 Emmanuel Faure
Emmanuel Faure Kelly Kwong
Kelly Kwong Dao Nguyen
Dao Nguyen- 1Department of Medicine, McGill University, Montreal, QC, Canada
- 2Research Institute of the McGill University Health Center, Montreal, QC, Canada
Bacteria that readily adapt to different natural environments, can also exploit this versatility upon infection of the host to persist. Pseudomonas aeruginosa, a ubiquitous Gram-negative bacterium, is harmless to healthy individuals, and yet a formidable opportunistic pathogen in compromised hosts. When pathogenic, P. aeruginosa causes invasive and highly lethal disease in certain compromised hosts. In others, such as individuals with the genetic disease cystic fibrosis, this pathogen causes chronic lung infections which persist for decades. During chronic lung infections, P. aeruginosa adapts to the host environment by evolving toward a state of reduced bacterial invasiveness that favors bacterial persistence without causing overwhelming host injury. Host responses to chronic P. aeruginosa infections are complex and dynamic, ranging from vigorous activation of innate immune responses that are ineffective at eradicating the infecting bacteria, to relative host tolerance and dampened activation of host immunity. This review will examine how P. aeruginosa subverts host defenses and modulates immune and inflammatory responses during chronic infection. This dynamic interplay between host and pathogen is a major determinant in the pathogenesis of chronic P. aeruginosa lung infections.
Introduction
Bacterial pathogens are most commonly studied for their ability to invade and injure the host, causing acute and invasive infections. In contrast, chronic infections present a distinct paradigm in infection pathogenesis which may challenge conventional notions of bacterial virulence and host defenses. To healthy individuals, Pseudomonas aeruginosa (PA) is a ubiquitous Gram-negative bacterium commonly encountered in the environment and readily cleared by host defenses. However, PA is also a formidable opportunistic pathogen that can cause invasive and fulminant infections, such as acute pneumonia or bloodstream infections, in immune compromised hosts. Remarkably, the same pathogen also causes chronic infections that persist for months to decades, such as the chronic lung infection in individuals with the genetic disease cystic fibrosis (CF). Chronic PA infections thus result from a dynamic and complex interplay between pathogen and host, where bacteria persist without causing overwhelming host injury, and where host defenses fail to eradicate the pathogen.
PA has a large genome (>6 Mb) that encodes many regulatory genes involved in sensing environmental signals, controlling expression of virulence factors, metabolism and resistance mechanisms. PA thus readily adapts to a wide range of environments and can exploit this versatility to enhance its long-term survival and persistence in the host. Importantly, host-pathogen interactions evolve over time and anatomical space, with the balance fluctuating between host recognition and vigorous activation of defense mechanisms, and immune evasion and tolerance by the host.
Chronic PA lung infections in individuals with CF persist for decades and provide a unique opportunity to examine how a bacterial pathogen can adapt to its host, modulate host responses and shift between different infection phenotypes. It is widely recognized that CF disease is associated with several intrinsic host defects, including impaired mucociliary clearance, and immune and inflammatory dysregulation. The implications of these host defects to the development of CF lung disease are beyond the scope of this review but may be found in excellent other ones (1–3). In this review, we will examine how PA defines the interactions central to the host immune and inflammatory response, and the bacterial adaptive strategies that promote bacterial persistence, and allow evasion and tolerance by the host during chronic infection. Specifically, we will highlight bacterial factors that undergo host-adaptation during chronic infections.
Bacterial Factors Involved in Host Interactions and Recognition
Flagellin and Flagellar Motility
PA possesses a single polar flagellum composed of polymerized flagellin, its major structural protein, and attached to a transmembrane motor complex. The flagellar-host interaction plays a major role in defining the immune and inflammatory outcomes of PA infection, as the flagellar complex interacts with immune and non-immune cells through its structural components and as well as motility function.
The flagellar-host interactions have been extensively characterized at the cellular and molecular level. Flagellin is best known as a pathogen-associated molecular pattern that binds to the extracellular Toll like receptor TLR5 (4) and intracellular NOD-like receptor (NLR) neuronal apoptosis-inhibitory protein (NAIP) (5), in human (6), leading to activation of the pro-inflammatory MyD88 pathway and the NLRC4-inflammasome, respectively (7). TLR5 mediates a major component of the epithelial cytokine and chemokine responses leading to neutrophil recruitment in PA lung infection (8–10), and contributes to the production of pro-IL-1ß in monocytes and macrophages (11). Flagellin is also translocated by the Type-3 secretion system (T3SS) in the cytoplasm of mammalian cells, thereby activating the NAIP-NLRC4-inflammasome and inducing mature IL-1ß secretion (12, 13). Notably, IL-1ß promotes phagocytosis through its autocrine and paracrine effects (11, 14). Interestingly both flagellin and a motile flagellum are required to activate the NAIP-NLRC4-inflammasome (5, 15–17), but how host cells sense flagellar motility remains unclear. Beyond its ability to activate host cell signaling pathways, the flagellum also promotes adherence and colonization of host surfaces, and various specific targets have been identified including MUC1 mucin (18), heparin sulfate (19), surfactant protein A (20), and asialoGM1 (21).
During chronic infection, PA uses multiple strategies to evade flagellum-mediated host recognition. Flagellin expression is under the complex regulation by several global transcriptional regulators (22–25). It is repressed in mucoid variants which over-produce the exopolysaccharide alginate (26), during biofilm growth (27), upon as well as in response to the host nutritional and inflammatory environment. Notably, flagellin is repressed in the presence of CF sputum and airway fluid (28) as well as neutrophil elastase released at sites of inflammation (29). PA also expresses the secreted bacterial proteases AprA and LasB which cleave extracellular flagellin, suggesting an intrinsic mechanism to shut down flagellin-mediated immune recognition (30). Finally, loss of flagellar motility is common in host-adapted PA strains from CF lung infections and is associated with increased bacterial burden and disease severity (31). Genome sequencing studies of longitudinal PA strains have revealed evidence of convergent evolution and genetic mutations in regulatory genes such as rpoN and fleQ which lead to downregulation of flagellar expression and motility (32, 33). In fact, PA isolates recovered from chronic CF lung infections fail to activate the inflammasome due to reduced expression of flagellin and T3SS (34).
Type 3 Secretion System (T3SS)
The type III secretion system (T3SS) is a complex needle-like secretion machinery found in gram-negative bacteria that allows the translocation of bacterial effectors directly into the cytoplasm of host cells, causing cytotoxicity, or subversion of host defenses (35). The T3SS causes tissue injury, promotes bacterial dissemination and has been implicated in the pathogenesis of acute and invasive infections, including pneumonia (36–38). Four T3SS-dependent effectors have been identified in PA, namely ExoS, ExoT, ExoY, and ExoU, and have been recently reviewed elsewhere (35). The T3SS effectors cause disruption of host cell cytoskeleton (ExoS, T, and U) and cleavage of phospholipases (ExoU), leading to cell death, a breach of epithelial and endothelial barriers and killing of phagocytes (39–41). ExoS also dampens phagocytosis by interfering with lysosome signaling in macrophages (42, 43).
Beyond its role in cytotoxicity, the T3SS activates innate immune responses through secretion of IL-1ß (44). The T3SS apparatus itself, independently of exotoxin, can activate the NLRC4-inflammasome through NAIP recognition (44–46), leading to pyroptotic cell death and the secretion of mature IL-1ß and IL-18. Whether inflammasome activation contributes to the effective immune response to control bacteria, or to the immunopathology associated with PA lung infections remains incompletely understood. On one hand, inflammasome activation and IL-1R signaling may be protective at early stages of infection (47, 48). On the other hand, NLRC4 activation is associated with reduced alveolar macrophages, reduced PA clearance and increased neutrophil recruitment, leading to greater lung immunopathology and mortality in a murine model of acute lung infection (49, 50).
Chronic infections appear to select against T3SS-expressing PA. Although many CF patients carry antibodies against T3SS effector proteins (51), suggesting that these effector proteins were secreted at some stage of the infection, most PA strains isolated from chronic infection are T3SS-negative (34, 52, 53). Loss of T3SS results in dampened inflammasome activation and lesser pyroptotic cell death in macrophages and neutrophils (34). CF isolates are rarely ExoU+ (54), also consistent with the notion that acute cytotoxicity, particularly when conferred by ExoU, is less compatible with chronic infection. As discussed later in this review, several mechanisms contribute to the loss of T3SS in CF-adapted PA strains.
Secreted Proteases
PA produces several secreted proteases, which include LasB (also known as PA elastase or pseudolysin), LasA, AprA, and protease IV. Secreted PA proteases interact with a wide range of host molecules, leading to diverse outcomes, from degradation of structural components to modulation of inflammatory responses. The PA proteases are most studied for their ability to cause direct tissue damage, and they are primarily known as virulence factors involved in the pathogenesis of acute infections. LasB, a broad specificity metallo-protease, degrades elastin (55), disrupts epithelial tight-junctions (56), and reduce endothelial barrier integrity (57, 58). As a consequence, LasB mutants are attenuated in virulence in experimental models of bacteremia (59), acute pneumonia (60), or burn wound model (61).
PA proteases also alter host responses by degrading secreted mediators, leading to a dampening of inflammatory and immune responses, which likely contributes to its ability to evade host defenses. In vitro studies have shown that PA proteases potently degrades secreted mediators such as cytokines (e.g., INF-γ, IL-6), chemokines (e.g., IL-8/CXCL1, MCP-1, CXCL-5, RANTES/CCL5) (62–66), host defense components such as immunoglobulins (67, 68), antimicrobial peptides (e.g., LL-37) (69), and membrane receptors (e.g., protease-activated receptor PAR-1,2 and 4) (70, 71). LasB helps PA subvert alveolar macrophage activity by down-regulating the oxidative burst and production of complement factors (72). LasB mediated degradation of surfactant proteins SP-A and SP-D also leads to phagocytosis resistance (73, 74). Proteolysis of thrombin by LasB releases an anti-inflammatory thrombin-derived peptide FYT21, which inhibits the activation of the transcription factors NF-κB and AP-1 (75). Finally, AprA and LasB can degrade flagellin monomers, and thus blunt TLR5-mediated responses (30) and inflammasome activation (76). Interestingly, the inflammasome activation is also dampened due to proteolytic degradation of extracellular inflammasome components by PA proteases (76).
Although most PA isolates recovered from environmental sources or acute infections produce secreted proteases, protease-deficient PA isolates are commonly isolated from patients with CF and chronic obstructive pulmonary disease (COPD) chronically colonized with PA (77, 78). In fact, loss of secreted protease activity occurs as part of the genetic adaptation of PA to the host environment (see section below) and is associated with chronic and more advanced lung disease (32, 79). As secreted proteases dampen inflammation, loss of protease activity in CF-adapted PA variants conversely can promote exaggerated inflammation and lung immunopathology, as observed in vitro, in vivo in murine models of chronic PA lung infections and in CF patients (80). The impact of secreted PA proteases on host responses and pathology thus varies in different infection settings, such as acute vs. chronic, invasive vs. localized, as the presence or loss of proteases promote disease through different mechanisms of host interactions.
Exopolysaccharides (EPS)
PA produces three extracellular polysaccharides (or exopolysaccharides), namely alginate, Psl, and Pel. They provide many protective properties and confer surface and self-adherence. They are constituents of the biofilm matrix, are involved in surface colonization and promote host immune evasion. A detailed review of these EPS and their distinct functions can be found elsewhere (81).
Mucoid PA overproduces the exopolysaccharide alginate and these strains are commonly associated with chronic CF lung infections and other chronic lung diseases (79, 82, 83). Alginate over-production (mucoidy) impairs host defenses and promotes bacterial persistence through several mechanisms. Alginate overproduction interferes with opsonophagocytosis and complement activation, scavenges ROS and inhibits phagocytic killing (82, 84, 85). It also confers resistance to host antimicrobials such as LL-37 and reactive oxygen species H2O2 (86). Whether mucoidy dampens host detection remains unclear. Mucoidy represses flagellar biosynthesis due to the co-regulation of flagellin and alginate (26), leading to reduced TLR5-dependent activation. However, mucoidy is linked with increases bacterial lipoproteins expression (87), which activates TLR2 in host airway epithelial cells (88), and is associated to greater resistance to the anti-inflammatory effects of corticosteroids (89).
Psl and Pel are exopolysaccharides which confer structural and aggregative properties to the biofilm matrix and contribute to the biofilm antibiotic tolerance (90, 91). Psl interferes with complement deposition and hinders neutrophil opsonophagocytosis and oxidative killing (92). Although its interactions with host cells are less well-characterized, Pel likely also contributes to resistance against neutrophil killing (93). PA genetic variants that overproduce Psl and/or Pel are found in chronic CF infections (94) and are associated with increased bacterial burden and host immune evasion (95).
Lipopolysaccharides (LPS)
LPS (also known as endotoxin) is a major component of the outer membrane of Gram negative bacteria. LPS is composed of three components: the lipid A and core oligosaccharides that form the outer leaflet of the bacterial outer membrane, and the O-antigen polysaccharide which interacts with the extracellular environment. LPS is recognized by the Toll like receptor 4 and myeloid differentiation factor 2 complex (TLR4-MD2). The O-antigen consists of highly variable and immunogenic oligosaccharide repeats which elicit a strong humoral response (96).
During chronic infection, the LPS undergoes important adaptive changes at the level of its synthesis and structure, leading to modification of the lipid A structure and loss of O antigen which likely promote immune evasion. Lipid A acylation patterns or addition of positively charged components, renders the outer membrane more resistant to host antimicrobial peptides (97–99), modulates TLR4-MD2 receptor recognition and dampens host inflammation (100). PA isolates from chronic infection commonly express little or no O-antigen (101, 102). Mutations in LPS and O-antigen biosynthesis are common (32, 103, 104) and appear to be a hotspot of genetic variation and adaptation during chronic CF infections (105). Finally, O-antigen biosynthesis is also modulated by cyclic-di-GMP, a second messenger involved in the switch from motile to adherent lifestyle of PA (106). A summary of the bacterial factors/complex involved in the host adaptation during chronic PA infections is provided in Table 1.
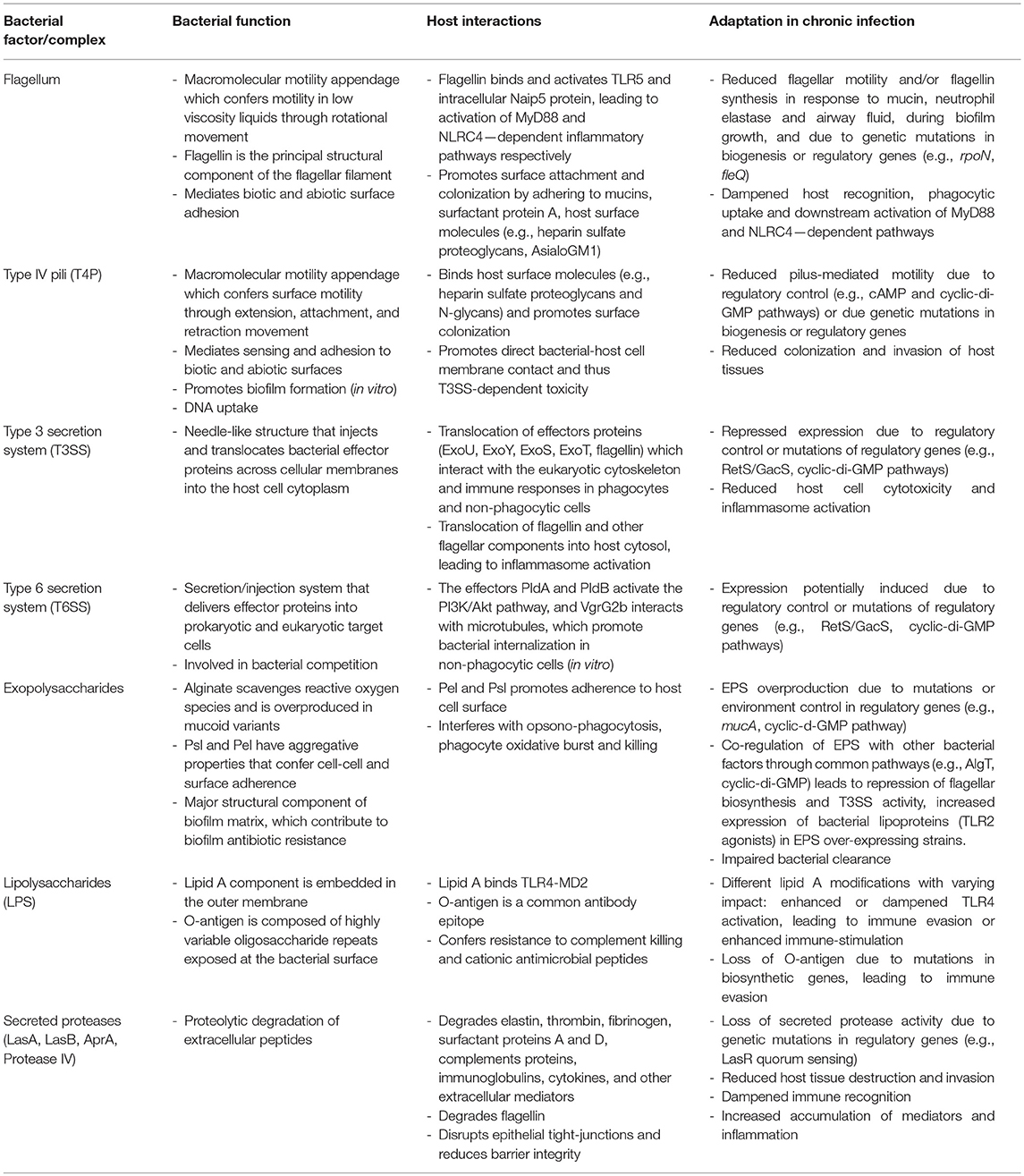
Table 1. Bacterial factors/complex involved in host-adaptation during chronic PA infections.
PA Phenotypic and Genetic Adaption to Host Environments
During the process of chronic infection, PA adapts to the host environment and undergoes changes which promote bacterial survival and evasion of host defenses. Certain adaptive processes occur at the phenotypic and regulatory level, while others occur through genetic mutations and evolution. We will review here the key regulatory and genetic adaptive processes that PA undergoes during chronic PA infection.
Biofilm Lifestyle
In contrast to the free-living bacterial lifestyle termed planktonic, PA can also grow in a multicellular and sessile form, termed biofilms. Biofilms are formed by self-aggregated or surface-adherent bacteria encased within an extracellular matrix. Biofilms cause many chronic and non-invasive human infections such as medical device associated infections, chronic CF lung infection and chronic wound infections. Our understanding of in vivo host responses to PA biofilms is limited by the lack of animal infection models that mimic human biofilm infections. Our insights are thus primarily drawn from in vitro studies that examine the response of various cell types to biofilm bacteria. Biofilm formation and its role in disease pathogenesis have been the subject of recent reviews (81, 107), and only aspects relevant to host-biofilm interactions are outlined here.
Host responses to PA biofilms are complex, as biofilms may both stimulate or suppress the immune system. Biofilms may be less immune-stimulatory than their free-living planktonic counterparts. For example, the expression of flagellin and T3SS is down-regulated (108, 109), and the complement system is less activated (110) during biofilm growth. Furthermore, bacterial factors involved in host interactions may be embedded within the biofilm matrix and not readily accessible for host recognition. Conversely, biofilms can induce a robust neutrophilic response where neutrophils are activated, undergo oxidative burst and degranulate, but are immobilized (111–113). Biofilm PA can also trigger necrotic cell death in neutrophils (113), leading to further inflammation and collateral tissue damage.
Importantly, innate immune responses are less effective against biofilm than planktonic PA. As described above, exopolysaccharides constitute the major components of the biofilm matrix and contribute to biofilm resistance against host antimicrobials defenses and phagocytic killing. Biofilm infections are thus associated with a smoldering immune response that is ineffective at clearing bacteria but remains active enough to cause tissue damage over long periods of time.
Regulatory Control to Switch Bacterial Lifestyle and Infection Strategy
PA is capable of phenotypically switching between its motile planktonic lifestyle and the sessile biofilm lifestyle through multiple and overlapping regulatory networks which include the RetS/GacS sensor pathway. Through the opposing functions of RetS and GacS and their signaling cascades, the RetS/GacS pathway converge on the regulator RsmA and is linked to the second messenger cyclic di-GMP. It coordinately controls the expression of motility, Pel and Psl exopolysaccharides, T3SS and Type VI secretion system (T6SS) -related gene (114, 115). Chronic infection is thus favored as PA represses its T3SS, motility and produces the exopolysaccharides that form the biofilm matrix. Interestingly, analysis of host-adapted PA strains from chronic CF infections identified genetic mutations in the RetS/GacS pathway, with the possibility that retS mutations promote a chronic infection state (116). Conversely, dysregulation of RetS/GacS pathway due to mutations in gacS or its regulator ladS can also cause excessive T3SS activity and cytotoxicity, leading to hyper-virulent PA strains that cause fulminant infections (117) or exacerbations during chronic CF infection (118).
Cyclic di-GMP is an intracellular bacterial secondary messenger that regulates multiple bacterial behaviors, most notably those involved in biofilm formation. The cellular level of c-di-GMP are modulated in response to environmental and intracellular signals, and affect expression of genes involved in flagellar and type IV pilus mediated motility, exopolysaccharide production and surface adhesion (115). Genetic variants that overproduce cyclic di-GMP display an auto-aggregative phenotype caused by the overproduction of Psl and Pel, have been recovered from chronic CF lung infections (94).
The RetS/GacS and sensor pathway, cyclic di-GMP signaling and other global regulators (e.g., quorum sensing, two component sensor regulators) allow PA to coordinately regulate numerous factors that define distinct bacterial infection strategies, namely acute and invasive disease, or chronic and localized disease. It is plausible that the ability of PA to phenotypically switch between acute and chronic virulence modes contributes to the complex disease phenotype it causes: the natural history of chronic PA lung infections is characterized by slowly progressive tissue pathology, but is also interrupted by periods of acute and more fulminant disease termed acute exacerbations. It is possible to speculate that exacerbation episodes may be caused in part by a phenotypic switch to acute virulence.
Genetic Adaptation During Chronic Infection
The bacterial genetic adaptation to host environments is a common theme during chronic infection. For PA, this has been best documented in chronic CF lung infection, and we suggest several excellent recent reviews (33, 116, 119) for a detailed discussion of the topic. In CF, factors that contribute to the mutagenesis of PA include the presence of hypermutator strains (120), and the pro-inflammatory environment of the CF lung rich in oxidative and nitrosative stresses (33).
During its long residence in the CF lung, PA populations show both genetic diversification as well as convergent evolution. On one hand, PA undergoes significant genetic and phenotypic diversification during chronic CF infection, a process likely attributable to the divergent evolution of clonally related PA inhabiting different regions and micro-environments of the lung (121). On the other hand, numerous studies have shown evidence of convergent evolution when comparing the PA genomes within patients over time, and across different patients (122). Genome sequence analyses show a strong positive selection for non-synonymous mutations in genes encoding or regulating virulence factors (e.g., T3SS, exotoxin A, quorum sensing), immunogenicity factors (e.g., O-antigen), motility (flagellar and T4P mediated motility), drug resistance (e.g., multidrug efflux pumps), and metabolism (e.g., iron uptake). Importantly, many of these mutations confer loss of function or secretion of extracellular factors (e.g., proteases, T3SS) and promote immune evasion (32, 123). For example, LasR quorum sensing and protease-deficient variants are observed in over a third of CF patients with chronic PA infections. This suggests that the host environment likely confers strong selective forces that shape host-pathogen interactions and drive the genetic adaptation of PA toward a state that promote bacterial survival and persistence in the face of host defenses.
Advances and Challenges in the Development of Alternative or Adjuvant Therapies for Chronic PA Infections
Alternative or adjuvant therapies that minimize direct bacterial damage to the host, that enhance protective host responses or subvert pathological ones, can improve infection outcomes (124). Such therapies are particularly needed in light of the alarming rise in drug resistance, and for drug tolerant chronic infections (125). The latter refers to the phenotypic state of slow growing and biofilm bacteria which are refractory to antibacterial killing even in the absence of drug resistance. Unfortunately, despite intense research efforts and many candidates in pre-clinical studies, the development of novel therapies in chronic PA infections has been arduous and met with very limited success so far.
Anti-virulence therapies target bacterial virulence without disrupting bacterial growth or viability. Although numerous PA targets (e.g., quorum sensing signaling, biofilm exopolysaccharides, T3SS complex, and effectors) and inhibitor molecules have been studied, very few have progressed past pre-clinical studies (126). Anti-virulence therapies face unique challenges due to the bacterial phenotypic heterogeneity and complex host interactions characteristic of chronic PA infections. First, many PA strains isolated from chronic infections do not express functional factors such as flagellum and T3SS, suggesting that these factors may not play as important a role in virulence during chronic infections as during acute PA infections. Furthermore, the genetic and phenotypic adaptation of PA to the host during chronic infection lead to extraordinary heterogeneity between different patients, as well as at different stages or anatomically distinct foci of disease within the same patient. Anti-virulence therapies may thus need to be tailored to specific patients and/or infection states (e.g., early infection or acute exacerbation) based on a more comprehensive microbiological profiling than currently available in the clinic.
Antibacterial antibodies can neutralize bacterial virulence factors, induce complement mediated lysis and enhance opsonophagocytic uptake and killing (127). Advances in antibody engineering and screening have accelerated antibody therapeutics, and a few anti-PA antibodies have reached clinical trials. Polyclonal anti-PA antibodies (PsAer-IgY) (128) are currently in Phase 3 clinical trials (NCT01455675) for the prevention of recurrent PA infections in CF patients. Monoclonal antibodies that target the exopolysaccharides alginate (AR-105, Aridis Pharmaceuticals) and Psl (129), the T3SS needle protein PcrV [MEDI3902, MedImmune (130); KB001 (131)], O11 serotype LPS [AR-101/KBPA101, Aridis Pharmaceuticals (132)], or combinations [e.g., bispecific anti Psl/PcrV MEDI3902, MedImmune (130)] are currently tested for the prevention or treatment of acute PA pneumonia but their utility in preventing or treating chronic infections remains to be determined (133).
Considering the intractable nature of chronic PA infection, an important strategy is also to prevent infection through approaches such as vaccine, antibody, enzyme or antibiotic-based treatments. Although several anti-PA vaccine targeting antigens such as LPS O-antigen, alginate, outer membrane or flagellar proteins showed promise in pre-clinical trials, their clinical efficacy in reducing the risk of chronic PA infection in susceptible individuals (such as CF patients) has been overall disappointing to date (134, 135).
Conclusion
Chronic PA infection illustrates a paradigm of chronic bacterial infections where pathogens dampen host defenses, adapt and evolve within the host to persist. Understanding the pathogenesis of chronic PA infection thus requires an intricate assessment of bacteria, host responses, and their interactions over time. Host-PA interactions are exceptionally complex in chronic infections, as they involve numerous host cell types and bacterial factors. These interactions are further complicated by the common co-existence of other pathogens or polymicrobial communities that interact with both host and PA, and by the potential changes in the host due to factors such as aging or environmental exposures. While decades of research have provided us with vast mechanistic data on host-PA interactions, integrating these mechanistic insights into a whole system understanding of chronic infection and translating this knowledge into effective treatments remain a major challenge. The development of better in vivo models of chronic PA infection and tools to simultaneously probe host and pathogen over time is critical in order to gain a more integrated understanding of chronic infections.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling Editor declared a shared affiliation, though no other collaboration, with all of the authors KK, EF, and DN.
Acknowledgments
We would like to acknowledge the vast amount of work relevant to this review and apologize for any that we could not cite due to space limitations. We would also like to thank Simon Rousseau for helpful review and discussions of the manuscript.
We would like to acknowledge funding from the Canadian Institutes of Health Research (PJT-148827 to DN), Cystic Fibrosis Canada (559985 and 2469 to DN) and salary support from Cystic Fibrosis Canada (KK), Fonds de Recherche Sante Quebec (DN), Meakins Christie Laboratories (KK and EF) and Edith and Richard Strauss Foundation (EF).
Abbreviations
PA, Pseudomonas aeruginosa; CF, cystic fibrosis; cyclic di-GMP, cyclic diguanylate; EPS, exopolysaccharide; IL, interleukin; LPS, lipopolysaccharide; NAIP, neuronal apoptosis-inhibitory protein; ROS, reactive oxygen species; T3SS, Type-3 secretion system; T4P, Type 4 pili; TLR, Toll like receptor.
References
1. Cohen TS, Prince A. Cystic fibrosis: a mucosal immunodeficiency syndrome. Nat Med. (2012) 18:509–19. doi: 10.1038/nm.2715
2. Ratner D, Mueller C. Immune responses in cystic fibrosis. Am J Respir Cell Mol Biol. (2012) 46:715–22. doi: 10.1165/rcmb.2011-0399RT
3. Stoltz DA, Meyerholz DK, Welsh MJ. Origins of cystic fibrosis lung disease. N Engl J Med. (2015) 372:351–62. doi: 10.1056/NEJMra1300109
4. Hayashi F, Smith KD, Ozinsky A, Hawn TR, Yi EC, Goodlett DR, et al. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature (2001) 410:1099–103. doi: 10.1038/35074106
5. Zhao Y, Yang J, Shi J, Gong Y-N, Lu Q, Xu H, et al. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature (2011) 477:596–600. doi: 10.1038/nature10510
6. Reyes Ruiz VM, Ramirez J, Naseer N, Palacio NM, Siddarthan IJ, Yan BM, et al. Broad detection of bacterial type III secretion system and flagellin proteins by the human NAIP/NLRC4 inflammasome. Proc Natl Acad Sci USA. (2017) 114:13242–7. doi: 10.1073/pnas.1710433114
7. Vijay-Kumar M, Carvalho FA, Aitken JD, Fifadara NH, Gewirtz AT. TLR5 or NLRC4 is necessary and sufficient for promotion of humoral immunity by flagellin. Eur J Immunol. (2010) 40:3528–34. doi: 10.1002/eji.201040421
8. Prince A. Flagellar activation of epithelial signaling. Am J Respir Cell Mol Biol. (2006) 34:548–51. doi: 10.1165/rcmb.2006-0022SF
9. Zhang Z, Reenstra W, Weiner DJ, Louboutin JP, Wilson JM. The p38 mitogen-activated protein kinase signaling pathway is coupled to toll-like receptor 5 to mediate gene regulation in response to Pseudomonas aeruginosa infection in human airway epithelial cells. Infect Immun. (2007) 75:5985–92. doi: 10.1128/IAI.00678-07
10. Beaudoin T, Lafayette S, Roussel L, Bérubé J, Desrosiers M, Nguyen D, et al. The Level of p38α mitogen-activated protein kinase activation in airway epithelial cells determines the onset of innate immune responses to planktonic and biofilm Pseudomonas aeruginosa. J Infect Dis. (2013). 207:1544–55. doi: 10.1093/infdis/jit059
11. Descamps D, Le Gars M, Balloy V, Barbier D, Maschalidi S, Tohme M, et al. Toll-like receptor 5 (TLR5), IL-1beta secretion, and asparagine endopeptidase are critical factors for alveolar macrophage phagocytosis and bacterial killing. Proc Natl Acad Sci USA. (2012) 109:1619–24. doi: 10.1073/pnas.1108464109
12. Wei H-L, Chakravarthy S, Worley JN, Collmer A. Consequences of flagellin export through the type III secretion system of Pseudomonas syringae reveal a major difference in the innate immune systems of mammals and the model plant Nicotiana benthamiana. Cell Microbiol. (2012) 15:601–18. doi: 10.1111/cmi.12059
13. Ince D, Sutterwala FS, Yahr TL. Secretion of flagellar proteins by the Pseudomonas aeruginosa type III secretion-injectisome system. J Bacteriol. (2015) 197:2003–11. doi: 10.1128/JB.00030-15
14. Amiel E, Lovewell RR, O'toole GA, Hogan DA, Berwin B. Pseudomonas aeruginosa evasion of phagocytosis is mediated by loss of swimming motility and is independent of flagellum expression. Infect Immun. (2010) 78:2937–45. doi: 10.1128/IAI.00144-10
15. Miao EA, Alpuche-Aranda CM, Dors M, Clark AE, Bader MW, Miller SI, et al. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1β via Ipaf. Nat Immunol. (2006) 7:569–75. doi: 10.1038/ni1344
16. Lightfield KL, Persson J, Trinidad NJ, Brubaker SW, Kofoed EM, Sauer JD, et al. Differential requirements for NAIP5 in activation of the NLRC4 inflammasome. Infect Immun. (2011) 79:1606–14. doi: 10.1128/IAI.01187-10
17. Patankar YR, Lovewell RR, Poynter ME, Jyot J, Kazmierczak BI, Berwin B. Flagellar motility is a key determinant for the magnitude of the inflammasome response to Pseudomonas aeruginosa. Infect Immun. (2013) 81:2043–52. doi: 10.1128/IAI.00054-13
18. Lillehoj EP, Kim BT, Kim KC. Identification of Pseudomonas aeruginosaflagellin as an adhesin for Muc1 mucin. Am J Physiol Lung Cell Mol Physiol. (2002) 282:L751–6. doi: 10.1152/ajplung.00383.2001
19. Bucior I, Pielage JF, Engel JN. Pseudomonas aeruginosa pili and flagella mediate distinct binding and signaling events at the apical and basolateral surface of airway epithelium. PLoS Pathog. (2012) 8:e1002616–1002618. doi: 10.1371/journal.ppat.1002616
20. Ketko AK, Lin C, Moore BB, Levine AM. Surfactant protein a binds flagellin enhancing phagocytosis and IL-1β production. PLoS ONE (2013) 8:e82680. doi: 10.1371/journal.pone.0082680
21. Adamo R, Sokol S, Soong G, Gomez MI, Prince A. Pseudomonas aeruginosa Flagella activate airway epithelial cells through asialoGM1 and toll-like receptor 2 as well as toll-like receptor 5. Am J Respir Cell Mol Biol. (2004) 30:627–34. doi: 10.1165/rcmb.2003-0260OC
22. Starnbach MN, Lory S. The filA (rpoF) gene of Pseudomonas aeruginosa encodes an alternative sigma factor required for flagellin synthesis. Mol Microbiol (1992) 6:459–469. doi: 10.1111/j.1365-2958.1992.tb01490.x
23. Arora SK, Ritchings BW, Almira EC, Lory S, Ramphal R. A transcriptional activator, FleQ, regulates mucin adhesion and flagellar gene expression in Pseudomonas aeruginosa in a cascade manner. J Bacteriol. (1997) 179:5574–81. doi: 10.1128/jb.179.17.5574-5581.1997
24. Garrett ES, Perlegas D, Wozniak DJ. Negative control of flagellum synthesis in Pseudomonas aeruginosa is modulated by the alternative sigma factor AlgT (AlgU). J Bacteriol. (1999) 181:7401–4.
25. Lo Y-L, Shen L, Chang C-H, Bhuwan M, Chiu C-H, Chang H-Y. Regulation of motility and phenazine pigment production by FliA is cyclic-di-GMP dependent in Pseudomonas aeruginosa PAO1. PLoS ONE (2016) 11:e0155397. doi: 10.1371/journal.pone.0155397
26. Tart AH, Blanks MJ, Wozniak DJ. The AlgT-dependent transcriptional regulator AmrZ (AlgZ) inhibits flagellum biosynthesis in mucoid, nonmotile Pseudomonas aeruginosa cystic fibrosis isolates. J Bacteriol. (2006) 188:6483–9. doi: 10.1128/JB.00636-06
27. Guttenplan SB, Kearns DB. Regulation of flagellar motility during biofilm formation. FEMS Microbiol Rev. (2013) 37:849–71. doi: 10.1111/1574-6976.12018
28. Wolfgang MC, Jyot J, Goodman AL, Ramphal R, Lory S. Pseudomonas aeruginosa regulates flagellin expression as part of a global response to airway fluid from cystic fibrosis patients. Proc Natl Acad Sci USA. (2004) 101:6664–8. doi: 10.1073/pnas.0307553101
29. Jyot J, Sonawane A, Wu W, Ramphal R. Genetic mechanisms involved in the repression of flagellar assembly by Pseudomonas aeruginosain human mucus. Mol Microbiol. (2007) 63:1026–38. doi: 10.1111/j.1365-2958.2006.05573.x
30. Casilag F, Lorenz A, Krueger J, Klawonn F, Weiss S, Häussler S. The LasB elastase of Pseudomonas aeruginosa acts in concert with alkaline protease AprA to prevent flagellin-mediated immune recognition. Infect Immun. (2015) 84:162–71. doi: 10.1128/IAI.00939-15
31. Luzar MA, Thomassen MJ, Montie TC. Flagella and motility alterations in Pseudomonas aeruginosa strains from patients with cystic fibrosis: relationship to patient clinical condition. Infect Immun. (1985) 50:577–82.
32. Smith EE, Buckley DG, Wu Z, Saenphimmachak C, Hoffman LR, D'Argenio DA, et al. Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. Proc. Natl. Acad. Sci. USA. (2006) 103:8487–92. doi: 10.1073/pnas.0602138103
33. Winstanley C, O'brien S, Brockhurst MA. Pseudomonas aeruginosa evolutionary adaptation and diversification in cystic fibrosis chronic lung infections. Trends Microbiol. (2016) 24:327–37. doi: 10.1016/j.tim.2016.01.008
34. Huus KE, Joseph J, Zhang L, Wong A, Aaron SD, Mah T-F, et al. Clinical isolates of Pseudomonas aeruginosa from chronically infected cystic fibrosis patients fail to activate the inflammasome during both stable infection and pulmonary exacerbation. J Immunol. (2016) 196:3097–108. doi: 10.4049/jimmunol.1501642
35. Hauser AR. The type III secretion system of Pseudomonas aeruginosa: infection by injection. Nat Rev Microbiol. (2009) 7:654–65. doi: 10.1038/nrmicro2199
36. Wiener-Kronish JP, Sakuma T, Kudoh I, Pittet JF, Frank D, Dobbs L, et al. Alveolar epithelial injury and pleural empyema in acute P. aeruginosa pneumonia in anesthetized rabbits. J Appl Physiol. (1993) 75:1661–9. doi: 10.1152/jappl.1993.75.4.1661
37. Shaver CM, Hauser AR. Relative contributions of Pseudomonas aeruginosa ExoU, ExoS, and ExoT to virulence in the lung. Infect Immun. (2004) 72:6969–77. doi: 10.1128/IAI.72.12.6969-6977.2004
38. Rangel SM, Diaz MH, Knoten CA, Zhang A, Hauser AR. The role of ExoS in dissemination of Pseudomonas aeruginosa during pneumonia. PLoS Pathog. (2015) 11:e1004945. doi: 10.1371/journal.ppat.1004945
39. Dacheux D, Attree I, Schneider C, Toussaint B. Cell death of human polymorphonuclear neutrophils induced by a Pseudomonas aeruginosa cystic fibrosis isolate requires a functional type III secretion system. Infect Immun. (1999) 67:6164–7.
40. Dacheux D, Toussaint B, Richard M, Brochier G, Croize J, Attree I. Pseudomonas aeruginosa cystic fibrosis isolates induce rapid, type III secretion-dependent, but ExoU-independent, oncosis of macrophages and polymorphonuclear neutrophils. Infect Immun. (2000) 68:2916–24. doi: 10.1128/IAI.68.5.2916-2924.2000
41. Garrity-Ryan L, Kazmierczak B, Kowal R, Comolli J, Hauser A, Engel JN. The arginine finger domain of ExoT contributes to actin cytoskeleton disruption and inhibition of internalization of Pseudomonas aeruginosa by epithelial cells and macrophages. Infect Immun. (2000) 68:7100–13. doi: 10.1128/IAI.68.12.7100-7113.2000
42. Zhang Y, Deng Q, Barbieri JT. Intracellular localization of type III-delivered Pseudomonas ExoS with endosome vesicles. J Biol Chem. (2007) 282:13022–32. doi: 10.1074/jbc.M606305200
43. Mustafi S, Rivero N, Olson JC, Stahl PD, Barbieri MA. Regulation of Rab5 function during phagocytosis of live Pseudomonas aeruginosa in macrophages. Infect Immun. (2013) 81:2426–36. doi: 10.1128/IAI.00387-13
44. Miao EA, Mao DP, Yudkovsky N, Bonneau R, Lorang CG, Warren SE, et al. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc Natl Acad Sci USA. (2010) 107:3076–80. doi: 10.1073/pnas.0913087107
45. Sutterwala FS, Mijares LA, Li L, Ogura Y, Kazmierczak BI, Flavell RA. Immune recognition of Pseudomonas aeruginosa mediated by the IPAF/NLRC4 inflammasome. J Exp Med. (2007) 204:3235–45. doi: 10.1084/jem.20071239
46. Yang J, Zhao Y, Shi J, Shao F. Human NAIP and mouse NAIP1 recognize bacterial type III secretion needle protein for inflammasome activation. Proc Natl Acad Sci USA. (2013) 110:14408–13. doi: 10.1073/pnas.1306376110
47. Franchi L, Stoolman J, Kanneganti T-D, Verma A, Ramphal R, Núñez G. Critical role for Ipaf in Pseudomonas aeruginosa-induced caspase-1 activation. Eur J Immunol. (2007) 37:3030–9. doi: 10.1002/eji.200737532
48. Iannitti RG, Napolioni V, Oikonomou V, De Luca A, Galosi C, Pariano M, et al. IL-1 receptor antagonist ameliorates inflammasome-dependent inflammation in murine and human cystic fibrosis. Nat Commun. (2016) 7:10791. doi: 10.1038/ncomms10791
49. Cohen TS, Prince AS. Activation of inflammasome signaling mediates pathology of acute P. aeruginosa pneumonia. J Clin Invest. (2013) 123:1630–7. doi: 10.1172/JCI66142
50. Faure E, Mear J-B, Faure K, Normand S, Couturier-Maillard A, Grandjean T, et al. Pseudomonas aeruginosa type-3 secretion system dampens host defense by exploiting the NLRC4-coupled inflammasome. Am J Respir Crit Care Med. (2014) 189:799–811. doi: 10.1164/rccm.201307-1358OC
51. Moss J, Ehrmantraut ME, Banwart BD, Frank DW, Barbieri JT. Sera from adult patients with cystic fibrosis contain antibodies to Pseudomonas aeruginosa type III apparatus. Infect Immun. (2001) 69:1185–8. doi: 10.1128/IAI.69.2.1185-1188.2001
52. Jain M, Ramirez D, Seshadri R, Cullina JF, Powers CA, Schulert GS, et al. Type III secretion phenotypes of Pseudomonas aeruginosa strains change during infection of individuals with cystic fibrosis. J Clin Microbiol. (2004) 42:5229–37. doi: 10.1128/JCM.42.11.5229-5237.2004
53. Jain M, Bar-Meir M, Mccolley S, Cullina J, Potter E, Powers C, et al. Evolution of Pseudomonas aeruginosa type III secretion in cystic fibrosis: a paradigm of chronic infection. Transl Res. (2008) 152:257–64. doi: 10.1016/j.trsl.2008.10.003
54. Feltman H, Schulert G, Khan S, Jain M, Peterson L, Hauser AR. Prevalence of type III secretion genes in clinical and environmental isolates of Pseudomonas aeruginosa. Microbiology (2001) 147:2659–69. doi: 10.1099/00221287-147-10-2659
55. Yang J, Zhao H-L, Ran L-Y, Li C-Y, Zhang X-Y, Su H-N, et al. Mechanistic insights into elastin degradation by pseudolysin, the major virulence factor of the opportunistic pathogen Pseudomonas aeruginosa. Nat Rev Cardiol. (2015) 5:9936. doi: 10.1038/srep09936
56. Nomura K, Obata K, Keira T, Miyata R, Hirakawa S, Takano K-I, et al. Pseudomonas aeruginosa elastase causes transient disruption of tight junctions and downregulation of PAR-2 in human nasal epithelial cells. Respir Res. (2014) 15:21. doi: 10.1186/1465-9921-15-21
57. Beaufort N, Corvazier E, Mlanaoindrou S, De Bentzmann S, Pidard D. Disruption of the endothelial barrier by proteases from the bacterial pathogen Pseudomonas aeruginosa: implication of matrilysis and receptor cleavage. PLoS ONE (2013) 8:e75708. doi: 10.1371/journal.pone.0075708
58. Golovkine G, Faudry E, Bouillot S, Voulhoux R, Attree I, Huber P. VE-cadherin cleavage by LasB protease from Pseudomonas aeruginosa facilitates type III secretion system toxicity in endothelial cells. PLoS Pathog. (2014) 10:e1003939. doi: 10.1371/journal.ppat.1003939
59. Tamura Y, Suzuki S, Sawada T. Role of elastase as a virulence factor in experimental Pseudomonas aeruginosa infection in mice. Microb Pathog. (1992) 12:237–44. doi: 10.1016/0882-4010(92)90058-V
60. Blackwood LL, Stone RM, Iglewski BH, Pennington JE. Evaluation of Pseudomonas aeruginosa exotoxin A and elastase as virulence factors in acute lung infection. Infect Immun. (1983) 39:198–201.
61. Pavlovskis OR, Wretlind B. Assessment of protease (elastase) as a Pseudomonas aeruginosa virulence factor in experimental mouse burn infection. Infect Immun. (1979) 24:181–7.
62. Horvat RT, Clabaugh M, Duval-Jobe C, Parmely MJ. Inactivation of human gamma interferon by Pseudomonas aeruginosa proteases: elastase augments the effects of alkaline protease despite the presence of alpha 2-macroglobulin. Infect Immun. (1989) 57:1668–74.
63. Parmely M, Gale A, Clabaugh M, Horvat R, Zhou WW. Proteolytic inactivation of cytokines by Pseudomonas aeruginosa. Infect Immun. (1990) 58:3009–14.
64. Leidal KG, Munson KL, Johnson MC, Denning GM. Metalloproteases from Pseudomonas aeruginosa degrade human RANTES, MCP-1, and ENA-78. J Interferon Cytokine Res. (2003) 23:307–18. doi: 10.1089/107999003766628151
65. Matheson NR, Potempa J, Travis J. Interaction of a novel form of Pseudomonas aeruginosa alkaline protease (aeruginolysin) with interleukin-6 and interleukin-8. Biol Chem. (2006) 387:911–5. doi: 10.1515/BC.2006.115
66. Saint-Criq V, Villeret B, Bastaert F, Kheir S, Hatton A, Cazes A, et al. Pseudomonas aeruginosa LasB protease impairs innate immunity in mice and humans by targeting a lung epithelial cystic fibrosis transmembrane regulator-IL-6-antimicrobial-repair pathway. Thorax (2018) 73:49–61. doi: 10.1136/thoraxjnl-2017-210298
67. Heck LW, Alarcon PG, Kulhavy RM, Morihara K, Russell MW, Mestecky JF. Degradation of IgA proteins by Pseudomonas aeruginosa elastase. J Immunol. (1990) 144:2253–7.
68. Hong YQ, Ghebrehiwet B. Effect of Pseudomonas aeruginosa elastase and alkaline protease on serum complement and isolated components C1q and C3. Clin Immunol Immunopathol. (1992) 62:133–8. doi: 10.1016/0090-1229(92)90065-V
69. Schmidtchen A, Frick I-M, Andersson E, Tapper H, Björck L. Proteinases of common pathogenic bacteria degrade and inactivate the antibacterial peptide LL-37. Mol Microbiol. (2002) 46:157–68. doi: 10.1046/j.1365-2958.2002.03146.x
70. Dulon S, Leduc D, Cottrell GS, D'Alayer J, Hansen KK, Bunnett NW, et al. Pseudomonas aeruginosa elastase disables proteinase-activated receptor 2 in respiratory epithelial cells. Am. J. Respir. Cell Mol. Biol. (2005) 32:411–9. doi: 10.1165/rcmb.2004-0274OC
71. Kida Y, Higashimoto Y, Inoue H, Shimizu T, Kuwano K. A novel secreted protease from Pseudomonas aeruginosa activates NF-κB through protease-activated receptors. Cell Microbiol. (2008) 10:1491–504. doi: 10.1111/j.1462-5822.2008.01142.x
72. Bastaert F, Kheir S, Saint-Criq V, Villeret B, Dang PM, El-Benna J, et al. Pseudomonas aeruginosa LasB subverts alveolar macrophage activity by interfering with bacterial killing through downregulation of innate immune defense, reactive oxygen species generation, and complement activation. Front Immunol. (2018) 9:1675. doi: 10.3389/fimmu.2018.01675
73. Mun JJ, Tam C, Kowbel D, Hawgood S, Barnett MJ, Evans DJ, et al. Clearance of Pseudomonas aeruginosa1 from a healthy ocular surface involves surfactant protein D and is compromised by bacterial elastase in a murine null-infection model. Infect Immun. (2009) 77:2392–8. doi: 10.1128/IAI.00173-09
74. Kuang Z, Hao Y, Hwang S, Zhang S, Kim E, Akinbi HT, et al. The Pseudomonas aeruginosa flagellum confers resistance to pulmonary surfactant protein-A by impacting the production of exoproteases through quorum-sensing. Mol Microbiol. (2011) 79:1220–35. doi: 10.1111/j.1365-2958.2010.07516.x
75. van der Plas MJ, Bhongir RK, Kjellström S, Siller H, Kasetty G, Mörgelin M, et al. (2016). Pseudomonas aeruginosa elastase cleaves a C-terminal peptide from human thrombin that inhibits host inflammatory responses. Nat. Commun. 7:11567. doi: 10.1038/ncomms11567
76. Yang J, Lee K-M, Park S, Cho Y, Lee E, Park J-H, et al. Bacterial Secretant from Pseudomonas aeruginosa dampens inflammasome activation in a quorum sensing-dependent manner. Front Immunol. (2017) 8:333. doi: 10.3389/fimmu.2017.00333
77. Le Berre R, Nguyen S, Nowak E, Kipnis E, Pierre M, Ader F, et al. Quorum-sensing activity and related virulence factor expression in clinically pathogenic isolates of Pseudomonas aeruginosa. Clin Microbiol Infect. (2008) 14:337–43. doi: 10.1111/j.1469-0691.2007.01925.x
78. Martinez-Solano L, Macia MD, Fajardo A, Oliver A, Martinez JL. Chronic Pseudomonas aeruginosa infection in chronic obstructive pulmonary disease. Clin Infect Dis. (2008) 47:1526–33. doi: 10.1086/593186
79. Mayer-Hamblett N, Rosenfeld M, Gibson RL, Ramsey BW, Kulasekara HD, Retsch-Bogart GZ, et al. Pseudomonas aeruginosa in vitro phenotypes distinguish cystic fibrosis infection stages and outcomes. Am J Respir Crit Care Med. (2014) 190:289–97. doi: 10.1164/rccm.201404-0681OC
80. Lafayette SL, Houle D, Beaudoin T, Wojewodka G, Radzioch D, Hoffman LR, et al. Cystic fibrosis–adapted Pseudomonas aeruginosa quorum sensing lasR mutants cause hyperinflammatory responses. Sci Adv. (2015) 1:e1500199. doi: 10.1126/sciadv.1500199
81. Mann EE, Wozniak DJ. Pseudomonasbiofilm matrix composition and niche biology. FEMS Microbiol Rev. (2012) 36:893–916. doi: 10.1111/j.1574-6976.2011.00322.x
82. Pier GB, Coleman F, Grout M, Franklin M, Ohman DE. Role of alginate O acetylation in resistance of mucoid Pseudomonas aeruginosa to opsonic phagocytosis. Infect Immun. (2001) 69:1895–901. doi: 10.1128/IAI.69.3.1895-1901.2001
83. Lyczak JB, Cannon CL, Pier GB. Lung infections associated with cystic fibrosis. Clin Microbiol Rev. (2002) 15:194–222. doi: 10.1128/CMR.15.2.194-222.2002
84. Learn DB, Brestel EP, Seetharama S. Hypochlorite scavenging by Pseudomonas aeruginosa alginate. Infect Immun. (1987) 55:1813–8.
85. Leid JG, Willson CJ, Shirtliff ME, Hassett DJ, Parsek MR, Jeffers AK. The exopolysaccharide alginate protects Pseudomonas aeruginosa biofilm bacteria from IFN- -mediated macrophage killing. J Immunol (2005) 175:7512–8. doi: 10.4049/jimmunol.175.11.7512
86. Malhotra S, Limoli DH, English AE, Parsek MR, Wozniak DJ. Mixed communities of mucoid and nonmucoid Pseudomonas aeruginosa exhibit enhanced resistance to host antimicrobials. MBio (2018) 9:e00275–18. doi: 10.1128/mBio.00275-18
87. Firoved AM, Ornatowski W, Deretic V. Microarray analysis reveals induction of lipoprotein genes in mucoid Pseudomonas aeruginosa: implications for inflammation in cystic fibrosis. Infect Immun. (2004) 72:5012–8. doi: 10.1128/IAI.72.9.5012-5018.2004
88. Beaudoin T, Lafayette S, Nguyen D, Rousseau S. Mucoid Pseudomonas aeruginosa caused by mucA mutations result in activation of TLR2 in addition to TLR5 in airway epithelial cells. Biochem Biophys Res Commun. (2012) 428:150–4. doi: 10.1016/j.bbrc.2012.10.030
89. Mizutani M, Bérubé J, Ahlgren HG, Bernier J, Matouk E, Nguyen D, et al. Corticosteroid-resistant inflammatory signalling in Pseudomonas-infected bronchial cells. ERJ Open Res. (2017) 3:1–7. doi: 10.1183/23120541.00144-2016
90. Colvin KM, Irie Y, Tart CS, Urbano R, Whitney JC, Ryder C, et al. The Pel and Psl polysaccharides provide Pseudomonas aeruginosa structural redundancy within the biofilm matrix. Environ Microbiol. (2011) 14:1913–28. doi: 10.1111/j.1462-2920.2011.02657.x
91. Tseng BS, Reichhardt C, Merrihew GE, Araujo-Hernandez SA, Harrison JJ, Maccoss MJ, et al. A biofilm matrix-associated protease inhibitor protects Pseudomonas aeruginosa from proteolytic attack. mBio (2018) 9:e00543–18. doi: 10.1128/mBio.00543-18
92. Mishra BB, Rathinam VAK, Martens GW, Martinot AJ, Kornfeld H, Fitzgerald KA, et al. and Sassetti CM. Nitric oxide controls the immunopathology of tuberculosis by inhibiting NLRP3 inflammasome-dependent processing of IL-1β. Nat Immunol. (2012) 14:52–60. doi: 10.1038/ni.2474
93. Baker P, Hill PJ, Snarr BD, Alnabelseya N, Pestrak MJ, Lee MJ, et al. Exopolysaccharide biosynthetic glycoside hydrolases can be utilized to disrupt and prevent Pseudomonas aeruginosa biofilms. Sci Adv. (2016) 2:e1501632. doi: 10.1126/sciadv.1501632
94. Starkey M, Hickman JH, Ma L, Zhang N, De Long S, Hinz A, et al. Pseudomonas aeruginosa rugose small-colony variants have adaptations that likely promote persistence in the cystic fibrosis lung. J Bacteriol. (2009) 191:3492–503. doi: 10.1128/JB.00119-09
95. Pestrak MJ, Chaney SB, Eggleston HC, Dellos-Nolan S, Dixit S, Mathew-Steiner SS, et al. Pseudomonas aeruginosa rugose small-colony variants evade host clearance, are hyper-inflammatory, and persist in multiple host environments. PLoS Pathog. (2018) 14:e1006842. doi: 10.1371/journal.ppat.1006842
96. Pier GB. Pseudomonas aeruginosa lipopolysaccharide: a major virulence factor, initiator of inflammation and target for effective immunity. Int J Med Microbiol. (2007) 297:277–95. doi: 10.1016/j.ijmm.2007.03.012
97. Ernst RK, Adams KN, Moskowitz SM, Kraig GM, Kawasaki K, Stead CM, et al. The Pseudomonas aeruginosa lipid a deacylase: selection for expression and loss within the cystic fibrosis airway. J Bacteriol. (2005) 188:191–201. doi: 10.1128/JB.188.1.191-201.2006
98. Moskowitz SM, Ernst RK. The Role of Pseudomonas Lipopolysaccharide in Cystic Fibrosis Airway Infection. Dordrecht: Springer Netherlands (2010).
99. Needham BD, Trent MS. Fortifying the barrier: the impact of lipid A remodelling on bacterial pathogenesis. Nat Rev Microbiol. (2013) 11:467–81. doi: 10.1038/nrmicro3047
100. Di Lorenzo F, Silipo A, Bianconi I, Lorè NI, Scamporrino A, Sturiale L, et al. Persistent cystic fibrosis isolate Pseudomonas aeruginosa strain RP73 exhibits an under-acylated LPS structure responsible of its low inflammatory activity. Mol Immunol. (2014) 63:166–75. doi: 10.1016/j.molimm.2014.04.004
101. Hancock RE, Mutharia LM, Chan L, Darveau RP, Speert DP, Pier GB. Pseudomonas aeruginosa isolates from patients with cystic fibrosis: a class of serum-sensitive, nontypable strains deficient in lipopolysaccharide O side chains. Infect Immun. (1983) 42:170–7.
102. King JD, Kocíncová D, Westman EL, Lam JS. Review: lipopolysaccharide biosynthesis in Pseudomonas aeruginosa. Innate Immun. (2009) 15:261–312. doi: 10.1177/1753425909106436
103. Cigana C, Curcurù L, Leone MR, Ieranò T, Lorè NI, Bianconi I, et al. Pseudomonas aeruginosa exploits lipid a and muropeptides modification as a strategy to lower innate immunity during cystic fibrosis lung infection. PLoS ONE (2009) 4:e8439. doi: 10.1371/journal.pone.0008439
104. Cramer N, Klockgether J, Wrasman K, Schmidt M, Davenport CF, Tümmler B. Microevolution of the major common Pseudomonas aeruginosa clones C and PA14 in cystic fibrosis lungs. Environ Microbiol. (2011) 13:1690–704. doi: 10.1111/j.1462-2920.2011.02483.x
105. Dettman JR, Rodrigue N, Aaron SD, Kassen R. Evolutionary genomics of epidemic and nonepidemic strains of Pseudomonas aeruginosa. Proc Natl Acad Sci USA. (2013) 110:21065–70. doi: 10.1073/pnas.1307862110
106. McCarthy RR, Mazon-Moya MJ, Moscoso JA, Hao Y, Lam JS, Bordi C, et al. Cyclic-di-GMP regulates lipopolysaccharide modification and contributes to Pseudomonas aeruginosa immune evasion. Nat Microbiol. (2017) 2:17027. doi: 10.1038/nmicrobiol.2017.27
107. Boisvert A-A, Cheng MP, Sheppard DC, Nguyen D. Microbial biofilms in pulmonary and critical care diseases. Ann Am Thorac Soc. (2016) 13:1615–23. doi: 10.1513/AnnalsATS.201603-194FR
108. Whiteley M, Bangera MG, Bumgarner RE, Parsek MR, Teitzel GM, Lory S, et al. Gene expression in Pseudomonas aeruginosa biofilms. Nature (2001) 413:860–4. doi: 10.1038/35101627
109. Sauer K, Camper AK, Ehrlich GD, Costerton JW, Davies DG. Pseudomonas aeruginosa displays multiple phenotypes during development as a biofilm. J Bacteriol. (2002) 184:1140–54. doi: 10.1128/jb.184.4.1140-1154.2002
110. Jensen ET, Kharazmi A, Garred P, Kronborg G, Fomsgaard A, Mollnes TE, et al. Complement activation by Pseudomonas aeruginosa biofilms. Microb Pathog. (1993) 15:377–88. doi: 10.1006/mpat.1993.1087
111. Jensen ET, Kharazmi A, Lam K, Costerton JW, Hoiby N. Human polymorphonuclear leukocyte response to Pseudomonas aeruginosa grown in biofilms. Infect Immun. (1990) 58:2383–5.
112. Jesaitis AJ, Franklin MJ, Berglund D, Sasaki M, Lord CI, Bleazard JB, et al. Compromised host defense on Pseudomonas aeruginosa biofilms: characterization of neutrophil and biofilm interactions. J Immunol. (2003) 171:4329–39. doi: 10.4049/jimmunol.171.8.4329
113. Jensen PO, Bjarnsholt T, Phipps R, Rasmussen TB, Calum H, Christoffersen L, et al. Rapid necrotic killing of polymorphonuclear leukocytes is caused by quorum-sensing-controlled production of rhamnolipid by Pseudomonas aeruginosa. Microbiology (2007) 153:1329–38. doi: 10.1099/mic.0.2006/003863-0
114. Moscoso JA, Mikkelsen H, Heeb S, Williams P, Filloux A. The Pseudomonas aeruginosa sensor RetS switches Type III and Type VI secretion via c-di-GMP signalling. Environ Microbiol. (2011) 13:3128–38. doi: 10.1111/j.1462-2920.2011.02595.x
115. Valentini M, Filloux A. Biofilms and Cyclic di-GMP (c-di-GMP) signaling: lessons from Pseudomonas aeruginosa and other bacteria. J Biol Chem. (2016) 291:12547–55. doi: 10.1074/jbc.R115.711507
116. Sommer LM, Molin SOR, Johansen HK, Marvig RL. Convergent evolution and adaptation of Pseudomonas aeruginosa within patients with cystic fibrosis Nat Genet. (2014) 47:57–64. doi: 10.1038/ng.3148
117. Mikkelsen H, McMullan R, Filloux A. The Pseudomonas aeruginosa reference strain PA14 displays increased virulence due to a mutation in ladS. PLoS ONE (2011) 6:e29113. doi: 10.1371/journal.pone.0029113
118. Sall KM, Casabona MG, Bordi C, Huber P, De Bentzmann S, Attree I, et al. A gacS deletion in Pseudomonas aeruginosa cystic fibrosis isolate CHA shapes its virulence. PLoS ONE (2014) 9:e95936. doi: 10.1371/journal.pone.0095936
119. Folkesson A, Jelsbak L, Yang L, Johansen HK, Ciofu O, Høiby N, et al. Adaptation of Pseudomonas aeruginosa to the cystic fibrosis airway: an evolutionary perspective. Nat Rev Microbiol. (2012) 10:841–51. doi: 10.1038/nrmicro2907
120. Oliver A, Canton R, Campo P, Baquero F, Blazquez J. High frequency of hypermutable Pseudomonas aeruginosa in cystic fibrosis lung infection. Science (2000) 288:1251–4. doi: 10.1126/science.288.5469.1251
121. Jorth P, Staudinger BJ, Wu X, Hisert KB, Hayden H, Garudathri J, et al. Regional isolation drives bacterial diversification within cystic fibrosis lungs. Cell Host Microbe (2015) 18:307–19. doi: 10.1016/j.chom.2015.07.006
122. Bragonzi A, Paroni M, Nonis A, Cramer N, Montanari S, Rejman J, et al. Pseudomonas aeruginosa microevolution during cystic fibrosis lung infection establishes clones with adapted virulence. Am J Respir Crit Care Med. (2009) 180:138–45. doi: 10.1164/rccm.200812-1943OC
123. Nguyen D, Singh PK. Evolving stealth: genetic adaptation of Pseudomonas aeruginosa during cystic fibrosis infections. Proc Natl Acad Sci USA. (2006) 103:8305–6. doi: 10.1073/pnas.0602526103
124. Czaplewski L, Bax R, Clokie M, Dawson M, Fairhead H, Fischetti VA, et al. Alternatives to antibiotics-a pipeline portfolio review. Lancet Infect Dis. (2016) 16:239–51. doi: 10.1016/S1473-3099(15)00466-1
125. Hauser AR, Mecsas J, Moir DT. Beyond antibiotics: new therapeutic approaches for bacterial infections. Clin Infect Dis. (2016) 63:89–95. doi: 10.1093/cid/ciw200
126. Maura D, Ballok AE, Rahme LG. Considerations and caveats in anti-virulence drug development. Curr Opin Microbiol. (2016) 33:41–6. doi: 10.1016/j.mib.2016.06.001
127. Digiandomenico A, Sellman BR. Antibacterial monoclonal antibodies: the next generation? Curr Opin Microbiol. (2015) 27:78–85. doi: 10.1016/j.mib.2015.07.014
128. Nilsson E, Larsson A, Olesen HV, Wejaker PE, Kollberg H. Good effect of IgY against Pseudomonas aeruginosa infections in cystic fibrosis patients. Pediatr Pulmonol. (2008) 43:892–9. doi: 10.1002/ppul.20875
129. Digiandomenico A, Warrener P, Hamilton M, Guillard S, Ravn P, Minter R, et al. Identification of broadly protective human antibodies to Pseudomonas aeruginosa exopolysaccharide Psl by phenotypic screening. J Exp Med. (2012) 209:1273–87. doi: 10.1084/jem.20120033
130. Digiandomenico A, Keller AE, Gao C, Rainey GJ, Warrener P, Camara MM, et al. A multifunctional bispecific antibody protects against Pseudomonas aeruginosa. Sci Transl Med. (2014) 6:262ra155. doi: 10.1126/scitranslmed.3009655
131. François B, Luyt C-E, Dugard A, Wolff M, Diehl J-L, Jaber S, et al. Safety and pharmacokinetics of an anti-PcrV PEGylated monoclonal antibody fragment in mechanically ventilated patients colonized with Pseudomonas aeruginosa: a randomized,double-blind, placebo-controlled trial. Crit Care Med. (2012) 40:2320–6. doi: 10.1097/CCM.0b013e31825334f6
132. Secher T, Fauconnier L, Szade A, Rutschi O, Fas SC, Ryffel B, et al. Anti-Pseudomonas aeruginosa serotype O11 LPS immunoglobulin M monoclonal antibody panobacumab (KBPA101) confers protection in a murine model of acute lung infection. J Antimicrob Chemother. (2011) 66:1100–9. doi: 10.1093/jac/dkr038
133. Milla CE, Chmiel JF, Accurso FJ, Vandevanter DR, Konstan MW, Yarranton G, et al. Anti-PcrV antibody in cystic fibrosis: a novel approach targeting Pseudomonas aeruginosa airway infection. Pediatr Pulmonol. (2014) 49:650–8. doi: 10.1002/ppul.22890
134. Grimwood K, Kyd JM, Owen SJ, Massa HM, Cripps AW. Vaccination against respiratory Pseudomonas aeruginosa infection. Hum Vaccin Immunother. (2015) 11:14–20. doi: 10.4161/hv.34296
Keywords: Pseudomonas aeruginosa, cystic fibrosis, immune evasion, chronic lung infection, host evasion, bacterial adaptation
Citation: Faure E, Kwong K and Nguyen D (2018) Pseudomonas aeruginosa in Chronic Lung Infections: How to Adapt Within the Host? Front. Immunol. 9:2416. doi: 10.3389/fimmu.2018.02416
Received: 01 June 2018; Accepted: 01 October 2018;
Published: 22 October 2018.
Edited by:
Maziar Divangahi, McGill University, CanadaReviewed by:
Loic Guillot, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceAndré Cantin, Université de Sherbrooke, Canada
Copyright © 2018 Faure, Kwong and Nguyen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dao Nguyen, ZGFvLm5ndXllbkBtY2dpbGwuY2E=