 Annika Reinhardt
Annika Reinhardt Immo Prinz
Immo Prinz- Institute of Immunology, Hannover Medical School, Hannover, Germany
γδ T cells, αβ T cells, and innate lymphoid cells (ILCs) are capable of producing interleukin (IL)-17A, IL-17F, and IL-22. Among these three families of lymphocytes, it is emerging that γδ T cells are, at least in rodents, the main source of these key pro-inflammatory cytokines. γδ T cells were implicated in multiple inflammatory and autoimmune diseases, including psoriasis, experimental autoimmune encephalomyelitis and uveitis, colitis, and rheumatoid arthritis. Recent findings pointed toward a central role of γδ T cells in the pathogenesis of spondyloarthritis (SpA), a group of inflammatory rheumatic diseases affecting the axial skeleton. SpA primarily manifests as inflammation and new bone formation at the entheses, which are connecting tendons or ligaments with bone. In SpA patients, joint inflammation is frequently accompanied by extra-articular manifestations, such as inflammatory bowel disease or psoriasis. In humans, genome-wide association studies could link the IL-23/IL-17 cytokine axis to SpA. Accordingly, antibodies targeting IL-23/IL-17 for SpA treatment already showed promising results in clinical studies. However, the contribution of IL-17-producing γδ T cells to SpA pathogenesis is certainly not an open-and-shut case. Indeed, the cell types that are chiefly involved in local inflammation in human SpA still remain largely unclear. Some studies focusing on blood or synovium from SpA patients reported augmented IL-17-producing and IL-23 receptor-expressing γδ T cells, but other cell types might contribute as well. Here, we summarize the current understanding of how γδ T cells, αβ T cells, and ILCs contribute to the pathogenesis of human and experimental SpA.
Introduction
Spondyloarthritis (SpA) encompasses a group of human rheumatic diseases that typically manifest as inflammation and new bone formation at axial joints, leading to severe lower back pain and impaired spinal mobility. Thereby, inflammation starts from entheses, the tendon to bone attachment sites. The family of SpA includes ankylosing spondylitis (AS), the SpA prototype (1), reactive arthritis, axial and undifferentiated SpA as well as psoriatic arthritis (PsA) and inflammatory bowel disease (IBD)-associated arthritis. Different SpA pathologies demonstrate similar disease patterns and similar genetic associations. First, the MHC class I molecule HLA-B27 was identified to confer susceptibility to SpA, and HLA-B27 is present in approximately 90% of AS patients in Europe (2). Over the last few years, more and more genome-wide association studies revealed a link between the interleukin (IL)-23/IL-17 axis and SpA susceptibility (3–6). Newly identified susceptibility genes comprise IL-12B, IL-1R, CARD9, TYK2, STAT3, and IL-23R, the gene encoding for the IL-23 receptor (IL-23R). The latter is particularly interesting, because single nucleotide polymorphisms in IL-23R were associated not only with AS (7) or PsA (8) but also with psoriasis (9) and IBD (10), hence pathologies that frequently accompany articular inflammation in SpA.
Nonsteroidal anti-inflammatory drugs and TNF inhibitors serve as first-line treatment for SpA. However, new treatment strategies emerged with the identification of the IL-23/IL-17 axis as putative key pathway associated with SpA. Most prominently, anti-IL-17A (receptor) treatment improved SpA disease symptoms (11–17). By contrast, IL-23 inhibition presented ambiguous results (18–21) (ClinicalTrials.gov number NCT02437162). If these drugs should completely replace old treatment modalities in the future, it still needs to be validated further (22–24).
Enthesitis (25), thus entheseal inflammation, represents a main characteristic of SpA. It was suggested that mechanical stress and local microdamage might initiate entheseal inflammation (26, 27), proposing the enthesis as primary lesion in SpA-associated joint inflammation (28–30). However, the link between host genetics, e.g., the IL-23/IL-17 axis, and local inflammation as well as new bone formation is not entirely clear. Strikingly, several SpA-focused studies suggested that the IL-23/IL-17 cytokine axis and innate immune activation might be of greater importance than classical autoreactivity of B or T cell receptors (6, 31, 32). Indeed, several albeit not all SpA patients demonstrated an increase in IL-23/IL-17 serum or synovial fluid levels (33–37). IL-17 cytokines are usually produced by lymphocytes, although earlier studies observed IL-17-producing mast cells (38), neutrophils, and myeloperoxidase-expressing cells (39) in SpA synovia. So, who does it? In the following, we summarize and discuss current data about human and experimental SpA and the three prime suspects of the IL-23/IL-17 axis: γδ T cells, αβ T cells, and innate lymphoid cells (ILCs).
γδ T Cells
Although pre-committed effector γδ T cells represent a major source of IL-17/IL-22 under steady-state conditions in rodents (40–42), data reporting IL-17/IL-22-producing γδ T cells in healthy human individuals are rare (42–44). However, γδ T cells are clearly associated with different infections and tumors as well as autoinflammatory and autoimmune diseases in humans (45, 46). First studies suggesting a possible connection between γδ T cells and SpA were published approximately 30 years ago, just shortly after the discovery of γδ T cells (47, 48). By now, a number of studies demonstrated a decrease of γδ T cells in blood (49–51), while others showed that γδ T cells were frequently present in SpA patients’ synovial fluid (52, 53), suggesting that γδ T cells might play a role in disease induction and/or persistence in humans.
In fact, a direct association of γδ T cells and IL-17/IL-22 secretion in human SpA was first described by Kenna and colleagues, demonstrating an enrichment of IL-23R+ IL-17-producing γδ T cells in blood of AS patients (54). Strikingly, this phenotype was absent in rheumatoid arthritis patients (54), suggesting specific involvement of IL-17-producing γδ T cells in SpA pathogenesis rather than in arthritic inflammation in general. Along the same line, the analysis of tissue samples from enthesitis-related arthritis (55), reactive arthritis or undifferentiated SpA (56) as well as juvenile idiopathic arthritis (JIA) patients (57) revealed an increase in blood and synovial fluid IL-17-producing γδ T cells. Notably, such increased numbers of IL-17-producing γδ T cells might be driven by a defined arthritic cytokine environment (57). Although IL-23 certainly represents the main driver cytokine inducing enhanced IL-17 secretion by different cell types, also IL-9-driven expansion of IL-17-producing γδ T cells in PsA synovial fluid was recently demonstrated (58).
γδ T cells were implicated not only in SpA and related diseases in humans but also in mice. In various mouse models for non-autoimmune arthritis, including non-autoimmune antigen-induced arthritis (59), mannan-induced arthritis (60), or CFA-injected IFN-γ−/− mice (61), γδ T cells were increased in numbers and were the main source of pathogenic IL-17 in inflamed tissues.
So, how do SpA-associated IL-17-producing γδ T cells get into inflamed sites in humans and mice? It is tempting to speculate that circulating and/or γδ T cells from distant tissues might leave their sites of origin and gather at the sites of crime, the inflamed joints. Accordingly, blood isopentenyl pyrophosphate-responsive Vγ9+ (53) or α4β7+ mucosal (γδ) T cells (52) might preferentially accumulate in JIA joints during an acute flare or a low acute-phase response, respectively. Likewise, CCR2+Vγ6+ IL-17-producing γδ T cells were recruited to joints by CCL2-inducing CD4+ T cells in arthritic Il-1rn−/− mice (62).
However, there is more to be considered than migration of γδ T cells into inflamed tissues when trying to solve the case of idiopathic local entheseal inflammation in SpA. Applying an IL-23-dependent mouse model resembling inflammation-driven bone destruction (63) and most features of human SpA (64), tissue-resident IL-23R+RORγt+CD3+CD4−CD8− lymphocytes were discovered in mouse entheses (64). Systemic IL-23 overexpression induced local inflammation in the enthesis by triggering resident IL-23R+RORγt+CD3+CD4−CD8− lymphocytes to secrete IL-17 and IL-22, ultimately leading to IL-17-dependent enthesitis, IL-22-dependent bone remodeling as well as aortic root inflammation and psoriasis (64). Based on this study, we could recently demonstrate that Vγ6+ γδ T cells reside within mouse entheses, where they constitute the large majority of IL-23R+RORγt+CD3+CD4−CD8− lymphocytes in steady state and increase in numbers during inflammation (65). Whether Tcrd−/− mice would thus be protected from IL-23-dependent entheseal inflammation is still a matter of investigation. Notably, aging male DBA mice still develop severe enthesitis and new bone formation in the absence of γδ T cells (66). However, whether this phenotype results from entheseal γδ T cell redundancy or an increased presence of enthesis-resident lymphocytes other than γδ T cells that functionally refills their empty niche (67) still remains an open question.
Strikingly, resident γδ T cells have just recently also been identified in human entheses (68). Thus, it appears likely that under steady-state conditions, entheseal γδ T cells reside in this very specific anatomical niche to control tissue homeostasis and possibly physiological bone remodeling after injury and exercise, while upon the elevation of IL-23 serum levels, they are driven to increase IL-17/IL-22 production and thus promote SpA (Figure 1).
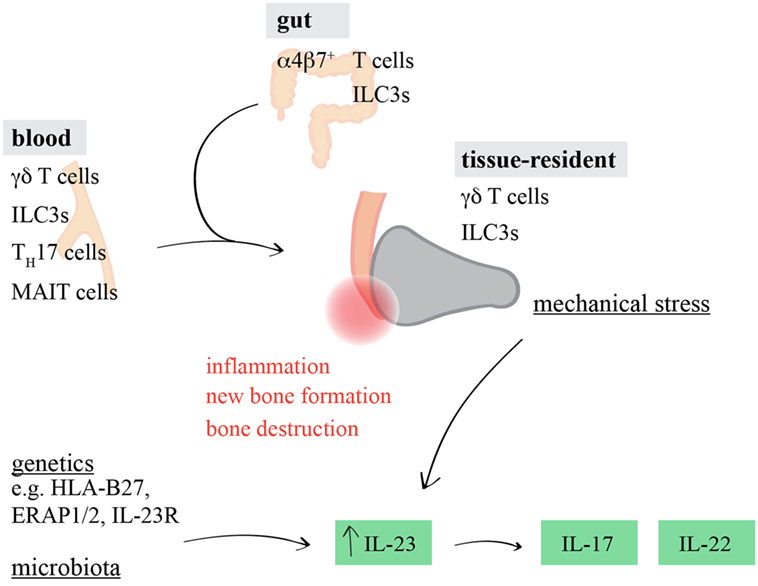
Figure 1. Involvement of interleukin (IL)-17/IL-22-producing lymphocytes in spondyloarthritis (SpA)-associated inflammation. Genetic and epigenetic predisposition, altered microbial composition, and entheseal microdamage can influence the induction and progression of tissue inflammation in SpA. In addition to tissue-resident γδ T cells and ILC3s, circulating and/or gut-derived γδ T cells, ILC3s, TH17 cells, or mucosal-associated invariant T (MAIT) cells might promote IL-23-driven joint inflammation by producing increased amounts of IL-17 and IL-22.
Innate Lymphoid cells
Innate lymphoid cells were identified around 10 years ago (69), but might have played important roles in joint inflammation even before adaptive immune cells developed over 450 million years ago. By now, it is clear that ILCs are crucially involved in the pathogenesis of a variety of inflammatory diseases, but also in tissue homeostasis (70, 71). For example, different ILC subsets were implicated in human and experimental rheumatic diseases (72). Recently, elevated numbers of ILC1s, ILC2s, and ILC3s were measured in blood samples from PsA patients, while only ILC3 numbers positively correlated with disease activity (73). However, circulating ILC3s in PsA patients displayed an immature phenotype, only produced moderate amounts of IL-17/IL-22, and did not express NKp44 (73). Interestingly, this further implies that final ILC3 maturation might only occur directly at the site of inflammation, presumably directly in the joints. However, as ILCs possess a high degree of plasticity (74), the transformation of inflammatory ILC1s or ILC2s into ILC3-like cells might also be possible (75, 76). In addition to an enrichment of NKp44− ILC3s in PsA blood, NKp44+CCR6+ IL-17- (77) or GM-CSF-producing (78) ILC3s were abundantly present in SpA synovial fluid. Here, circulating NKp44+ILC3 numbers inversely correlated with disease activity (77), possibly resulting from the migration of circulating (immature) ILC3s to target tissues.
Association between SpA and intestinal inflammation is well established (79), and at least 50% of SpA patients suffer from (sub)clinical gut inflammation (80). A connection between ILCs and SpA-associated gut inflammation was described by different studies. In blood from entheropathic SpA patients, levels of IL-17-producing ILC3s were significantly higher as compared to IBD patients and healthy controls (81). While intestinal CD4+ T cells represented the main source of local IL-22 in Crohn’s disease patients, IL-22-producing NKp44+ ILCs were predominant in the gut of AS patients (82), highlighting differences between the etiology of IBD and SpA-associated intestinal inflammation. Surprisingly, particularly α4β7+IL-23R+ IL-17/IL-22-producing ILC3s were increased in gut, blood, bone marrow, and synovial fluid from AS patients with intestinal inflammation, suggesting that gut-derived functionally mature ILC3s might emigrate from intestinal tissues to α4β7 ligand-expressing joints, promoting local SpA-associated inflammation (83). SpA-associated α4β7+ synovial T cells were also already described before (84). Based on these results, ILC3s were proposed to function as “cytokine shuttles from gut” to extra-intestinal tissues (85). Strikingly, IL-17/IL-22-producing ILC3s appeared to be RORγt− but Tbet+ (83), possibly reflecting a particular developmental stage (86).
Although tissue residency is not well established for human ILCs, there is good evidence that ILCs in mice are generally tissue-resident (87, 88). Thus, the migration of gut-derived ILCs into SpA joints appears surprising at first glance. However, photoconversion experiments in mice could reveal CCR7-dependent ILC3 migration from gut to mesenteric lymph nodes (89), supporting the notion that intestinal ILCs can, at least to some extent, traffic to distant sites. The expression of a particular chemokine/cytokine signature, homing receptors, and respective ligands might promote their traveling. For instance, NKp44 ligand was shown to be expressed by chondrocytes, even in non-inflamed joints (90).
The migration of peripheral ILC3s into inflamed tissues in the context of SpA appears to be an interesting hint. However, the presence of resident NKp44+ ILC3s in non-inflamed human spinal entheses has just been reported (68). Further, that study demonstrated that, in consistence with mouse entheseal tissues (64, 65), human entheses responded to stimulation with IL-23/IL-1β by increasing IL-17/IL-22 production (68). Thus, in addition to γδ T cells, enthesis-resident ILC3s represent another candidate of innate lymphocytes that might be involved in the induction and/or progression of SpA (Figure 1).
αβ T Cells
Although recent studies point toward a chief contribution of innate lymphocytes to SpA, it is inevitable to include αβ T cells into the inner circle of prime suspects.
The “arthritogenic peptide theory” suggested that CD8+ T cells specific for HLA-B27-presented peptides might be involved in SpA disease pathogenesis (91, 92). Some immunohistological analyses indeed demonstrated that CD8+ T cells were predominant in human entheseal infiltrates (93, 94). However, studies in an experimental SpA model, HLA-B27-transgenic rats, did not support this theory (95, 96). Notably, HLA-B27 molecules can also be recognized as B27 β2-microglobulin-free heavy chains by killer immunoglobulin-like receptors (KIRs) (97, 98). HLA-B27/KIR3DL2 binding can induce RORγt expression in CD4+ αβ T cells and thus a T helper 17 (TH17) cell phenotype (99). Accordingly, although not confirmed in early axial SpA (100), KIR3DL2+ TH17 cells were increased in AS patients, suggesting that these cells might represent a therapeutic target for SpA treatment (101). HLA-B27 also appears to be associated with dysbiosis (102–105), possibly resulting from HLA-B27 misfolding-induced (106, 107) upregulation of the IL-23/IL-17 axis (108), innate immune activation, and intestinal TH17 cell expansion (109) early in life (110).
In fact, several albeit not all (39, 54, 111) SpA studies observed increased amounts of TH17 cells in blood and/or synovial fluid (100, 112–117), possibly in a sex-dependent manner (118). While IL-17 is the most representative cytokine ascribed to TH17 cells, GM-CSF-producing TH17 cells were also elevated in SpA patients (78). However, the increase in GM-CSF-producing lymphocytes was not specific for TH17 cells, as IL-17-producing CD8+ T cells, γδ T cells, and ILC3s co-producing GM-CSF were similarly expanded (78).
Supposing that increased numbers of TH17 cells promote SpA—how to keep these cells in check? In fact, miR-10b-5p (119) and IL-10-producing B cells (120) were recently proposed as putative negative regulators trying to control TH17 cells from SpA patients. Although increases in regulatory T cells in gut (121), blood, and synovial fluid (122, 123) from SpA patients also hinted toward an unsuccessful reaction to suppress autoinflammation, these cells might be functionally defect (124), demonstrating an imbalance in IL-10/IL-17 production (125).
Finally, unconventional/innate IL-17/IL-22-producing αβ T cell subsets might be associated with SpA. While neither human nor experimental SpA-associated IL-17-producing invariant natural killer T cells were identified so far, IL-17-producing CD8+ T cells (78, 126) or mucosal-associated invariant T (MAIT) cells (127, 128) were described. Surprisingly, increased IL-17 production by MAIT cells derived from AS patients was IL-23-independent, but rather promoted by IL-7 (128)—similar findings were recently reported in multiple sclerosis patients (129). Indeed, IL7R polymorphisms are associated with AS (4), and it was proposed that mechanical stress-induced IL-7 secretion by synovial fibroblasts could induce MAIT cell activation and thus IL-17 secretion during SpA pathogenesis (130). However, enthesis-resident MAIT cells have not been described so far.
Although seemingly plenty of studies described an association of TH17 cells and SpA, CD4+ cell depletion did not protect from IL-23-dependent inflammation (64). Along the same line, aging male DBA Tcrb−/− mice did still develop enthesitis and new bone formation (66), and γδ T cells, but not αβ T cells, dominated among pathogenic IL-17-producing enthesis-resident lymphocytes in mice (65). Together, this indicates that TH17 cells might rather not mediate the first line of action when local entheseal immune cells are provoked by the various environmental triggers, such as mechanical stress, dysbiosis, or genetic and epigenetic predisposition (131) (Figure 1).
Concluding Remarks: Whodunit?
After lining up the usual and a few unusual suspects, it seems that although strong arguments point toward an important contribution of the IL-23/IL-17 axis mediating SpA, it still remains difficult to pinpoint which of the above-described cell types are major players (132). Overall, recent studies collectively favor innate and innate-like immune cell involvement rather than adaptive T cells (6, 31, 32). As opposed to conventional B and T cells, innate and innate-like lymphocytes are commonly enriched in non-lymphoid tissues, and thus association with autoinflammatory diseases affecting particular tissues appears feasible (133). In this respect, it is worth considering the differential effects that genetics and environmental factors might elicit in innate versus adaptive immune traits (134).
Still, the relative contributions of tissue-resident cells, i.e., γδ T cells and ILC3s, versus recruited cells to SpA pathologies are not entirely clear. The “mechanical stress and entheseal microdamage hypothesis” (26, 27) supports the idea of inflammation-promoting enthesis-resident cells. In this regard, one might hypothesize that mechanical stress triggers resident immune cell activation—either directly or indirectly via stromal cell activation. Indeed, mechanical stress was shown to support enthesitis and new bone formation in TNFΔARE and aging male DBA mice, whereas remarkable experiments involving hind limb unloading significantly reduced disease symptoms (135). Since human tissues are generally difficult to obtain, various animal studies experimentally addressed immune pathways associated with SpA. However, it should be noted that many animal models only work in a specific genetic background, and vast differences exist between individual SpA models. While some models strongly depend on IL-23, others are based on TNF dysregulation (136, 137). Consequently, experimental data might be controversial: while Rag2−/− mice did not develop pathologies upon IL-23 overexpression (64), arguing against a role for innate lymphocytes in disease induction, enthesitis in Rag1−/− TNFΔARE mice was unaffected (135).
In the human system, many traits originate from analysis of circulating immune cell populations. However, such data remain inherently difficult to interpret: an increase in a particular circulating cell population does not unequivocally suggest their increased migration to joint tissues, while a decrease cannot unambiguously imply these cells already relocated from blood into distant sites. And why should otherwise tissue-resident ILC3s, TH17 cells, MAIT cells, or γδ T cells, leave the intestine and migrate into axial sites and distant tissues? Indeed, increased levels of CCL20, the chemokine attracting CCR6+ cells, were detected in SpA joints, albeit not as prominent as in rheumatoid arthritis (138). Altered gut epithelial and vascular barrier integrity in SpA patients might further promote intestinal immune cell emigration (139). Importantly, the trafficking of intestinal IL-17-producing γδ T cells to an entirely different tissue, the leptomeninges, was described in a mouse model for stroke (140, 141). Notably, a gut/joint axis in SpA might also exist for antigen-presenting cells carrying bacterial antigens from gut to axial sites, thus contributing to the induction of a local immune response (142). Whether SpA-associated joint-infiltrating lymphocytes enter tissues as already activated and functionally mature cells also remains an open question. Relatedly, CXCL4 was recently identified as a novel potent inducer of human TH17 cells enriched in PsA joints, and CXCL4 levels also positively correlated with disease severity, thus suggesting a CXCL4-driven local boost of TH17 cells (143).
In conclusion, there is growing evidence that innate and tissue-resident IL-17-producing γδ T cells and perhaps also ILC3s might be locally primed by genetic and epigenetic predisposition, mechanical stress as well as by increased systemic inflammation caused by intestinal dysbiosis. However, future studies will need to elaborate prevailing theories about the SpA-associated sequence of events. No matter whether ILC3s, γδ T cells, or any other innate lymphocytes primarily promote inflammation in SpA, in the end, the pathogenic action of all these cell types can be collectively targeted via the IL-23/IL-17 axis.
Author Contributions
All the authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer GG and handling Editor declared their shared affiliation.
Funding
Research in our group is supported by Deutsche Forschungsgemeinschaft, grants PR727/4-1 and PR727/8-1 to IP.
References
1. Ranganathan V, Gracey E, Brown MA, Inman RD, Haroon N. Pathogenesis of ankylosing spondylitis—recent advances and future directions. Nat Rev Rheumatol (2017) 13:359–67. doi:10.1038/nrrheum.2017.56
2. Stolwijk C, Boonen A, van Tubergen A, Reveille JD. Epidemiology of spondyloarthritis. Rheum Dis Clin North Am (2012) 38:441–76. doi:10.1016/j.rdc.2012.09.003
3. Reveille JD. Genetics of spondyloarthritis—beyond the MHC. Nat Rev Rheumatol (2012) 8:296–304. doi:10.1038/nrrheum.2012.41
4. Cortes A, Hadler J, Pointon JP, Robinson PC, Karaderi T, Leo P, et al. Identification of multiple risk variants for ankylosing spondylitis through high-density genotyping of immune-related loci. Nat Genet (2013) 45:730–8. doi:10.1038/ng.2667
5. Sherlock JP, Buckley CD, Cua DJ. The critical role of interleukin-23 in spondyloarthropathy. Mol Immunol (2014) 57:38–43. doi:10.1016/j.molimm.2013.06.010
6. Lubberts E. The IL-23-IL-17 axis in inflammatory arthritis. Nat Rev Rheumatol (2015) 11:562. doi:10.1038/nrrheum.2015.128
7. Rueda B, Orozco G, Raya E, Fernandez-Sueiro JL, Mulero J, Blanco FJ, et al. The IL23R Arg381Gln non-synonymous polymorphism confers susceptibility to ankylosing spondylitis. Ann Rheum Dis (2008) 67:1451–4. doi:10.1136/ard.2007.080283
8. Popadic S, Ramic Z, Medenica L, Pravica V, Popadic D. IL-23R gene polymorphism rs2201841 is associated with psoriatic arthritis. Int J Immunogenet (2014) 41:335–7. doi:10.1111/iji.12127
9. Cargill M, Schrodi SJ, Chang M, Garcia VE, Brandon R, Callis KP, et al. A large-scale genetic association study confirms IL12B and leads to the identification of IL23R as psoriasis-risk genes. Am J Hum Genet (2007) 80:273–90. doi:10.1086/511051
10. Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science (2006) 314:1461–3. doi:10.1126/science.1135245
11. Baeten D, Baraliakos X, Braun J, Sieper J, Emery P, van der Heijde D, et al. Anti-interleukin-17A monoclonal antibody secukinumab in treatment of ankylosing spondylitis: a randomised, double-blind, placebo-controlled trial. Lancet (2013) 382:1705–13. doi:10.1016/S0140-6736(13)61134-4
12. Mease PJ, Genovese MC, Greenwald MW, Ritchlin CT, Beaulieu AD, Deodhar A, et al. Brodalumab, an anti-IL17RA monoclonal antibody, in psoriatic arthritis. N Engl J Med (2014) 370:2295–306. doi:10.1056/NEJMoa1315231
13. McInnes IB, Mease PJ, Kirkham B, Kavanaugh A, Ritchlin CT, Rahman P, et al. Secukinumab, a human anti-interleukin-17A monoclonal antibody, in patients with psoriatic arthritis (FUTURE 2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet (2015) 386:1137–46. doi:10.1016/S0140-6736(15)61134-5
14. Baeten D, Sieper J, Braun J, Baraliakos X, Dougados M, Emery P, et al. Secukinumab, an interleukin-17A inhibitor, in ankylosing spondylitis. N Engl J Med (2015) 373:2534–48. doi:10.1056/NEJMoa1505066
15. Raychaudhuri SP, Raychaudhuri SK. Mechanistic rationales for targeting interleukin-17A in spondyloarthritis. Arthritis Res Ther (2017) 19:51. doi:10.1186/s13075-017-1249-5
16. Mease PJ, van der Heijde D, Ritchlin CT, Okada M, Cuchacovich RS, Shuler CL, et al. Ixekizumab, an interleukin-17A specific monoclonal antibody, for the treatment of biologic-naive patients with active psoriatic arthritis: results from the 24-week randomised, double-blind, placebo-controlled and active (adalimumab)-controlled period of the phase III trial SPIRIT-P1. Ann Rheum Dis (2017) 76:79–87. doi:10.1136/annrheumdis-2016-209709
17. Cheung PP. Anti-IL17A in axial spondyloarthritis-where are we at? Front Med (2017) 4:1. doi:10.3389/fmed.2017.00001
18. Poddubnyy D, Hermann KG, Callhoff J, Listing J, Sieper J. Ustekinumab for the treatment of patients with active ankylosing spondylitis: results of a 28-week, prospective, open-label, proof-of-concept study (TOPAS). Ann Rheum Dis (2014) 73:817–23. doi:10.1136/annrheumdis-2013-204248
19. Ritchlin C, Rahman P, Kavanaugh A, McInnes IB, Puig L, Li S, et al. Efficacy and safety of the anti-IL-12/23 p40 monoclonal antibody, ustekinumab, in patients with active psoriatic arthritis despite conventional non-biological and biological anti-tumour necrosis factor therapy: 6-month and 1-year results of the phase 3, multicentre, double-blind, placebo-controlled, randomised PSUMMIT 2 trial. Ann Rheum Dis (2014) 73:990–9. doi:10.1136/annrheumdis-2013-204655
20. Kavanaugh A, Puig L, Gottlieb AB, Ritchlin C, You Y, Li S, et al. Efficacy and safety of ustekinumab in psoriatic arthritis patients with peripheral arthritis and physician-reported spondylitis: post-hoc analyses from two phase III, multicentre, double-blind, placebo-controlled studies (PSUMMIT-1/PSUMMIT-2). Ann Rheum Dis (2016) 75:1984–8. doi:10.1136/annrheumdis-2015-209068
21. Chimenti MS, Ortolan A, Lorenzin M, Triggianese P, Talamonti M, Costa L, et al. Effectiveness and safety of ustekinumab in naive or TNF-inhibitors failure psoriatic arthritis patients: a 24-month prospective multicentric study. Clin Rheumatol (2018) 37(2):397–405. doi:10.1007/s10067-017-3953-6
22. Sieper J. New treatment targets for axial spondyloarthritis. Rheumatology (Oxford) (2016) 55:ii38–42. doi:10.1093/rheumatology/kew349
23. Poddubnyy D, Sieper J. What is the best treatment target in axial spondyloarthritis: tumour necrosis factor alpha, interleukin 17, or both? Rheumatology (Oxford) (2017). doi:10.1093/rheumatology/kex361
24. Deodhar A, Yu D. Switching tumor necrosis factor inhibitors in the treatment of axial spondyloarthritis. Semin Arthritis Rheum (2017) 47:343–50. doi:10.1016/j.semarthrit.2017.04.005
25. Schett G, Lories RJ, D’Agostino MA, Elewaut D, Kirkham B, Soriano ER, et al. Enthesitis: from pathophysiology to treatment. Nat Rev Rheumatol (2017) 13:731–41. doi:10.1038/nrrheum.2017.188
26. Benjamin M, Toumi H, Suzuki D, Redman S, Emery P, McGonagle D. Microdamage and altered vascularity at the enthesis-bone interface provides an anatomic explanation for bone involvement in the HLA-B27-associated spondyloarthritides and allied disorders. Arthritis Rheum (2007) 56:224–33. doi:10.1002/art.22290
27. Jacques P, McGonagle D. The role of mechanical stress in the pathogenesis of spondyloarthritis and how to combat it. Best Pract Res Clin Rheumatol (2014) 28:703–10. doi:10.1016/j.berh.2014.10.009
28. Kehl AS, Corr M, Weisman MH. Review: enthesitis: new insights into pathogenesis, diagnostic modalities, and treatment. Arthritis Rheumatol (2016) 68:312–22. doi:10.1002/art.39458
29. Benjamin M, McGonagle D. The anatomical basis for disease localisation in seronegative spondyloarthropathy at entheses and related sites. J Anat (2001) 199:503–26. doi:10.1046/j.1469-7580.2001.19950503.x
30. Benjamin M, Moriggl B, Brenner E, Emery P, McGonagle D, Redman S. The “enthesis organ” concept: why enthesopathies may not present as focal insertional disorders. Arthritis Rheum (2004) 50:3306–13. doi:10.1002/art.20566
31. Al-Mossawi MH, Ridley A, Kiedel S, Bowness P. The role of natural killer cells, gamma delta T-cells and other innate immune cells in spondyloarthritis. Curr Opin Rheumatol (2013) 25:434–9. doi:10.1097/BOR.0b013e3283620163
32. Venken K, Elewaut D. IL-23 responsive innate-like T cells in spondyloarthritis: the less frequent they are, the more vital they appear. Curr Rheumatol Rep (2015) 17:30. doi:10.1007/s11926-015-0507-2
33. Wendling D, Cedoz JP, Racadot E, Dumoulin G. Serum IL-17, BMP-7, and bone turnover markers in patients with ankylosing spondylitis. Joint Bone Spine (2007) 74:304–5. doi:10.1016/j.jbspin.2006.11.005
34. Wang X, Lin Z, Wei Q, Jiang Y, Gu J. Expression of IL-23 and IL-17 and effect of IL-23 on IL-17 production in ankylosing spondylitis. Rheumatol Int (2009) 29:1343–7. doi:10.1007/s00296-009-0883-x
35. Mei Y, Pan F, Gao J, Ge R, Duan Z, Zeng Z, et al. Increased serum IL-17 and IL-23 in the patient with ankylosing spondylitis. Clin Rheumatol (2011) 30:269–73. doi:10.1007/s10067-010-1647-4
36. Andersen T, Rasmussen TK, Hvid M, Holm CK, Madsen KJ, Jurik AG, et al. Increased plasma levels of IL-21 and IL-23 in spondyloarthritis are not associated with clinical and MRI findings. Rheumatol Int (2012) 32:387–93. doi:10.1007/s00296-010-1655-3
37. Appel H, Maier R, Bleil J, Hempfing A, Loddenkemper C, Schlichting U, et al. In situ analysis of interleukin-23- and interleukin-12-positive cells in the spine of patients with ankylosing spondylitis. Arthritis Rheum (2013) 65:1522–9. doi:10.1002/art.37937
38. Noordenbos T, Yeremenko N, Gofita I, van de Sande M, Tak PP, Canete JD, et al. Interleukin-17-positive mast cells contribute to synovial inflammation in spondyloarthritis. Arthritis Rheum (2012) 64:99–109. doi:10.1002/art.33396
39. Appel H, Maier R, Wu P, Scheer R, Hempfing A, Kayser R, et al. Analysis of IL-17(+) cells in facet joints of patients with spondyloarthritis suggests that the innate immune pathway might be of greater relevance than the Th17-mediated adaptive immune response. Arthritis Res Ther (2011) 13:R95. doi:10.1186/ar3370
40. Sutton CE, Mielke LA, Mills KH. IL-17-producing gammadelta T cells and innate lymphoid cells. Eur J Immunol (2012) 42:2221–31. doi:10.1002/eji.201242569
41. Haas JD, Ravens S, Duber S, Sandrock I, Oberdorfer L, Kashani E, et al. Development of interleukin-17-producing gammadelta T cells is restricted to a functional embryonic wave. Immunity (2012) 37:48–59. doi:10.1016/j.immuni.2012.06.003
42. Prinz I, Silva-Santos B, Pennington DJ. Functional development of gammadelta T cells. Eur J Immunol (2013) 43:1988–94. doi:10.1002/eji.201343759
43. Caccamo N, La Mendola C, Orlando V, Meraviglia S, Todaro M, Stassi G, et al. Differentiation, phenotype, and function of interleukin-17-producing human Vgamma9Vdelta2 T cells. Blood (2011) 118:129–38. doi:10.1182/blood-2011-01-331298
45. Lawand M, Dechanet-Merville J, Dieu-Nosjean MC. Key features of gamma-delta T-cell subsets in human diseases and their immunotherapeutic implications. Front Immunol (2017) 8:761. doi:10.3389/fimmu.2017.00761
46. Papotto PH, Reinhardt A, Prinz I, Silva-Santos B. Innately versatile: gammadelta17 T cells in inflammatory and autoimmune diseases. J Autoimmun (2017) 87:26–37. doi:10.1016/j.jaut.2017.11.006
47. Meliconi R, Pitzalis C, Kingsley GH, Panayi GS. Gamma/delta T cells and their subpopulations in blood and synovial fluid from rheumatoid arthritis and spondyloarthritis. Clin Immunol Immunopathol (1991) 59:165–72. doi:10.1016/0090-1229(91)90090-W
48. Keystone EC, Rittershaus C, Wood N, Snow KM, Flatow J, Purvis JC, et al. Elevation of a gamma delta T cell subset in peripheral blood and synovial fluid of patients with rheumatoid arthritis. Clin Exp Immunol (1991) 84:78–82.
49. Wouters CH, Ceuppens JL, Stevens EA. Different circulating lymphocyte profiles in patients with different subtypes of juvenile idiopathic arthritis. Clin Exp Rheumatol (2002) 20:239–48.
50. Spadaro A, Scrivo R, Moretti T, Bernardini G, Riccieri V, Taccari E, et al. Natural killer cells and gamma/delta T cells in synovial fluid and in peripheral blood of patients with psoriatic arthritis. Clin Exp Rheumatol (2004) 22:389–94.
51. Macaubas C, Nguyen K, Deshpande C, Phillips C, Peck A, Lee T, et al. Distribution of circulating cells in systemic juvenile idiopathic arthritis across disease activity states. Clin Immunol (2010) 134:206–16. doi:10.1016/j.clim.2009.09.010
52. Black AP, Bhayani H, Ryder CA, Pugh MT, Gardner-Medwin JM, Southwood TR. An association between the acute phase response and patterns of antigen induced T cell proliferation in juvenile idiopathic arthritis. Arthritis Res Ther (2003) 5:R277–84. doi:10.1186/ar791
53. Bendersky A, Marcu-Malina V, Berkun Y, Gerstein M, Nagar M, Goldstein I, et al. Cellular interactions of synovial fluid gammadelta T cells in juvenile idiopathic arthritis. J Immunol (2012) 188:4349–59. doi:10.4049/jimmunol.1102403
54. Kenna TJ, Davidson SI, Duan R, Bradbury LA, McFarlane J, Smith M, et al. Enrichment of circulating interleukin-17-secreting interleukin-23 receptor-positive gamma/delta T cells in patients with active ankylosing spondylitis. Arthritis Rheum (2012) 64:1420–9. doi:10.1002/art.33507
55. Gaur P, Misra R, Aggarwal A. Natural killer cell and gamma delta T cell alterations in enthesitis related arthritis category of juvenile idiopathic arthritis. Clin Immunol (2015) 161:163–9. doi:10.1016/j.clim.2015.07.012
56. Chowdhury AC, Chaurasia S, Mishra SK, Aggarwal A, Misra R. IL-17 and IFN-gamma producing NK and gammadelta-T cells are preferentially expanded in synovial fluid of patients with reactive arthritis and undifferentiated spondyloarthritis. Clin Immunol (2017) 183:207–12. doi:10.1016/j.clim.2017.03.016
57. Kessel C, Lippitz K, Weinhage T, Hinze C, Wittkowski H, Holzinger D, et al. Proinflammatory cytokine environments can drive interleukin-17 overexpression by gamma/delta T cells in systemic juvenile idiopathic arthritis. Arthritis Rheumatol (2017) 69:1480–94. doi:10.1002/art.40099
58. Guggino G, Ciccia F, Di Liberto D, Lo Pizzo M, Ruscitti P, Cipriani P, et al. Interleukin (IL)-9/IL-9R axis drives gammadelta T cells activation in psoriatic arthritis patients. Clin Exp Immunol (2016) 186:277–83. doi:10.1111/cei.12853
59. Cornelissen F, Mus AM, Asmawidjaja PS, van Hamburg JP, Tocker J, Lubberts E. Interleukin-23 is critical for full-blown expression of a non-autoimmune destructive arthritis and regulates interleukin-17A and RORgammat in gammadelta T cells. Arthritis Res Ther (2009) 11:R194. doi:10.1186/ar2893
60. Khmaladze I, Kelkka T, Guerard S, Wing K, Pizzolla A, Saxena A, et al. Mannan induces ROS-regulated, IL-17A-dependent psoriasis arthritis-like disease in mice. Proc Natl Acad Sci U S A (2014) 111:E3669–78. doi:10.1073/pnas.1405798111
61. Avau A, Mitera T, Put S, Put K, Brisse E, Filtjens J, et al. Systemic juvenile idiopathic arthritis-like syndrome in mice following stimulation of the immune system with Freund’s complete adjuvant: regulation by interferon-gamma. Arthritis Rheumatol (2014) 66:1340–51. doi:10.1002/art.38359
62. Akitsu A, Ishigame H, Kakuta S, Chung SH, Ikeda S, Shimizu K, et al. IL-1 receptor antagonist-deficient mice develop autoimmune arthritis due to intrinsic activation of IL-17-producing CCR2(+)Vgamma6(+)gammadelta T cells. Nat Commun (2015) 6:7464. doi:10.1038/ncomms8464
63. Adamopoulos IE, Tessmer M, Chao CC, Adda S, Gorman D, Petro M, et al. IL-23 is critical for induction of arthritis, osteoclast formation, and maintenance of bone mass. J Immunol (2011) 187:951–9. doi:10.4049/jimmunol.1003986
64. Sherlock JP, Joyce-Shaikh B, Turner SP, Chao CC, Sathe M, Grein J, et al. IL-23 induces spondyloarthropathy by acting on ROR-gammat+ CD3+CD4-CD8- entheseal resident T cells. Nat Med (2012) 18:1069–76. doi:10.1038/nm.2817
65. Reinhardt A, Yevsa T, Worbs T, Lienenklaus S, Sandrock I, Oberdorfer L, et al. Interleukin-23-dependent gamma/delta T cells produce interleukin-17 and accumulate in the enthesis, aortic valve, and ciliary body in mice. Arthritis Rheumatol (2016) 68:2476–86. doi:10.1002/art.39732
66. Corthay A, Hansson AS, Holmdahl R. T lymphocytes are not required for the spontaneous development of entheseal ossification leading to marginal ankylosis in the DBA/1 mouse. Arthritis Rheum (2000) 43:844–51. doi:10.1002/1529-0131(200004)43:4<844::AID-ANR15>3.0.CO;2-B
67. Jameson JM, Cauvi G, Witherden DA, Havran WL. A keratinocyte-responsive gamma delta TCR is necessary for dendritic epidermal T cell activation by damaged keratinocytes and maintenance in the epidermis. J Immunol (2004) 172:3573–9. doi:10.4049/jimmunol.172.6.3573
68. Cuthbert RJ, Fragkakis EM, Dunsmuir R, Li Z, Coles M, Marzo-Ortega H, et al. Brief report: group 3 innate lymphoid cells in human enthesis. Arthritis Rheumatol (2017) 69:1816–22. doi:10.1002/art.40150
69. Artis D, Spits H. The biology of innate lymphoid cells. Nature (2015) 517:293–301. doi:10.1038/nature14189
70. Klose CS, Artis D. Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis. Nat Immunol (2016) 17:765–74. doi:10.1038/ni.3489
71. Ebbo M, Crinier A, Vely F, Vivier E. Innate lymphoid cells: major players in inflammatory diseases. Nat Rev Immunol (2017) 17:665–78. doi:10.1038/nri.2017.86
72. Wenink MH, Leijten EFA, Cupedo T, Radstake T. Review: innate lymphoid cells: sparking inflammatory rheumatic disease? Arthritis Rheumatol (2017) 69:885–97. doi:10.1002/art.40068
73. Soare A, Weber S, Maul L, Rauber S, Gheorghiu AM, Luber M, et al. Cutting edge: homeostasis of innate lymphoid cells is imbalanced in psoriatic arthritis. J Immunol (2018) 200(4):1249–54. doi:10.4049/jimmunol.1700596
74. Gronke K, Kofoed-Nielsen M, Diefenbach A. Innate lymphoid cells, precursors and plasticity. Immunol Lett (2016) 179:9–18. doi:10.1016/j.imlet.2016.07.004
75. Bernink JH, Krabbendam L, Germar K, de Jong E, Gronke K, Kofoed-Nielsen M, et al. Interleukin-12 and -23 control plasticity of CD127(+) group 1 and group 3 innate lymphoid cells in the intestinal lamina propria. Immunity (2015) 43:146–60. doi:10.1016/j.immuni.2015.06.019
76. Huang Y, Guo L, Qiu J, Chen X, Hu-Li J, Siebenlist U, et al. IL-25-responsive, lineage-negative KLRG1(hi) cells are multipotential ‘inflammatory’ type 2 innate lymphoid cells. Nat Immunol (2015) 16:161–9. doi:10.1038/ni.3078
77. Leijten EF, van Kempen TS, Boes M, Michels-van Amelsfort JM, Hijnen D, Hartgring SA, et al. Brief report: enrichment of activated group 3 innate lymphoid cells in psoriatic arthritis synovial fluid. Arthritis Rheumatol (2015) 67:2673–8. doi:10.1002/art.39261
78. Al-Mossawi MH, Chen L, Fang H, Ridley A, de Wit J, Yager N, et al. Unique transcriptome signatures and GM-CSF expression in lymphocytes from patients with spondyloarthritis. Nat Commun (2017) 8:1510. doi:10.1038/s41467-017-01771-2
79. Wendling D. The gut in spondyloarthritis. Joint Bone Spine (2016) 83:401–5. doi:10.1016/j.jbspin.2016.02.017
80. Mielants H, Veys EM, Cuvelier C, De Vos M, Goemaere S, De Clercq L, et al. The evolution of spondyloarthropathies in relation to gut histology. III. Relation between gut and joint. J Rheumatol (1995) 22:2279–84.
81. Triggianese P, Conigliaro P, Chimenti MS, Biancone L, Monteleone G, Perricone R, et al. Evidence of IL-17 producing innate lymphoid cells in peripheral blood from patients with enteropathic spondyloarthritis. Clin Exp Rheumatol (2016) 34:1085–93.
82. Ciccia F, Accardo-Palumbo A, Alessandro R, Rizzo A, Principe S, Peralta S, et al. Interleukin-22 and interleukin-22-producing NKp44+ natural killer cells in subclinical gut inflammation in ankylosing spondylitis. Arthritis Rheum (2012) 64:1869–78. doi:10.1002/art.34355
83. Ciccia F, Guggino G, Rizzo A, Saieva L, Peralta S, Giardina A, et al. Type 3 innate lymphoid cells producing IL-17 and IL-22 are expanded in the gut, in the peripheral blood, synovial fluid and bone marrow of patients with ankylosing spondylitis. Ann Rheum Dis (2015) 74:1739–47. doi:10.1136/annrheumdis-2014-206323
84. Elewaut D, De Keyser F, Van Den Bosch F, Lazarovits AI, De Vos M, Cuvelier C, et al. Enrichment of T cells carrying beta7 integrins in inflamed synovial tissue from patients with early spondyloarthropathy, compared to rheumatoid arthritis. J Rheumatol (1998) 25:1932–7.
85. Neerinckx B, Elewaut D, Lories RJ. Spreading spondyloarthritis: are ILCs cytokine shuttles from base camp gut? Ann Rheum Dis (2015) 74:1633–5. doi:10.1136/annrheumdis-2015-207735
86. Klose CS, Kiss EA, Schwierzeck V, Ebert K, Hoyler T, d’Hargues Y, et al. A T-bet gradient controls the fate and function of CCR6-RORgammat+ innate lymphoid cells. Nature (2013) 494:261–5. doi:10.1038/nature11813
87. Gasteiger G, Fan X, Dikiy S, Lee SY, Rudensky AY. Tissue residency of innate lymphoid cells in lymphoid and nonlymphoid organs. Science (2015) 350:981–5. doi:10.1126/science.aac9593
88. Fan X, Rudensky AY. Hallmarks of tissue-resident lymphocytes. Cell (2016) 164:1198–211. doi:10.1016/j.cell.2016.02.048
89. Mackley EC, Houston S, Marriott CL, Halford EE, Lucas B, Cerovic V, et al. CCR7-dependent trafficking of RORgamma(+) ILCs creates a unique microenvironment within mucosal draining lymph nodes. Nat Commun (2015) 6:5862. doi:10.1038/ncomms6862
90. Bialoszewska A, Baychelier F, Niderla-Bielinska J, Czop A, Debre P, Vieillard V, et al. Constitutive expression of ligand for natural killer cell NKp44 receptor (NKp44L) by normal human articular chondrocytes. Cell Immunol (2013) 285:6–9. doi:10.1016/j.cellimm.2013.08.005
91. Fiorillo MT, Maragno M, Butler R, Dupuis ML, Sorrentino R. CD8(+) T-cell autoreactivity to an HLA-B27-restricted self-epitope correlates with ankylosing spondylitis. J Clin Invest (2000) 106:47–53. doi:10.1172/JCI9295
92. Faham M, Carlton V, Moorhead M, Zheng J, Klinger M, Pepin F, et al. Discovery of T cell receptor beta motifs specific to HLA-B27-positive ankylosing spondylitis by deep repertoire sequence analysis. Arthritis Rheumatol (2017) 69:774–84. doi:10.1002/art.40028
93. Laloux L, Voisin MC, Allain J, Martin N, Kerboull L, Chevalier X, et al. Immunohistological study of entheses in spondyloarthropathies: comparison in rheumatoid arthritis and osteoarthritis. Ann Rheum Dis (2001) 60:316–21. doi:10.1136/ard.60.4.316
94. Appel H, Kuhne M, Spiekermann S, Ebhardt H, Grozdanovic Z, Kohler D, et al. Immunohistologic analysis of zygapophyseal joints in patients with ankylosing spondylitis. Arthritis Rheum (2006) 54:2845–51. doi:10.1002/art.22060
95. May E, Dorris ML, Satumtira N, Iqbal I, Rehman MI, Lightfoot E, et al. CD8 alpha beta T cells are not essential to the pathogenesis of arthritis or colitis in HLA-B27 transgenic rats. J Immunol (2003) 170:1099–105. doi:10.4049/jimmunol.170.2.1099
96. Taurog JD, Dorris ML, Satumtira N, Tran TM, Sharma R, Dressel R, et al. Spondyloarthritis in HLA-B27/human beta2-microglobulin-transgenic rats is not prevented by lack of CD8. Arthritis Rheum (2009) 60:1977–84. doi:10.1002/art.24599
97. Allen RL, O’Callaghan CA, McMichael AJ, Bowness P. Cutting edge: HLA-B27 can form a novel beta 2-microglobulin-free heavy chain homodimer structure. J Immunol (1999) 162:5045–8.
98. Kollnberger S, Bird L, Sun MY, Retiere C, Braud VM, McMichael A, et al. Cell-surface expression and immune receptor recognition of HLA-B27 homodimers. Arthritis Rheum (2002) 46:2972–82. doi:10.1002/art.10605
99. Ridley A, Hatano H, Wong-Baeza I, Shaw J, Matthews KK, Al-Mossawi H, et al. Activation-induced killer cell immunoglobulin-like receptor 3DL2 binding to HLA-B27 licenses pathogenic T cell differentiation in spondyloarthritis. Arthritis Rheumatol (2016) 68:901–14. doi:10.1002/art.39515
100. Jansen DT, Hameetman M, van Bergen J, Huizinga TW, van der Heijde D, Toes RE, et al. IL-17-producing CD4+ T cells are increased in early, active axial spondyloarthritis including patients without imaging abnormalities. Rheumatology (Oxford) (2015) 54:728–35. doi:10.1093/rheumatology/keu382
101. Bowness P, Ridley A, Shaw J, Chan AT, Wong-Baeza I, Fleming M, et al. Th17 cells expressing KIR3DL2+ and responsive to HLA-B27 homodimers are increased in ankylosing spondylitis. J Immunol (2011) 186:2672–80. doi:10.4049/jimmunol.1002653
102. Lin P, Bach M, Asquith M, Lee AY, Akileswaran L, Stauffer P, et al. HLA-B27 and human beta2-microglobulin affect the gut microbiota of transgenic rats. PLoS One (2014) 9:e105684. doi:10.1371/journal.pone.0105684
103. Costello ME, Ciccia F, Willner D, Warrington N, Robinson PC, Gardiner B, et al. Brief report: intestinal dysbiosis in ankylosing spondylitis. Arthritis Rheumatol (2015) 67:686–91. doi:10.1002/art.38967
104. Breban M, Tap J, Leboime A, Said-Nahal R, Langella P, Chiocchia G, et al. Faecal microbiota study reveals specific dysbiosis in spondyloarthritis. Ann Rheum Dis (2017) 76:1614–22. doi:10.1136/annrheumdis-2016-211064
105. Manasson J, Shen N, Garcia Ferrer HR, Ubeda C, Iraheta I, Heguy A, et al. Gut microbiota perturbations in reactive arthritis and postinfectious spondyloarthritis. Arthritis Rheumatol (2018) 70(2):242–54. doi:10.1002/art.40359
106. DeLay ML, Turner MJ, Klenk EI, Smith JA, Sowders DP, Colbert RA. HLA-B27 misfolding and the unfolded protein response augment interleukin-23 production and are associated with Th17 activation in transgenic rats. Arthritis Rheum (2009) 60:2633–43. doi:10.1002/art.24763
107. Ciccia F, Accardo-Palumbo A, Rizzo A, Guggino G, Raimondo S, Giardina A, et al. Evidence that autophagy, but not the unfolded protein response, regulates the expression of IL-23 in the gut of patients with ankylosing spondylitis and subclinical gut inflammation. Ann Rheum Dis (2014) 73:1566–74. doi:10.1136/annrheumdis-2012-202925
108. Gill T, Asquith M, Brooks SR, Rosenbaum JT, Colbert RA. Effects of HLA-B27 on gut microbiota in experimental spondyloarthritis implicate an ecological model of dysbiosis. Arthritis Rheumatol (2017) 70(4):555–65. doi:10.1002/art.40405
109. Viladomiu M, Kivolowitz C, Abdulhamid A, Dogan B, Victorio D, Castellanos JG, et al. IgA-coated E. coli enriched in Crohn’s disease spondyloarthritis promote TH17-dependent inflammation. Sci Transl Med (2017) 9:eaaf9655. doi:10.1126/scitranslmed.aaf9655
110. Asquith MJ, Stauffer P, Davin S, Mitchell C, Lin P, Rosenbaum JT. Pertur-bed mucosal immunity and dysbiosis accompany clinical disease in a rat model of spondyloarthritis. Arthritis Rheumatol (2016) 68:2151–62. doi:10.1002/art.39681
111. Bautista-Caro MB, Arroyo-Villa I, Castillo-Gallego C, de Miguel E, Peiteado D, Puig-Kroger A, et al. Decreased Th17 and Th1 cells in the peripheral blood of patients with early non-radiographic axial spondyloarthritis: a marker of disease activity in HLA-B27(+) patients. Rheumatology (Oxford) (2013) 52:352–62. doi:10.1093/rheumatology/kes267
112. Jandus C, Bioley G, Rivals JP, Dudler J, Speiser D, Romero P. Increased numbers of circulating polyfunctional Th17 memory cells in patients with sero-negative spondyloarthritides. Arthritis Rheum (2008) 58:2307–17. doi:10.1002/art.23655
113. Nistala K, Moncrieffe H, Newton KR, Varsani H, Hunter P, Wedderburn LR. Interleukin-17-producing T cells are enriched in the joints of children with arthritis, but have a reciprocal relationship to regulatory T cell numbers. Arthritis Rheum (2008) 58:875–87. doi:10.1002/art.23291
114. Shen H, Goodall JC, Gaston JS. Frequency and phenotype of T helper 17 cells in peripheral blood and synovial fluid of patients with reactive arthritis. J Rheumatol (2010) 37:2096–9. doi:10.3899/jrheum.100146
115. Xueyi L, Lina C, Zhenbiao W, Qing H, Qiang L, Zhu P. Levels of circulating Th17 cells and regulatory T cells in ankylosing spondylitis patients with an inadequate response to anti-TNF-alpha therapy. J Clin Immunol (2013) 33:151–61. doi:10.1007/s10875-012-9774-0
116. Benham H, Norris P, Goodall J, Wechalekar MD, FitzGerald O, Szentpetery A, et al. Th17 and Th22 cells in psoriatic arthritis and psoriasis. Arthritis Res Ther (2013) 15:R136. doi:10.1186/ar4317
117. Fiocco U, Stramare R, Martini V, Coran A, Caso F, Costa L, et al. Quantitative imaging by pixel-based contrast-enhanced ultrasound reveals a linear relationship between synovial vascular perfusion and the recruitment of pathogenic IL-17A-F(+)IL-23(+) CD161(+) CD4(+) T helper cells in psoriatic arthritis joints. Clin Rheumatol (2017) 36:391–9. doi:10.1007/s10067-016-3500-x
118. Gracey E, Yao Y, Green B, Qaiyum Z, Baglaenko Y, Lin A, et al. Sexual dimorphism in the Th17 signature of ankylosing spondylitis. Arthritis Rheumatol (2016) 68:679–89. doi:10.1002/art.39464
119. Chen L, Al-Mossawi MH, Ridley A, Sekine T, Hammitzsch A, de Wit J, et al. miR-10b-5p is a novel Th17 regulator present in Th17 cells from ankylosing spondylitis. Ann Rheum Dis (2017) 76:620–5. doi:10.1136/annrheumdis-2016-210175
120. Mavropoulos A, Varna A, Zafiriou E, Liaskos C, Alexiou I, Roussaki-Schulze A, et al. IL-10 producing Bregs are impaired in psoriatic arthritis and psoriasis and inversely correlate with IL-17- and IFNgamma-producing T cells. Clin Immunol (2017) 184:33–41. doi:10.1016/j.clim.2017.04.010
121. Ciccia F, Accardo-Palumbo A, Giardina A, Di Maggio P, Principato A, Bombardieri M, et al. Expansion of intestinal CD4+CD25(high) Treg cells in patients with ankylosing spondylitis: a putative role for interleukin-10 in preventing intestinal Th17 response. Arthritis Rheum (2010) 62:3625–34. doi:10.1002/art.27699
122. Cao D, Borjesson O, Larsson P, Rudin A, Gunnarsson I, Klareskog L, et al. FOXP3 identifies regulatory CD25bright CD4+ T cells in rheumatic joints. Scand J Immunol (2006) 63:444–52. doi:10.1111/j.1365-3083.2006.001755.x
123. Appel H, Wu P, Scheer R, Kedor C, Sawitzki B, Thiel A, et al. Synovial and peripheral blood CD4+FoxP3+ T cells in spondyloarthritis. J Rheumatol (2011) 38:2445–51. doi:10.3899/jrheum.110377
124. Guo H, Zheng M, Zhang K, Yang F, Zhang X, Han Q, et al. Functional defects in CD4(+) CD25(high) FoxP3(+) regulatory cells in ankylosing spondylitis. Sci Rep (2016) 6:37559. doi:10.1038/srep37559
125. Araujo LM, Fert I, Jouhault Q, Labroquere K, Andrieu M, Chiocchia G, et al. Increased production of interleukin-17 over interleukin-10 by Treg cells implicates inducible costimulator molecule in experimental spondyloarthritis. Arthritis Rheumatol (2014) 66:2412–22. doi:10.1002/art.38737
126. Lau MC, Keith P, Costello ME, Bradbury LA, Hollis KA, Thomas R, et al. Genetic association of ankylosing spondylitis with TBX21 influences T-bet and pro-inflammatory cytokine expression in humans and SKG mice as a model of spondyloarthritis. Ann Rheum Dis (2017) 76:261–9. doi:10.1136/annrheumdis-2015-208677
127. Hayashi E, Chiba A, Tada K, Haga K, Kitagaichi M, Nakajima S, et al. Involvement of mucosal-associated invariant T cells in ankylosing spondylitis. J Rheumatol (2016) 43:1695–703. doi:10.3899/jrheum.151133
128. Gracey E, Qaiyum Z, Almaghlouth I, Lawson D, Karki S, Avvaru N, et al. IL-7 primes IL-17 in mucosal-associated invariant T (MAIT) cells, which contribute to the Th17-axis in ankylosing spondylitis. Ann Rheum Dis (2016) 75:2124–32. doi:10.1136/annrheumdis-2015-208902
129. Willing A, Jager J, Reinhardt S, Kursawe N, Friese MA. Production of IL-17 by MAIT cells is increased in multiple sclerosis and is associated with IL-7 receptor expression. J Immunol (2018) 200:974–82. doi:10.4049/jimmunol.1701213
130. Debusschere K, Lories RJ, Elewaut D. MAIT cells: not just another brick in the wall. Ann Rheum Dis (2016) 75:2057–9. doi:10.1136/annrheumdis-2016-209695
131. Paine A, Ritchlin CT. Targeting the interleukin-23/17 axis in axial spondyloarthritis. Curr Opin Rheumatol (2016) 28:359–67. doi:10.1097/BOR.0000000000000301
132. Venken K, Elewaut D. New immune cells in spondyloarthritis: key players or innocent bystanders? Best Pract Res Clin Rheumatol (2015) 29:706–14. doi:10.1016/j.berh.2016.02.002
133. Exley MA, Tsokos GC, Mills KH, Elewaut D, Mulhearn B. What rheumatologists need to know about innate lymphocytes. Nat Rev Rheumatol (2016) 12:658–68. doi:10.1038/nrrheum.2016.140
134. Mangino M, Roederer M, Beddall MH, Nestle FO, Spector TD. Innate and adaptive immune traits are differentially affected by genetic and environmental factors. Nat Commun (2017) 8:13850. doi:10.1038/ncomms13850
135. Jacques P, Lambrecht S, Verheugen E, Pauwels E, Kollias G, Armaka M, et al. Proof of concept: enthesitis and new bone formation in spondyloarthritis are driven by mechanical strain and stromal cells. Ann Rheum Dis (2014) 73:437–45. doi:10.1136/annrheumdis-2013-203643
136. Breban M, Araujo LM, Chiocchia G. Animal models of spondyloarthritis: do they faithfully mirror human disease? Arthritis Rheumatol (2014) 66:1689–92. doi:10.1002/art.38636
137. Vieira-Sousa E, van Duivenvoorde LM, Fonseca JE, Lories RJ, Baeten DL. Review: animal models as a tool to dissect pivotal pathways driving spondyloarthritis. Arthritis Rheumatol (2015) 67:2813–27. doi:10.1002/art.39282
138. Melis L, Vandooren B, Kruithof E, Jacques P, De Vos M, Mielants H, et al. Systemic levels of IL-23 are strongly associated with disease activity in rheumatoid arthritis but not spondyloarthritis. Ann Rheum Dis (2010) 69:618–23. doi:10.1136/ard.2009.107649
139. Ciccia F, Guggino G, Rizzo A, Alessandro R, Luchetti MM, Milling S, et al. Dysbiosis and zonulin upregulation alter gut epithelial and vascular barriers in patients with ankylosing spondylitis. Ann Rheum Dis (2017) 76:1123–32. doi:10.1136/annrheumdis-2016-210000
140. Benakis C, Brea D, Caballero S, Faraco G, Moore J, Murphy M, et al. Commensal microbiota affects ischemic stroke outcome by regulating intestinal gammadelta T cells. Nat Med (2016) 22:516–23. doi:10.1038/nm.4068
141. Arunachalam P, Ludewig P, Melich P, Arumugam TV, Gerloff C, Prinz I, et al. CCR6 (CC chemokine receptor 6) is essential for the migration of detrimental natural interleukin-17-producing gammadelta T cells in stroke. Stroke (2017) 48:1957–65. doi:10.1161/STROKEAHA.117.016753
142. Berthelot JM, Claudepierre P. Trafficking of antigens from gut to sacroiliac joints and spine in reactive arthritis and spondyloarthropathies: mainly through lymphatics? Joint Bone Spine (2016) 83:485–90. doi:10.1016/j.jbspin.2015.10.015
Keywords: interleukin-23, interleukin-17, γδ T cells, TH17 cells, innate lymphoid cells, spondyloarthritis
Citation: Reinhardt A and Prinz I (2018) Whodunit? The Contribution of Interleukin (IL)-17/IL-22-Producing γδ T Cells, αβ T Cells, and Innate Lymphoid Cells to the Pathogenesis of Spondyloarthritis. Front. Immunol. 9:885. doi: 10.3389/fimmu.2018.00885
Received: 23 February 2018; Accepted: 10 April 2018;
Published: 25 April 2018
Edited by:
Francesco Dieli, Università degli Studi di Palermo, ItalyReviewed by:
Camilla Jandus, Université de Lausanne, SwitzerlandGiuliana Guggino, Università degli Studi di Palermo, Italy
Copyright: © 2018 Reinhardt and Prinz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Annika Reinhardt, YW5uaWthLnJlaW5oYXJkdEBraS5zZQ==
†Present address: Annika Reinhardt, Department of Medicine Solna, Immunology and Allergy Unit, Karolinska Institutet, Stockholm, Sweden