 Charles Arama1*
Charles Arama1* Jaclyn E. Quin2
Jaclyn E. Quin2 Bourèma Kouriba1
Bourèma Kouriba1 Ann-Kristin Östlund Farrants2
Ann-Kristin Östlund Farrants2 Marita Troye-Blomberg2*
Marita Troye-Blomberg2* Ogobara K. Doumbo1†
Ogobara K. Doumbo1†
- 1Malaria Research and Training Centre, Department of Epidemiology of Parasitic Diseases, International Center of Excellence in Research, University of Sciences, Technique and Technology of Bamako, Bamako, Mali
- 2Department of Molecular Biosciences, The Wenner-Gren Institute, Stockholm University, Stockholm, Sweden
A better understanding of stable changes in regulation of gene expression that result from epigenetic events is of great relevance in the development of strategies to prevent and treat infectious diseases. Histone modification and DNA methylation are key epigenetic mechanisms that can be regarded as marks, which ensure an accurate transmission of the chromatin states and gene expression profiles over generations of cells. There is an increasing list of these modifications, and the complexity of their action is just beginning to be understood. It is clear that the epigenetic landscape plays a fundamental role in most biological processes that involve the manipulation and expression of DNA. Although the molecular mechanism of gene regulation is relatively well understood, the hierarchical order of events and dependencies that lead to protection against infection remain largely unknown. In this review, we propose that host epigenetics is an essential, though relatively under studied, factor in the protection or susceptibility to malaria.
Introduction
Complex infectious diseases such as malaria, in which environmental and clinical features along with genetic susceptibility factors contribute significantly to the pathology of the disease, pose a great challenge for the identification of relevant biomarkers for protection against the disease. In the human host, the disease outcome is determined by a complex relationship between the host, the parasite, and the environment (1). For example, parasite virulence, infection burden, the route of inoculation, host’s immunity and susceptibility to infection, nutrition and gut microbiota, previous exposure to antimalarial drugs (2), or coinfection, such as with helminthes, virus, and bacteria (3) may influence disease severity. The contribution of immunogenetic factors to resistance against malaria has been thoroughly investigated (4, 5). However, immunogenetic biomarkers that significantly correlate with a more protective immune response against malaria are not yet available. Recent studies generating large sequencing and immune variable data sets have begun to provide valuable insights (6). However, to date, a lack of data has hampered the identification of functional genomic features as well as the discovery of specific roles of genes in malaria.
Our understanding of the role of epigenetics in complex disease is rapidly emerging. Epigenetics play an important role in hematopoiesis, for example, proliferation of hematopoietic stem cells, as well as the successive stages of differentiation into more committed progenitors, are regulated at the transcriptional level through epigenetic modifications (7). Although many works in epigenetics have been conducted in the field of hematopoietic cancers and autoimmune diseases, there is still a gap of knowledge of how these epigenetics mechanisms contribute to the susceptibility or resistance to infectious diseases such as malaria.
In this review, we present and discuss the potential role of epigenetic factors in the protection/susceptibility to diseases. Particularly, we reviewed how the current knowledge on how epigenetic regulation contributes to protective immunity and susceptibility to disease can be applied to address the challenge of malaria.
Genetic Variations and Susceptibility to Malaria
Knowledge of the host genetic susceptibility to malaria is key to understanding the complexity of the host immune response and its interaction with parasite infection. Plasmodium has been a major cause of morbidity and mortality throughout human history. As a result of this, malaria is believed to have exerted evolutionary pressure on the human genome by selecting genetic polymorphisms that provide protection against severe disease (8). Hence, many studies have attempted to assess host genetic factors involved in both the host immune response to malaria and the disease outcome. The best examples of such diseases are hemoglobinopathies, in which hemoglobin S was the first described human host genetic factor associated with protection against malaria (9). Studies have shown that HbAS has a 90% protective effect against severe and lethal malaria (10) and 50% protective effect against mild clinical cases (11). In addition, the carriage of HbAS was associated with a significant delay in the time to first malaria clinical episode (12). Other hemoglobinopathies, which may have important role in the protection against malaria, include homozygote and heterozygote α-thalassemia. These genetic modifications showed a decreased risk of severe malaria in a systematic review and meta-analysis study (13). Genetic variations such as polymorphism in hemoglobin, intracellular enzymes, red-blood cell (RBC) channels, RBC-surface markers, and proteins impacting the RBC cytoskeleton and RBC morphology have also been shown to attenuate malaria pathogenesis (14). The RBC surface protein Duffy antigen receptor for chemokines (DARC) gene is one of the most compelling pieces of evidence for RBC evolution against Plasmodim vivax malaria. Mutation of the DARC gene is common among individuals in West and Central Africa and confers protection against P. vivax (15). However, recent evidence shows that P. vivax infects DARC negative individuals (16, 17). Therefore, understanding the molecular basis of genetic variations arising from selective pressure by malaria in different ethnic groups may offer insight into protective mechanisms against malaria pathogenesis.
Epigenome-Wide Association Studies (EWAS) for Common Human Diseases
Epigenetics study the mechanisms that determine and/or perpetuate genomic functions without changes in DNA sequence (18). It consists of the collective changes in phenotype due to processes that arise independently of primary DNA sequence (18).
During the past decades, genome-wide association studies have incrementally provided evidence of the association between genetic variations at a whole genome level and susceptibility to human diseases (19). However, genetic variation alone has not been able to give a clear explanation of the complex interaction between the genomic expression and the outcome of certain diseases. The impact of environmental factors on manifestation of disease may be the reason for these limitations. Particularly, different environmental conditions can result in the establishment of different epigenetic states responsible for mediating gene expression patterns and other genomic responses. This makes the epigenome an especially intriguing and interesting target to study. Recent technological advances in high-throughput genomic analysis have improved the genome-wide examination of epigenetic modifications such as DNA methylation and histone modification, collectively referred to as EWAS. These have enabled unprecedented systematic large-scale association testing in correlation with disease phenotypes. Importantly, EWAS have begun to establish the link between variation in epigenetic regulation and susceptibility to disease, including autoimmune diseases such as rheumatoid arthritis (20) and type I diabetes (21).
The difficulty in EWAS arises in the interpretation of the findings. For example, vastly different epigenetic patterns exist in distinct cell types, and thus, cell subtype effects account for a major proportion of the epigenetic changes associated with disease phenotypes (22). To date, EWAS represent an important contribution toward a better understanding of the etiological role of epigenetic variations in autoimmune diseases; however, more evidence is needed to establish the relationship with more complex infectious diseases. However, in malaria, researchers have only just begun to perform genome wide examination of epigenetic variations and protection from malaria in different ethnic groups (23). It is the ambition that further advances will help account for our gap in knowledge of what underlies the differences in clinical phenotypes of certain complex infectious diseases including malaria, tuberculosis, and AIDS.
Epigenetics Mechanisms and Acquisition of Protective Immunity
Epigenetic changes underlie both the differentiation and activation of immune cells, which are regulated by precise spatial and temporal control of gene expression (7). For example, hematopoietic stem cell proliferation and differentiation into different immune cell types requires changes in chromatin structures and nuclear architecture, which depend on complex epigenetic regulation (24–26). The importance of epigenetic processes for the function of the immune system is illustrated by the prevalence of mutations in hematopoietic epigenetic regulators in leukemia and lymphoma (27–30), as well as identification of somatic mutations of epigenetic regulators in autoimmune diseases and other immune-based disorders (31–36). Environmental exposures throughout the life span induce genetic and epigenetic alterations, particularly in susceptible populations (37). This indicates that epigenetics may also hold the key to a larger understanding of the contributing factors of human health, where early life events shape later susceptibility to disease.
The major chromatin changes in immune cells occur by DNA methylation and histone modification, but also by rearranging chromatin structure. The primary DNA modification is 5-methylcytosine (5meC) DNA methylation, arising from transfer of a methyl moiety from S-adenosylmethionine to the 5-position of cytosine in certain CpG dinucleotides with the help of the DNA methyltransferases (DNMTs). Other epigenetic DNA modifications consist of the conversion of 5-methylcytosine to 5-hydroxymethylcytosine, and adenine methylation (38). The DNA methylation pattern changes often during hematopoiesis to silence some genes by introducing CpG methylations and activate others by removing DNA methylations. Posttranslational modifications of histones such as acetylation and methylation are important in regulating the transcriptional activity of cells. They occur in a site-specific manner that influences the binding and activities of other proteins and chromatin organization (39). “Writers” such as histone acetyltransferases and methyltransferases (HMTs) catalyze histone acetylation and methylation while “erasers” such as the histone deacetylases (HDACs) and histone demethylases result in the removal modifications within the chromatin. The epigenetic modification of chromatin is precisely regulated via mediating the activity and recruitment of these enzymes to specific loci, resulting in specific changes in gene expression, chromatin organization, and other DNA regulatory processes, for example, via establishing co-regulatory transcription programs or specialized functional domains within the nucleus. These are also regulated by chromatin remodeling complexes that alter the density of chromatin (40).
Recently, it has been shown that epigenetic modifications regulate the expression of key immune system genes, underlying both the innate and adaptive immune responses (41, 42). In the adaptive immune response, the changes in phenotype that accompany T- and B-cell activation and differentiation are mediated through acquired transcriptional regulatory mechanisms, including epigenetic modifications resulting in distinct DNA methylation and histone modification patterns (43–46). For example, genome-wide DNA methylation analysis of T- and B-cells reveals distinct differences during the transition from naive to effector cells (47, 48). In memory T-cells, histone modifications epigenetically mark genes and prime them for rapid and robust transcription following exposure to specific antigens (45). In different T-cell populations, specific regions are differentially methylated, for example, the CD4 gene was hypermethylated in CD8+ T-cells and hypomethylated in CD4+ T-cells (48), while differential methylation of interferon gamma (IFN-γ) mediates differentiation of Th1 and Th2 cells (49). In contrast, in B-cell activation, DNA is predominantly hypomethylated (50). Epigenetic mechanisms also enable somatic hypermutation and class switch DNA recombination (51), thereby mediating antibody responses. Understanding the molecular mechanisms of these epigenetic changes in the memory responses of T- and B-cells may offer new areas in the development of safer and more effective vaccines (43).
In addition to classical adaptive immune memory, the innate immune system also has a memory, which manifests as a previous challenge driving an increased (“trained”) or decreased (“tolerized”) response to a second challenge in comparison to naïve cells. This altered state can persist for weeks to months following the initial stimuli and results in cells of the innate immune system, including monocytes and macrophages, being more or less capable of producing inflammatory cytokines, and/or phagocytizing and killing microorganisms, in response to a second unrelated stimuli. On one side, “tolerance” can arise following high bacterial burden, preventing responsiveness to an additional challenge, or the immunosuppressive phenotype observed in late sepsis and is viewed as a strategy to limit inflammation (52). At the other end of the spectrum, “trained” immunity can be induced following certain live vaccinations (BCG vaccination is the best characterized example), infectious stimuli, or metabolites, and is characterized by a change in cellular metabolism from oxidative phosphorylation toward aerobic glycolysis, an increased proinflammatory response, and resistance to infection (53, 54). The main mechanism by which innate cells develop a memory is through long-term epigenetic reprogramming (54, 55). Tolerance and trained immunity are associated with distinct and opposing epigenomic states (56). For example, monocytes tolerized by LPS treatments are associated with H3K4 monomethyaltion and a failure to accumulate H3K27 acetylation and active histone marks at the promoters of tolerized genes, such as in the lipid metabolism and phagocytic pathways, during a second challenge (57, 58). On the other hand, monocytes trained by exposure to β-glucan are associated with H3K4 trimethylation and H3K27 acetylation at the promoters of genes, enabling higher transcriptional levels in genes such as pathogen-recognition receptors, signaling molecules, and proinflammatory cytokines (57, 59).
Epigenetics and Malaria
Despite understanding that host epigenetics underlie differentiation and activation of immune cells, as well as the regulation of key genes in both the innate and adaptive immune responses, there is a vast gap of knowledge of role of epigenetic factors in the protection from or susceptibility to malaria. A systematic database search of all relevant publications illustrates how under studied the role of host epigenetics in malaria has been until very recently (Figure 1). Only 231 publications were identified that address both malaria and epigenetics, and of these, the vast majority address the role of epigenetics in gene regulation in the malaria parasite. Current knowledge of the role of epigenetics in Plasmodium biology, and how this may be exploited to combat malaria, has been comprehensively covered by recent reviews, and will not be discussed in depth here (60, 61).
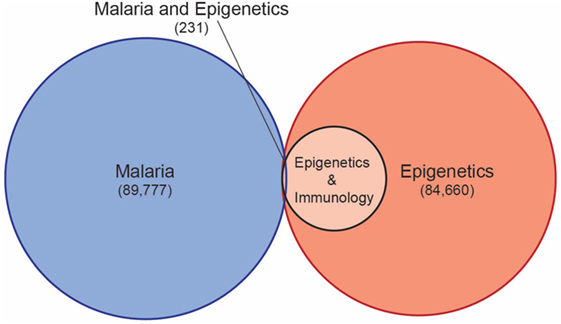
Figure 1. A database search of the Web of Science Core Collection for all publications with the topic of malaria (search term malaria*), epigenetics (search term epigen*), and epigenetics and immunology (search term epigen* and immun*), identified 12,484 publications addressing epigenetics and immunology, but only 231 publications addressing epigenetics and malaria (search term malaria* and epigen*) (https://www.webofknowledge.com accessed 20 June 2018).
The few research articles that address epigenetics of the infected host are not comprehensive but apply current knowledge of epigenetics and infection to specific questions in malaria. They investigated the epigenetic regulation of specific host genes that provide resistance to malaria (62) or mediate the immune response to malaria (e.g., promoter DNA methylation of TLR6 (63) and ABCB1 (64), and H3K4me3 at TNF, IL-6, and mTOR promoters (65)). Recently, Lessard et al. utilized epigenomic profiling to determine functionally relevant genomic sequences at a loci associated with protection from malaria (66). Others discussed the role of epigenetics in generating specific immune cell populations that contribute to susceptibility to or protection from malaria, particularly, CD4+ T-cells (67), and CD8+ T-cells (68), or the mechanisms by which testosterone (69) or nutrition (70) can confer susceptibility to or protection from malaria. Kumari et al. investigated the effect of the antimalarial drug artemisinin on epigenetic modifiers (71). Others examined global changes in miRNA expression (72) or DNA methylation in specific cell or tissue types of malaria-infected individuals (23, 64, 73). The majority of these research articles have been published within the last 2 years, hopefully indicating an interest in applying the lessons we have learned regarding the important role of epigenetics in other diseases to the challenge of malaria.
Epigenetics and Protection from Malaria
Despite the few studies that directly address the role of host epigenetics in protection from or susceptibility to malaria, a number of recent studies indicate that immune responses that are controlled by epigenetic changes are important for protection from the disease. There is growing evidence that Plasmodium can induce a state of trained innate immunity. Stimulation of human peripheral blood mononuclear cells (PBMCs) with Plasmodium falciparum, both in vivo and in vitro, results in subsequent toll-like receptor specific stimuli driving significantly higher proinflammatory responses (74, 75). The hyper-responsiveness of PBMCs that have been exposed to either P. falciparum-infected RBCs or hemozoin is associated with increased H3K4 trimethylation at specific immuno-metabolic promoters (65). This trained immunity may be associated with protection from malaria. In P. falciparum malaria, patients who display the production of malaria-specific IFN-γ by PBMCs have significantly lower rates of reinfection (76, 77). Further, a recent study of the Fulani, an ethnic group with lower susceptibility to malaria, found that following P. falciparum infection, the Fulani displayed characteristics suggestive of trained immunity, with more transcriptionally reactive monocytes and a more pro-inflammatory response relative to a sympatric ethnic group (72). Finally, in mice BCG vaccination, which is known to induce trained innate immunity, results in reduced parasitemia during a subsequent challenge with malaria parasites (78). Conversely, malaria may induce innate immune tolerance, particularly, after multiple infections or in patients in which the infection progresses without treatment (79). Individuals historically infected with malaria as therapy for neurosyphilis exhibited depressed responses to a subsequent challenge with heat-killed Salmonella (80). The parasite burden that initiates symptomatic malaria increases with multiple infections, with individuals in endemic areas infected with P. falciparum for weeks to months while remaining apparently healthy (81, 82). Therefore, the dynamics of epigenetic regulation of innate immune memory, to either a “trained” or “tolerized” phenotype, has implications for the outcome of the disease.
A role for epigenetics in protection from malaria may also extend to nutrition, microbiome, and other factors that affect the metabolome. The role of dietary nutrients and gut microbiota in influencing immune function has now been well established (83–85). For example, probiotic commensal bacteria can dampen immune activation, protecting against allergy development (86, 87). The different activation of immune cells is underscored by epigenetic changes (88–90). In the context of malaria, gut microbiota has a potential role in resistance of individuals to malaria (91). Particularly, the role of dietary advanced glycation end-products in the modulation of immune responses through chronic oxidative stress, which mediates epigenetic and transcriptional programs, has been established, which also seems to play a role in the natural protection against malaria (92).
Whole blood genomic DNA studies of global DNA methylation in individuals infected with P. falciparum have shown that global DNA methylation levels are inversely proportional to parasitemia, with reduced 5meC levels in infected compared to uninfected individuals (64). However, currently, it is unclear whether this is due to a strategy of the host immune system to mediate the immune response, or alternatively whether the parasite and/or its by-products can change methylation levels in the host genome to provide itself with a survival advantage.
Epigenetics and Susceptibility to Malaria: The other Side of the Coin
Immunity to malaria is short-lived, despite repeated parasite exposure in endemic areas, and the established strong selective force of malaria on human populations. Malaria parasites have evolved to escape the immune system of the human host, utilizing a number of mechanisms including allelic variation and modification of host cell phenotype (93). Recently, the ability of the parasite to impact host immune response through modulating epigenetic and transcriptional pathways has been proposed. For example, malaria patients display reduced numbers of circulating dendritic cells (DCs) and an accumulation of immature DCs (94), and further studies have implicated the malaria pigment hemozoin in partially preventing DC maturation and capacity to activate T-cells (95–98). However, so far, the role of epigenetic mechanisms have not been established, only implied. For example, the malaria parasite and its by-products drive DNA hypomethylation and increased expression of the ABCB1 gene (99), and the hypothesis that this multidrug resistance transporter protein is regulated by malaria infection to eliminate hemoglobin degradation products is being investigated (64). In support of this, other parasites have demonstrated ability to co-opt host epigenetic mechanisms to orchestrate changes in host gene expression (100). For example, Mycobacterium tuberculosis inhibits IFN-γ-induced expression of several immune genes through histone acetylation, contributing to the persistence of long-term chronic tuberculosis infections in some patients (101–103). Other protozoan parasites specifically, including Leishmania and Toxoplasma, employ a variety of strategies to actively modulate host immune epigenome and transcriptome. For example, Leishmania parasite replicates in the macrophages of its mammalian host, where it efficiently inhibits activation of innate immune reponses, such as antigen presentation, IFN-γ, and activation of cytokines and chemokines. Infection of macrophages with Leishmania donovani, compared to a heat-killed control, results in global changes in DNA methylation, including at genes involved in macrophage activation (104, 105). The intracellular parasite Toxoplasma gondii can prevent chromatin remodeling and association of transcription factors at the TNF-α promoter, and mediate levels of DNA methylation at the arginine vasopressin (Avp) gene promoter (103, 106, 107).
The other side of the coin is also related to the role of immune response in the pathology of malaria as a disease. Severe pathophysiological events during malaria infection include erythrocyte destruction and ineffective erythropoiesis, adhesion of Plasmodium-infected red blood cells to capillary veins of vital host organs, and excessive production and the release of proinflammatory cytokines (108). These symptoms are driven by high levels of proinflammatory immune responses. In escaping the host immunity, the parasite may also prevent the development of symptoms of severe malaria infection. Thus, understanding the epigenetic mechanisms by which the parasite stimulates or evades immune response in the human host may shed light into how this complex host–parasite interaction results in the pathology of the disease.
Conclusion
Despite long lasting efforts to control and eliminate malaria infection, the disease remains a public health concern in sub-Saharan Africa. According the World Health Organization (WHO), malaria remains a major cause of morbidity and mortality, causing an estimated 445,000 deaths globally in 2016 (109). Sub-Saharan Africa is the most affected region with 92% of global malaria death. Among these, 88% occurs in children under 5 years of age (109). Challenges in malaria include difficulties in estimating the changing burden of disease due to limitations in health reporting systems in many African countries, inequality in malaria intervention coverage in countries with slow growth or a large baseline inequality (110), emergence of mosquitos resistant to insecticides, and the spread of artemisinin-resistant malaria parasites in Southeast Asia (111). Consequently, progress against malaria has stalled, with increased worldwide incidence of malaria reported for the first time in recent history (109).
The development of new strategies that will explore new research avenues and hypotheses on biological factors of malaria susceptibility or resistance are urgently needed. This should be a priority if the goal of eradication is to be achieved by 2030.
Interestingly, growing evidence suggests that epigenetics play a key role to multiple levels of this complex disease, including immune evasion by the parasite, tolerance, training, and adaptive responses. The tools and frameworks are readily available to investigate the impact of epigenetics in the protection from or susceptibility to malaria in more depth. In other fields, drugs targeting epigenetic enzymes and processes have already advanced to clinic. For example, the DNMT inhibitors and HDAC inhibitors have proven efficacious in the treatment of cancer, particularly hematopoietic cancers (29, 112, 113). Inhibitors of chromatin modifying enzymes that target the Plasmodium parasite have already been considered as an antimalarial strategy (61). Understanding how epigenetic mechanisms in the human host impact the disease outcome during malaria, through either driving or dampening immune responses, may offer a relatively achievable approach to develop new strategies that can be applied to the treatment of the disease.
There are several challenges that epigenetic epidemiological studies have to address to elucidate fully the role of epigenetics in susceptibility or protection to malaria. One of these challenges is to understand how the parasite alters the host immune responses by exerting a strong selective pressure on population genetics in endemic regions. A better understanding of the mechanisms that underlie the chromatin and DNA methylation changes are topics of scientific interest. Such studies will generate more evidence in the role of epigenetics in the acquisition of protective immunity against infectious disease such as malaria. As we gain insight into the functional significance of changes in DNA methylation events and other epigenetic mechanisms, there will be a push to manipulate these processes, as tools and strategies to develop vaccines and target for therapeutic discoveries.
Author Contributions
CA: conceptualization, writing—original draft, writing—review and editing; BK and JQ: writing—review and editing; MT-B, A-KF, and OD: conceptualization, writing—review and editing. All authors read and approved the final manuscript and agreed to be accountable for all aspects of the work.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported through the DELTAS Africa Initiative (grant 107740/Z/15/Z). The DELTAS Africa Initiative is an independent funding scheme of the African Academy of Sciences (AAS)’s Alliance for Accelerating Excellence in Science in Africa (AESA) and supported by the New Partnership for Africa’s Development Planning and Coordinating Agency (NEPAD Agency) with funding from the Wellcome Trust (grant #PD00217ML) and the UK government. The views expressed in this publication are those of the author(s) and not necessarily those of AAS, NEPAD Agency, Wellcome Trust, or the UK government. We thank Dr. Amed Ouattara for the critical reading and encouragement for this review.
Funding
The Developing Excellence in Leadership and Genetics Training for Malaria Elimination in sub-Saharan Africa (DELGEME) program (grant #PD00217ML) sponsored by the Wellcome Trust Developing Excellence in Leadership, Training, and Science Africa (DELTAS Africa) initiative.
References
1. Miller LH, Baruch DI, Marsh K, Doumbo OK. The pathogenic basis of malaria. Nature (2002) 415:673–9. doi:10.1038/415673a
3. Troye-Blomberg M, Berzins K. Immune interactions in malaria co-infections with other endemic infectious diseases: implications for the development of improved disease interventions. Microbes Infect (2008) 10:948–52. doi:10.1016/j.micinf.2008.07.014
4. Hill AV, Allsopp CE, Kwiatkowski D, Anstey NM, Twumasi P, Rowe PA, et al. Common west African HLA antigens are associated with protection from severe malaria. Nature (1991) 352:595–600. doi:10.1038/352595a0
5. Modiano D, Luoni G, Sirima BS, Lanfrancotti A, Petrarca V, Cruciani F, et al. The lower susceptibility to Plasmodium falciparum malaria of Fulani of Burkina Faso (west Africa) is associated with low frequencies of classic malaria-resistance genes. Trans R Soc Trop Med Hyg (2001) 95:149–52. doi:10.1016/S0035-9203(01)90141-5
6. Steri M, Orrù V, Idda ML, Pitzalis M, Pala M, Zara I, et al. Overexpression of the cytokine BAFF and autoimmunity risk. N Engl J Med (2017) 376:1615–26. doi:10.1056/NEJMoa1610528
7. Sharma S, Gurudutta G. Epigenetic regulation of hematopoietic stem cells. Int J Stem Cells (2016) 9:36–43. doi:10.15283/ijsc.2016.9.1.36
8. Gong L, Parikh S, Rosenthal PJ, Greenhouse B. Biochemical and immunological mechanisms by which sickle cell trait protects against malaria. Malar J (2013) 12:317. doi:10.1186/1475-2875-12-317
9. Allison AC. Protection afforded by sickle-cell trait against subtertian malarial infection. Br Med J (1954) 1:290–4. doi:10.1136/bmj.1.4857.290
10. Aidoo M, Terlouw DJ, Kolczak MS, McElroy PD, ter Kuile FO, Kariuki S, et al. Protective effects of the sickle cell gene against malaria morbidity and mortality. Lancet (2002) 359:1311–2. doi:10.1016/S0140-6736(02)08273-9
11. Williams TN, Mwangi TW, Wambua S, Alexander ND, Kortok M, Snow RW, et al. Sickle cell trait and the risk of Plasmodium falciparum malaria and other childhood diseases. J Infect Dis (2005) 192:178–86. doi:10.1086/430744
12. Crompton PD, Traore B, Kayentao K, Doumbo S, Ongoiba A, Diakite SAS, et al. Sickle cell trait is associated with a delayed onset of malaria: implications for time-to-event analysis in clinical studies of malaria. J Infect Dis (2008) 198:1265–75. doi:10.1086/592224
13. Taylor SM, Parobek CM, Fairhurst RM. Impact of haemoglobinopathies on the clinical epidemiology of malaria: a systematic review and meta-analysis. Lancet Infect Dis (2012) 12:457–68. doi:10.1016/S1473-3099(12)70055-5
14. Goheen MM, Campino S, Cerami C. The role of the red blood cell in host defence against falciparum malaria: an expanding repertoire of evolutionary alterations. Br J Haematol (2017) 179:543–56. doi:10.1111/bjh.14886
15. Miller LH, Mason SJ, Clyde DF, McGinniss MH. The resistance factor to Plasmodium vivax in blacks. The Duffy-blood-group genotype, FyFy. N Engl J Med (1976) 295:302–4. doi:10.1056/NEJM197608052950602
16. Niangaly A, Gunalan K, Ouattara A, Coulibaly D, Sá JM, Adams M, et al. Plasmodium vivax infections over 3 years in duffy blood group negative Malians in Bandiagara, Mali. Am J Trop Med Hyg (2017) 97:744–52. doi:10.4269/ajtmh.17-0254
17. Gunalan K, Niangaly A, Thera MA, Doumbo OK, Miller LH. Plasmodium vivax infections of duffy-negative erythrocytes: historically undetected or a recent adaptation? Trends Parasitol (2018) 34:420–9. doi:10.1016/j.pt.2018.02.006
18. Goldberg AD, Allis CD, Bernstein E. Epigenetics: a landscape takes shape. Cell (2007) 128:635–8. doi:10.1016/j.cell.2007.02.006
19. Quintana-Murci L. Genetic and epigenetic variation of human populations: an adaptive tale. C R Biol (2016) 339:278–83. doi:10.1016/j.crvi.2016.04.005
20. Liu Y, Aryee MJ, Padyukov L, Fallin MD, Hesselberg E, Runarsson A, et al. Epigenome-wide association data implicate DNA methylation as an intermediary of genetic risk in rheumatoid arthritis. Nat Biotechnol (2013) 31:142–7. doi:10.1038/nbt.2487
21. Rakyan VK, Beyan H, Down TA, Hawa MI, Maslau S, Aden D, et al. Identification of type 1 diabetes–associated DNA methylation variable positions that precede disease diagnosis. PLoS Genet (2011) 7:e1002300. doi:10.1371/journal.pgen.1002300
22. Jaffe AE, Irizarry RA. Accounting for cellular heterogeneity is critical in epigenome-wide association studies. Genome Biol (2014) 15:R31. doi:10.1186/gb-2014-15-2-r31
23. Quin JE, Bujila I, Chérif M, Sanou GS, Qu Y, Vafa Homann M, et al. Major transcriptional changes observed in the Fulani, an ethnic group less susceptible to malaria. Elife (2017) 6:e29156. doi:10.7554/eLife.29156
24. Sun D, Luo M, Jeong M, Rodriguez B, Xia Z, Hannah R, et al. Epigenomic profiling of young and aged HSCs reveals concerted changes during aging that reinforce self-renewal. Cell Stem Cell (2014) 14:673–88. doi:10.1016/j.stem.2014.03.002
25. Farlik M, Halbritter F, Müller F, Choudry FA, Ebert P, Klughammer J, et al. DNA methylation dynamics of human hematopoietic stem cell differentiation. Cell Stem Cell (2016) 19:808–22. doi:10.1016/j.stem.2016.10.019
26. Avgustinova A, Benitah SA. Epigenetic control of adult stem cell function. Nat Rev Mol Cell Biol (2016) 17:643–58. doi:10.1038/nrm.2016.76
27. Hu D, Shilatifard A. Epigenetics of hematopoiesis and hematological malignancies. Genes Dev (2016) 30:2021–41. doi:10.1101/gad.284109.116
28. Stahl M, Kohrman N, Gore SD, Kim TK, Zeidan AM, Prebet T. Epigenetics in cancer: a hematological perspective. PLoS Genet (2016) 12:e1006193. doi:10.1371/journal.pgen.1006193
29. Gallipoli P, Huntly BJP. Novel epigenetic therapies in hematological malignancies: current status and beyond. Semin Cancer Biol (2017). doi:10.1016/j.semcancer.2017.07.005
30. Goyama S, Kitamura T. Epigenetics in normal and malignant hematopoiesis: an overview and update 2017. Cancer Sci (2017) 108:553–62. doi:10.1111/cas.13168
31. Iliopoulos D, Malizos KN, Tsezou A. Epigenetic regulation of leptin affects MMP-13 expression in osteoarthritic chondrocytes: possible molecular target for osteoarthritis therapeutic intervention. Ann Rheum Dis (2007) 66:1616–21. doi:10.1136/ard.2007.069377
32. Reinius LE, Acevedo N, Joerink M, Pershagen G, Dahlén S-E, Greco D, et al. Differential DNA methylation in purified human blood cells: implications for cell lineage and studies on disease susceptibility. PLoS One (2012) 7:e41361. doi:10.1371/journal.pone.0041361
33. Bjanesoy TE, Andreassen BK, Bratland E, Reiner A, Islam S, Husebye ES, et al. Altered DNA methylation profile in Norwegian patients with autoimmune addison’s disease. Mol Immunol (2014) 59:208–16. doi:10.1016/j.molimm.2014.02.018
34. Lei W, Luo Y, Lei W, Luo Y, Yan K, Zhao S, et al. Abnormal DNA methylation in CD4+ T cells from patients with systemic lupus erythematosus, systemic sclerosis, and dermatomyositis. Scand J Rheumatol (2009) 38:369–74. doi:10.1080/03009740902758875
35. Zhang Z, Zhang R. Epigenetics in autoimmune diseases: pathogenesis and prospects for therapy. Autoimmun Rev (2015) 14:854–63. doi:10.1016/j.autrev.2015.05.008
36. Mehta S, Cronkite DA, Basavappa M, Saunders TL, Adiliaghdam F, Amatullah H, et al. Maintenance of macrophage transcriptional programs and intestinal homeostasis by epigenetic reader SP140. Sci Immunol (2017) 2:eaag3160. doi:10.1126/sciimmunol.aag3160
37. Marsit CJ. Influence of environmental exposure on human epigenetic regulation. J Exp Biol (2015) 218:71–9. doi:10.1242/jeb.106971
38. Breiling A, Lyko F. Epigenetic regulatory functions of DNA modifications: 5-methylcytosine and beyond. Epigenetics Chromatin (2015) 8:24. doi:10.1186/s13072-015-0016-6
39. Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res (2011) 21:381–95. doi:10.1038/cr.2011.22
40. Chen T, Dent SYR. Chromatin modifiers: regulators of cellular differentiation. Nat Rev Genet (2014) 15:93–106. doi:10.1038/nrg3607
41. Lim PS, Li J, Holloway AF, Rao S. Epigenetic regulation of inducible gene expression in the immune system. Immunology (2013) 139:285–93. doi:10.1111/imm.12100
42. Obata Y, Furusawa Y, Hase K. Epigenetic modifications of the immune system in health and disease. Immunol Cell Biol (2015) 93:226–32. doi:10.1038/icb.2014.114
43. Youngblood B, Hale JS, Ahmed R. T-cell memory differentiation: insights from transcriptional signatures and epigenetics. Immunology (2013) 139:277–84. doi:10.1111/imm.12074
44. Álvarez-Errico D, Vento-Tormo R, Sieweke M, Ballestar E. Epigenetic control of myeloid cell differentiation, identity and function. Nat Rev Immunol (2015) 15:7–17. doi:10.1038/nri3777
45. Dunn J, McCuaig R, Tu WJ, Hardy K, Rao S. Multi-layered epigenetic mechanisms contribute to transcriptional memory in T lymphocytes. BMC Immunol (2015) 16:27. doi:10.1186/s12865-015-0089-9
46. Zan H, Casali P. Epigenetics of peripheral B-cell differentiation and the antibody response. Front Immunol (2015) 6:631. doi:10.3389/fimmu.2015.00631
47. Lai AY, Mav D, Shah R, Grimm SA, Phadke D, Hatzi K, et al. DNA methylation profiling in human B cells reveals immune regulatory elements and epigenetic plasticity at Alu elements during B-cell activation. Genome Res (2013) 23:2030–41. doi:10.1101/gr.155473.113
48. Sellars M, Huh JR, Day K, Issuree PD, Galan C, Gobeil S, et al. Regulation of DNA methylation dictates Cd4 expression during the development of helper and cytotoxic T cell lineages. Nat Immunol (2015) 16:746–54. doi:10.1038/ni.3198
49. Dong J, Chang H-D, Ivascu C, Qian Y, Rezai S, Okhrimenko A, et al. Loss of methylation at the IFNG promoter and CNS-1 is associated with the development of functional IFN-γ memory in human CD4(+) T lymphocytes. Eur J Immunol (2013) 43:793–804. doi:10.1002/eji.201242858
50. Shaknovich R, Cerchietti L, Tsikitas L, Kormaksson M, De S, Figueroa ME, et al. DNA methyltransferase 1 and DNA methylation patterning contribute to germinal center B-cell differentiation. Blood (2011) 118:3559–69. doi:10.1182/blood-2011-06-357996
51. Xue LQ, Stacey KJ, Horne-Debets JM, Cridland JA, Fischer K, Narum D, et al. Malaria infection alters the expression of B cell activating factor resulting in diminished memory antibody responses and survival. Eur J Immunol (2012) 42:3291–301. doi:10.1002/eji.201242689
52. Medzhitov R, Schneider DS, Soares MP. Disease tolerance as a defense strategy. Science (2012) 335:936–41. doi:10.1126/science.1214935
53. Netea MG, Latz E, Mills KHG, O’Neill LAJ. Innate immune memory: a paradigm shift in understanding host defense. Nat Immunol (2015) 16:675–9. doi:10.1038/ni.3178
54. Rusek P, Wala M, Druszczyńska M, Fol M. Infectious agents as stimuli of trained innate immunity. Int J Mol Sci (2018) 19:456. doi:10.3390/ijms19020456
55. Cris¸an TO, Netea MG, Joosten LAB. Innate immune memory: implications for host responses to damage-associated molecular patterns. Eur J Immunol (2016) 46:817–28. doi:10.1002/eji.201545497
56. Glass CK, Natoli G. Molecular control of activation and priming in macrophages. Nat Immunol (2015) 17:26–33. doi:10.1038/ni.3306
57. Saeed S, Quintin J, Kerstens HHD, Rao NA, Aghajanirefah A, Matarese F, et al. Epigenetic programming during monocyte to macrophage differentiation and trained innate immunity. Science (2014) 345:1251086. doi:10.1126/science.1251086
58. Novakovic B, Habibi E, Wang S-Y, Arts RJW, Davar R, Megchelenbrink W, et al. β-Glucan reverses the epigenetic state of LPS-induced immunological tolerance. Cell (2016) 167:1354–68.e14. doi:10.1016/j.cell.2016.09.034
59. Quintin J, Saeed S, Martens JHA, Giamarellos-Bourboulis EJ, Ifrim DC, Logie C, et al. Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe (2012) 12:223–32. doi:10.1016/j.chom.2012.06.006
60. Batugedara G, Lu XM, Bunnik EM, Le Roch KG. The role of chromatin structure in gene regulation of the human malaria parasite. Trends Parasitol (2017) 33:364–77. doi:10.1016/j.pt.2016.12.004
61. Cortés A, Deitsch KW. Malaria epigenetics. Cold Spring Harb Perspect Med (2017) 7:a025528. doi:10.1101/cshperspect.a025528
62. Qidwai T, Jamal F, Singh S. Exploring putative molecular mechanisms of human pyruvate kinase enzyme deficiency and its role in resistance against Plasmodium falciparum malaria. Interdiscip Sci (2014) 6:158–66. doi:10.1007/s12539-013-0025-8
63. Al-Quraishy S, Dkhil MA, Abdel-Baki AAS, Delic D, Santourlidis S, Wunderlich F. Genome-wide screening identifies Plasmodium chabaudi-induced modifications of DNA methylation status of Tlr1 and Tlr6 gene promoters in liver, but not spleen, of female C57BL/6 mice. Parasitol Res (2013) 112:3757–70. doi:10.1007/s00436-013-3565-2
64. Gupta H, Chaudhari S, Rai A, Bhat S, Sahu PK, Hande MH, et al. Genetic and epigenetic changes in host ABCB1 influences malaria susceptibility to Plasmodium falciparum. PLoS One (2017) 12:e0175702. doi:10.1371/journal.pone.0175702
65. Schrum JE, Crabtree JN, Dobbs KR, Kiritsy MC, Reed GW, Gazzinelli RT, et al. Cutting edge: Plasmodium falciparum induces trained innate immunity. J Immunol (2018) 200:1243–8. doi:10.4049/jimmunol.1701010
66. Lessard S, Gatof ES, Beaudoin M, Schupp PG, Sher F, Ali A, et al. An erythroid-specific ATP2B4 enhancer mediates red blood cell hydration and malaria susceptibility. J Clin Invest (2017) 127:3065–74. doi:10.1172/JCI94378
67. Burel JG, Apte SH, Groves PL, Klein K, McCarthy JS, Doolan DL. Reduced Plasmodium Parasite burden associates with CD38+ CD4+ T cells displaying cytolytic potential and impaired IFN-γ production. PLoS Pathog (2016) 12:e1005839. doi:10.1371/journal.ppat.1005839
68. Förster M, Boora RK, Petrov JC, Fodil N, Albanese I, Kim J, et al. A role for the histone H2A deubiquitinase MYSM1 in maintenance of CD8+ T cells. Immunology (2017) 151:110–21. doi:10.1111/imm.12710
69. Al-Quraishy S, Dkhil MA, Abdel-Baki A-AS, Araúzo-Bravo MJ, Delic D, Wunderlich F. Testosterone persistently dysregulates hepatic expression of Tlr6 and Tlr8 induced by Plasmodium chabaudi malaria. Parasitol Res (2014) 113:3609–20. doi:10.1007/s00436-014-4026-2
70. Ackland ML, Michalczyk AA. Zinc and infant nutrition. Arch Biochem Biophys (2016) 611:51–7. doi:10.1016/j.abb.2016.06.011
71. Kumari K, Keshari S, Sengupta D, Sabat SC, Mishra SK. Transcriptome analysis of genes associated with breast cancer cell motility in response to Artemisinin treatment. BMC Cancer (2017) 17:858. doi:10.1186/s12885-017-3863-7
72. Dkhil MA, Al-Quraishy SA, Abdel-Baki A-AS, Delic D, Wunderlich F. Differential miRNA expression in the liver of Balb/c mice protected by vaccination during crisis of Plasmodium chabaudi blood-stage malaria. Front Microbiol (2016) 7:2155. doi:10.3389/fmicb.2016.02155
73. Al-Quraishy S, Dkhil MA, Abdel-Baki A-AS, Ghanjati F, Erichsen L, Santourlidis S, et al. Protective vaccination and blood-stage malaria modify DNA methylation of gene promoters in the liver of Balb/c mice. Parasitol Res (2017) 116:1463–77. doi:10.1007/s00436-017-5423-0
74. McCall MBB, Hopman J, Daou M, Maiga B, Dara V, Ploemen I, et al. Early interferon-gamma response against Plasmodium falciparum correlates with interethnic differences in susceptibility to parasitemia between sympatric Fulani and Dogon in Mali. J Infect Dis (2010) 201:142–52. doi:10.1086/648596
75. Franklin BS, Parroche P, Ataíde MA, Lauw F, Ropert C, de Oliveira RB, et al. Malaria primes the innate immune response due to interferon-γ induced enhancement of toll-like receptor expression and function. Proc Natl Acad Sci U S A (2009) 106:5789–94. doi:10.1073/pnas.0809742106
76. Luty AJ, Lell B, Schmidt-Ott R, Lehman LG, Luckner D, Greve B, et al. Interferon-gamma responses are associated with resistance to reinfection with Plasmodium falciparum in young African children. J Infect Dis (1999) 179:980–8. doi:10.1086/314689
77. Dodoo D, Omer FM, Todd J, Akanmori BD, Koram KA, Riley EM. Absolute levels and ratios of proinflammatory and anti-inflammatory cytokine production in vitro predict clinical immunity to Plasmodium falciparum malaria. J Infect Dis (2002) 185:971–9. doi:10.1086/339408
78. Parra M, Liu X, Derrick SC, Yang A, Tian J, Kolibab K, et al. Molecular analysis of non-specific protection against murine malaria induced by BCG vaccination. PLoS One (2013) 8:e66115. doi:10.1371/journal.pone.0066115
79. Boutlis CS, Yeo TW, Anstey NM. Malaria tolerance – for whom the cell tolls? Trends Parasitol (2006) 22:371–7. doi:10.1016/j.pt.2006.06.002
80. Heyman A, Beeson PB. Influence of various disease states upon the febrile response to intravenous injection of typhoid bacterial pyrogen; with particular reference to malaria and cirrhosis of the liver. J Lab Clin Med (1949) 34:1400–3.
81. Gatton ML, Cheng Q. Evaluation of the pyrogenic threshold for Plasmodium falciparum malaria in naive individuals. Am J Trop Med Hyg (2002) 66:467–73. doi:10.4269/ajtmh.2002.66.467
82. Portugal S, Tran TM, Ongoiba A, Bathily A, Li S, Doumbo S, et al. Treatment of chronic asymptomatic Plasmodium falciparum infection does not increase the risk of clinical malaria upon reinfection. Clin Infect Dis (2017) 64:645–53. doi:10.1093/cid/ciw849
83. Kamada N, Seo S-U, Chen GY, Núñez G. Role of the gut microbiota in immunity and inflammatory disease. Nat Rev Immunol (2013) 13:321–35. doi:10.1038/nri3430
84. Pickard JM, Zeng MY, Caruso R, Núñez G. Gut microbiota: role in pathogen colonization, immune responses, and inflammatory disease. Immunol Rev (2017) 279:70–89. doi:10.1111/imr.12567
85. Claycombe KJ, Brissette CA, Ghribi O. Epigenetics of inflammation, maternal infection, and nutrition. J Nutr (2015) 145:1109S–15S. doi:10.3945/jn.114.194639
86. Johansson MA, Sjögren YM, Persson J-O, Nilsson C, Sverremark-Ekström E. Early colonization with a group of Lactobacilli decreases the risk for allergy at five years of age despite allergic heredity. PLoS One (2011) 6:e23031. doi:10.1371/journal.pone.0023031
87. Johansson MA, Saghafian-Hedengren S, Haileselassie Y, Roos S, Troye-Blomberg M, Nilsson C, et al. Early-life gut bacteria associate with IL-4-, IL-10- and IFN-γ production at two years of age. PLoS One (2012) 7:e49315. doi:10.1371/journal.pone.0049315
88. Canani RB, Costanzo MD, Leone L, Bedogni G, Brambilla P, Cianfarani S, et al. Epigenetic mechanisms elicited by nutrition in early life. Nutr Res Rev (2011) 24:198–205. doi:10.1017/S0954422411000102
89. Indrio F, Martini S, Francavilla R, Corvaglia L, Cristofori F, Mastrolia SA, et al. Epigenetic matters: the link between early nutrition, microbiome, and long-term health development. Front Pediatr (2017) 5:178. doi:10.3389/fped.2017.00178
90. Krautkramer KA, Kreznar JH, Romano KA, Vivas EI, Barrett-Wilt GA, Rabaglia ME, et al. Diet-microbiota interactions mediate global epigenetic programming in multiple host tissues. Mol Cell (2016) 64:982–92. doi:10.1016/j.molcel.2016.10.025
91. Yilmaz B, Portugal S, Tran TM, Gozzelino R, Ramos S, Gomes J, et al. Gut microbiota elicits a protective immune response against malaria transmission. Cell (2014) 159:1277–89. doi:10.1016/j.cell.2014.10.053
92. Traoré K, Arama C, Médebielle M, Doumbo O, Picot S. Do advanced glycation end-products play a role in malaria susceptibility? Parasite (2016) 23:15. doi:10.1051/parasite/2016015
93. Lüder CGK, Stanway RR, Chaussepied M, Langsley G, Heussler VT. Intracellular survival of apicomplexan parasites and host cell modification. Int J Parasitol (2009) 39:163–73. doi:10.1016/j.ijpara.2008.09.013
94. Pinzon-Charry A, Woodberry T, Kienzle V, McPhun V, Minigo G, Lampah DA, et al. Apoptosis and dysfunction of blood dendritic cells in patients with falciparum and vivax malaria. J Exp Med (2013) 210:1635–46. doi:10.1084/jem.20121972
95. Skorokhod OA, Alessio M, Mordmüller B, Arese P, Schwarzer E. Hemozoin (malarial pigment) inhibits differentiation and maturation of human monocyte-derived dendritic cells: a peroxisome proliferator-activated receptor-γ-mediated effect. J Immunol (2004) 173:4066–74. doi:10.4049/jimmunol.173.6.4066
96. Millington OR, Di Lorenzo C, Phillips RS, Garside P, Brewer JM. Suppression of adaptive immunity to heterologous antigens during Plasmodium infection through hemozoin-induced failure of dendritic cell function. J Biol (2006) 5:5. doi:10.1186/jbiol34
97. Giusti P, Urban BC, Frascaroli G, Albrecht L, Tinti A, Troye-Blomberg M, et al. Plasmodium falciparum-infected erythrocytes and β-hematin induce partial maturation of human dendritic cells and increase their migratory ability in response to lymphoid chemokines. Infect Immun (2011) 79:2727–36. doi:10.1128/IAI.00649-10
98. Bujila I, Schwarzer E, Skorokhod O, Weidner JM, Troye-Blomberg M, Östlund Farrants A-K. Malaria-derived hemozoin exerts early modulatory effects on the phenotype and maturation of human dendritic cells. Cell Microbiol (2016) 18:413–23. doi:10.1111/cmi.12521
99. Fyrberg A, Peterson C, Kågedal B, Lotfi K. Induction of fetal hemoglobin and ABCB1 gene expression in 9-β-D-arabinofuranosylguanine-resistant MOLT-4 cells. Cancer Chemother Pharmacol (2011) 68:583–91. doi:10.1007/s00280-010-1524-5
100. Busslinger M, Tarakhovsky A. Epigenetic control of immunity. Cold Spring Harb Perspect Biol (2014) 6:a019307. doi:10.1101/cshperspect.a019307
101. Pennini ME, Pai RK, Schultz DC, Boom WH, Harding CV. Mycobacterium tuberculosis 19-kDa lipoprotein inhibits IFN-γ-induced chromatin remodeling of MHC2TA by TLR2 and MAPK signaling. J Immunol (2006) 176:4323–30. doi:10.4049/jimmunol.176.7.4323
102. Gómez-Díaz E, Jordà M, Peinado MA, Rivero A. Epigenetics of host–pathogen interactions: the road ahead and the road behind. PLoS Pathog (2012) 8:e1003007. doi:10.1371/journal.ppat.1003007
103. Cheeseman K, Weitzman JB. Host–parasite interactions: an intimate epigenetic relationship. Cell Microbiol (2015) 17:1121–32. doi:10.1111/cmi.12471
104. Marr AK, MacIsaac JL, Jiang R, Airo AM, Kobor MS, McMaster WR. Leishmania donovani infection causes distinct epigenetic DNA methylation changes in host macrophages. PLoS Pathog (2014) 10:e1004419. doi:10.1371/journal.ppat.1004419
105. Robert McMaster W, Morrison CJ, Kobor MS. Epigenetics: a new model for intracellular parasite–host cell regulation. Trends Parasitol (2016) 32:515–21. doi:10.1016/j.pt.2016.04.002
106. Leng J, Denkers EY. Toxoplasma gondii inhibits covalent modification of histone H3 at the IL-10 promoter in infected macrophages. PLoS One (2009) 4:e7589. doi:10.1371/journal.pone.0007589
107. Hari Dass SA, Vyas A. Toxoplasma gondii infection reduces predator aversion in rats through epigenetic modulation in the host medial amygdala. Mol Ecol (2014) 23:6114–22. doi:10.1111/mec.12888
108. Kalantari P. The emerging role of pattern recognition receptors in the pathogenesis of malaria. Vaccines (Basel) (2018) 6:13. doi:10.3390/vaccines6010013
110. Galactionova K, Smith TA, de Savigny D, Penny MA. State of inequality in malaria intervention coverage in sub-Saharan African countries. BMC Med (2017) 15:185. doi:10.1186/s12916-017-0948-8
111. Ashley EA, Dhorda M, Fairhurst RM, Amaratunga C, Lim P, Suon S, et al. Spread of artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med (2014) 371:411–23. doi:10.1056/NEJMoa1314981
112. Pfister SX, Ashworth A. Marked for death: targeting epigenetic changes in cancer. Nat Rev Drug Discov (2017) 16:241–63. doi:10.1038/nrd.2016.256
Keywords: epigenetic, immunity, malaria, falciparum, protection, susceptibility
Citation: Arama C, Quin JE, Kouriba B, Östlund Farrants A-K, Troye-Blomberg M and Doumbo OK (2018) Epigenetics and Malaria Susceptibility/Protection: A Missing Piece of the Puzzle. Front. Immunol. 9:1733. doi: 10.3389/fimmu.2018.01733
Received: 08 May 2018; Accepted: 12 July 2018;
Published: 03 August 2018
Edited by:
Juarez Antonio Simões Quaresma, Instituto Evandro Chagas, BrazilReviewed by:
Pius Sedowhe Fasinu, Campbell University, United StatesRajeev Kumar Mehlotra, Case Western Reserve University, United States
Copyright: © 2018 Arama, Quin, Kouriba, Östlund Farrants, Troye-Blomberg and Doumbo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Charles Arama, Y2hhcmFtYUBpY2VybWFsaS5vcmc=;
Marita Troye-Blomberg, bWFyaXRhLnRyb3llYmxvbWJlcmdAc3Uuc2U=
†In memorium: Prof. Ogobara K. Doumbo passed away June 9, 2018 at the age of 62 years.