 Benjamin G. Chousterman1,2*
Benjamin G. Chousterman1,2* Marine Arnaud2
Marine Arnaud2
- 1Département d’Anesthésie-Réanimation-SMUR, Hôpitaux Universitaires Lariboisière – Saint-Louis, AP-HP, Paris, France
- 2INSERM U1160, Hôpital Saint-Louis, Paris, France
Sepsis is a complex syndrome characterized by simultaneous activation of pro- and anti-inflammatory processes. After an inflammatory phase, patients present signs of immunosuppression and possibly persistent inflammation. Hematopoietic growth factors (HGFs) are glycoproteins that cause immune cells to mature and/or proliferate. HGFs also have a profound effect on cell functions and behavior. HGFs play crucial role in sepsis pathophysiology and were tested in several clinical trials without success to date. This review summarizes the role played by HGFs during sepsis and their potential therapeutic role in the Management of sepsis-related immune disturbances.
Introduction
Infectious diseases are a major cause of death and morbidity worldwide and especially in intensive care units (ICUs) (1, 2). Decades of basic and clinical research led to the observation that most of the infections can lead to an uncontrolled response to the pathogen, the sepsis. Sepsis was recently defined by an international consortium of experts as a life-threatening organ dysfunction due to a dysregulated host response to infection (3). This definition points out that it is not only the virulence of the germ or the damages caused directly to the infected organ(s) that are responsible for the disseminated consequences on the body, it is the “host response” that is causing severe troubles. Host response is mainly mediated by the immune system. After recognition of specific patterns [from the germ, the pathogen-associated molecular patterns (PAMPs) or from the damaged cells, damage-associated molecular patterns (DAMPS)] by Pattern Recognition Receptors (PRRs), a chain reaction will lead to an auto-amplifying cytokine storm that will in turn remotely activate immune and endothelial cells (4). Organs will suffer from this friendly fire aiming to combat the initial insult. Over the last 20 years, the prognosis of septic patients has drastically improved (5). Nevertheless, there is still no specific treatment of inflammation during sepsis.
Sepsis is a complex double-face syndrome. Once having crossed the defensive barriers of the body (skin, mucus, complement, …), pathogens will activate the innate immune system and induce inflammation. As seen in most of biological processes, inflammation is well balanced by a counter-inflammation process driven by cellular reprogramming and anti-inflammatory cytokines (6, 7). The most prominent actors of the innate immunity involved in sepsis are the neutrophils and the monocytes/macrophages/dendritic cell (DC) system. Although T and B lymphocytes, usually described as part of the adaptive immune system, are also involved, they appear to play a major role in the second immunosuppressive state (sepsis-induced immunosuppression, SIS).
Indeed, after the initial phase of cell activation, the immune system appears to be “blunted” by the assault and patients face an immunosuppressive state. Poor response to infection, lymphopenia, and decreased reparative properties of immune cells are observed. Some authors termed this phase a persistent inflammation, immunosuppression, and catabolism syndrome (PICS) (8, 9). There is a debate whether the second phase clinical presentation is mostly due to inflammation or immunosuppression (10–14).
This whole sequence is compartmentalized in space and time. Immune disturbances and time variations are observed in the whole body, the hematopoietic organs (bone marrow, spleen), the blood and the tissues.
During sepsis, immune cells undergo profound phenotypic modifications in their activation state, response to stimuli, localization, and numbers. These phenomena are finely regulated by various cytokines and hematopoietic growth factors (HGFs).
An HGF is defined as a relatively stable, secreted, or membrane-bound glycoprotein that causes immune cells to mature and/or proliferate. They also have profound effects on cell functions and behaviors.
Hematopoietic growth factors are deeply involved in sepsis pathophysiology both in the initial phase and the late phase. They were naturally identified as potential therapeutic targets to treat septic patients. However, until now, there is no evidence of clinical benefit for the use of HGFs during sepsis.
In this review, we will detail how the most studied HGFs are involved in sepsis, explore the findings from clinical trials, and discuss the perspectives for HGF-mediated immunotherapy of sepsis.
Granulocyte Macrophage – Colony Stimulating Factor (GM-CSF)
Granulocyte macrophage – colony stimulating factor (also known as CSF-2) was discovered after observing that a factor present in lung-conditioned medium was able to induce the formation of granulocytes and macrophages (15, 16). GM-CSF is coded by the Csf2 gene located on chromosome 5 in humans. GM-CSF is a monomeric glycoprotein composed of 144 amino acids [22 kilodaltons (kDa)]. GM-CSF is produced at low level during steady state and is dramatically increased during inflammatory conditions (17); it is secreted by a wide variety of cell such as monocytes-macrophages, T and B cells, mast cells, fibroblasts, and epithelial cells. The biological activity of GM-CSF is mediated through a heterodimeric cell receptor (GM-CSF-receptor, GM-CSF-R) composed of a GM-CSF specific subunit (major binding subunit GM-Rα) and a subunit chain that is common to interleukin (IL)-5-and IL-3-receptors (common signaling subunit βc). GM-CSF-R signal is mediated via Extracellular signal-regulated kinase (ERK) 1/2, phosphoinositide-3-kinase (PI3K), mitogen-activated protein (MAP) kinase, and Janus kinase (JAK) – signal transducer and activator of transcription (STAT) pathways. Forty years after its discovery, it appears that this protein action is far more complex than just a proliferative agent; it has pleiotropic effects ranging from cell activation, survival, differentiation, chemotaxis (18), and proliferation (19). GM-CSF is frequently prescribed in patients treated by chemotherapy in order to reduce the duration of the granulocytopenia.
Until recently, most of literature regarding the role of GM-CSF during sepsis was focused on the early inflammatory phase. In murine/rat models and human cells culture, GM-CSF modulation through antibody mediated blockade, genetic invalidation, or protein supplementation helped to understand its functions in the host response to infections.
Granulocyte macrophage – colony stimulating factor role in host defense against infection is highly complex since it acts at different phases of the host response. GM-CSF deficiency is protective in models of lethal endotoxemia (20). In contrast, in infection models using full pathogens, the absence of GM-CSF appears clearly to be detrimental. In models of bacterial (21), fungal, parasitic, or viral infections (22), the absence of GM-CSF is shown to increase mortality and tissue lesions. Alveolar macrophages from GM-CSF−/− mice have reduced abilities to phagocyte and kill pathogens, have reduced Fcγ receptors (FcγR) expression, and have lower membrane expression of TLRs and subsequent lipopolysaccharide (LPS) or peptidoglycan-induced tumor necrosis factor alpha (TNFα) release. GM-CSF-deficient alveolar macrophages have markedly reduced reactive oxygen species (ROS) production and adenovirus-elicited Interferon (IFN)γ, IL-18, and IL-12 production. GM-CSF also increases the expression of scavenger receptors such as macrophage receptor with collagenous structure (MARCO) and other class A scavenger receptors (SR-As) (23, 24). These scavenger receptors interact with TLRs and are shown to limit the TLR4 response in case of infection (25). Most of these pro-inflammatory and germ-killing GM-CSF effects are mediated by the transcription factor PU.1 which is essential for GM-CSF signaling during inflammation.
In caecal ligation and puncture (CLP) model, recombinant murine (rm)GM-CSF treatment improves survival and reduces bacterial translocation (26).
Interestingly, in some studies, injection of GM-CSF or genetic invalidation did not impact mice survival after CLP whether due to a timing of administration or dose issues. Inflammatory cytokines levels are higher when GM-CSF is present or enhanced (27, 28). In a model of type-A influenza infection, in which GM-CSF global deficiency is detrimental, mice with specific expression of GM-CSF only in the lung were found to have a better outcome than wild-type mice. Overexpression of GM-CSF is associated with tissue damage revealing the need for an adequate modulation (i.e., timely compartmentalization) of GM-CSF production (29).
Regarding the late phase, proliferative capacities of monocytes during sepsis in response to GM-CSF are reduced in a time-dependent manner. Early myeloid-derived suppressor cells (MDSCs) obtained 3 days after CLP procedures produced more macrophages and DCs after GM-CSF stimulation than late MDSCs obtained 12 days after CLP (30).
Granulocyte macrophage – colony stimulating factor is shown to promote type-1 pro-inflammatory cytokines production and downregulate anti-inflammatory cytokines (IL-10, IL-4) (31). GM-CSF also promotes T-cell proliferation (32) and communication with myeloid cells in the tissues (33). During the late phase of sepsis, DCs are shown to secrete less IL-12, a pivotal cytokine necessary to induce a T-Helper (TH)1 response. During sepsis, GM-CSF and IFNγ treatment can restore IL-12 production by splenic DCs. Mayuzumi et al. (34) showed that IL-33 promotes the generation of DC in the bone marrow through induction of GM-CSF production. GM-CSF is therefore efficient to restore TH1 response during the late phase of sepsis.
Granulocyte macrophage – colony stimulating factor has also pro-angiogenic effects and promotes endothelial cells proliferation (35–37); thus, GM-CSF could protect endothelial cells during sepsis.
The GM-CSF receptor is downregulated in human monocytes during sepsis (38) and in human neutrophils during endotoxemia (39).
The source of GM-CSF during sepsis was unknown and thought to be mainly due to macrophages activation. In 2012, a study conducted in Swirski Lab in Boston (MA, USA) tackled this question. It appears that the main source of GM-CSF following abdominal sepsis is a new cell originating from a B1-type B cells (40). This new cell type, named innate response activator B cell (IRA B cell), appears after relocalization of peritoneal B cells into the spleen where they acquire IRA B cell features. Specific depletion of GM-CSF production in B cell using a complex model of chimeric mice demonstrated the crucial role of IRA B cells in cytokine production, bacterial clearance, organ damage, and survival. In 2014, we demonstrated that GM-CSF production by IRA B cell plays a central role in the activation of a GM-CSF-IgM axis that serves as a front line of defense against pneumonia (41). Relocalization of IRA B cells and in situ GM-CSF production demonstrates the importance of the spatial and temporal organization of this growth factor secretion.
It appears that a lack of GM-CSF is responsible for an immunosuppressed status which in turn could be associated with a worst outcome. Treatment with GM-CSF does not appear suitable for the initial cytokine storm-associated phase. However, immunosuppression features can be reversed by GM-CSF making this protein a potential candidate as an immune therapy for SIS.
During the immunosuppressive phase, GM-CSF was mainly tested to restore monocyte functions or monocytes-granulocytes numbers. A study by Williams et al. (42) showed that GM-CSF is able to restore, in vitro, monocytes functions in septic patients. GM-CSF treatment increases respiratory burst activity, integrin, and CD14 expressions. Same results are observed when AIDS patients with Mycobacterium Avium Complex bacteremia are treated with GM-CSF (43).
Reduced human leukocyte antigen (HLA)-DR in a common feature of SIS, GM-CSF was shown to be effective to increase monocyte (m)HLA-DR expression. Injection of GM-CSF is more efficient than G-CSF to restore HLA-DR levels in septic neonates (44). Intraperitoneal injection of GM-CSF in peritoneal dialysis patients induces an increase in peritoneal macrophages number, integrin expression, and cytokines and chemokines production (such as IL-6 or CCL2/MCP-1) (45). Sepsis-associated neutropenia in neonates is reverted by subcutaneous injection of 5 μg/kg/day for seven consecutive days with a direct impact on mortality (46).
A clinical trial conducted by Presneill et al. (47) showed that GM-CSF treatment improves lung function in sepsis patients treated with the growth factor compared with eight controls. In a randomized trial published in 2005, GM-CSF treatment did not improve mortality but enhanced clinical and microbial resolution of infection as well as markers of monocytes and neutrophils functions (48); of note in this trial, patients with septic shock were excluded and significant proportion of patient had an organ transplant. In a trial of 58 patients, adjunction of GM-CSF to antibiotic treatment in abdominal sepsis led to reduced length of hospitalization, infectious complications, and hospital costs (49).
The most famous trial regarding the use of GM-CSF in sepsis was conducted by Meisel et al. (50). Thirty-eight septic patients with reduced mHLA-DR expression (under 8000 AB/C) for 2 days were treated with GM-CSF (4 μg/kg/d) or placebo. After 24 h, in the GM-CSF group HLA-DR significantly increases, cytokine production related to LPS stimulation increases as well. Immune cell number (neutrophils, monocytes, T cells) increases significantly compared with the control group. Duration of mechanical ventilation is shortened in the GM-CSF treatment group but no effect on mortality could be observed. This was a key study in the field of sepsis immunotherapy since the selection of patients was not only based on the diagnosis of sepsis but also on the estimation of immunosuppression through monocyte (m)HLA-DR expression measurement. In a second paper derived from these data, Schefold et al. (51) showed that GM-CSF decreases indoleamine 2,3-dioxygenase (IDO) activity and reduces kynurenine pathway activity without affecting tryptophan levels. In a remarkable study on healthy volunteers subjected to endotoxemia, GM-CSF at a dose of 4 μg/kg/day was shown to be less effective than IFNγ to reverse feature of endotoxin tolerance such as a reduced TNFα production and increased IL-10 production after LPS stimulation (52).
Bo et al. (53) conducted a meta-analysis on GM-CSF and G-CSF treatment for sepsis. Among 12 RCT identified, only four were GM-CSF-related studies (n = 147 patients). Mortality was consistently found to be unchanged.
A multicenter prospective randomized controlled trial is now ongoing in France (NCT02361528).
Use of GM-CSF in sepsis studies is characterized by a small number of patients and high heterogeneity of diagnosis and clinical presentation.
To date, results of clinical trials show no benefit of GM-CSF treatment during sepsis. At least, there are no major adverse events observed after nearly 20 years of clinical use. It is insufficient to conclude in a lack of safety threat but still there is no big signal of risk and there are potential benefits. There is no definitive answer to the appropriateness of GM-CSF as an immunotherapy for certain subgroups of septic patients. The timing and the phenotype of the patients that could benefit from such treatment is to be elucidated. Due to the finely regulated GM-CSF response to sepsis in time and space, intravenous or subcutaneous injections could have reached negative results because of mixed benefic and adverse effects that could be ameliorated by adding GM-CSF in specific tissues or organs and/or timepoints.
Granulocyte – Colony Stimulating Factor (G-CSF)
Granulocyte – colony stimulating factor, also known as CSF-3, is a 25-kDa glycoprotein coded by the Csf3 gene that is located on chromosome 17 in humans. G-CSF levels are low in steady state and rise after inflammatory stimuli (54). TNFα, IL-1, or LPS stimulation of macrophages or epithelial cells induces high levels of G-CSF production (55). T cells can also induce G-CSF production through IL-17 release. The main effects of G-CSF are to induce proliferation and differentiation but also survival of cells in the neutrophils lineage. It has effects on early progenitors such as hematopoietic stem cells and in all intermediate cells up to the mature neutrophils. G-CSF-induced neutrophil production and function has been extensively studied (56–58). G-CSF plays a central role in response to infections and in situations of aplasia or neutropenia. G-CSF also enhances neutrophils production of cytokines, production of ROS, and phagocytosis when added to other stimulati. Actions of G-CSF are mediated through its receptor, the G-CSF-R. G-CSF-R requires its homodimerization in order to be fully functional. The binding of G-CSF on G-CSF-R activates a JAK-STAT phosphorylation cascade pathway. It also involves PI3K, Akt, and MAPK. Suppressor of cytokine signaling 3 SOCS3 acts as a negative regulator.
Granulocyte – colony stimulating factor is now widely used in routine to treat or prevent chemotherapy-induced neutropenia. G-CSF treatment is recommended by experts in various clinical scenarios (59–61).
Granulocyte – colony stimulating factor exerts proliferative effects but also enhances mobilization of neutrophils in a direct and indirect chemotactic effect. Intravenous or subcutaneous injection of G-CSF is rapidly followed by a marked neutrophilia together with a release of progenitors and immature cells. G-CSF has also an impact of the generation of regulatory DCs and indirectly on T-cell populations.
Granulocyte – colony stimulating factor is also shown to finely modulate the neutrophil response to infection by reducing responsiveness of neutrophils to the chemokine CXCL2 by reducing the CXCR2 mediated intracellular signaling (62). Thus, G-CSF, a mobilizing cytokine, prevents overwhelming neutrophils invasion during infections. Genetic invalidation of the G-CSF gene in mice results in neutropenia and the subsequent increased risk of developing bacterial or fungal infections and a weakened host response to infection. The impact of G-CSF loss was tested in several mice or rat models of pneumonia or abdominal sepsis. In a mice model of P. aeruginosa infection, G-CSF-deficient mice have decreased survival and augmented neutrophils apoptosis while local production of cytokine remains unchanged (63).
Granulocyte – colony stimulating factor is able to correct the defect of neutrophils chemotaxis to the lung in a double-hit model of LPS instillation after CLP (64). Pretreatment with G-CSF before pneumonia induction after CLP leads to increased bacterial clearance.
Granulocyte – colony stimulating factor can partially correct the impeded host response to Klebsiella pneumoniae infection in MCP-1/CCL2-deficient mice (65). Mice pretreated with G-CSF before CLP have improved survival (66).
Liu et al. tried to modulate the excessive inflammation related to G-CSF treatment by blocking the increase of LPS binding protein after intraperitoneal injection of feces. This leads to a reduced neutrophils infiltration into the peritoneum but with an augmented bacterial clearance ability of local neutrophils (67).
Tanaka et al. studied the levels of G-CSF after sepsis and trauma. They found elevated G-CSF levels in both conditions except that in sepsis these increased levels remained high for a larger duration (68). Ishikawa et al. studied septic patients with relative neutropenia. High levels of G-CSF at baseline were associated with poor outcome and small/no response to G-CSF treatment (69).
Most of the prospective clinical studies of G-CSF focused on patients with pneumonia. The first randomized control trial of G-CSF in severe infections was conducted by Nelson et al. Nearly 760 patients with severe community acquired pneumonia were included. G-CSF does not have an impact on mortality or length of stay but effectively increases neutrophils counts and diminishes the rate of serious complications such as ARDS or pleural empyema (70). The same authors reproduced the study with patients having multilobar pneumonia (n = 480) (71). The treatment does not impact mortality and shows a possible effect on patients having bacteremia. In the second largest RCT conducted on G-CSF treatment, Root et al. show that during severe sepsis secondary to pneumonia, G-CSF treatment does not improve patients’ outcome without any significant adverse event (72).
Another trial focused on patient having nosocomial pneumonia showed no clinical benefit of G-CSF treatment but was associated with a trend lower apparition of sepsis features compared with placebo (73). A study published by Stephens et al. (74) confirms the risks associated with the use of G-CSF. This randomized controlled trial included 166 septic shock patients and allocated them to be treated with G-CSF (n = 83) or placebo (n = 83). Mortality does not differ between groups but the rate of liver dysfunction and elevation of troponin raises in the G-CSF group. Thus, G-CSF appears not only to be non-beneficial but detrimental in septic patients. These results point out the risk of increasing inflammation without precise guidance.
In the meta-analysis of the impact of G-CSF during sepsis conducted by Bo et al. (53), G-CSF appears to have overall no effects on mortality and is not associated with a significant rate of adverse events. One trial is ongoing (NCT01913938) evaluating the impact of G-CSF treatment on septic patients with cytopenia with a special interest on the occurrence of hemophagocytosis.
The limits of the described trials are nearly the same for G-CSF than for GM-CSF. The main difference is that GM-CSF is a better candidate to the reversal of various features of SIS, while G-CSF is only effective on neutrophils.
Macrophage – Colony Stimulation Factor (M-CSF)
Macrophage – colony stimulation factor, also termed CSF-1, is an 85-kDa glycoprotein in its homodimeric (secreted) form. M-CSF also exists as a membrane bound protein. The M-CSF receptor (M-CSF-R) is coded by the c-fms protooncogene. M-CSF-R has a ligand-inducible tyrosine kinase activity. Binding of M-CSF to M-CSF-R induces a dimerization, auto-phosphorylation, and activation of the kinase activity. M-CSF is also essential during pregnancy for the development and biology of the placenta.
Macrophage – colony stimulation factor increases monocyte production of G-CSF, GM-CSF, IL-6, and TNFα after LPS stimulation (75). TNFα and GM-CSF induce M-CSF production by human monocytes (76). M-CSF is also produced by several cell types, especially endothelial cells and fibroblasts.
Macrophage – colony stimulation factor plays a fundamental role in bone homeostasis; mice lacking M-CSF are osteoporotic. Using CLP model, Ogiku et al. found that the deficit of M-CSF is associated with a decreased survival, reduced phagocytosis, and increased HMGB1 levels (77).
Several studies showed that M-CSF contributes to monocytes- and macrophages-mediated immune response and bacterial clearance in response to various pathogens (78–81).
In a model of K. pneumoniae pneumonia, M-CSF increases locally in the lung, promotes monocytes and macrophages survival in the lung and the liver, and enhances bacterial killing (82).
Macrophage – colony stimulation factor selectively expands CD16 + monocytes in human and primates (83).
Macrophage – colony stimulation factor added to culture of murine macrophages enhances the macrophages response to TLR4 agonists while lowering the response to TLR9 agonists (CpG) (84).
Macrophage – colony stimulation factor-elevated levels during sepsis are associated with the occurrence of hemophagocytosis and thrombopenia (85).
Macrophage – colony stimulation factor can also control DC production (86). Recently, M-CSF was found to be responsible for reduced monocytes ability to convert into DC and to respond properly to inflammatory stimuli. These effects are mediated through an epigenetic regulation of the PU.1 transcription factor (87).
To date, there was no clinical trial testing the impact of M-CSF treatment during sepsis.
Interleukin-3 (IL-3)
Interleukin-3, also named multicolony-stimulating factor (MSF), contributes to leukocyte production, proliferation, and survival (88). IL-3 stimulates the differentiation of multipotent hematopoietic stem cells into myeloid progenitor cells or, with the addition of IL-7, into lymphoid progenitor cells. IL-3 gene is located on the chromosome 5 near the GM-CSF (Csf2) gene. It is deeply involved in the pathogenesis of asthma, allergy, or blood malignancies. Until recently, the role of IL-3 in sepsis was not investigated.
In steady state, the main sources of IL-3 are activated T-helper cells. IL-3 has important effects on macrophages-DC and mastocytes in synergy with IFNγ (89).
We recently published that published that IL-3 has a crucial role in the pathogenesis of the early phase of sepsis (90). We showed that mice lacking IL-3 were partially protected from sepsis lethality induced by a CLP.
Mechanistically, we showed that IL-3 contributed to the emergency myelopoiesis that induces a rapid increase of inflammatory (Ly6Chigh) monocytes and neutrophils in blood and increases inflammation. Surprisingly, the sources of IL-3 during sepsis are the IRA B cell making these cells a producer of two crucial HGFs during sepsis (GM-CSF and IL-3) (40).
Importantly, we showed that during human sepsis, high levels of plasma IL-3 at admission were correlated with a better survival at 28 days after sepsis in two independent cohorts of patients. IL-3 levels are associated with responsiveness to corticosteroid therapy during septic shock (91).
The study of the role of IL-3 during sepsis is at its very beginning. The roles of IL-3 during the reparative phase of sepsis in mice and human are to be elucidated. As a potent DC function regulating cytokine (92), IL-3 could be involved in functional features of SIS.
Interleukin-7 (IL-7)
Interleukin-7is a 25-kDa glycoprotein (152 amino acids in humans) mainly produced by stroma epithelial cells of the thymus and the bone marrow. IL-7 gene is located on chromosome 8.
Interleukin-7 receptor (IL-7R) is composed of two subunits: IL-7Rα (CD127) and the common gamma chain (γc) (CD132). IL-7R is expressed on the lymphoid lineage. IL-7 effects are mediated through JAK3 activation and STAT1,2,3,5, and PI3K pathways.
The main effects of IL-7 aim to maintain lymphocytes survival. It has been recognized as a potential treatment of an HIV-related lymphopenia in a phase-II trial (93).
Sepsis is associated with a lymphopenia. All types of lymphocytes, except for the regulatory T cells, see their numbers reduced in blood and tissues. The remaining lymphocytes, essentially the T cells, present signs of immunosuppression (called T-cell exhaustion). Markers of apoptosis are elevated, while the ability to proliferate is reduced together with reduced cytokines productions.
Unsinger et al. (94) used CLP model to test impact of recombinant human (rh)IL-7 treatment. rhIL7 improved mice survival, reduced drastically lymphocytes apoptosis, and improved cytokine production, especially IFNγ. LFA-1 and VLA-4, two adhesion markers, have their expression on lymphocytes increased. Il-7 is also able to improve neutrophil mobilization and recruitment through an IL-17 and CXCL1-mediated mechanism (95). IL-7 treatment can revert lethality in a model of fungemia following abdominal sepsis in mice (96). In the same model, Shindo et al. compared the effects of IL-7 and anti-PD-1 treatments (97). IL-7 is efficient at reversing T-cell exhaustion features, while anti-PD-1 increases HLA-DR expression on macrophages and DCs. These interesting results unveil a possible role for combination of immunotherapy agents during sepsis.
Recently, Terashima et al. identified osteoblasts as a major source of IL-7 during sepsis (98). Depletion of osteoblasts or suppression of osteoblasts production of IL-7 recapitulates a lymphopenic phenotype. Parathyroid hormone, an osteoblasts activator, is effective at correcting sepsis-associated lymphopenia. In a two-hit model, CLP followed by P. Aeruginosa infection, and IL-7 improves host response and survival (99).
A human clinical study shows that IL-7 gene expression is reduced during sepsis but remains surprisingly normal during bacteremia; however, IL-7 level is unchanged (100). Another study found reduced circulating IL-7 during sepsis (101). Boomer et al. noticed a reduction in IL-7R expression on lymphocytes during sepsis (102). Soluble IL-7R (sIL-7R or sCD127) levels are higher in non-surviving septic shock patients (103). Venet et al. demonstrated the potential of IL-7 to treat T-cell exhaustion during sepsis (104). In this study, IL-7 levels are augmented in septic shock patients, while it is not correlated to survival or ICU acquired secondary infections. sCD127 levels are, there again, correlated with survival but also with nosocomial infections. IL-7 is highly efficient to promote stimulated T-cell proliferation and IFNγ production. The first “proof of concept” clinical trials on IL-7 during sepsis are conducted in USA and Europe. The two studies are twins as they share the same design. The results are much awaited to evaluate IL-7 potential as a future tool for sepsis immunotherapy (IRIS-7-A and B trials, NCT02797431 and NCT02640807).
Erythropoietin (EPO)
Erythropoietin is a 30-KDa glycoprotein mainly secreted by the peritubular interstitial fibroblasts in the kidneys. EPO gene is located on chromosome 7. EPO binding to its receptor (Epo-R that shares the common beta-chain with the IL-3, IL-5, and GM-CSF receptors) activates JAK2 signaling and increase erythropoiesis. EPO is well known for its impact on acute and chronic anemia, especially during chronic kidney disease, hematologic disease, or after chemotherapy. EPO was originally seen as potential treatment of sepsis-associated anemia. EPO levels are usually low in critically ill patients (105) but were shown to elevate in sepsis patients (106).
It appears that EPO effects on the vascular tone and its anti-apoptotic properties could also be beneficial.
Several studies have shown anti-apoptotic effects of EPO during inflammation.
In CLP model, EPO reduces renal and pulmonary damages in mice (107). EPO is also capable to correct hypotension related to sepsis by reducing endothelial nitric oxide synthase (eNOS) synthesis and inducible (i)NOS function, and preserving G-protein Receptor Kinase (GRK)2 and alpha1D receptor expressions and functions (108). EPO demonstrates cardioprotective effects in rat model of abdominal sepsis (109). Kao et al. found that EPO activates eNOS and protect skeletal muscle microvasculature (110). EPO exerts protective effect on sepsis-associated encephalopathy (111, 112).
During endotoxemia, EPO reduces AKI through decreased apoptosis (113) and activation of the beta-common receptor (114). Other group did not find such protective effects in pigs (115) possibly due to the low dose used. EPO at low dose are detrimental in endotoxinic shock (116). This may be related to dose related response to EPO and variable sensitivity of the target cells.
Erythropoietin impedes lymphoid cell apoptosis after CLP without major effect on mortality in rats (117).
The effects of EPO treatment on critical illness-associated anemia and especially sepsis-associated anemia are debated. Two major clinical studies show conflicting results (118, 119) and a recent meta-analysis concludes that the effect on anemia is small (120). Pearl discusses the negative results of EPO in trials and suggests that the doses are insufficient (121). However, when used in brain injury patients, EPO is associated with increased thrombo-embolic events (122) that could counterbalance any beneficial effects.
One clinical trial is ongoing to test the effect of EPO on microcirculatory alterations (NCT1087450) during sepsis (Table 1).
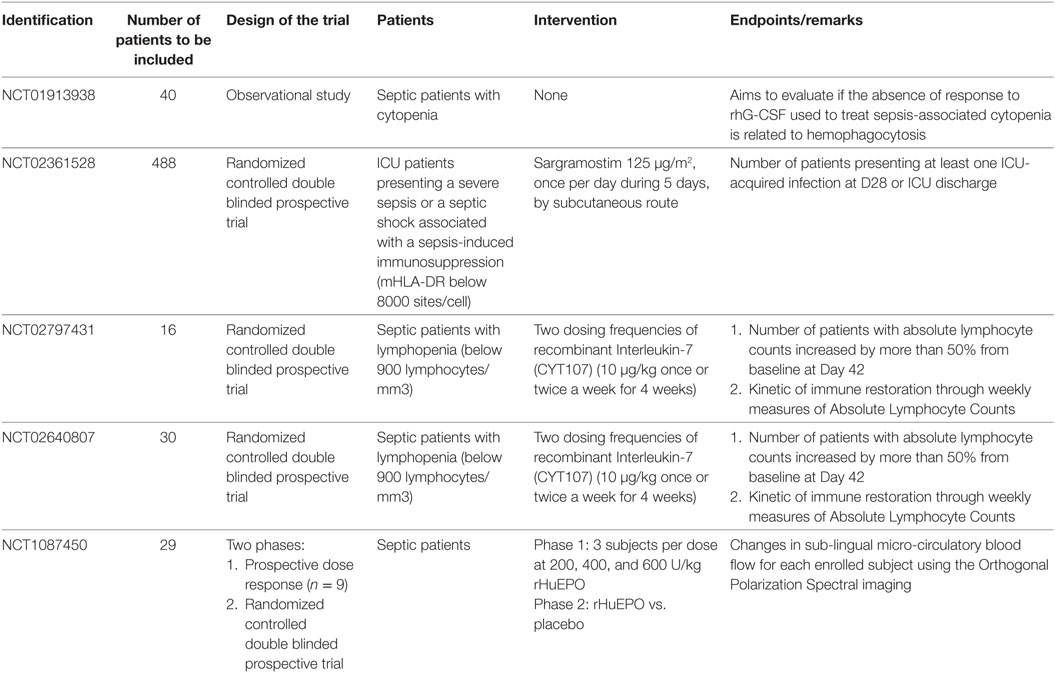
Table 1. Ongoing trials on the use of hematopoietic growing factors during sepsis.
Figures 1 and 2 summarize the main effects of hematopoietic growth factors during sepsis.
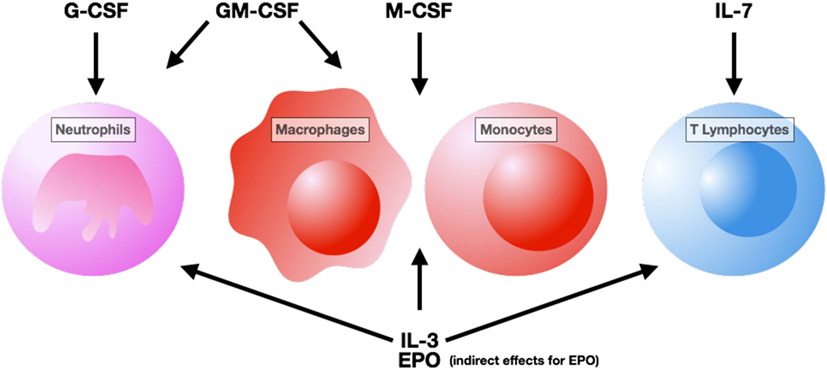
Figure 1. Cells targeted by hematopoietic growth factor therapy. Abbreviation: G-CSF, granulocyte – colony stimulating factor; GM-CSF, granulocyte macrophage – colony stimulating factor; M-CSF, macrophage – colony stimulating factor; IL, interleukin; EPO, erythropoietin.
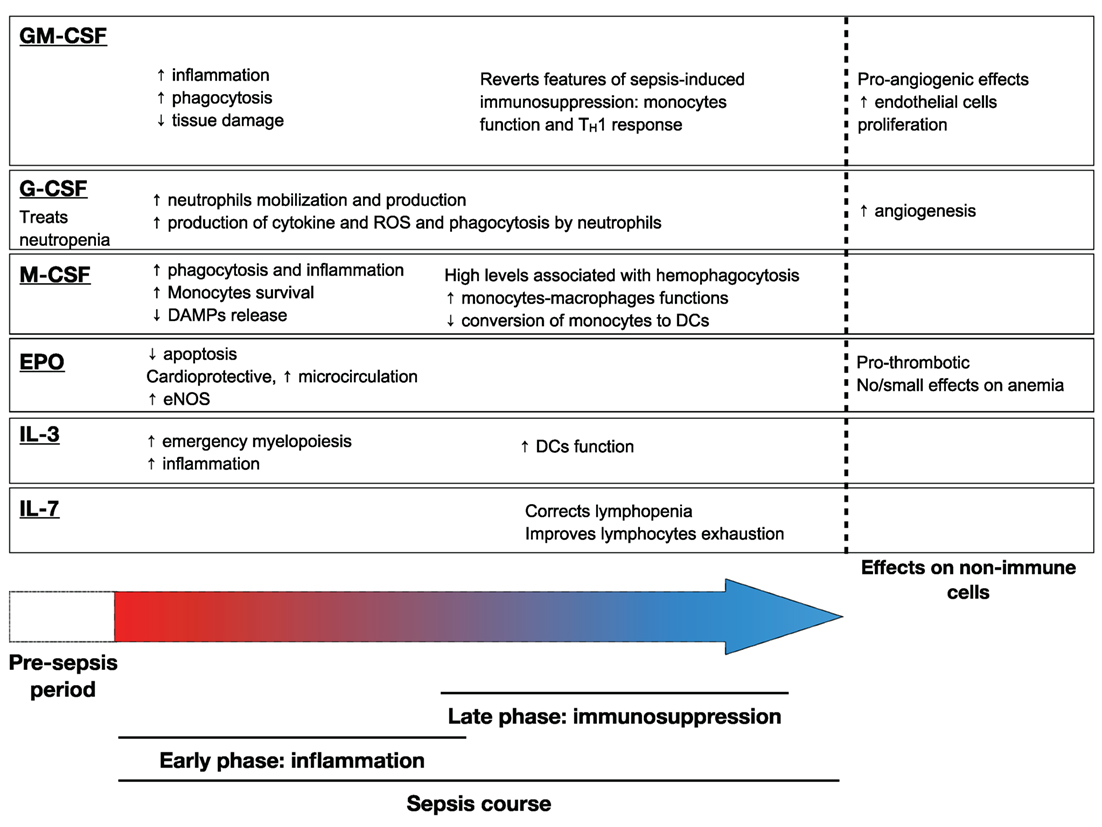
Figure 2. Main effects of hematopoietic growth factors during sepsis. Abbreviation: GM-CSF, granulocyte macrophage – colony stimulating factor; EPO, erythropoietin; IL-3, interleukin-3; IL-7, interleukin-7; DAMP, damage-associated molecular pattern; DC, dendritic cell; ROS, reactive oxygen species; eNOS, endothelial Nitric oxide synthase.
Conclusion
The understanding of sepsis is still limited. Tremendous efforts have been made in order to decipher its complex pathophysiology. HGFs play a crucial in both early and late phases of sepsis but there is to date no positive clinical trial regarding their use. The various pathways involved and the wide range of clinical presentation may explain partly the negatives results of clinical trials. As emphasized earlier, included patients in clinical trials are highly heterogeneous. Therefore, a potential benefit of some HGFs could have not been seen because of its futility in some clusters of patients. In addition, HGF effects are compartmentalized in space and time. Therefore, the timing of administration and the route of administration are crucial and require more developments. Most of the efforts regarding the use of HGFs during sepsis are now concentrated on the immunosuppressed patients. One important underlying question is that of the appropriateness of reintroducing inflammation in these patients. The boosting of the immune system could be exaggerated and at the end detrimental. The undergoing trial of GM-CSF and IL-7 during sepsis will help to have a better idea of their utility in this indication. EPO and G-CSF treatments seem to be deleterious during sepsis.
Anyway, there is a crucial need to be able to identify the “endotype” of sepsis patients. The biological effects of HGFs are incompletely understood and require further investigations. Next generation of sepsis trials will use this advanced knowledge and will be biomarker guided trials as recommended by experts (123). Fundamental research and clinical trial learn from each other and are complementary. In conclusion, there is no benefit with the systematic use of HGFs during sepsis, and clusters of patients that could beneficiate of such treatments are to be identified.
Author Contributions
Both authors contributed to the literature search and the writing of the review.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work is supported by a public grant overseen by the French National Research Agency (ANR) (Project “CMOS,” ANR-15-CE15-0019) and a grant from the European Society of Intensive Care Medicine (ESICM, Basic science research grant).
References
1. Vincent JL, Rello J, Marshall J, Silva E, Anzueto A, Martin CD, et al. International study of the prevalence and outcomes of infection in intensive care units. JAMA (2009) 302(21):2323–9. doi:10.1001/jama.2009.1754
2. Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the global burden of disease study 2010. Lancet (2012) 380(9859):2095–128. doi:10.1016/S0140-6736(12)61728-0
3. Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The third international consensus definitions for sepsis and septic shock (sepsis-3). JAMA (2016) 315(8):801–10. doi:10.1001/jama.2016.0287
4. Chousterman BG, Swirski FK, Weber GF. Cytokine storm and sepsis disease pathogenesis. Semin Immunopathol (2017) 39(5):517–28. doi:10.1007/s00281-017-0639-8
5. Kaukonen KM, Bailey M, Suzuki S, Pilcher D, Bellomo R. Mortality related to severe sepsis and septic shock among critically ill patients in Australia and New Zealand, 2000–2012. JAMA (2014) 311(13):1308–16. doi:10.1001/jama.2014.2637
6. Hotchkiss RS, Moldawer LL, Opal SM, Reinhart K, Turnbull IR, Vincent JL. Sepsis and septic shock. Nat Rev Dis Primers (2016) 2:16045. doi:10.1038/nrdp.2016.45
7. Hotchkiss RS, Monneret G, Payen D. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol (2013) 13(12):862–74. doi:10.1038/nri3552
8. Gentile LF, Cuenca AG, Efron PA, Ang D, Bihorac A, McKinley BA, et al. Persistent inflammation and immunosuppression: a common syndrome and new horizon for surgical intensive care. J Trauma Acute Care Surg (2012) 72(6):1491–501. doi:10.1097/TA.0b013e318256e000
9. Mira JC, Gentile LF, Mathias BJ, Efron PA, Brakenridge SC, Mohr AM, et al. Sepsis pathophysiology, chronic critical illness, and persistent inflammation-immunosuppression and catabolism syndrome. Crit Care Med (2017) 45(2):253–62. doi:10.1097/CCM.0000000000002074
10. Boomer JS, To K, Chang KC, Takasu O, Osborne DF, Walton AH, et al. Immunosuppression in patients who die of sepsis and multiple organ failure. JAMA (2011) 306(23):2594–605. doi:10.1001/jama.2011.1829
11. Hoover DB, Brown TC, Miller MK, Schweitzer JB, Williams DL. Loss of sympathetic nerves in spleens from patients with end stage sepsis. Front Immunol (2017) 8:1712. doi:10.3389/fimmu.2017.01712
12. Valdes-Ferrer SI, Rosas-Ballina M, Olofsson PS, Lu B, Dancho ME, Ochani M, et al. HMGB1 mediates splenomegaly and expansion of splenic CD11b+ Ly-6C(high) inflammatory monocytes in murine sepsis survivors. J Intern Med (2013) 274(4):381–90. doi:10.1111/joim.12104
13. Valdes-Ferrer SI, Rosas-Ballina M, Olofsson PS, Lu B, Dancho ME, Li J, et al. High-mobility group box 1 mediates persistent splenocyte priming in sepsis survivors: evidence from a murine model. Shock (2013) 40(6):492–5. doi:10.1097/SHK.0000000000000050
14. Angus DC, Yang L, Kong L, Kellum JA, Delude RL, Tracey KJ, et al. Circulating high-mobility group box 1 (HMGB1) concentrations are elevated in both uncomplicated pneumonia and pneumonia with severe sepsis. Crit Care Med (2007) 35(4):1061–7. doi:10.1097/01.CCM.0000259534.68873.2A
15. Sheridan JW, Metcalf D. A low molecular weight factor in lung-conditioned medium stimulating granulocyte and monocyte colony formation in vitro. J Cell Physiol (1973) 81(1):11–23. doi:10.1002/jcp.1040810103
16. Sheridan JW, Metcalf D, Stanley ER. Further studies on the factor in lung-conditioned medium stimulating granulocyte and monocyte colony formation in vitro. J Cell Physiol (1974) 84(1):147–58. doi:10.1002/jcp.1040840117
17. Hamilton JA. GM-CSF in inflammation and autoimmunity. Trends Immunol (2002) 23(8):403–8. doi:10.1016/S1471-4906(02)02260-3
18. O’Brien AD, Standiford TJ, Christensen PJ, Wilcoxen SE, Paine R III. Chemotaxis of alveolar macrophages in response to signals derived from alveolar epithelial cells. J Lab Clin Med (1998) 131(5):417–24. doi:10.1016/S0022-2143(98)90142-1
19. Ushach I, Zlotnik A. Biological role of granulocyte macrophage colony-stimulating factor (GM-CSF) and macrophage colony-stimulating factor (M-CSF) on cells of the myeloid lineage. J Leukoc Biol (2016) 100(3):481–9. doi:10.1189/jlb.3RU0316-144R
20. Basu S, Dunn AR, Marino MW, Savoia H, Hodgson G, Lieschke GJ, et al. Increased tolerance to endotoxin by granulocyte-macrophage colony-stimulating factor-deficient mice. J Immunol (1997) 159(3):1412–7.
21. LeVine AM, Reed JA, Kurak KE, Cianciolo E, Whitsett JA. GM-CSF-deficient mice are susceptible to pulmonary group B streptococcal infection. J Clin Invest (1999) 103(4):563–9. doi:10.1172/JCI5212
22. Sever-Chroneos Z, Murthy A, Davis J, Florence JM, Kurdowska A, Krupa A, et al. GM-CSF modulates pulmonary resistance to influenza A infection. Antiviral Res (2011) 92(2):319–28. doi:10.1016/j.antiviral.2011.08.022
23. Shibata Y, Berclaz PY, Chroneos ZC, Yoshida M, Whitsett JA, Trapnell BC. GM-CSF regulates alveolar macrophage differentiation and innate immunity in the lung through PU.1. Immunity (2001) 15(4):557–67. doi:10.1016/S1074-7613(01)00218-7
24. Paine R III, Preston AM, Wilcoxen S, Jin H, Siu BB, Morris SB, et al. Granulocyte-macrophage colony-stimulating factor in the innate immune response to Pneumocystis carinii pneumonia in mice. J Immunol (2000) 164(5):2602–9. doi:10.4049/jimmunol.164.5.2602
25. Mukhopadhyay S, Varin A, Chen Y, Liu B, Tryggvason K, Gordon S. SR-A/MARCO-mediated ligand delivery enhances intracellular TLR and NLR function, but ligand scavenging from cell surface limits TLR4 response to pathogens. Blood (2011) 117(4):1319–28. doi:10.1182/blood-2010-03-276733
26. Gennari R, Alexander JW, Gianotti L, Eaves-Pyles T, Hartmann S. Granulocyte macrophage colony-stimulating factor improves survival in two models of gut-derived sepsis by improving gut barrier function and modulating bacterial clearance. Ann Surg (1994) 220(1):68–76. doi:10.1097/00000658-199407000-00010
27. Toda H, Murata A, Oka Y, Uda K, Tanaka N, Ohashi I, et al. Effect of granulocyte-macrophage colony-stimulating factor on sepsis-induced organ injury in rats. Blood (1994) 83(10):2893–8.
28. Spight D, Trapnell B, Zhao B, Berclaz P, Shanley TP. Granulocyte-macrophage-colony-stimulating factor-dependent peritoneal macrophage responses determine survival in experimentally induced peritonitis and sepsis in mice. Shock (2008) 30(4):434–42. doi:10.1097/SHK.0b013e3181673543
29. Lang RA, Metcalf D, Cuthbertson RA, Lyons I, Stanley E, Kelso A, et al. Transgenic mice expressing a hemopoietic growth factor gene (GM-CSF) develop accumulations of macrophages, blindness, and a fatal syndrome of tissue damage. Cell (1987) 51(4):675–86. doi:10.1016/0092-8674(87)90136-X
30. Brudecki L, Ferguson DA, McCall CE, El Gazzar M. Myeloid-derived suppressor cells evolve during sepsis and can enhance or attenuate the systemic inflammatory response. Infect Immun (2012) 80(6):2026–34. doi:10.1128/IAI.00239-12
31. Flohe SB, Agrawal H, Flohe S, Rani M, Bangen JM, Schade FU. Diversity of interferon gamma and granulocyte-macrophage colony-stimulating factor in restoring immune dysfunction of dendritic cells and macrophages during polymicrobial sepsis. Mol Med (2008) 14(5–6):247–56. doi:10.2119/2007-00120.Flohe
32. Eksioglu EA, Mahmood SS, Chang M, Reddy V. GM-CSF promotes differentiation of human dendritic cells and T lymphocytes toward a predominantly type 1 proinflammatory response. Exp Hematol (2007) 35(8):1163–71. doi:10.1016/j.exphem.2007.05.001
33. Becher B, Tugues S, Greter M. GM-CSF: from growth factor to central mediator of tissue inflammation. Immunity (2016) 45(5):963–73. doi:10.1016/j.immuni.2016.10.026
34. Mayuzumi N, Matsushima H, Takashima A. IL-33 promotes DC development in BM culture by triggering GM-CSF production. Eur J Immunol (2009) 39(12):3331–42. doi:10.1002/eji.200939472
35. Wang QR, Wang F, Zhu WB, Lei J, Huang YH, Wang BH, et al. GM-CSF accelerates proliferation of endothelial progenitor cells from murine bone marrow mononuclear cells in vitro. Cytokine (2009) 45(3):174–8. doi:10.1016/j.cyto.2008.12.002
36. Bussolino F, Ziche M, Wang JM, Alessi D, Morbidelli L, Cremona O, et al. In vitro and in vivo activation of endothelial cells by colony-stimulating factors. J Clin Invest (1991) 87(3):986–95. doi:10.1172/JCI115107
37. Soldi R, Primo L, Brizzi MF, Sanavio F, Aglietta M, Polentarutti N, et al. Activation of JAK2 in human vascular endothelial cells by granulocyte-macrophage colony-stimulating factor. Blood (1997) 89(3):863–72.
38. Pangault C, Le Tulzo Y, Tattevin P, Guilloux V, Bescher N, Drénou B. Down-modulation of granulocyte macrophage-colony stimulating factor receptor on monocytes during human septic shock. Crit Care Med (2006) 34(4):1193–201. doi:10.1097/01.CCM.0000207339.11477.62
39. Hollenstein U, Homoncik M, Stohlawetz PJ, Marsik C, Sieder A, Eichler HG, et al. Endotoxin down-modulates granulocyte colony-stimulating factor receptor (CD114) on human neutrophils. J Infect Dis (2000) 182(1):343–6. doi:10.1086/315659
40. Rauch PJ, Chudnovskiy A, Robbins CS, Weber GF, Etzrodt M, Hilgendorf I, et al. Innate response activator B cells protect against microbial sepsis. Science (2012) 335(6068):597–601. doi:10.1126/science.1215173
41. Weber GF, Chousterman BG, Hilgendorf I, Robbins CS, Theurl I, Gerhardt LM, et al. Pleural innate response activator B cells protect against pneumonia via a GM-CSF-IgM axis. J Exp Med (2014) 211(6):1243–56. doi:10.1084/jem.20131471
42. Williams MA, White SA, Miller JJ, Toner C, Withington S, Newland AC, et al. Granulocyte-macrophage colony-stimulating factor induces activation and restores respiratory burst activity in monocytes from septic patients. J Infect Dis (1998) 177(1):107–15. doi:10.1086/513802
43. Kemper CA, Bermudez LE, Deresinski SC. Immunomodulatory treatment of Mycobacterium avium complex bacteremia in patients with AIDS by use of recombinant granulocyte-macrophage colony-stimulating factor. J Infect Dis (1998) 177(4):914–20. doi:10.1086/515249
44. Drossou-Agakidou V, Kanakoudi-Tsakalidou F, Sarafidis K, Tzimouli V, Taparkou A, Kremenopoulos G, et al. In vivo effect of rhGM-CSF And rhG-CSF on monocyte HLA-DR expression of septic neonates. Cytokine (2002) 18(5):260–5. doi:10.1006/cyto.2002.1037
45. Selgas R, Fernandez de Castro M, Jimenez C, Carcamo C, Contreras T, Bajo MA, et al. Immunomodulation of peritoneal macrophages by granulocyte-macrophage colony-stimulating factor in humans. Kidney Int (1996) 50(6):2070–8. doi:10.1038/ki.1996.531
46. Bilgin K, Yaramis A, Haspolat K, Tas MA, Gunbey S, Derman O. A randomized trial of granulocyte-macrophage colony-stimulating factor in neonates with sepsis and neutropenia. Pediatrics (2001) 107(1):36–41. doi:10.1542/peds.107.1.36
47. Presneill JJ, Harris T, Stewart AG, Cade JF, Wilson JW. A randomized phase II trial of granulocyte-macrophage colony-stimulating factor therapy in severe sepsis with respiratory dysfunction. Am J Respir Crit Care Med (2002) 166(2):138–43. doi:10.1164/rccm.2009005
48. Rosenbloom AJ, Linden PK, Dorrance A, Penkosky N, Cohen-Melamed MH, Pinsky MR. Effect of granulocyte-monocyte colony-stimulating factor therapy on leukocyte function and clearance of serious infection in nonneutropenic patients. Chest (2005) 127(6):2139–50. doi:10.1378/chest.127.6.2139
49. Orozco H, Arch J, Medina-Franco H, Pantoja JP, Gonzalez QH, Vilatoba M, et al. Molgramostim (GM-CSF) associated with antibiotic treatment in nontraumatic abdominal sepsis: a randomized, double-blind, placebo-controlled clinical trial. Arch Surg (2006) 141(2):150–3; discussion 4. doi:10.1001/archsurg.141.2.150
50. Meisel C, Schefold JC, Pschowski R, Baumann T, Hetzger K, Gregor J, et al. Granulocyte-macrophage colony-stimulating factor to reverse sepsis-associated immunosuppression: a double-blind, randomized, placebo-controlled multicenter trial. Am J Respir Crit Care Med (2009) 180(7):640–8. doi:10.1164/rccm.200903-0363OC
51. Schefold JC, Zeden JP, Pschowski R, Hammoud B, Fotopoulou C, Hasper D, et al. Treatment with granulocyte-macrophage colony-stimulating factor is associated with reduced indoleamine 2,3-dioxygenase activity and kynurenine pathway catabolites in patients with severe sepsis and septic shock. Scand J Infect Dis (2010) 42(3):164–71. doi:10.3109/00365540903405768
52. Leentjens J, Kox M, Koch RM, Preijers F, Joosten LA, van der Hoeven JG, et al. Reversal of immunoparalysis in humans in vivo: a double-blind, placebo-controlled, randomized pilot study. Am J Respir Crit Care Med (2012) 186(9):838–45. doi:10.1164/rccm.201204-0645OC
53. Bo L, Wang F, Zhu J, Li J, Deng X. Granulocyte-colony stimulating factor (G-CSF) and granulocyte-macrophage colony stimulating factor (GM-CSF) for sepsis: a meta-analysis. Crit Care (2011) 15(1):R58. doi:10.1186/cc10031
54. Panopoulos AD, Watowich SS. Granulocyte colony-stimulating factor: molecular mechanisms of action during steady state and ‘emergency’ hematopoiesis. Cytokine (2008) 42(3):277–88. doi:10.1016/j.cyto.2008.03.002
55. Roberts AW. G-CSF: a key regulator of neutrophil production, but that’s not all! Growth Factors (2005) 23(1):33–41. doi:10.1080/08977190500055836
56. Eyles JL, Hickey MJ, Norman MU, Croker BA, Roberts AW, Drake SF, et al. A key role for G-CSF-induced neutrophil production and trafficking during inflammatory arthritis. Blood (2008) 112(13):5193–201. doi:10.1182/blood-2008-02-139535
57. Roberts AW, Zaiss M, Boyd AW, Nicola NA. G-CSF-mobilized peripheral blood progenitor cells: in vitro growth pattern and hematopoietic growth factor receptor profile. Exp Hematol (1997) 25(4):298–305.
58. Bendall LJ, Bradstock KF. G-CSF: from granulopoietic stimulant to bone marrow stem cell mobilizing agent. Cytokine Growth Factor Rev (2014) 25(4):355–67. doi:10.1016/j.cytogfr.2014.07.011
59. Smith TJ, Khatcheressian J, Lyman GH, Ozer H, Armitage JO, Balducci L, et al. 2006 update of recommendations for the use of white blood cell growth factors: an evidence-based clinical practice guideline. J Clin Oncol (2006) 24(19):3187–205. doi:10.1200/JCO.2006.06.4451
60. Aapro MS, Bohlius J, Cameron DA, Dal Lago L, Donnelly JP, Kearney N, et al. 2010 update of EORTC guidelines for the use of granulocyte-colony stimulating factor to reduce the incidence of chemotherapy-induced febrile neutropenia in adult patients with lymphoproliferative disorders and solid tumours. Eur J Cancer (2011) 47(1):8–32. doi:10.1016/j.ejca.2010.10.013
61. Clark OA, Lyman GH, Castro AA, Clark LG, Djulbegovic B. Colony-stimulating factors for chemotherapy-induced febrile neutropenia: a meta-analysis of randomized controlled trials. J Clin Oncol (2005) 23(18):4198–214. doi:10.1200/JCO.2005.05.645
62. Bajrami B, Zhu H, Kwak HJ, Mondal S, Hou Q, Geng G, et al. G-CSF maintains controlled neutrophil mobilization during acute inflammation by negatively regulating CXCR2 signaling. J Exp Med (2016) 213(10):1999–2018. doi:10.1084/jem.20160393
63. Gregory AD, Hogue LA, Ferkol TW, Link DC. Regulation of systemic and local neutrophil responses by G-CSF during pulmonary Pseudomonas aeruginosa infection. Blood (2007) 109(8):3235–43. doi:10.1182/blood-2005-01-015081
64. Attalah HL, Azoulay E, Yang K, Lasclos C, Jouault H, Soussy CJ, et al. Granulocyte colony-stimulating factor enhances host defenses against bacterial pneumonia following peritonitis in nonneutropenic rats. Crit Care Med (2002) 30(9):2107–14. doi:10.1097/00003246-200209000-00026
65. Balamayooran G, Batra S, Theivanthiran B, Cai S, Pacher P, Jeyaseelan S. Intrapulmonary G-CSF rescues neutrophil recruitment to the lung and neutrophil release to blood in Gram-negative bacterial infection in MCP-1-/- mice. J Immunol (2012) 189(12):5849–59. doi:10.4049/jimmunol.1200585
66. O’Reilly M, Silver GM, Greenhalgh DG, Gamelli RL, Davis JH, Hebert JC. Treatment of intra-abdominal infection with granulocyte colony-stimulating factor. J Trauma (1992) 33(5):679–82. doi:10.1097/00005373-199211000-00014
67. Liu A, Weiss S, Fang H, Claus RA, Rodel J, Dirsch O, et al. Lipopolysaccharide-binding protein (LBP) blockade augments the protective effect of granulocyte colony-stimulating factor (G-CSF) in a rat sepsis model. Shock (2015) 43(5):497–503. doi:10.1097/SHK.0000000000000338
68. Tanaka H, Ishikawa K, Nishino M, Shimazu T, Yoshioka T. Changes in granulocyte colony-stimulating factor concentration in patients with trauma and sepsis. J Trauma (1996) 40(5):718–25; discussion 25–6. doi:10.1097/00005373-199605000-00006
69. Ishikawa K, Tanaka H, Nakamori Y, Hosotsubo H, Ogura H, Nishino M, et al. Difference in the responses after administration of granulocyte colony-stimulating factor in septic patients with relative neutropenia. J Trauma (2000) 48(5):814–24; discussion 24–5. doi:10.1097/00005373-200005000-00004
70. Nelson S, Belknap SM, Carlson RW, Dale D, DeBoisblanc B, Farkas S, et al. A randomized controlled trial of filgrastim as an adjunct to antibiotics for treatment of hospitalized patients with community-acquired pneumonia. CAP study group. J Infect Dis (1998) 178(4):1075–80. doi:10.1086/515694
71. Nelson S, Heyder AM, Stone J, Bergeron MG, Daugherty S, Peterson G, et al. A randomized controlled trial of filgrastim for the treatment of hospitalized patients with multilobar pneumonia. J Infect Dis (2000) 182(3):970–3. doi:10.1086/315775
72. Root RK, Lodato RF, Patrick W, Cade JF, Fotheringham N, Milwee S, et al. Multicenter, double-blind, placebo-controlled study of the use of filgrastim in patients hospitalized with pneumonia and severe sepsis. Crit Care Med (2003) 31(2):367–73. doi:10.1097/01.CCM.0000048629.32625.5D
73. Hartmann P, Lammertink J, Mansmann G, Hübel K, Salzberger B, Stützer H, et al. A randomized, placebo-controlled study of the use of filgrastim in non neutropenic patients with nosocomial pneumonia. Eur J Med Res (2005) 10(1):29–35.
74. Stephens DP, Thomas JH, Higgins A, Bailey M, Anstey NM, Currie BJ, et al. Randomized, double-blind, placebo-controlled trial of granulocyte colony-stimulating factor in patients with septic shock. Crit Care Med (2008) 36(2):448–54. doi:10.1097/01.CCM.0B013E318161E480
75. Asakura E, Hanamura T, Umemura A, Yada K, Yamauchi T, Tanabe T. Effects of macrophage colony-stimulating factor (M-CSF) on lipopolysaccharide (LPS)-induced mediator production from monocytes in vitro. Immunobiology (1996) 195(3):300–13. doi:10.1016/S0171-2985(96)80047-7
76. Gruber MF, Gerrard TL. Production of macrophage colony-stimulating factor (M-CSF) by human monocytes is differentially regulated by GM-CSF, TNF alpha, and IFN-gamma. Cell Immunol (1992) 142(2):361–9. doi:10.1016/0008-8749(92)90297-3
77. Ogiku M, Kono H, Ishii K, Hosomura N, Fujii H. Role of macrophage colony-stimulating factor in polymicrobial sepsis according to studies using osteopetrotic (op/op) mice. J Surg Res (2011) 169(1):106–16. doi:10.1016/j.jss.2009.10.023
78. Roilides E, Lyman CA, Mertins SD, Cole DJ, Venzon D, Pizzo PA, et al. Ex vivo effects of macrophage colony-stimulating factor on human monocyte activity against fungal and bacterial pathogens. Cytokine (1996) 8(1):42–8. doi:10.1006/cyto.1996.0006
79. Gioulekas E, Goutzioulis M, Farmakis C, Drossou V, Kremenopoulos G, Tsiouris J, et al. Effects of macrophage colony-stimulating factor on antifungal activity of neonatal monocytes against Candida albicans. Biol Neonate (2001) 80(4):251–6. doi:10.1159/000047152
80. Gonzalez CE, Lyman CA, Lee S, Del Guercio C, Roilides E, Bacher J, et al. Recombinant human macrophage colony-stimulating factor augments pulmonary host defences against Aspergillus fumigatus. Cytokine (2001) 15(2):87–95. doi:10.1006/cyto.2001.0889
81. Roilides E, Sein T, Holmes A, Chanock S, Blake C, Pizzo PA, et al. Effects of macrophage colony-stimulating factor on antifungal activity of mononuclear phagocytes against Aspergillus fumigatus. J Infect Dis (1995) 172(4):1028–34. doi:10.1093/infdis/172.4.1028
82. Bettina A, Zhang Z, Michels K, Cagnina RE, Vincent IS, Burdick MD, et al. M-CSF mediates host defense during bacterial pneumonia by promoting the survival of lung and liver mononuclear phagocytes. J Immunol (2016) 196(12):5047–55. doi:10.4049/jimmunol.1600306
83. Munn DH, Bree AG, Beall AC, Kaviani MD, Sabio H, Schaub RG, et al. Recombinant human macrophage colony-stimulating factor in nonhuman primates: selective expansion of a CD16+ monocyte subset with phenotypic similarity to primate natural killer cells. Blood (1996) 88(4):1215–24.
84. Sweet MJ, Campbell CC, Sester DP, Xu D, McDonald RC, Stacey KJ, et al. Colony-stimulating factor-1 suppresses responses to CpG DNA and expression of toll-like receptor 9 but enhances responses to lipopolysaccharide in murine macrophages. J Immunol (2002) 168(1):392–9. doi:10.4049/jimmunol.168.1.392
85. Francois B, Trimoreau F, Vignon P, Fixe P, Praloran V, Gastinne H. Thrombocytopenia in the sepsis syndrome: role of hemophagocytosis and macrophage colony-stimulating factor. Am J Med (1997) 103(2):114–20. doi:10.1016/S0002-9343(97)00136-8
86. Fancke B, Suter M, Hochrein H, O’Keeffe M. M-CSF: a novel plasmacytoid and conventional dendritic cell poietin. Blood (2008) 111(1):150–9. doi:10.1182/blood-2007-05-089292
87. Lapko N, Zawadka M, Polosak J, Worthen GS, Danet-Desnoyers G, Puzianowska-Kuznicka M, et al. Long-term monocyte dysfunction after sepsis in humanized mice is related to persisted activation of macrophage-colony stimulation factor (M-CSF) and demethylation of PU.1, and it can be reversed by blocking M-CSF in vitro or by transplanting naive autologous stem cells in vivo. Front Immunol (2017) 8:401. doi:10.3389/fimmu.2017.00401
88. Williams GT, Smith CA, Spooncer E, Dexter TM, Taylor DR. Haemopoietic colony stimulating factors promote cell survival by suppressing apoptosis. Nature (1990) 343(6253):76–9. doi:10.1038/343076a0
89. Frendl G. Interleukin 3: from colony-stimulating factor to pluripotent immunoregulatory cytokine. Int J Immunopharmacol (1992) 14(3):421–30. doi:10.1016/0192-0561(92)90172-H
90. Weber GF, Chousterman BG, He S, Fenn AM, Nairz M, Anzai A, et al. Interleukin-3 amplifies acute inflammation and is a potential therapeutic target in sepsis. Science (2015) 347(6227):1260–5. doi:10.1126/science.aaa4268
91. Bentzer P, Fjell C, Walley KR, Boyd J, Russell JA. Plasma cytokine levels predict response to corticosteroids in septic shock. Intensive Care Med (2016) 42(12):1970–9. doi:10.1007/s00134-016-4338-z
92. Lutz MB. IL-3 in dendritic cell development and function: a comparison with GM-CSF and IL-4. Immunobiology (2004) 209(1–2):79–87. doi:10.1016/j.imbio.2004.03.001
93. Thiebaut R, Jarne A, Routy JP, Sereti I, Fischl M, Ive P, et al. Repeated cycles of recombinant human interleukin 7 in HIV-infected patients with low CD4 T-cell reconstitution on antiretroviral therapy: results of 2 phase II multicenter studies. Clin Infect Dis (2016) 62(9):1178–85. doi:10.1093/cid/ciw065
94. Unsinger J, McGlynn M, Kasten KR, Hoekzema AS, Watanabe E, Muenzer JT, et al. IL-7 promotes T cell viability, trafficking, and functionality and improves survival in sepsis. J Immunol (2010) 184(7):3768–79. doi:10.4049/jimmunol.0903151
95. Kasten KR, Prakash PS, Unsinger J, Goetzman HS, England LG, Cave CM, et al. Interleukin-7 (IL-7) treatment accelerates neutrophil recruitment through gamma delta T-cell IL-17 production in a murine model of sepsis. Infect Immun (2010) 78(11):4714–22. doi:10.1128/IAI.00456-10
96. Unsinger J, Burnham CA, McDonough J, Morre M, Prakash PS, Caldwell CC, et al. Interleukin-7 ameliorates immune dysfunction and improves survival in a 2-hit model of fungal sepsis. J Infect Dis (2012) 206(4):606–16. doi:10.1093/infdis/jis383
97. Shindo Y, Unsinger J, Burnham CA, Green JM, Hotchkiss RS. Interleukin-7 and anti-programmed cell death 1 antibody have differing effects to reverse sepsis-induced immunosuppression. Shock (2015) 43(4):334–43. doi:10.1097/SHK.0000000000000317
98. Terashima A, Okamoto K, Nakashima T, Akira S, Ikuta K, Takayanagi H. Sepsis-induced osteoblast ablation causes immunodeficiency. Immunity (2016) 44(6):1434–43. doi:10.1016/j.immuni.2016.05.012
99. Shindo Y, Fuchs AG, Davis CG, Eitas T, Unsinger J, Burnham CD, et al. Interleukin 7 immunotherapy improves host immunity and survival in a two-hit model of Pseudomonas aeruginosa pneumonia. J Leukoc Biol (2017) 101(2):543–54. doi:10.1189/jlb.4A1215-581R
100. White M, Mahon V, Grealy R, Doherty DG, Stordeur P, Kelleher DP, et al. Post-operative infection and sepsis in humans is associated with deficient gene expression of gammac cytokines and their apoptosis mediators. Crit Care (2011) 15(3):R158. doi:10.1186/cc10293
101. Andreu-Ballester JC, Cuellar C, Garcia-Ballesteros C, Perez-Griera J, Amigo V, Peiro-Gomez A, et al. Deficit of interleukin 7 in septic patients. Int Immunopharmacol (2014) 23(1):73–6. doi:10.1016/j.intimp.2014.08.015
102. Boomer JS, Shuherk-Shaffer J, Hotchkiss RS, Green JM. A prospective analysis of lymphocyte phenotype and function over the course of acute sepsis. Crit Care (2012) 16(3):R112. doi:10.1186/cc11404
103. Demaret J, Villars-Mechin A, Lepape A, Plassais J, Vallin H, Malcus C, et al. Elevated plasmatic level of soluble IL-7 receptor is associated with increased mortality in septic shock patients. Intensive Care Med (2014) 40(8):1089–96. doi:10.1007/s00134-014-3346-0
104. Venet F, Foray AP, Villars-Mechin A, Malcus C, Poitevin-Later F, Lepape A, et al. IL-7 restores lymphocyte functions in septic patients. J Immunol (2012) 189(10):5073–81. doi:10.4049/jimmunol.1202062
105. Rogiers P, Zhang H, Leeman M, Nagler J, Neels H, Melot C, et al. Erythropoietin response is blunted in critically ill patients. Intensive Care Med (1997) 23(2):159–62. doi:10.1007/s001340050310
106. Abel J, Spannbrucker N, Fandrey J, Jelkmann W. Serum erythropoietin levels in patients with sepsis and septic shock. Eur J Haematol (1996) 57(5):359–63. doi:10.1111/j.1600-0609.1996.tb01393.x
107. Heitrich M, Garcia DM, Stoyanoff TR, Rodriguez JP, Todaro JS, Aguirre MV. Erythropoietin attenuates renal and pulmonary injury in polymicrobial induced-sepsis through EPO-R, VEGF and VEGF-R2 modulation. Biomed Pharmacother (2016) 82:606–13. doi:10.1016/j.biopha.2016.05.045
108. Kandasamy K, Choudhury S, Singh V, Addison MP, Darzi SA, Kasa JK, et al. Erythropoietin reverses sepsis-induced vasoplegia to norepinephrine through preservation of alpha1D-adrenoceptor mRNA expression and inhibition of GRK2-mediated desensitization in mouse aorta. J Cardiovasc Pharmacol Ther (2016) 21(1):100–13. doi:10.1177/1074248415587968
109. Zhang X, Dong S, Qin Y, Bian X. Protective effect of erythropoietin against myocardial injury in rats with sepsis and its underlying mechanisms. Mol Med Rep (2015) 11(5):3317–29. doi:10.3892/mmr.2015.3155
110. Kao RL, Martin CM, Xenocostas A, Huang W, Rui T. Erythropoietin improves skeletal muscle microcirculation through the activation of eNOS in a mouse sepsis model. J Trauma (2011) 71(5 Suppl 1):S462–7. doi:10.1097/TA.0b013e318232e7a2
111. Gao R, Tang YH, Tong JH, Yang JJ, Ji MH, Zhu SH. Systemic lipopolysaccharide administration-induced cognitive impairments are reversed by erythropoietin treatment in mice. Inflammation (2015) 38(5):1949–58. doi:10.1007/s10753-015-0175-4
112. Comim CM, Cassol OJ Jr, Abreu I, Moraz T, Constantino LS, Vuolo F, et al. Erythropoietin reverts cognitive impairment and alters the oxidative parameters and energetic metabolism in sepsis animal model. J Neural Transm (Vienna) (2012) 119(11):1267–74. doi:10.1007/s00702-012-0774-2
113. Stoyanoff TR, Todaro JS, Aguirre MV, Zimmermann MC, Brandan NC. Amelioration of lipopolysaccharide-induced acute kidney injury by erythropoietin: involvement of mitochondria-regulated apoptosis. Toxicology (2014) 318:13–21. doi:10.1016/j.tox.2014.01.011
114. Coldewey SM, Khan AI, Kapoor A, Collino M, Rogazzo M, Brines M, et al. Erythropoietin attenuates acute kidney dysfunction in murine experimental sepsis by activation of the beta-common receptor. Kidney Int (2013) 84(3):482–90. doi:10.1038/ki.2013.118
115. Solling C, Christensen AT, Nygaard U, Krag S, Frokiaer J, Wogensen L, et al. Erythropoietin does not attenuate renal dysfunction or inflammation in a porcine model of endotoxemia. Acta Anaesthesiol Scand (2011) 55(4):411–21. doi:10.1111/j.1399-6576.2011.02396.x
116. Wu WT, Hu TM, Lin NT, Subeq YM, Lee RP, Hsu BG. Low-dose erythropoietin aggravates endotoxin-induced organ damage in conscious rats. Cytokine (2010) 49(2):155–62. doi:10.1016/j.cyto.2009.11.002
117. Koroglu TF, Yilmaz O, Gokmen N, Tugyan K, Baskin H, Egrilmez MY. Erythropoietin prevents lymphoid apoptosis but has no effect on survival in experimental sepsis. Pediatr Res (2013) 74(2):148–53. doi:10.1038/pr.2013.86
118. Corwin HL, Gettinger A, Pearl RG, Fink MP, Levy MM, Shapiro MJ, et al. Efficacy of recombinant human erythropoietin in critically ill patients: a randomized controlled trial. JAMA (2002) 288(22):2827–35. doi:10.1001/jama.288.22.2827
119. Corwin HL, Gettinger A, Fabian TC, May A, Pearl RG, Heard S, et al. Efficacy and safety of epoetin alfa in critically ill patients. N Engl J Med (2007) 357(10):965–76. doi:10.1056/NEJMoa071533
120. Zarychanski R, Turgeon AF, McIntyre L, Fergusson DA. Erythropoietin-receptor agonists in critically ill patients: a meta-analysis of randomized controlled trials. CMAJ (2007) 177(7):725–34. doi:10.1503/cmaj.071055
121. Pearl RG. Erythropoietin and organ protection: lessons from negative clinical trials. Crit Care (2014) 18(5):526. doi:10.1186/s13054-014-0526-9
122. Nichol A, French C, Little L, Presneill J, Cooper DJ, Haddad S, et al. Erythropoietin in traumatic brain injury: study protocol for a randomised controlled trial. Trials (2015) 16:39. doi:10.1186/s13063-014-0528-6
Keywords: hematopoietic growth factors, G-CSF, GM-CSF, M-CSF, IL-7, IL-3, EPO, sepsis
Citation: Chousterman BG and Arnaud M (2018) Is There a Role for Hematopoietic Growth Factors During Sepsis? Front. Immunol. 9:1015. doi: 10.3389/fimmu.2018.01015
Received: 30 November 2017; Accepted: 23 April 2018;
Published: 21 June 2018
Edited by:
Tobias Schuerholz, Universitätsmedizin Rostock, GermanyReviewed by:
Oreste Gualillo, Servicio Gallego de Salud, SpainSergio Iván Valdés-Ferrer, Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán, Mexico
Copyright: © 2018 Chousterman and Arnaud. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Benjamin G. Chousterman, YmVuamFtaW4uY2hvdXN0ZXJtYW5AYXBocC5mcg==