi:10.1016/j.yexmp.2017.06.010
57. Allez M, Skolnick BE, Wisniewska-Jarosinska M, Petryka R, Overgaard RV. Anti-NKG2D monoclonal antibody (NNC0142-0002) in active Crohn’s disease: a randomised controlled trial. Gut (2017) 66(11):1918–25. doi:10.1136/gutjnl-2016-311824
58. Lindfors K, Blomqvist T, Juuti-Uusitalo K, Stenman S, Venäläinen J, Mäki M, et al. Live probiotic Bifidobacterium lactis bacteria inhibit the toxic effects induced by wheat gliadin in epithelial cell culture. Clin Exp Immunol (2008) 152(3):552–8. doi:10.1111/j.1365-2249.2008.03635.x
59. Adlercreutz EH, Weile C, Larsen J, Engkilde K, Agardh D, Buschard K, et al. A gluten-free diet lowers NKG2D and ligand expression in BALB/c and non-obese diabetic (NOD) mice. Clin Exp Immunol (2014) 177(2):391–403. doi:10.1111/cei.12340
60. Espinoza JL, Elbadry MI, Nakao S. An altered gut microbiota may trigger autoimmune-mediated acquired bone marrow failure syndromes. Clin Immunol (2016) 171:62–4. doi:10.1016/j.clim.2016.08.008
61. Green ED, Watson JD, Collins FS. Human genome project: twenty-five years of big biology. Nature (2015) 526(7571):29–31. doi:10.1038/526029a
62. Espinoza JL, Kotecha R, Nakao S. Microbe-induced inflammatory signals triggering acquired bone marrow failure syndromes. Front Immunol (2017) 8:186. doi:10.3389/fimmu.2017.00186
63. Png CW, Lindén SK, Gilshenan KS, Zoetendal EG, McSweeney CS, Sly LI, et al. Mucolytic bacteria with increased prevalence in IBD mucosa augment in vitro utilization of mucin by other bacteria. Am J Gastroenterol (2010) 105(11):2420–8. doi:10.1038/ajg.2010.281
64. Lerner A, Aminov R, Matthias T. Dysbiosis may trigger autoimmune diseases via inappropriate post-translational modification of host proteins. Front Microbiol (2016) 7:84. doi:10.3389/fmicb.2016.00084
65. Belizário JE, Napolitano M. Human microbiomes and their roles in dysbiosis, common diseases, and novel therapeutic approaches. Front Microbiol (2015) 6:1050. doi:10.3389/fmicb.2015.01050
66. Tieng V, Le Bouguénec C, du Merle L, Bertheau P, Desreumaux P, Janin A, et al. Binding of Escherichia coli adhesin AfaE to CD55 triggers cell-surface expression of the MHC class I-related molecule MICA. Proc Natl Acad Sci U S A (2002) 99(5):2977–82. doi:10.1073/pnas.032668099
67. Borchers MT, Harris NL, Wesselkamper SC, Zhang S, Chen Y, Young L, et al. The NKG2D-activating receptor mediates pulmonary clearance of Pseudomonas aeruginosa. Infect Immun (2006) 74(5):2578–86. doi:10.1128/IAI.74.5.2578-2586.2006
68. Montalban-Arques A, Wurm P, Trajanoski S, Schauer S, Kienesberger S, Halwachs B, et al. Propionibacterium acnes overabundance and natural killer group 2 member D system activation in corpus-dominant lymphocytic gastritis. J Pathol (2016) 240(4):425–36. doi:10.1002/path.4782
69. Nielsen JA, Roberts CA, Lager DJ, Putcha RV, Jain R, Lewin M. Lymphocytic gastritis is not associated with active Helicobacter pylori infection. Helicobacter (2014) 19(5):349–55. doi:10.1111/hel.12139
70. Wu TT, Hamilton SR. Lymphocytic gastritis: association with etiology and topology. Am J Surg Pathol (1999) 23(2):153–8. doi:10.1097/00000478-199902000-00003
71. Polydorides AD. Pathology and differential diagnosis of chronic, noninfectious gastritis. Semin Diagn Pathol (2014) 31(2):114–23. doi:10.1053/j.semdp.2014.02.008
72. Andresen L, Hansen K, Jensen H, Pedersen S, Stougaard P, Hansen H, et al. Propionic acid secreted from propionibacteria induces NKG2D ligand expression on human-activated T lymphocytes and cancer cells. J Immunol (2009) 183(2):897–906. doi:10.4049/jimmunol.0803014
73. Das H, Groh V, Kuijl C, Sugita M, Morita CT, Spies T, et al. MICA engagement by human Vgamma2Vdelta2 T cells enhances their antigen-dependent effector function. Immunity (2001) 15(1):83–93. doi:10.1016/S1074-7613(01)00168-6
74. Kawashima T, Kosaka A, Yan H, Guo Z, Uchiyama R, Fukui R, et al. Double-stranded RNA of intestinal commensal but not pathogenic bacteria triggers production of protective interferon-β. Immunity (2013) 38(6):1187–97. doi:10.1016/j.immuni.2013.02.024
75. Frisan T, Cortes-Bratti X, Chaves-Olarte E, Stenerlöw B, Thelestam M. The Haemophilus ducreyi cytolethal distending toxin induces DNA double-strand breaks and promotes ATM-dependent activation of RhoA. Cell Microbiol (2003) 5(10):695–707. doi:10.1046/j.1462-5822.2003.00311.x
76. Fox JG, Rogers AB, Whary MT, Ge Z, Taylor NS, Xu S, et al. Gastroenteritis in NF-kappaB-deficient mice is produced with wild-type Campylobacter jejuni but not with C. jejuni lacking cytolethal distending toxin despite persistent colonization with both strains. Infect Immun (2004) 72(2):1116–25. doi:10.1128/IAI.72.2.1116-1125.2004
77. Saliba AM, de Assis MC, Nishi R, Raymond B, Marques EA, Lopes UG, et al. Implications of oxidative stress in the cytotoxicity of Pseudomonas aeruginosa ExoU. Microbes Infect (2006) 8(2):450–9. doi:10.1016/j.micinf.2005.07.011
78. Herrtwich L, Nanda I, Evangelou K, Nikolova T, Horn V, Sagar , et al. DNA damage signaling instructs polyploid macrophage fate in granulomas. Cell (2016) 167(5):1264–80.e18. doi:10.1016/j.cell.2016.09.054
79. Hamerman JA, Ogasawara K, Lanier LL. Cutting edge: toll-like receptor signaling in macrophages induces ligands for the NKG2D receptor. J Immunol (2004) 172(4):2001–5. doi:10.4049/jimmunol.172.4.2001
80. Antonangeli F, Soriani A, Cerboni C, Sciumè G, Santoni A. How mucosal epithelia deal with stress: role of NKG2D/NKG2D ligands during inflammation. Front Immunol (2017) 8:1583. doi:10.3389/fimmu.2017.01583
81. Bryant JC, Dabbs RC, Oswalt KL, Brown LR, Rosch JW, Seo KS, et al. Pyruvate oxidase of Streptococcus pneumoniae contributes to pneumolysin release. BMC Microbiol (2016) 16(1):271. doi:10.1186/s12866-016-0881-6
82. Rai P, Parrish M, Tay IJ, Li N, Ackerman S, He F, et al. Streptococcus pneumoniae secretes hydrogen peroxide leading to DNA damage and apoptosis in lung cells. Proc Natl Acad Sci U S A (2015) 112(26):E3421–30. doi:10.1073/pnas.1424144112
83. Li Z, Zhang C, Zhou Z, Zhang J, Tian Z. Small intestinal intraepithelial lymphocytes expressing CD8 and T cell receptor γδ are involved in bacterial clearance during Salmonella enterica serovar Typhimurium infection. Infect Immun (2012) 80(2):565–74. doi:10.1128/IAI.05078-11
84. Buc E, Dubois D, Sauvanet P, Raisch J, Delmas J, Darfeuille-Michaud A, et al. High prevalence of mucosa-associated E. coli producing cyclomodulin and genotoxin in colon cancer. PLoS One (2013) 8(2):e56964. doi:10.1371/journal.pone.0056964
85. Nougayrède JP, Homburg S, Taieb F, Boury M, Brzuszkiewicz E, Gottschalk G, et al. Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science (2006) 313(5788):848–51. doi:10.1126/science.1127059
86. Putze J, Hennequin C, Nougayrède JP, Zhang W, Homburg S, Karch H, et al. Genetic structure and distribution of the colibactin genomic island among members of the family Enterobacteriaceae. Infect Immun (2009) 77(11):4696–703. doi:10.1128/IAI.00522-09
87. Crnigoj M, Podlesek Z, Budič M, Zgur-Bertok D. The Escherichia coli uropathogenic-specific-protein-associated immunity protein 3 (Imu3) has nucleic acid-binding activity. BMC Microbiol (2014) 14:16. doi:10.1186/1471-2180-14-16
88. Zaw MT, Yamasaki E, Yamamoto S, Nair GB, Kawamoto K, Kurazono H. Uropathogenic specific protein gene, highly distributed in extraintestinal uropathogenic Escherichia coli, encodes a new member of H-N-H nuclease superfamily. Gut Pathog (2013) 5(1):13. doi:10.1186/1757-4749-5-13
89. Nipič D, Podlesek Z, Budič M, Črnigoj M, Žgur-Bertok D. Escherichia coli uropathogenic-specific protein, Usp, is a bacteriocin-like genotoxin. J Infect Dis (2013) 208(10):1545–52. doi:10.1093/infdis/jit480
90. Lee K, Jeong JE, Kim IH, Kim KS, Ju BG. Cyclo(phenylalanine-proline) induces DNA damage in mammalian cells via reactive oxygen species. J Cell Mol Med (2015) 19(12):2851–64. doi:10.1111/jcmm.12678
91. Rai P, He F, Kwang J, Engelward BP, Chow VT. Pneumococcal pneumolysin induces DNA damage and cell cycle arrest. Sci Rep (2016) 6:22972. doi:10.1038/srep22972
92. Kidane D, Chae WJ, Czochor J, Eckert KA, Glazer PM, Bothwell AL, et al. Interplay between DNA repair and inflammation, and the link to cancer. Crit Rev Biochem Mol Biol (2014) 49(2):116–39. doi:10.3109/10409238.2013.875514
93. Dutilh BE, Backus L, van Hijum SA, Tjalsma H. Screening metatranscriptomes for toxin genes as functional drivers of human colorectal cancer. Best Pract Res Clin Gastroenterol (2013) 27(1):85–99. doi:10.1016/j.bpg.2013.03.008
94. Bhatt AP, Redinbo MR, Bultman SJ. The role of the microbiome in cancer development and therapy. CA Cancer J Clin (2017) 67(4):326–44. doi:10.3322/caac.21398
95. Wroblewski LE, Peek RM, Coburn LA. The role of the microbiome in gastrointestinal cancer. Gastroenterol Clin North Am (2016) 45(3):543–56. doi:10.1016/j.gtc.2016.04.010
96. Chumduri C, Gurumurthy RK, Zadora PK, Mi Y, Meyer TF. Chlamydia infection promotes host DNA damage and proliferation but impairs the DNA damage response. Cell Host Microbe (2013) 13(6):746–58. doi:10.1016/j.chom.2013.05.010
97. Guerra N, Pestal K, Juarez T, Beck J, Tkach K, Wang L, et al. A selective role of NKG2D in inflammatory and autoimmune diseases. Clin Immunol (2013) 149(3):432–9. doi:10.1016/j.clim.2013.09.003
98. Chen F, Evans A, Pham J, Plosky B. Cellular stress responses: a balancing act. Mol Cell (2010) 40(2):175. doi:10.1016/j.molcel.2010.10.008
99. Nakad R, Schumacher B. DNA damage response and immune defense: links and mechanisms. Front Genet (2016) 7:147. doi:10.3389/fgene.2016.00147
100. Yin X, Lu X, Xiuwen Z, Min Z, Xiao R, Mao Z, et al. Role of NKG2D in cytokine-induced killer cells against lung cancer. Oncol Lett (2017) 13(5):3139–43. doi:10.3892/ol.2017.5800
101. Hayashi T, Imai K, Morishita Y, Hayashi I, Kusunoki Y, Nakachi K. Identification of the NKG2D haplotypes associated with natural cytotoxic activity of peripheral blood lymphocytes and cancer immunosurveillance. Cancer Res (2006) 66(1):563–70. doi:10.1158/0008-5472.CAN-05-2776
102. Imai K, Matsuyama S, Miyake S, Suga K, Nakachi K. Natural cytotoxic activity of peripheral-blood lymphocytes and cancer incidence: an 11-year follow-up study of a general population. Lancet (2000) 356(9244):1795–9. doi:10.1016/S0140-6736(00)03231-1
103. Hall AB, Tolonen AC, Xavier RJ. Human genetic variation and the gut microbiome in disease. Nat Rev Genet (2017) 18(11):690–9. doi:10.1038/nrg.2017.63
104. Castanys-Muñoz E, Martin MJ, Vazquez E. Building a beneficial microbiome from birth. Adv Nutr (2016) 7(2):323–30. doi:10.3945/an.115.010694
105. Ruff WE, Kriegel MA. Autoimmune host-microbiota interactions at barrier sites and beyond. Trends Mol Med (2015) 21(4):233–44. doi:10.1016/j.molmed.2015.02.006
106. Blaser MJ. The microbiome revolution. J Clin Invest (2014) 124(10):4162–5. doi:10.1172/JCI78366
Keywords: natural killer group 2 member D ligands, microbiota, dysbiosis, bacterial genotoxin, immunosurveillance, inflammatory bowel disease
Citation: Espinoza JL and Minami M (2018) Sensing Bacterial-Induced DNA Damaging Effects via Natural Killer Group 2 Member D Immune Receptor: From Dysbiosis to Autoimmunity and Carcinogenesis. Front. Immunol. 9:52. doi: 10.3389/fimmu.2018.00052
Received: 06 November 2017; Accepted: 09 January 2018;
Published: 25 January 2018
Edited by:
Leticia A. Carneiro, Universidade Federal do Rio de Janeiro, BrazilReviewed by:
Marit Inngjerdingen, Oslo University Hospital, NorwayJue Hou, Virginia Mason Medical Center, United States
Guntram A. Grassl, Hannover Medical School, Germany
Copyright: © 2018 Espinoza and Minami. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: J. Luis Espinoza, bHVpc0BtZWQua2luZGFpLmFjLmpw