 Soumi Sadhu
Soumi Sadhu Dipendra Kumar Mitra
Dipendra Kumar Mitra- 1Department of Transplant Immunology, All India Institute of Medical Sciences (AIIMS), New Delhi, India
- 2Department of Immunogenetics, All India Institute of Medical Sciences (AIIMS), New Delhi, India
Leprosy is a chronic intracellular infection caused by the acid-fast bacillus, Mycobacterium leprae. The disease chiefly affects the skin, peripheral nerves, mucosa of the upper respiratory tract, and the eyes. The damage to peripheral nerves results in sensory and motor impairment with characteristic deformities and disability. Presently, the disease remains concentrated in resource-poor countries in tropical and warm temperate regions with the largest number of cases reported from India. Even though innate immunity influences the clinical manifestation of the disease, it is the components of adaptive immune system which seem to tightly correlate with the characteristic spectrum of leprosy. M. leprae-specific T cell anergy with bacillary dissemination is the defining feature of lepromatous leprosy (LL) patients in contrast to tuberculoid leprosy (TT) patients, which is characterized by strong Th1-type cell response with localized lesions. Generation of Th1/Th2-like effector cells, however, cannot wholly explain the polarized state of immunity in leprosy. A comprehensive understanding of the role of various regulatory T cells, such as Treg and natural killer T cells, in deciding the polarized state of T cell immunity is crucial. Interaction of these T cell subsets with effector T cells like Th1 (IFN-γ dominant), Th2 (interluekin-4 dominant), and Th17 (IL-17+) cells through various regulatory cytokines and molecules (programmed death-1/programmed death ligand-1) may constitute key events in dictating the state of immune polarization, thus controlling the clinical manifestation. Studying these important components of the adaptive immune system in leprosy patients is essential for better understanding of immune function, correlate(s) the immunity and mechanism(s) of its containment.
Introduction
Leprosy is regarded as a stigmatized disease even today. Even though prevalence has fallen substantially in the past few decades, its transmission continues and the disease remains a major public health problem, especially in many third world countries. The chronic infectious disease is caused by the acid-fast, rod-shaped Bacillus, Mycobacterium leprae. It results in extensive damage to the skin, eyes, mucosa of the upper respiratory tract, and peripheral nerves, in some cases leading to sensory and motor impairment with characteristic deformities and disability (1). Worldwide, two to three million people are estimated to be permanently disabled because of leprosy (2). India has the largest number of cases, with Brazil second, and Burma third (2). Although the reported number of registered cases worldwide has declined in the past two decades, the number of new cases registered each year has remained almost same (3). For the immunologists, however, leprosy still garners a lot of attention mainly because M. leprae infection which evokes distinct polarized T cell responses in humans, which correlates with the clinical manifestations. The two polar forms of leprosy, known as tuberculoid type (TT) and lepromatous leprosy (LL), have clinical, microbiological, and immunological linkage [(4, 5), Figure 1]. TT is characterized by fewer skin lesions, low numbers of bacteria in lesions, and histologically well-formed granulomas containing abundant CD4+ T cells. On the other hand, LL is characterized by numerous infiltrative skin lesions, large numbers of bacteria in lesions, and poorly formed granulomas with fewer lymphocytes (6). However, most leprosy patients display a pathogenesis somewhere in between and are classified as either borderline tuberculoid (BT) or borderline lepromatous (BL) (4). Leprosy reactions known as type 1 reactions (T1R) (Figure 1) are common in these immunologically unstable borderline groups and involve an upregulation of the host response to M. leprae antigens (5). In patients with the disseminated LL, a reaction known as erythema nodosum leprosum (ENL) or type 2 reactions (T2R) is frequent, being observed in almost half of these patients receiving antimicrobial therapy (1).
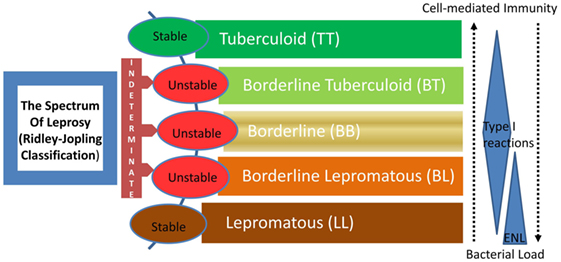
Figure 1. The spectrum of leprosy: Ridley–Jopling classification and the relationship with host immunity. ENL, erythema nodosum leprosum or type 2 reaction.
Polarized Immunity in Leprosy: Possible Causes
Several factors may be involved in regulating the polarization of newly activated naïve T cells into mature Th1 or Th2-like effector cells (7): viz., the local cytokine milieu; the presence of immunologically active hormones; the dose and route of antigen administration; the type of antigen-presenting cell stimulating T cells; and the “strength of signal” of the T-cell receptor for the MHC-antigen complex. The most important among these is the cytokine milieu surrounding the newly activated T cell. In the context of leprosy, reciprocal changes in cytokine expression in TT vs. LL along with complex cytokine regulatory networks have been evidenced at the site of infection (8). However, the important question is: which of these factors serve as the initial determinant of the polar immune responses to M. leprae? Given the extremely high relatedness of leprosy bacilli genomes worldwide (9), bacterial diversity is unlikely. This leaves differential host responses as the most likely mechanism.
Polarized T cell response (Th1/Th2 biased) to M. leprae is believed to be a critical element in the pathogenesis of leprosy and its varied clinical manifestations (8). The generation of Th1 effector cells chiefly producing the cytokine interferon-gamma (IFN-γ) vs. Th2 effector cells producing interluekin-4 (IL-4) have been held primarily responsible for the polarized state of immunity. During reversal reaction, lesional sites have demonstrated presence of CD4+ M. leprae-responsive T cells with a polarized type 1-like phenotype (10). However, the immune response manifested at the pathologic site(s) of leprosy is an extremely complex process, particularly in the light of recently evidenced remarkable heterogeneity of T cell subsets (11). Proportional enrichment of selective T cell subsets, particularly at the pathologic sites, determines the bulk T cells response (12). The major focus of the review is on the functionality of various relatively infrequent, yet significant lymphocyte subsets, which subsequently regulate the host’s cellular immune response and consequently disease pathogenesis. Numerous smaller subsets of lymphocytes have been identified in the past few decades which play critical roles in shaping the host immunity via their T cell response. These include natural killer T cells (NKT), regulatory T (Treg) cells, γδ T cells, and the very recently identified regulatory B cells. These cells have been demonstrated to exert regulatory influences on the generation of various effector T cells, such as Th1, Th17, and Th9-like cells (13–15).
The NKT Cells
First described in 1987 (16, 17), NKT cells are a unique subset of mature T cells co-expressing a semi-invariant Vα24Jα18 T cell antigen receptor (TCR)α chain and surface markers characteristic of NK cells. The semi-invariant TCR on iNKT cells recognizes glycolipids bound to monomorphic CD1d molecules. The most prominent and characteristic function of NKT cells is the early rapid production of immune-regulatory cytokines, such as IL-4, IFN-γ, and TNF-α upon their activation (18).
Earlier studies on the tissue origin and developmental pathway of iNKT cells from Taniguchi’s group (19) have suggested that iNKT cells develop extra-thymically, particularly in the liver. However, others have demonstrated that the majority of iNKT cells, like conventional T cells, are generated in the thymus (20, 21). The finding that not only peptides, but also glycolipids can serve as a source of antigen recognized by these NKT cells opened up new vistas in the study of antigen processing and presentation (22). The ability of nonpolymorphic CD1 molecules to present structurally diverse glycolipids to T cells has generated interest on these fascinating lipid–protein interactions. Since, NKT cells exercise a determining influence on a variety of immune responses in mice, ranging from autoimmunity to tumors and infections (23–25), significant interest has been generated to study their roles in human diseases as well.
Invariant iNKT cells (26, 27), which have a limited diversity of their TCR chains recognize glycolipid antigens from certain bacteria that are presented by CD1d, a nonpolymorphic antigen-presenting molecule (25). CD1-restricted T cells appear to play a major role in immune responses to mycobacteria. However, results of studies in mouse models are inconsistent. For example, although CD1d-deficient mice did not differ significantly in susceptibility to Mycobacterium tuberculosis (28), NKT cells predominate in the granulomatous reaction to M. tuberculosis cell wall preparations, and such granulomas do not form in NKT cell-deficient Jα2812/2 mice (29). Furthermore, NKT cells of normal mice respond to mycobacterial infection by decreasing IL-4 and increasing IFN-γ production (30), changes that aid the host response to mycobacteria, since IFN-γ plays a critical role in pathogen clearance.
Leprosy-specific studies on NKT cells (31) have shown mycobacterium-reactive double-negative T-cell lines derived from skin lesion of a leprosy patient responded to subcellular fractions of mycobacteria in the presence of CD1-expressing antigen-presenting cells (APCs). However, lipoarabinomannan-depleted soluble cell wall fraction did not induce detectable T-cell proliferation. Recognition of purified lipoarabinomannan from M. leprae was restricted by CD1b, and T cells lysed lipoarabinomannan-pulsed monocytes in a CD1b-restricted manner. Lipoarabinomannan also induced these T cells to secrete large amounts of IFN-γ. Upon examination of leprosy patients, they found few CD1+ cells in LL leprosy lesions. In contrast, there was a strong upregulation of CD1+ cells in the granulomatous lesions of patients with TT leprosy or reversal reaction (32). These cells were also CD83+, a marker for dendritic cells, indicating a strong correlation between CD1 expression and cell-mediated immunity in leprosy. Interestingly, administration of granulocyte-macrophage colony-stimulating factor, a cytokine which can promote dendritic-cell activation, to LL leprosy patients induced infiltration of CD1+ cells into the lesions (33, 34). NKT cells were also found in T-cell-reactive leprosy, but when compared with the granulomas in cutaneous sarcoidosis these cells were undetectable (35). They studied the TCR Vα repertoire and found that all patients with T-cell-reactive leprosy showed a very restricted T-cell-reactive Vα repertoire with a strong bias toward the use of the Vα6 and Vα14 segments. Unpublished data from our laboratory have given clear indications that NKT cell-derived cytokines control the ensuing effector T cell responses on activation with lipid antigens and further help in dictating the overall T cell response and manifestation of the disease. All of these studies strongly suggest that NKT cells play a determining role in regulating the varied type of immune responses as evidenced in leprosy affected individuals.
The Treg Cells
Regulatory T cells, on the other hand, are essential for maintaining peripheral tolerance, preventing autoimmune diseases, and limiting chronic inflammatory diseases (36). However, in case of chronic infections, they also limit such beneficial effect by suppressing the host immunity. During an infection, immune regulation is the result of the host’s response to the infection in a bid to maintain or restore a homeostatic environment and/or it can be actively induced by the pathogen to promote pathogen survival, like in the case of M. leprae (37). The presence of T cells with suppressive or anergic activity was discovered a long time back when they were known as suppressor T cells (38, 39). These same cells were shown to produce IL-10 and generated in vivo during infection (40). Recently, it has emerged that there are several specialized subsets of Treg cells, which contribute to the elaborate regulatory network within the infected host.
Based on their origin, generation, and mechanism of action, two main subsets of Treg cells have been identified: one is the naturally occurring CD4+CD25+ Treg cells (natural Treg cells), which mainly develop in the thymus and regulate self-reactive T cells in the periphery (41). Others are the inducible Treg cells, which develop in the periphery from conventional CD4+ T cells after exposure to signals, such as regulatory cytokines, immunosuppressive drugs, or APCs conditioned by microbial products (42). Both types of Treg cells, by virtue of their capacity to control the intensity of effector responses have been shown to have a major role in infection (12, 43). Treg cells mediate their suppressive capacity on inflammatory effector T cells, such as Th1, Th17, and Th9 cells both by contact dependent as well as contact-independent manner (36). From a functional perspective, Treg cells can be grouped into four basic “modes of action:” the various potential suppression mechanisms used by these include suppression by inhibitory cytokines, suppression by cytolysis, suppression by metabolic disruption, and suppression by modulation of dendritic cell maturation or function (44). Inhibitory cytokines, IL-10 and TGF-β, have been the focus of considerable attention as mediators of Treg cell induced suppression (45–47).
Differential trafficking of these Treg cells to the diseased sites are thought to be under the influence of tissue chemokine response elicited at the site of lepromatous lesions. This in turn is believed to determine the local immunity of BT/TT and BL/LL forms of leprosy. The tissue chemokine response at the lepromin DTH site and lesions of various forms of leprosy determines the recruitment of effector T cells at the lesional levels in leprosy patients (48). Therefore, subset composition of T cells infiltrating the pathologic/lesional site(s) of leprosy patients appears to be the key element in deciding the local immunity in leprosy, which may dictate the clinical manifestation of the disease. Some of these subsets have been demonstrated to be hierarchy in nature and known to exert significant influence on the effector T cells, and thus regulate the immune response at the pathologic site(s) of various chronic infectious diseases, including leprosy. These include the FoxP3 positive Treg cells as one of the most potent hierarchic cell type suppressing the effector T cell function with eventual regulation of immune response elicited by the host during intracellular infections, such as tuberculosis and leishmaniasis (49). Over representation of Treg cells either in peripheral compartment or more particularly at the pathologic site(s) has been shown to be of critical importance in determining the local immunity, thus dictating the outcome of the disease among patients suffering from various forms of tuberculosis (12). In leprosy as well, works have suggested that Tregs are present in increased numbers in LL patients, and they may have a pathogenic role in leprosy patients harboring uncontrolled bacillary multiplication (50). CD25+ Treg cells have also been shown to play a role in M. leprae-induced Th1 unresponsiveness in LL (51). FoxP3+ inducible Tregs producing the immunosuppressive cytokine TGF-β may also downregulate the T cell responses leading to antigen-specific anergy associated with LL (52).
Recent studies have revealed (53) that T2R or ENL patients have significantly lower number of circulating and in situ Tregs than T1R patients and controls with concomitant increase in pro-inflammatory cytokines such as TNF-α and IFN-γ produced by Th1 lymphocytes.
The Th17 Cells
Very recently, a third subset of T helper cells, Th17 cells, has been identified based on their cytokine production profile. These cells produce IL-17A (also referred to as IL-17), IL-17F, and IL-22, cytokines involved in neutrophilia, tissue remodeling and repair, and production of antimicrobial proteins. Th17 cells differentiate in response to the STAT3-activating cytokines IL-6, IL-21, and IL-23 along with TGF-β and IL-1β (54). They are abundant at mucosal interfaces, where they contain infection with pathogenic bacteria and fungi (55). Skin-homing T helper cells that produce IL-22, but not IL-17, have also been described in humans, and they may represent a new T cell subset with distinct effector functions (56). It is believed that the differentiation of CD4+ T cells that produce IL-17 and IL-22 is influenced by the composition of the intestinal microbiota and by the presence of innate immune cells mainly the neutrophils that amplify the Th17 cell response.
For long, Th1 cells were considered to be the major effectors in multiple autoimmune diseases, while Th2 cells were involved in atopy and asthma. In recent times, however, Th17 cells have been implicated as culprits in a plethora of autoimmune and other inflammatory diseases in mice and humans. Many diseases that were previously associated with Th1 cells, e.g., experimental autoimmune encephalomyelitis (EAE, a model for multiple sclerosis), collagen-induced arthritis, and some forms of colitis, were shown to be caused by IL-23-dependent Th17 cells or other IL-17-producing lymphoid cell types (57–59). Conversely, defects in the Th17 cell differentiation axis may predispose the host to bacterial and fungal infections at mucosal surfaces (60). Th17 cells mediate their pro-inflammatory function by (i) recruiting neutrophils, (ii) activating macrophages, and (iii) enhancing Th1 effector cells (54). Much of the inflammatory damage previously ascribed to type 1 response is now thought to depend on IL-17 and IL-23 (the cytokine responsible for supporting Th17 response in vivo) (58).
CD4+ Th17 cells have been recently identified in borderline cases of leprosy (61), which highlighted their importance in infectious diseases as well. A persistent and very relevant concept is that an imbalance between Th17 and Treg cell function may be critical in the immunopathogenesis of many disease states (62). This concept is highlighted in a leprosy-specific study, where IL-10+ produced by Treg cells in BL/LL patients correlates significantly with polarized immunity highlighted by lesser IL-17 by CD4+ T cells in the same group. Blocking of IL-10/TGF-β resulted in the reversal of effector immune response (IL-17) in BL/LL with higher frequency of Th17 cells (63). This indicates that by negating the influence of suppressive cytokines we can successfully gain back immune responsiveness. The presence of Th17 cytokines (IL-6, IL-17, and IL-23) in vitro results in reduction of FoxP3 expression on Tregs simultaneously, possibly leading to increase in IL-17-producing CD4+ cells in BL/LL (63). This further suggests that the generation of antigen-specific Treg cells is very much dependent on the environment of cytokines they are exposed to. Hence, these cells may be targeted for reversal of effector response in BL/LL patients proving to be an important mode of immune modulation in the immunocompromised hosts to revive the immune response.
An imbalance in Treg and Th17 populations has also been observed in patients with leprosy reactions (53, 64). Studies done in biopsies from T2R patients showed a decrease in Tregs and associated cytokines, TGF-β and increase in cells producing IL-6, IL-21, and IL-17. On the other hand, T1R patients are showing the opposite trend with increased Tregs and reduced IL-17+ cells. This increase in inflammatory cytokines along with downregulation of Tregs may be responsible for the lesional inflammation characterizing T2R reactions.
The Programmed Death-1(PD)-1-Programmed Death Ligand-1 (PD-L1) Pathway
T cell responses during parasitic infections are tightly controlled by co-stimulatory or co-inhibitory molecules. It is well known that interactions between PD-1 and its ligand, PD-L1 can inhibit the effector functions, such as proliferation, cytokine production, and survival of the T cells, thus balancing the tolerance, autoimmunity, infection, and immunopathology (65, 66). On infection with M. tuberculosis, protective T cells are generated in the infected host. However, T-cell-mediated immunity does not easily eradicate these bacteria because they have evolved effective strategies to overcome the host defense mechanisms (67). Studies have identified various virulence-associated genes and intracellular survival mechanisms of mycobacteria (68). The PD-1 signaling pathway is activated during persistent infection with various microorganisms and contributes to the impairment of protective immunity (69–71). A recent study showed that in vitro blockade of PD-1 signaling with the specific antibody enhanced IFN-γ production by T cells of TB patients on stimulation with M. tuberculosis antigen (72). In pulmonary TB patients, inhibiting this signaling pathway rescues M. tuberculosis-specific IFN-γ producing T cells from apoptosis (73). Similarly, persisting infection with pathogens like Helicobacter pylori and Porphyromonas gingivalis, showed elevated expression of PD-L1 on gastric epithelial cells and monocytes, suggesting a potential involvement of PD-L1 in promoting chronic infections (74).
Leprosy-specific studies show reduced expression of the positive signaling co-stimulatory molecules, CD28 and CD86 on T-cells, consistent with the LL anergy, in contrast to TT patients which displayed increased expression of the negative signaling molecules CD152 and PD-1 (75). This may represent a probable means of modulating an exacerbated immune response and avoiding immunopathology. However, another recent study in leprosy reveals elevated surface expression of PD-1 on T cells, NKT, and Treg cells and its ligand PD-L1 on APCs, such as monocytes and B cells, in BL/LL as compared to BT/TT leprosy patients (63). The authors have demonstrated that the PD-1/PD-L1 pathway preferentially suppress IFN-γ against TNF-α in BL/LL which is touted as the designate cytokine for generating protective immune response in the immunosuppressed host. This may also be one of the contact-dependent mechanisms utilized by Treg cells for immune suppression of effector T cells. These findings raise the possibility that the antigen-specific T-cell response is impaired by several inhibitory mechanism(s), thereby allowing mycobacterial persistence.
Conclusion
However, it needs to be emphasized that no single mechanism of suppression can account for the kind of M. leprae-specific T cell anergy evidenced in LL. The diversity of effector mechanisms characteristic of NKT affords versatility capable of restraining diverse types of inflammatory responses in different tissues. Likewise, both Tregs and Th17 cells can exert beneficial as well as pathogenic effects depending on the physiology of the infected host. Other cell subsets, such as Th9 (76) or γδ T cells, have also been identified in leprosy patients, but their exact roles have not been defined till date. The intricate mechanisms governing differentiation and functions of these pro- and anti-inflammatory cells are yet to be discerned and pose major challenges ahead. Therefore, in conclusion, we can state that as seen in other chronic granulomatous diseases, NKT and Treg cells along with Th17 and the PD-1-PD-L1 pathway play crucial roles in the outcome of the host–parasite interactions in leprosy (Figure 2). Providing a balanced level of function for these cell subsets is the key to achieving an appropriate level of parasite control without inducing immunopathology. This would be a major goal in the management of this still-challenging infectious disease.
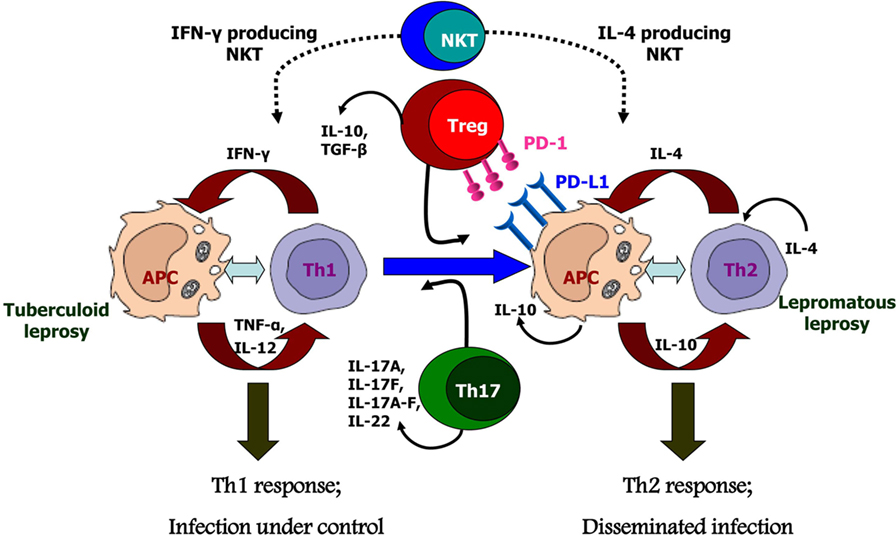
Figure 2. Possible causes for polarized host immunity in tuberculoid type (TT) vs. lepromatous leprosy questioning the well-established Th1–Th2 paradigm. Natural killer T cells which are initial responders producing either Th1 cytokines like IFN-γ or Th2 cytokines like interluekin-4 depending on the basal cytokine response of the host. Tregs cells which are predominantly suppressive in nature and produce cytokines IL-10 and TGF-β, along with increased expression of programmed death-1 and its ligand, programmed death ligand-1 on antigen-presenting cells; these cells are found in significant numbers in lepromatous leprosy patients. Th17 or T helper 17 cells which produce the cytokines IL-17 and IL-22 demonstrate inflammatory phenotype and are, therefore, found in increased numbers in TT leprosy patients.
Author Contributions
Author has planned and structured the review article.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
The authors are grateful to the funding agency, Indian Council of Medical Research, Delhi and to the All India Institute of Medical Sciences, Delhi for supporting this work.
References
1. Scollard DM, Adams LB, Gillis TP, Krahenbuhl JL, Truman RW, Williams DL. The continuing challenges of leprosy. Clin Microbiol Rev (2006) 19:338–81. doi:10.1128/CMR.19.2.338-381.2006
3. WHO. Global leprosy update, 2015: time for action, accountability and inclusion. Wkly Epidemiol Rec (2016) 91:405–20.
4. Ridley DS, Jopling WH. Classification of leprosy according to immunity. A five-group system. Int J Lepr Other Mycobact Dis (1966) 34:255–73.
5. Ridley DS. Histological classification and the immunological spectrum of leprosy. Bull World Health Organ (1974) 51:451–65.
6. Modlin RL. Th1-Th2 paradigm: insights from leprosy. J Invest Dermatol (1994) 102(6):828–32. doi:10.1111/1523-1747.ep12381958
7. Constant SL, Bottomly K. Induction of Th1 and Th2 CD4+ T cell responses: the alternative approaches. Annu Rev Immunol (1997) 15:297–322. doi:10.1146/annurev.immunol.15.1.297
8. Mitra DK, De Rosa SC, Luke A, Balamurugan A, Khaitan BK, Tung J, et al. Differential representations of memory T cell subsets are characteristic of polarized immunity in leprosy and atopic diseases. Int Immunol (1999) 11(11):1801–10. doi:10.1093/intimm/11.11.1801
9. Monot M, Honoré N, Garnier T, Zidane N, Sherafi D, Paniz-Mondolfi A, et al. Comparative genomic and phylogeographic analysis of Mycobacterium leprae. Nat Genet (2009) 41:1282–9. doi:10.1038/ng.477
10. Verhagen CE, Wierenga EA, Buffing AA, Chand MA, Faber WR, Das PK. Reversal reaction in borderline leprosy is associated with a polarized shift to type 1-like Mycobacterium leprae T cell reactivity in lesional skin: a follow-up study. J Immunol (1997) 159(9):4474–83.
11. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature (2006) 441:235–8. doi:10.1038/nature04753
12. Sharma PK, Saha PK, Singh A, Sharma SK, Ghosh B, Mitra DK. FoxP3+ regulatory T cells suppress effector T-cell function at pathologic site in miliary tuberculosis. Am J Respir Crit Care Med (2009) 179(11):1061–70. doi:10.1164/rccm.200804-529OC
13. Littman DR, Rudensky AY. Th17 and regulatory T cells in mediating and restraining inflammation. Cell (2010) 140:845–58. doi:10.1016/j.cell.2010.02.021
14. Nowak EC, Weaver CT, Turner H, Begum-Haque S, Becher B, Schreiner B, et al. IL-9 as a mediator of Th17-driven inflammatory disease. J Exp Med (2009) 206:1653–60. doi:10.1084/jem.20090246
15. Yang M, Rui K, Wang S, Lu L. Regulatory B cells in autoimmune diseases. Cell Mol Immunol (2013) 10(2):122–32. doi:10.1038/cmi.2012.60
16. Budd RC, Miescher GC, Howe RC, Lees RK, Bron C, MacDonald HR. Developmentally regulated expression of T cell receptor beta chain variable domains in immature thymocytes. J Exp Med (1987) 166(2):577–82. doi:10.1084/jem.166.2.577
17. Fowlkes BJ, Kruisbeek AM, Ton-That H, Weston MA, Coligan JE, Schwartz RH, et al. A novel population of T-cell receptor alpha beta-bearing thymocytes which predominantly expresses a single V beta gene family. Nature (1987) 329(6136):251–4. doi:10.1038/329251a0
18. Kawano T, Cui J, Koezuka Y, Toura I, Kaneko Y, Motoki K, et al. CD1d restricted and TCR-mediated activation of Vα14 NKT cells by glycosylceramides. Science (1997) 278:1626. doi:10.1126/science.278.5343.1626
19. Makino Y, Yamagata N, Sasho T, Adachi Y, Kanno R, Koseki H, et al. Extrathymic development of V alpha 14-positive T cells. J Exp Med (1993) 177(5):1399–408. doi:10.1084/jem.177.5.1399
20. Coles MC, Raulet DH. NK1.1+ T cells in the liver arise in the thymus and are selected by interactions with class I molecules on CD4+CD8+ cells. J Immunol (2000) 164:2412–8. doi:10.4049/jimmunol.164.5.2412
21. Tilloy F, Di Santo JP, Bendelac A, Lantz O. Thymic dependence of invariant Vα14+ natural killer-T cell development. Eur J Immunol (1999) 29:3313–8. doi:10.1002/(SICI)1521-4141(199910)29:10<3313::AID-IMMU3313>3.0.CO;2-8
22. Brigl M, Brenner MB. CD1: antigen presentation and T cell function. Annu Rev Immunol (2004) 22:817–90. doi:10.1146/annurev.immunol.22.012703.104608
23. Kronenberg M, Gapin L. The unconventional lifestyle of NKT cells. Nat Rev Immunol (2002) 2:557–68. doi:10.1038/nri854
24. Kronenberg M. Toward an understanding of NKT cell biology: progress and paradoxes. Annu Rev Immunol (2005) 23:877–900. doi:10.1146/annurev.immunol.23.021704.115742
25. Kinjo Y, Kronenberg M. V alpha14 iNKT cells are innate lymphocytes that participate in the immune response to diverse microbes. J Clin Immunol (2005) 25(6):522–33. doi:10.1007/s10875-005-8064-5
26. Yu KO, Porcelli SA. The diverse functions of CD1d-restricted NKT cells and their potential for immunotherapy. Immunol Lett (2005) 100:42–55. doi:10.1016/j.imlet.2005.06.010
27. Godfrey DI, MacDonald HR, Kronenberg M, Smyth MJ, Van Kaer L. NKT cells: what’s in a name? Nat Rev Immunol (2004) 4(3):231–7. doi:10.1038/nri1309
28. Behar SM, Dascher CC, Grusby MJ, Wang CR, Brenner MB. Susceptibility of mice deficient in CD1D or TAP1 to infection with Mycobacterium tuberculosis. J Exp Med (1999) 189(12):1973–80. doi:10.1084/jem.189.12.1973
29. Apostolou I, Takahama Y, Belmant C, Kawano T, Huerre M, Marchal G, et al. Murine natural killer T (NKT) cells contribute to the granulomatous reaction caused by mycobacterial cell walls. Proc Natl Acad Sci U S A (1999) 96:5141–6. doi:10.1073/pnas.96.9.5141
30. Emoto M, Emoto Y, Buchwalow IB, Kaufmann SHE. Induction of IFN-γ producing CD4+ natural killer T cells by Mycobacterium bovis bacillus Calmette-Guerin. Eur J Immunol (1999) 29:650–9. doi:10.1002/(SICI)1521-4141(199902)29:02<650::AID-IMMU650>3.0.CO;2-M
31. Sieling PA, Chatterjee D, Porcelli SA, Prigozy TI, Mazzaccaro RJ, Soriano T, et al. CD1-restricted T cell recognition of microbial lipoglycan antigens. Science (1995) 269:227–30. doi:10.1126/science.7542404
32. Rosat JP, Grant EP, Beckman EM, Dascher CC, Sieling PA, Frederique D, et al. CD1-restricted microbial lipid antigen-specific recognition found in the CD8+ alpha beta T cell pool. J Immunol (1999) 162(1):366–71.
33. Sieling PA, Jullien D, Dahlem M, Tedder TF, Rea TH, Modlin RL, et al. CD1 expression by dendritic cells in human leprosy lesions: correlation with effective host immunity. J Immunol (1999) 162:1851–8.
34. Sampaio EP, Moreira AL, Sarno EN, Malta AM, Kaplan G. Prolonged treatment with recombinant interferon gamma induces erythema nodosum leprosum in lepromatous leprosy patients. J Exp Med (1992) 175:1729–37. doi:10.1084/jem.175.6.1729
35. Mempel M, Flageul B, Suarez F, Ronet C, Dubertret L, Kourilsky P, et al. Comparison of the T cell patterns in leprous and cutaneous sarcoid granulomas. Presence of Valpha24-invariant natural killer T cells in T cell-reactive leprosy together with a highly biased T cell receptor Valpha repertoire. Am J Pathol (2000) 157:509–23. doi:10.1016/S0002-9440(10)64562-2
36. Sakaguchi S, Sakaguchi N, Shimizu J, Yamazaki S, Sakihama T, Itoh M, et al. Immunologic tolerance maintained by CD25+ CD4+ regulatory T cells: their common role in controlling autoimmunity, tumor immunity, and transplantation tolerance. Immunol Rev (2001) 182:18–32. doi:10.1034/j.1600-065X.2001.1820102.x
37. Shevach EM, DiPaolo RA, Andersson J, Zhao DM, Stephens GL, Thornton AM. The lifestyle of naturally occurring CD4+ CD25+ Foxp3+ regulatory T cells. Immunol Rev (2006) 212:60–73. doi:10.1111/j.0105-2896.2006.00415.x
39. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor α-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol (1995) 155:1151–64.
40. Mahanty S, Mollis SN, Ravichandran M, Abrams JS, Kumaraswami V, Jayaraman K, et al. High levels of spontaneous and parasite antigen-driven interleukin-10 production are associated with antigen-specific hyporesponsiveness in human lymphatic filariasis. J Infect Dis (1996) 173:769–73. doi:10.1093/infdis/173.3.769
41. Belkaid Y, Rouse BT. Natural regulatory T cells in infectious disease. Nat Immunol (2005) 6(4):353–60. doi:10.1038/ni1181
42. O’Garra A, Vieira PL, Vieira P, Goldfeld AE. IL-10-producing and naturally occurring CD4+ Tregs: limiting collateral damage. J Clin Invest (2004) 114:1372–8. doi:10.1172/JCI23215
43. Belkaid Y. Regulatory T cells and infection: dangerous necessity. Nat Rev Immunol (2007) 7:875–88. doi:10.1038/nri2189
44. Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol (2008) 8(7):523–32. doi:10.1038/nri2343
45. Hawrylowicz CM, O’Garra A. Potential role of interleukin-10-secreting regulatory T cells in allergy and asthma. Nat Rev Immunol (2005) 5:271–83. doi:10.1038/nri1589
46. Annacker O, Asseman C, Read S, Powrie F. Interleukin-10 in the regulation of T cell-induced colitis. J Autoimmun (2003) 20:277–9. doi:10.1016/S0896-8411(03)00045-3
47. Fahlén L, Read S, Gorelik L, Hurst SD, Coffman RL, Flavell RA, et al. T cells that cannot respond to TGF-β escape control by CD4+CD25+ regulatory T cells. J Exp Med (2005) 201:737–46. doi:10.1084/jem.20040685
48. Mitra DK, Joshi B, Dinda AK, Rai AK, Girdhar BK, Katoch K, et al. Induction of lepromin reactivity in cured lepromatous leprosy patients: impaired chemokine response dissociates protective immunity from delayed type hypersensitivity. Microbes Infect (2009) 11(14–15):1122–30. doi:10.1016/j.micinf.2009.08.006
49. Mendez S, Reckling SK, Piccirillo CA, Sacks D, Belkaid Y. Role for CD4(+) CD25(+) regulatory T cells in reactivation of persistent leishmaniasis and control of concomitant immunity. J Exp Med (2004) 200(2):201–10. doi:10.1084/jem.20040298
50. Palermo ML, Pagliari C, Trindade MA, Yamashitafuji TM, Duarte AJ, Cacere CR, et al. Increased expression of regulatory T cells and down-regulatory molecules in lepromatous leprosy. Am J Trop Med Hyg (2012) 86:878–83. doi:10.4269/ajtmh.2012.12-0088
51. Bobosha K, Wilson L, van Meijgaarden KE, Bekele Y, Zewdie M, Jolien J, et al. T-cell regulation in lepromatous leprosy. PLoS Negl Trop Dis (2014) 8(4):e2773. doi:10.1371/journal.pntd.0002773
52. Saini C, Ramesh V, Nath I. Increase in TGF-β secreting CD4+CD25+ FOXP3+ T regulatory cells in anergic lepromatous leprosy patients. PLoS Negl Trop Dis (2014) 8(1):e2639. doi:10.1371/journal.pntd.0002639
53. Vieira AP, Trindade MAB, Pagliari C, Avancini J, Sakai-Valente NY, Benard G, et al. Development of type 2, but not type 1, leprosy reactions is associated with a severe reduction of circulating and in situ regulatory T-cells. Am J Trop Med Hyg (2016) 94(4):721–7. doi:10.4269/ajtmh.15-0673
54. Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 cells. Annu Rev Immunol (2009) 27:485–517. doi:10.1146/annurev.immunol.021908.132710
55. Weaver CT, Hatton RD, Mangan PR, Harrington LE. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu Rev Immunol (2007) 25:821–52. doi:10.1146/annurev.immunol.25.022106.141557
56. Duhen T, Geiger R, Jarrossay D, Lanzavecchia A, Sallusto F. Production of interleukin 22 but not interleukin 17 by a subset of human skin-homing memory T cells. Nat Immunol (2009) 10:857–63. doi:10.1038/ni.1767
57. Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature (2003) 421(6924):744–8. doi:10.1038/nature01355
58. Ahern PP, Izcue A, Maloy KJ, Powrie F. The interleukin-23 axis in intestinal inflammation. Immunol Rev (2008) 226:147–59. doi:10.1111/j.1600-065X.2008.00705.x
59. McKenzie BS, Kastelein RA, Cua DJ. Understanding the IL-23-IL-17 immune pathway. Trends Immunol (2006) 27:17–23. doi:10.1016/j.it.2005.10.003
60. Ouyang W, Kolls JK, Zheng Y. The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity (2008) 28(4):454–67. doi:10.1016/j.immuni.2008.03.004
61. Saini C, Ramesh V, Nath I. CD4+ Th17 cells discriminate clinical types and constitute a third subset of non Th1, Non Th2 T cells in human leprosy. PLoS Negl Trop Dis (2013) 7(7):e2338. doi:10.1371/journal.pntd.0002338
62. Eisenstein EM, Williams CB. The Treg/Th17 cell balance: a new paradigm for autoimmunity. Pediatr Res (2009) 65(5 Pt 2):26R–31R. doi:10.1203/PDR.0b013e31819e76c7
63. Sadhu S, Khaitan BK, Joshi B, Sengupta U, Nautiyal AK, Mitra DK. Reciprocity between regulatory T cells and Th17 cells: relevance to polarized immunity in leprosy. PLoS Negl Trop Dis (2016) 10(1):e0004338. doi:10.1371/journal.pntd.0004338
64. Saini C, Siddiqui A, Ramesh V, Nath I. Leprosy reactions show increased Th17 cell activity and reduced FOXP3+ Tregs with concomitant decrease in TGF-β and increase in IL-6. PLoS Negl Trop Dis (2016) 10(4):e0004592. doi:10.1371/journal.pntd.0004592
65. Dong HD, Zhu GF, Tamada K, Chen LP. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med (1999) 5:1365–9. doi:10.1038/70932
66. Keir ME, Freeman GJ, Sharpe AH. PD-1 regulates self-reactive CD8+ T cell responses to antigen in lymph nodes and tissues. J Immunol (2007) 179(8):5064–70. doi:10.4049/jimmunol.179.8.5064
67. Rohde K, Yates RM, Purdy GE, Russell DG. Mycobacterium tuberculosis and the environment within the phagosome. Immunol Rev (2007) 219:37–54. doi:10.1111/j.1600-065X.2007.00547.x
68. Pieters J. Mycobacterium tuberculosis and the macrophage: maintaining a balance. Cell Host Microbe (2008) 3(6):399–407. doi:10.1016/j.chom.2008.05.006
69. Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature (2006) 439:682–7. doi:10.1038/nature04444
70. Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature (2006) 443:350–4. doi:10.1038/nature05115
71. Urbani S, Amadei B, Tola D, Massari M, Schivazappa S, Missale G, et al. PD-1 expression in acute hepatitis C virus (HCV) infection is associated with HCV-specific CD8 exhaustion. J Virol (2006) 80(22):11398–403. doi:10.1128/JVI.01177-06
72. Jurado JO, Alvarez IB, Pasquinelli V, Martínez GJ, Quiroga MF, Abbate E, et al. Programmed death (PD)-1: PD-ligand 1/PD-ligand 2 pathway inhibits T cell effector functions during human tuberculosis. J Immunol (2008) 181(1):116–25. doi:10.4049/jimmunol.181.1.116
73. Singh A, Mohan A, Dey AB, Mitra DK. Inhibiting PD-1 pathway rescues M. tuberculosis specific IFN-γ producing T cells from apoptosis among pulmonary tuberculosis patients. J Infect Dis (2013) 208:603–15. doi:10.1093/infdis/jit206
74. Das S, Suarez G, Beswick EJ, Sierra JC, Graham DY, Reyes VE. Expression of B7-H1 on gastric epithelial cells: its potential role in regulating T cells during Helicobacter pylori infection. J Immunol (2006) 176:3000–9. doi:10.4049/jimmunol.176.5.3000
75. Palermo Mde L, Trindade MÂ, Duarte AJ, Cacere CR, Benard G. Differential expression of the costimulatory molecules CD86, CD28, CD152 and PD-1 correlates with the host-parasite outcome in leprosy. Mem Inst Oswaldo Cruz (2012) 107(Suppl 1):167–73. doi:10.1590/S0074-02762012000900024
Keywords: polarized immunity, natural killer T cells, regulatory T cells, Th 17, programmed death-1-programmed death ligand-1
Citation: Sadhu S and Mitra DK (2018) Emerging Concepts of Adaptive Immunity in Leprosy. Front. Immunol. 9:604. doi: 10.3389/fimmu.2018.00604
Received: 22 September 2017; Accepted: 09 March 2018;
Published: 09 April 2018
Edited by:
Pranab Kumar Das, University of Birmingham, United KingdomReviewed by:
Roberta Olmo Pinheiro, Fundação Oswaldo Cruz (Fiocruz), BrazilMirian Nacagami Sotto, Universidade de São Paulo, Brazil
Copyright: © 2018 Sadhu and Mitra. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dipendra Kumar Mitra, c2FsaWxtaXRyYTJAZ21haWwuY29t